
Abstract
Medulloblastoma (MB) is the most common malignant brain tumor occurring in children, and although high long-term survival rates have been reached with current therapeutic protocols, several neurological injuries are still observed among survivors. It has been shown that the development of MB is highly dependent on the microenvironment surrounding it and that the CXCL12 chemokine and its receptor, CXCR4 and the Sonic Hedgehog (SHH) pathway are crucial for cerebellar development, coordinating proliferation and migration of embryonic cells and malfunctions in these axes can lead to MB development. Indeed, the concomitant overactivation of these axes was suggested to define a new MB molecular subgroup. New molecules are being studied, aiming to inhibit either CXCR4 or the SHH pathways and have been tested in preclinical settings for the treatment of cancers. The use of these molecules could improve MB treatment and save patients from aggressive surgery, chemotherapy and radiotherapy regimens, which are responsible for severe neurological consequences. This review aims to summarize current data about the experimental inhibition of CXCR4 and SHH pathways in MB and its potential implications in treatment of this cancer.
Introduction
Central nervous system (CNS) tumors are the most common pediatric solid neoplasm and are the major causes of deaths from pediatric cancers [Citation1]. Among these, medulloblastoma (MB) is one of the most frequent and is recognized as a fast-growing tumor that origins from brain cells in the cerebellum. More than 70% of MB cases are diagnosed in children between 5 and 7 years of age, and 80% of patients are younger than 15 years [Citation2]; the incidence of medulloblastoma is approximately 0.7 per 100,000 children per year [Citation3] and is more common in male than female children. MB is a rare disease among adults, with an annual incidence rate of 0.05 per 100,000 per year, which represents fewer than 3% of all the primary neoplasms of the CNS [Citation4]. With surgery, radiotherapy, and high-dose chemotherapy, the 5-year survival rate for MB reaches 75% to 85% [Citation5] but the patients’ survival depends on several factors, including the disease stage, molecular subgroup and age at diagnosis [Citation6].
According to the World Health Organization classification [Citation7] and the International Medulloblastoma Working Group [Citation8], histological variants of MB are recognized as including the classic, the desmoplastic/nodular medulloblastoma with extensive nodularity and the large cell/anaplastic MB subtypes.
Risk stratification is based on the patient's age, metastasis, extent of resected tissue and anaplasia on histology. Patients older than 3 years with localized lesions, no anaplasia, no metastasis and residual tumor below 1.5 cm2 have good predicted outcome and are classified as the standard risk group. The high-risk group with poor outcome prognosis consists of very young children with dispersed metastasis and unresectable or residual tumor after surgery over 1.5 cm2.
Currently, the treatment is risk-adapted. Maximum surgical resection is recommended, always followed by chemotherapy and neuroaxis radiotherapy. Although high 5-year survival rates have been reached through current therapeutic protocols, MB patients are at high risk of relapse or developing secondary malignancies, have low long-term survival and often develop neurological sequelae resulting from the tumor itself or from the treatment [Citation9]. Furthermore, currently, the impact of intratumoral heterogeneity driving MB evolution, metastasis and resistance to cytotoxic chemotherapy is only beginning to be understood [Citation10]. Thus, patients’ stratification for treatment should include, beyond progression risk classification, measures of the disease aggressiveness and assessment of intrinsic markers governing MB development. This highlights the importance of molecular research which leads to MB subgroup classification and targets identification [Citation11].
Gene expression profile data indicates that MB comprises distinct molecular subgroups that are likely to have different cellular origins and driving-mutations, namely: wingless (WNT; group 1), sonic hedgehog (SHH; group 2), group 3 (v-myc avian myelocytomatosis viral oncogene homolog [MYC] amplified) and group 4. Patient demographics, histology, DNA copy-number aberrations and prognosis generally differ between these 4 molecular subgroups [Citation12].
Molecular subgroups are better determinants of the prognosis of MB patients than histopathological types and may be used to guide the therapy of MB [Citation13]. In a study performed by Jiang, Zhang et al. (2017), MB in Chinese patients aged between 3 to 18 years, the nonmetastasized group 4 had an excellent prognosis and chemotherapy maintenance did not significantly affect these patients. The SHH group and group 3 patients had worse prognosis suggesting that these patients should receive more intensive treatment, while WNT MB had the most favorable prognosis compared to the other subgroups and SHH MB had an intermediate prognosis.
In this review, we will focus on the SHH molecular subgroup, that accounts for ∼30% of all MBs and is characterized by activated Hedgehog (HH) signaling [Citation14]. Individuals with germline mutations in the gene encoding the SHH receptor Patched (PTCH) and the SHH inhibitor suppressor of fused (SUFU) are predisposed to childish MB [Citation15,Citation16]. Furthermore, somatic mutations in the genes encoding SHH co-receptor and Smoothened (SMO), as well as amplification of the genes encoding the Glioma-Associated 1 and 2 (GLI1 and GLI2) transcription factors have been found in sporadic MB [Citation14].
During the development of the nervous system, populations of progenitor cells that eventually become neurons and glia face the complex task of finding their way from their place of birth to their final destinations. Chemokines and their receptors are of crucial importance in directing this process, coordinating the proliferation and migration of immature neurons, glia and their precursors. Furthermore, chemokine system continues to be important in driving neuroplasticity throughout adult life, and chemokines participate in various brain disorders, neuroinflammatory diseases and tumors [Citation17].
Diverse chemokine and chemokine receptors were shown to be active in human CNS [Citation18], among them, the CXCL12/CXCR4 axis plays important roles coordinating migration of embryonal cells [Citation19–21] and axonal elongation [Citation22]. Thus, malfunction in this pathway may lead to cerebellar development diseases, including MB. Indeed, the synergistic activation of CXCR4 and SHH was suggested to define a new MB subgroup with poor prognosis [Citation23].
Based on their importance in the pathogenesis of different tumors, CXCR4 and SHH pathways were pointed to as promising targets for cancer therapy, and many therapeutic strategies targeting them are currently under development [Citation24,Citation25]. The use of these new therapies could improve MB treatment and save patients from aggressive surgery, radio and chemotherapy. Therefore, the present review aims to compile data regarding the potential usefulness of these novel therapies in MB.
SHH signaling overview
Hedgehog (HH) gene was initially discovered in 1980 in genetic screenings in Drosophila melanogaster as an important morphogen affecting embryonic segmentation and polarization [Citation26]. Soon after, three HH homologs were identified in vertebrates, namely Indian Hedgehog (IHH), Desert Hedgehog (DHH) and Sonic Hedgehog (SHH) [Citation27–30].
HH production depends on multiple steps of post-translational transformations. HH proteins undergo the secretory pathway during their production and once in endoplasmic reticulum (ER) they undergo an autoproteolytic cleavage catalyzed by its C-terminal portion, resulting in the attachment of a cholesterol molecule to the carboxyl terminus of N-terminal portion of the protein. Subsequently, the Skinny Hedgehog (SKI) acyltransferase attaches a palmitic acid group to the most N-terminal cysteine residue of this newly-generated peptide [Citation31].
The double lipidation of the N-terminal HH portion permits its attachment to ER membrane and subsequently to the plasma membrane, were it remains attached until its release either as a monomer by the action of Dispatched (DISP) and Signal peptide-CUB-EGF domain-containing protein 2 (SCUBE2); as a soluble multimer; in association with lipoprotein particles; or on the membrane of exovesicles. Either of these possible secretion modes influence HH diffusion into tissues, and have crucial importance in coordinating its’ actions as a morphogen [Citation32].
In vertebrates, the key components of hedgehog signaling are the HH ligands, the receptors Patched 1 (PTCH1) and Smoothened (SMO), which are enriched in primary cilia, and the final effectors Glioma-associated gene homologs (GLIs) transcription factors. In the absence of HH ligands, the 12-transmembrane protein PTCH1 represses the activity of the seven-transmembrane protein SMO by a mechanism that is yet unclear [Citation32]. Recent evidence suggests that PTCH1 is a transporter that uses osmotic gradient of Na+ to deprive SMO from inner membrane-bound cholesterol, which is essential for its’ activation, or to pump inside the cell an yet unknown SMO inhibitor [Citation33].
There are three GLI isoforms in vertebrates, namely GLI1, GLI2 and GLI3 [Citation34]. All GLI transcription factor poses the same DNA-binding specificity and have a C-terminal activation domain. GLI2 and GLI3 also have a N-terminal repressor domain and may function either as positive or negative transcriptional regulators depending on post-transcriptional and post-translational processing [Citation35]. In the absence of SMO signaling, the SUFU protein retains GLI transcription factor attached to microtubules, thus inhibiting its signaling to the nucleus [Citation36–38]. Furthermore, GLI2 and GLI3 are phosphorylated in multiple sites on its’ C-terminal domain and proteolytically processed to their repressor forms [Citation39–43].
Upon binding of HH ligands to PTCH1 this protein exit primary cilia base, is internalized and degraded, releasing SMO signaling. Despite SMO is a member of the G protein-coupled receptor (GPCR) family, there is still controversies on whether G proteins activation is required for classical SMO signaling [Citation32].
Once released for signalization by HH inhibition of PTCH1, SMO becomes enriched in primary cilia, where it is phosphorylated by GPCR kinase 2 (GRK2) and casein kinase 1 alpha 1 (CSNK1A1) and gated to cilia associated with β-arrestin and the kinesin superfamily protein (KIF) 3A, leading to retention of SUFU and GLI2 and GLI3 dissociation of SUFU-GLI complex, and translocation of full length active form of GLIs to the nucleus mediated by the KIF3. In cell nucleus, GLI transcription factors activate the expression of antiapoptotic genes (such as BCL2) and genes involved in proliferation (such as Cyclins D1, D2 and E and MYCN) [Citation44,Citation45], angiogenesis (such as ANGPT1, ANGPT2 and VEGFA) [Citation46,Citation47], epithelial-to-mesenchymal transition (such as SNAIL1) [Citation48,Citation49] and stem cell maintenance (such as NANOG and SOX2) [Citation50].
Besides cytoplasmic regulation, GLI function can be also regulated by post-translational modifications in the nucleus. The nuclear-localized serine/threonine kinase Dyrk1 phosphorylates GLI1 on multiple sites enhancing its’ transcriptional activity and retention in the nucleus () [Citation51]. Moreover, it has been shown that GLIs can also be regulated through acetylation, which reduces GLI attachment to chromatin and prevents the upregulation of key genes, and that histone deacetylases HDAC1 and HDAC2 counteracts this regulatory mechanism, eliciting stronger GLI responses by direct deacetylation of GLI proteins () [Citation52–54].
Figure 1. CXCR4 and SHH pathways crosstalk and druggable targets. SHH and CXCR4 signaling pathways, their crosstalk, as well as molecular targets of drugs described in the text are shown. The intricate connection among these pathways with multiple crossing points among them, suggests that compensatory mechanisms may be involved in therapeutic resistance for treatments targeting each of them, and makes the dual targeting of both routes an attractive therapeutic strategy. AC: Adenylate cyclase; AKT: Protein kinase B; AMP: adenosine monophosphate; cAMP: Cyclic adenosine monophosphate; CXCL12: CXC chemokine ligand 12; CXCR4: CXC chemokine receptor 4; Dyrk1: Dual Specificity Tyrosine-(Y)-Phosphorylation Regulated Kinase 1; ER: Endoplasmic reticulum; GLI: Glioma-associated gene homolog; Gi: Guanine nucleotide binding protein from I family; HH: Hedgehog; HDAC1/2: Histone deacetylase 1 and 2; IP3: Inositol 1,4,5-triphosphate; JAK1/2: Janus kinase 1 and 2; MK: Mitogen activated protein kinase (MAPK); MKK: MAPK kinase (MAPKK); MKKK: MAPKK kinase; PI3K: Phosphatidylinositol 3-kinase; PiP2: Phosphatidylinositol 4,5-biphosphate; PiP3: Phosphatidylinositol 3,4,5-triphosphate; PKA: Protein kinase A; PKC: Protein kinase C; PLC: Phospholipase C; PTCH1: Patched 1 receptor; RAS: Rat sarcoma virus oncogene homolog protein; SMO: smoothened receptor; STAT: Signal transducer and activator of transcription; SUFU: Suppressor of fused homolog; TF: transcription factor.
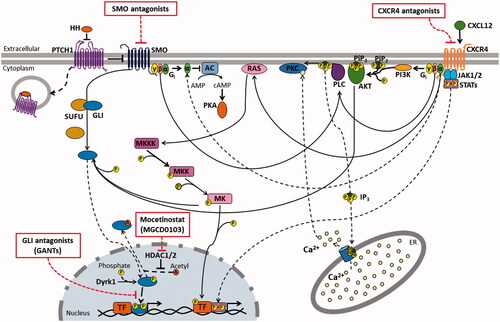
Beyond the above-described classical HH pathway, alternative pathways related to this protein might be activated, which may relate to SMO-independent activation of GLIs [Citation55] (I), to GLI-independent actions of SMO (II) and to PTCH1 actions beyond SMO inhibition (III), adding much more complexity to the spectrum of HH-signaling effects [Citation24,Citation56]:
(I) GLI transcription factors might be activated by diverse pathways beyond that mediated by SMO [Citation24,Citation55], such as K-RAS [Citation57,Citation58] and PI3K/AKT [Citation59–61] (); and p53 has been shown to be a negative regulator of GLIs [Citation62,Citation63].
(II) SMO has also been shown to activate multiple processes that do not end in the activation of GLIs, such as the formation of stress fibers and migration stimulus through the activation of Rho-family small GTPases [Citation64,Citation65] and arachidonic acid metabolism [Citation66], the activation of Src-kinase family members [Citation67,Citation68], and the control of intracellular Ca2+ levels through a mechanism that involves a Gi protein, phospholipase C-γ and inositol triphosphate (IP3) generation () [Citation69].
(III) Ultimately, PTCH1 is capable to induce apoptosis and restrain cell cycle progression independently of SMO regulation through its’ cytoplasmic C-terminal domain [Citation70,Citation71] and by directly binding activated Cyclin B1/CDK1 complexes retaining them in the cytoplasm [Citation72–74], respectively.
CXCR4 pathway
CXCR4 is a GPCR and belongs to the C-X-C family of chemokine receptors, and upon the binding of its ligand, CXCL12, it activates several pathways related to chemotaxis, survival, proliferation and apoptosis in a cell- and context-dependent manner [Citation75].
As a GPCR, CXCR4 actions are primarily mediated by trimeric G-proteins, composed by Gα, Gβ and Gγ subunits; and as a chemokine receptor, CXCR4 acts mainly through the Gi class of G-proteins, in which Gαi subunit functions to inhibit the production of cyclic AMP (cAMP) by adenylate cyclase, while Gβγi dimer mainly activates Phospholipase C-β (PLC-β), which generates IP3 and DAG from the plasma membrane, releasing Ca2+ from intracellular stores, and ultimately resulting in the activation of signaling and motor proteins in cellular cytoplasm, favoring cell motility () [Citation75].
PI3K is also activated by CXCR4 signaling through both Gi-protein Gαi subunit or Gβγi dimer and activates focal adhesion components, involved in cell migration, such as focal adhesion kinase, proline-rich kinase-2 (Pyk-2), and Crk, which is an adapter protein that activates the JNK MAPK [Citation76]. PI3K activation also leads to the activation of other kinases such as AKT, which is related to survival processes [Citation77]. Other proteins are involved in the chemotactic process elicited through CXCR4 activation, such as PKC, which is activated by PLC-β-generated DAG [Citation76], and Erk1/2 MAPKs, which are activated by Gαi () [Citation77].
There are also CXCR4-activated pathways that are independent of Gi protein. It has been suggested that calcium mobilization may also be a result of CXCR4 activation of Gs-, Go- and Gq-proteins [Citation78]. Furthermore, other pathways may be also activated by CXCR4 beyond those mediated by G-proteins. JAK2 and JAK3 were shown to associate with CXCR4 and to activate multiple STAT transcription factors () [Citation79]. Other kinases involved in lymphocyte activation were also shown to be activated by CXCR4 signaling, among them kinases Lyn [Citation80], Fyn, Src, Lck [Citation81], and ZAP-70 [Citation82,Citation83].
Thus, a plethora of signaling pathways may be activated by CXCR4, and the context- and cell-dependent pattern of their activation is an important issue to be evaluated in future studies, holding promising clues to the understanding of diverse physiological and pathological processes.
Of note, many molecules and pathways activated by CXCR4 overlap with those from SHH pathway, indicating that these pathways may have synergistic effects and that their crossing-points might have a compensatory role in therapeutic resistance in treatments targeting either of them ().
Sonic hedgehog (SHH) pathway and CXCL12/CXCR4 axis in MB
The SHH subgroup is the most studied and well defined among the subtypes of MB [Citation84]. Most infant and adult MBs are SHH-driven tumors [Citation85]. The SHH pathway exerts various roles in a developing embryo, acting mainly on skeleton, intestine and lung development [Citation86]. It is involved in the development of the nervous system as well, acting specifically on oligodendrocytes and in the control of axial growing [Citation87].
During cerebellar development, Purkinje cell-derived SHH induces proliferation of an external granule layer (EGL) [Citation88]. In the postnatal period, SHH enter a state of down-regulation in the central nervous system, being activated only for homeostasis of other systems. Regulation failures can lead to uncontrolled activation of the SHH pathway, leading to tumor formation, such as in MB [Citation89,Citation90]. In this context, this pathway is a promising target for MB treatment.
During the embryonic period, CXCR4 is expressed in cerebellar progenitor cells [Citation91] and promotes the maximal proliferation of these cells and migration during the postnatal period [Citation92,Citation93].
Studies have shown that CXCR4 is overexpressed in MB [Citation94,Citation95]. The maximum proliferation rate of granular neuron precursors in MB is associated with overexpression of both SHH and CXCR4, which contribute to each other in a synergistic manner, increasing the severity of the disease, and leading to higher mortality [Citation96].
In a study conducted by Sengupta, Dubuc [Citation23] a new MB molecular subgroup which was characterized by the coactivation of SHH and CXCR4 pathways was described. Therefore, the use of CXCR4 inhibitors may also be valuable in MB treatment.
Treatments targeting CXCL12/CXCR4 and/or SHH
MB shows great variability in clinical outcome among its molecular subgroups and treatments focusing on molecular targets specific for each subgroup could potentially increase the survival rates of MB patients [Citation97]. Therefore, knowledge about the molecular basis of the disease is extremely important.
CXCR4 and SHH pathways are therapeutic targets validated in many cancer types, including MB. Ward, Warrington [Citation98] verified that inhibition of the SHH and CXCR4 pathways exerts potent antitumor effects in a murine model of SHH-MB subgroup. Moreover, this study provides a rationale to evaluate combinatorial inhibition of SHH and CXCR4 in patients with MB as well as other cancers driven by SHH that co-express high levels of CXCR4. The shows some SHH and CXCR4 pathway inhibitors evaluated in both clinical and preclinical studies focusing on MB treatment.
Table 1. SHH and CXCR4 inhibitors in clinical and preclinical trials phases for MB.
Dysfunction of the CXCL12/CXCR4 axis contributes to several human pathologies, including cancer and inflammatory diseases. Consequently, inhibition of this axis is suggested as an attractive target for therapeutic intervention. There are several CXCR4 inhibitors, such as AMD3100 (plerixafor, nonpeptidic CXCR4 antagonist), AMD3465 (non-peptidic CXCR4 antagonist) and CTCE-9908 (CXCL12 peptidic analogue) [Citation119]. Although these molecules act by direct inhibition of CXCR4 () and show efficacy when compared to other chemotherapeutic agents, their toxic effects are not well understood yet [Citation98].
AMD3100 is widely recognized as having broad applications in clinics, such as a bone marrow stem cell mobilizer in transplantation protocols, for which this compound already holds Food and Drug Administration U.S. (FDA) approval. Diverse other clinical trials are currently in course for oncology applications, and small clinical studies have shown its efficacy in combination with other chemotherapeutics for some cancers [Citation120]. In preclinical settings both AMD3100 and AMD3465 showed promising results against models of cerebral tumors, such as glioblastoma [Citation121] and MBs [Citation117,Citation118].
On the other hand, there are promising data about MB treatments targeting the SHH pathway [Citation122]. Since the discovery of the association between SHH hyperactivation and oncogenesis, tumor progression, and drug resistance, pharmacological inhibitors of this pathway have been developed, such as SMO inhibitors and GLI protein antagonists () [Citation123].
Currently, there are numerous SMO inhibitors being tested as potential anticancer agents [Citation24], some of which have been reported to have some efficacy against MB. However, there is still a need for convincing results regarding SHH inhibitors on this disease [Citation124–126].
Among promising inhibitors, GDC-0449/Vismodegib and NVP-LDE225/Erismodegib were approved by the FDA for the treatment of basal cell carcinoma [Citation127]. However, SHH-MB patients treated with GDC-0449 have shown rapid development of drug resistance and tumor relapse due to novel SMO mutations [Citation128]. Moreover, a subset of patients did not respond to GDC-0449 since mutations were localized downstream of SMO [Citation103].
Compounds with a direct binding ability and GLI activity inhibition, such as GANT61 [Citation129], ATO [Citation130,Citation131] and GlaB [Citation132] have shown efficacy against SHH-MB growth in preclinical models. However, toxicity and specificity are still being investigated for these compounds and further pharmacological studies are still required before clinical trials can be started [Citation133]. GANT61 have already demonstrated promising results in many cancer cell lines, such as squamous lung cancer [Citation134], alveolar rhabdomyosarcoma [Citation135], neuroblastoma [Citation136], MB [Citation114], glioma [Citation137], breast cancer [Citation138], myeloid leukemia [Citation139] and renal carcinoma [Citation61] in which it has been tested in preclinical research.
Alternatively, indirect inhibitors, mostly affecting GLI post-translational modifications, or inhibitors of key pathways regulated by HH/GLI, could be used for the same purpose [Citation140,Citation141]. In mouse models, selective inhibition histone deacetylases (HDAC1 and HDAC2) counteracts SHH-MB growth. These two deacetylases are upregulated in tumor and their knockdown inhibits HH signaling and decreases tumor growth. A selective HDAC1/HDAC2 inhibitor, Mocetinostat (MGCD0103), is a potent HH inhibitor that enhances GLI1 acetylation at K518, inhibiting GLI1 accession to the chromatin and promoting nuclear exclusion (), resulting in reduced proliferation and increased apoptosis of tumor cells and prolonged survival in a murine model of MB. Collectively, these data demonstrate the preclinical efficacy of targeting the downstream HDAC1/2-GLI1 acetylation in the treatment of SHH-MB [Citation53].
Therefore, further studies are needed to understand the mechanisms of resistance to SMO inhibitors, and how to overcome them. Case reports from MB patients treated with GDC-0449 indicate two specific SMO mutations that promotes treatment resistance [Citation128]. The SMO G497W mutation results in a partial blocking of the drug entry site while the SMO D437H mutation totally disrupts hydrogen bond stability in the binding site in patients with basal cell carcinoma [Citation142,Citation143]. Further development of drugs such as MRT-92, which binds multiple sites on SMO and shows activity against the SMO D473H mutant, is crucial to overcome treatment-induced resistance [Citation144].
Recently, Ward, Warrington [Citation98] reported that dual inhibition of SHH and CXCR4 pathways with GDC-0449 and AMD3100, respectively, showed synergistic effects in a murine model of SHH-subgroup of MB, with more potent antitumor activity than each inhibition alone. This finding is of great importance given that cancers treated with GDC-0449 alone often develop resistance due to mutations in SMO or other SHH signaling molecules that render it insensitive to this agent [Citation128,Citation142,Citation145] or due to unknown causes [Citation103], which might be attributable to overactivation of other compensatory pro-oncogenic pathways, such as CXCR4. Moreover, the recent identification of an MB poor prognosis subtype based on SHH and CXCR4 hyperactivation [Citation23] may indicate a subgroup of patients that might benefit from this therapeutic strategy.
Conclusion and future perspectives
Identification of SHH and CXCR4 as oncogenic drivers of MB pathophysiology has opened new avenues for the development of new treatments for this cancer, which still faces severe challenges concerning patients’ survival rate, long-term, overall and disease-free survival and life quality after treatment through current therapeutic protocols, especially for poor-prognosis MB subgroups. The development of new therapies targeting these pathways may fill this gap by enhancing treatment effectiveness while diminishing its aggressiveness. The implementation of these therapies in clinical practice will depend on the results of basic research, pre-clinical studies and clinical trials for development and assessment of efficacy and toxicity and dose refinement of these newly described agents.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Eberhart CG, Kepner JL, Goldthwaite PT, et al. Histopathologic grading of medulloblastomas. A Pediatric Oncology Group Study. Cancer. 2002;94:552–560.
- Hershatter BW, Halperin EC, Cox EB. Medulloblastoma: the Duke University Medical Center experience. Int J Radiat Oncol Biol Phys. 1986;12:1771–1777.
- PG F. Embryonal tumors. New York: Springer; 2004. (Gupta N BA, Haas-Kogan D, editor. Pediatric CNS Tumors).
- Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109.
- Gajjar A, Chintagumpala M, Ashley D, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7:813–820.
- ASCO ASoCO. Medulloblastoma – Childhood: Statistics. CancerNet Editorial Board. 2016.
- Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131:803–820.
- Gottardo NG, Hansford JR, McGlade JP, et al. Medulloblastoma Down Under 2013: a report from the third annual meeting of the International Medulloblastoma Working Group. Acta Neuropathol. 2014;127:189–201.
- Crawford JR, MacDonald TJ, Packer RJ. Medulloblastoma in childhood: new biological advances. Lancet Neurol. 2007;6:1073–1085.
- Ramaswamy V, Taylor MD. Medulloblastoma: From Myth to Molecular. J Clin Oncol. 2017;35:2355–2363.
- Gilbertson RJ. Medulloblastoma: signalling a change in treatment. Lancet Oncol. 2004;5:209–218.
- McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of hedgehog signaling. Curr Top Development Biol. 2003;53:1–114.
- Yu J, Zhao R, Shi W, et al. Risk factors for the prognosis of pediatric medulloblastoma: a retrospective analysis of 40 cases. Clinics. 2017;72:294–304.
- Northcott PA, Hielscher T, Dubuc A, et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011;122:231–240.
- Taylor MD, Liu L, Raffel C, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002;31:306–310.
- Taylor MD, Mainprize TG, Rutka JT. Molecular insight into medulloblastoma and central nervous system primitive neuroectodermal tumor biology from hereditary syndromes: a review. Neurosurgery. 2000;47:888–901.
- Tran PB, Miller RJ. Chemokine receptors: signposts to brain development and disease. Nat Rev Neurosci. 2003;4:444–455.
- de Haas AH, van Weering HR, de Jong EK, et al. Neuronal chemokines: versatile messengers in central nervous system cell interaction. Mol Neurobiol. 2007;36:137–151.
- Bagri A, Gurney T, He X, et al. The chemokine SDF1 regulates migration of dentate granule cells. Development. 2002;129:4249–4260.
- Zhu Y, Yu T, Zhang XC, et al. Role of the chemokine SDF-1 as the meningeal attractant for embryonic cerebellar neurons. Nat Neurosci. 2002;5:719–720.
- Zou YR, Kottmann AH, Kuroda M, et al. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–599.
- Ohshima Y, Kubo T, Koyama R, et al. Regulation of axonal elongation and pathfinding from the entorhinal cortex to the dentate gyrus in the hippocampus by the chemokine stromal cell-derived factor 1 alpha. J Neurosci. 2008;28:8344–8353.
- Sengupta R, Dubuc A, Ward S, et al. CXCR4 activation defines a new subgroup of Sonic hedgehog-driven medulloblastoma. Cancer Res. 2012;72:122–132.
- Rimkus TK, Carpenter RL, Qasem S, et al. Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers (Basel). 2016;8. DOI: https://doi.org/10.3390/cancers8020022.
- Wang Y, Xie Y, Oupický D. Potential of CXCR4/CXCL12 Chemokine Axis in Cancer Drug Delivery. Curr Pharmacol Rep. 2016;2:1–10.
- Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801.
- Chang DT, Lopez A, von Kessler DP, et al. Products, genetic linkage and limb patterning activity of a murine hedgehog gene. Development. 1994;120:3339–3353.
- Echelard Y, Epstein DJ, St-Jacques B, et al. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–1430.
- Riddle RD, Johnson RL, Laufer E, et al. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;75:1401–1416.
- Roelink H, Augsburger A, Heemskerk J, et al. Floor plate and motor neuron induction by vhh-1, a vertebrate homolog of hedgehog expressed by the notochord. Cell. 1994;76:761–775.
- Chen X, Tukachinsky H, Huang CH, et al. Processing and turnover of the Hedgehog protein in the endoplasmic reticulum. J Cell Biol. 2011;192:825–838.
- Briscoe J, Thérond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14:416–429.
- Myers BR, Neahring L, Zhang Y, et al. Rapid, direct activity assays for Smoothened reveal Hedgehog pathway regulation by membrane cholesterol and extracellular sodium. Proc Natl Acad Sci USA. 2017;114:E11141–E11150.
- Ruppert JM, Kinzler KW, Wong AJ, et al. The GLI-Kruppel family of human genes. Mol Cell Biol. 1988;8:3104–3113.
- Sasaki H, Nishizaki Y, Hui C, et al. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1999;126:3915–3924.
- Cheng SY, Bishop JM. Suppressor of Fused represses Gli-mediated transcription by recruiting the SAP18-mSin3 corepressor complex. Proc Natl Acad SciUSA. 2002;99:5442–5447.
- Kogerman P, Grimm T, Kogerman L, et al. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999;1:312–319.
- Paces-Fessy M, Boucher D, Petit E, et al. The negative regulator of Gli, Suppressor of fused (Sufu), interacts with SAP18, Galectin3 and other nuclear proteins. The. Biochem J. 2004;378:353–362.
- Jia J, Amanai K, Wang G, et al. Shaggy/GSK3 antagonizes hedgehog signalling by regulating cubitus interruptus. Nature. 2002;416:548–552.
- Price MA, Kalderon D. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by glycogen synthase kinase 3 and casein kinase 1. Cell. 2002;108:823–835.
- Wang B, Li Y. Evidence for the direct involvement of {beta}TrCP in Gli3 protein processing. Proc Natl Acad Sci USA. 2006;103:33–38.
- Tempe D, Casas M, Karaz S, et al. Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3 ubiquitination by SCFbetaTrCP. Mol Cell Biol. 2006;26:4316–4326.
- Kaesler S, Luscher B, Ruther U. Transcriptional activity of GLI1 is negatively regulated by protein kinase A. Biol Chem 2000;381:545–551.
- Kenney AM, Rowitch DH. Sonic hedgehog promotes G1 cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol. 2000;20:9055–9067.
- Kenney AM. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130:15–28.
- Chen W, Tang T, Eastham-Anderson J, et al. Canonical hedgehog signaling augments tumor angiogenesis by induction of VEGF-A in stromal perivascular cells. Proc Natl Acad Sci. 2011;108:9589–9594.
- Pola R, Ling LE, Silver M, et al. The morphogen Sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat Med. 2001;7:706–711.
- Heiden KB, Williamson AJ, Doscas ME, et al. The sonic hedgehog signaling pathway maintains the cancer stem cell self-renewal of anaplastic thyroid cancer by inducing snail expression. J Clin Endocrinol Metabol. 2014;99:E2178–E2187.
- Li X, Deng W, Lobo-Ruppert SM, et al. Gli1 acts through Snail and E-cadherin to promote nuclear signaling by β-catenin. Oncogene. 2007;26:4489–4498.
- Po A, Ferretti E, Miele E, et al. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. Embo J. 2010;29:2646–2658.
- Mao J, Maye P, Kogerman P, et al. Regulation of Gli1 transcriptional activity in the nucleus by Dyrk1. J Biol Chem. 2002;277:35156–35161.
- Castresana JS, Coni S, Antonucci L, et al. Gli2 acetylation at lysine 757 regulates hedgehog-dependent transcriptional output by preventing its promoter occupancy. PLoS One. 2013;8:e65718.
- Coni S, Mancuso AB, Di Magno L, et al. Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH Medulloblastoma. Sci Rep. 2017;7:44079.
- Canettieri G, Di Marcotullio L, Greco A, et al. Histone deacetylase and Cullin3–RENKCTD11 ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010;12:132–142.
- Stecca B, Ruiz i Altaba A. Context-dependent regulation of the GLI code in cancer by HEDGEHOG and Non-HEDGEHOG signals. J Mol Cell Biol. 2010;2:84–95.
- Brennan D, Chen X, Cheng L, et al. Noncanonical Hedgehog signaling. Vitam Horm. 2012;88:55–72.
- Seto M, Ohta M, Asaoka Y, et al. Regulation of the hedgehog signaling by the mitogen-activated protein kinase cascade in gastric cancer. Mol Carcinog. 2009;48:703–712.
- Rajurkar M, De Jesus-Monge WE, Driscoll DR, et al. The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc Natl Acad Sci.. 2012;109:E1038–E1047.
- Ramaswamy B, Lu Y, Teng K, et al. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res. 2012;72:5048–5059.
- Ke Z, Caiping S, Qing Z, et al. Sonic hedgehog–Gli1 signals promote epithelial–mesenchymal transition in ovarian cancer by mediating PI3K/AKT pathway. Med Oncol. 2015;32:368.
- Zhou J, Zhu G, Huang J, et al. Non-canonical GLI1/2 activation by PI3K/AKT signaling in renal cell carcinoma: A novel potential therapeutic target. Cancer Lett. 2016;370:313–323.
- Yoon JW, Lamm M, Iannaccone S, et al. p53 modulates the activity of the GLI1 oncogene through interactions with the shared coactivator TAF9. DNA Repair. 2015;34:9–17.
- Stecca B, Ruiz i Altaba A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. Embo J. 2009;28:663–676.
- Chinchilla P, Xiao L, Kazanietz MG, et al. Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle. 2010;9:570–579.
- Polizio AH, Chinchilla P, Chen X, et al. Heterotrimeric Gi proteins link Hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J Biol Chem. 2011;286:19589–19596.
- Bijlsma MF, Borensztajn KS, Roelink H, et al. Sonic hedgehog induces transcription-independent cytoskeletal rearrangement and migration regulated by arachidonate metabolites. Cell Signal. 2007;19:2596–2604.
- Yam PT, Langlois SD, Morin S, et al. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron. 2009;62:349–362.
- Chang H, Li Q, Moraes RC, et al. Activation of Erk by sonic hedgehog independent of canonical hedgehog signalling. Int J Biochem Cell Biol. 2010;42:1462–1471.
- Belgacem YH, Borodinsky LN. Sonic hedgehog signaling is decoded by calcium spike activity in the developing spinal cord. Proc Natl Acad Sci USA. 2011;108:4482–4487.
- Mille F, Thibert C, Fombonne J, et al. The patched dependence receptor triggers apoptosis through a DRAL-caspase-9 complex. Nat Cell Biol. 2009;11:739–746.
- Thibert C, Teillet MA, Lapointe F, et al. Inhibition of neuroepithelial patched-induced apoptosis by sonic hedgehog. Science. 2003;301:843–846.
- Adolphe C, Hetherington R, Ellis T, et al. Patched1 functions as a gatekeeper by promoting cell cycle progression. Cancer Res. 2006;66:2081–2088.
- Jenkins D, Winyard PJ, Woolf AS. Immunohistochemical analysis of Sonic hedgehog signalling in normal human urinary tract development. J Anatomy. 2007;211:620–629.
- Jiang X, Yang P, Ma L. Kinase activity-independent regulation of cyclin pathway by GRK2 is essential for zebrafish early development. Proc Natl Acad Sci USA. 2009;106:10183–10188.
- Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927–2931.
- Wang JF, Park IW, Groopman JE. Stromal cell-derived factor-1alpha stimulates tyrosine phosphorylation of multiple focal adhesion proteins and induces migration of hematopoietic progenitor cells: roles of phosphoinositide-3 kinase and protein kinase C. Blood. 2000;95:2505–2513.
- Barbero S, Bonavia R, Bajetto A, et al. Stromal cell-derived factor 1alpha stimulates human glioblastoma cell growth through the activation of both extracellular signal-regulated kinases 1/2 and Akt. Cancer Res. 2003;63:1969–1974.
- Rubin JB. Chemokine signaling in cancer: One hump or two? Semin Cancer Biol. 2009;19:116–122.
- Vila-Coro AJ, Rodriguez-Frade JM, Martin De AA, et al. The chemokine SDF-1alpha triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. 1999;13:1699–1710.
- Nakata Y. Integrin inhibition through Lyn-dependent cross talk from CXCR4 chemokine receptors in normal human CD34+ marrow cells. Blood. 2006;107:4234–4239.
- Ghosh MC, Baatar D, Collins G, et al. Dexamethasone augments CXCR4-mediated signaling in resting human T cells via the activation of the Src kinase Lck. Blood. 2008;113:575–584.
- Ottoson NC, Pribila JT, Chan ASH, et al. Cutting Edge: T Cell Migration Regulated by CXCR4 Chemokine Receptor Signaling to ZAP-70 Tyrosine Kinase. J Immunol. 2001;167:1857–1861.
- Wu C-Y, Tsai Y-Y, Chen S-Y, et al. Interaction of Zap70 and CXCR4 receptor at lamellipodia that determines the directionality during Jurkat T cells chemotaxis. Mol Immunol. 2017;90:245–254.
- Markant SL, Esparza LA, Sun J, et al. Targeting sonic hedgehog-associated medulloblastoma through inhibition of Aurora and Polo-like kinases. Cancer Res. 2013;73:6310–6322.
- Robinson G, Parker M, Kranenburg TA, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–48.
- Riobo NA, Lu K, Emerson CP Jr. Hedgehog signal transduction: signal integration and cross talk in development and cancer. Cell Cycle. 2006;5:1612–1615.
- Heussler HS, Suri M. Sonic hedgehog. Mol Pathol: 2003;56:129–131.
- Roussel MF, Hatten ME. Cerebellum development and medulloblastoma. Curr Top Develop Biol. 2011;94:235–282.
- Sahebjam S, Siu LL, Razak AA. The utility of hedgehog signaling pathway inhibition for cancer. The Oncologist. 2012;17:1090–1099.
- Xie J, Bartels CM, Barton SW, et al. Targeting hedgehog signaling in cancer: research and clinical developments. OncoTargets Ther. 2013;6:1425–1435.
- Zhu Y, Yu T, Rao Y. Temporal regulation of cerebellar EGL migration through a switch in cellular responsiveness to the meninges. Develop Biol. 2004;267:153–164.
- Klein RS, Rubin JB, Gibson HD, et al. SDF-1 alpha induces chemotaxis and enhances Sonic hedgehog-induced proliferation of cerebellar granule cells. Development. 2001;128:1971–1981.
- Ozawa PM, Ariza CB, Ishibashi CM, et al. Role of CXCL12 and CXCR4 in normal cerebellar development and medulloblastoma. Int J Cancer. 2016;138:10–13.
- Akamandisa MP, Nahta R, Castellino RC. PPM1D/WIP1 in medulloblastoma. Oncoscience. 2016;3:186–187.
- Buss MC, Remke M, Lee J, et al. The WIP1 oncogene promotes progression and invasion of aggressive medulloblastoma variants. Oncogene. 2015;34:1126–1140.
- Huang SY, Yang JY. Targeting the Hedgehog pathway in pediatric medulloblastoma. Cancers (Basel). 2015;7:2110–2123.
- Ellison DW, Dalton J, Kocak M, et al. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011;121:381–396.
- Ward SA, Warrington NM, Taylor S, et al. Reprogramming medulloblastoma-propagating cells by a combined antagonism of sonic Hedgehog and CXCR4. Cancer Res. 2017;77:1416–1426.
- A Phase II Study of Oral LDE225 in Patients With Hedge-Hog (Hh)-Pathway Activated Relapsed Medulloblastoma (MB) [Internet]. 2012. Available from: https://clinicaltrials.gov/ct2/show/study/NCT01708174
- NCT01125800. A Phase I Dose Finding and Safety Study of Oral LDE225 in Children and a Phase II Portion to Assess Preliminary Efficacy in Recurrent or Refractory MB. ClinicalTrialsgov [Internet] Bethesda (MD): National Library of Medicine (US). 2010.
- NCT00880308. ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000 Feb 29. ClinicalTrialsgov [Internet] Bethesda (MD): National Library of Medicine (US) 2000 Feb 29. 2009.
- NCT01208831. Phase I, multicenter, open-label, dose-escalation study of sonidegib in Asian patients with advanced solid tumors. ClinicalTrialsgov [Internet] Bethesda (MD): National Library of Medicine (US) 2016.
- Robinson GW, Orr BA, Wu G, et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog–subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. JCO. 2015;33:2646–2654.
- NCT00822458. GDC-0449 in Treating Young Patients With Medulloblastoma That is Recurrent or Did Not Respond to Previous Treatment. ClinicalTrialsgov [Internet] Bethesda (MD): National Library of Medicine (US) 2000 Feb 29. 2009.
- Meyers-Needham M, Lewis JA, Gencer S, et al. Off-target function of the Sonic hedgehog inhibitor cyclopamine in mediating apoptosis via nitric oxide-dependent neutral sphingomyelinase 2/ceramide induction. Mol Cancer Ther. 2012;11:1092–1102.
- Lu X, Peng Y, Wang C, et al. Design, synthesis, and biological evaluation of optimized phthalazine derivatives as hedgehog signaling pathway inhibitors. Eur J Med Chem. 2017;138:384–395.
- Lee MJ, Hatton BA, Villavicencio EH, et al. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc Natl Acad SciUSA. 2012;109:7859–7864.
- Ge X, Milenkovic L, Suyama K, et al. Phosphodiesterase 4D acts downstream of Neuropilin to control Hedgehog signal transduction and the growth of medulloblastoma. eLife. 2015;4. DOI: https://doi.org/10.7554/eLife.07068.
- Wang J, Peng Y, Liu Y, et al. Berberine, a natural compound, suppresses Hedgehog signaling pathway activity and cancer growth. BMC Cancer. 2015;15:595.
- Wang X, Wang N, Li H, et al. Up-Regulation of PAI-1 and down-regulation of uPA are involved in suppression of invasiveness and motility of hepatocellular carcinoma cells by a natural compound berberine. IJMS. 2016;17:577.
- Li B, Fei DL, Flaveny CA, et al. Pyrvinium attenuates Hedgehog signaling downstream of smoothened. Cancer Res. 2014;74:4811–4821.
- Rohner A, Spilker ME, Lam JL, et al. Effective targeting of Hedgehog signaling in a medulloblastoma model with PF-5274857, a potent and selective smoothened antagonist that penetrates the blood-brain barrier. Mol Cancer Ther. 2012;11:57–65.
- Lu W, Liu Y, Ma H, et al. Design, synthesis, and structure-activity relationship of tetrahydropyrido[4,3-d]pyrimidine derivatives as potent smoothened antagonists with in vivo activity. ACS Chem Neurosci. 2017;8:1980–1994.
- Lin Z, Li S, Sheng H, et al. Suppression of GLI sensitizes medulloblastoma cells to mitochondria-mediated apoptosis. J Cancer Res Clin Oncol. 2016;142:2469–2478.
- Lin Z, Sheng H, You C, et al. Inhibition of the CyclinD1 promoter in response to sonic hedgehog signaling pathway transduction is mediated by Gli1. Exp Ther Med. 2017;13:307–314.
- Bao X, Peng Y, Lu X, et al. Synthesis and evaluation of novel benzylphthalazine derivatives as hedgehog signaling pathway inhibitors. Bioorganic Med Chem Lett. 2016;26:3048–3051.
- Rubin JB, Kung AL, Klein RS, et al. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci USA. 2003;100:13513–13518.
- Yang L, Jackson E, Woerner BM, et al. Blocking CXCR4-mediated cyclic AMP suppression inhibits brain tumor growth in vivo. Cancer Res. 2007;67:651–658.
- Van Hout A, D'Huys T, Oeyen M, et al. Comparison of cell-based assays for the identification and evaluation of competitive CXCR4 inhibitors. PLoS One. 2017;12:e0176057.
- Domanska UM, Kruizinga RC, Nagengast WB, et al. A review on CXCR4/CXCL12 axis in oncology: No place to hide. Eur J Cancer. 2013;49:219–230.
- Redjal N, Chan JA, Segal RA, et al. CXCR4 Inhibition synergizes with cytotoxic chemotherapy in gliomas. Clin Cancer Res. 2006;12:6765–6771.
- Jager T, Ocker M, Kiesslich T, et al. Thoughts on investigational hedgehog pathway inhibitors for the treatment of cancer. Expert Opin Investig Drugs. 2017;26:133–136.
- Yang L, Xie G, Fan Q, et al. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene. 2010;29:469–481.
- Rodon J, Tawbi HA, Thomas AL, et al. A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clin Cancer Res. 2014;20:1900–1909.
- Rudin CM, Hann CL, Laterra J, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–1178.
- Gajjar A, Stewart CF, Ellison DW, et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clin Cancer Res. 2013;19:6305–6312.
- Khatra H, Bose C, Sinha S. Discovery of hedgehog antagonists for cancer therapy. Curr Med Chem. 2017;24:2033–2058.
- Yauch RL, Dijkgraaf GJP, Alicke B, et al. Smoothened mutation confers resistance to a hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572–574.
- Lauth M, Bergstrom A, Shimokawa T, et al. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci USA. 2007;104:8455–8460.
- Kim J, Lee JJ, Kim J, et al. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci USA. 2010;107:13432–13437.
- Beauchamp EM, Ringer L, Bulut G, et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J Clin Invest. 2011;121:148–160.
- Infante P, Mori M, Alfonsi R, et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. Embo J. 2015;34:200–217.
- Infante P, Alfonsi R, Botta B, et al. Targeting GLI factors to inhibit the Hedgehog pathway. Trends in Pharmacol Sci. 2015;36:547–558.
- Huang L, Walter V, Hayes DN, et al. Hedgehog-GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin Cancer Res. 2014;20:1566–1575.
- Srivastava RK, Kaylani SZ, Edrees N, et al. GLI inhibitor GANT-61 diminishes embryonal and alveolar rhabdomyosarcoma growth by inhibiting Shh/AKT-mTOR axis. Oncotarget 2014;5:12151–12165.
- Wang J, Gu S, Huang J, et al. Inhibition of autophagy potentiates the efficacy of Gli inhibitor GANT-61 in MYCN-amplified neuroblastoma cells. BMC Cancer. 2014;14:768.
- Li J, Cai J, Zhao S, et al. GANT61, a GLI inhibitor, sensitizes glioma cells to the temozolomide treatment. J Exp Clin Cancer Res. 2016;35:184.
- Benvenuto M, Masuelli L, De Smaele E, et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget. 2016;7:9250–9270.
- Wellbrock J, Latuske E, Kohler J, et al. Expression of Hedgehog Pathway Mediator GLI Represents a Negative Prognostic Marker in Human Acute Myeloid Leukemia and Its Inhibition Exerts Antileukemic Effects. Clin Cancer Res. 2015;21:2388–2398.
- Di Magno L, Coni S, Di Marcotullio L, et al. Digging a hole under Hedgehog: downstream inhibition as an emerging anticancer strategy. Biochimica Et Biophysica Acta. 2015;1856:62–72.
- D'Amico D, Canettieri G. Translating Hedgehog in cancer: controlling protein synthesis. Trends Mol Med. 2016;22:851–862.
- Sharpe HJ, Pau G, Dijkgraaf GJ, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell. 2015;27:327–341.
- Pricl S, Cortelazzi B, Dal Col V, et al. Smoothened (SMO) receptor mutations dictate resistance to vismodegib in basal cell carcinoma. Mol Oncol. 2015;9:389–397.
- Hoch L, Faure H, Roudaut H, et al. MRT-92 inhibits Hedgehog signaling by blocking overlapping binding sites in the transmembrane domain of the Smoothened receptor. FASEB J. 2015;29:1817–1829.
- Atwood Scott X, Sarin Kavita Y, Whitson Ramon J, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342–353.