
Abstract
Background
Ulceration is an independent adverse prognostic factor in cutaneous malignant melanoma (CMM). There is, however, a need for additional prognostic markers to identify patients with ulcerated stage I–II CMM who have a high-risk for recurrence. The aim of this study was to examine the prognostic impact of BRAF mutation, proliferation and presence of tumour infiltrating lymphocytes (TILs) in primary ulcerated CMM.
Material and methods
We have used a consecutive cohort consisting of 71 primary ulcerated CMM (T1b–T4b). BRAF mutation was detected using Cobas test and pyrosequencing. Protein expression of the proliferation marker Ki67 was analysed using immunohistochemistry. Presence of TILs was evaluated in representative hematoxylin-eosin stained formalin-fixed paraffin-embedded tumour sections.
Results
Proportion of BRAF mutated alleles, proliferation and presence of TILs all had a statistically significant impact on recurrence free survival in univariate analyses (HR 2.44, 95% CI 1.23–4.84, p = 0.011; HR 2.66, 95% CI 1.32–5.35, p = 0.006 respectively HR 0.48, 95% CI 0.24–0.98, p = 0.045). A trend test found a statistically significant decrease in the proportion of recurrence by including the three favourable factors (BRAF wildtype/low proportion of BRAF mutated alleles, low proliferation and high presence of TILs) (p = 0.0004). When at least two out of three factors were present there was a statistically significant association with longer recurrence free survival in the multivariate analysis (HR 0.30, 95% CI 0.15–0.61, p = 0.001) when adjusted for Breslow thickness, an established independent prognostic marker for CMM.
Conclusion
Thus, this panel of markers could be an interesting novel concept for predicting the clinical outcome in patients with high-risk stage I–II ulcerated CMM.
Background
There are several established clinicopathological prognostic markers in cutaneous malignant melanoma (CMM), which are included in the AJCC staging system for CMM [Citation1]. For primary tumours Breslow thickness and presence of ulceration are established independent prognostic markers [Citation1].
The presence of ulceration in CMM impairs the prognosis remarkably. This is illustrated by classification of ulcerated primary tumours in a more advanced sublevel of the tumour stage (T1b–T4b) in the TNM system than non-ulcerated primary tumours (T1a–T4a) [Citation1]. Ulceration of CMM is based on pathological microscopic examination and is defined as loss of epidermis in combination with a host reaction of inflammatory cells (neutrophils and/or fibrin deposition) in the absence of trauma [Citation2].
Ulceration is more often present in CMMs with a higher Breslow thickness and in nodular CMM [Citation3]. Higher expression of genes/proteins associated with tumour-related inflammation, proliferation, and angiogenesis, has been shown in ulcerated CMM [Citation4–7].
There are several studies correlating elevated expression of genes/proteins related to proliferation, including Ki67, with a worse clinical outcome in CMMs [Citation6–10]. It has been implied that higher tumour cell proliferation may be an underlying or a contributing factor to ulceration in CMM [Citation11]. A suggested precursor of ulceration has been associated with increased proliferation of CMM cells and consumption of the epidermis, without increased inflammatory response, indicating this as an early event [Citation12].
In general, immunological host response is considered to be prognostically favourable. Several studies have demonstrated that presence of tumour infiltrating lymphocytes (TILs) is associated with a better outcome and may promote spontaneous tumour regression [Citation13,Citation14]. There seems to be a difference in immune microenvironment between ulcerated and non-ulcerated CMMs. It has been shown that presence of TILs is associated with a better clinical outcome only in ulcerated CMM [Citation15].
Several studies have investigated the prognostic value of activating BRAF V600 mutations in CMM. Some studies have found an association of BRAF mutation with disease-free or melanoma-specific overall survival (OS) [Citation16–18], while other studies have not [Citation19–21]. In a recent meta-analysis, a significant association of BRAF mutation to OS was found, but not for stage I–II cohorts [Citation22]. The impact of BRAF mutation on outcome remains unclear and has also been associated with presence of ulceration [Citation23]. In a previous publication from our research group we described that ulceration was associated with poor outcome only when the primary CMM carried a BRAF mutation [Citation24].
It is well-known that intra- and inter-tumour heterogeneity is commonly observed in CMM [Citation25]. The presence of sub-clones with different BRAF genotypes has been demonstrated in primary CMM [Citation26,Citation27]. The quantitative proportion of BRAF mutated alleles in a primary tumour may therefore be relevant for prognosis.
The primary aim of the present study was to validate our previous results regarding the prognostic impact of BRAF mutation on ulcerated primary CMM in a large consecutive cohort of ulcerated primary CMM. The secondary aim of our study was to examine the prognostic impact of a panel of markers including BRAF mutation, Ki67 expression and presence of TILs in ulcerated primary CMM, since we have in two previous studies [Citation28,Citation29] demonstrated that a panel of prognostic markers is stronger than any single marker in stage III CMM.
Materials and methods
Patient cohort
A consecutive cohort of 120 ulcerated primary CMM diagnosed during 2004–2006 was identified from the Stockholm-Gotland Regional Melanoma Registry. From this cohort we collected 76 primary tumour samples, pT1b–pT4b, from pathology departments in Stockholm. The specimens have been re-evaluated by a senior melanoma pathologist (L.K) to verify ulceration status. Three samples were excluded after re-evaluation due to lack of ulceration or suitable specimen for examination. In total, 73 ulcerated tumours were included in further analyses in this study. The tumours were classified as stage IB (n = 5), IIA (n = 13), IIB (n = 26) and IIC (n = 29) according to the AJCC version 8.
Data on pathological parameters and follow-up information was obtained from the Regional Melanoma Registry data base, patient records and pathology files. Sentinel node biopsies were not performed, as that was not a standard procedure at the time of diagnosis. The first CMM specific event, such as local, regional or distant recurrence was registered. Two subjects have moved to another region in Sweden, hence follow-up information on recurrence was lost.
This study has been approved by the Research Ethics Committee of Karolinska Insitutet and they waived the requirement for informed consent. The study was performed in accordance with the Declaration of Helsinki. No individual patient data is available for sharing.
For additional information, see supplementary material and methods.
Results
Patient characteristics
Overall, 76 CMM patients were included in this study but five were excluded from the final analyses, three cases where ulceration could not be confirmed and two patients with lack of follow-up data. Details about 71 patients and pathological characteristics are shown in . The median follow-up time was 61 months (range 2–148 months). Recurrence at any site (local, regional lymph nodes or distant) was registered in 48% (34/71) of the subjects. Of these, 59% (20/34) had loco-regional relapse and 41% (14/34) had disseminated disease as their first recurrence. Among patients with locoregional recurrence 70% (14/20) later developed disseminated disease. Among the 34 subjects with recurrence 79% died of their disease. The median time of recurrence-free survival (RFS) was 15.5 months (range 2–70) among the subjects with recurrence. There was a shorter median RFS among the subjects with loco-regional relapse compared to the subjects with disseminated disease as their first recurrence, 9.5 months (range 2–61) and 29.5 months (range 8–70), respectively.
Table 1. Patient and pathological characteristics of 71 cases with ulcerated primary CMM.
Breslow thickness was a statistically significant adverse prognostic factor (p < 0.001; ) while there were no significant correlations to recurrence regarding gender, age at diagnosis and histological subtype.
Higher presence of TILs in tumours from recurrence-free CMM patients
Presence of TILs was evaluated () in the 71 ulcerated primary CMM. Thirty-four of 70 (49%) tumours had TILs infiltration, one was not evaluable. There was a statistically significantly higher presence of TILs within the dermal CMM clusters in tumours from patients without recurrence (HR 0.48, 95% CI 0.24–0.98, p = 0.045) ().
Figure 1. Representative hematoxylin-eosin slides of primary ulcerated CMMs demonstrating presence of TILs (a) and representative expression of protein Ki67 with immunohistochemistry demonstrating high proportion (b) and low proportion (c). Twenty-seven percent (10/37) of the recurrence-free patients and 61% (20/33) of the patients with a recurrence had primary tumours with high Ki67 staining (cut-off level 10%). Scale bar = 50 µm.
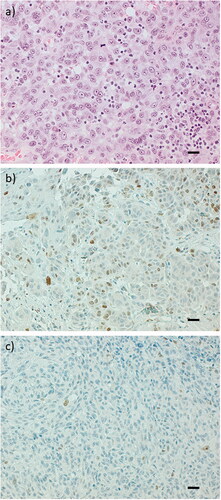
Table 2. Cox regression analysis.
Longer RFS among CMM cases with BRAF wild type/low proportion of BRAF mutated alleles
BRAF mutation screening was performed using both the Cobas test and pyrosequencing on all primary tumour samples (n = 71). Overall, a BRAF mutation was detected in 35/71 (49%) of the cases, all of them detected by pyrosequencing, while the Cobas test identified a BRAF mutation in 29/71(41%) of tumour samples. Hence six samples (17%) with BRAF mutation were missed by the Cobas test. Sixty-nine % (n = 24) of the BRAF mutated tumours were codon V600E and 20% (n = 7) were codon V600K. Four samples (11%) had a BRAF mutation in exon 11(codon G464E, G466E, G469A and G469E) which could not be detected by Cobas. However, two of the BRAF mutations missed by Cobas were, V600K and a rare complex mutation subtype of V600E (V600E2), respectively.
For the samples where no mutations were detected by Cobas or pyrosequencing or only by one of these methods (n = 42), Sanger sequencing was performed to detect mutations at exons 11 and 15 of the BRAF gene. Sanger sequencing confirmed the mutations detected by pyrosequencing as well as detecting a rare 3 base pair TGA deletion (VK600-601E) that Cobas and pyrosequencing did not.
The proportion of BRAF mutated alleles in the tumour specimens was assessed by pyrosequencing. There was a range from 7% to 83% of BRAF mutated alleles, with a median of 35%. When necessary dissection was performed to enrich tumour cells but the differences in proportion of BRAF mutated alleles could partially be due to contamination of normal cells. However, examination of the assessed areas of the hematoxylin-eosin stained slides indicated that this could not be the full explanation ().
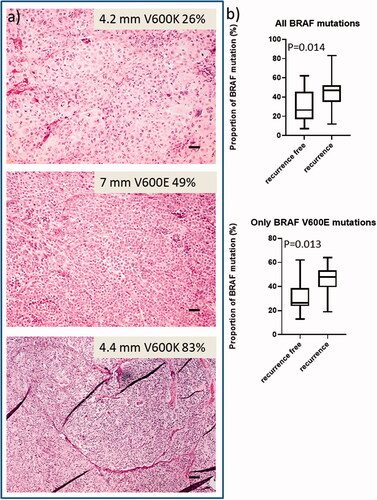
Figure 2. Representative hematoxylin-eosin slides of ulcerated primary CMMs showing the proportion of tumour cell content in relation to proportion of BRAF mutated alleles (a). Box plots demonstrate the distribution of proportion of all BRAF mutated alleles (recurrence free n = 16; recurrence n = 19) and BRAF V600E only (recurrence free n = 10; recurrence n = 13), with no respect to BRAF wildtype samples and not either to non-BRAF V600E in the bottom graph. There is a statistically significant difference in the distribution between patients who had recurrence and those who were recurrence-free (b). (t-test p = 0.014 and p = 0.013 respectively). Scale bar = 250 µm.
In this study cohort we did not observe any correlation between RFS and presence of BRAF mutation/deletion (HR 1.14, 95% CI 0.58–2.24, p = 0.71). We also grouped cases with BRAF wildtype and low proportion of BRAF mutated alleles ≤35% (median) together as a BRAF wildtype/low proportion of BRAF mutated alleles group and compared it to cases with a high proportion of BRAF mutated alleles >35%. Seventy-four % (52/70) of the tumours were assigned to the group with BRAF wild type/low proportion of BRAF mutated alleles, and 26% (18/70) to the group with high proportion of BRAF mutated alleles, respectively, not including the case with the deletion, since it could not be detected/quantitated by pyrosequencing. We found a statistically significantly shorter RFS among cases with a high proportion of BRAF mutated alleles than among cases with BRAF wild type/low proportion of BRAF mutated alleles t (HR 2.44, 95% CI 1.23–4.84, p = 0.011). Also, we could demonstrate that the distribution of proportion of BRAF mutated alleles statistically significantly differ between patients who had recurrence and those who were recurrence-free which is shown in for all BRAF mutations and BRAF V600E mutations only (t-test p = 0.014 respectively p = 0.013).
Lower Ki67 expression among recurrence-free CMM subjects
We assessed tumour proliferation by Ki67 in 71 tumours using immunohistochemistry. One sample was not evaluable. Representative images of tumour samples with high or low Ki67 staining are shown in . Higher Ki67 expression was associated with shorter RFS (HR 2.65, 95% CI 1.32–5.35; p = 0.006).
Ki67 expression was statistically significantly lower in the group of BRAF wildtype/low proportion of BRAF mutated alleles (35%, 18/52) compared with the group of high proportion of BRAF mutated alleles (65%, 11/17) (Fisher’s exact test, p = 0.046). There was a higher proportion of high Ki67 expression among subjects with loco-regional relapse (70%) compared to those with disseminated disease as their first relapse (33%), (p = 0.068).
The combination of presence of TILs, BRAF wildtype/low proportion of BRAF mutated alleles and low Ki67 expression in a panel is a better prognostic biomarker for recurrence of CMM than any factor alone
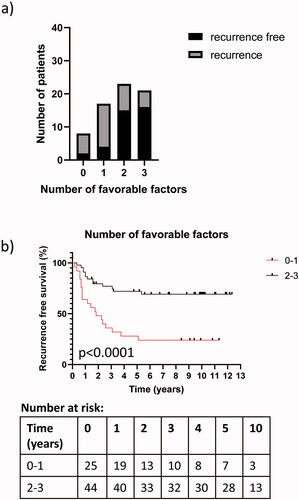
We wanted to investigate if a panel of potentially favourable prognostic factors that were significant in univariate analysis; including presence of TILs, low expression of Ki67 and BRAF wildtype/low proportion of BRAF mutated alleles, has a higher prognostic impact compared to any factor alone. We performed a test of trend and found a statistically significant decrease in the proportion of recurrence by including the potentially favourable factors in the analysis (p < 0.0004) (), supporting the relevance of using a panel of multiple prognostic factors. The combination of BRAF wildtype/low proportion of BRAF mutated alleles, low expression of Ki67 and presence of TILs in univariate analysis became statistically significant when at least two out of three factors were present (HR 0.28, 95% CI 0.14–0.56, p < 0.001). Presence of at least two factors was associated with longer RFS (). Hence this prognostic panel of three factors was stronger compared to any single factor alone ().
Figure 3. The prognostic power of a panel consisting of potentially three favourable prognostic factors; including BRAF wildtype/low proportion of BRAF mutated alleles, low expression of Ki67 and presence of TILs. A statistically significant increase in the proportion of patients with no recurrence in association with the increase from one to three factors was observed (a). (Chi-square test for trend; p = 0.0004). Kaplan-Meier curves showing a statistically significantly longer RFS for patients with presence of at least two favourable prognostic factors (b) (Log-rank (Mantel-Cox) test; p < 0.0001) and number at risk. The marks on the lines represent censored points.
The three factor panel is an independent prognostic biomarker for recurrence of CMM
The factors statistically significantly associated with RFS in univariate analysis were included in a multivariate analysis; i.e., Breslow thickness, TILs and BRAF wildtype/low proportion of BRAF mutated alleles and low Ki67 expression. Only Ki67 expression (HR 2.21, 95% CI 1.02–4.77, p = 0.043) and Breslow thickness (HR 1.15, 95% CI 1.05–1.26, p = 0.003) remained statistically significant ().
When at least two out of three factors were present, it remained statistically significant in the multivariate analysis when adjusted for Breslow (HR 0.30, 95% CI 0.15–0.61, p = 0.001). Also Breslow thickness remained statistically significant (HR 1.13, 95% CI 1.03–1.23, p = 0.008).
Discussion
We have validated our previous findings regarding the association of ulceration with an adverse outcome in tumours carrying BRAF mutation [Citation24], but could only confirm the association when considering cases with a high proportion, >35%, of BRAF mutated alleles. We have also demonstrated that a panel of favourable prognostic markers was an independent positive predictor of RFS in ulcerated stage I–II CMM and was stronger than any single marker, in agreement with our two previous studies on stage III CMM [Citation28,Citation29].
There is to our knowledge no previous publication investigating the prognostic impact of the proportion of BRAF mutated alleles in primary CMM. This is a newer concept of describing in a semi-quantitative manner BRAF mutational status in CMM in relation to outcome. Satzger et al. made an attempt to correlate allele frequencies of V600E mutation in CMM to outcome of BRAF inhibitor therapy [Citation30]. They found no significant correlation but used a cut-off of 18% around the detection limit of 15–20% for Sanger sequencing. Moreover, they demonstrated the problem of tumour heterogeneity in metastatic CMM, since matched primary and metastatic tumours differed > three-fold regarding proportion of mutated alleles in 23% of the cases. It is reasonable to assume that differences in proportion of BRAF mutated alleles could have some impact on the tumour biology. There was a wide range of the proportion of BRAF mutated alleles (7–83%) in our study, which has also been observed by others using deep NGS or pyrosequencing technology [Citation27,Citation30] suggesting that gain or loss of the mutant or wildtype alleles may occur frequently in CMM. It has been demonstrated that amplification of BRAF V600E can lead to resistance to BRAF inhibitors [Citation31]. In addition, several publications have described inter-tumour heterogeneity regarding BRAF mutation [Citation26,Citation32,Citation33], while other publications have demonstrated that BRAF mutation inter-tumour heterogeneity is rare [Citation30,Citation34,Citation35]. Yancovitz et al. showed high intra-tumour heterogeneity regarding BRAF V600E mutations, 4/9 primary CMM tumours showed pronounced differing proportions of BRAF mutation in the different subclones [Citation25]. In a meta-analysis including 22 studies a discrepancy between primary and metastatic lesions in melanoma was 13.4% [Citation36]. Hence, a BRAF mutated clone in primary CMM may not always be the driver of the metastatic process.
The contradictory results between different studies regarding the prognostic value of BRAF mutations in CMM may be dependent on several different factors such as stage, other genetic alterations etc. and whether the endpoint was RFS or OS. It has been demonstrated that BRAF mutated primary tumours are associated with a distinct clinical phenotype, with a higher proportion of ulceration and regional lymph node metastases [Citation20], features known to correlate with an aggressive tumour behaviour. However, several studies have not shown that BRAF mutation in stage I–IV CMM is associated with a worse outcome [Citation20,Citation37–39]. These studies differed regarding type of patient cohorts (with mixed samples from different tumour stages) and whether the study was prospective or retrospective, which may affect the results. It has been shown that BRAF-V600E expression in primary tumours, analysed by IHC, was an independent prognostic factor associated with a reduced survival, whereas BRAF mutation status by itself was not significant [Citation40]. Twelve % of the BRAF V600E mutant tumours were false negative for IHC. It may be due to low abundance of BRAF V600E mutation. In another study low allele frequency of BRAF V600E mutation could be an explanation for some of the observed discordant results between pyrosequencing and IHC [Citation41] which suggests that the proportion of BRAF V600E mutation may have an impact. The proportion of BRAF mutant allele has also been demonstrated to have an impact on progression free survival in melanoma treated with MAPK inhibitors [Citation42]. There are also data indicating that the prognostic impact of BRAF mutation is dependent on co-existence with additional factors in primary tumours, for example TERT-promoter mutations [Citation43].
We have used different methods for detecting BRAF mutation in this study. The Cobas assay is primarily designed to detect BRAF V600E mutation, with a sensitivity of 5% mutation rate in a wildtype background of tumour DNA (Cobas®4800 BRAF V600 Mutation test kit manual). This PCR method is not designed for analysis of V600K and other rare BRAF mutations, although there is a detection of BRAF mutations in the same codon (V600 K and D) to some extent.
There is an estimated cross-reactivity of the Cobas test of 30% for V600K mutation samples with more than 50% of tumoural tissue content according to the manufacturer (Cobas®4800 BRAF V600 Mutation test kit manual). Therefore, it was not surprising that one of the six mutations that were missed by Cobas was a V600K. For the complex mutation V600E2 the detection level is higher than 68%. It is of interest to assess the different BRAF mutation genotypes as there are data supporting that tumours with other genotypes than V600E, like V600K and V600R, may also benefit from therapy with BRAF inhibitors [Citation44,Citation45] but also other more rare mutations [Citation46,Citation47], which may have implications also in an adjuvant setting at diagnosis. Our results showing prognostic impact of the proportion of BRAF mutated alleles give support for using pyrosequencing as the method for BRAF mutation detection. Pyrosequencing has been shown to be the most efficient method also in other studies when comparing different methods for BRAF mutation detection [Citation48].
There are differences in the study design between our previous and present studies which may contribute to the different results regarding impact of BRAF mutation on clinical outcome. The previous study by Jovanovic et al. had a small set of ulcerated primary CMM tumours (28%; 16/57) in the total set of cases [Citation24]. The cases were selected regarding Breslow (T1–T2 tumours) and outcome. Our present study was performed on a larger, consecutive and unselected cohort of only ulcerated primary T1–T4 tumours where the majority was T3–T4.
Presence of TILs was associated with a favourable outcome. It has been considered that the inflammatory microenvironment may be a contributing factor for increased activity of TILs, hence there may be a greater prognostic impact of TILs in ulcerated than non-ulcerated CMM [Citation15]. Benefit from adjuvant interferon-alpha therapy was reported in high-risk CMM patients with ulcerated tumours [Citation49] supporting the role of the immune microenvironment in ulcerated CMM.
Ki67 is associated with adverse outcome in several malignancies, but is not used routinely in CMM, although there are several publications in stage I–II CMM demonstrating an adverse prognostic impact [Citation6,Citation7,Citation50]. We have both in a previous and the present study support that Ki67 is an important prognostic marker when predicting the prognosis in stage I–III CMM.
A number of different factors play a role for the development of metastatic disease. A strategy to improve the identification of T1b–T4b patients with high risk of recurrence could be to use a combination of prognostic variables from multiple signalling pathways and biological functions of importance for the CMM disease, including clinicopathological variables, rather than single genes or proteins, as illustrated in this study.
The strength of our study is that we used a large and consecutive cohort of ulcerated primary CMMs. The absence of a non-ulcerated control group is a limitation and is recommended to be used in a further validation study. Although we confirm the favourable impact of TIL on outcome we lack information regarding the value of different TIL phenotypes by not staining for CD8, CD3, FOXP3 etc. Sentinel node biopsy (SNB) was not part of clinical routine during 2004–2006 but today SNB is routinely used as a staging tool for confirmed primary tumours. Some of the tumours in our cohort are expected to have positive SNB and be classified as IIIA CMM if SNB had been performed at the time of diagnoses. That lack of prognostic information is considered to be a limitation of our study. Adjuvant therapy with PD-1 inhibitors or combination of BRAF and MEK inhibitors have recently been introduced for stage III CMM. Today adjuvant therapy is not established for stage IB-IIC CMM, but a phase III study (KEYNOTE-716) will be run to evaluate the clinical benefit of PD-1 inhibitor in high-risk stage II CMM. Thus, this panel of markers may contribute to identify patients with high-risk stage I–II ulcerated CMM who may benefit from receiving adjuvant therapy.
In conclusion, determination of the proportion of BRAF mutated alleles in ulcerated primary CMM could be an interesting novel concept for predicting the clinical outcome that needs to be studied further. We have also demonstrated that a panel of prognostic markers including TILs, proportion of BRAF mutated alleles and Ki67 performs better than any single marker alone in primary ulcerated CMM, which is in line with our previous studies on stage III CMM. To our knowledge there is no previous paper presenting these factors in combination in a cohort of ulcerated primary CMM. A follow-up study to validate these findings in an independent cohort of ulcerated primary CMM is necessary to confirm the impact of these markers on risk for recurrence.
| Abbreviations | ||
| 95% CI | = | 95% confidence interval |
| CMM | = | Cutaneous melanoma |
| SSM | = | superficial spreading melanoma |
| NM | = | nodular melanoma |
| LMM | = | lentigo maligna melanoma |
| TILs | = | Tumour infiltrating lymphocytes |
| RFS | = | recurrence-free survival |
| IHC | = | immunohistochemistry |
Supplemental Material
Download MS Word (23.4 KB)Acknowledgements
We thank pathologists Göran Elmberger and Lorand Kis for valuable cooperation and Marianne Frostvik-Stolt for immunohistochemical analysis.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Gershenwald JE, Scolyer RA, Hess KR, et al. Melanoma staging: evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J Clin. 2017;67(6):472–492.
- Spatz A, Cook MG, Elder DE, et al. Interobserver reproducibility of ulceration assessment in primary cutaneous melanomas. Eur J Cancer. 2003;39(13):1861–1865.
- Warycha MA, Christos PJ, Mazumdar M, et al. Changes in the presentation of nodular and superficial spreading melanomas over 35 years. Cancer. 2008;113(12):3341–3348.
- Jewell R, Elliott F, Laye J, et al. The clinicopathological and gene expression patterns associated with ulceration of primary melanoma. Pigment Cell Melanoma Res. 2015;28(1):94–104.
- Rakosy Z, Ecsedi S, Toth R, et al. Integrative genomics identifies gene signature associated with melanoma ulceration. PLoS One. 2013;8(1):e54958.
- Straume O, Sviland L, Akslen LA. Loss of nuclear p16 protein expression correlates with increased tumor cell proliferation (Ki-67) and poor prognosis in patients with vertical growth phase melanoma. Clin Cancer Res. 2000;6(5):1845–1853.
- Ladstein RG, Bachmann IM, Straume O, et al. Ki-67 expression is superior to mitotic count and novel proliferation markers PHH3, MCM4 and mitosin as a prognostic factor in thick cutaneous melanoma. BMC Cancer. 2010;10:140.
- Jonsson G, Busch C, Knappskog S, et al. Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin Cancer Res. 2010;16(13):3356–3367.
- Bogunovic D, O’Neill DW, Belitskaya-Levy I, et al. Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc Natl Acad Sci USA. 2009;106(48):20429–20434.
- Winnepenninckx V, Lazar V, Michiels S, et al. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J Natl Cancer Inst. 2006;98(7):472–482.
- Scolyer RA, Shaw HM, Thompson JF, et al. Interobserver reproducibility of histopathologic prognostic variables in primary cutaneous melanomas. Am J Surg Pathol. 2003;27(12):1571–1576.
- Bonnelykke-Behrndtz LM, Schmidt H, Damsgaard TE, et al. Consumption of the epidermis: a suggested precursor of ulceration associated with increased proliferation of melanoma cells. Am J Dermatopathol. 2015;37(11):841–845.
- Clemente CG, Mihm MC Jr, Bufalino R, et al. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 1996;77(7):1303–1310.
- Thomas NE, Busam KJ, From L, et al. Tumor-infiltrating lymphocyte grade in primary melanomas is independently associated with melanoma-specific survival in the population-based genes, environment and melanoma study. J Clin Oncol. 2013;31(33):4252–4259.
- de Moll EH, Fu Y, Qian Y, et al. Immune biomarkers are more accurate in prediction of survival in ulcerated than in non-ulcerated primary melanomas. Cancer Immunol Immunother. 2015;64(9):1193–1203.
- Mar VJ, Liu W, Devitt B, et al. The role of BRAF mutations in primary melanoma growth rate and survival. Br J Dermatol. 2015;173(1):76–82.
- Thomas NE, Edmiston SN, Alexander A, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncol. 2015;1(3):359–368.
- Nagore E, Requena C, Traves V, et al. Prognostic value of BRAF mutations in localized cutaneous melanoma. J Am Acad Dermatol. 2014;70(5):858–862.e1–2.
- Heppt MV, Siepmann T, Engel J, et al. Prognostic significance of BRAF and NRAS mutations in melanoma: a German study from routine care. BMC Cancer. 2017;17(1):536.
- Ellerhorst JA, Greene VR, Ekmekcioglu S, et al. Clinical correlates of NRAS and BRAF mutations in primary human melanoma. Clin Cancer Res. 2011;17(2):229–235.
- Akslen LA, Angelini S, Straume O, et al. BRAF and NRAS mutations are frequent in nodular melanoma but are not associated with tumor cell proliferation or patient survival. J Invest Dermatol. 2005;125(2):312–317.
- Ny L, Hernberg M, Nyakas M, et al. BRAF mutational status as a prognostic marker for survival in malignant melanoma: a systematic review and meta-analysis. Acta Oncol. 2020;59(7):833–844.
- Bombonato C, Ribero S, Pozzobon FC, et al. Association between dermoscopic and reflectance confocal microscopy features of cutaneous melanoma with BRAF mutational status. J Eur Acad Dermatol Venereol. 2017;31(4):643–649.
- Jovanovic B, Krockel D, Linden D, et al. Lack of cytoplasmic ERK activation is an independent adverse prognostic factor in primary cutaneous melanoma. J Invest Dermatol. 2008;128(11):2696–2704.
- Harbst K, Staaf J, Masback A, et al. Multiple metastases from cutaneous malignant melanoma patients may display heterogeneous genomic and epigenomic patterns. Melanoma Res. 2010;20(5):381–391.
- Yancovitz M, Litterman A, Yoon J, et al. Intra- and inter-tumor heterogeneity of BRAF(V600E))mutations in primary and metastatic melanoma. PLoS One. 2012;7(1):e29336.
- Helias-Rodzewicz Z, Funck-Brentano E, Baudoux L, et al. Variations of BRAF mutant allele percentage in melanomas. BMC Cancer. 2015;15(1):497.
- Falkenius J, Lundeberg J, Johansson H, et al. High expression of glycolytic and pigment proteins is associated with worse clinical outcome in stage III melanoma. Melanoma Res. 2013;23(6):452–460.
- Falkenius J, Johansson H, Tuominen R, et al. Presence of immune cells, low tumor proliferation and wild type BRAF mutation status is associated with a favourable clinical outcome in stage III cutaneous melanoma. BMC Cancer. 2017;17(1):584.
- Satzger I, Marks L, Kerick M, et al. Allele frequencies of BRAFV600 mutations in primary melanomas and matched metastases and their relevance for BRAF inhibitor therapy in metastatic melanoma. Oncotarget. 2015;6(35):37895–37905.
- Rizos H, Menzies AM, Pupo GM, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20(7):1965–1977.
- Colombino M, Capone M, Lissia A, et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J Clin Oncol. 2012;30(20):2522–2529.
- Heinzerling L, Baiter M, Kuhnapfel S, et al. Mutation landscape in melanoma patients clinical implications of heterogeneity of BRAF mutations. Br J Cancer. 2013;109(11):2833–2841.
- Nielsen LB, Dabrosin N, Sloth K, et al. Concordance in BRAF V600E status over time in malignant melanoma and corresponding metastases. Histopathology. 2018;72(5):814–825.
- Omholt K, Platz A, Kanter L, et al. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9(17):6483–6488.
- Valachis A, Ullenhag GJ. Discrepancy in BRAF status among patients with metastatic malignant melanoma: a meta-analysis. Eur J Cancer. 2017;81:106–115.
- Edlundh-Rose E, Egyhazi S, Omholt K, et al. NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: a study based on mutation screening by pyrosequencing. Melanoma Res. 2006;16(6):471–478.
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239–1246.
- Rutkowski P, Gos A, Jurkowska M, et al. Molecular alterations in clinical stage III cutaneous melanoma: correlation with clinicopathological features and patient outcome. Oncol Lett. 2014;8(1):47–54.
- Hugdahl E, Kalvenes MB, Puntervoll HE, et al. BRAF-V600E expression in primary nodular melanoma is associated with aggressive tumour features and reduced survival. Br J Cancer. 2016;114(7):801–808.
- Pearlstein MV, Zedek DC, Ollila DW, et al. Validation of the VE1 immunostain for the BRAF V600E mutation in melanoma. J Cutan Pathol. 2014;41(9):724–732.
- Stagni C, Zamuner C, Elefanti L, et al. BRAF gene copy number and mutant allele frequency correlate with time to progression in metastatic melanoma patients treated with MAPK inhibitors. Mol Cancer Ther. 2018;17(6):1332–1340.
- Macerola E, Loggini B, Giannini R, et al. Coexistence of TERT promoter and BRAF mutations in cutaneous melanoma is associated with more clinicopathological features of aggressiveness. Virchows Arch. 2015;467(2):177–184.
- Klein O, Clements A, Menzies AM, et al. BRAF inhibitor activity in V600R metastatic melanoma. Eur J Cancer. 2013;49(5):1073–1079.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323–332.
- Busser B, Leccia MT, Gras-Combe G, et al. Identification of a novel complex BRAF mutation associated with major clinical response to vemurafenib in a patient with metastatic melanoma. JAMA Dermatol. 2013;149(12):1403–1406.
- Trudel S, Odolczyk N, Dremaux J, et al. The clinical response to vemurafenib in a patient with a rare BRAFV600DK601del mutation-positive melanoma. BMC Cancer. 2014;14:727.
- Colomba E, Helias-Rodzewicz Z, Von Deimling A, et al. Detection of BRAF p.V600E mutations in melanomas: comparison of four methods argues for sequential use of immunohistochemistry and pyrosequencing. J Mol Diagn. 2013;15(1):94–100.
- Eggermont AM, Suciu S, Testori A, et al. Ulceration and stage are predictive of interferon efficacy in melanoma: results of the phase III adjuvant trials EORTC 18952 and EORTC 18991. Eur J Cancer. 2012;48(2):218–225.
- Gimotty PA, Van Belle P, Elder DE, et al. Biologic and prognostic significance of dermal Ki67 expression, mitoses, and tumorigenicity in thin invasive cutaneous melanoma. J Clin Oncol. 2005;23(31):8048–8056.