
Abstract
Background
The diagnostic work-up and treatment of patients with gastroenteropancreatic (GEP) neuroendocrine neoplasms (NEN) has undergone major advances and new methods are introduced. Furthermore, an update of the WHO classification has resulted in a new nomenclature for GEP-NEN that is implemented in the clinic.
Aim
These Nordic guidelines summarise the Nordic Neuroendocrine Tumour Group’s current view on how to diagnose and treat GEP-NEN patients and aims to be useful in the daily practice for clinicians.
Epidemiology
Neuroendocrine neoplasms (NEN) account for 1.0–1.5% of all gastroenteropancreatic (GEP) neoplasms [Citation1]. Data from Norway, which is comparable to those found in other studies from Europe and the USA, show an increasing incidence of GEP-NENs and was 4.01/100,000 per year in 2006–2010 [Citation2]. This increase may be due to better diagnostic methods, improved classifications and higher awareness of the disease. The prevalence has been reported as high as 35/100,000 [Citation1] as neuroendocrine tumour (NET) patients have a long survival. At the time of diagnosis, approximately 50% of all NENs are localised and 40% have distant metastases [Citation2].
Pathology and classification
A histological biopsy or surgical specimen should be used for diagnosis. A fine needle aspirate is insufficient for specific diagnosis and calculation of Ki-67 proliferation index.
The NEN diagnosis is based on the growth pattern, the uniformity of tumour cells, and confirmed by immunohistochemical (IHC) staining with the general neuroendocrine markers synaptophysin and chromogranin A (CgA). Calculation of Ki-67 index is mandatory and should be assessed in hot spots. The index is counted with 500 tumour cells as a reference. Other IHC markers (e.g., thyroid transcription factor-1 (TTF-1), serotonin, CDX2, islet1) may indicate primary tumour site.
GEP-NENs are classified according to the WHO Classification of Digestive System Tumours 2019 () and IUCC’s TNM classification of Malignant Tumours or ENETS TNM classification. GEP-NENs are divided into well-differentiated NETs Grades 1, 2 and 3 and poorly differentiated neuroendocrine carcinomas, NECs. Small-cell NECs are morphologically similar to small-cell lung cancer. Large-cell NECs usually have large pleomorphic cells and areas of necrosis but may be deceptively gland forming resembling an adenocarcinoma. Therefore, in cases with poorly differentiated GEP adenocarcinomas, we advocate generous use of staining for synaptophysin and CgA [Citation3,Citation4]. It is not always easy to separate NET G3 from NEC and additional morphological criteria can be helpful [Citation5]. Genetic alterations in TP53 and/or Rb1 are frequently seen in poorly differentiated NENs, whereas mutations in ATRX or DAXX are only seen in well-differentiated pancreatic tumours [Citation6]. Furthermore, IHC staining for p53, Rb1 and in pancreatic NENs, additional staining for SMAD4, ATRX/DAXX and MEN1 may be helpful in deciding whether a tumour is well or poorly differentiated [Citation7]. This distinction is important for selection of management strategy. Next-generation sequencing or other methods may be helpful in selected cases where genomic profiling may have a therapeutic or prognostic impact as for instance in colon NECs where BRAF mutations may be checked if BRAF/MEK inhibitor treatment is considered. Furthermore, sequencing of TP53 and Rb1 may prove useful to distinguish between NET G3 and NEC. However, genomic analysis is not yet recommended for all NEN patients.
Table 1. The WHO 2019 Classification for neuroendocrine neoplasms (NEN) of the digestive system.
Biochemical markers
Serum CgA (s-CgA) is the most commonly used biomarker in blood and elevated in the majority of patients with residual NETs. The serum concentration at baseline is a predictor of patient outcome [Citation8]. S-CgA should not be used for screening since the specificity is hampered by proton pump inhibitors, kidney, liver and heart failure, chronic atrophic gastritis, as well as several non-neuroendocrine malignancies. Furthermore, GEP-NENs may have normal s-CgA levels. The value of s-CgA for therapy monitoring and surveillance has been investigated in retrospective studies showing that s-CgA is a specific and sensitive marker of tumour progression and that basal s-CgA levels can predict overall survival [Citation8,Citation9]. However, in a recent prospective multicenter study, there was only a weak association between change in s-CgA and change in tumour burden [Citation10]. Overall, s-CgA as a single plasma biomarker is inadequate to predict tumour progression and must be used in association with imaging follow-up. The role of emerging biomarker panels such as the NETestTM, has to be determined [Citation11]. Depending on the primary tumour and symptoms of the patient, measurement of specific markers, such as gastrin, insulin, c-peptide, pro-insulin, glucagon, VIP, somatostatin, ACTH or calcitonin should be performed. In small intestinal NETs (SI-NETs), 5-hydroxyindoleacetic acid (5HIAA) is often elevated and should be measured, preferably in blood [Citation12]. N-terminal pro-brain natriuretic peptide (NT-pro-BNP) can be useful for detection of carcinoid heart disease (CHD) [Citation13].
Radiology
Computed tomography (CT) is the basic radiological modality for NEN imaging. However, magnetic resonance imaging (MRI) allows for better visualisation and characterisation of lesions in liver, pancreas, rectum, bone and brain [Citation14], and is useful before surgery or when reduction in the radiation exposure is wanted, e.g., for long-term follow-up in younger patients. Proper use of intravenous contrast media for CT (‘triple phase CT’), MRI and ultrasonography (US) is fundamental to visualise and characterise hyper- and hypovascular NET lesions. Endoscopic US (EUS) is highly sensitive for visualisation and loco-regional staging for tumours in the pancreas, duodenum and stomach. Intraoperative US is mandatory in hepato-biliary-pancreatic surgery for detection and localisation of small pancreatico-duodenal tumours and metastatic disease. The radiology report should include Response Evaluation Criteria in Solid Tumours (RECIST 1.1) information. Due to the slow-growing nature of GEP-NETs it is important to compare lesion size over a longer period of time (6–12 months) to detect changes in tumour size.
Somatostatin receptor imaging (SRI)
SRI is used for tumour staging, diagnosis of recurrent disease, and to evaluate eligibility for peptide receptor radionuclide therapy (PRRT). Small tumours and tumours with no or low density of somatostatin receptors (SSTRs) may not be visualised. SRI should be performed as a whole-body investigation by positron emission tomography (PET) with 68Ga or 64Cu coupled to a SSA [Citation15,Citation16] and combined with a fully diagnostic CT or MRI if needed and available.
Other types of positron emission tomography (PET)
18FDG-PET and SRI provide complementary information regarding the different biological characteristics of the lesions. Prognosis is generally worse if a tumour is 18FDG positive and SRI negative and vice versa [Citation17–19]. 18FDG-PET should be performed in localised NET G3 as well as in NECs and may be valuable in G1-2 NETs for prognostication and therapy planning. GLP-1-PET ligands for insulinomas, e.g., 68Ga-Exendin [Citation20], are of value, while other PET tracers such as 18F-DOPA and 11C-5-hydroxy-tryptophan are available as problem-solving tools in single centres.
Treatment overview
Surgery
All NEN patients should be considered for surgery. Patients with insulinomas, appendix and rectal NETs are often cured by surgery alone. However, curative surgery is probably possible in less than 30% of all NET patients and recurrences are common. Resection of the primary tumour and/or debulking surgery may be beneficial for control of local and endocrine symptoms even if R0 resection cannot be achieved [Citation21, Citation22].
Chemotherapy or PRRT may, in a few GEP-NEN cases, be used for downstaging the tumour to obtain resectability. Intravenous infusion of SSA should be used to prevent excess hormone release and carcinoid crisis during interventional procedures or surgery of SI-NET patients. Patients referred for a needle biopsy do not need preventive SSA treatment unless the patient suffers from a severe carcinoid syndrome.
Chemotherapy
Many studies on chemotherapy are difficult to interpret, as they report data from heterogeneous populations of NEN patients with a mixture of different primary tumour sites and rarely reports the proliferation index. Tumours with an initial low Ki-67 index may develop a more aggressive phenotype over time, making re-biopsy advisable with reassessment of Ki-67 index as these patients could then benefit from treatment with chemotherapy.
There are basically two indications for chemotherapy established in clinical practice: advanced pancreatic NET (panNET) and GEP-NET G3/NEC. Streptozocin (STZ)/5-fluorouracil (5FU) or temozolomide (TEM)/capecitabine (CAP) are often used as first-line treatment for advanced panNET. For STZ/5FU, progression-free survival (PFS) of 23 months and overall survival (OS) of almost 52 months have been reported [Citation23]. TEM may be used as first- or second-line treatment for advanced panNET. TEM or TEM/CAP treatment show similar PFS of 11-20 months with a slightly higher tumour size response rate for the combination [Citation24,Citation25]. Toxicity is usually mild with nausea and haematological toxicity. Cis- or carboplatin plus etoposide is used as adjuvant treatment for NEC and is the preferred first-line regimen in metastatic disease [Citation3, Citation26]. Second-line options include TEM/CAP, FOLFOX and FOLFIRI although only small cohort studies have been published [Citation26].
Somatostatin analogues
More than 90% of NETs express SSTRs. The SSAs octreotide and lanreotide are used for symptomatic and anti-proliferative treatment. Similar symptomatic and biochemical response is observed in NET patients using either one of the SSAs. SSA may have an effect even if the uptake on SRI is low. The anti-proliferative effect of SSA treatment in NET-patients has been demonstrated in randomised trials [Citation27,Citation28]. In the PROMID study including SI-NET patients, median PFS was significantly longer using octreotide LAR compared to placebo (14.3 vs. 6 months). In the CLARINET study including non-functioning NETs of different origins, median PFS was significantly longer using lanreotide autogel compared to placebo (38.5 vs 18 months). SSA treatment is therefore recommended in metastatic GEP-NET disease with a low Ki-67 (<10%). The dose may be increased or the interval between injections shortened to achieve symptomatic or disease control. In the NETTER-1 study, SI-NET patients progressing on standard SSA dose were randomised to PRRT or SSA dose escalation. Although PRRT was superior, the higher dose of SSA resulted in a further 9 months stabilisation of disease [Citation29]. Common side effects of SSAs include abdominal pain, diarrhoea, flatulence, nausea, subcutaneous nodules at the injection site and development of bile stones. Steatorrhoea caused by reduced pancreatic enzyme secretion should be treated with pancreatic enzyme replacement.
Tryptophan hydroxylase inhibitors
Telostristat ethyl (TE) inhibits tryptophan hydroxylase, a major enzyme in the production of serotonin from tryptophan. In two placebo controlled randomised trials, SI-NET patients with carcinoid syndrome inadequately controlled by SSA, were treated with TE that significantly decreased the number of bowel movements and 5HIAA levels while flushing was unaffected [Citation30,Citation31]. The most common side effect is nausea and elevated liver enzymes.
Peptide receptor radionuclide therapy (PRRT)
The main indication for PRRT is inoperable or metastatic NET demonstrating a high tumour uptake on SRI. The recent NETTER-I study in SI-NETs compared PRRT (4 cycles of 177Lu-DOTATATE) with high dose SSA therapy. At 20 months, 65% of patients in the PRRT arm were without progression compared to 11% in the SSA arm [Citation29]. Previous cohort studies on GEP-NETs including >2000 patients demonstrated similar results including improvements in QoL, symptom control and hormone secretion in the majority of patients [Citation32]. Patients with progression after previous response to PRRT may be re-treated [Citation33]. PRRT is effective not only in G1 and G2 tumours, but also in G3 tumours [Citation34,Citation35]. Most centres administer PRRT as four cycles of 177Lu-DOTATATE/DOTATOC, with 8 weeks intervals. In current protocols, long-acting SSA treatment is interrupted 4–6 weeks before PRRT in order not to block the SSTRs. Short-acting analogues can be used until the day before PRRT. New data suggests that combining PRRT with long-acting SSA in between cycles may improve therapy results [Citation36]. Chemotherapy and treatment with molecular targeted agents should be stopped 4 weeks before PRRT. Side effects include nausea, vomiting, pain and bone marrow depression, which is usually reversible. Impairment of renal function is usually Grades I–II and reversible. Patients with extensive liver or bone metastases need careful evaluation prior to PRRT because of the risk of severe toxicity. Myelodysplastic syndrome and leukaemia have been reported (1–4%) [Citation37].
Molecular targeted therapy
Everolimus, a specific inhibitor of the mTOR pathway, has demonstrated efficacy in NET patients. The RADIANT 3 study [Citation38] including panNET patients, showed a significantly longer median PFS for everolimus of 11.4 vs. 5.4 months for placebo. In the RADIANT 4 study that included GI- and lung-NET patients, median PFS was significantly longer for the treatment arm compared to placebo (11.0 vs. 3.9 months) for the whole group, but for SI-NETs there was no difference [Citation39, Citation40]. The main side effects of everolimus are stomatitis, rash, diarrhoea, hyperglycaemia, fatigue and infections. Dose reduction or temporary interruptions are frequently needed. A serious side effect is non-infectious pneumonitis. Everolimus is recommended for progressing panNET. Its exact place in the treatment of SI-NETs has still to be clarified [Citation39,Citation40].
Sunitinib is an oral multi-targeted tyrosine kinase inhibitor that mainly targets the VEGF receptor. In a multicentre randomised phase III trial with panNET patients, sunitinib resulted in a significantly longer PFS of 11.4 vs. 5.5 months for placebo [Citation41]. Major side effects include rash and hypertension. Dose reduction or temporary interruptions are frequently needed. Sunitinib is recommended for treatment of progressing panNET.
Specific part
This part refers mainly to G1 and G2 NET tumours while NET G3 and NEC are described in separate sections at the end. The length of follow-up described for each tumour group should be related to the patients’ age, comorbidity and available treatment options.
Inherited syndromes with associated panNETs
Pancreatico-duodenal NETs can be part of a familial syndrome. Most common are multiple endocrine neoplasia type 1 (MEN-1) and von Hippel-Lindau disease (VHL). Patients with Carneys complex and neurofibromatosis type 1 (NF1) may also develop NETs [Citation42]. Twenty-five per cent of all gastrinoma patients have MEN-1. S-Ca, PTH and anterior pituitary hormones should be measured and family history recorded. Genetic screening for MEN-1 should be done in young patients (<35 years) with panNETs, if there is a family history indicating MEN-1 or in patient presenting with at least two manifestations of the syndrome (lesion in pancreas, pituitary or parathyroid). VHL disease gives rise to multiple non-functioning panNETs and other tumours such as pheochromocytomas. NF1 is associated with somatostatinomas located at the ampulla of Vater.
Oesophageal NETs
Oesophageal NENs are extremely rare (<1% of all NENs) with less than 1% of them being well differentiated NETs and the rest poorly differentiated NECs, see this section [Citation43].
Clinical presentation: There is a male predominance (6:1). Dysphagia and weight loss can be seen, as well as hoarseness and pain.
Diagnostic procedures: In the rare case of an oesophageal NET the most common setting is incidental finding during endoscopy.
Treatment: Endoscopic or surgical removal of the primary tumour is recommended. Due to the rarity of this tumour entity there are no data to base recommendations for systemic treatment on.
Follow-up and prognosis: Repeated endoscopies should be performed every 6–12 months. CT/MRI and SRI should only be performed when tumour growth or metastases are suspected. There is no survival data available for this tumour entity.
Gastric NETs
Type I and II gastric NETs
Clinical presentation: Most type I and II gastric NETs are incidental findings at gastroscopy. They comprise 85% of gastric NETs and result from hypergastrinemia. In type I, the cause of hypergastrinemia is chronic atrophic gastritis [Citation43]. Type II develops in patients with gastrin-producing NETs.
Diagnostic procedures: Gastric NETs are usually multiple, <2 cm and have a Ki-67 index ≤5%. Multiple biopsies are recommended. S-CgA and s-gastrin are usually highly elevated but the clinical value of repeated measurements is questionable. In type I, gastric acid secretion is low or not measurable (high pH), while in type II acid secretion is elevated (low pH). EUS with evaluation of tumour invasion and regional lymph nodes is mandatory.
Treatment: Patients with tumours, single or multiple, <1 cm should only undergo surveillance, since the lesions usually are benign. Tumours >1cm should be locally resected by endoscopic mucosal resection (EMR), or endoscopic submucosal dissection (ESD) or by surgery depending on the number of lesions, and whether or not there appears to be invasion into the muscularis propria.
Follow-up and prognosis: Repeated endoscopies with 6–12 months interval should be performed. EUS, CT/MRI and SRI should only be performed when tumour growth or metastases are suspected. In type I gastric NETs, lymph node metastases are rare and liver metastases almost never seen. In type II gastric NETs, regional metastases are found in 5–10% and liver metastases in <2%. The prognosis is good with a 5-year survival of 100% in type I and 95% in type II gastric NETs.
Type III gastric NETs
Clinical presentation: Type III gastric NETs are sporadic, not associated with hypergastrinemia and constitute 10–20% of all gastric NETs [Citation43]. Patients may present with symptoms of GI bleeding, dyspepsia and gastric obstruction. Local or liver metastases are seen in >50% of the patients at diagnosis.
Diagnostic procedures: The tumour is usually a solitary NET G2 and >2 cm in diameter. There are no specific biomarkers but s-CgA may be elevated. EUS, CT/MRI and SRI should be performed for tumour staging.
Treatment: Surgical resection with lymph node dissection should be performed when possible similar to gastric adenocarcinoma. Although firm clinical data is missing, for disseminated cases, SSA may be used if Ki-67 < 10%, while chemotherapy is an option if Ki-67 > 10%.
Follow-up and prognosis: Measurement of s-CgA, if elevated initially, gastroscopy and imaging with CT/MRI should be performed every 3–6 months. Five-year survival is 50% [Citation43].
Duodenal NETs
Clinical presentation: The most common tumours are gastrinomas (40%) and somatostatinomas (30%) while gangliocytic paragangliomas and calcitonin or serotonin-producing tumours are rare.
Diagnostic procedures: Histological examination is crucial to distinguish between the different types. S-gastrin and s-CgA should be measured and, if indicated by symptoms, u/p-5HIAA and p-calcitonin. CT/MRI, EUS and SRI are recommended for staging.
Treatment
Surgery: EMR or ESD may be possible in selected non-periampular NETs <1–2 cm. Otherwise, local resection(s), pancreaticoduodenectomy or pancreatic sparing duodenectomy with reimplantation of the ampulla of Vater may be performed. Intraoperative US of the liver and pancreas is mandatory. Surgical treatment of gastrinomas in patients with MEN-1 is controversial, since these tumours are generally multiple and rarely metastasise to the liver. If operated, duodenotomy with a thorough search for multiple tumours in the entire duodenum and pancreas is preferred and intraoperative endoscopy or EUS performed if necessary.
Systemic treatment: Patients with gastrinomas should be treated with proton pump inhibitors, often in high doses, since even very small duodenal gastrinomas may give profound acid related symptoms. Functional NETs may benefit from SSA treatment. Metastatic disease should be treated as panNETs.
Follow-up and prognosis: Measurement of s-CgA, and tumour-specific hormones, endoscopy, EUS and CT/MRI should be done every 3–12 months dependent on the malignant potential of the tumour. Median survival is >100 months for local and regionally metastasised tumours. For tumours with distant metastases the 5-year survival around 60% [Citation1].
Pancreatic NETs
Clinical presentation: PanNETs account for 1–2% of all pancreatic neoplasms. Apart from insulinomas, where less than 10% have metastasised, panNETs have a significant malignant potential. Non-functioning tumours constitute approximately 70–80% and the remaining 20–30% are functioning tumours. Of these, 50% produce insulin, 40% gastrin, and the rest produce a variety of hormones, such as glucagon, somatostatin and VIP. Small tumours of a few mm may give rise to debilitating symptoms. Metastases are usually seen in the liver and regional lymph nodes. Small panNETs (<1–2 cm) discovered on imaging performed for other indications are referred to as ‘incidentalomas’ and are usually asymptomatic with a benign disease course.
Diagnostic procedures: IHC examination for specific hormones is advocated. Measurement of s-CgA, s-insulin, s-C-peptide, s-proinsulin, s-gastrin, p-VIP, p-glucagon, s-calcitonin, s-pancreatic polypeptide and p-somatostatin should be considered depending on symptoms. For the diagnosis of insulinoma a 72-h fasting test is mandatory.
CT/MRI and SRI should be performed before a surgical decision is taken. EUS with biopsy can confirm the diagnosis. Insulinomas are often negative on SRI, and PET with specific tracers may localise the tumour. Intra-operative US of the pancreas and liver is often useful.
Treatment
Surgery: Incidentalomas <2 cm in size usually have no or minimal growth for years and may be managed with surveillance [Citation44]. However, surgery could be considered in younger patients and when the tumour grows. Surgery is usually recommended for larger tumours and for functional tumours irrespective of size.
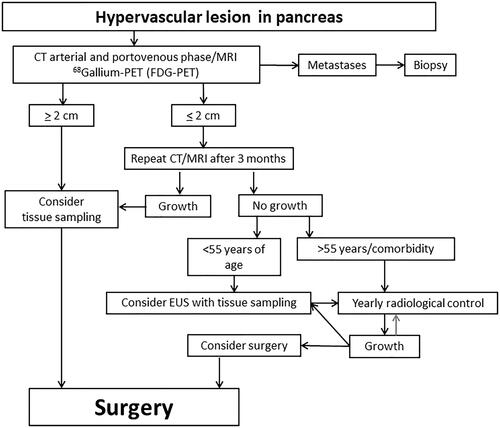
Since most insulinomas have not metastasised, enucleation or simple resection can often be performed laparoscopically. For other panNETs, a pancreaticoduodenectomy or distal/left pancreatectomy, including a splenectomy may be combined with regional lymph node dissection, hepatic surgery and/or radiofrequency ablation of liver metastases. Total pancreatectomy may be performed to achieve radical resection of NETs infiltrating major parts of the pancreas or in the case of multiple tumours in the gland. Involvement of the portal and mesenteric veins or adjacent organs is not a contraindication for surgery. Debulking procedures may be considered to reduce severe endocrine symptoms. However, the benefit of resecting the pancreatic primary tumour in advanced non-curable disease is controversial and blind resections should be avoided. See a suggested algorithm for the surgical management of panNET in .
Figure 1. Treatment algorithm for the surgical treatment of pancreatic neuroendocrine tumours. CT: computed tomography; EUS: endoscopic ultrasonography; MRI: magnetic resonance imaging; PET: positron emission tomography.
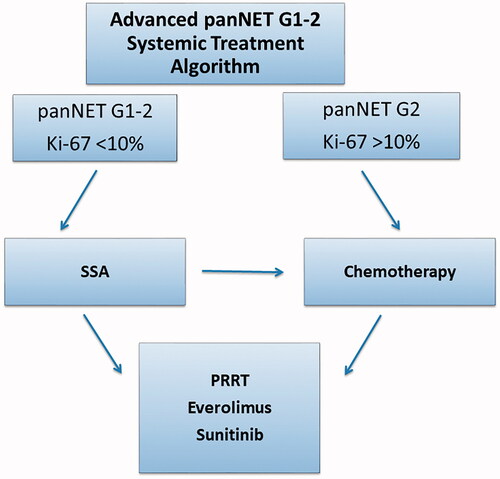
Systemic treatment: Palliative chemotherapy (STZ/5FU, TEM or TEM/CAP), or SSA when Ki-67 < 10%, are frequently used as first-line treatment. To reduce tumour load, chemotherapy could be more effective than SSA and should be chosen. PRRT is considered as second or third-line treatment. Everolimus and sunitinib are used as second or third-line treatment in patients not eligible for PRRT. See a suggested algorithm for systemic treatment of panNETs in .
Figure 2. Treatment algorithm for the systemic treatment of pancreatic neuroendocrine tumours. * indicates treatment regimens with either streptozotocin + 5-fluourouracil or temozolomide ± capecitabine. Debulking treatment such as surgery, radiofrequency ablation and liver embolisation are not included in the algorithm. G: grade; SSA: somatostatin analogue; panNET: pancreatic neuroendocrine tumour; PRRT: peptide receptor radionuclide treatment.
Symptomatic treatment: In gastrinoma patients, proton pump inhibitors can control the symptoms of increased acid secretion. Everolimus is effective in treating hypoglycaemia, diazoxide, SSA and continuous glucose infusion are other options. Glucocorticoids is only recommended when other treatments have failed, since the protective counter-regulatory mechanism is blocked in case of hypoglycaemic attacks. SSAs reduce VIP-induced symptoms like diarrhoea and skin lesions in glucagonomas.
Follow-up and prognosis: Patients with radically resected insulinomas have a good prognosis and one postoperative clinical and biochemical follow-up is usually sufficient. Other radically operated patients should be followed with biochemical markers and CT/MRI initially every 4–12 months, depending on proliferation index, later every 1–2 years for 5–10 years. Patients with residual disease should be monitored, initially every 3–6 months, with biochemical markers, CT/MRI and, when indicated, SRI. The 5- and 10-year survival was 94% and 31% for localised disease and 31% and 18% for patients with distant metastases in a large Norwegian cohort [Citation2].
Small intestinal NETs (SI-NET)
Clinical presentation: Approximately 25–30% of GEP-NENs are SI-NETs, most originating in the distal ileum. Most primary tumours are small (1–2 cm) and may be multiple. Approximately 60% have metastasised at diagnosis mainly in the mesenteric and para-aortal lymph nodes and the liver. The primary tumour, mesenteric lymph node metastases and tumour-induced fibrosis may cause bowel obstruction and vascular encasement accompanied by abdominal pain, diarrhoea and weight loss. Carcinoid syndrome with flushing and diarrhoea is present in less than 20% of the patients at diagnosis. In <5% CHD with tricuspid valve insufficiency and pulmonary valve stenosis cause right-sided heart failure.
Hereditary SI-NETs: SI-NETs are usually sporadic tumours but may rarely be inherited [Citation45].
Diagnostic procedures: The tumours stain positive for serotonin and Ki-67 is usually 1–5%. S-CgA and u/p-5HIAA should be measured. CT/MRI and SRI should always be performed. In patients with metastatic disease, echocardiography and pro-BNP should be done initially and when symptoms occur to evaluate possible CHD.
Treatment
Surgery: If an R0/R1 resection can be obtained patients should be considered for surgery independent of tumour stage. Intra-operative palpation of the entire small intestine is recommended to identify multiple tumours. In case of acute surgery, extensive central mesenteric dissection should be avoided due to the high risk of damage to the mesenteric vessels resulting in short bowel syndrome. In patients with stage IV disease, surgery is considered when there are symptoms of bowel obstruction or ischaemia. A recent study did not demonstrate any survival benefit after primary tumour resection in asymptomatic patients with metastatic disease. Instead, frequent need for reoperations for bowel obstruction after initial early surgery was observed [Citation46]. Surgery of liver metastases may be considered if R0 resection can be obtained or if needed to reduce endocrine symptoms. Patients with symptomatic CHD, should be offered valve replacement with biological valves, preferably before hepatic surgery, to reduce the blood pressure in the hepatic veins.
Systemic treatment: SSA is the primary treatment for patients with disseminated disease. TE can be used for SSA refractory serotonin-induced diarrhoea.
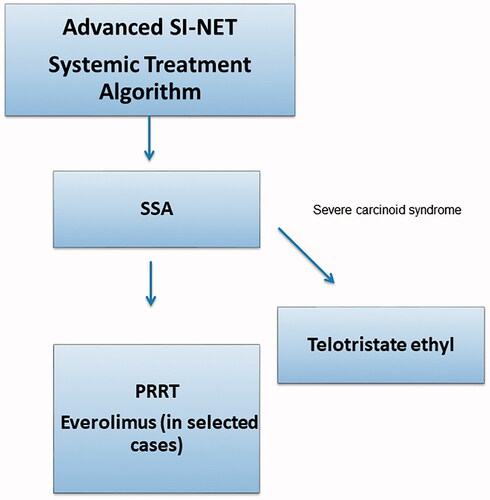
PRRT is recommended for patients who progress on SSA therapy [Citation29]. There is insufficient data to support the general use of everolimus in SI-NETs but it could be considered in selected patients [Citation39,Citation40]. Chemotherapy is generally not recommended in low proliferating SI-NETs. See a suggested algorithm for systemic treatment of SI-NETs in .
Figure 3. Treatment algorithm for the systemic treatment of small intestinal neuroendocrine tumours. Debulking treatment such as surgery, radiofrequency ablation and liver embolisation are not included in the algorithm. G: grade; PRRT: peptide receptor radionuclide treatment; SSA: somatostatin analogue; SI-NET: small intestinal neuroendocrine tumour.
Follow-up and prognosis: After radical surgery, follow-up is recommended every 3–12 months and later every 24 months for at least 5 years and according to institutional preferences, with imaging including SRI and s-CgA, since late recurrence may occur. Patients with metastatic disease should be examined every 3–12 months with CT/MRI and relevant biomarkers. SRI should be performed when PRRT is considered and when CT/MRI findings, biochemistry and the patients clinical status are contradictive. The 5- and 10-year survival was 77% and 63% for localised disease and 59% and 39% for patients with distant metastases in a large Norwegian cohort [Citation2].
Appendix NETs
Clinical presentation
Tumours are mostly incidental findings in surgical specimens after appendectomy due to acute appendicitis. More than 60% are <1 cm and G1 tumours.
Diagnostic procedures: Most appendix NETs (85%) are morphologically similar to SI-NETs and serotonin immunoreactive. Some (<20%) are small tubular carcinoids negative for serotonin but positive for glucagon. Biochemical markers (s-CgA and u/p-5HIAA) are usually normal.
Treatment: Appendectomy is sufficient treatment in >90% of cases. Right-sided hemicolectomy is not indicated for R0 resected tumours <1cm regardless of the presence of risk factors. Risk factors for malignant disease include invasion of mesoappendix >3 mm, vascular invasion, lymphatic invasion and Ki-67 > 10% [Citation47]. R0 resected tumours 1–2 cm with the presence of two or more risk factors is an indication for right-sided hemicolectomy with lymph node dissection [Citation48]. If the tumour size is >2 cm, SRI is recommended, followed by right-sided hemicolectomy. Right-sided hemicolectomy should be performed if the tumour involves resection margins, or has spread to regional lymph nodes irrespective of size. In metastatic disease, medical treatment is similar to that for SI-NETs.
Follow-up and prognosis: In patients where appendectomy is sufficient, no further follow-up is required. After right-sided hemicolectomy, follow-up is only necessary if lymph node metastases or residual tumour is present. For patients who have undergone right-sided hemicolectomy with findings of lymph node metastases and in patients with advanced disease, follow-up should be identical to that of SI-NETs. The 5-year survival is close to 100%.
Colon NETs
Clinical presentation: NETs in the colon comprise <1% of colonic malignancies. Most colon NETs originating in the caecum, are G1-2 tumours and resemble SI-NET [Citation49]. Tumours in the remaining part of the colon are usually NECs, see this section.
Diagnostic procedures: S-CgA should be measured in all patients and u/p-5HIAA should be measured in NETs originating in the right colon. Colonoscopy with biopsy is mandatory. CT/MRI and SRI are performed for staging.
Treatment: Surgical treatment includes bowel resection with lymph node dissection. The medical treatment for G1-2 NET is as for SI-NET.
Follow-up and Prognosis: See SI-NETs [Citation2].
Rectal NETs
Clinical presentation: Rectal NETs comprise approximately 20% of GEP-NENs and are usually small, benign polyps found incidentally at routine endoscopy. Hormone-related symptoms are never seen.
Diagnostic procedures: Tumours show a trabecular growth pattern with positive synaptophysin, CgA and glucagon immunohistochemistry but negative for serotonin. S-CgA and u/p-5HIAA are usually normal. In G1 tumours ≥1 cm and in G2 tumours, irrespectively of size, CT/MRI and SRI are recommended.
Treatment: Rectal NETs <2 cm can often be radically resected by endoscopic procedures. Before attempts on endoscopic resection, EUS is recommended in tumours >0.5–1 cm to investigate the depth of tumour growth. Invasion into the muscularis propria is a contraindication for endoscopic removal. If endoscopic removal is not possible, the tumour should be surgically treated according to the same principles as for rectal adenocarcinomas. Medical treatment is as for SI-NET.
Follow-up and prognosis: Patients with radically resected G1 polyps <1 cm do not need further surveillance. Those with radically resected larger or higher grade lesions, or with lymph node metastases, should be followed as SI-NET [Citation50]. If NETs G1-3 are incompletely resected, flex-endoscopy with biopsies should be performed immediately and then yearly for 3 years, and thereafter every 2–3 years for at least 10 years. Five-year survival for patients with G1 and G2 tumours with localised disease is >90%, regional disease 50% and distant disease 30–40%.
Management of NET liver metastases
Single or few hepatic metastases may be resected with radical intent. Palliative debulking surgery and, if surgery is not possible, RF/microwave ablation can be indicated to reduce severe hormonal or local symptoms, but the impact on survival is uncertain [Citation51,Citation52]. Larger tumour burden confined to the liver may be treated by embolisation of the hepatic artery or its branches. Beneficial effects on hormone-induced symptoms as well as reduction of tumour size is seen in >50% of the patients with a duration of 10–24 months.
Radio-embolisation with intrahepatic infusion of 90Y-coated resin or glass particles has shown promising results. In systematic reviews, there has not been shown any differences in effect between bland- chemo- and radioembolization of the liver [Citation53]. Orthotopic liver transplantation is rarely indicated, but may be considered in carefully selected young patients without extrahepatic disease and with a Ki-67 index <10%. Approximately 20% are recurrence free after 5 years and the 5-year survival rate may be as high as 90% [Citation54].
Neuroendocrine tumour G3 (NET G3)
Clinical presentation: Epidemiological data on this relatively recently defined subtype is limited. NET G3 accounts for 10–15% of GEP-NEN G3. The primary tumour is frequently in the pancreas [Citation55]. Metastatic disease is often present at diagnosis and functional tumours are seen in up to 25% of cases [Citation55].
Diagnostic procedures: NET G3 is morphologically well differentiated, but may be difficult to distinguish from the poorly differentiated NEC in some cases [Citation5]. The Ki-67 index is usually 21–50%, and very rarely higher. CT/MRI and SRI should be performed for initial staging and 18FDG-PET if radical surgery or definitive radiotherapy is planned.
Treatment
Surgery: Treatment of loco-regional disease is poorly documented. In principle, surgery for NET G3 should be considered as for NET G2 [Citation56]. Surgery for distant metastases may be considered.
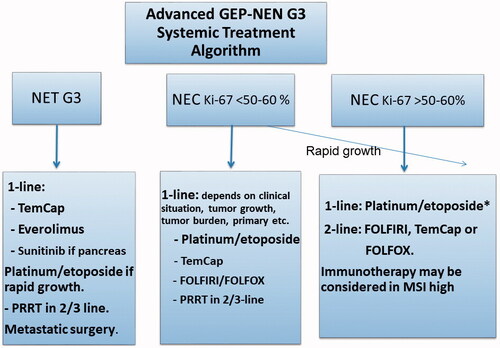
Systemic treatment: No adjuvant treatment is recommended and there is too little data to provide solid advice on optimal first-line palliative care [Citation57]. Response rates (0–24%) and PFS (2.4–5 months) after platinum/etoposide treatment are worse than for NEC. Awaiting further data, TEM/CAP, everolimus and sunitinib have all shown efficacy in small studies [Citation57]. There is little experience with the use of SSA, but it may be considered in NET G3 with lower Ki-67. PRRT appears to have a good effect on NET G3 [Citation34]. Platinum/etoposide may be appropriate if there is a relatively high Ki-67 and clinical evidence of rapid progression. See a suggested algorithm for systemic treatment of NEN with Ki67 > 20% in .
Figure 4. Treatment algorithm for the systemic treatment of neuroendocrine tumours with Ki-67 > 20%. *Colo-rectal carcinoma primaries have low RR/PFS/OS, other schedules could be considered. GEP-NEN: Gastroenteropancreatic neuroendocrine neoplasias; G: grade; MSI: microsatellite instability; NET: neuroendocrine tumour; NEC: neuroendocrine carcinoma; PRRT: peptide receptor radionuclide treatment; TemCap: temozolomid + capecitabine.
Follow-up and prognosis: Radically operated patients should be examined with CT/MRI every 3–6 months for 5–10 years. Patients on active treatment should be monitored every third month. Prognosis for NET G3 is better than for NEC, but worse than for NET G2. Median survival in metastatic disease is 33–41 months [Citation57].
Neuroendocrine carcinoma (NEC)
Clinical presentation: GEP-NECs account for approximately 10–20% of GEP-NEN. The primary site is usually in the oesophagus, stomach, pancreas, colon or rectum [Citation3, Citation58] with metastatic disease at diagnosis in 60–70%. Hormone-induced symptoms are rare.
Diagnostic procedures: GEP-NECs are characterised by poorly differentiated morphology and immunoreactivity for synaptophysin and less frequently CgA [Citation3]. In some cases, separation between NET G3 and NEC is problematic [Citation5,Citation57]. Ki-67 index is by definition >20%, but usually >50%. CT/MRI should be performed for staging. Common metastatic sites are lymph nodes, liver, lung and bone. CT/MRI of the brain should only be performed if brain metastases are suspected. 18FDG-PET should be performed before surgery of localised disease [Citation59]. SRI has a high uptake in 30–50% of NEC cases [Citation34,Citation57] and is relevant when PRRT is considered. Before consideration of PRRT both 18FDG-PET and SRI should be done, to visualise any possible discordant uptake [Citation18,Citation35].
Treatment: Radical surgery should be considered for loco-regional disease. For oesophagus stage 3, chemoradiation seems better than surgery alone [Citation60]. Surgery for distant metastases is generally not recommended, however could be considered for highly selected cases [Citation61].
Adjuvant treatment: The aggressive behaviour of GEP-NECs warrants consideration of adjuvant therapy after surgery. Therefore, although definite data are missing, 4–6 cycles of cis/carboplatin and etoposide is recommended [Citation62].
Palliative treatment: For patients with metastatic GEP-NECs, rapid consideration of chemotherapy is essential. Median survival is only 1 month for patients who are not given chemotherapy [Citation3]. In retrospective studies, treatment with cis/carboplatin and etoposide show PFS of 4 months and median survival of 11–13 months [Citation3, Citation63]. There is no difference of effect comparing cis- vs carboplatin-based regimes [Citation3]. Patients with Ki-67 index <55% are less responsive to platinum-based chemotherapy, but have a significantly longer survival compared to those with higher Ki-67 levels. Consequently, patients with a Ki-67 > 55% should be treated with platinum-based chemotherapy (however, benefit for colo-rectal primaries seems limited) while patients with a Ki-67 index <55%, could be considered for other regimens, such as TEM-based therapy. Second-line regimens used are TEM/CAP, FOLFOX and FOLFIRI, however PFS (< 3 m) and OS (< 6 months) are generally short [Citation64, Citation65]. There is no data to support the use of SSA in NECs. Immunotherapy with check-point inhibitors is currently under investigation in extrapulmonary NEC, however monotherapy studies have reported response rates less than 5% [Citation66]. Dual anti-CTLA4 and PD1 inhibition reported interesting data with two responders among the eight gastrointestinal NEC patients [Citation67]. At present, monotherapy with check-point inhibitors should only be considered for patients with microsatellite instability (MSI). PRRT may be an option for patients with a high uptake on SRI and a Ki-67 < 55% [Citation35]. See a suggested algorithm for systemic treatment of NEN with Ki-67 > 20% in .
Follow-up and prognosis: Radically operated patients should be monitored with CT every three months initially. If there is no recurrence within 5 years, the patient can be considered as cured. Patients with metastatic disease should be followed with CT every 2–3 months. Five-year survival is <10% for patients with metastatic disease.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Yao JC, Hassan M, Phan A, et al. One hundred years after "carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063–3072.
- Boyar Cetinkaya R, Aagnes B, Myklebust TA, et al. Survival in neuroendocrine neoplasms; A report from a large Norwegian population-based study. Int J Cancer. 2018;142(6):1139–1147.
- Sorbye H, Welin S, Langer SW, et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): the NORDIC NEC study. Ann Oncol. 2013;24(1):152–160.
- Jepsen DNM, Fiehn AK, Garbyal RS, et al. Immunohistochemical staining with neuroendocrine markers is essential in the diagnosis of neuroendocrine neoplasms of the esophagogastric junction. Appl Immunohistochem Mol Morphol. 2021. DOI:https://doi.org/10.1097/PAI.0000000000000906
- Elvebakken H, Perren A, Scoazec JY, et al. A consensus developed morphological re-evaluation of 196 high-grade gastroenteropancreatic neuroendocrine neoplasms and its clinical correlations. Neuroendocrinology. 2020. DOI:https://doi.org/10.1159/000511905
- Yao J, Garg A, Chen D, et al. Genomic profiling of NETs: a comprehensive analysis of the RADIANT trials. Endocr Relat Cancer. 2019;26(4):391–403.
- Nielsen K, Binderup T, Langer SW, et al. P53, Somatostatin receptor 2a and Chromogranin A immunostaining as prognostic markers in high grade gastroenteropancreatic neuroendocrine neoplasms. BMC Cancer. 2020;20(1):27.
- Papantoniou D, Gronberg M, Landerholm K, et al. Assessment of hormonal levels as prognostic markers and of their optimal cut-offs in small intestinal neuroendocrine tumours grade 2. Endocrine. 2020. DOI:https://doi.org/10.1007/s12020-020-02534-8
- Jensen KH, Hilsted L, Jensen C, et al. Chromogranin A is a sensitive marker of progression or regression in ileo-cecal neuroendocrine tumors. Scand J Gastroenterol. 2013;48(1):70–77.
- Dam G, Grønbaek H, Sorbye H, et al. Prospective study of chromogranin A as a predictor of progression in patients with pancreatic, small-intestinal, and unknown primary neuroendocrine tumors. Neuroendocrinology. 2020;110(3-4):217–224.
- Malczewska A, Kos-Kudla B, Kidd M, et al. The clinical applications of a multigene liquid biopsy (NETest) in neuroendocrine tumors. Adv Med Sci. 2020;65(1):18–29.
- Adaway JE, Dobson R, Walsh J, et al. Serum and plasma 5-hydroxyindoleacetic acid as an alternative to 24-h urine 5-hydroxyindoleacetic acid measurement. Ann Clin Biochem. 2016;53(Pt 5):554–560.
- Bhattacharyya S, Toumpanakis C, Caplin ME, et al. Usefulness of N-terminal pro-brain natriuretic peptide as a biomarker of the presence of carcinoid heart disease. Am J Cardiol. 2008;102(7):938–942.
- Sundin A, Arnold R, Baudin E, Antibes Consensus Conference participants, et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: radiological, nuclear medicine & hybrid imaging. Neuroendocrinology. 2017;105(3):212–244.
- Sandstrom M, Velikyan I, Garske-Roman U, et al. Comparative biodistribution and radiation dosimetry of 68Ga-DOTATOC and 68Ga-DOTATATE in patients with neuroendocrine tumors. J Nucl Med. 2013;54(10):1755–1759.
- Johnbeck CB, Knigge U, Loft A, et al. Head-to-head comparison of 64Cu-DOTATATE and 68Ga-DOTATOC PET/CT: a prospective study of 59 patients with neuroendocrine tumors . J Nucl Med. 2017;58(3):451–457.
- Majala S, Seppanen H, Kemppainen J, et al. Prediction of the aggressiveness of non-functional pancreatic neuroendocrine tumors based on the dual-tracer PET/CT. EJNMMI Res. 2019;9(1):116.
- Binderup T, Knigge U, Johnbeck CB, et al. F-FDG-PET is superior to WHO grading as prognostic tool in neuroendocrine neoplasms and useful in guiding peptide receptor radionuclide therapy: a prospective 10-year follow-up study of 166 patients. J Nucl Med. 2020. DOI:https://doi.org/10.2967/jnumed.120.244798
- Carlsen EA, Johnbeck CB, Binderup T, et al. 64Cu-DOTATATE PET/CT and prediction of overall and progression-free survival in patients with neuroendocrine neoplasms. J Nucl Med. 2020;61(10):1491–1497.
- Antwi K, Fani M, Nicolas G, et al. Localization of hidden insulinomas with (6)(8)Ga-DOTA-exendin-4 PET/CT: a pilot study. J Nucl Med. 2015;56(7):1075–1078.
- Norlen O, Stalberg P, Oberg K, et al. Long-term results of surgery for small intestinal neuroendocrine tumors at a tertiary referral center. World J Surg. 2012;36:1419–1431.
- Capurso G, Bettini R, Rinzivillo M, et al. Role of resection of the primary pancreatic neuroendocrine tumour only in patients with unresectable metastatic liver disease: a systematic review. Neuroendocrinology. 2011;93(4):223–229.
- Clewemar Antonodimitrakis P, Sundin A, Wassberg C, et al. Streptozocin and 5-fluorouracil for the treatment of pancreatic neuroendocrine tumors: efficacy, prognostic factors and toxicity. Neuroendocrinology. 2016;103(3-4):345–353.
- de Mestier L, Walter T, Evrard C, et al. Temozolomide alone or combined with capecitabine for the treatment of advanced pancreatic neuroendocrine tumor. Neuroendocrinology. 2020;110(1-2):83–91.
- Bongiovanni A, Liverani C, Foca F, et al. Temozolomide alone or combined with capecitabine for the treatment of metastatic neuroendocrine neoplasia: a "Real World" data analysis. Neuroendocrinology. 2020. DOI:https://doi.org/10.1159/000513218
- Sorbye H, , Baudin E, Borbath I, ENETS 2016 Munich Advisory Board Participants, et al. Unmet Needs in High-Grade Gastroenteropancreatic Neuroendocrine Neoplasms (WHO G3). Neuroendocrinology. 2019;108(1):54–62.
- Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656–4663.
- Caplin ME, , Pavel M, Phan AT, CLARINET Investigators, et al. Lanreotide autogel/depot in advanced enteropancreatic neuroendocrine tumours: final results of the CLARINET open-label extension study. Endocrine. 2021;71(2):502–513.
- Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of (177)Lu-dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125–135.
- Kulke MH, Horsch D, Caplin ME, et al. Telotristat ethyl, a tryptophan hydroxylase inhibitor for the treatment of carcinoid syndrome. J Clin Oncol. 2017;35(1):14–23.
- Pavel M, Gross DJ, Benavent M, et al. Telotristat ethyl in carcinoid syndrome: safety and efficacy in the TELECAST phase 3 trial. Endocr Relat Cancer. 2018;25(3):309–322.
- Fross-Baron K, Garske-Roman U, Welin S, et al. 177Lu-DOTATATE therapy of advanced pancreatic neuroendocrine tumors heavily pretreated with chemotherapy: analysis of outcome, safety, and their determinants. Neuroendocrinology. 2021;111(4):330–343.
- Zacho MD, Iversen P, Villadsen GE, et al. Clinical efficacy of first and second series of peptide receptor radionuclide therapy in patients with neuroendocrine neoplasm: a cohort study. Scand J Gastroenterol. 2021;56(3):289–297.
- Carlsen EA, Fazio N, Granberg D, et al. Peptide receptor radionuclide therapy in gastroenteropancreatic NEN G3: a multicenter cohort study. Endocr Relat Cancer. 2019;26(2):227–239.
- Sorbye H, Kong G, Grozinsky-Glasberg S. PRRT in high-grade gastroenteropancreatic neuroendocrine neoplasms (WHO G3). Endocr Relat Cancer. 2020;27(3):R67–R77.
- Yordanova A, Wicharz MM, Mayer K, et al. The role of adding somatostatin analogues to peptide receptor radionuclide therapy as a combination and maintenance therapy. Clin Cancer Res. 2018;24(19):4672–4679.
- Goncalves I, Burbury K, Michael M, et al. Characteristics and outcomes of therapy-related myeloid neoplasms after peptide receptor radionuclide/chemoradionuclide therapy (PRRT/PRCRT) for metastatic neuroendocrine neoplasia: a single-institution series. Eur J Nucl Med Mol Imaging. 2019;46(9):1902–1910.
- Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514–523.
- Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968–977.
- Singh S, Carnaghi C, Buzzoni R, RAD001 in Advanced Neuroendocrine Tumors, Fourth Trial (RADIANT-4) Study Group, et al. Everolimus in neuroendocrine tumors of the gastrointestinal tract and unknown primary. Neuroendocrinology. 2018;106(3):211–220.
- Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501–513.
- Mafficini A, Scarpa A. Genetics and epigenetics of gastroenteropancreatic neuroendocrine neoplasms. Endocr Rev. 2019;40(2):506–536.
- Mastracci L, Rindi G, Grillo F, et al. Neuroendocrine neoplasms of the esophagus and stomach. Pathologica. 2021;113(1):5–11.
- Partelli S, Mazza M, Andreasi V, et al. Management of small asymptomatic nonfunctioning pancreatic neuroendocrine tumors: limitations to apply guidelines into real life. Surgery. 2019;166(2):157–163.
- Dumanski JP, Rasi C, Bjorklund P, et al. A MUTYH germline mutation is associated with small intestinal neuroendocrine tumors. Endocr Relat Cancer. 2017;24(8):427–443.
- Daskalakis K, Karakatsanis A, Hessman O, et al. Association of a prophylactic surgical approach to stage IV small intestinal neuroendocrine tumors with survival. JAMA Oncol. 2018;4(2):183–189.
- Holmager P, Willemoe GL, Nielsen K, et al. Neuroendocrine neoplasms of the appendix: characterization of 335 patients referred to the Copenhagen NET Center of Excellence. Eur J Surg Oncol. 2021. DOI:https://doi.org/10.1016/j.ejso.2021.02.005
- Brighi N, La Rosa S, Rossi G, et al. Morphological factors related to nodal metastases in neuroendocrine tumors of the appendix: a multicentric retrospective study. Ann Surg. 2020;271(3):527–533.
- Volante M, Grillo F, Massa F, et al. Neuroendocrine neoplasms of the appendix, colon and rectum. Pathologica. 2021;113(1):19–27.
- Wei G, Feng X, Wang W, et al. Analysis of risk factors of lymph node metastasis in rectal neuroendocrine neoplasms using multicenter data. Future Oncol. 2018;14(18):1817–1823.
- Eriksson J, Stalberg P, Nilsson A, et al. Surgery and radiofrequency ablation for treatment of liver metastases from midgut and foregut carcinoids and endocrine pancreatic tumors. World J Surg. 2008;32(5):930–938.
- Knigge U, Hansen CP. Surgery for GEP-NETs. Best Pract Res Clin Gastroenterol. 2012;26(6):819–831.
- Kanabar R, Barriuso J, McNamara MG, et al. Liver embolisation for patients with neuroendocrine neoplasms: systematic review. Neuroendocrinology. 2021;111(4):354–369.
- Mazzaferro V, Battiston C, Sposito C. Pro (with caution): extended oncologic indications in liver transplantation. Liver Transpl. 2018;24(1):98–103.
- Coriat R, Walter T, Terris B, et al. Gastroenteropancreatic well-differentiated grade 3 neuroendocrine tumors: review and position statement. Oncologist. 2016;21(10):1191–1199.
- Merola E, Rinke A, Partelli S, et al. Surgery with radical intent: is there an indication for g3 neuroendocrine neoplasms? Ann Surg Oncol. 2020;27(5):1348–1355.
- Sorbye H, Baudin E, Perren A. The problem of high-grade gastroenteropancreatic neuroendocrine neoplasms: well-differentiated neuroendocrine tumors, neuroendocrine carcinomas, and beyond. Endocrinol Metab Clin North Am. 2018;47(3):683–698.
- Dasari A, Mehta K, Byers LA, et al. Comparative study of lung and extrapulmonary poorly differentiated neuroendocrine carcinomas: a SEER database analysis of 162,983 cases. Cancer. 2018;124(4):807–815.
- Binderup T, Knigge U, Loft A, et al. Functional imaging of neuroendocrine tumors: a head-to-head comparison of somatostatin receptor scintigraphy, 123I-MIBG scintigraphy, and 18F-FDG PET. J Nucl Med. 2010;51(5):704–712.
- Meng MB, Zaorsky NG, Jiang C, et al. Radiotherapy and chemotherapy are associated with improved outcomes over surgery and chemotherapy in the management of limited-stage small cell esophageal carcinoma. Radiother Oncol. 2013;106(3):317–322.
- Galleberg RB, Knigge U, Tiensuu Janson E, et al. Results after surgical treatment of liver metastases in patients with high-grade gastroenteropancreatic neuroendocrine carcinomas. Eur J Surg Oncol. 2017;43(9):1682–1689.
- Garcia-Carbonero R, Rinke A, Valle JW, Antibes Consensus Conference participants, et al. ENETS consensus guidelines for the standards of care in neuroendocrine neoplasms. systemic therapy 2. Neuroendocrinology. 2017;105(3):281–294.
- Heetfeld M, Chougnet CN, Olsen IH, et al. Characteristics and treatment of patients with G3 gastroenteropancreatic neuroendocrine neoplasms. Endocr Relat Cancer. 2015;22(4):657–664.
- Hentic O, Hammel P, Couvelard A, et al. FOLFIRI regimen: an effective second-line chemotherapy after failure of etoposide-platinum combination in patients with neuroendocrine carcinomas grade 3. Endocr Relat Cancer. 2012;19(6):751–757.
- Welin S, Sorbye H, Sebjornsen S, et al. Clinical effect of temozolomide-based chemotherapy in poorly differentiated endocrine carcinoma after progression on first-line chemotherapy. Cancer. 2011;117(20):4617–4622.
- Vijayvergia N, Dasari A, Deng M, et al. Pembrolizumab monotherapy in patients with previously treated metastatic high-grade neuroendocrine neoplasms: joint analysis of two prospective, non-randomised trials. Br J Cancer. 2020;122(9):1309–1314.
- Patel SP, Othus M, Chae YK, et al. A phase II basket trial of dual anti-CTLA-4 and anti-PD-1 blockade in rare tumors (DART SWOG 1609) in patients with nonpancreatic neuroendocrine tumors. Clin Cancer Res. 2020;26(10):2290–2296.