
Abstract
Background
Metastatic colorectal cancer (mCRC) is a complex and heterogeneous disease with few standard and targeted treatment options. Next-generation sequencing of tumor tissue was performed to identify cancer driver mutations to discover possible personalized treatment options, as targeted treatment possibilities are limited for this patient population. Results of genomic sequencing in patients with treatment-refractory mCRC are described in this retrospective analysis.
Material and methods
Clinico-pathological characteristics and genomic sequence results of consecutive patients with refractory mCRC, referred to the Experimental Cancer Therapy Unit (ECTU) at Department of Oncology, Herlev & Gentofte Hospital in the period from 1 October 2015 to 14 December 2018 were reviewed in this retrospective analysis. Tumor tissue from the patients was analyzed by next-generation sequencing using the Oncomine Comprehensive primer panel to detect actionable variants of cancer driver mutations and microsatellite instability status. From August 2018 tumor mutational burden was also analyzed.
Results
A total of 80 patients with treatment-refractory mCRC and in a fairly good performance were referred to the ECTU during this period. Genomic sequencing of tumor tissue was performed for all 80 patients and a cancer driver mutation was identified in 90% (n = 72) of the patients. A total of 31.3% (n = 25) of the patients received therapy either as targetable therapy outside an available trial (n = 2), FDA approved therapy (n = 2), or treatment in phase 1 or 2 trials, independent of the genomic signature 26.3% (n = 21).
Conclusion
Most mCRC patients refractory to standard anti-neoplastic therapies, presented with a cancer driver mutation, however, only a few of these mutations gave rise to matched therapies as only 2.5% of the patients from this period received targeted therapy.
Background
The genomic landscape has been examined in several tumor types [Citation1] and a genomic signature may give rise to matched therapies, which is a fast-growing field in oncology [Citation2,Citation3]. Several trials have been conducted to identify patient-specific, targetable genetic mutations with the aim to improve patient outcomes with current and novel therapies [Citation4–6]. Colorectal cancer (CRC) is the third most common cancer worldwide accounting for 1.8 million new cases in 2018 [Citation7]. Despite some advances in the cytotoxic and targeted therapies, metastatic CRC (mCRC) is still one of the leading causes of cancer-related death, as targeted treatment possibilities are limited and heterogeneity is a challenge [Citation8]. The testing of RAS and BRAFV600E genotyping as well as mismatch repair deficiency are recommended in Denmark for patients with CRC before initiation of therapy to guide the treatment strategy. Current chemotherapy options include 5-fluoropyrimidine (5-FU) or 5-FU based therapy, such as capecitabine often in combination with irinotecan and/or oxaliplatin. Targeted agents used for the treatment of mCRC include the epidermal growth factor receptor (EGFR) inhibitors cetuximab or panitumumab for patients with RAS wild type tumors [Citation9,Citation10] and inhibitors of angiogenesis by the vascular epidermal growth factor (VEGF) inhibitor bevacizumab, as well as ramucirumab, aflibercept and regorafenib [Citation11,Citation12]. Patients with microsatellite instable high (MSI-high)/deficient mismatch repair (dMMR) mCRC are likely to respond to immunotherapy by the program death-1 (PD-1) inhibitors nivolumab or pembrolizumab as single agents and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) inhibitor ipilimumab in combination with nivolumab [Citation13–17]. In Denmark, the standard targeted therapies of mCRC are limited to cetuximab, panitumumab, and bevacizumab. Patients harboring a BRAF V600E mutation are associated with a poor prognosis and aggressive upfront treatment with 5-FU, oxaliplatin, and irinotecan (Folfoxiri) with or without bevacizumab may be considered for these patients [Citation18]. A subsequent option for patients with a BRAFV600E mutated mCRC is a double blockade using cetuximab and the BRAFV600E inhibitor encorafenib [Citation19,Citation20]. This treatment combination was just approved for use in Denmark 27 January 2021. In this study, Next Generation Sequencing (NGS) in mCRC patients refractory to standard anti-neoplastic therapies were performed at Herlev & Gentofte Hospital, for the assessment of personalized therapy options. An overview of the identified cancer driver mutations is reported here along with the therapy options selected for these patients.
Aim and hypothesis
The aim of this work was to present the genomic sequence of cancer cells from mCRC patients based on the oncomine comprehensive panel to assess and improve treatment possibilities. The hypothesis was that drugable genomic variants would have the potential to improve patient outcomes in mCRC patients refractory to standard therapies.
Material and methods
This is a retrospective analysis of genomic sequencing of tumor tissue from consecutive patients with mCRC refractory to standard therapies, who were referred to the Experimental Cancer Therapy Unit at the Department of Oncology, Herlev & Gentofte Hospital. Patients were referred for the assessment of experimental therapeutic options during the period from 1 October 2015 to 14 December 2018. After clinical assessment and informed consent, NGS using the Oncomine Comprehensive panel version 1 in period 1 October 2015 to 30 November 2017 and Oncomine Comprehensive panel version 3 in period 7 December 2017 to 14 December 2018, was performed. From 1 October 2015 to 30 November 2017, thirteen patients had an Oncomine Focus panel performed. The genomic sequencing and all other molecular analyses were performed at the Department of Pathology at Herlev & Gentofte Hospital, Copenhagen, Denmark. The study was approved by the health authority, Danish Patient Safety Authority (ref: 3-3013-2793/1).
Targeted therapies available
All CRC patients, who had a genomic sequence performed had their genomic signature reviewed by a local molecular tumor board including experts in oncology, pathology, and molecular biology, for the evaluation of possible targeted therapies. Targeted therapy was possible if drugs were available and clinical evidence of the drug was applicable. BRAF directed therapy was not proved to be efficient in mCRC at this timepoint, hence this therapy option was not used [Citation19,Citation21,Citation22]. For patients with MSI-high mCRC, the use of checkpoint inhibitors was an upcoming and promising therapy, which was FDA approved in 2017.
Biospecimens
Fresh biopsies were performed by experienced interventional radiologists under ultrasonographic guidance using a 1.2 mm (18 gauge) biopsy system for histology. The number of biopsies (needle passes) per procedure was 1–3 dependent on the macroscopic outcome of each needle pass. Sections from archival metastatic tumor samples (41.3%, n = 33) or a newly collected tumor biopsy (57.5%, n = 46) were stained with hematoxylin and eosin (H&E). An area of interest with a minimum of 20% tumor cells and a minimum of 20% non-neoplastic cells, was identified by a specialized pathologist and punched out to ensure optimal material for both NGS and MSI analyses. The limit for detection of genetic changes in allele was 5% corresponding to the minimum presence of 10% of tumor cells in analyzed tissue. Tumor tissue was mainly from liver metastases (56.4%, n = 44). Tissue slides or punched single 1 mm cores were taken from each formalin-fixed paraffin-embedded (FFPE) block for DNA extraction using the Maxwell Rapid Sample Concentrator (RSC) DNA FFPE kit (AS1450) with the automated Maxwell RSC Instrument (Promega, Sweden) according to the manufacturer's instructions. The DNA concentrations were calculated using the Qubit® dsDNA HS (High Sensitivity) Assay Kit with the Qubit® Fluorometer (ThermoFisher Scientific, Denmark). Subsequently, a second H&E slide was performed to verify the area of interest.
Next-generation sequencing analysis
The DNA sequencing of tumor tissue was performed by NGS using an Ion Torrent S5XL (ThermoFisher Scientific, Denmark) [Citation23], with a primer panel including all known targets for biological treatment of cancer, the Oncomine Comprehensive Assay (ThermoFisher Scientific, Denmark) version 1, including 143 oncogenes [Citation24] and the Oncomine Comprehensive version 3 [Citation25], including 161 oncogenes. The Oncomine Focus panel was performed for a few patients (n = 13) for technical reasons for a short period of time. The Oncomine™ Tumor Mutation Load (TML) Assay was used from August 2018 in addition to the Oncomine Comprehensive v3 panel for fourteen patients, as the TML analysis became available. The NGS Library preparation was carried out using the Ion AmpliSeq™ Library kit 2.0 (ThermoFisher Scientific, Denmark), Ion Xpress™ Barcode Adapters for multiplexing (ThermoFisher Scientific, Denmark), and Ion Library Equalizer™ Kit to standardize the Library concentration (ThermoFisher Scientific, Denmark) following the manufacturer's protocols. The NGS chip was loaded using an Ion CHEF system (ThermoFisher Scientific, Denmark).
Sequencing results of the Ion Torrent S5XL runs were examined by an experienced molecular biologist to verify that the NGS runs were performed as expected. The analyses included both statistics and quality metrics for the actual Ion Torrent run. Coverage statistics using the Ion Torrent Suite Coverage Analysis (ThermoFisher Scientific, Denmark) were used to evaluate the quality of each library in the run. Binary Alignment Map (BAM) data files with aligned sequences to the human genome reference sequence Hg19 were uploaded to the IonReporter Software (ThermoFisher Scientific, Denmark) where the analysis using Oncomine Comprehensive v1-540-w2.3.1—DNA and Fusions—Single Sample workflow 5.0–5.6 and interpretation assignment was performed. Ion Reporter Software analysis and variant calling help to focus on variants of interest supported by full annotation from >25 public and proprietary databases, among these are, e.g., dbSNP, OncoKB, Varsome, COSMIC, OMIM, GeneModel, GenePanel, SIFT, PolyPhen-2 scores, Grantham scores, PhyloP, Gene Ontology, Pfam, FATHMM, ClinVar, DGV, 5000Exomes, University of Santa Cruz (UCSC) Common SNPs, ExAC, DRA, DrugBank. Variants annotated were graphically visualized using the software Integrative Genomics Viewer (IGV) [Citation26]. Genes were reported as mutated if relevant mutations were identified at a minor allele frequency (MAF) of 5% and reading depth >250× for somatic gene variants to ensure consistent and reproducible results. The variants with allele frequencies above 5% and structural variants of potential clinical significance were sequenced. Actionable target genes were uploaded to the Oncomine Knowledgebase Reporter which provides global clinical trial information from 69 countries with summary information including contact information for enrollment [Citation25].
The Ion Reporter analysis workflow for Oncomine™ Tumor Mutation Load (TML) Assay utilizes a custom variant calling and germline variant filtering algorithm to accurately estimate somatic variants described in the genelist (https://assets.thermofisher.com/TFS-Assets/CSD/Flyers/oncomine-tumor-mutation-load-assay-flyer.pdf) in cancer research samples [Citation27,Citation28]. Mutation load was defined as the number of non-synonymous variants [missense and nonsense single nucleotide variants (SNVs)], insertion and deletion variants (INDELs), splice site variants, synonymous not located in UCSC Common SNPs genome browser (www.genome.ucsc.edu), and multiple nucleotide variants (MNVs) detected per megabase (Mb) of exonic sequence (1.656 MB) [Citation29,Citation30]. TML <10 was defined as tumor mutational burden (TMB) low, TML between 10 and 20 as TMB intermediate, and TML higher than 20 was categorized as TMB high.
MSI analysis
The Promega MSI Multiplex System Version 1.1 (Promega, Sweden), is a fluorescent PCR-based assay for the detection of microsatellite instability. It consists of five nearly monomorphic mononucleotide markers (BAT-25, BAT-26, NR-21, NR-24, and MONO-27) for MSI determination and two polymorphic pentanucleotide markers (Penta C and Penta D) for sample identification. MSI analysis was performed according to the manufacturer’s directions (Promega Corp., Sweden). Fragments were analyzed by capillary electrophoresis using an ABI 3130xl Genetic Analyzer (Applied Biosystems), microsatellite instability at ≥2 mononucleotide loci was defined as MSI-high, instability at a single mononucleotide locus as MSI-low, and no instability at any of the loci tested as microsatellite stable (MSS) (https://www.pentabase.com/molecular-diagnostics/plentiplex-msi).
Results
In the period from 1 October 2015 to 14 December 2018, NGS was performed for 80 consecutive patients with mCRC refractory to standard therapies at Herlev & Gentofte Hospital, Copenhagen, Denmark. Of those, 41.3% (n = 33) were women and 58.7% (n = 47) men. The median age for men was 62 years (range 31–77 years) and for women 66 years (range 34–77 years), respectively. Patients referred for experimental therapy needed to be in a good performance (PS), which was the case for most patients, see patient characteristics in . Most patients (91.3%) had received at least two lines of palliative standard therapy before the NGS analysis ().
Table 1. Clinical characteristics.
Genomic sequencing results
In 90% (n = 72) of the patients, a minimum of one cancer driver mutation or amplification was identified. Of the patients with cancer driver mutations, 16.3% of the patients (n = 13) had one mutation and 73.8% of the patients (n = 59) had two or more mutations in the analyzed tumor tissue. In six patients (7.5%) cancer driver mutations were not identified and for two patients the genomic sequencing results were inconclusive.
The most prevalent oncogenic driver mutations were the tumor suppressor genes TP53 (61.2%, n = 41 out of 67 tested) and APC (48.9%, n = 23 out of 47 tested). Of the total patients tested, the most prominent affected signaling pathway was the EGFR pathway with at mutation in KRAS in 51.3% (n = 41 out of 80 patients tested), a BRAF mutation was observed in 11.3% (n = 9 out of 80 tested) of the patients. EGFR mutations were observed in 10.4% of the patients (n = 7 out of 67 tested) and an ERBB2 amplification was only observed in one patient (copy number value: 54). Out of 67 patients tested, FLT3 mutation was detected in one patient and MAP2K1 mutation was detected in two patients. MET mutation was detected in two patients and one patient had a MET amplification. Mutations in the PIK3/AKT/mTOR pathway were reported with PIK3CA mutation in 11.3% (n = 9 out of 80 tested) and mTOR/TSC1/TSC2 mutations in 7.5% (n = 5 in 67 tested). FBXW7 gene mutation was observed in 11.9% (n = 8 out of 67 patients tested). Mutation in the PTEN tumor suppressor gene was only observed in three patients (4.5%) out of 67 patients tested. Changes in genes encoding components of the DNA Damage Repair (DDR) pathway [Citation31] were observed in a total of 11.9% (n = 8 out of 67 tested) of the patients. All of those were ATM mutations. For the TGFβ-SMAD4 pathway, a SMAD4 mutation was observed in 10.4% (n = 7 out of 67 tested). Furthermore, variants in the FGFR gene were observed in 4.5% of the patients (n = 3 of 67 tested). Mutations and amplifications observed are listed in and . Additionally, the genomic signature for right-sided and left-sided colon cancer and rectal cancer are listed in the Supplementary table. illustrates the major genomic changes observed in right-sided, left-sided, and rectal cancer.
Figure 1. Genomic sequencing results for right- and left-sided colon cancer, as well as for rectal cancer. Results of microsatellite instability and stability based on primary tumor location are added.
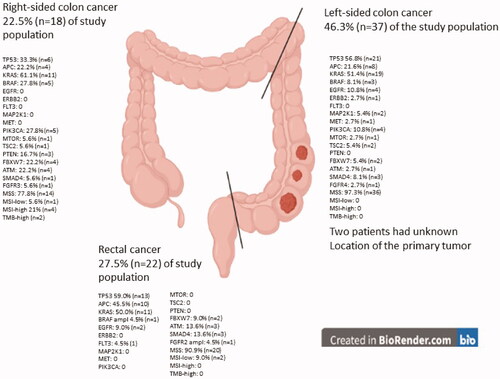
Table 2. Mutations.
Table 3. Mutations according to primary tumor location.
Four patients had an MSI-high tumor (5%) and three patients had an MSI-low tumor (3.8%). Analysis of tumor mutational load, which was converted to tumor mutational burden (TMB), was performed in fourteen patients from August to December 2018. TMB low was observed in six patients, TMB intermediate and TMB high in two patients each. In concordance, the two patients with TMB-high were also MSI-high. The two patients with TMB-intermediate and six patients with TMB low were all reported as MSS, see .
Table 4. MSI-TMB table.
Targeted therapy based on the tumor genomic sequencing
Two patients (2.5%) received targeted therapy based on the genomic sequencing results. One patient received targeted therapy based on the MAP2K1 mutation. Besides the MAP2K1 mutation, the patient had a TP53 mutation, SMAD4 mutation, FBXW7 mutation, and no KRAS mutation. This patient received a MEK inhibitor, trametinib, and an EGFR inhibitor, panitumumab. Treatment was discontinued after 2 cycles due to fatigue, skin toxicity, dyspnea, and nausea. The patient received a high dose of prednisolone due to skin toxicity. Computed tomography (CT) scan after the two cycles of panitumumab and trametinib showed stable disease. The second patient received trastuzumab and lapatinib based on ERBB2 amplification confirmed by HER2 positive expression in immunohistochemistry. In addition to ERBB2 amplification (CNV 54.7), the patient had these cancer driver mutations: CDK6 amplification (CNV 4.9), EGFR amplification (CNV 4.9), BRAF amplification (CNV 4.8), and no KRAS mutation. CT scan revealed progressive disease after only two cycles and the patient discontinued the treatment combination.
Four patients had an MSI-high tumor and two of these patients had also a TMB-high. The two patients with MSI-high and TMB-high received immunotherapy with pembrolizumab as this was an FDA-approved therapy. One MSI-high patient had a PS 2 and was too poor for further therapies and the fourth MSI-high patient received experimental therapy in a first-in-human trial, independent of the genomic signature. Twenty-one patients (26.3%) were treated in phase 1 and phase 2 clinical trials. The trials were either investigator-initiated or company driven trials, such as adoptive T-cell therapy trial for ‘all solid cancer’ (EudraCT 2017-002323-25), oral irinotecan plus capecitabine trial (EudraCT 2014-005584-32, NCT03295084), Foxy 5 trial (EudraCT 2012-004200-35, NCT02020291 and EudraCT 2015-004767-36, NCT02655952), TAS-102 trial (EudraCT 2016-005241-23) and hepatic arterial infusion (HAI) trial (EudraCT 2011-000273-31, NCT01511146), all independent from the genomic sequencing results. Overall, a total of 25 patients (31.3%) received systemic therapy, either matched therapy, treatment in clinical trials, or as an FDA-approved therapy.
Discussion
The patients referred to the ECTU are only a small fraction of the CRC patients at our hospital. Several criteria need to be fulfilled to have genomic sequencing performed, such as no major co-morbidity, preferable PS 0–1, and refractory to available standard therapies. Even though about 500 CRC patients are referred to our department of oncology every year only a few patients fulfill these criteria. Our data demonstrate that genomic sequencing by NGS enables detection of cancer driver mutations in most of the patients and that there is a reasonable potential for precision medicine in CRC as ∼90% of the treatment-refractory patients harbor at least one driver mutation. For the time being, however, only a few mutations in CRC are matched with targeted drugs and therefore a unique demand for more biological treatments is needed to improve treatment and prognosis for mCRC. We acknowledge that only two patients harbored an actionable driver mutation and received targeted therapy. The first patient with the MAP2K1 mutation stopped therapy after only two cycles of panitumumab and trametinib, due to skin toxicity and fatigue, even though his CT scan demonstrated stable disease. It is furthermore known that MAP2K1 may be a resistance marker of EGFR inhibitors [Citation32]. Inhibition of the EGFR pathway with an EGFR inhibitor and MEK inhibitor was investigated in a phase 2 trial for RAS/RAF wild type mCRC, where the objective response rate was found to be 38%, but the tolerability of the treatment combination was poor [Citation33], which was in concordance with our patient. The second patient with an ERBB2 amplification, who progressed after only two cycles of HER2-directed therapy, had additionally a BRAF amplification and this could potentially represent a resistance mechanism to the HER2 inhibition [Citation34]. Heterogeneity of the tumor could potentially be another possible resistance mechanism [Citation35].
In the IMPACT trial (n = 1144) patients given matched targeted therapy had a significantly longer overall survival in comparison to non-matched patients (OS; 13.4 vs. 9.0 months, p = 0.017) [Citation36,Citation37]. However, the trial results are not comparable to the results of this retrospective analysis, as patients in the IMPACT trial received a variety of regimes, such as AKT and mTOR inhibitors for PIK3CA mutations and PTEN loss and MEK inhibitor to patients with a GNAQ, RAS, or BRAF mutation. In the French Moscato 01 trial, a survival benefit was also observed for patients who received targeted therapy, with the most frequent actionable targets reported to be PIK3CA, ERBB2, PTEN, FGFR1, EGFR, NOTCH 1/2/3/4 [Citation38]. The Copenhagen Precision Medicine (CoPPO) trial reported that 25% of the CRC patients were identified with an actionable mutation [Citation39]. A discrepancy between an identified potential actionable target mutation, real actionable driver mutations, and institutional relevant mutations was described. It is therefore difficult to compare results from matched therapy trials, as the definition and various possibilities of targeted therapy vary at different sites. Today genomic variants are assessed in accordance with the Tier classification [Citation40] and the ESMO Scale for Clinical Actionability of molecular Targets (ESCAT) classification [Citation41], which is a great help for the assessment of genomic variants and the possibility for targeted drugs.
Promising targets for mCRC are the kinase fusions NTRK-, ALK-, and ROS fusions, however, these alterations are only observed in 0.5–2.0% of the mCRC patients [Citation42,Citation43]. No NTRK fusions were observed in our patient population during this period. ERBB2 amplification and HER2 overexpression are observed in ∼2% of the CRC patients and are described in the Heracles trials and MyPathway trial as a possible druggable target for CRC [Citation44–49]. Furthermore, KRASG12C inhibition has been investigated in a phase 1 trial, where a response rate of 7.1%, was described for heavily pretreated mCRC and NSCLC patients [Citation50,Citation51]. Homologous recombinant DNA damage repair (HR-DDR) deficiencies are identified in mCRC, but no HR-DRR directed therapies have yet been approved for mCRC [Citation52]. Genomic sequencing is a need for the development, understanding, and improvement of personalized medicine. In Denmark, a national trial of targeted therapy, the ProTarget trial (EudraCT 2019-004771-40, NCT04341181), has been initiated to enable targeted therapies to cancer patients with specific drugable genomic variants. The trial is inspired by the Dutch DRUP trial, EudraCT 2015-004398-33, and the TAPUR trial, NCT0263535 [Citation53].
There might be a great potential of targeted therapies for mCRC as 90% of the patients harbor at minimum one cancer driver mutation, however, the balance between sensitivity and resistance is complex. In a Norwegian study (n = 26) genomic sequence results were reviewed and genetic variants were assessed in relation to targeted drug sensitivity and resistance [Citation52]. Eight out of 22 patients with a genomic driver mutation did not receive targeted therapy due to the presence of resistance variants, why resistance mechanisms are important to identify for the optimal targeted therapy. The interpretation of genomic variants for precision medicine and enrollment of patients with a specific mutation in early clinical trials is needed for a strong validation of new targeted drugs. In CRC, four subtypes have been identified, based on distinct molecular and microenvironmental features of the tumor [Citation54,Citation55]. Such subtype grouping of tumors may increase the understanding of cancer biology, targeted therapies, and resistance mechanisms in CRC, promoting new drug development for precision medicine [Citation56–58]. Harmonization of interpretation of genomic variants for precision medicine and enrollment of patients with specific mutations in clinical trials is needed for a strong validation of new matched therapies. Maybe genomic sequencing should be performed earlier in the disease course, to ensure that patients with drugable mutations could be enrolled into trials at an earlier time point, when they are in a good condition and not too weak from their cancer disease.
Conclusion
Most mCRC patients refractory to standard anti-neoplastic therapies, presented with a cancer driver mutation, however, only a few of these mutations gave rise to matched therapies as only 2.5% of the patients from this period received targeted therapy.
Author contributions
R.L.E., S.T., D.N., and E.H.: provided design of the work, planning eCRF for data, and interpretation of data. R.L.E., K.S.S., and P.G.: collection of data. R.L.E. and I.M.C.: drafted the paper. K.L., T.L., T.S.P., and E.H.: drafted the methods section of the paper. All authors have contributed to the interpretation of data, contributed to the revision of the paper, and approved the submitted manuscript.
Supplemental Material
Download MS Excel (16.1 KB)Disclosure statement
RLE and IMC are members of an expert tumorboard organised by Amgen.
Related Research Data
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421.
- Sicklick JK, Kato S, Okamura R, et al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med. 2019;25(5):744–750.
- Jardim DL, Schwaederle M, Wei C, et al. Impact of a biomarker-based strategy on oncology drug development: a meta-analysis of clinical trials leading to FDA approval. J Natl Cancer Inst. 2015;107(11):djv253.
- Moscow JA, Fojo T, Schilsky RL. The evidence framework for precision cancer medicine. Nat Rev Clin Oncol. 2018;15(3):183–192.
- Kumar-Sinha C, Chinnaiyan AM. Precision oncology in the age of integrative genomics. Nat Biotechnol. 2018;36(1):46–60.
- Yurkovich JT, Tian Q, Price ND, et al. A systems approach to clinical oncology uses deep phenotyping to deliver personalized care. Nat Rev Clin Oncol. 2020;17(3):183–194.
- Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
- Molinari C, Marisi G, Passardi A, et al. Heterogeneity in colorectal cancer: a challenge for personalized medicine? Int J Mol Sci. 2018;19(12):3733.
- Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351(4):337–345.
- Douillard JY, Siena S, Cassidy J, et al. Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol. 2014;25(7):1346–1355.
- Saltz LB, Clarke S, Diaz-Rubio E, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26(12):2013–2019.
- Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):303–312.
- Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–1191.
- Overman MJ, Lonardi S, Wong KYM, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36(8):773–779.
- Andre T, Shiu KK, Kim TW, et al. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N Engl J Med. 2020;383(23):2207–2218.
- Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520.
- Le DT, Kim TW, Van Cutsem E, et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: KEYNOTE-164. J Clin Oncol. 2020;38(1):11–19.
- Cremolini C, Loupakis F, Antoniotti C, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015;16(13):1306–1315.
- Kopetz S, Grothey A, Yaeger R, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med. 2019;381(17):1632–1643.
- Kopetz S, Grothey A, Van Cutsem E, et al. BEACON CRC: a randomized, 3-Arm, phase 3 study of encorafenib and cetuximab with or without binimetinib vs. choice of either irinotecan or FOLFIRI plus cetuximab in BRAF V600E-mutant metastatic colorectal cancer. Ann Oncol. 2019;30(Suppl 4):iv154.
- Kopetz S, Desai J, Chan E, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33(34):4032–4038.
- Corcoran RB, Atreya CE, Falchook GS, et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J Clin Oncol. 2015;33(34):4023–4031.
- Hovelson DH, McDaniel AS, Cani AK, et al. Development and validation of a scalable next-generation sequencing system for assessing relevant somatic variants in solid tumors. Neoplasia. 2015;17(4):385–399. Apr
- Dehghani M, Rosenblatt KP, Li L, et al. Validation and clinical applications of a comprehensive next generation sequencing system for molecular characterization of solid cancer tissues. Front Mol Biosci. 2019;6(82):82.
- Sakai K, Takeda M, Shimizu S, et al. A comparative study of curated contents by knowledge-based curation system in cancer clinical sequencing. Sci Rep. 2019;9(1):11340.
- Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26.
- Chaudhary R, Quagliata L, Martin JP, et al. A scalable solution for tumor mutational burden from formalin-fixed, paraffin-embedded samples using the oncomine tumor mutation load assay. Transl Lung Cancer Res. 2018;7(6):616–630.
- Alborelli I, Leonards K, Rothschild SI, et al. Tumor mutational burden assessed by targeted NGS predicts clinical benefit from immune checkpoint inhibitors in non-small cell lung cancer. J Pathol. 2020;250(1):19–29.
- Karolchik D, Barber GP, Casper J, et al. The UCSC genome browser database: 2014 update. Nucleic Acids Res. 2014;42(Database issue):D764–D770.
- Lee C, Bae JS, Ryu GH, et al. A method to evaluate the quality of clinical gene-panel sequencing data for single-nucleotide variant detection. J Mol Diagn. 2017;19(5):651–658.
- Mauri G, Arena S, Siena S, et al. The DNA damage response pathway as a land of therapeutic opportunities for colorectal cancer. Ann Oncol. 2020;31(9):1135–1147.
- Sveen A, Kopetz S, Lothe RA. Biomarker-guided therapy for colorectal cancer: strength in complexity. Nat Rev Clin Oncol. 2020;17(1):11–32.
- Alshammari K, Aung KL, Zhang T, et al. Phase II trial of trametinib and panitumumab in RAS/RAF wild type metastatic colorectal cancer. Clin Colorectal Cancer. 2021. DOI:10.1016/j.clcc.2021.07.004
- Pohlmann PR, Mayer IA, Mernaugh R. Resistance to trastuzumab in breast cancer. Clin Cancer Res. 2009;15(24):7479–7491.
- Marusyk A, Janiszewska M, Polyak K. Intratumor heterogeneity: the Rosetta stone of therapy resistance. Cancer Cell. 2020;37(4):471–484.
- Tsimberidou AM, Iskander NG, Hong DS, et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res. 2012;18(22):6373–6383.
- Tsimberidou AM, Hong DS, Ye Y, et al. Initiative for molecular profiling and advanced cancer therapy (IMPACT): an MD Anderson precision medicine study. J Clin Oncol Precis Oncol. 2017;2017:1–18.
- Massard C, Michiels S, Ferte C, et al. High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: results of the MOSCATO 01 trial. Cancer Discov. 2017;7(6):586–595.
- Tuxen IV, Rohrberg KS, Oestrup O, et al. Copenhagen prospective personalized oncology (CoPPO)-clinical utility of using molecular profiling to select patients to phase I trials. Clin Cancer Res. 2019;25(4):1239–1247.
- Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19(1):4–23.
- Mateo J, Chakravarty D, Dienstmann R, et al. A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO scale for clinical actionability of molecular targets (ESCAT). Ann Oncol. 2018;29(9):1895–1902.
- Pietrantonio F, Di Nicolantonio F, Schrock AB, et al. ALK, ROS1, and NTRK rearrangements in metastatic colorectal cancer. J Natl Cancer Inst. 2017;109(12):djx089.
- Ricciuti B, Genova C, Crino L, et al. Antitumor activity of larotrectinib in tumors harboring NTRK gene fusions: a short review on the current evidence. OTT. 2019;12:3171–3179.
- Greally M, Kelly CM, Cercek A. HER2: an emerging target in colorectal cancer. Curr Probl Cancer. 2018;42(6):560–571.
- Sartore-Bianchi A, Trusolino L, Martino C, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17(6):738–746.
- Sartore-Bianchi A, Lonardi S, Martino C, et al. Pertuzumab and trastuzumab emtansine in patients with HER2-amplified metastatic colorectal cancer: the phase II HERACLES-B trial. ESMO Open. 2020;5(5):e000911.
- Meric-Bernstam F, Hurwitz H, Raghav KPS, et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): an updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019;20(4):518–530.
- Sartore-Bianchi A, Amatu A, Porcu L, et al. HER2 positivity predicts unresponsiveness to EGFR-targeted treatment in metastatic colorectal cancer. Oncologist. 2019;24(10):1395–1402.
- Valtorta E, Martino C, Sartore-Bianchi A, et al. Assessment of a HER2 scoring system for colorectal cancer: results from a validation study. Mod Pathol. 2015;28(11):1481–1491.
- Hong DS, Fakih MG, Strickler JH, et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med. 2020;383(13):1207–1217.
- Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–223.
- Ree AH, Nygaard V, Boye K, et al. Molecularly matched therapy in the context of sensitivity, resistance, and safety; patient outcomes in end-stage cancer – the MetAction study. Acta Oncol. 2020;59(7):733–740.
- Mangat PK, Halabi S, Bruinooge SS, et al. Rationale and design of the targeted agent and profiling utilization registry (TAPUR) study. J Clin Oncol Precis Oncol. 2018;2018. DOI:10.1200/PO.18.00122
- Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21(11):1350–1356.
- Stintzing S, Wirapati P, Lenz HJ, et al. Consensus molecular subgroups (CMS) of colorectal cancer (CRC) and first-line efficacy of FOLFIRI plus cetuximab or bevacizumab in the FIRE3 (AIO KRK-0306) trial. Ann Oncol. 2019;30(11):1796–1803.
- Joung JG, Oh BY, Hong HK, et al. Tumor heterogeneity predicts metastatic potential in colorectal cancer. Clin Cancer Res. 2017;23(23):7209–7216.
- Schwaederle M, Zhao M, Lee JJ, et al. Impact of precision medicine in diverse cancers: a meta-analysis of phase II clinical trials. J Clin Oncol. 2015;33(32):3817–3825.
- Von Hoff DD, Stephenson JJ Jr., Rosen P, et al. Pilot study using molecular profiling of patients' tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol. 2010;28(33):4877–4883.