
Abstract
Background: Oxygen (O2) homeostasis is an indispensable requirement of eukaryotes. O2 concentration in cellular milieu is defined as normoxia (∼21% O2), physoxia (∼1–13% O2) or hypoxia (∼0.1–1% O2). Hypoxia, a striking micro-environmental feature in tumorigenesis, is countered by tumor cells via induction of O2 governed transcription factor, hypoxia inducible factor-1 (HIF-1). Post discovery, HIF-1 has emerged as a promising anticancer therapeutic target during the last two decades. Recent reports have highlighted that enhanced levels of HIF-1 correlate with tumor metastasis leading to poor patient prognosis.
Material and methods: A systematic search in PubMed and SciFinder for the literature on HIF-1 biology and therapeutic importance in cancer was carried out.
Results: This review highlights the initial description as well as the recent insights into HIF-1 biology and regulation. We have focused on emerging data regarding varied classes of HIF-1 target genes affecting various levels of crosstalk among tumorigenic pathways. We have emphasized on the fact that HIF-1 acts as a networking hub coordinating activities of multiple signaling molecules influencing tumorigenesis. Emerging evidences indicate role of many HIF-induced proteomic and genomic alterations in malignant progression by mediating a myriad of genes stimulating angiogenesis, anaerobic metabolism and survival of cancer cells in O2-deficient microenvironment.
Conclusions: Better understanding of the crucial role of HIF-1 in carcinogenesis could offer promising new avenues to researchers and aid in elucidating various open issues regarding the use of HIF-1 as an anticancer therapeutic target. In spite of large efforts in this field, many questions still remain unanswered. Hence, future investigations are necessary to devise, assess and refine methods for translating previous research efforts into novel clinical practices in cancer treatment.
Introduction
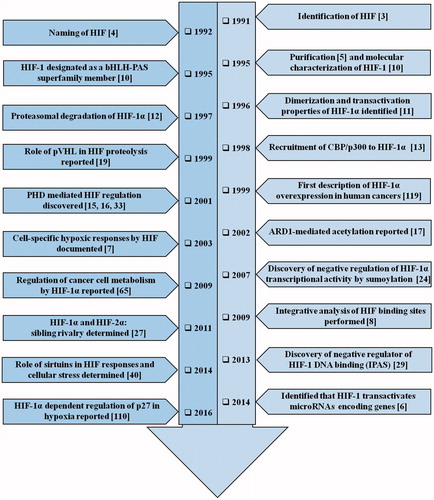
Oxygen (O2) homeostasis is a critically important factor for the survival of vertebrates [Citation1]. The transcriptional induction of a multitude of genes engaged in angiogenesis, glucose metabolism, cell proliferation/survival and the regulation of physiological responses occurring at low O2 levels is brought about by hypoxia inducible factor-1 (HIF-1), an O2-labile DNA binding transcriptional activator [Citation1]. Hypoxia abundantly occurs in core of the tumors owing to high proliferation rates of cancer cells coupled with the presence of abnormal vasculature [Citation2]. Studies on the molecular mechanisms of hematopoietic growth hormone erythropoietin (EPO) induction, the most salient response to hypoxia, set the course for recognition of HIF-1 which operates ubiquitously in mammalian cells [Citation3]. It was later purified and designated as HIF-1 [4]. Since its discovery [Citation4], biochemical purification and molecular characterization [Citation5], there has been an exponential rise in the interest to study the importance of HIF-1 in tumor biology in the past two decades, which has been depicted in a timeline form in .
Figure 1. The timeline of HIF-1 research. The year-wise sequence of path breaking events in HIF-1 research are indicated with references. Events include the initial identification of HIF-1 up to the work being carried out in this field recently. The list is non-exhaustive and only a few major events are highlighted in the flowchart.
DNA sequencing in combination with genome-wide chromatin immunoprecipitation (ChIP-seq) and microarray analysis projected many directly transactivated HIF target genes and microRNAs (miRNAs) that promote acclimatization to acute and chronic hypoxia [Citation6]. However, within a cell only a fragment of this vast spectrum of genes is activated, causing cell-specific hypoxic responses [Citation7]. Apart from this, gene expression is also indirectly regulated by HIFs through transactivating genes that encode microRNAs [Citation6] and chromatin-modulatory enzymes [Citation8]. The target genes triggered by HIF-1 includes those involved in angiogenesis, glucose metabolism, cell proliferation/viability, invasion and metastasis [Citation9]. Proteins encoded by HIF target genes fall into two prominent categories, proteins increasing O2 delivery and those decreasing O2 consumption thus making HIF-mediated pathways critical in the development, physiology and disease. Notably, majority of human tumors have an overexpression of a wide plethora of these gene products [reviewed in 9], thus emphasizing the relevance of HIF-1 based transcriptome modulations in tumor pathophysiology. In this review, our focus will be more on the recent insights into the understanding of HIF-1 expression, activation and functions during cellular responses. We will outline emerging reports on various levels of crosstalk among HIF-1 and its metabolic regulators to summarize the role of HIF-1 downstream genes in tumorigenesis.
Structure and molecular biology of HIF-1: the master regulator of hypoxia
HIF-1 transcription factor is a heterodimer with one of the three hypoxia-induced alpha subunits (HIF-1α/2α/3α) and a stable constitutively expressing beta subunit (HIF-1β, also known as aryl hydrocarbon receptor nuclear translocator (ARNT)). HIF-1α, a basic-helix-loop-helix (bHLH)-PER (Period)-ARNT-SIM (single minded) (PAS) superfamily member is the O2-labile subunit and is ubiquitously expressed in hypoxia [Citation10]. Amino (NH2−) terminus half (amino acids 1-390) of HIF-1α contains bHLH and PAS domains vital for DNA binding and dimerization. On the other hand, the carboxy (COOH−) terminus possesses oxygen-dependent degradation domain (ODDD), which confers O2-dependent instability and also two autonomous transactivation domains (N-TAD and C-TAD) in addition to an inhibitory domain (ID, negative regulator of the transactivation domains) [Citation11]. ODDD is a proline-serine-threonine rich protein stabilization domain (PSTD) extending from amino acids 429 to 608 of HIF-1α [Citation12]. Nuclear localization signal (NLS) domains extend from residues 17–33 (NLS-N) and 718–721 (NLS-C) [Citation13] ().
Figure 2. Structure and fate of the hypoxic master regulator. Figure presents the schematic representation of HIF-1α structure. N-terminal includes nuclear localization signal (NLS), followed by a basic helix-loop-helix (bHLH) domain that precedes two Per-ARNT-Sim homology domains (PAS-A and PAS-B). In addition to it, inhibitory domain (ID) separates N- and C-terminal transactivation domains (N-TAD and C-TAD). Figure also portrays the fate of HIF-1 through the sequence of events that occur during normoxic and hypoxic cellular microenvironment. During normoxia, certain post translational modifications lead to HIF-1α-pVHL binding and blocking of HIF-1α-p300/CBP interaction thereby causing HIF-1α degradation leading to the inhibition of HIF-1α-mediated transcription. During hypoxia, HIF-1α-pVHL binding does not occur, while p300/CBP can interact with HIF-1α, leading to transcriptional activation of target genes. Numbers highlights the amino-acid residues of various domains.
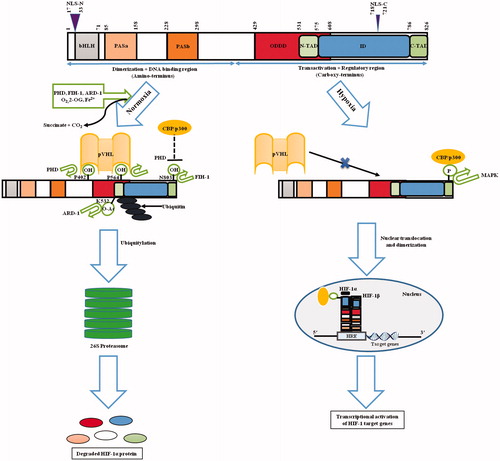
During hypoxia, N-TAD stabilizes HIF-1α, whereas C-TAD modulates transcription of HIF-1α target genes in association with CREB-binding protein (CBP)/p300 co-activator. Low O2 level stabilizes the α-subunit causing its nuclear translocation (where it dimerizes with HIF-1β) and further assembly of transcriptional co-activators [Citation13]. HIF-1 target genes contain hypoxia responsive element (HRE, containing 5′-(A/G)CGTG-3′ consensus sequence) where these heterodimers bind to an enhancer domain, activating their transcription [Citation13].
Under normoxia, two proline residues (P402 and P564 in humans) located in the LXXLAP motif [Citation14] in ODDD of HIF-1α are hydroxylated by prolyl-4-hydroxylases (PHDs) [Citation15]. PHD enzymes are dioxygenases that are 2-oxoglutarate (2-OG) and O2-dependent, requiring Fe2+ and ascorbate as cofactors in a reaction yielding succinate and carbon dioxide as by-products. It is widely known that the activity of PHDs depends on O2 as a direct substrate. Hence, PHDs have been accurately projected as O2 sensors coupling HIF-mediated molecular responses to cellular O2 levels [Citation16]. Lysine-532 (K532), another ODDD residue is acetylated by an acetyl transferase enzyme, arrest defective-1 (ARD-1) [Citation17]. Collectively, these alterations lead to 26S proteasomal degradation of HIF-1α subunit by tethering to the β-domain of von Hippel-Lindau protein (pVHL), an E3 ubiquitin ligase at Leucine-574 (L574) [Citation18]. Maxwell et al. [Citation19] first demonstrated a central role of pVHL in HIF-1α regulation in 1999, and during the next decade mechanistic specifications of pVHL-HIF pathway and regulation of proteasomal degradation and polyubiquitylation were elucidated. Further, it was found that asparagine-803 (N803) residue of HIF-1α is hydroxylated by asparginyl hydroxylase, factor inhibiting HIF-1 (FIH-1) in an O2-dependent way [Citation20] (). This drives the blocking of interaction between C-TAD and CBP/p300 domains and further abrogation of gene transcription by HIF-1α [Citation20]. Similar to PHDs, FIH-1 acts as a second O2 sensor and is a 2-OG-dependent dioxygenase, requiring ascorbate and Fe2+ as cofactors [Citation21].
Furthermore, S-nitrosation of HIF-1α on cysteine-800 (C800) has been demonstrated to enhance its transactivation by interacting with CBP/p300 [Citation22]. Later it was reported that the domain from 531 to 826 in HIF-1α gets phosphorylated and regulates its expression [Citation23]. It was also revealed that sumoylation on three lysine residues (K391, K477 and K532 on HIF-1α) negatively regulates transcriptional activity of HIF-1α [Citation24].
HIF-1 isoforms
Homology searches in databases and cloning efforts led to the discovery of HIF-1 isoforms, HIF-2α {HIF-1α like factor or endothelial PAS domain protein 1 (EPAS1)} and HIF-3α, which are other representatives of bHLH-PAS superfamily. The influence of HIF family on gene expression may vary among numerous cell types. Similar to HIF-1α, HIF-2α is also stabilized by hypoxia and dimerizes with HIF-1β. HIF-2α, which has a restricted expression mainly in renal, endothelial, hepatic and lung cells, has 48% amino acid sequence similarity with HIF-1α, unlike HIF-3α [reviewed in 25]. Though both HIF-1α and HIF-2α stimulate target genes by linking to same HRE, they are functionally non-redundant [Citation26]. HIF-1α regulates glycolytic gene expression and preferentially drives apoptotic pathways not addressed by HIF-2α, whereas HIF-2α encourages tumor growth and angiogenesis [Citation27].
Co-relation between HIF-1α and HIF-2α levels with tumor growth, angiogenesis and metastasis has been established in both animal as well as clinical investigations [Citation1]. It is worth mentioning that HIF-1α plays a critical role in initial responses (2–24 h) to severe hypoxia (<0.1% O2) whereas HIF-2α impacts chronic hypoxia (48–72 h) [Citation28]. Very little was known about the newest isoform HIF-3α until the identification of its spliced variant, inhibitory PAS (IPAS) which in a dominant negative way regulates HIF-1 DNA binding ability [Citation29]. Recent reports have indicated that HIF-3α is hypoxia-activated and promotes a distinct transcriptional response in zebrafish embryos [Citation30]. HIF-3α, unlike the other two isoforms has only one TAD, a unique LXXLL motif and a leucine zipper domain [Citation30].
To date significant developments have occurred towards deciphering HIF’s roles under both pathophysiological and physiological conditions. During physiological settings, interplay of HIF-1α with HIF-2α and HIF-3α would provide immense understanding of HIF(s). Likewise, the interaction of these transcription factors with other important proteins and cascades will help in setting the future trends of investigation on HIF biology and its implications.
Regulation of HIF-1 pathway
In general, HIF-1α is synthesized and constitutively transcribed by a cascade involving a series of growth factors and signaling events (). Normoxia leads to quick degradation of HIF-1α transcript (t1/2 ∼ 5 min) [Citation31]. Conversely, hypoxic condition results in stability and transcriptional activity of HIF-1α (as discussed above), through several pathways and post-translational modifications (hydroxylation, acetylation, ubiquitylation, phosphorylation, sumoylation, S-nitrosation) [Citation32]. Under normal O2 tension, the PHDs become active and cause prolyl hydroxylation in HIF-1, leading to a recognition indicator for pVHL binding, ubiquitylation and subsequent degradation of HIF-1α [Citation33]. Hypoxia ceases enzymatic activity, terminating proline modification and pVHL-HIF binding, causing HIF-1α stabilization, nuclear translocation and further aggregation. The important modulators of HIF-1 are summarized in .
Figure 3. HIF-1 signaling cascade. Synthesis and constitutive expression of HIF-1α by a cascade involving a series of growth factors and signaling events are indicated. The major differences among the hypoxic and normoxic signaling and sequence of events is also depicted clearly in the flowchart. Normoxia leads to HIF-1α protein degradation whereas hypoxia leads to HIF-1α-regulated target gene expression. The downstream sequence of events leading to tumorigenesis is also portrayed.
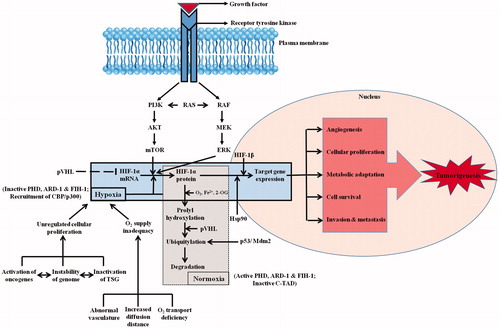
Table 1. Critical regulators of HIF-1 activity.
Reports have shown that activation of mechanistic target of rapamycin (mTOR) [Citation34] and phosphatidyl inositol-4,5-bisphosphate-3-kinase (PI3K) [Citation35] can upregulate HIF-1α translation. In addition, some growth factors activating RAS consequently stimulates RAS/RAF/MEK/ERK kinase pathway. The co-activator CBP/p300 is phosphorylated by ERK increasing the formation of HIF-1α/p300 complex, thus in turn stimulating its transcriptional activation trait [Citation35]. Recently HIF-1α has been reported to be activated at physiological O2 levels in a p42/44 mitogen-activated protein kinase (MAPK)-dependent manner determining increased cellular proliferation commonly found in these conditions [Citation36]. Apart from tumor suppressor genes reviewed ahead, certain growth factors and cytokines also have reasonable effects on the HIF-1 network. These include platelet-derived growth factor (PDGF) [Citation37], insulin [Citation38] and insulin-like growth factors (IGF) [Citation39], which induces HIF-1α translation ().
Recently a stress-responsive family of histone deacetylases, sirtuins is reported to influence DNA repair, transcription metabolism and metastasis [Citation40]. These nicotinamide adenine dinucleotide (NAD+)-dependent enzymes have exhibited potential to regulate HIF activity, thereby strengthening the association between HIF responses and cellular stress [Citation40]. During hypoxia, cellular NAD+ is decreased which reduces sirtuin 1 (Sirt1) levels, enhances HIF-1α acetylation, and hence promotes HIF-1α target gene expression. Poly (ADP-ribose) polymerase 1 (PARP1) is one more NAD+-dependent enzyme, which was reported to interact with and promote HIF-1α transactivation [Citation41]. Additionally, it has been reported that elevated nitric oxide (NO) levels and enhanced expression of nitric oxide synthase (NOS) isoforms increases HIF-1α protein stability in human oral squamous cell carcinoma [Citation42] ().
HIF-1 in tumors
A proper O2 homeostasis is essential for mammalian cells in order to fulfill their energy requirement and aerobic metabolism. However, cancer cells become hypoxic due to decreased O2 delivery and diffusion, combined with increased O2 consumption. Tumors have high impairment in their cellular O2 balance due to inadequate and chaotic vascularization. Hypoxia, a prominent characteristic feature of the tumor milieu drives aggressiveness of a growing tumor [Citation43]. About 60% of solid tumors have <1% O2 leading to intra-tumoral hypoxia. Tumor cells counter these alterations in O2 tension by delegating HIF-1α for orchestrating numerous of its essential functions. Upregulation of HIF-1α activates many crucial cancer hallmarks such as angiogenesis, glucose metabolism, cell proliferation/viability, invasion and metastasis and has a crucial role in tumor survival and progression as discussed in the following sections. provides a list of genes targeted by HIF-1 that regulates various tumorigenic pathways. Manalo et al. [Citation44] documented through DNA microarray experiments that greater than 2% of all genes in humans are HIF-1 regulated (directly or indirectly). In clinical settings, immunohistochemistry (IHC) analysis of patient’s biopsy specimens exhibited dramatic HIF-1 overexpression in common human cancers, which leads to increased metastasis and mortality rates (). clearly lists the localization of HIF-1α expression in common human cancers along with the details of antibodies used for the immunostaining.
Figure 4. HIF-1 in the driver seat of tumorigenesis. The list of HIF-1 regulated genes (with corresponding references highlighted in bracket) having an impact on tumorigenic pathway is showcased. The HIF-1 target genes presented here are involved in processes such as cellular metabolism, proliferation and survival, angiogenesis, invasion and metastasis of tumor cells. The list is intended to be an illustration rather than being comprehensive.
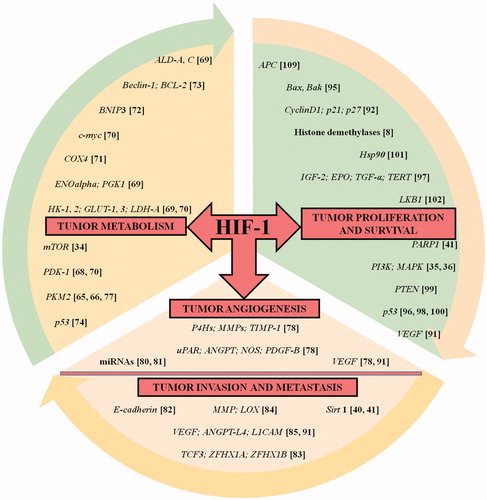
Table 2. Human cancers exhibiting increased levels of HIF-1α protein.
HIF-1 and reprograming of tumor metabolism
Increased glycolytic rate during hypoxia is a critical step in meeting cellular energy demands. Metastatic tumor cells have a remarkably increased glucose uptake rate in comparison to normal cells. This is such a reliable indication that it acts as a basis for diagnosis of tumor in patients by 18-F-fluorodeoxyglucose-positron emission tomography (FDG-PET) to ascertain metastases by imaging of FDG tracer [Citation63]. HIF-1 serves as an important moderator of the Warburg effect, the tumor-related metabolic switch, which helps cancer cells to create energy largely by disintegration of glucose in a non-oxidative manner rather than typical oxidative phosphorylation [Citation64]. HIF-1 stimulates the expression and activation of glycolytic enzyme isoforms differing from those found in normal cells, thereby supporting the Warburg effect by potentiating macromolecular biosynthesis and energy production pathways in human cancers [Citation65]. For example, pyruvate kinase isoform M2 (PKM2) expression is imperative for the Warburg effect. Instead of PKM1, PKM2 physically joins HIF-1 stimulating its activity [Citation66]. Additionally, recent reports point to mTOR and PKM2 as critical determinants of the Warburg effect, which to a certain degree can be attributed to their effect on HIF-1α [Citation34]. Furthermore, critical fallout of this glycolytic switch is tumor microenvironment acidosis. Collectively, the acidic milieu and metabolic transformation provides plenty of metabolic intermediates that spur progression and aggressiveness of tumors [Citation67].
Interestingly, HIF-1α induces glycolysis and actively restrains mitochondrial function and O2 utilization by inducing pyruvate dehydrogenase kinase-1 (PDK-1) activity [Citation68]. In the glycolytic pathway, HIF-1 targets include hexokinase 1 and 2 (HK1, HK2) enzymes, glucose transporters 1 and 3 (GLUT1, GLUT3), lactate dehydrogenase-A (LDH-A), aldolase A and C (ALD-A, ALD-C), phosphofructokinase liver type (PFK-L), 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB-3), enolase alpha (ENOalpha) and phosphoglycerate kinase 1 (PGK1) [Citation69]. Out of these GLUT1, LDH-A and HK2 are direct targets of oncogenic transcription factor MYC. HIF-1α inhibits c-MYC activity under physiological conditions but during stress c-MYC works together with HIF-1 to induce expression of PDK-1 and HK2 leading to angiogenesis and aerobic glycolysis [reviewed in Citation70]. Also it has been seen that under hypoxic condition, HIF-1α manipulates cytochrome c oxidase subunit 4 (COX4) switch, a homeostatic response optimizing respiration efficacy at varied O2 concentrations [Citation71]. BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) is a HIF-1 target gene correlated to autophagy [Citation72] through disruption of interaction between Beclin-1 (a highly conserved autophagy inducer protein) and B-cell lymphoma 2 (Bcl-2) or B-cell lymphoma-extra-large (Bcl-xL) [Citation73]. Recent reports have exhibited that p53 is stimulated by hypoxia and acts as a negative regulator of glycolysis [Citation74] ().
HIF-1-regulated switch in tumor angiogenesis
The angiogenic switch in hypoxic tumor microenvironment may be attributed to enhanced O2 consumption and curtailed O2 delivery as a result of increased diffusion distance. The discovery that hypoxia induces vascular endothelial growth factor (VEGF), served as a crucial link between hypoxia and angiogenesis [Citation75]. Lately, it was reported that copper induces HIF-1α and VEGF expression in hepatic and breast cancer cells [Citation76]. Recently, it has come to light that in pancreatic cancer, PKM2 interferes in the activation of HIF-1α that further triggers secretion of VEGF and subsequent formation of blood vessels [Citation77]. Angiogenesis an intricate multistep and ordered process is essential for tumor formation and involves numerous genes, regulators and pathways [Citation78]. HIF-1α has been described to portray a vital part in angiogenesis, thus, silencing/inhibiting this pathway might result in reduced ability of cells to muster a strong vasculature and colonize at distant sites, even in the presence of an invasive phenotype.
The angiogenic induction results in an increased vascular density and decreased diffusion distance of O2. However, local flow of blood under pathophysiological setting is regulated by vascular tone modulation through HIF-1α-mediated direct induction of various genes encoding angiogenic growth regulators. This includes urokinase type plasminogen activator receptor (uPAR) that modulates matrix metabolism, angiopoietin 1 and 2 (ANGPT), vascular tone governing NOS, VEGF and PDGF-B (). Additionally, HIF-1α signaling pathways regulate factors such as collagen prolyl-4-hydroxylases (P4Hs), matrix metalloproteinases (MMPs) and tissue inhibitors of matrix metalloproteinases (TIMP-1) [Citation78]. In vivo investigations revealed that targeting HIF-1 function significantly inhibits tumor vascularization [Citation79]. Previous studies have demonstrated number of miRNAs induced during hypoxia and they play critical roles in angiogenesis [Citation80]. miR-429 is induced during early stage of hypoxia in tumor cells and regulates HIF-1 mRNA levels [Citation81]. Therefore, it is evident that HIF-1 antes up angiogenesis by a largely complex mechanisms rather than straightforward VEGF induction.
HIF-1 at the nexus of tumor invasion and metastasis
Metastasis, the primary reason of cancer-related mortality, takes place in a series of well-defined events. These encompass local invasion of tumor cells followed by intravasation into the blood stream, survival of circulating tumor cells, extravasation to far-off sites and finally proliferation that leads to colonization. The invasive and metastasizing property of tumor cells is unleashed by hypoxia. Stimulation of genes regulated by HIF has been related with enhanced metastasis in multiple tumors [Citation9]. HIF isoforms have been reported to show divergent effects on invasion and metastasis [Citation27]. Many studies have pointed towards the detection of HIFs in multiple cancers but their role in regulating extracellular matrix (ECM) degeneration, glucose metabolism, invasion and metastasis remains largely unexplored.
One of the most striking hallmarks of invasive cells is epithelial-to-mesenchymal transition (EMT), which is symbolized by impairment of epithelial cell-to-cell contact and the attainment of mesenchymal traits and motility. HIF-1 directs the expression of many EMT regulators thereby acting as one of the contributing factor in tumor metastasis. Many reports have revealed that HIF-1α expression in cancer increases invasion and induces the loss of E-cadherin [Citation82]. In addition to this, HIF-1 drives transcriptional activation of genes encoding repressors of E-cadherin expression such as TCF3, ZFHX1A and ZFHX1B as well as other such proteins that are involved in providing a rigid cytoskeleton and cell-to-cell adhesion [Citation83].
HIF-1 is engaged in the hypoxia-intervened modulation of many genes engrossed intricately in tumor growth and advancement [Citation9]. The extent of tumor invasiveness and metastasis depends on HIF-1 regulated transcriptional activation of MMP and lysyl oxidase (LOX) that degrades and remodels ECM, respectively [Citation84]. Further, HIF-1 target genes include permeability factors such as VEGF that promote cancer cells intravasation into blood vessels. In addition to this, secreted ANGPT-L4 and cell surface L1 cell adhesion molecule (L1CAM) proteins cause extravasation of malignant cells into metastatic sites [Citation85] (). Furthermore, silencing of HIF-2α was found to be more effective in decreasing ECM degradation and reducing metastasis under hypoxia [Citation27].
Experimental investigations on mouse models revealed that foundation of a premetastatic niche is needed at the metastatic site before the arrival of cancer cells. Tumor-secreted factors are the initiators of this process and act by mobilizing the bone-marrow-derived cells and their subsequent recruitment to the metastatic sites. These factors are further responsible for the modification of ECM thereby generating a niche required for extravasation and colonization of cancer cells [Citation86]. A small subset of cancer cells capable of unlimited self-renewal are called cancer stem cells (CSCs) commonly known as tumor initiating cells. Many HIF inhibitors have been reported to target CSCs in an attempt to improve the effects of chemotherapy and angiogenesis inhibitors in in vivo models. CSCs offer unlimited proliferation potential and are responsible for metastatic tumors [Citation87].
Many recent reports suggested the role of HIF-1 in CSCs maintenance in various cancer types [Citation88]. Hypoxic exposure to cancer cell lines induces a HIF-1-dependent expression of certain genes that promote pluripotency and represses differentiation [Citation88]. Breast cancer mouse model having knockdown of HIF-1α led to a reduction in the percentage of breast cancer stem cells [Citation89]. A major cause that limits the efficacy of cytotoxic chemotherapy in certain tumors is the enrichment of CSCs following treatment. CSCs have been reported to enhance the expression of genes that increase neoplastic cell survival by mediating cellular efflux of chemotherapeutic drugs [Citation90]. It is quite possible that a strong positive selection occurs for severe O2 gradient generating cancer cells due to the linked hypoxia-induced proliferation of CSCs. In a nutshell, these reports validate the role of intratumoral hypoxia and HIF-1 as the critical regulators of CSCs maintenance by transactivating genes promoting CSC phenotype.
HIF-1: the playmaker in tumor proliferation and survival
The first differentiating change between neoplastic and normal cells is the enhanced cellular proliferation rate and a diminished cell death rate caused due to increased expression of survival and growth factors. In the HIF pathway, VEGF is a noteworthy transcription target with involvement in cellular proliferation and metastasis in tumors [Citation91]. Emerging evidences document the potential role of HIF-1 in preventing cell death, stimulating cellular proliferation or even induction of apoptosis [Citation92] as discussed below. The putative target genes of HIFs that can modify cell-cycle progression are cyclin D1, p21 and p27 [reviewed in 92]. The part that HIF pathway portrays in cell death is dubious and it depends on the O2 concentration to determine whether apoptosis will occur or not. 0–0.5% O2 level in cells induces apoptosis whereas if O2 levels are in 1–3% range apoptosis does not occur [Citation93].
The key determinant of apoptosis in cells is ATP; during hypoxia abundant glycolytic ATP leads to apoptosis [Citation94]. Moreover, O2 deprivation leads to inhibition of electron transport chain thereby causing a reduction in membrane potential at the mitochondria. This sequence of events causes release of cytochrome C into the cytosol of hypoxic cells through activation of BCL2-associated X protein (Bax) or BCL2-antagonist/killer (Bak), leading to activation of caspase-9-mediated apoptosis [Citation95]. In addition to it, p53, an important apoptosis regulator, can induce Bak and Bax proteins thereby stimulating the apoptosis cascade through cytochrome C [Citation96]. The growth/survival agents encoded by genes regulated by HIF in various tumors include VEGF, EPO, IGF-2 and transforming growth factor-α (TGF-α). These genes are the controlling hub of all the important tumor progression pathways, including angiogenesis, proliferation, invasion and colonization of far-off sites. Additionally, the expression of telomerase reverse transcriptase (TERT) causes the immortalization of tumor cells [reviewed in 97] ().
Tumor suppressor genes and HIF-1
Tumor suppressor genes (TSGs) p53 [Citation98] and phosphatase and tensin homolog (PTEN) [Citation99] influence HIF-1α by suppressing its hypoxic induction and target activation, probably via AKT modulation (). Literature suggests association of p53 loss of function with enhanced levels of HIF-1α in certain tumors. In normoxia, HIF-1α interacts with p53 allowing mouse double minute 2 homolog (Mdm2) governed HIF-1α ubiquitylation and proteasomal degradation [Citation100]. The chaperone heat shock protein 90 (Hsp90) interacts with HIF-1α directly and is shown to induce certain conformational changes making it fit to couple with HIF-1β, hence initiating transactivation pathway [Citation101]. Interestingly, exposure to elevated temperature strongly upregulates HIF-1α in mice model, suggesting a unique mechanism that stabilizes HIF-1α under normoxia [Citation101]. Many of the predominant driver mutations observed in malignant cells alter tumor metabolism as part of their mode of action. Recent data implicate the tumor suppressor gene liver kinase B1 (LKB1) as a central regulator of tumor metabolism and growth control through the regulation of HIF-1α-dependent metabolic reprograming [Citation102]. Similarly, loss of PTEN can promote increased glucose uptake through elevated PI3K/Akt/mTOR signaling [Citation103] while loss of the VHL also promotes a similar metabolic phenotype through stabilization of the HIF-1α [Citation104].
Experimental data suggests that HIF-1α can itself serve a tumor suppressor role in certain solid tumors [Citation105]. In clear renal cell carcinoma, where VHL loss of function leads to an accumulation of HIF-1/2α, HIF-1α is lost during tumor progression, indicating its tumor suppressor role in the later stages of tumorigenesis [Citation106]. The importance of HIF-1α in leukemia has been investigated in different experimental models, which proposes that HIF-1α has vital role in self-renewal and proliferation of leukemic cells. HIF1α deletion impairs the proliferation of chronic myeloid leukemia by inhibiting progression of cell-cycle and induction of apoptosis in leukemia stem cells [Citation107]. Similar observations have been made for the requirement of HIF-1/2α in human AML using echinomycin inhibition [Citation108]. Deletion of HIF-1α resulted in faster development of the disease and an enhanced leukemia phenotype in some of the investigated models in acute myelogenous leukemia (AML) thus validating the tumor suppressor role of HIF-1α [Citation105]. Furthermore, hypoxia promotes a reduction in adenomatous polyposis coli (APC) mRNAs and protein in colorectal tumor via HIF-1α-dependent mechanism, suggesting that suppression of APC by hypoxia can contribute to increased survival in hypoxic tumors [Citation109]. It was also observed that loss of p53 in certain type of tumors is associated with elevated levels of HIF-1α. In hypoxic tumors, loss or mutations in p53 revokes any chances for Mdm2-mediated degradation of HIF-1α [Citation100]. The role of HIF-1α in regulating events that are involved in cell cycle arrest has been unclear. However, it is seen that hypoxia could cause HIF-1α dependent increase in the expression of p27 leading to cell cycle arrest in G0/G1 phase [Citation110].
Targeting HIF-1 for therapy: preclinical strategies
It is quite evident till now that HIF-1 regulation is an exceedingly convoluted network comprising several overlapping mechanisms and signaling cascades. Each and every step of this complex process has offered immense potential for research and therapeutic benefits. There has been huge curiosity in unraveling the intricate biology and processes of HIF-1 pathways. HIF-1 has shown profound impact on cancer progression, thus, there has been great enthusiasm in developing therapeutics that could target HIF-1. But, the utter complexity of HIF-1 pathway regulation has made this a very challenging task.
In an effort to discern the physiological importance of HIF (a key stress-responsive transcription factor), many recent studies have been performed on HIF-1α knockout models both in vitro and in vivo. Recently, HIF-1α−/− in mouse embryos has been reported to be lethal due to vascular regression and cardiac malformations. Also, HIF-1α± mice have been shown to develop normally but have impaired hypoxic and ischemic responses. Furthermore, studies were performed on HIF-1α−/− mice confirming that HIF-1α maintains O2 homeostasis [Citation111]. Various reports also suggested that PHD inhibition stabilizes HIF and augments gene expression which along with FIH inhibition induces a constellation of target genes [reviewed in Citation112]. HIF-1α loss-of-function mutation has been shown to modulate longevity and stress tolerance in Caenorhabditis elegans by different pathways. It has advanced our knowledge about the regulatory networks linking oxygen homeostasis and ageing [Citation113]. In a recent study, HIF-1α deletion led to enhanced leukemia phenotype in mice model [Citation105]. Knockdown of HIF-1α expression has been reported to cause complete inhibition of the hypoxic induction in breast cancer stem cells [Citation89].
Targeting HIF-1 for therapy: current clinical settings
To date, inhibitors of HIF-1 have been classified by their inhibitory mechanisms yet none of them appears to inhibit the HIF-1 pathway as their specific target [Citation114]. Hence, design of selective HIF-1 targeting molecules is highly likely to be the scope of future research. The regulation of HIF-1α comprises very complex cascades hence designing the selective and rational inhibitors of HIF-1α becomes highly challenging task. It is evident that the success of this research importantly depends on the sensitive and specific screening methods available. Targeting HIF-1 has become focus of cancer therapy research and quite a few HIF-1 suppressing agents are in the discovery pipeline.
Compounds with anti-HIF-1 activity are mainly classified on their modes of action as direct or indirect HIF-1 inhibitors. Direct HIF-1 inhibitors prevent transactivation, DNA binding and transcriptional activity of HIF-1α, on the other hand indirect HIF-1 inhibitors work by blocking the HIF-1α transcription or translation or by promoting HIF-1α protein degradation [Citation25] (). Inhibiting HIF-1 offers a novel avenue for modulating the tumor niche and may have promising clinical outcomes. Currently available HIF-1 inhibitors suffer a huge bottleneck due to their nonspecific mode of action. Where on one hand, majority of HIF-1 inhibitors reported thus far show antitumor action by HIF-1α inhibitory mechanism, which includes HIF-1 but is not limited to it. While on the other hand, some HIF-1 inhibitors have a complex HIF-1 inhibition mechanism which possibly involves blocking several points in the HIF-1 pathway unselectively.
Table 3. Major chemical inhibitors of HIF-1 activity.
Many small molecules have been reported as HIF-1α inhibitors [Citation114]. Till date, no clinically approved selective HIF-1α inhibitor has been reported, which may be partially due to the need of specifically targeting protein–protein interactions while escaping other pathways. The chemical inhibitors of HIF-1 can be classified as inhibitors of HIF-1α transcriptional activity, protein translation, protein degradation and protein and nucleic acid inhibitors of HIF-1 [reviewed in 115]. The major chemical inhibitors of HIF-1 activity along with their modes of action have been listed in .
It is worth mentioning that majority of the potential HIF-1 targeting therapeutics were primarily discovered to target other endogenous molecules. Better understanding of the HIF-1 in consortium with empirical testing later led to the identification of the HIF-1 inhibiting activity of these molecules [Citation114]. This could be the reason that currently there are no specific HIF-1 targeting potential therapeutic drugs in the market. This poses a great challenge in hypoxic tumor settings that demands for specific HIF-1 targeting drugs, which are currently being administered at lower doses so as to maintain prolonged inhibition of HIF activity [Citation97]. Furthermore, it was found that combination treatment using HIF inhibitors and anti-angiogenic agents might improve outcome efficacy, reduce the likelihood of side-effects and effectively lower drug doses as shown in mouse models [Citation116,Citation117]. HIF targeting molecules possess the potential to combat cancer as well as cancer-associated inflammation and hence can be efficient future therapeutics. Traditional chemotherapy drugs might be more proficient when administered along with an HIF inhibitor. Interestingly, the therapeutic effects of HIF-1 inhibition by small molecules have already been exhibited in preclinical mouse models [Citation118]. More competent understanding of structure and molecular biology of HIF-1 domains will unquestionably result in unearthing therapeutic compounds, which further could be explored for preclinical trials. In a nutshell, the development of specific HIF-1 inhibitors is of great importance.
Conclusion and future prospects
Seventeen years have passed since first description of HIF-1α overexpression in human cancers [Citation119], and during this time a large amount of experimental data has emerged delineating the molecular mechanisms and outcomes of enhanced HIF-1 expression in tumorigenesis (). Despite numerous recent advances in discerning molecular mechanisms of HIF pathways in response to hypoxic condition, many critical questions still remain unanswered. Investigating such queries could yield novel insights into interplay between various HIF-1 post-translational modifications (PTMs), better understanding of HIF-1 paralogs’ functionality and the linkage among HIF-1 and other oncogenic pathways. The main challenge in front of the research fraternity today is the identification of malignancies in which HIF-1 portrays a crucial part in disease pathogenesis in patients. Close association of laboratory investigators and clinical oncologists is necessary to devise, assess and refine methods for translating investigative outcomes into effective and safer cancer therapies.
In the present review, physiological adaptations of HIF pathways along with its importance have been discussed. It has been highlighted that the role of HIF varies under diverse settings. Various studies have showcased that HIFs are upregulated in tumor cells. HIF-1 portrays a pleiotropic part by boosting various molecular processes linked with tumorigenesis. Apart from this, HIFs have been reported to orchestrate integral roles in several crucial aspects of tumor biology such as microRNAs and epigenetic alterations. These findings accentuate the need to explore the feasibility of targeting HIF-1 directly or indirectly to clarify the enigma behind tumorigenesis. Therapeutic effects of HIF-1 inhibition have been determined in vivo. In a nutshell, this research area assures momentous advances in the days to come.
Future research should attempt to unearth the missing links between HIF and cancer metabolism. With the rapid advancement of molecular biology and emerging strategies in efficiently disrupting protein–protein interactions, it is very promising that selective HIF-1 inhibitors can be developed in the future. Future investigations should be aimed toward better understanding of therapeutic modulation of HIF-1 by deciphering molecular structure of domains and mechanisms mediating critical HIF-1 functions. Considering the rapid advancements in the field of molecular biology and presently emerging strategies it would soon be possible to efficiently study protein–protein interactions that are the cornerstones in understanding HIF-1 regulation. This will not only present supplementary research avenues, but will also help to answer complex questions that will undoubtedly boost our appreciation for therapeutic applicability of HIF pathways in malignancy. Finally, HIF-1, the play maker in the hypoxic world could be the next big player in cancer research and be the much needed answer to the enigma of tumorigenesis.
Acknowledgments
The authors would like to acknowledge the Director, CSIR-IHBT, Palampur for his support. Mr Sourabh Soni would like to thank CSIR for Senior Research Fellowship and AcSIR, New Delhi, India for PhD registration.
Disclosure statement
The authors have no conflicts of interest. The authors are solely responsible for the content and writing of the manuscript.
Additional information
Funding
Related Research Data
References
- Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol. 2014;9:47–71.
- Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. 2016;352:175–180.
- Semenza GL, Nejfelt MK, Chi SM, et al. Hypoxia inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc Natl Acad Sci USA. 1991;88:5680–5684.
- Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454.
- Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–1237.
- Camps C, Saini HK, Mole DR, et al. Integrated analysis of microRNA and mRNA expression and association with HIF binding reveals the complexity of microRNA expression regulation under hypoxia. Mol Cancer. 2014;13:28–48. doi:10.1186/1476-4598-13-28
- Kelly BD, Hackett SF, Hirota K, et al. Cell type–specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in non-ischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. 2003;93:1074–1081.
- Xia X, Lemieux ME, Li W, et al. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc Natl Acad Sci USA. 2009;106:4260–4265.
- Balamurugan K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int J Cancer. 2016;138:1058–1066.
- Wang GL, Jiang BH, Rue EA, et al. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514.
- Jiang BH, Rue E, Wang GL, et al. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem. 1996;271:17771–17778.
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997;272:22642–22647.
- Kallio PJ, Okamoto K, O’brien S, et al. Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. Embo J.1998;17:6573–6586.
- Chowdhury R, McDonough MA, Mecinovic J, et al. Structural basis for binding of hypoxia-inducible factor to the oxygen-sensing prolyl hydroxylases. Structure. 2009;17:981–989.
- Epstein AC, Gleadle JM, McNeill LA, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54.
- Jewell UR, Kveitikova I, Scheid A, et al. Induction of Hif-1alpha in response to hypoxia is instantaneous. FASEB J. 2001;15:1312–1314.
- Jeong JW, Bae MK, Ahn MY, et al. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell. 2002;111:709–720.
- Huang LE, Pete EA, Schau M, et al. Leu-574 of HIF-1alpha is essential for the von Hippel-Lindau (VHL)-mediated degradation pathway . J Biol Chem. 2002;277:41750–41755.
- Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275.
- McNeill LA, Hewitson KS, Claridge TD, et al. Hypoxia-inducible factor asparaginyl hydroxylase (FIH-1) catalyses hydroxylation at the β-carbon of asparagine-803. Biochem J. 2002;367:571–575.
- Dayan F, Roux D, Brahimi-Horn MC, et al. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1α. Cancer Res. 2006;66:3688–3698.
- Yasinska IM, Sumbayev VV. S-nitrosation of Cys-800 of HIF-1alpha protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett. 2003;549:105–109.
- Mazure NM, Brahimi-Horn MC, Berta MA, et al. HIF-1: master and commander of the hypoxic world. A pharmacological approach to its regulation by siRNAs. Biochem Pharmacol. 2004;68:971–980.
- Berta MA, Mazure N, Hattab M, et al. SUMOylation of hypoxia-inducible factor-1α reduces its transcriptional activity. Biochem Biophys Res Commun. 2007;360:646–652.
- Masoud GN, Li W. HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B. 2015;5:378–389.
- Doonachar A, Gallo MD, Doukas D, et al. Differential effects of HIF-α isoforms on apoptosis in renal carcinoma cell lines. Cancer Cell Int. 2015;15:1.
- Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12:9–22.
- Holmquist-Mengelbier L, Fredlund E, Lofstedt T, et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell. 2006;10:413–423.
- Torii S, Sakaki K, Otomo M, et al. Nucleocytoplasmic shuttling of IPAS by its unique nuclear import and export signals unshared with other HIF-3α splice variants. J Biochem. 2013;154:561–567.
- Zhang P, Yao Q, Lu L, et al. Hypoxia-inducible factor 3 is an oxygen-dependent transcription activator and regulates a distinct transcriptional response to hypoxia. Cell Rep. 2014;6:1110–1121.
- Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol. 2006;70:1469–1480.
- Kuschel A, Simon P, Tug S. Functional regulation of HIF‐1α under normoxia-is there more than post‐translational regulation? J Cell Physiol. 2012;227:514–524.
- Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing . Science. 2001;292:464–468.
- Sun Q, Chen X, Ma J, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type m2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci USA. 2011;108:4129–4134.
- Agani F, Jiang BH. Oxygen-independent regulation of HIF-1: novel involvement of PI3K/AKT/mTOR pathway in cancer. Curr Cancer Drug Targets. 2013;13:245–251.
- Carrera S, Senra J, Acosta MI, et al. The role of the HIF-1α transcription factor in increased cell division at physiological oxygen tensions. PLoS One. 2014;9:e97938.
- Lau CK, Yang ZF, Ho DW, et al. An Akt/hypoxia-inducible factor-1α/platelet-derived growth factor-BB autocrine loop mediates hypoxia-induced chemoresistance in liver cancer cells and tumorigenic hepatic progenitor cells. Clin Cancer Res. 2009;15:3462–3471.
- Biswas S, Mukherjee R, Tapryal N, et al. Insulin regulates hypoxia-inducible factor-1α transcription by reactive oxygen species sensitive activation of Sp1 in 3T3-L1 preadipocyte. PLoS One. 2013;8:e62128.
- Sinha S, Koul N, Dixit D, et al. IGF-1 induced HIF-1α-TLR9 cross talk regulates inflammatory responses in glioma. Cell Signal. 2011;23:1869–1875.
- Yoon H, Shin SH, Shin DH, et al. Differential roles of Sirt1 in HIF-1α and HIF-2α mediated hypoxic responses. Biochem Biophys Res Commun. 2014;444:36–43.
- Elser M, Borsig L, Hassa PO, et al. Poly(ADP-ribose) polymerase 1 promotes tumor cell survival by coactivating hypoxia-inducible factor-1-dependent gene expression. Mol Cancer Res. 2008;6:282–290.
- Quintero M, Brennan PA, Thomas GJ, et al. Nitric oxide is a factor in the stabilization of hypoxia-inducible factor-1alpha in cancer: role of free radical formation. Cancer Res. 2006;66:770–774.
- Vaupel P, Mayer A. Hypoxia in tumors: pathogenesis-related classification, characterization of hypoxia subtypes, and associated biological and clinical implications. Adv Exp Med Biol. 2014;812:19–24.
- Manalo DJ, Rowan A, Lavoie T, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669.
- Huang Z, Xu R, Lv C, et al. A chronic obstructive pulmonary disease negatively influences the prognosis of patients with bladder urothelial carcinoma via hypoxia inducible factor-1alpha. Int J Clin Exp Med. 2014;7:3344–3353.
- Maroni P, Matteucci E, Drago L, et al. Hypoxia induced E-cadherin involving regulators of Hippo pathway due to HIF-1α stabilization/nuclear translocation in bone metastasis from breast carcinoma. Exp Cell Res. 2015;330:287–299.
- Haugland HK, Vukovic V, Pintilie M, et al. Expression of hypoxia-inducible factor-1α in cervical carcinomas: correlation with tumor oxygenation. Int J Radiat Oncol Biol Phys. 2002;53:854–861.
- Jo JO, Kim SR, Bae MK, et al. Thymosin β4 induces the expression of vascular endothelial growth factor (VEGF) in a hypoxia-inducible factor (HIF)-1α-dependent manner. BBA-Mol Cell Res. 2010;1803:1244–1251.
- Malfettone A, Silvestris N, Paradiso A, et al. Overexpression of nuclear NHERF1 in advanced colorectal cancer: association with hypoxic microenvironment and tumor invasive phenotype. Exp Mol Pathol. 2012;92:296–303.
- Horree N, Gort EH, Van der Groep P, et al. Hypoxia‐inducible factor 1α is essential for hypoxic p27 induction in endometrioid endometrial carcinoma. J Pathol. 2008;214:38–45.
- Chen Y, Lu Y, Lu C, et al. Beclin-1 expression is a predictor of clinical outcome in patients with esophageal squamous cell carcinoma and correlated to hypoxia-inducible factor (HIF)-1α expression. Pathol Oncol Res. 2009;15:487–493.
- Takahashi R, Tanaka S, Hiyama T, et al. Hypoxia-inducible factor 1α expression and angiogenesis in gastrointestinal stromal tumor of the stomach. Oncol Rep. 2003;10:797–802.
- Hermansen SK, Nielsen BS, Aaberg-Jessen C, et al. miR-21 is linked to glioma angiogenesis-a co-localization study. J Histochem Cytochem. 2016;64:138–148.
- Jokilehto T, Rantanen K, Luukkaa M, et al. Overexpression and nuclear translocation of hypoxia-inducible factor prolyl hydroxylase PHD2 in head and neck squamous cell carcinoma is associated with tumor aggressiveness. Clin Cancer Res. 2006;12:1080–1087.
- Wu XH, Lu YF, Hu XD, et al. Expression of hypoxia inducible factor-1α and its significance in laryngeal carcinoma. J Int Med Res. 2010;38:2040–2046.
- Yasuda S, Arii S, Mori A, et al. Hexokinase II and VEGF expression in liver tumors: correlation with hypoxia-inducible factor 1 alpha and its significance. J Hepatol. 2004;40:117–123.
- Aquino-Galvez A, Gonzalez-Avila G, Delgado-Tello J, et al. Effects of 2-methoxyestradiol on apoptosis and HIF-1α and HIF-2α expression in lung cancer cells under normoxia and hypoxia. Oncol Rep. 2016;35:577–583.
- Slominski A, Kim TK, Brozyna AA, et al. The role of melanogenesis in regulation of melanoma behavior: Melanogenesis leads to stimulation of HIF-1α expression and HIF-dependent attendant pathways. Arch Biochem Biophys. 2014;563:79–93.
- Abraham S, Hu N, Jensen R. Hypoxia-inducible factor-1-regulated protein expression and oligodendroglioma patient outcome: comparison with established biomarkers and preoperative UCSF low-grade scoring system. J Neurooncol. 2012;108:459–468.
- Zhou ZL, Luo ZG, Yu B, et al. Increased accumulation of hypoxia-inducible factor-1α with reduced transcriptional activity mediates the antitumor effect of triptolide. Mol Cancer. 2010;9:268–278.
- Zapatero A, Morente M, de Vidales CM, et al. HIF1A expression in localized prostate cancer treated with dose escalation radiation therapy. Cancer Biomark. 2015;15:41–46.
- Klatte T, Seligson DB, Riggs SB, et al. Hypoxia-inducible factor 1α in clear cell renal cell carcinoma. Clin Cancer Res. 2007;13:7388–7393.
- Fletcher JW, Djulbegovic B, Soares HP, et al. Recommendations on the Use of 18F-FDG PET in Oncology. J Nucl Med. 2008;49:480–508.
- Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496:238–242.
- Marin-Hernandez A, Gallardo-Perez JC, Ralph SJ, et al. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. MRMC. 2009;9:1084–1101.
- Luo W, Semenza GL. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget. 2011;2:551–556.
- Parks SK, Chiche J, Pouyssegur J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat Rev Cancer. 2013;13:611–623.
- He GD, Jiang Y, Zhang B, et al. The effect of HIF-1α on glucose metabolism, growth and apoptosis of pancreatic cancerous cells. Asia Pac J Clin Nutr. 2014;23:174–180.
- Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol. 2011;76:347–353.
- Dang CV, Kim JW, Gao P, et al. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008;8:51–56.
- Fukuda R, Zhang H, Kim JW, et al. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122.
- Walls KC, Ghosh AP, Ballestas ME, et al. bcl-2/Adenovirus E1B 19-kd interacting protein 3 (BNIP3) regulates hypoxia-induced neural precursor cell death. J Neuropathol Exp Neurol. 2009;68:1326–1338.
- Ranjan K, Pathak C. Expression of cFLIPL Determines the Basal Interaction of Bcl‐2 With Beclin‐1 and Regulates p53 Dependent Ubiquitination of Beclin‐1 During Autophagic Stress. J Cell Biochem. 2016;117:1757–1768.
- Zhang C, Liu J, Wu R, et al. Tumor suppressor p53 negatively regulates glycolysis stimulated by hypoxia through its target RRAD. Oncotarget. 2014;5:5535–5546.
- Morfoisse F, Kuchnio A, Frainay C, et al. Hypoxia induces VEGF-C expression in metastatic tumor cells via a HIF-1α-independent translation-mediated mechanism. Cell Rep. 2014;6:155–167.
- Rigiracciolo DC, Scarpelli A, Lappano R, et al. Copper activates HIF-1α/GPER/VEGF signalling in cancer cells. Oncotarget. 2015;6:34158–34177.
- Azoitei N, Becher A, Steinestel K, et al. PKM2 promotes tumor angiogenesis by regulating HIF-1α through NF-κB activation. Mol Cancer. 2016;15:3–17.
- Rey S, Semenza GL. Hypoxia-inducible factor-1- dependent mechanisms of vascularization and vascular remodelling. Cardiovasc Res. 2010;86:236–242.
- Lee K, Zhang H, Qian DZ, et al. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci USA. 2009;106:17910–17915.
- Greco S, Martelli F. MicroRNAs in Hypoxia Response. Antioxid Redox Signal. 2014;21:1164–1166.
- Bartoszewska S, Kochan K, Piotrowski A, et al. The hypoxia-inducible miR-429 regulates hypoxia-inducible factor-1α expression in human endothelial cells through a negative feedback loop. Faseb J. 2015;29:1467–1479.
- Zhang S, Zhou X, Wang B, et al. Loss of VHL expression contributes to epithelial-mesenchymal transition in oral squamous cell carcinoma. Oral Oncol. 2014;50:809–817.
- Krishnamachary B, Zagzag D, Nagasawa H, et al. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006;66:2725–2731.
- Wong CC, Gilkes DM, Zhang H, et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci USA. 2011;108:16369–16374.
- Zhang H, Wong CC, Wei H, et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene. 2012;31:1757–1770.
- Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009;9:285–293.
- Oskarsson T, Batlle E, Massague J. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell. 2014;14:306–321.
- Peng G, Liu Y. Hypoxia-inducible factors in cancer stem cells and inflammation. Trends Pharmacol Sci. 2015;36:374–383.
- Conley SJ, Gheordunescu E, Kakarala P, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA. 2012;109:2784–2789.
- Samanta D, Gilkes DM, Chaturvedi P, et al. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci USA. 2014;111:5429–5438.
- Barak V, Pe’er J, Kalickman I, et al. VEGF as a biomarker for metastatic uveal melanoma in humans. Curr Eye Res. 2011;36:386–390.
- Kumar H, Choi DK. Hypoxia Inducible Factor Pathway and Physiological Adaptation: A Cell Survival Pathway? Mediators Inflamm. 2015;2015:584758.
- Santore MT, McClintock DS, Lee VY, et al. Anoxia-induced apoptosis occurs through a mitochondria-dependent pathway in lung epithelial cells. Am J Physiol Lung Cell Mol. Physiol. 2002;282:L727–L734.
- McClintock DS, Santore MT, Lee VY, et al. Bcl-2 family members and functional electron transport chain regulate oxygen deprivation-induced cell death. Mol Cell Biol. 2002;22:94–104.
- Yoo BH, Wu X, Derouet M, et al. Hypoxia-induced downregulation of autophagy mediator Beclin 1 reduces the susceptibility of malignant intestinal epithelial cells to hypoxia-dependent apoptosis. Autophagy. 2009;5:1166–1179.
- Li F, Chen X, Xu B, et al. Curcumin induces p53-independent necrosis in H1299 cells via a mitochondria-associated pathway. Mol Med Rep. 2015;12:7806–7814.
- Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33:207–214.
- Sermeus A, Michiels C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011;2:e164.
- Muh CR, Joshi S, Singh AR, et al. PTEN status mediates 2ME2 anti-tumor efficacy in preclinical glioblastoma models: role of HIF1α suppression. J Neurooncol. 2014;116:89–97.
- Kamat CD, Green DE, Warnke L, et al. Mutant p53 facilitates pro-angiogenic, hyperproliferative phenotype in response to chronic relative hypoxia. Cancer Lett. 2007;249:209–219.
- Katschinski DM, Le L, Schindler SG, et al. Interaction of the PAS B domain with HSP90 accelerates hypoxia-inducible factor-1α stabilization. Cell Physiol Biochem. 2004;14:351–360.
- Faubert B, Vincent EE, Griss T, et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc Natl Acad Sci USA. 2014;111:2554–2559.
- Pan JG, Mak TW. Metabolic targeting as an anticancer strategy: dawn of a new era? Sci STKE. 2007;381:e14.
- Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med. 2011;365:537–547.
- Velasco-Hernandez T, Hyrenius-Wittsten A, Rehn M, et al. HIF-1α can act as a tumor suppressor gene in murine acute myeloid leukemia. Blood. 2014;124:3597–3607.
- Maranchie JK, Vasselli JR, Riss J, et al. The contribution of VHL substrate binding and HIF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell. 2002;1:247–255.
- Zhang H, Li H, Xi HS, et al. HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood. 2012;119:2595–2607.
- Wang Y, Liu Y, Malek SN, et al. Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell. 2011;8:399–411.
- Näthke I, Rocha S. Antagonistic crosstalk between APC and HIF-1α. Cell Cycle. 2011;10:1545–1547.
- Kumar S, Vaidya M. Hypoxia inhibits mesenchymal stem cell proliferation through HIF1α-dependent regulation of P27. Mol Cell Biochem. 2016;415:29–38.
- Cai Z, Luo W, Zhan H, et al. Hypoxia-inducible factor 1 is required for remote ischemic preconditioning of the heart. Proc Natl Acad Sci USA. 2013;110:17462–17467.
- Karuppagounder SS, Ratan RR. Hypoxia-inducible factor prolyl hydroxylase inhibition: robust new target or another big bust for stroke therapeutics? J Cereb Blood Flow Metab. 2012;32:1347–1361.
- Zhang Y, Shao Z, Zhai Z, et al. The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PLoS One. 2009;4:6348–6356.
- Wigerup C, Påhlman S, Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther. 2016;164:152–169.
- Hu Y, Liu J, Huang H. Recent agents targeting HIF‐1α for cancer therapy. J Cell Biochem. 2013;114:498–509.
- Falchook GS, Wheler JJ, Naing A, et al. Targeting hypoxia-inducible factor-1α (HIF-1α) in combination with antiangiogenic therapy: a phase I trial of bortezomib plus bevacizumab. Oncotarget. 2014;5:10280–10292.
- Ban HS, Uto Y, Won M, et al. ( 2016;). Hypoxia-inducible factor (HIF) inhibitors: a patent survey (2011-2015). Expert Opin Ther Pat. 26:309–322.
- Shay JE, Imtiyaz HZ, Sivanand S, et al. Inhibition of hypoxia-inducible factors limits tumor progression in a mouse model of colorectal cancer. Carcinogenesis. 2014;35:1067–1077.
- Zhong H, D, Marzo AM, Laughner E, et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999;59:5830–5835.