
Abstract
Introduction: In a recent phase I trial in a heterogeneous group of carcinoma patients with advanced disease, we did not observe objective responses by CT at 8 weeks in patients treated with either the anti-EpCAM immunotoxin MOC31PE alone or administered in combination with the immunosuppressor cyclosporin (CsA). We have now assessed overall survival (OS) data for the two groups to reveal potential differences, and to elucidate putative underlying mechanisms.
Material and methods: The OS time of MOC31PE monotherapy (34 patients) and MOC31PE in combination with CsA (23 patients), was assessed. Pre- and post-treatment patient sera were analyzed in a multiplex immunoassay, and the immunogenic effects of MOC31PE were studied in vitro and in a dendritic cell maturation assay.
Results: When the data were analyzed for all treated patients regardless of cancer type, the MOC31PE alone group had a median OS of 12.7 months (95% CI = 5.6–19.8 months) compared to 6.2 months (95% CI = 5.6–6.8 months) (p=.066) for the patients treated with MOC31PE + CsA group. For the subgroup of patients with colorectal cancer, the median OS survival was 16.3 months (95% CI = 5.6–27.0) for the MOC31PE only cohort (n = 15), compared to 6.0 months (CI = 5.8–6.2) (p < .001) for the combination group. The cytokine profile in patient sera and the in vitro immunological studies indicate that MOC31PE induced an immunogenic response leading to T-cell activation; a response that was suppressed in patients treated with MOC31PE + CsA.
Conclusions: The results reveal a promising clinical benefit of anti-EpCAM immunotoxin treatment in patients with advanced disease, an effect apparently explained by a previously unknown immunogenic effect of MOC31PE.
Introduction
One of the hallmarks of cancer is to escape anti-tumor immune surveillance, and mechanisms suppressing immune activation attenuate therapeutic efficacy [Citation1]. Various approaches to overcome cancer immune resistance are applied clinically in several tumor types [Citation1–3], but in spite of examples of durable responses, only a fraction of patients respond, and many develop resistance to the treatment. Hence, new drugs with different mechanisms of immune stimulation are needed.
Our EpCAM-targeting immunotoxin MOC31PE is composed of the full murine MOC31 IgG1 monoclonal anti-EpCAM antibody covalently linked to the complete Pseudomonas exotoxin A (PE). After binding to EpCAM-expressing cancer cells, it is internalized, and the toxin effector moiety triggers cell death by irreversible catalytic inactivation of protein synthesis and by inducing apoptosis [Citation4–6].
Previously, we reported the results of a phase I clinical trial with the EpCAM-targeting immunotoxin MOC31PE given intravenously, with and without concurrent administration of the immunosuppressor CsA [Citation7], and determined the safety and maximum tolerable doses (MTDs) in patients with EpCAM-positive tumors. Of grade 3–4 toxicities, only reversible increase in transaminases as dose-limiting liver toxicity was observed. As expected [Citation8], the concurrent administration of CsA delayed the emergence of anti-MOC31PE antibodies in patients’ sera [Citation7].
In the present study, we investigated the survival data from the phase I trial, also examining more closely the data for different patient subgroups. Unexpectedly, patients treated with MOC31PE alone had a markedly prolonged overall survival (OS) time compared to patients treated with the combination of MOC31PE and CsA. Evidence is provided for the promising OS effect of MOC31PE as a result of an immunotoxin-induced strong immunogenic stimulation.
Material and methods
Patients and study drug
The MOC31PE phase I trial comprised two dose escalation regimens, the first with MOC31PE alone in 34 patients, and the second in 23 patients’ receiving MOC31PE and concomitant administration of Sandimmune® (CsA) [Citation7]. In both treatment arms, MOC31PE was administered every second week up to four times (days 1, 14, 28 and 42). Patients in the combination group were given CsA one day before MOC31PE treatment and then for four subsequent days.
The inclusion criteria were EpCAM-positive metastatic carcinoma (at least 10% positive cells), age 18 years or older, with ECOG performance status 0–2. Prior chemotherapy and/or radiation should be completed at least four weeks prior to study enrollment. The study drug MOC31PE has previously been described [Citation7]. All tumors showed more than 80% positive EpCAM staining.
Ethics
The clinical phase I study was approved by the Norwegian Medicines Agency, Norwegian Regional Ethical Committee (REC), the institutional review board and registered at clinicaltrial.gov (NCT01061645). Before inclusion and study related investigations, the patients signed a written informed consent.
Cytokine measurement
Serum samples were taken prior to treatment, stored at –70 °C and analyzed in a multiplex biometric immunoassay (Bioplex, Bio-Rad Lab., Inc., Hercules, CA, USA). The cytokines measured included: interleukin-1β (IL-1β), interleukin-2 (IL-2), interleukin-6 (IL-6), interleukin-12 (IL-12), granulocyte-macrophage colony-stimulating factor (GM-CSF), tumor necrosis factor α (TNF-α) and interferon-γ (IFN-γ).
Determination of the immunogenic cell death markers; HMGB1 and ATP
Colon cancer cell lines HCT116 and SW480 were cultured by standard procedure (ATCC, Rockville, MD, USA). The Cell Titer 96 AqueousOne solution assay (MTS, Promega, Madison, WI, USA) was used to determine cell viability [Citation5].
Tumor cells were treated for 24 h, and conditioned media from cells treated with MOC31PE (10 and 100 ng/ml), mAb MOC31 (100 ng/ml) or vehicle (PBS, 0.1% HSA) were centrifuged. Equal volumes of cell supernatant were separated by NuPAGE Bis-Tris gel (Invitrogen, Carlsbad, CA, USA), and subsequently transferred by electrophoresis to Immobilon membrane (Millipore, Bedford, MA, USA). The membrane was analyzed for non-histone chromatin-binding nuclear high-mobility group box 1 protein (HMGB1) with rabbit anti-HMGB1 (#3935S, Cell Signaling Technology, Boston, MA, USA).
Generation of dendritic cells
Immature dendritic cells (immDCs) were generated as described in Subklewe et al. [Citation9]. Briefly, human monocytes obtained from leukapheresis products (REC project no.: 2013/624-15) were cultured for two days with GM-CSF and interleukin-4 (IL-4) in ultra-low attachment cell culture flasks. Cancer cell lines were treated for 24 h with MOC31PE, 9.2.27PE [Citation10] and mAb MOC31 at indicated concentrations.
The immDCs were then matured for either 24 h or 48 h with MOC31PE-treated colon cancer cell line supernatants in 96-well round-bottom plates, while cytokines facilitating maturation were used as a positive control as previously described [Citation11]. ImmDCs cultured with IL-4 and GM-CSF were used as negative control. The mature DC phenotype was evaluated by flow cytometry.
Flow cytometry
Cells were washed in staining buffer consisting of PBS containing 2% FCS before staining with CD9-BV510 (M-L13, BD Biosciences, San Jose, CA, USA), CD14-FITC (6ID3, Thermo Fisher Scientific Inc, Waltham, MA, USA) and CD86-APC (Invitrogen, Carlsbad, CA, USA). Finally, cells were resuspended in staining buffer containing 1% paraformaldehyde. Samples were run on an LSR II flow cytometer and the data were analyzed using FlowJo software (Treestar Inc., Ashland, OR, USA).
Statistical analysis
Survival was assessed by the Kaplan–Meier method and survival curves compared using the log-rank test. Univariate analysis was conducted by the Cox proportional hazards regression.
The non-parametric two-tailed Mann–Whitney’s test was used to compare the median values of cytokine variables in the pre- and post-treatment groups, and statistical analysis was performed using the SPSS statistical package (version 21, SPSS, Chicago, IL, USA). The level of statistical significance was set at p<.05.
All statistical analyses for DC maturation were performed using GraphPad Prism® (GraphPad Software, Inc., La Jolla, CA, USA). Paired t-tests were used for comparison of DC maturation between conditions and all p values given are two-tailed values. The level of statistical significance was set to p<.05.
Results
Overall survival
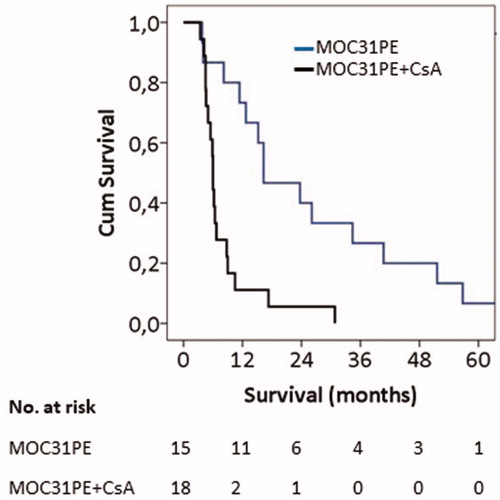
In the phase I trial, the toxicity of systemic therapy with MOC31PE alone and in combination with CsA was modest, and the maximal tolerated dose (MTD) was determined [Citation7]. No objective tumor response was observed as determined by CT scans eight weeks after the first MOC31PE infusion [Citation7]. However, as this might have been too early to capture an eventual effect of the treatment [Citation12], we have now investigated the OS for all patients treated with MOC31PE and compared the results with those receiving MOC31PE combined with CsA. The results for all patients regardless of cancer type showed that the MOC31PE alone group had a median OS of 12.7 months (95% CI = 5.6–19.8 months), as compared to 6.2 months (95% CI = 5.6–6.8 months) (p=.066) for the MOC31PE + CsA group. The MOC31PE cohort consisted of a heterogeneous group of patients with few cases of different tumor type, except for the group with colorectal cancer. For this group, the median OS was as 16.3 months (95% CI = 5.6–27.0) for the MOC31PE only cohort (n = 15), compared to 6.0 months (CI = 5.8–6.2) (p<.001) for the patients in the combination group (n = 18) (). Even after excluding five colorectal cancer patients in the MOC31PE monotherapy group who were given additional therapy after MOC31PE treatment, of whom two received only local radiotherapy for bone metastases, the median OS was 12 months (not shown). In univariate analysis, HR with 95% credible interval (CrI) was calculated for monotherapy vs. combination (HR = 0.248, 95% CrI: 0.109–0.564).
Figure 1. Overall survival: Kaplan–Meier’s plot. Overall survival of patients with EpCAM-positive metastatic colorectal cancer treated with MOC31PE (n = 15, median 16.3 months) or with MOC31PE plus CsA (n = 18, median 6.0 months). MOC31PE treated group (blue line) and MOC31PE plus CsA (black line). The significance of differences in survival between MOC31PE and MOC31PE plus CsA patients was determined by the log-rank test (p<.001).
Induction of cytokines
We hypothesized that MOC31PE had induced a strong immunogenic anti-tumor effect, which was inhibited by the immunosuppression caused by concomitant CsA. Hence, we examined serum samples from CRC patients obtained pretreatment and two weeks after the first dose of immunotoxin for the levels of pro-inflammatory Th1-type cytokines IL-2, IL-6, IL-12, IFN-γ and TNF-α, and the Th1 related IL-1β and GM-CSF.
With the exception of IL-6, the levels of all cytokines increased significantly in serum from MOC31PE treated patients relative to the corresponding pretreatment levels (p<.01) (). Also, IL-2 (p<.05), IL-12 (p<.01) and GM-CSF (p<.01) levels were significantly higher in the serum from MOC31PE treated patients compared to the combination group (). The median, minimum and maximum ranges in cytokine concentration are presented in the Supplementary Table 1. The results demonstrate that in most patients MOC31PE induced a significant Th1 cytokine response, which was almost absent in the combination group.
Figure 2. The box plot showing the cytokine level in patient serum. The levels (pg/ml) of indicated cytokines in serum of metastatic colorectal cancer patients; pretreatment (pre, n = 20), two weeks after MOC31PE (post MOC31PE, n = 9) and MOC31PE plus CsA (post MOC31PE + CsA, n = 12) treatment measured by multiplex cytokine ELISA assay. The box plots show median values (horizontal lines), interquartile ranges (the box lengths), extreme values (x) and outliers (o).
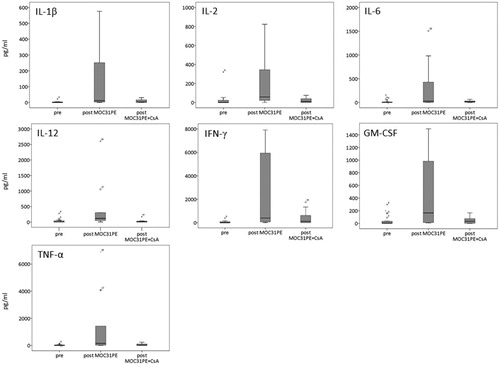
A large fraction of the pretreatment samples had cytokine levels below the detection sensitivity of the assay. One patient in the monotherapy group (11%) and three in the combination group (25%) had all cytokine levels close to zero, the reason for which is unknown
In vitro studies: immunogenic cell death markers and dendritic cell maturation
Next, we studied putative effects of MOC31PE treatment of colon cancer cell lines on the levels of the immunogenic cell death (ICD) marker HMGB1 [Citation13]. MOC31PE treatment decreased the viability of the HCT116 and SW480 tumor cells in a dose-dependent manner, with ID50 values of 100 ng/ml for both cell lines (not shown). The supernatants from MOC31PE-treated cells contained HMGB1, whereas the supernatants from cells treated with 100 ng/ml of unconjugated MOC31 antibody did not ().
Figure 3. Immunogenic cell death markers and dendritic cell maturation. (a) Western blot analysis of high mobility group box 1 protein (HMGB1) in supernatant from HCT116 and SW480 cells, untreated (vehicle control), treated with MOC31PE immunotoxin (10, 100 and 1000 ng/ml) and MOC31 monoclonal antibody (100 ng/ml) for 24 h. Equal volumes of supernatants were run on SDS-PAGE gel and stained with anti-HMGB1 antibody. Results are representative of two independent experiments. ab = monoclonal antibody. (b) ATP extracellular analysis in supernatant from HCT116 cells, untreated (vehicle control), treated with immunotoxin MOC31PE (100 ng/ml) for 24 h. Equal volumes of supernatants were analyzed. Results are representative of two independent experiments performed in duplicates. (c) Mean fluorescence intensity (MFI) decrease in CD14 expression analyzed by flow cytometry. Immature DCs treated with conditioned medium from MOC31PE-treated colorectal cancer cells (HCT116 (green) or SW480 (blue)) show a significant decrease in CD14 expression levels compared to immature DCs + conditioned medium from non-treated cells. Concentrations of MOC31PE were as indicated, 100 or 1000 ng/ml. The result is representative of triplicates. The assay was repeated three times with similar outcome. ***p<.0003, **p<.0057, *p<.186.
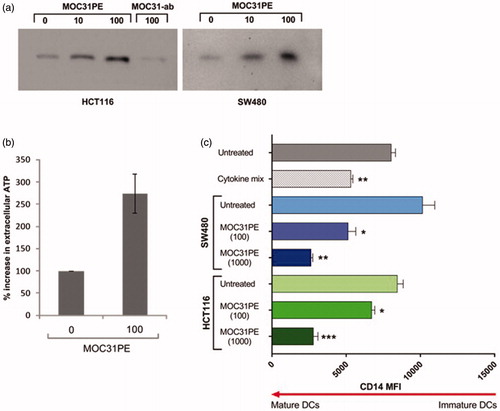
To determine if ICD factors in medium from MOC31PE-treated CRC cells could activate antigen-presenting cells, we generated immDCs ex vivo and incubated them with conditioned medium from treated HCT116 and SW480 cells. The CD14 protein level in immDCs is known to decrease during DC maturation, and in our ex vivo system, the cell surface expression of CD14 decreased significantly (p<.0186) in DCs cultured with conditioned medium, to an extent similar to that of the cytokine treated positive control (). The effect was dose-dependent, with decreasing CD14 expression with increasing concentrations of MOC31PE. Neither, the irrelevant immunotoxin 9.2.27PE (1000 ng/ml) (recognizing antigen HMW-MAA, a melanoma-associated antigen not expressed by colorectal cancer cells), nor the unconjugated MOC31 antibody killed the CRC cells, and their supernatants had no maturation effect on immDCs (not shown).
We then measured the levels of co-stimulatory receptor CD86 in the DCs. CD86 is required for efficient T cell activation and priming, and its expression is a marker of mature DCs. DCs were matured for 48 h for this measurement, and the cytokine maturation cocktail was not used as positive control in this assay as incubation was too long. The expression of CD86 increased nearly twofold in DCs cultured with conditioned medium from MOC31PE-treated HCT116 and SW480 CRC cells compared to that from untreated control cells, and also from oxaliplatin-treated (25 µM) cells (Supplementary Figure 1). Together, the results demonstrate that MOC31PE treatment of colorectal cancer cells induces the release of ICD stimulating factors that mature immDCs.
Discussion
Patients with metastatic CRC are treated with conventional chemotherapeutic drugs and radiotherapy until appearance of unacceptable toxicity or progressive disease on the treatment regimen. The last lines of treatment are commonly Regorafenib and or TAS 102, which are reported to yield an increase in OS of 1.4–1.8 months in randomized studies compared to best supportive care, which for these patients is about 5–7 months [Citation14,Citation15]. Median OS in CRC patients treated with TAS-102 outside of studies (‘real life data’) has been reported to be 6.6 months [Citation16]. The main finding in the present study is that in this group of CRC patients MOC31PE treatment resulted in a median OS of 16.3 months, and as previously reported the immunotoxin treatment resulted in only minor subjective side effects to the patients [Citation7]. In the study protocol, only a CT scan was performed at week 8, with no further systematic CT-scans later on. Despite no radiological response according to RECIST criteria, median OS was unexpectedly long. Stable disease or progressive disease at CT-scans at week 8 may in some patients represent pseudo-progression of the measured lesions as described with immune check-point inhibitors in different non-CRC tumor types [Citation17,Citation18].
For CRC patients, the OS for the combination group is comparable to that of patients with progressive disease on last line of standard chemotherapy [Citation14,Citation15], whereas the OS for patients treated with MOC31PE is highly superior. CsA seems to inhibit the suggested immune stimulated increased OS in CRC patients. A limitation of the phase I study regarding OS is that no specific prior lines of chemotherapy was stated in the protocol, and patients included had different types of tumors.
The difference between the OS data for the CRC patients treated with MOC31PE with and without CsA was unexpected, as in preclinical studies the addition of CsA to MOC31PE was synergistic both in vitro and in human tumor xenograft models [Citation8]. The prolonged OS of patients treated with MOC31PE alone compared to current standard of care (Regorafenib/TAS-102), and the lack of this effect when CsA was added, made us hypothesize that MOC31PE had induced a previously unknown immunogenic anti-tumor effect, which was inhibited by the immunosuppression caused by concomitant CsA.
It is well known that the immunogenicity of immunotoxins leads to generation of anti-immunotoxin antibodies in animals and in man [Citation19]. In preclinical experiments, we have demonstrated that combination therapy with CsA circumvents this problem by delaying anti-immunotoxin antibody production, also observed in our clinical phase I study [Citation7,Citation8]. In animal experiments, the combination also increased anti-tumor efficacy, whereas in the clinic the long OS in MOC31PE treated patients was abolished when combined with CsA. Hence, we hypothesized that MOC31PE induces an immunostimulatory effect of potentially major clinical importance, and present results strongly support this notion.
The immunogenic effect seems to depend on specific binding of the immunotoxin to tumor cells expressing its target antigen, followed by killing of accessible tumor cells. One might speculate that tumor cell killing of tumor cells in this way will cause release of tumor antigens, which is taken up by antigen presenting cell (DCs) in vivo. Immunogenic cell death of tumor cells also releases a plethora of strongly immunogenic molecules stimulating maturation of DCs that have taken up antigen. These DCs will go to the lymph nodes where antigen-specific T-cells can be primed. Activated T cells will then be able to leave the lymph nodes, attacking the tumor in the periphery [Citation20]. The induction of inflammation is supported by the presence and increased levels of several cytokines in the serum of MOC31PE treated patients. We found increased levels of IL-12, required for the induction of IFNγ- and TNFα-producing Th1 cells and necessary for promoting potent cytotoxic T cells accountable for antitumor immunity [Citation21]. IL-2, another cytokine induced by MOC31PE, has been shown to be crucial to the expansion of CD8+ T cells and particularly important for the functional maturation of activated T cells. The cytokine response could be caused by MOC31PE-initiated ICD factors, as supported by the observed release of (extracellular) HMGB1 from MOC31PE treated colon cancer cells [Citation13].
The success of checkpoint inhibitors in several cancer types in the clinic has been limited by aberrant immune checkpoint inhibition and/or absence of appropriate co-stimulation, adverse side effects and development of resistance, and only a subset of patients respond and experience long-term disease-free or OS. These limitations have prompted the need for new immunogenic drugs, as well as for combination studies with other inhibitors or with chemo- and radiotherapy [Citation22–24]. MOC31PE has a different mechanism of action compared to most anticancer agents, inhibiting protein synthesis and inducing apoptosis [Citation4,Citation5,Citation8], and has also an attractive safety profile with only minor clinical side effects and an asymptomatic and spontaneous reversible increase in liver transaminases. We conclude that the MOC31PE immunotoxin may represent a promising targeted immunostimulant, and pending validation of the observed long survival in further studies, the compound may bring new hope to colorectal cancer patients with advanced disease.
Author contributors
YA, EMI, GK, SD, and OF designed this study. YA, TMH and EMI did laboratory experimentation, analysis and prepared figures. SD, OE, OF and YA collected patient data and provided clinical interpretations. All authors contributed to and approved the final manuscript.
Supplemental Material
Download Zip (140.6 KB)Acknowledgments
The authors are grateful for the assistance from Dr. Hans Christian Aass with cytokine analyses. The authors thank Dr. Kjetil Boye for assistance with SSPS and critical reading.
Disclosure statement
The authors declare no conflicts of interest.
Additional information
Funding
References
- Gnjatic S, Bronte V, Brunet LR, et al. Identifying baseline immune-related biomarkers to predict clinical outcome of immunotherapy. J Immunother Cancer. 2017;5:44.
- Kamta J, Chaar M, Ande A, et al. Advancing cancer therapy with present and emerging immuno-oncology approaches. Front Oncol. 2017;7:64.
- Dempke WCM, Fenchel K, Uciechowski P, et al. Second- and third-generation drugs for immuno-oncology treatment—the more the better? Eur J Cancer. 2017;74:55–72.
- Andersson Y, Juell S, Fodstad O. Downregulation of the antiapoptotic MCL-1 protein and apoptosis in MA-11 breast cancer cells induced by an anti-epidermal growth factor receptor-Pseudomonas exotoxin A immunotoxin. Int J Cancer. 2004;112:475–483.
- Andersson Y, Le H, Juell S, et al. AMP-activated protein kinase protects against anti-epidermal growth factor receptor-Pseudomonas exotoxin A immunotoxin-induced MA11 breast cancer cell death. Mol Cancer Ther. 2006;5:1050–1059.
- Risberg K, Fodstad O, Andersson Y. Immunotoxins: a promising treatment modality for metastatic melanoma? Ochsner J. 2010;10:193–199.
- Andersson Y, Engebraaten O, Juell S, et al. Phase I trial of EpCAM-targeting immunotoxin MOC31PE, alone and in combination with cyclosporin. Br J Cancer. 2015;113:1548–1555.
- Andersson Y, Engebraaten O, Fodstad O. Synergistic anti-cancer effects of immunotoxin and cyclosporin in vitro and in vivo. Br J Cancer. 2009;101:1307–1315.
- Subklewe M, Sebelin-Wulf K, Beier C, et al. Dendritic cell maturation stage determines susceptibility to the proteasome inhibitor bortezomib. Hum Immunol. 2007;68:147–155.
- Risberg K, Fodstad O, Andersson Y. The melanoma specific 9.2.27PE immunotoxin efficiently kills melanoma cells in vitro. Int J Cancer. 2009;125:23–33.
- Spranger S, Javorovic M, Bürdek M, et al. Generation of Th1-polarizing dendritic cells using the TLR7/8 agonist CL075. J Immunol. 2010;185:738–747.
- Wolchok JD, Hoos A, O'Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–7420.
- Kepp O, Senovilla L, Vitale I, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3:e955691.
- Grothey A, Cutsem EV, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:303–312.
- Mayer RJ, Van Cutsem E, Falcone A, et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med. 2015;372:1909–1919.
- Andersen SE, Andersen IB, Jensen BV, et al. A systematic review of observational studies of trifluridine/tipiracil (TAS-102) for metastatic colorectal cancer. Acta Oncol. 2019;58:1149–1157.
- Zhao L, Yang Y, Li W, et al. Pseudoprogression: an indicator for cure in combined immunotherapy? Immunotherapy. 2019;11:1087–1093.
- Beer L, Hochmair M, Haug AR, et al. Comparison of RECIST, iRECIST, and PERCIST for the evaluation of response to PD-1/PD-L1 blockade therapy in patients with non-small cell lung cancer. Clin Nucl Med. 2019;44:535–543.
- Pastan I, Hassan R, FitzGerald DJ, et al. Immunotoxin therapy of cancer. Nat Rev Cancer. 2006;6:559–565.
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10.
- Garg AD, De Ruysscher D, Agostinis P. Immunological metagene signatures derived from immunogenic cancer cell death associate with improved survival of patients with lung, breast or ovarian malignancies: a large-scale meta-analysis. Oncoimmunology. 2016;5:e1069938.
- Lohmueller J, Finn OJ. Current modalities in cancer immunotherapy: immunomodulatory antibodies, CARs and vaccines. Pharmacol Ther. 2017;178:31–47.
- Kuusk T, Albiges L, Escudier B, et al. Antiangiogenic therapy combined with immune checkpoint blockade in renal cancer. Angiogenesis. 2017;20:205–215.
- Esposito A, Criscitiello C, Curigliano G. Immune checkpoint inhibitors with radiotherapy and locoregional treatment: synergism and potential clinical implications. Curr Opin Oncol. 2015;27:445–451.