
Abstract
Background: Medulloblastoma (MB) and supratentorial primitive neuroectodermal tumor of the central nervous system (CNS-PNET) are among the most common pediatric brain tumors. The diagnosis, treatment, and outcome of MB/CNS-PNET patients treated during the last four decades at Oslo University Hospital (OUH) are described.
Material and methods: All patients younger than 20 years of age diagnosed and treated for MB/CNS-PNET at OUH between 1 January 1974 and 31 December 2013 were identified.
Results: We found 175 patients. In 13 of them, the diagnosis was changed upon histopathological review and in 4 patients part of the treatment was administered at other hospitals. Thus, 158 patients were included for further analysis. Eight patients did not receive adjuvant therapy because of a dismal clinical condition. The overall 5-year survival rate for MB and CNS-PNET was 54%, for MB 57%, and for CNS-PNET 41%. Gross total resection (GTR) was achieved in 118 patients and 5-year overall survival for patients with GTR versus those with non-GTR differed significantly with 64% versus 22%. Cytological examination of the cerebrospinal fluid was performed in 52 patients. A total of 126 patients received radiotherapy as part of the primary treatment and 24 did not due to young age. Median time from surgery to start of radiotherapy was 33 days. Duration of radiotherapy was more than 48 days in 22% of patients. At the time of analysis, 63 patients were alive and disease-free, one alive with disease, and 94 patients were deceased; 84 of these due to MB/CNS-PNET and 10 due to supposed late effects from the treatment.
Conclusions: Survival was comparable to data from other population-based studies. The importance of GTR for survival was corroborated. Reporting real-world data remains crucial to know the true outcome of patients treated outside clinical trials.
Introduction
Medulloblastoma (MB) and supratentorial primitive neuroectodermal tumor (CNS-PNET) are two of the most common malignant brain tumors occurring in childhood [Citation1]. These closely related neoplastic entities make up 26% of all primary brain tumors in children and they are both considered curable disease [Citation2]. 25–35% of children with MB are younger than 3 years of age when diagnosed and the median age at diagnosis is 6 years [Citation3,Citation4]. Knowledge of molecular mechanisms in MB/CNS-PNET is rapidly increasing and molecular parameters are now clinically used for diagnostic purposes, as important prognostic factors and are also promising guide to optimal, individualized based treatment [Citation5–12]. This development is reflected in the updated 2016 version of the 2007 4th edition of the WHO classification of tumors of the central nervous system (CNS). In this revised classification, the use of molecular parameters in addition to histology is of paramount importance to define different neoplastic entities, including MB and CNS-PNET [Citation12].
Standard risk MB patients, in contrast to high risk patients, have traditionally been regarded to include those older than 3 years of age, without metastatic disease, and with a near gross total resection with less than 1.5 cm2 of residual tumor postoperatively [Citation9,Citation12,Citation13]. Both MB and CNS-PNET have a marked propensity to metastasize via the cerebrospinal fluid (CSF) and evidence of metastatic spread is present in up to 35% of cases at the time of primary diagnosis [Citation3]. Treatment of MB and CNS-PNET is multimodal and current therapy includes surgery aiming at gross total resection (GTR), craniospinal radiation with a boost to the tumor bed and chemotherapy. Several diagnostic and therapeutic improvements have been made since the introduction of postoperative radiotherapy some 50 years ago [Citation14–16]. Also, after realizing the detrimental late effects of radiotherapy, this therapeutic modality has been omitted in the youngest children although such a strategy reduces chances of cure [Citation17,Citation18]. Five-year overall survival (OS) for standard risk MB patients included in clinical trials is reported to be ∼80% [Citation19,Citation20]. Outcome for high risk MB and CNS-PNET patients is worse despite intensified adjuvant postoperative therapy. Historically, five-year OS for the first patient group has been around 40% [Citation21]. However, more recent data show a 5-year OS of 55–76% for high risk MB patients, depending on the definition of high risk MB [Citation20–22]. For CNS-PNET patients, 5-year OS has recently been reported to be as high as 60% [Citation23,Citation24].
Epidemiological data have suggested that survival for Norwegian MB/CNS-PNET patients has been poorer than expected compared to international data [Citation25]. The primary objective of this retrospective observational study was to determine the outcome for children with MB/CNS-PNET treated at Oslo University Hospital (OUH) and to identify possible changes in outcome during the study period.
Material and methods
Patients
The study was designed as a retrospective observational study. Inclusion criteria were patient age younger than 20 years at primary diagnosis, a histopathologically confirmed MB or CNS-PNET, all treatment administered by or at least governed by OUH, and primary diagnosis between January 1974 and December 2013. Patients were identified using the archives of Department of Pathology, surgical protocols at Department of Neurosurgery, and data from the Cancer Registry of Norway. The year 1974 was chosen as starting point because the first international trial on the use of adjuvant chemotherapy in children with medulloblastoma (International Society of Paediatric Oncology [SIOP] I) was opened that year, wherein Norwegian patients were included. A systematic review of medical records was performed with registration of detailed clinical data. 1 October 2016 was defined as the date of last follow-up for surviving patients.
Histopathological review
Biopsy specimens from all patients were reviewed and classified according to the World Health Organization (WHO) 2007 classification of brain tumors by author BKK. In some of the oldest cases, the diagnosis was based solely on morphological characteristics in hematoxylin and eosin stained as well as reticulin stained sections. When necessary for diagnosis, supplementary immunohistochemical staining was performed with antibodies against glial fibrillary acidic protein (GFAP), synaptophysin, NeuN, chromogranin A, neurofilament protein, INI-1, Ki-67, actin, desmin, smooth muscle actin and epithelial membrane antigen (EMA). Patients where the revised histopathological diagnosis was different from MB or CNS-PNET were excluded from further analysis.
Radiological review
Author PDT reviewed radiological imaging and reports. Diagnostically, cerebral angiography and air ventriculography were regularly used until 1979, at which time point computed tomography (CT) came into use at OUH. CT was gradually replaced by magnetic resonance imaging (MRI), with the first cerebral MRI performed in 1988 and the first craniospinal MRI in March 1993. Until 2003, all radiological imaging performed at OUH was routinely condemned 10 years after the imaging procedure. For many of the patients included in this study (i.e., those primarily treated between 1974 and 2003), detailed review of radiological imaging was therefore not possible. In these cases, radiological reports and patient records including surgical notes were used to evaluate the presence or absence of any residual tumor and/or metastatic disease. In all cases with documented recurrence, the site of recurrence was determined and radiologically reviewed as for the primary tumor.
Risk group allocation of individual patients
Patients under the age of three years (because they did not receive radiotherapy), with postoperative residual tumor exceeding 1.5 cm2, and/or documented metastatic disease at the time of primary diagnosis were considered high risk, whereas patients where the latter two characteristics could be properly evaluated and deemed not present were defined as standard risk. If available data were insufficient for determining the presence or not of residual tumor and/or metastatic disease, the patient was classified as uncertain risk. The presence or absence of residual tumor was determined based on radiological imaging when available, radiological reports, and surgical notes. Metastatic disease was defined as positive lumbar CSF cytology and/or radiological evidence of multifocal disease. MYC status, histological subgroup, beta-catenin status, and molecular subgroup were not used to define patient risk status. Authors ES and PB categorized the patients.
Statistical methods for survival analysis
Time of recurrence was defined as date of imaging procedure where recurrent tumor was confirmed, or date of first patient record note with information on recurrence. To estimate event-free survival (EFS) time from date of surgery to date of death, date of recurrence, or to date of administrative censoring (1 October 2016), whichever came first, was used. For overall survival (OS) estimates, the same procedure was used, however, the date of recurrence was not relevant. Both OS and EFS were estimated using the traditional Kaplan–Meier method, and differences in survival were tested using log-rank tests. p Values less than or equal to .05 were considered statistically significant. Survival estimates are presented in tables and as standard survival curves. OS and EFS was estimated for the total patient material, MB patients alone, CNS-PNET patients alone, patients treated according to the HIT-SIOP PNET 4 protocol (standard risk patients treated between 2005 and 2013), for different MB risk categories, for patients with and without gross total resection (GTR), and for different decades. Stata14 (StataCorp LP, 4905 Lakeway Drive, College Station, TX) was used for statistical analysis.
Ethics
Regional Committees for Medical and Health Research Ethics of the South-Eastern Norway Regional Health Authority (2013/1959/REK sør-øst D) and the Data Protection Officer at OUH were notified about the study in writing and had no objections to the study that was considered a quality assurance study of high importance.
Results
Patients
A total of 175 patients treated for MB/CNS-PNET at OUH between 1 January 1974 and 31 December 2013 were identified. Fourteen of these patients were only registered in OUH internal registries and three only in the Cancer Registry of Norway. Four of the 175 patients received either surgery or at least parts of their adjuvant treatment at other hospitals and were therefore excluded from further analysis. Twelve patients whose revised histological diagnosis was not MB or CNS-PNET (see below) were also excluded, as was a patient with a preexisting pontine glioma. Thus, 158 patients were included for further analysis (). No patients were lost to follow-up. Survival data for the whole cohort include patients who succumbed peri- or early postoperatively before adjuvant therapy was started. Seventeen patients were not treated according to any protocol; these were mostly teenagers/young adults. 73 patients (46%) underwent both pre- and postoperative MRI. Three patients had congenital genetic conditions that may have contributed to disease progression during the treatment and/or to poor tolerance to treatment; one of these had Fanconi anemia, one had Nijmegen Breakage Syndrome, and one was retrospectively suspected to have Gorlin syndrome because three out of five siblings in the patient’s family were diagnosed with MB.
Table 1. Clinical characteristics of the 158 included MB and CNS-PNET patients.
Histopathological review
In 12 of the initially identified 175 patients (7.4%), histopathological revision revealed a diagnosis different from MB or CNS-PNET. The revised diagnoses were atypical teratoid/rhabdoid tumor (AT/RT) in four cases, glioblastoma in three, anaplastic astrocytoma in two, germinoma in two, and unspecified malignant glial/glioneuronal tumor in one case. The remaining cohort of 158 patients consisted of 123 cases of MB and 35 cases of CNS-PNET ().
Surgery
All patients underwent surgery at the time of initial diagnosis. Median age at primary surgery was 7.1 years (range 2 days to 19 years). GTR was achieved in 118 patients (75%), subtotal resection (STR) in 21 (13%), biopsy only was performed in five patients (3.2%) and in 12 patients (7.6%) the grade of resection was uncertain. Eight of the 158 patients (5.1%), all in a dismal clinical condition, died without initiation of adjuvant therapy. Of these eight, there were two perioperative deaths, four other postoperative deaths (<30 days), and another two patients died after 36 and 91 days, respectively. Six of these eight children were younger than 3 years of age. The cause of death was uncontrollable perioperative bleeding in two patients, medulla oblongata infarction in two, postoperative bleeding in one, supratentorial hydrocephalus in one, a combination of meningitis, pneumonia, and liver steatosis in one, and one with disseminated MB. One of these eight deaths occurred in the 1970s, three in the 1980s, three in the 1990s and one after year 2000.
Radiotherapy
A total of 150 patients received adjuvant therapy. Of these, 126 patients (84%) received radiotherapy in the primary diagnosis setting, whereas 24 (16%) did not due to young age (below 3–5 years). All 126 but four patients received craniospinal radiotherapy. Median time from surgery to start of radiotherapy was 33 days (range 5–320 days); including those 16 patients (13%) who were treated according to the UKCCSG/SIOP CNS-PNET3 protocol with sandwich chemotherapy prior to radiotherapy. Thirty-eight patients (30%) started radiotherapy more than 40 days following surgery. Median duration of radiotherapy was 43 days (range 26–76 days). Radiotherapy duration was below 48 days for 93 patients (74%), 48 days for 5 patients (4.0%), and more than 48 days for 28 patients (22%). For the patients with a relatively long radiotherapy duration (> =48 days, n = 33), the cause for interruption of radiotherapy was not documented in the patient records; however, thrombocytopenia and/or neutropenia seemed to be the most frequent causes. Sixteen of the 28 patients with radiotherapy duration more than 48 days are deceased, 12 are alive. Also, six other patients, all with total radiotherapy duration less than 48 days, had radiotherapy interruptions. Of the ones with a documented reason for radiotherapy delay or interruption, three patients (2.4%) were delayed because of intercurrent disease and 10 patients (7.9%) were paused because of cytopenia. Details of protocol interruption are summarized in Supplementary Table 1. Fifty-six (44%) were treated in line with their designated radiation therapy protocol, whereas 70 patients (56%) were not. The main deviations were prescription of a too low dose to the brain, boost, or CNS axis, or wrong fraction dose. Forty-one of the latter 70 are deceased, 29 are alive. Protocol interruptions and deviations did not significantly affect survival. In 121 patients (96%), radiotherapy with photons was used, in two (1.6%) a combination of photons and electrons, in one (0.80%) electrons, in one (0.80%) protons and one patient (0.80%) was treated with cobalt-60. Of the 55 patients who experienced tumor recurrence, 14 patients (25%) received radiotherapy at the time of recurrence, whereas 41 patients (75%) did not.
Chemotherapy
Forty-eight patients (32%) did not have chemotherapy according to protocol guidelines (Supplementary Table 1). For most of these patients, records did not clearly state why, but neutropenia and/or thrombocytopenia seemed to be the main reasons for interruption of chemotherapy.
Risk group allocation
We considered all CNS-PNET patients as high risk and the risk group allocation pertains to the MB patients. Risk group allocation was seriously hampered because of two factors. First, the institutional policy of condemnation of radiological imaging 10 years from the imaging procedure made it impossible to retrospectively review imaging for the majority of patients. Nonetheless, diagnostic imaging procedures were considered optimal in 92 patients (58%) and suboptimal in 66 patients (42%). Second, CSF cytology as part of disease staging was performed in only 52 cases (33%), not in 100 patients (63%), and for six patients (4.0%) it seems to have been performed, but the result is not documented. From 2002 to 2005, CSF sampling for cytological analysis was done in a growing percentage of patients, but only from 2005 CSF cytology was routinely performed in all MB/CNS-PNET patients. In total, of the 123 MB patients, 31 patients (25%) were defined as standard risk, 37 (30%) as high risk and 55 (45%) as uncertain risk.
Outcome
Survival estimates are shown in Supplementary Table 2. At the time of analysis, 64 of 158 patients (41%) were alive (). For MB and CNS-PNET seen together 5-year OS was 54% (). Five-year OS for MB was significantly better (57%) than for CNS-PNET (41%, p = .027, , Supplementary Table 3), as was 5-year EFS (p = .007, Supplementary Table 3). Five-year EFS for MB was 52%, for CNS-PNET 32%, and for MB and CNS – PNET seen together 48% (Supplementary Table 2). Notably, several late recurrences (more than 5 years from diagnosis) were seen. For standard risk patients treated according to the HIT-SIOP PNET 4 protocol, five-year OS and EFS were both 77% (Supplementary Table 2). Five-year OS for patients with GTR versus those with non-GTR differed significantly with 64% versus 22% (p < .001, , Supplementary Table 3). Survival analyses did not show significant changes between different MB risk groups or between decades (Supplementary Figure 1, Supplementary Figure 2 and Supplementary Table 3). Twenty-four patients of those 79 who had tumor recurrence experienced progression during adjuvant treatment, whereas 55 patients were diagnosed with recurrence after adjuvant treatment. Amongst the latter 55 patients, most had a local recurrence or a combined local and distant recurrence (). Three of the 55 patients with recurrent disease were alive at the time of last follow-up; two of them without evidence of disease. Eleven patients developed second primary tumors; three papillary thyroid carcinoma, three multiple basal cell carcinomas, two cerebellar glioblastoma, one acute myeloid leukemia, one acute lymphoblastic leukemia, and in one patient a squamous cell carcinoma at the left ear and a rectal adenocarcinoma was diagnosed. At the time of analysis, three patients had died from their second primary tumor and a total of seven patients died from another cause than MB/CNS-PNET and without evidence of recurrent MB/CNS-PNET (). Also, six patients are known to have been diagnosed with meningioma, but the completeness of meningioma registration is not optimal as MRI has not recently been performed in all patients. Ten patients who did not receive radiotherapy as part of the primary treatment are still alive. Those 10 patients were treated according to Baby Brain-UKCCSG Study CNS 9204 (n = 2) or HIT 2000 (n = 8). Three of them recurred and received radiotherapy at the time of recurrence. Radiotherapy variables did not affect outcome significantly. For the 33 patients with radiotherapy duration of 48 days or longer, 19 patients were dead and 14 alive at the time of last follow-up. Three of the four patients receiving only local radiotherapy are deceased, the fourth was treated two years ago and is alive with no evidence of disease.
Figure 1. OS and EFS for all 158 MB and CNS-PNET patients.
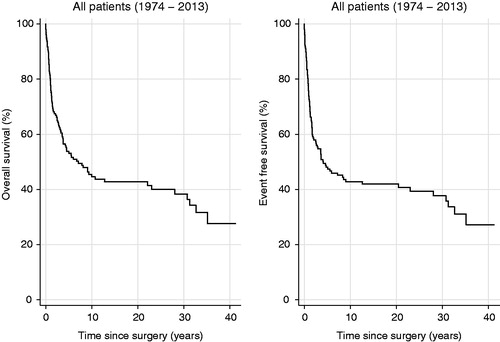
Figure 2. OS and EFS for MB (N = 123) and CNS-PNET (N = 35) patients divided by entity.
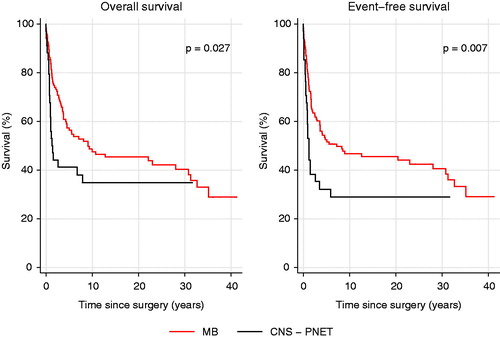
Figure 3. OS and EFS for MB and CNS-PNET patients by resection grade, gross total resection (GTR, N = 118) and non-GTR (N = 38).
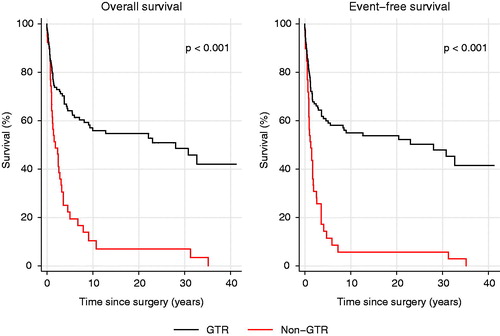
Table 2. Outcome.
Table 3. Patients deceased of other causes than recurrence of MB/CNS-PNET.
Discussion
The herein presented MB/CNS-PNET patient cohort is heterogeneous with respect to many parameters including tumor location, age at primary diagnosis, treatment protocol and strategy for adjuvant treatment. Furthermore, this is a retrospective review covering four decades which of course is a challenge in terms of data collection and analysis. On the other hand, we believe that this non-selected complete and consecutive series has a large value as it includes all children diagnosed with and treated for MB/CNS-PNET at OUH from 1974 to 2013. We have used three independent sources to identify patients rendering us confident that we have a non-selected and consecutive series of MB/CNS-PNET patients. Survival figures and outcome data are thereby more realistic than similar data from patient studies with strict inclusion criteria. Importantly, no patient was lost to follow-up.
A weakness of the study is the occasionally suboptimal quality of patient records, particularly from the earlier years of the study period. This pertains to clinical patient record data as well as to radiological imaging and made the retrospective review difficult, and it is exemplified by our risk category distribution. In most unselected series from the literature, ∼70% of MB patients would be classified as standard risk, whereas in the present series only 25% of the MB patients were standard risk. The category called MB uncertain risk in this material was, however, quite large and with a relatively good outcome. We therefore suspect that most of these cases were in fact standard risk patients, which would give a relative distribution between standard and high risk patients fitting well with literature data. Also, given this large MB uncertain risk category, the most important survival data are the ones for the whole patient cohort. It should be noticed though, that the patients who were categorized as MB uncertain risk did worse with time, and it might be that some of these were high risk patients who were undertreated (Supplementary Table 2). Another challenge with the review of the present material is that the radiological imaging for a large part was condemned and that image descriptions were suboptimal. An evaluation on the presence/absence of residual tumor was not found routinely in the radiological reports before the early 2000s. Furthermore, in only 75 patients both preoperative and postoperative MRI was performed.
The 5-year overall survival of 57% for MB in this material is inferior to a Canadian study that showed a 5-year OS for MB patients of 69%, however, the Canadian study did not include patients from 1974 to 1990 [Citation26]. From another Norwegian study looking at survival for MB from 1960 to 1997, 5-year OS was 53% for those treated after 1974 [Citation27]. In a North American study based on US Surveillance Epidemiology and End Results Program (SEER) data, 5-year survival of MB/CNS-PNET increased from 44% in 1974–1978 to 63% in 1999–2003, which is in line with our data. Outcome in the US, Canadian, and present study is, not surprisingly, inferior to results from clinical studies like the HIT 91 protocol [Citation28] and the HIT-SIOP PNET 4 trial [Citation17]. Patients in this material treated according to the HIT- SIOP PNET 4 protocol had, as expected, a relatively good outcome (5-year EFS and OS 77%, Supplementary Table 2). Solheim et al. [Citation25] described significant survival differences between Norwegian hospitals for MB/CNS-PNET patients treated between 1988 and 2008, in favor of patients treated at smaller university hospitals. We did not find significant survival differences between the decades and the 5-year OS in the total material is better than reported by Solheim et al. This is explained by the fact that herein, as opposed to the aforementioned study, a thorough review of histopathological diagnosis, surgery, adjuvant therapy, and patient data was performed. It is interesting and very important to recognize that our data confirm the existence of late recurrences, i.e., beyond 5 years from diagnosis (). Furthermore, it is also worth noticing that seven patients are long-term survivors without having received radiotherapy.
Over the last decades, GTR has been regarded to be a favorable prognostic factor. This is corroborated in the present material; survival was significantly better in those patients were GTR was achieved when compared to those with non-GTR (, p < .001). CSF cytological examination was not systematically performed before the HIT-SIOP PNET 4 protocol was opened at OUH in 2005. There seems to have been some local skepticism towards the procedure in early years [Citation29,Citation30]. A likely consequence of this suboptimal staging is that stratification for postoperative treatment has not been based on satisfactory grounds, and this might have led to both over- and under-treatment. Potentially, this improper diagnostic work-up might have influenced outcome.
Another factor that might have influenced outcome is radiotherapeutic management. It is well known that radiotherapy duration more than 48 days is prognostically unfavorable and in 23% of patients included in this study radiotherapy duration did exceed 48 days. However, survival figures for these patients were not significantly lower than the rest of the patient population. It would also have been very interesting to look at the radiotherapy fields for the patients experiencing recurrences; did they receive adequate doses to the location of the recurrence or not? Unfortunately, this was not feasible to investigate as we do not have adequate radiotherapy documentation from early years. In total, as much as 43% of patients were not treated according to the protocol they were supposed to be treated in/according to. This should not be surprising though, because chemotherapy as used in protocols for MB/CNS-PNET patients is often associated with toxicity and chemotherapy modifications are therefore often necessary. Also, a large proportion of our patients (n = 24) had treatment-resistant disease and progressed during primary treatment. Another important finding is the occurrence of second primary tumors in eleven patients; all suspected to be second malignant neoplasms (SMN) related to treatment for their MB/CNS-PNET. This is similar to other studies [Citation31]. The latter observation reminds us that survival data should never be the only parameter looked upon when the success of cancer treatment is measured; functional outcome and quality of life in long-term survivors is also of immense importance.
Overall, this retrospective study shows that treatment in several cases and particularly in the earliest periods was based on suboptimal staging and contained a relatively high percentage of therapeutic modifications not always documented in the patient records. This might have influenced outcome for the patient group as a whole in a negative manner, and emphasizes the importance of adherence to protocols and guidelines. Nonetheless, patient outcome seems to have improved in the later years of the study period, although not reaching statistical significance. The importance of GTR for survival was confirmed, as was the existence of tumor recurrences beyond five years from diagnosis.
IONC_1301679_Supplemental_information.zip
Download Zip (41.5 KB)Acknowledgments
We wish to thank Dr Lars Eikvar, Dr Olav Røise, Dr Terje Rootwelt, and Dr Sigbjørn Smeland for supporting this work.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
Related Research Data
References
- Patel S, Bhatnagar A, Wear C, et al. Are pediatric brain tumors on the rise in the USA? Significant incidence and survival findings from the SEER database analysis. Childs Nerv Syst. 2014;30:147–154.
- Kaatsch P, Rickert CH, Kuhl J, et al. Population-based epidemiologic data on brain tumors in German children. Cancer. 2001;92:3155–3164.
- Packer RJ, Sutton LN, Elterman R, et al. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J Neurosurg. 1994;81:690–698.
- Rickert CH, Paulus W. Epidemiology of central nervous system tumors in childhood and adolescence based on the new WHO classification. Childs Nerv Syst. 2001;17:503–511.
- Northcott PA, Dubuc AM, Pfister S, et al. Molecular subgroups of medulloblastoma. Expert Rev Neurother. 2012;12:871–884.
- Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:465–472.
- Northcott PA, Korshunov A, Pfister SM, et al. The clinical implications of medulloblastoma subgroups. Nat Rev Neurol. 2012;8:340–351.
- Northcott PA, Shih DJ, Peacock J, et al. Subgroup-specific structural variation across 1000 medulloblastoma genomes. Nature. 2012;488:49–56.
- Ramaswamy V, Remke M, Bouffet E, et al. Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol. 2013;14:1200–1207.
- Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109.
- Sturm D, Orr BA, Toprak UH, et al. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell. 2016;164:1060–1072.
- Louis DH, Ohgaki H, Wiestler OD, et al., editors. WHO classification of tumours of the central nervous system revised. 4th ed. Lyon: IARC; 2016.
- Zeltzer PM, Boyett JM, Finlay JL, et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children's Cancer Group 921 randomized phase III study. J Clin Oncol. 1999;17:832–845.
- Rutkowski S, von Hoff K, Emser A, et al. Survival and prognostic factors of early childhood medulloblastoma: an international meta-analysis. J Clin Oncol. 2010;28:4961–4968.
- Tait DM, Thornton-Jones H, Bloom HJ, et al. Adjuvant chemotherapy for medulloblastoma: the first multi-centre control trial of the International Society of Paediatric Oncology (SIOP I). Eur J Cancer. 1990;26:464–469.
- Bailey CC, Gnekow A, Wellek S, et al. Prospective randomised trial of chemotherapy given before radiotherapy in childhood medulloblastoma. International Society of Paediatric Oncology (SIOP) and the (German) Society of Paediatric Oncology (GPO): SIOP II. Med Pediatr Oncol. 1995;25:166–178.
- Lannering B, Rutkowski S, Doz F, et al. Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: results from the randomized multicenter HIT-SIOP PNET 4 trial. J Clin Oncol. 2012;30:3187–3193.
- Packer RJ, Sposto R, Atkins TE, et al. Quality of life in children with primitive neuroectodermal tumors (medulloblastoma) of the posterior fossa. Pediatr Neurosci. 1987;13:169–175.
- Hoppe-Hirsch E, Renier D, Lellouch-Tubiana A, et al. Medulloblastoma in childhood: progressive intellectual deterioration. Child's Nerv Syst. 1990;6:60–65.
- Packer RJ, Gajjar A, Vezina G, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24:4202–4208.
- Gajjar A, Chintagumpala M, Ashley D, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7:813–820.
- Taylor RE, Bailey CC, Robinson KJ, et al. Outcome for patients with metastatic (M2-3) medulloblastoma treated with SIOP/UKCCSG PNET-3 chemotherapy. Eur J Cancer. 2005;41:727–734.
- Tarbell NJ, Friedman H, Polkinghorn WR, et al. High-risk medulloblastoma: a pediatric oncology group randomized trial of chemotherapy before or after radiation therapy (POG 9031). J Clin Oncol. 2013;31:2936–2941.
- Jakacki RI, Burger PC, Kocak M, et al. Outcome and prognostic factors for children with supratentorial primitive neuroectodermal tumors treated with carboplatin during radiotherapy: a report from the Children's Oncology Group. Pediatr Blood Cancer. 2015;62:776–783.
- Solheim O, Salvesen O, Cappelen J, et al. The impact of provider surgical volumes on survival in children with primary tumors of the central nervous system–a population-based study. Acta Neurochir. 2011;153:1219–1229. discussion 29.
- Johnston DL, Keene D, Kostova M, et al. Survival of children with medulloblastoma in Canada diagnosed between 1990 and 2009 inclusive. J Neurooncol. 2015;124:247–253.
- Helseth E, Due-Tonnessen B, Wesenberg F, et al. Posterior fossa medulloblastoma in children and young adults (0–19 years): survival and performance. Childs Nerv Syst. 1999;15:451–455. discussion 6.
- von Hoff K, Hinkes B, Gerber NU, et al. Long-term outcome and clinical prognostic factors in children with medulloblastoma treated in the prospective randomised multicentre trial HIT'91. Eur J Cancer. 2009;45:1209–1217.
- Neidhardt MK, Bailey CC. Prospective randomized cooperative medulloblastoma trial (MED 84) of the International Society of Paediatric Oncology (SIOP) and of the (German) Society of Paediatric Oncology (GPO). Childs Nerv Syst. 1987;3:70–73.
- Hovind KH. Editorial comment. Child's Nerv Syst. 1987;3:73.
- Goldstein AM, Yuen J, Tucker MA. Second cancers after medulloblastoma: population-based results from the United States and Sweden. Cancer Causes Control. 1997;8:865–871.