
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
In the 1960s only 1/3 of children with soft-tissue sarcomas survived, however with improved treatments survival today has reached 70%. Given the previous poor survival and the rarity of soft-tissue sarcomas, the risk of somatic late effects in a large cohort of Nordic soft-tissue sarcoma survivors has not yet been assessed.
Methods
In this population-based cohort study we identified 985 five-year soft-tissue sarcoma survivors in Nordic nationwide cancer registries and late effects in national hospital registries covering the period 1964–2012. Information on tumour site and radiotherapy was available for Danish and Finnish survivors (N = 531). Using disease-specific rates of first-time hospital contacts for somatic diseases in survivors and in 4,830 matched comparisons we calculated relative rates (RR) and rate differences (RD).
Results
Survivors had a RR of 1.5 (95% CI 1.4–1.7) and an absolute RD of 23.5 (17.7–29.2) for a first hospital contact per 1,000 person-years. The highest risks in both relative and absolute terms were of endocrine disorders (RR = 2.5; RD = 7.6), and diseases of the nervous system (RR = 1.9; RD = 6.6), digestive organs (RR = 1.7; RD = 5.4) and urinary system (RR = 1.7; RD = 5.6). By tumour site, excess risk was lower after extremity tumours. Irradiated survivors had a 2.6 (1.2–5.9) times higher risk than non-irradiated.
Conclusions
Soft-tissue sarcoma survivors have an increased risk of somatic late effects in 5 out of 10 main diagnostic groups of diseases, and the risk remains increased up to 40 years after cancer diagnosis. Risks were slightly lower for those treated for tumours in the extremities, and radiotherapy increased the risk by more than two-fold.
Introduction
Soft-tissue sarcomas (STS) in childhood are rare accounting for about 8% of all childhood cancers [Citation1,Citation2]. The tumours represent a diverse group of malignancies with rhabdomyosarcomas (RMS) constituting almost 50% of all STS’s in children [Citation2]. The remaining sub-types of STSs are classified as non-rhabdomyosarcoma soft-tissue sarcomas (NRSTS) comprising all other STSs including a variety of rare subtypes [Citation1].
Rhabdomyosarcoma is an aggressive tumour and in the 1960s, when treatment consisted of surgery and radiotherapy alone, only 1/3 of children survived. However, with the addition of multidrug chemotherapy survival has now reached about 70%, although with variations according to age at diagnosis, tumour site and histology [Citation3–6]. Chemotherapy was introduced as part of the treatment of STS in children in the late 1970s. At first, it was not standardised but based on the use of vincristine and actinomycin D. From the mid-eighties, the Nordic countries used international protocols: Denmark followed the SIOP-MMT protocols: SIOP MMT-84, −89, −95 and the EpSSG 2005 protocol [Citation7–9], Sweden followed the CWS protocols CWS-81, −86, −91, −96 − 08 [Citation10–12] and Finland used either European or US protocols [Citation13–15].
Chemotherapy regimens in these different protocols were rather similar, combining vincristine, actinomycin, cyclophosphamide or ifosfamide and doxorubicin along with other drugs as etoposide, carboplatin or topotecan in some cases. Treatment was given for 24– 46 weeks depending on tumour classification and protocol. Radiotherapy indications, doses and administration varied between protocols using hyperfractionated accelerated or intensity modulated radiotherapy in more recent years. Treatment-induced adverse medical conditions following aggressive multimodal therapy may be the long-term price with many late effects becoming clinically evident up to years after the children have been cured [Citation16–24]. The rarity of STS and previous poor survival, especially for high-risk disease, explains the very sparse information about late effects in this group of survivors [Citation1,Citation25,Citation26].
To better understand the health risks associated with the disease and its treatment, we studied 5-year survivors of STS in four Nordic countries identified through nationwide, population-based cancer registries. Late effects were identified as medically verified discharge diagnoses in the national hospital registries. Using matched population comparisons as reference, we assessed the survivors’ relative rates and rate differences of first hospital contacts for somatic diseases.
Material and methods
The cohort of STS survivors is a sub-cohort of the Nordic Adult Life after Childhood Cancer in Scandinavia (ALiCCS) cohort (www.aliccs.org). ALiCCS comprises 33,576 individuals in Denmark, Finland, Iceland and Sweden in whom cancer was diagnosed before age 20 years since the start of the cancer registries in the 1940s and 1950s through 2008 (Finland, 1971–2008). Patients from Norway were not included, as complete hospitalisation histories with all diseases included in the present study were not available. The ALiCCS study design is described in detail elsewhere [Citation27]. In brief, five comparisons without childhood cancer who were alive in the year of cancer diagnosis of the corresponding survivor and matched on sex, birth year and country of residence were randomly selected from national population registries [Citation27]. Of 33,576 childhood cancer patients, 2,150 had soft-tissue sarcoma.
After exclusions, 985 five-year STS survivors and 4,830 directly matched comparisons were available for analyses (Supplementary Figure 1). The population-based and nationwide hospital registries contain information on all hospital admissions for somatic disease (in Sweden since 1964, Finland, 1975, Denmark, 1977, Iceland, 1999), including outpatient visits since 1995 in Denmark and 2001 in Sweden. Registration is mandatory and is recorded by the treating physician. For both survivors and comparisons, we retrieved all hospital admissions and outpatient visits (combined referred to as hospital contacts) with a primary or supplementary diagnosis (not including second malignant neoplasms, see footnote in ). We grouped diagnoses into 10 main diagnostic groups and 104 more disease-specific subcategories according to the International Classification of Diseases (ICD), with ICD-7, 9, and 10 codes adapted to ICD-8 codes to the extent possible (). For a sub-cohort, including the vast majority of survivors from Denmark and Finland (483/531), we were able to retrieve information on radiotherapy (See footnote in ) and tumour site from the cancer registries (referred to as clinical sub-cohort) enabling us to study the effect of these clinical variables on the risk of developing late effects. We grouped tumour site into four main sites; i.e., head/neck, extremity, genitourinary and other. Radiotherapy was included on a yes/no level (). Unfortunately, we were unable to investigate the effects of specific chemotherapeutic agents, as this information is not available systematically in the Nordic cancer registries.
Table 1. Relative rates (RR) and Rate differences (RD) of first hospital contacts in 985 five-year survivors of soft-tissue sarcomas in the 10 main diagnostic groups of diseases and in each of the 104 specific disease categories for which there were at least five observations (leaving 79 disease categories) including diagnoses for both in- and outpatient hospital visitsTable Footnotea (descriptive numbers for in-patient visits only given in parentheses).
Table 2. Clinical characteristics of the cohort of 985 five-year soft-tissue sarcoma survivors, a sub-cohort of 531 5-year survivors for whom information on radiotherapy and tumour site was available, and the directly matched comparisons from 4 Nordic countries.
Statistical analyses
Follow-up for hospital contacts started 5 years after the date of cancer diagnosis and the corresponding date for comparisons. Follow-up ended on the date of death, emigration or end of study (Iceland: 31 December 2008; Sweden: 31 December 2009; Denmark: 31 October 2010; Finland: 31 December 2012). If an individual had more than one hospital contact for a disease within a specific disease category, we only counted the first contact recorded, except in the analysis of recurrent events including re-admissions.
We estimated the incidence rate of first hospital contacts overall, within each of the 10 main diagnostic groups, and in 104 more disease-specific sub-categories, by dividing the number of observed incident events by the total amount of person-years in the survivor cohort and comparison cohort. The relative rates (RR) and rate differences (RD) were estimated and compared by a Poisson model and reported per 1,000 person-years of follow-up [Citation30]. All analyses included diagnoses from both in- and outpatient visits. For descriptive purposes, the number of diagnoses from ‘inpatient visits only’ was reported in .
We estimated the cumulative incidence of first hospital contacts using the Aalen-Johansen estimator considering death as a competing event with time since diagnosis as the underlying time scale. To investigate the disease burden in survivors, we used the method of mean cumulative count (MCC), which allows a summary of all events that occur in a population by a given time and not just the first event in each subject [Citation31]. The MCC was calculated as expected numbers for all first-time diagnoses in the 104 disease sub-categories and for all diagnoses, including readmissions. We estimated the cumulative incidence and MCC for all survivors as well as for the clinical sub-cohort according to tumour site. Differences in the cumulative incidence function were tested by the Grey test [Citation32] and differences in mean values for MCC were tested 25 years after diagnosis by t-test or anova and 95% confidence intervals calculated using the Lin and Ghosh standard errors [Citation33].
The excess cumulative risk of first-time hospital contacts in survivors was estimated as the difference between the risk of survivors and comparisons by time since cancer diagnosis using a new statistical approach, taking the matching structure of the data into account resulting in naturally age-adjusted estimates. The advantage of this approach is to estimate the risk only attributable to childhood cancer since we eliminate the potential influence of the factors by which the survivors and comparisons are matched, i.e., the effects of sex, age, and country of residence, on the risk estimates [Citation34]. First, we predicted the cumulative excess risk according to sub-type of soft-tissue sarcoma, age at cancer diagnosis, tumour site and radiotherapy. Second, we investigated the effect of sub-type of soft-tissue sarcoma, age at diagnosis, sex, tumour site and radiotherapy on the excess hazard for a first hospital contact using a multivariable regression model. The model assumes proportional hazards and is similar to the Cox model, with hazard ratios (HR) interpreted in a standard way as the mean effect of the covariate during follow-up. To take into account that the length of follow-up time strictly depends on the period of diagnosis, we stratified the baseline cumulative excess hazard according to period of cancer diagnosis. Missing information on radiotherapy for 48 survivors was included by multiple imputation using 10 imputed datasets and following the multivariate imputation by chained equations algorithm [Citation35]. We used the statistical software R version 3.5.1.
Results
Clinical characteristics of 985 five-year survivors of STS, the sub-cohort of 531 survivors with information on tumour site and radiotherapy, and 4,830 matched comparisons are given in .
The 5-year survivors were followed in the national hospital registries for a median of 13.9 years (range 0–45.5), yielding 14,889 person-years of observation. In total, 601 survivors had a first hospital contact, giving a first-time hospitalisation rate of 67.95 per 1,000 person-years of follow-up with 44.50 expected, resulting in a relative rate (RR) of 1.53 (95% CI 1.40 − 1.67). In absolute terms, the hospitalisation rate was 23.5 per 1,000 person-years (95% CI 17.7 − 29.2) higher in survivors; thus, an excess ofapproximately two hospital contacts for a new disease per 100 5-year survivors. gives an overview of relative and absolute risk estimates in the 10 main diagnostic groups of diseases and in the more disease-specific subcategories with 5 or more observations (79/104). In 8 out of 10 main diagnostic disease groups, the RR’s were statistically significantly increased. The highest RR’s were for diseases of the blood (RR 2.84; 95% CI 1.91 − 4.21), endocrine disorders (RR 2.49; 95% CI 2.08 − 2.98) and diseases of the skin (RR 2.06; 95% CI 1.68 − 2.51). However, considering both relative and absolute numbers, the highest risks were for endocrine disorders, and diseases of the nervous system and sensory organs, digestive organs, and the genito-urinary system. Outpatient visits accounted for the same proportion of first-time hospital contacts in survivors and comparisons (46% and 47%, respectively). A sensitivity analysis excluding outpatient visits, resulted in a slightly increased overall RR of 1.60 (95% CI 1.43 − 1.80).
shows the cumulative incidence of first hospital contacts for the overall cohort of STS survivors and of matched comparisons and in the clinical sub-cohort divided by tumour site. Overall, 59% (95% CI 56.0–62.9) of 5-year survivors and 47% (95% CI 45.1–48.3) of comparisons had at least one hospital contact within 20 years after the date of cancer diagnosis (). Taking tumour site into account (), the cumulative incidence of survivors of head/neck tumours was 60% (95% CI 50.2–70.6) within the first 20 years after cancer diagnosis, 53% (95% CI 44.0 − 61.) for extremity tumours, 64% (95% CI 52.6–76.4) for genitourinary site and 61% (95% CI 52.0–70.0) for other sites, however, the difference did not reach statistical significance (p-value = .12).
Figure 1. Cumulative incidence of first diagnoses (%) (A + D) and mean cumulative count as expected numbers for all first-time hospital contacts (B + E), and for all hospital contacts, including readmissions (C + F), for all 985 survivors of soft-tissue sarcomas and for the sub-cohort of survivors with available information on tumour site, respectively, and for matched comparisons, with time since cancer diagnosis as the underlying time scale.
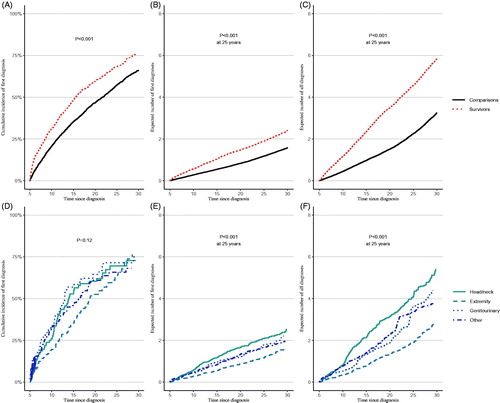
shows that at any given time survivors could expect to have a higher number of hospital contacts (MCC) for different disorders than comparisons. Specifically, within 25 years from cancer diagnosis survivors could on average expect 1.9 (95% CI 1.7–2.1) hospital contacts and comparisons 1.2 (95% CI 1.1–1.2) with the difference being statistically significant (p ≤ .001). The analyses including readmissions () illustrated the same pattern, however with a more pronounced difference between survivors and comparisons with an average of 4.6 hospital contacts in survivors (95% CI 3.9–5.2) and 2.3 (95% CI 2.1–2.4) in comparisons. The expected numbers of both first hospital contacts and all hospital contacts were significantly different by tumour site, with the highest expected numbers in survivors of head/neck tumours, followed by genitourinary and other sites, and lastly extremities ().
The graphs in illustrate the excess risk of first hospital contacts in survivors by time since cancer diagnosis and according to sub-type of STS, age at cancer diagnosis, tumour site, and radiotherapy and can be interpreted as the difference between proportions of first-time hospital contacts in survivors and comparisons over time. Among survivors, the excess risk was highest within the first 10–15 years of follow-up and did not increase notably thereafter. The excess risk 20 years after cancer diagnosis was 21% (95% CI 13–29) for survivors of RMS and 7% (95% CI 2–12) for NRSTS, but did not seem to be affected by age at cancer diagnosis, before or after 10 years of age. However, the risk differed according to tumour site with similar risks in survivors after genitourinary site (19%; 95% CI 2–35) and head/neck tumours (18%; 95% CI 5–30) followed by other sites (11%; 95% CI −1 to 23) but was slightly lower for survivors after tumours of extremities (8%; 95% CI −3 to 19). Survivors treated with radiotherapy had an increased excess risk of 24% (95% CI 12–35) compared with 7% (95% CI −1 to 14) in non-irradiated survivors.
Figure 2. Excess risk of first hospital contacts by subtype of soft-tissue sarcoma, age at diagnosis, tumour site, and radiotherapy for all 985 survivors of soft-tissue sarcomas and for the sub-cohorts of survivors with available information on tumour site and radiotherapy, respectively, and for matched comparisons, with time since cancer diagnosis as the underlying time scale and with shaded areas representing 95% confidence intervals.
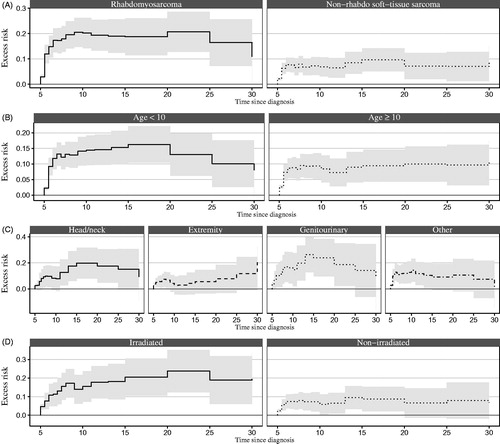
The multivariable model estimating the excess hazard of first hospital contacts () displayed a statistically significant effect of radiotherapy, i.e., irradiated survivors had a 2.6 times higher risk (95% CI 1.2 − 5.9; p = .02) of having a first hospital contact at any given time compared with non-irradiated survivors, regardless of the radiotherapy exposure site (data not shown).
Table 3. Multivariable model estimating the effect of subtype of soft-tissue sarcoma, age at cancer diagnosis, sex, tumour site and radiotherapy on the excess hazard of a first hospital contact in the sub-cohort of 531 survivors of soft-tissue sarcomas with information on radiotherapy and tumour site available, stratified according to the period of diagnosisa and with multiple imputation of missing radiotherapy informationb.
Discussion
Survivors of soft-tissue sarcomas had a 53% increased rate of first hospital contacts for somatic diseases. Thus, for each year of follow-up approximately two of every 100 five-year survivors had a hospital contact for a new excess disease beyond the disease level in comparisons. Diseases within the endocrine system, nervous system, digestive organs and urinary system constituted 63% of all excess hospital contacts. The cumulative risk of having at least one somatic disease within 20 years after the date of the cancer diagnosis was 59% in survivors and 47% in comparisons and the risk remained increased up to 30 years after cancer diagnosis. At any given time, survivors could expect to have a higher number of hospital contacts than comparisons and the more pronounced difference when including re-admissions further indicates a higher burden of each disorder in survivors. However, the risk varied by tumour site, and was slightly lower for survivors after tumours of extremities, whereas radiotherapy treatment increased the risk more than two-fold.
In an analysis of CCSS data, Kurt el al. found a hospitalisation rate in STS survivors that was 1.7 (95% CI 1.6–1.9) times higher than in the background population [Citation36] which is quite consistent with the relative rate of first hospital contacts in our study of 1.53. In another CCSS study focussing on RMS survivors, an increased risk of endocrine impairments, eye disturbances, cardiopulmonary disorders, and neurosensory and neuromotor impairments was reported [Citation21].
We found that survivors had a RR of 2.5 and a RD of 7.6 for endocrine disorders and that 18% of all survivors were registered with an endocrine disorder 5 years or more from cancer diagnosis. Some of the most frequent endocrine disorders were diseases of the thyroid gland and pituitary hypofunction (). The main risk factor for developing endocrine disorders is radiotherapy to the head, which affects the function of the pituitary gland, and may lead to growth hormone deficiency, hypothyroidism and delayed puberty [Citation37]. This was also evident in our study where 10 out of 12 survivors diagnosed with pituitary hypofunction in the clinical sub-cohort had head/neck tumours of whom 8 were treated with radiotherapy.
We found an increased risk of visual disturbances with approximately 10% of survivors diagnosed with an ophthalmological disorder, including a RR for cataract of 4.3 based on 21 cases. A cataract was diagnosed in 10 individuals in the clinical sub-cohort of whom 9 had a tumour in the head/neck region and 8 were treated with radiotherapy, thus supporting the association between radiotherapy to the head and visual disturbances. We also found an increased risk of hearing loss and deafness, which could be due to tumours in the ears, such as of pars petrosa, and localised radiotherapy treatment. Diseases of the nerves and peripheral ganglia, including neuropathies, were observed in 42 survivors supporting the findings of an increased risk of abnormalities of the sensory systems reported in CCSS, which may be related to the frequent use of vincristine. The risk of developing epilepsy (16 cases) was also increased and might be due to radiotherapy to the head/neck region as 6 out of 7 individuals with epilepsy in the clinical sub-cohort were survivors of head/neck tumours with 5 treated with radiotherapy. The increased risk of diseases of the digestive organs may becaused by surgical procedures or radiotherapy, except for diseases of teeth and supporting structures found in 55 survivors more likely being side effects of radiotherapy and chemotherapy. We also found an increased risk of diseases of the circulatory system, with a particularly high risk of heart failure diagnosed, however, only in 10 survivors. Unfortunately, information on chemotherapeutic agents was not available, but it is known that treatment regimens for soft-tissue sarcomas have included anthracyclines over the past several decades, known for their potential cardiotoxic effects [Citation4].
Endocrine disorders and diseases within the nervous system and sensory organs are mainly treated in outpatient care, which is illustrated by the number of patients registered with an inpatient visit only, versus the number of in- and outpatient visits combined. This underscores the need for including outpatient visits in a study aiming to provide an overview of somatic late effects in childhood cancer survivors. As information on outpatient visits was only available in more recent periods, and only for Denmark and Sweden, the risk estimates for diseases mainly treated in outpatient care, such as endocrine disorders, may be underestimated. In a sensitivity analysis only including inpatient visits, the RR for the main group of endocrine disorders was slightly higher (RR = 2.8; 95% CI 2.1–3.8) indicating that the risk of developing more serious disorders requiring in-patient care is more pronounced in survivors. Excluding outpatient visits did not affect risk estimates in the other 9 main groups of diseases (data not shown). ‘Note also that the use of hospital-based diagnoses limits us from reporting on conditions treated at the primary care physicians, although we anticipate that the vast majority of survivors will be further referred to a hospital’. We cannot exclude the possibility that childhood cancer survivors might be under closer medical surveillance than comparisons, leading to earlier detection of health care problems than in the general population. This might partly explain the relatively large number of survivors diagnosed with disorders such as inflammatory diseases of the eyes and ears – conditions mainly treated in outpatient care. Still, since we only include conditions serious enough to require a hospital contact in the tax-supported hospitals of the Nordic countries, the effect of surveillance bias is likely to be limited.
Due to the large number of outcomes investigated in this study, including 10 main diagnostic groups of disease and 104 sub-categories, the risk of chance findings (false positive associations) should be taken into account when interpreting the individual results. However, as this study is descriptive and aimed at presenting an overview of the disease burden experienced by survivors of soft-tissue sarcomas rather than being hypothesis testing, we decided not to adjust for multiple testing.
In conclusion, our study shows that 5-year survivors of soft-tissue sarcomas in childhood treated according to Nordic protocols have an increased risk of somatic late effects with the most prevalent disorders involving the endocrine system, nervous system, digestive organs and the urinary system. The risk remained increased up to 30 years after the cancer diagnosis and survivors of tumours of the extremities displayed a slightly more moderate risk than survivors of tumours in other sites. Still, irradiated survivors, independent of tumour site, had a more than two-fold increased risk of late effects compared with non-irradiated survivors. Continued systematic medical surveillance with more frequent visits up to 15 years post cancer diagnosis is recommended to ensure the best possible long-term health in survivors of soft-tissue sarcomas in childhood.
Ethical Approvals
The design of the ALiCCS study was approved by national bioethics committees, data protection authorities, or the national institutes for health and welfare in each country (Denmark: 2010-41-4334, Finland: THL/520/5.05.00/2016, Iceland: VSN 10-041 & VSN 12-084-V1 and Sweden: Ö 10-2010, 2011/19).
Author contributions
FNN and CB contributed to the study design, design of analyses, interpretation of results, and drafted the manuscript. CR, ASH, LT, L-MMH, HS, THS, HH, JFW and KKA contributed to the study design and interpretation of results. CR, HH, JFW and KKA contributed to the study design, design of analyses and interpretation of results. CB contributed to the design of the analyses and conducted statistical analyses. All authors revised the manuscript for intellectual content and approved the final version.
Supplemental Material
Download PDF (53.8 KB)Supplemental Material
Download PDF (179.4 KB)Acknowledgements
We thank the ALiCCS board (Finn Wesenberg, Catherine Rechnitzer, Kirsi Jahnukainen, Anna Sällfors Holmqvist and Nea Malila) for their valuable help and guidance.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
To request registry data from the four countries used in this study, contact the following authorities: Denmark: Research services at Sundhedsdatastyrelsen ([email protected]); Finland: TLH National Institute for Health and Welfare in Finland (Service telephone for research authorisation applications: tel. +358 29 524 6677); Iceland: The Icelandic Cancer Registry ([email protected]); The Directorate of Health ([email protected]); Statistics Iceland www.statice.is/services ([email protected]); and Sweden: The National Board of Health and Welfare ([email protected]). The ALiCCS study group welcomes collaboration with other researchers in using our registry data. Study protocols can be planned in collaboration with us, and the study material can be analysed accordingly at the Danish Cancer Society Research Centre in Copenhagen, Denmark. For further information regarding collaborative ALiCCS projects, please contact Professor Jeanette Falck Winther, MD, DMSc. ([email protected]).
Additional information
Funding
References
- Bisogno GA. Solid tumors of childhood – soft tissue sarcoma. In: Estlin EJ GR, Wynn RF, ed. Pediatric hematology and oncology: scientific principles and clinical practice. 1st ed. Oxford, UK: Blackwell Publishing Ltd; 2010. p. 216–233.
- M. Fatih Okcu ASP, John Hicks, Lynn Millon, Richard J. Andrassy, and Sheri L. Spunt. The NonRhabdymyosarcoma soft tissue sarcomas. In: Poplack PAPaDG, ed. Principles and practice of pediatric oncology. Philadelphia: Wolters Kluwer; 2011. p. 954–988.
- Gatta G, Botta L, Rossi S, et al. Childhood cancer survival in Europe 1999–2007: results of EUROCARE-5–a population-based study. Lancet Oncol. 2014;15(1):35–47.
- Green DM, Kun LE, Matthay KK, et al. Relevance of historical therapeutic approaches to the contemporary treatment of pediatric solid tumors. Pediatr Blood Cancer. 2013;60(7):1083–1094.
- Punyko JA, Mertens AC, Baker KS, et al. Long-term survival probabilities for childhood rhabdomyosarcoma. A population-based evaluation. Cancer. 2005;103(7):1475–1483.
- Affinita MC, Ferrari A, Milano GM, et al. Long-term results in children with head and neck rhabdomyosarcoma: a report from the Italian Soft Tissue Sarcoma Committee. Pediatr Blood Cancer. 2018;65(3):e26876.
- Bisogno G, Jenney M, Bergeron C, et al. Addition of dose-intensified doxorubicin to standard chemotherapy for rhabdomyosarcoma (EpSSG RMS 2005): a multicentre, open-label, randomised controlled, phase 3 trial. Lancet Oncol. 2018;19(8):1061–1071.
- Oberlin O, Rey A, Sanchez de Toledo J, et al. Randomized comparison of intensified six-drug versus standard three-drug chemotherapy for high-risk nonmetastatic rhabdomyosarcoma and other chemotherapy-sensitive childhood soft tissue sarcomas: long-term results from the International Society of Pediatric Oncology MMT95 study. J Clin Oncol. 2012;30(20):2457–2465.
- Stevens MC, Rey A, Bouvet N, et al. Treatment of nonmetastatic rhabdomyosarcoma in childhood and adolescence: third study of the International Society of Paediatric Oncology-SIOP Malignant Mesenchymal Tumor 89. J Clin Oncol. 2005;23(12):2618–2628.
- Dantonello TM, Int-Veen C, Harms D, et al. Cooperative trial CWS-91 for localized soft tissue sarcoma in children, adolescents, and young adults. J Clin Oncol. 2009;27(9):1446–1455.
- Koscielniak E, Harms D, Henze G, et al. Results of treatment for soft tissue sarcoma in childhood and adolescence: a final report of the German Cooperative Soft Tissue Sarcoma Study CWS-86. J Clin Oncol. 1999;17(12):3706–3719.
- Koscielniak E, Jurgens H, Winkler K, et al. Treatment of soft tissue sarcoma in childhood and adolescence. a report of the German Cooperative Soft Tissue Sarcoma Study. Cancer. 1992;70(10):2557–2567.
- Arndt CA, Stoner JA, Hawkins DS, et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: children’s oncology group study D9803. J Clin Oncol. 2009;27(31):5182–5188.
- Crist W, Gehan EA, Ragab AH, et al. The third intergroup rhabdomyosarcoma study. J Clin Oncol. 1995;13(3):610–630.
- Crist WM, Anderson JR, Meza JL, et al. Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease. J Clin Oncol. 2001;19(12):3091–3102.
- Robison LL, Hudson MM. Survivors of childhood and adolescent cancer: life-long risks and responsibilities. Nat Rev Cancer. 2014;14(1):61–70.
- Norris RE, Adamson PC. Challenges and opportunities in childhood cancer drug development. Nat Rev Cancer. 2012;12(11):776–782.
- Oeffinger KC, Robison LL. Childhood cancer survivors, late effects, and a new model for understanding survivorship. JAMA. 2007;297(24):2762–2764.
- Oeffinger KC, Mertens AC, Sklar CA, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med. 2006;355(15):1572–1582.
- de Fine Licht S, Winther JF, Gudmundsdottir T, et al. Hospital contacts for endocrine disorders in Adult Life after Childhood Cancer in Scandinavia (ALiCCS): a population-based cohort study. Lancet. 2014;383(9933):1981–1989.
- Punyko JA, Mertens AC, Gurney JG, et al. Long-term medical effects of childhood and adolescent rhabdomyosarcoma: a report from the childhood cancer survivor study. Pediatr Blood Cancer. 2005;44(7):643–653.
- Norsker FN, Rechnitzer C, Cederkvist L, et al. Somatic late effects in 5-year survivors of neuroblastoma: a population-based cohort study within the Adult Life after Childhood Cancer in Scandinavia study. Int J Cancer. 2018;143(12):3083–3096.
- Gonzalez CD, Randall RL, Wright J, et al. Long-term survivors of childhood bone and soft tissue sarcomas are at risk of hospitalization. Pediatr Blood Cancer. 2017;64(6):e26371.
- Gibson TM, Mostoufi-Moab S, Stratton KL, et al. Temporal patterns in the risk of chronic health conditions in survivors of childhood cancer diagnosed 1970–99: a report from the Childhood Cancer Survivor Study cohort. Lancet Oncol. 2018;19(12):1590–1601.
- Fromm M, Littman P, Raney RB, et al. Late effects after treatment of twenty children with soft tissue sarcomas of the head and neck. Experience at a single institution with a review of the literature. Cancer. 1986;57(10):2070–2076.
- Nag S, Tippin D, Ruymann FB. Long-term morbidity in children treated with fractionated high-dose-rate brachytherapy for soft tissue sarcomas. J Pediatr Hematol Oncol. 2003;25(6):448–452.
- Asdahl PH, Winther JF, Bonnesen TG, et al. The Adult Life After Childhood Cancer in Scandinavia (ALiCCS) study: design and characteristics. Pediatr Blood Cancer. 2015;62(12):2204–2210.
- Boshuizen H, Feskens E. Fitting additive Poisson models. Epidemiol Perspect Innov. 2010;7:4.
- Dong H, Robison LL, Leisenring WM, Martin LJ, et al. Estimating the burden of recurrent events in the presence of competing risks: the method of mean cumulative count. Am J Epidemiol. 2015;181(7):532–540.
- Gray RJ. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Statist. 1988;16(3):1141–1154.
- Ghosh D, Lin DY. Nonparametric analysis of recurrent events and death. Biometrics. 2000;56(2):554–562.
- Boschini C, Andersen KK, Scheike TH. Excess risk estimation for matched cohort survival data. Stat Methods Med Res. 2019;28(10–11):3451–3465.
- Buuren S, Groothuis-Oudshoorn K. MICE: multivariate imputation by chained equations in R. J Stat Soft. 2011;45(3):1–67.
- Kurt BA, Nolan VG, Ness KK, et al. Hospitalization rates among survivors of childhood cancer in the Childhood Cancer Survivor Study cohort. Pediatr Blood Cancer. 2012;59(1):126–132.
- Nandagopal R, Laverdiere C, Mulrooney D, et al. Endocrine late effects of childhood cancer therapy: a report from the Children’s Oncology Group. Horm Res. 2008;69(2):65–74.
- Ingimarsdottir IJ, Rusch E, Engholm G, et al. Quality assessment of prostate cancer reports to the Danish Cancer Registry. Acta Oncol. 2016;55(1):24–29.
- Korhonen P, Malila N, Pukkala E, et al. The Finnish Cancer Registry as follow-up source of a large trial cohort-accuracy and delay. Acta Oncol. 2002;41(4):381–388.