
Introduction
The introduction of immunomodulatory drugs (IMiDs) such as thalidomide, and later lenalidomide, together with proteasome inhibitors (PI), has revolutionized the management of multiple myeloma (MM) compared to classical chemotherapy improving the overall survival from a median of 3 up to 8 years [Citation1,Citation2]. Lenalidomide is one of the essential agents in MM treatment since it is used in the induction phase and maintenance therapy. However, it is alleged to increase the risk of second primary malignancy (SPM), especially of hematopoietic origin such as myelodysplastic syndromes, Hodgkin's lymphoma, and acute myeloid leukemia [Citation3–5], which has been confirmed in a recent meta-analysis of seven trials [Citation6]. The rates of hematological malignancies were 3.1% in lenalidomide treated patients compared to 1.4% in patients treated without lenalidomide. However, there are only a few cases reporting therapy-related acute lymphoblastic leukemia (tr-ALL) diagnosed during lenalidomide treatment in patients with MM. Development of ALL after another B-cell originating neoplasm may suggest a clonal evolution from MM to ALL; however, recently published data challenged this hypothesis [Citation7]. The clonal relationship between primary MM and tr-ALL has not been clearly confirmed. It is also unclear if prior exposure to MM treatment may render the ALL cells resistant to the standard ALL treatment. It is also unknown if patients eligible for allogeneic hematopoietic cell transplantation (allo-HCT) can benefit long-term from this procedure. Here, we present a patient with MM and therapy-related B-cell ALL following lenalidomide maintenance successfully treated with standard ALL induction regimen and allogeneic hematopoietic cell transplantation. We also review all other published cases of tr-ALL to MM in order to assess the potential role of allo-HCT in the treatment plan.
Methods
To identify all case reports of patients with tr-ALL after lenalidomide maintenance in addition to our case, we used PubMed to search for a combination of words ‘Lymphoblastic + Lenalidomide’, ‘therapy-related lymphoblastic leukemia’; ‘secondary lymphoblastic leukemia’ and ‘Lymphoblastic leukemia and myeloma’. Only patients with sufficient pre-defined data were included. Patients with tr-ALL after thalidomide maintenance were excluded since this type of maintenance is no longer used.
In the presented case report immunophenotyping at the diagnosis was performed with antibodies included in Supplementary materials. Minimal residual disease (MRD) was monitored with multiparameter flow cytometry (before transplant) and next generation flow (after transplant). Karyotype changes were confirmed in classical cytogenetics, FISH panels were not used. Fusion genes were detected with RT-PCR. All data were analyzed retrospectively after receiving the informed consent of the described patient.
Case description
A 55-year-old man was diagnosed with MM, stage DS-IIIA ISS II, with hyperdiploid karyotype (52, XY, +3, +5, +7, t(14;22;20)(q11.2;q13;q13), +15, +18, +19[3]/46, XY[17]) in January 2017. He met two myeloma-defining events (bone lesions with pathological fracture of L2 vertebra body and hypercalcemia). Bone marrow examination revealed 24% infiltration by pathological plasmacyte population with the following immunophenotype: CD45−, CD38+high, CD138+, CD56+, CD19−, CD20−, CD27+, CD117+, CD71+, CD81+, CD28−, CD200+, CD28±. Total protein level was elevated up to 87 g/l with 32 g/l of monoclonal protein IgG kappa and FLC κ/λ index of 41.5.
The patient started a standard Bortezomib, Thalidomide, Dexamethasone (VTD) treatment. After four cycles, he achieved a partial response, and bone marrow infiltration with MRD of 0.6%. The treatment was consolidated with autologous hematopoietic cell transplantation (AHCT) in August 2017, followed by maintenance with lenalidomide at doses of 15–25 mg. In August 2020, still being on maintenance, he presented with fatigue, profound night sweating, and hemorrhagic diathesis. Morphology showed pancytopenia with 38% blasts, and 88% infiltration of blasts in medium-cellular bone marrow was present. Molecular analyses excluded presence of BCR/ABL(−), KMT2A-AF4(−), TCF3-PBX1(−), ETV6-RUNX1(−); immunophenotyping confirmed diagnosis of acute lymphoblastic leukemia B-common with phenotype: CD45+, CD117−, CD123±, CD71+, HLA-DR+, CD11b−, CD11c−, CD13−, CD15−, CD33−, CD4−, CD14−, CD64−, CD19+, CD20+, CD10+, CD38+, CD56−, CD52+, CD3−, CD5−, CD7−, MPO−, cytCD3−, CD79a+, TdT+. Cytograms from the diagnosis are presented in . Cytogenetics showed low-hypodiploid karyotype in six metaphases (33, XY, +1, +5, +6, −7, +11, +18, +20, +21, +mar1, +mar2, +mar3), in two metaphases there was triploid karyotype and in 12 normal. There was no CNS involvement. At the ALL diagnosis, his MM remained in complete response (CR) with both negative MRD and immunofixation.
Figure 1. Cytograms from the diagnosis of ALL.
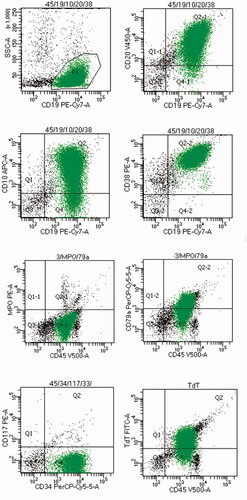
He was started on induction chemotherapy according to the Polish Adult Leukemia Group (PALG) protocol for patients above 55 years old (dexamethasone, vincristine, daunorubicin, peg-asparaginase, rituximab), followed by two consolidations mini-FLAM (fludarabine, cytarabine, mitoxantrone) with rituximab intermittent with intermediate-dose methotrexate, cytarabine, and rituximab. He achieved CR with a negative ALL-MRD of 0.003%. but with a residual very small population of abnormal plasmocytes – MM-MRD of 0.05%. The patient was referred for allo-HCT from a matched related donor (sister, 55 years old, two pregnancies, minor ABO incompatibility). He was conditioned with a reduced regimen (thiotepa 10 mg/kg, busulfan 8 × 0.8 mg/kg, and fludarabine 150 mg/m2) together with anti-thymocyte globulin 2.5 mg/kg, and received standard post-transplant immunosuppression with cyclosporine and a short course of methotrexate. Engraftment was reached on day 18. Routine bone marrow examination at day 30 was normal with full donor chimerism and MRD 0% for ALL and 0.016% for MM. Immunofixation remained negative. Immunosuppressive treatment was discontinued 6 months after transplant. At the last follow-up 6 months later (>18 months from tr-ALL diagnosis), the patient remains in deep remission of both diseases with a good performance status without any severe transplant complications.
Other cases
We found additional 14 case reports eligible for the study − 13 including MM and 1 report of systemic amyloidosis. There were five another cases of tr-ALL after lenalidomide maintenance [Citation8]. However, they were excluded from further analysis, since there was insufficient data regarding treatment and response. The characteristics, cytogenetic changes, treatment protocols of 14 patients included in the study are summarized in [Citation9–18]: tr-ALL occurred across all age groups – both in younger and elderly patients; males prevailed (by 60%). The median age at tr-ALL diagnosis was 64 years (40–82 years old) and the median time to development of tr-ALL was 5.3 years (range 1, 6–15 years) since the primary diagnosis of MM. The lenalidomide maintenance dose varied from 5 to even 25 mg, although most patients received low doses (5–10 mg). At least 50% were in CR for MM at the time of ALL diagnosis based on the bone marrow examination and/or immunofixation. All patients were BCR/ABL negative. The cytogenetic results are unknown in two patients. In the remaining 13, only 1 had a normal karyotype; the rest had cytogenetic aberrations. In the 15 reviewed cases, only three patients (20%) did not receive melphalan as a part of MM treatment, and the majority (10/15; 66%) were conditioned with myeloablative doses of melphalan before AHCT. The follow-up was longer than 12 months in all treated patients that survived induction therapy. Information about treatment response was present in 14 cases: 10 patients reached CR and 1 PR, one was not treated, two died during induction, four patients relapsed after primary treatment. Consolidation in the form of AHCT or allo-HCT was performed in 1 and 3 patients, respectively.
Table 1. The characteristics of the study group, cytogenetic changes, and treatment protocols.
Discussion
Secondary acute leukemia refers to patients with either therapy-related or disease progressing from an antecedent hematologic disorder typically a myelodysplastic syndrome or a myeloproliferative neoplasm. Neither MM is considered a typical neoplasm that leads to secondary hematopoietic neoplasms nor ALL is a typical secondary hematopoietic malignancy. Lymphoblasts and plasmocytes are cells both originating from lymphopoiesis; however, their proliferating potential is very different. Plasma cells represent the final differentiation stage, nevertheless, there are hypotheses that somatic mutations occurring in younger precursors affect further stages of lymphopoiesis, and the final oncogenic events take place in secondary lymphoid organs [Citation19,Citation20]. The hereditary component of MM susceptibility was reported many years ago; however, increased risk for other B-cell originating neoplasms was also noticed in MM-risk families [Citation1,Citation8]. Interestingly tr-ALL, which accounts for up to 9% of all ALL, occurs mainly in women after chemotherapy for breast cancer [Citation21]. Regardless, the second most common cause of tr-ALL is MM [Citation22].
The exact mechanism of tr-ALL development is still unknown and remains speculative. One study suggests that the etiology is due to the initial treatment of MM and ALL is a second primary malignancy following MM, not clonally related [Citation7]. Results from other studies propose that the etiology is multifactorial involving advanced patient age, a high-risk subtype of multiple myeloma, and the use of lenalidomide and melphalan [Citation6,Citation23,Citation24]. Immunomodulatory properties of lenalidomide prolong disease-free survival by suppressing indolent cells but may promote the development of secondary neoplasms allowing the expansion of a more aggressive clone that evolves into acute leukemia [Citation25,Citation26]. Interestingly, tr-ALL was also observed in patients with del 5q myelodysplastic syndrome treated with lenalidomide, which might confirm that hypothesis [Citation27]. There are also a few cases of tr-ALL after maintenance therapy with thalidomide which also supports the role of IMIDS in the development of tr-ALL [Citation12,Citation28,Citation29].
There is not much data for the prognosis of tr-ALL after MM, since most published data refer to the whole population of therapy-related malignancies with different diagnoses and different types of first-line treatment, varying from radiotherapy to systemic polychemotherapy [Citation8,Citation22,Citation30]. Our patient, except for age and karyotype, didn`t have other adverse clinical factors: he had no leukocytosis or molecular abnormalities (unfortunately data on TP53 mutation is not available) or central nervous system involvement, and had a good response to steroid pretreatment and negative MRD after induction therapy. However, the prognosis is mainly driven by molecular and cytogenetics aberrations. Therapy-related ALLs have different molecular and cytogenetic presentations compared to de novo ALL [Citation30]. Even within tr-ALL group, the pattern of molecular aberrations and time of onset varies. The time from primary malignancy to tr-ALL ranges from months to even several years, with higher KMT2A occurrence in the early period (up to 2 years) and hypodiploid aberrations in the late period (median 7.1 years) of observation. KMT2A aberrations occur in older patients whereas hypodiploid aberrations in younger patients [Citation8,Citation30]. Nevertheless, both aforementioned genetic aberrations together with complex karyotype assigned patients to the high-risk group [Citation8,Citation22,Citation31]. Biallelic loss of TP53 is observed more often in the population of tr-ALL than in de novo ALL (42% vs. 23%) predominantly in cases with hypodiploid karyotype or KMT2A rearrangements [Citation8,Citation32,Citation33]. Deletion of 17p or mutation of TP53 were also reported in the presented other cases. For tr-ALL hypodiploid karyotype, such as in our case, is more prevalent, although in some cases, hyperdiploidy was reported, which has, at least in pediatric populations, a favorable prognosis. In adult settings, however, hyperdiploidy is often accompanied by additional structural changes that confer a worse prognosis [Citation33]. In our patient additional structural chromosomal changes within low-hypodiploidy (33 chromosomes) were present. Interestingly, in two metaphases near-triploid karyotype was also found which most likely resulted from endoduplication of hypodiploid clone with chromosome doubling, but the copy number analysis was not possible to perform. Besides, the presence of putative doubled clone does not affect the poor prognosis of hypodiploid ALL [Citation32].
Currently, ALL therapy follows a combination of treatments usually integrating high-dose chemotherapy, hematopoietic cell transplantation, and targeted therapy. Most of the presented cases received intensive induction chemotherapy according to local protocols. Therapy-related ALL patients are less likely to achieve CR, and the MRD positive status is often observed at the end of the treatment, while relapse occurs in more than 50% of cases in the presence of adverse cytogenetics [Citation22,Citation30,Citation31]. In presented patients more than 70% of patients that survived induction therapy reached CR, however, data concerning MRD status are missing. Although these results are encouraging, long-lasting remissions were experienced in only half of the patients. Similar results were presented by others: 80% of tr-ALL reached CR(22), alas, overall survival was inferior compared to de novo ALL [Citation8]. The treatments of the published cases () varied, however, 12 of 14 treated patients received intensive anthracycline-based chemotherapy regimens the same as for de novo ALL but with 15% early mortality (2/13 died due to infection). Three other patients died due to disease progression. Surprisingly most of the patients achieved CR. However, based on the clinical presentation and the presence of adverse cytogenetics we referred our patient for allo-HCT. The role of allo-HCT has been not established yet, some data indicate that allo-HCT may abrogate the poor prognosis associated with unfavorable cytogenetic changes in this group [Citation22,Citation30]. In the presented group only 3 patients received allo-HCT as consolidation therapy. The median age at the tr-ALL diagnosis was 64.5 years old, which may impact the low percentage of transplanted patients in this population, however, the introduction of reduced-intensity conditioning regimens seems to be an option for the elderly with acceptable non-relapse mortality.
In conclusion, although the incidence of tr-ALL following maintenance therapy with lenalidomide is low, the number of published cases is gradually increasing. The mechanism for the development of tr-ALL to MM during lenalidomide maintenance therapy is not clear, but it may be connected to a combination of IMiDs and melphalan. Nowadays, since tr-ALL has a poor prognosis and the relapse rate is high, allo-HCT remains the only curative option that may overcome adverse cytogenetic prognosis and should be offered to every patient with tr-ALL eligible for transplant. In the future, CAR-T cells directed against common antigens for both diseases might be the valid option, especially in the elderly group.
Supplemental Material
Download MS Word (14.9 KB)Disclosure statement
The authors report there are no competing interests to declare
Data availability statement
The data that support the findings of this study are openly available https://pubmed.ncbi.nlm.nih.gov/. Reference number: DOI:10.3109/10428194.2013.786072, DOI:10.1002/hon.2248, DOI:10.1155/2018/9052314, DOI:10.5301/tj.5000377, PMID: 23694793, DOI:10.1093/AJCP/AQAA109, DOI:10.1186/s12885-019-6286-9, PMID: 24966987, DOI:10.7759/cureus.6009, DOI:10.1182/blood.2020009141.
References
- Hemminki K, Försti A, Houlston R, et al. Epidemiology, genetics and treatment of multiple myeloma and precursor diseases. Int J Cancer. 2021;149(12):1980–1996.
- Blimark CH, Turesson I, Genell A, et al. Outcome and survival of myeloma patients diagnosed 2008-2015. Real-world data on 4904 patients from the Swedish myeloma registry. Haematologica. 2018;103(3):506–513.
- McCarthy PL, Owzar K, Hofmeister CC, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366(19):1770–1781.
- Attal M, Lauwers-Cances V, Marit G, et al. Lenalidomide maintenance after Stem-Cell transplantation for multiple myeloma. N Engl J Med. 2012;366(19):1782–1791.
- Palumbo A, Hajek R, Delforge M, et al. Continuous lenalidomide treatment for newly diagnosed multiple myeloma. N Engl J Med. 2012;366(19):1759–1769.
- Palumbo A, Bringhen S, Kumar SK, et al. Second primary malignancies with lenalidomide therapy for newly diagnosed myeloma: a meta-analysis of individual patient data. Lancet Oncol. 2014;15(3):333–342.
- Aldoss I, Capelletti M, Park J, et al. Acute lymphoblastic leukemia as a clonally unrelated second primary malignancy after multiple myeloma. Leukemia. 2019;33(1):266–270.
- Barnea Slonim L, Gao J, Burkart M, et al. Therapy-related B-cell acute lymphoblastic leukemia in adults has unique genetic profile with frequent loss of TP53 and inferior outcome. Leukemia. 2021;35(7):2097–2101.
- García-Muñoz R, Robles-de-Castro D, Muñoz-Rodríguez A, et al. Acute lymphoblastic leukemia developing during maintenance therapy with lenalidomide in a patient with multiple myeloma. Leuk Lymphoma. 2013;54(12):2753–2755.
- Tan M, Fong R, Lo M, et al. Lenalidomide and secondary acute lymphoblastic leukemia: a case series. Hematol Oncol. 2017;35(1):130–134.
- Khan AM, Muzaffar J, Murthy H, et al. Acute lymphoblastic leukemia following lenalidomide maintenance for multiple myelome; two cases with unexpected presentation and good prognostic features. Case Rep Hematol. 2018;2018:1–5.
- Junxun L, Junru L, Meilan C, et al. Three patients with multiple myeloma developing secondary lymphoblastic leukemia: case reports and review of the literature. Tumori 2016;102(Suppl 2):131–136.
- Gonzalez MM, Kidd L, Quesada J, et al. Acute myelofibrosis and acute lymphoblastic leukemia in an elderly patient with previously treated multiple myeloma. Ann Clin Lab Sci. 2013;43(2):176–180.
- Germans SK, Kulak O, Koduru P, et al. Lenalidomide-associated secondary B-lymphoblastic leukemia/lymphoma – a unique entity. Am J Clin Pathol. 2020;154(6):816–827.
- Sinit RB, Hwang DG, Vishnu P, et al. B-cell acute lymphoblastic leukemia in an elderly man with plasma cell myeloma and long-term exposure to thalidomide and lenalidomide: a case report and literature review. BMC Cancer. 2019;19(1):1–9.
- Sharma N, Hassoun H, Hatem J, et al. Cardiac ALL: Most unusual occurrence of lenalidomide-associated acute lymphoblastic leukemia with subsequent cardiac involvement. Cureus. 2019;11(10):e600–9.
- Nair R, Gheith S, Popescu D, et al. A rare case of acute lymphoblastic leukemia in a patient with light chain (AL) amyloidosis treated with lenalidomide. Int J Clin Exp Pathol. 2014;7(5):2683–2689.
- Tashakori M, Khoury JD. B acute lymphoblastic leukemia arising during maintenance therapy for multiple myeloma. Blood. 2020;136(23):2720.
- Corre J, Munshi N, Avet-Loiseau H. Genetics of multiple myeloma: another heterogeneity level? Blood. 2015;125(12):1870–1876.
- Mitchell JS, Li N, Weinhold N, et al. Genome-wide association study identifies multiple susceptibility loci for multiple myeloma. Nat Commun. 2016;7:12050.
- Østgård LSG, Medeiros BC, Sengeløv H, et al. Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: a national population-based cohort study. J Clin Oncol. 2015;33(31):3641–3649.
- Saygin C, Kishtagari A, Cassaday RD, et al. Therapy-related acute lymphoblastic leukemia is a distinct entity with adverse genetic features and clinical outcomes. Blood Adv. 2019;3(24):4228–4237.
- Mahindra A, Raval G, Mehta P, et al. New cancers after autotransplantations for multiple myeloma. Biol Blood Marrow Transplant. 2015;21(4):738–745.
- Thomas A, Mailankody S, Korde N, et al. Second malignancies after multiple myeloma: from 1960s to 2010s. Blood. 2012;119(12):2731–2737.
- Jones JR, Cairns DA, Gregory WM, et al. Second malignancies in the context of lenalidomide treatment: an analysis of 2732 myeloma patients enrolled to the myeloma XI trial. Blood Cancer J. 2016;6(12):e506.
- Musto P, Anderson KC, Attal M, et al. Second primary malignancies in multiple myeloma: an overview and IMWG consensus. Ann Oncol. 2017;28(2):228–245.
- Agostino NM, Ahmed B, Popescu D, et al. Transformation of the 5q- syndrome to acute lymphoblastic leukemia: a report of two cases and review of the literature. Int J Clin Exp Pathol. 2011;4(3):322–326.
- Li J, Zhan J, Zhang FAN, et al. Secondary lymphoblastic leukemia occurring 38 months after the primary diagnosis of multiple myeloma: a case report. Oncol Lett. 2016;12(2):847–856.
- Usmani SZ, Sexton R, Hoering A, et al. Second malignancies in total therapy 2 and 3 for newly diagnosed multiple myeloma: influence of thalidomide and lenalidomide during maintenance. J Am Soc Hematol. 2012;120(8):1597–1600.
- Abdel Rahman ZH, Parrondo RD, Heckman MG, et al. Comparative study of therapy-related and de novo adult b-cell acute lymphoblastic leukaemia. Br J Haematol. 2022;196(4):963–966.
- Moorman AV, Barretta E, Butler ER, et al. Prognostic impact of chromosomal abnormalities and copy number alterations in adult B-cell precursor acute lymphoblastic leukaemia: a UKALL14 study. Leukemia. 2022;36(3):625–636.
- Safavi S, Paulsson K. Near-haploid and low-hypodiploid acute lymphoblastic leukemia: two distinct subtypes with consistently poor prognosis. Blood. 2017;129(4):420–423.
- Chen Z, Sun Y, Xie W, et al. Is hyperdiploidy a favorable cytogenetics in adults with B-lymphoblastic leukemia? Cancer Med. 2019;8(9):4093–4099.