
Abstract
Background
While circulating tumour (ct)DNA is an indicator of minimal residual disease and negative prognostic factor in stage II-III colon cancer, no study has ever analysed the value of this biomarker in colon cancer patients treated with neoadjuvant chemotherapy. We sought to fill this gap by using prospectively collected plasma samples from 80 stage III colon cancer patients, receiving one cycle of neoadjuvant FOLFOX followed by surgery +/− adjuvant FOLFOX in the PePiTA trial.
Material and Methods
Samples were collected at baseline, 2 weeks and surgery. NPY and WIF1 were selected as universal methylation markers for ctDNA, and analysed with ddPCR technology. ROC curves were applied for cut-off points, and outcome measures included 5-year disease-free survival (DFS) and 6-year overall survival (OS).
Results
After a median follow-up of 52.5 months, baseline circulating-free (cf) DNA was an independent prognostic factor for DFS (HR 3.35, 95% CI: 1.15–9.77, p = .03), and a trend towards a similar association was observed for relative cfDNA changes between baseline and surgery (HR 2.57, 95% CI: 0.94–7.05, p = .07). Among 60 ctDNA assessable patients, 25 (42%) had detectable ctDNA at baseline. While detection of ctDNA at any pre-operative timepoint was not associated with outcome, patients with ctDNA increase (change of the worst trending methylation marker ≥11%, or mean ctDNA change of NPY and WIF1 ≥ 0%) between baseline and surgery showed a trend towards worse 5-year DFS (HR 3.66, 95% CI: 0.81–16.44, p = .09).
Conclusion
This is the first study of ctDNA in the neoadjuvant setting of early-stage colon cancer. Results are hypothesis-generating and should be confirmed in larger series.
Background
Oxaliplatin-based adjuvant chemotherapy is a standard treatment for stage III, and an option for high-risk stage II colon cancer patients, who undergo curative-intent resection of their primary tumour [Citation1]. This treatment, however, is beneficial only in 5%–23% of cases, the vast majority of people being unnecessarily treated and exposed to short- and long-term toxicities, which may have a negative impact on their quality of life [Citation2,Citation3]. Better criteria than pathological tumour stage are needed to assess individual patient risk and optimise treatment decisions, ultimately reducing overtreatment to a minimum. While a number of prognostic factors have been identified, all the attempts to validate predictive biomarkers for adjuvant chemotherapy have been unsuccessful. The only biomarker routinely used in routine practice is microsatellite instability or mismatch repair deficiency, which allows identifying patients with pathological stage II tumours who do not benefit from adjuvant single agent fluoropyrimidine therapy [Citation4].
Circulating tumour (ct)DNA after surgery or completion of adjuvant chemotherapy is an indicator of minimal residual disease, and a strong predictive factor for tumour recurrence in stage II and III colon cancer [Citation5–7]. In this setting, many trials are ongoing to assess prospectively the value of ctDNA analysis as a tool to guide the post-operative management, and de-escalation and intensification strategies are generally proposed for ctDNA negative and ctDNA positive patients, respectively [Citation8–11]. Not surprisingly given the current standards of care including upfront surgery followed by adjuvant chemotherapy, no study has ever analysed the prognostic value of ctDNA in early-stage colon cancer patients who receive neoadjuvant chemotherapy.
Based on these premises, we sought to evaluate the prognostic value of baseline and early, on-treatment changes of circulating-free (cf)DNA and ctDNA in stage II-III colon cancer patients who were treated with one cycle of neoadjuvant FOLFOX followed by surgery plus or minus adjuvant chemotherapy within the context of a prospective clinical trial.
Material and methods
Study population
PePiTA was an academic, multicentre, single-arm, phase II trial sponsored by the Institut Jules Bordet, and registered at ClinicalTrials.gov (NCT00994864) [Citation12]. This trial was run between January 2010 and January 2016, at 21 sites across Belgium. The primary objective was to evaluate the association between in vivo tumour chemosensitivity to one cycle of neoadjuvant FOLFOX (as assessed by metabolic response on 18F-FDG PET/CT scan on day 14) and 3-year disease-free survival (DFS) in clinical stage III colon cancer.
Inclusion criteria and study procedures were previously discussed in detail [Citation12]. In short, eligibility was restricted to patients 18 years or older, diagnosed with adenocarcinoma of the colon compatible at the endoscopy examination with a stage III cancer (i.e., invasion of more than one third of the circumferential colonic lumen), and considered amenable to curative (R0) resection. All eligible patients were treated with one cycle of neoadjuvant FOLFOX chemotherapy followed by surgery and adjuvant FOLFOX chemotherapy (for a maximum of 12 cycles in total). For patients with pathological stage I-II tumours, the decision about adjuvant chemotherapy was left to the investigators’ discretion.
Patients who were enrolled in PePiTA, and had available plasma samples were eligible for this retrospective analysis.
Plasma sample collection and circulating nucleic acid analysis
Since June 2013, plasma samples were prospectively collected for all patients enrolled in the PePiTA trial at pre-defined time points including baseline, after 2 weeks (i.e., after one cycle of neoadjuvant FOLFOX), and before surgery.
Clarified plasma was prepared from each sampling by a first centrifugation step at 1600×g for 10 min at 4 °C, and a second centrifugation at 10,000×g for 10 min, within one hour of blood collection. Samples were then stored at −80 °C for downstream applications. cfDNA was isolated using the QIAmp Circulating Nucleic Acids Kit (Qiagen, Hilden, Germany) from 3 mL of plasma in an elution volume of 30 µL, and quantified with the Qubit fluorometer (Life-Technologies, Carlsbad, USA). cfDNA samples were bisulphite converted using the EZ DNA Methylation-Gold™ Kit (Zymo Research, Irvine, USA). Polymerase-chain reaction (PCR) was used to test the bisulphite conversion process with primers provided by the manufacturer (Zymo Research, Irvine, USA).
The NPY and WIF1 genes were selected as universal methylation markers for ctDNA, and digital droplet (dd)PCR technology according to previously reported methods was used [Citation13]. Commercially available genomic DNA (Promega, Madison, USA) and hypermethylated DNA (Zymo Research, Irvine, USA) were each diluted to a final concentration of 2 ng/µL, bisulphite converted, and used as negative and positive controls, respectively. The maximum number of false positive droplets in negative controls was 4 for WIF1 (n = 10) and 1 for NPY (n = 18), with mean false positive droplets being 1.89 (standard deviation [SD] 0.99) and 0.17 (SD 0.37), respectively. The calculated limit of blank was 4.95 for WIF1 and 1.64 for NPY. Samples with ≥5 positive droplets for WIF1 and/or ≥2 positive droplets for NPY were considered as ctDNA positive. Samples with <10,000 droplets were not considered assessable.
All primers and probes were manifactured by Applied Biosystems (Thermo Fisher Scientific, Waltham, USA). Probe sequences are detailed in Supplementary Table 1. ddPCR conditions for WIF1 were: Step 1: 2′ at 50 °C. Step 2: 10′ at 95 °C. Step 3: 44 cycles at 95 °C for 15″ and 57.7 °C for 1′ 2 °C/second ramp rate. Step 4: 10′ at 98 °C. ddPCR conditions for NPY were: Step 1: 2′ at 50 °C. Step 2: 10′ at 95 °C. Step 3: 44 cycles at 95 °C for 15″ and 55.5 °C for 1′ 1.5 °C/second ramp rate. Step 4: 10′ at 98 °C.
Table 1. Multivariable analysis for disease-free survivala.
Data were processed with the QuantaSoft V1.6 software (Bio-Rad, Hercules, USA).
Pathological analysis
Assessment of tumour regression on samples from the resection specimens was retrospectively carried out by an expert gastrointestinal pathologist, who was blinded to the cf/ctDNA results and other clinical data. Tumour regression grade (TRG) was scored according to the College of American Pathologists (CAP) – modified Ryan schema [Citation14].
Statistical analysis
Levels of cfDNA were expressed as ng/ml, while changes of cfDNA after neoadjuvant chemotherapy were analysed as relative variations compared with baseline. Levels of ctDNA were expressed as methylated copies/ml, while changes of ctDNA after neoadjuvant chemotherapy were analysed as relative variations compared with baseline, using worst and mean relative variations of NPY and WIF1 for patients with methylation of both genes.
Outcome measures were 5-year DFS and 6-year overall survival (OS). ROC curves of 5-year DFS were applied for all cut-off points for survival analyses, while Kaplan-Meier method, Cox proportional hazards models and log-rank tests were used to estimate survival and to test for associations. When feasible based on the number of assessable patients, multivariable analyses were performed using logistic regression, and including all variables from the univariable analyses with a p-value of < .2. P-values less than 0.05 were considered statistically significant. Statistical analyses were carried out with the SPSS for MacOS version 25.0 (SPSS Inc., Chicago, USA).
Regulatory approval
The PePiTA trial was approved by the Institutional Review Board at the Institute Jules Bordet and by an Ethics Committee (CE1669). All patients provided a written informed consent.
Data ava ilability statement
Raw data are available upon request.
Results
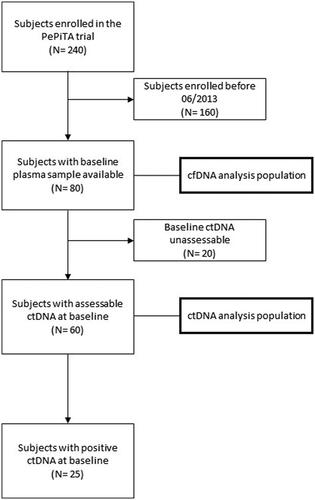
Eighty out of the 240 PePiTA trial patients (33.3%) were eligible for this retrospective study (). Baseline characteristics are summarised in Supplementary Table 2. Median age was 63 years, and the primary tumour was localised in the left colon or rectum in 56% of cases. All patients had received one cycle of neoadjuvant FOLFOX chemotherapy, followed by an R0 resection. Following analysis of the surgical samples, 36 (45%) were found to have pathological stage III tumours, all except one receiving adjuvant FOLFOX chemotherapy for a maximum of 12 cycles. Only one of the remaining 44 (55%) patients with pathological stage I or II tumours was given adjuvant chemotherapy. At the time of this analysis, 11 patients had suffered tumour recurrence and 10 had died. After a median follow-up of 52.5 months, 5-year DFS was 68% (95% confidence intervals [CI]: 52–84) and 6-year OS 84% (95% CI: 74–94) (Supplementary Figure 1).
Figure 1. Study flow diagram.
Median time from the start of neoadjuvant chemotherapy to the collection of baseline plasma samples was −1 days (range −21−0), and median cfDNA value was 1.080 ng/ml (range 0−3.220). Patients with high baseline cfDNA (i.e., ≥1.100 ng/ml) had numerically worse 5-year DFS (HR 2.62, 95% CI: 0.93–7.34, p = .07) and 6-year OS (HR 3.24, 95% CI: 0.67–15.61, p = .14) than those with low baseline cfDNA (). Plasma samples for the 2-week and surgery timepoints were collected at a median of 14 (range 8−22) and 20 (range 8−33) days, respectively, after the start of neoadjuvant chemotherapy. At the same timepoints, median cfDNA values were 1.390 ng/ml (range 0−7.820), and 1.310 ng/ml (range 0−9.360). Median relative changes (from baseline) of cfDNA at 2 weeks and before surgery were +20.8% (range −100%–+853.7%) and +18.4% (range −100%–+410.6%), respectively. At neither timepoint, these changes predicted DFS (at 2 weeks: HR 0.96, 95% CI: 0.38–2.43, p = .92; at surgery: HR 2.04, 95% CI: 0.78–5.30, p = .15) or OS (at 2 weeks: HR 0.62, 95% CI: 0.16–2.50, p = .50; at surgery: HR 1.65, 95% CI: 0.44–6.18, p = .46) ( and Supplementary Figure 2). In a multivariable analysis including ECOG Performance Status (PS), baseline CEA, baseline cfDNA, relative cfDNA changes between baseline and surgery, and pathological TNM stage, baseline cfDNA was the only factor independently associated with DFS (HR 3.35, 95% CI: 1.15–9.77, p = .03). A trend towards statistically significance was observed for pathological stage (HR 2.69, 95% CI: 0.92–7.83, p = .07), and relative cfDNA changes between baseline and surgery (HR 2.57, 95% CI: 0.94–7.04, p = .07) ().
Figure 2. Disease-free survival (A) and overall survival (B) by baseline cfDNA.
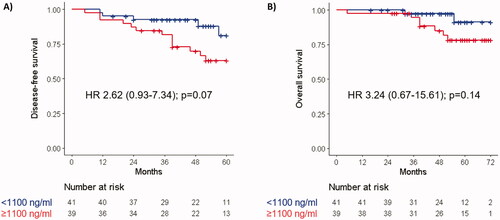
Figure 3. Disease-free survival by cfDNA (A) and ctDNA (B) relative changes from baseline to surgery.
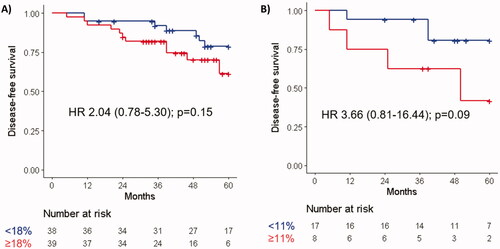
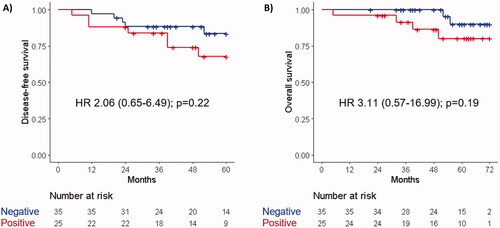
Sixty patients were technically assessable for ctDNA at baseline. Of these, 18 (30%) were found to have methylation of NPY (median 16.85 copies/ml, range 7.50−65.90), 18 (30%) of WIF 1 (median 9.30 copies/ml, range 2.70−59.10), 11 (18%) of both NPY and WIF1, and 25 (42%) of either NPY or WIF1. The outcome of patients with detectable ctDNA (as defined by the presence of methylation of either NPY or WIF1) at baseline was not significantly different in terms of DFS (HR 2.06, 95% CI: 0.65–6.49, p = .22) or OS (HR 3.11, 95% CI: 0.57–16.99, p = .19) than that of patients with undetectable ctDNA (). Similar results were observed when detection of ctDNA at any timepoint before surgery was considered (DFS: HR 1.65, 95% CI: 0.54–5.04, p = .38; OS: HR 2.80, 95% CI: 0.54–14.44, p = .22). Patients with a ctDNA change of the worst trending methylation marker ≥11%, or those with a mean ctDNA change of NPY and WIF1 ≥ 0% between baseline and surgery, had worse 5-year DFS (HR 3.66, 95% CI: 0.81–16.44, p = .09 for either analysis) (). In contrast, relative changes of ctDNA between baseline and week 2 were not associated with outcome (data not shown).
Figure 4. Disease-free survival (A) and overall survival (B) by baseline ctDNA.
Samples from the resection specimens were available for 76 (95%) patients. Of these, 7 (9%) were found to have some evidence of tumour regression (TRG 2), while the remaining 69 (91%) were classified as having poor or no response (TRG 3). A DFS event occurred in 1/7 (14%) patients with TRG 2 and in 15/69 (22%) patients with TRG 3. Only 2 patients with TRG 2 were also assessable for ctDNA changes before surgery, and in both cases a mean ctDNA reduction ≥30% was observed, versus 9 out of 16 patients with a TRG 3.
Discussion
Strong interest has recently emerged for the investigation of neoadjuvant treatment strategies in early-stage colon cancer. The FOxTROT trial showed that administering three cycles of oxaliplatin-based chemotherapy before surgery increases the chances of clear surgical margins, reduces the risk of post-operative complications, and is associated with a numerically lower risk of recurrence for patients with clinical stage II-III tumours [Citation15]. Due to the lack of statistical significance for the primary endpoint, however, neoadjuvant chemotherapy remains an investigational treatment, being generally considered a suitable option only for locally advanced, borderline resectable tumours. Beyond the potential beneficial effects on clinical outcome measures, a strategy of delivering neoadjuvant chemotherapy to resectable colon cancer patients provides an opportunity window for the in vivo assessment of tumour chemosensitivity, and implementation of individualised, peri-operative treatment approaches. In this setting, blood-based biomarkers that can be easily sampled and rapidly analysed represent optimal candidates as decision-guiding tools.
While several studies on circulating DNA in the neoadjuvant setting of rectal cancer have already been conducted [Citation16–18], this is to our knowledge the first study investigating circulating DNA in colon cancer patients who are treated with neoadjuvant chemotherapy. Although evidence on the value of cf/ctDNA has rapidly accumulated over the last few years, this has mostly been limited to settings such as post-resection of early-stage or metastatic tumours (i.e., for the detection of minimal residual disease) [Citation7,Citation19,Citation20] and palliative treatment of unresectable disease (i.e., for the longitudinal assessment of response to treatment and tracking of resistant clones) [Citation21,Citation22]. For this study, we took advantage of serial plasma samples, which were prospectively collected within the context of a phase II clinical trial with a unique design [Citation12].
We found that cfDNA at baseline was an independent prognostic factor, high levels of this being associated with a higher risk of tumour recurrence or death following curative-intent resection of the primary tumour. These data are in line overall with studies in metastatic colorectal cancer showing an association between cfDNA, disease burden and prognosis [Citation23,Citation24]. Further studies are needed to confirm our results, and especially address whether patients with high levels of baseline cfDNA are most likely to benefit from intensified post-operative treatments (with regard to type and duration of adjuvant chemotherapy) or alternative management strategies such as early target of micrometastases with neoadjuvant chemotherapy. Notably, and in contrast with the value of cfDNA at baseline, only a trend towards a statistically significant association was observed between early on-treatment changes of this biomarker and outcome. This apparent inconsistency could possibly be due to the confounding effect of other conditions including chemotherapy-induced toxicity, causing increased release of non-tumoural cfDNA and eventually balancing reductions of tumour-derived cfDNA in chemotherapy-responding patients [Citation25]. Unfortunately, we could not account for this potential source of bias. While contamination by genomic DNA in our study should have been reduced by isolating plasma within one hour after venesection, the fluorometric method used for DNA quantification does not allow discriminating the origin of DNA [Citation26,Citation27].
To overcome these limitations, we also analysed ctDNA in the same population. Based on previous data showing high concordance between mutation- and epigenetic-based ctDNA detection techniques, we used universal methylation markers such as NPY and WIF1, which are especially convenient for early-stage tumours that may have not undergone yet genomic profiling [Citation22,Citation28]. The main advantage of this approach is that it does not require prior knowledge of the genomic characteristics of the tumour, this resulting in rapid turnaround time and reduced costs. In our study, only 42% of patients were found to have detectable ctDNA at baseline, which is overall in line with previous studies especially considering the high proportion of pathological stage I-II patients in our series [Citation13,Citation18,Citation22]. Notably, baseline ctDNA detection was not useful in terms of prognostication, whereas increased ctDNA levels after one cycle of neoadjuvant chemotherapy and before surgery appeared to be associated, although non-statistically significantly, with worse outcome. We acknowledge that these data should be interpreted with a lot of caution especially bearing in mind the substantial proportion (i.e., 25%) of non-assessable patients due to technical failures, and the small number of patients who could be followed up for changes of ctDNA after neoadjuvant chemotherapy. While these caveats may account for the lack of statistical significance, they could have increased as well the risk of random associations. Larger studies are needed to confirm the intriguing hypothesis that early ctDNA dynamics during pre-operative chemotherapy can be used as a marker of treatment response and stratification factor to select patients who benefit from delivery of further systemic treatment, either in the neoadjuvant or adjuvant setting.
In addition to the abovementioned flaws, other limitations should be considered when interpreting the results of our analysis. Unfortunately, prospective collection of blood samples was not included in the initial version of the PePiTA trial protocol, but it was added as an amendment when approximately two thirds of patients had already been recruited, this reducing substantially the proportion of patients who were eligible for this retrospective study. Furthermore, very few events were observed, partly due to the high proportion of patients with pathological stage I-II tumours. While tumour downstaging after one cycle of chemotherapy is theoretically possible, such an unexpectedly high proportion coupled with the results of the tumour regression analysis rather suggest some clinical overstaging at study entry. Finally, the lack of a control group makes our assumptions regarding the potential relevance of the investigated biomarkers in relation to the use of neoadjuvant or adjuvant chemotherapy highly speculative. In particular, it is unknown whether baseline cfDNA and pre-operative changes of cfDNA/ctDNA are simply prognostic, or also predict benefit from administration of further chemotherapy.
Taking all this into account, our study still represents a first step towards understanding the potential of circulating DNA in the developing field of neoadjuvant treatment for colon cancer. Future efforts are needed to build on our preliminary results, and to fine-tune the possible clinical applications of circulating DNA-based biomarkers in this setting.
Supplemental Material
Download MS Word (738.8 KB)Acknowledgements
This Research Project was supported by ESMO through a Research Fellowship granted to GB to attend the Institut Jules Bordet. Any views, opinions, findings, conclusions or recommendations expressed in this material are those solely of the Authors and do not necessarily reflect those of ESMO. FS is supported by the Fondation Contre le Cancer.
Disclosure statement
AD - Travel grants: Amgen, Ipsen. AH - Consultancy, advisory role, honoraria: Amgen, Bayer, Eli Lilly, Merck, Pierre Fabre, Servier, Sirtex. Research funding: Amgen, AstraZeneca, Ipsen, Leo Pharma, Merck, Roche, Sanofi, Teva Pharma. Travel grant: Merck, Roche, Sirtex. FS. Consultancy, advisory role, honoraria: Amal Therapeutics, Bayer, BMS, Dragonfly Therapeutics, Nordic Pharma, Roche. Research funding: Amgen (inst), AstraZeneca (Inst), Bayer (Inst), BMS (Inst), Roche (Inst), Sanofi (Inst). Travel grants: Amgen, Bayer, Lilly. Leadership role: Co-Chair EORTC Colon, Rectum, Anal Task Force. All other authors do not have any conflicts of interests.
References
- Argilés G, Tabernero J, Labianca R, et al. Localised colon cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020;31(10):1291–1305.
- André T, Boni C, Navarro M, et al. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III Colon cancer in the MOSAIC trial. J Clin Oncol. 2009;27(19):3109–3116.
- Shah MA, Renfro LA, Allegra CJ, et al. Impact of patient factors on recurrence risk and time dependency of oxaliplatin benefit in patients with colon cancer: analysis from modern-era adjuvant studies in the adjuvant colon cancer end points (ACCENT) database. J Clin Oncol. 2016;34(8):843–853.
- Sargent DJ, Marsoni S, Monges G, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28(20):3219–3226.
- Tie J, Wang Y, Tomasetti C, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II Colon cancer. Sci Transl Med. 2016;8:346ra92.
- Tie J, Cohen JD, Wang Y, et al. Circulating tumor DNA analyses as markers of recurrence risk and benefit of adjuvant therapy for stage III Colon cancer. JAMA Oncol. 2019;5(12):1710–1717.
- Tie J, Cohen JD, Lo SN, et al. Prognostic significance of postsurgery circulating tumor DNA in nonmetastatic colorectal cancer: individual patient pooled analysis of three cohort studies. Int J Cancer. 2021;148(4):1014–1026.
- Verbus EA, Rossi AJ, Luna AJ, et al. Circulating tumor DNA as a predictive biomarker in adjuvant chemotherapy for patients with stage 2A colon cancer (COBRA). Ann Surg Oncol. 2021;28(8):4095–4097.
- Taniguchi H, Nakamura Y, Kotani D, et al. CIRCULATE-Japan: Circulating tumor DNA-guided adaptive platform trials to refine adjuvant therapy for colorectal cancer. Cancer Sci. 2021;112(7):2915–2920.
- Circulating tumour DNA analysis informing adjuvant chemotherapy in stage III colon cancer: a multicentre phase II/III randomised controlled study (DYNAMIC-III). Available at: https://www.australianclinicaltrials.gov.au/anzctr/trial/ACTRN12617001566325. (accessed on 27.01.2022).
- A study to evaluate the use of circulating tumour DNA to guide adjuvant chemotherapy on recurrence-free survival in patients with stage II colon or rectal cancer. Available at: https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?ACTRN=12615000381583. (accessed on 27.01.2022).
- Hendlisz A, Golfinopoulos V, Deleporte A, et al. Preoperative chemosensitivity testing as predictor of treatment benefit in adjuvant stage III colon cancer (PePiTA): protocol of a prospective BGDO (belgian group for digestive oncology) multicentric study. BMC Cancer. 2015;15:173.
- Garrigou S, Perkins G, Garlan F, et al. A study of hypermethylated circulating tumor DNA as a universal colorectal cancer biomarker. Clin Chem. 2016;62(8):1129–1139.
- Ryan R, Gibbons D, Hyland JM, et al. Pathological response following long-course neoadjuvant chemoradiotherapy for locally advanced rectal cancer. Histopathology. 2005;47(2):141–146.
- Seymour MT, Morton D. FOxTROT: an international randomised controlled trial in 1052 patients (pts) evaluating neoadjuvant chemotherapy (NAC) for Colon cancer. J Clin Oncol. 2019;37(15_suppl):3504–3504.
- Tie J, Cohen JD, Wang Y, et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: a prospective biomarker study. Gut. 2019;68(4):663–671.
- Khakoo S, Carter PD, Brown G, et al. MRI tumor regression grade and circulating tumor DNA as complementary tools to assess response and guide therapy adaptation in rectal cancer. Clin Cancer Res. 2020;26(1):183–192.
- Sclafani F, Chau I, Cunningham D, et al. KRAS and BRAF mutations in circulating tumour DNA from locally advanced rectal cancer. Sci Rep. 2018;8(1):1445.
- Tie J, Wang Y, Cohen J, et al. Circulating tumor DNA dynamics and recurrence risk in patients undergoing curative intent resection of colorectal cancer liver metastases: a prospective cohort study. PLoS Med. 2021;18(5):e1003620.
- Loupakis F, Sharma S, Derouazi M, et al. Detection of molecular residual disease using personalized circulating tumor DNA assay in patients with colorectal cancer undergoing resection of metastases. J Clin Oncol Precis Oncol. 2021;5:PO.21.00101.
- Siravegna G, Mussolin B, Buscarino M, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21(7):795–801.
- Garlan F, Laurent-Puig P, Sefrioui D, et al. Early evaluation of circulating tumor DNA as marker of therapeutic efficacy in metastatic colorectal cancer patients (PLACOL study). Clin Cancer Res. 2017;23(18):5416–5425.
- Spindler KG, Boysen AK, Palisgaard N, et al. A systematic review and Meta-analysis of the prognostic value of total cell-free DNA quantification in metastatic colorectal cancer. Ann Oncol. 2016;27(Supplement 6):vi149–vi206.
- Hamfjord J, Guren TK, Dajani O, et al. Total circulating cell-free DNA as a prognostic biomarker in metastatic colorectal cancer before first-line oxaliplatin-based chemotherapy. Ann Oncol. 2019;30(7):1088–1095.
- Mouliere F, Thierry AR. The importance of examining the proportion of circulating DNA originating from tumor, microenvironment and normal cells in colorectal cancer patients. Expert Opin Biol Ther. 2012;12(sup1):S209–S215.
- Norton SE, Lechner JM, Williams T, et al. A stabilizing reagent prevents cell-free DNA contamination by cellular DNA in plasma during blood sample storage and shipping as determined by digital PCR. Clin Biochem. 2013;46(15):1561–1565.
- Alcaide M, Cheung M, Hillman J, et al. Evaluating the quantity, quality and size distribution of cell-free DNA by multiplex droplet digital PCR. Sci Rep. 2020;10(1):12564.
- Thomsen CB, Andersen RF, Lindebjerg J, et al. Correlation between tumor-specific mutated and methylated DNA in colorectal cancer. J Clin Oncol Precis Oncol. 2019;3:1–8.