
Abstract
Background
This study aimed to investigate the distribution and frequency of concurrent alterations in different cancers across KRAS subtypes and in different KRAS subtypes across cancers, and to identify potentially actionable targets and patients who received targeted treatment matched to their genomic profile (GP).
Materials and Methods
In this descriptive and single-center study, we included 188 patients with solid tumors harboring KRAS mutations in codon 12, 13, 61, 117, or 146, referred to the Phase 1 Unit, Rigshospitalet, Copenhagen, Denmark from mid-2016 to 2020. Genomic co-alterations were detected with whole-exome sequencing, RNA sequencing, SNP array, and mRNA expression array on fresh biopsies. The study is part of the Copenhagen Prospective Personalized Oncology study (NCT02290522).
Results
The majority of patients had colorectal cancer (60.1%), non-small cell lung cancer (11.2%), or pancreatic cancer (10.6%). Most tumors were KRAS-mutated in codon 12 or 13 (93.7%) including G12D (27.1%), G12V (26.6%), G12C (11.7%), and G13D (11.2%). A total of 175 different co-alterations were found, most frequently pathogenic APC and TP53 mutations (55.9% and 46.4%, respectively) and high expression of CEACAM5 (73.4%). Different cancers and KRAS subtypes showed different patterns of co-alterations, and 157 tumors (83.5%) had potentially actionable targets with varying evidence of targetability (assessed using ESMO Scale for Clinical Actionability of molecular Targets). Of the 188 patients included in the study, 15 (7.4%) received treatment matched to their GP (e.g., immunotherapy and synthetic lethality drugs), of whom one had objective partial response according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1.
Conclusion
Performing extensive genomic analysis in patients with known KRAS-mutated solid tumors may contribute with information to the genomic landscape of cancers and identify targets for immunotherapy or synthetic lethality drugs, but currently appears to have overall limited clinical impact, as few patients received targeted therapy matched to their GP.
Introduction
The KRAS gene is mutated in approximately 22% of all tumors [Citation1], predominantly in pancreatic, colorectal, and lung carcinoma [Citation2] with a mutation frequency of 90%, 40%, and 30%, respectively [Citation3–6]. The KRAS protein is active when GTP-bound and mainly operates in the MAPK and PI3K pathways, which are involved in cell proliferation, growth, and survival [Citation6].
The oncogenic activity of active GTP-KRAS comes from increased rate of nucleotide dissociation and/or decreased rate of hydrolysis of GTP to GDP and the most common mutations (at codon 12, 13, 61, 117, and 146) cluster around the nucleotide binding pocket [Citation7,Citation8].
Novel drugs targeting the KRAS mutation subtype G12C (e.g., adagrasib and sotorasib) work by locking KRAS in its inactive GDP-state [Citation9]. Apart from G12C-selective inhibitors there are no effective KRAS targeted therapies available. Drug resistance to upstream and downstream inhibitors [Citation10], such as anti-EGFR or anti-HER2 therapy [Citation11–13] means few gain-of-function co-mutations can be targeted. This study aimed to investigate the potential benefits of extensive genomic analysis in patients with KRAS-mutated solid tumors. We examined the distribution and frequency of concurrent pathogenic mutations and other genomic alterations found in different KRAS mutation subtypes across histological cancer diagnoses and in different cancer diagnoses across KRAS subtypes. We also identified potentially actionable targets and patients who received targeted therapy matched to their genomic profile (GP).
Methods and materials
Study design
This is a descriptive single-center study conducted at the Phase 1 Unit at the Department of Oncology at Rigshospitalet, Copenhagen University Hospital, Denmark.
Patients with advanced solid tumors, who have exhausted standard treatment options, can be referred to the Phase 1 Unit for extensive genomic tumor profiling. The GP is discussed at a multidisciplinary national tumor board meeting and potential treatment options are suggested based on targetable aberrations.
All patients with a GP from 1 July 2016 to 31 December 2020 harboring a KRAS-mutated tumor with subtype G12X, G13X, Q61X, K117X, or A146X were included. A total of 188 patients, corresponding to approx. 17% of all referrals in the same time period, were identified.
This study is a part of the Copenhagen Prospective Personalized Oncology (CoPPO, NCT02290522 [Citation14]) study, which is conducted in accordance with the Declaration of Helsinki and has been approved by an institutional review board and the Regional Ethics Committee (Danish Ethical Committee, file number: 1300530) [Citation15]. Written informed consent has been obtained from all study participants.
Clinical and genomic data
Study data were managed using REDCap electronic data capture tools [Citation16,Citation17] version 10.3.3. The genomic analyses were performed on either biopsy samples (n = 170, 90.4%), circulating tumor DNA (ctDNA) (n = 14, 7.5%), both ctDNA and biopsy (n = 3, 1.6%), or archived formalin-fixed, paraffin-embedded (FFPE) tissue (n = 1, 0.5%). Individual genomic reports based on analysis results from whole-exome sequencing (WES), RNA sequencing, SNP array, and mRNA expression array were provided by the Center for Genomic Medicine, Rigshospitalet, Denmark. The tumor specific variants were called and annotated by the software used at the time (either CLC workbench or GATK). Variants with an allele frequency down to 10% were assessed and used for further curation. Methods regarding genomic data processing have been reported in previous CoPPO articles [Citation15,Citation18]. The turnaround time from tumor sample to report was 4–6 weeks.
All reports were evaluated for co-alterations and actionability by board certified clinical oncologists at the Phase 1 Unit and were then recorded in REDCap. The recorded genomic alterations comprised all: (I) cancer-associated mutations (classified as either variant of unknown significance (VUS) or pathogenic) incl. high Tumor Mutational Burden (TMB) defined as >10 mut/Mb as defined by the Center for Genomic Medicine, corresponding to approx. 300 mutations (not exclusively missense mutations included) [Citation19]; (II) fusions; (III) amplifications; (IV) biallelic deletions; (V) homologous recombination deficiency (HRD) [Citation20]; and (VI) high expression of CEACAM5. All other expression findings, loss of heterozygosity (LOH), and deletions were not recorded due to potential inconsistency over time and low possibility of targetability. However, we included the PTEN gene if deletion and/or LOH occurred concomitantly with a mutation, corresponding to PTEN loss (biallelic deletion). In the case of multiple genomic reports showing alterations co-occurring with a KRAS mutation in the same patient, all alterations (I–VI) were recorded, but the same alteration would only be recorded once to prevent duplicate entries.
Potentially actionable targets were assessed from a present-day perspective due to the ever-evolving understanding and definition of targetability. The term ‘actionable’ refers to the ability of the alteration to be targeted by an agent based on clinical or preclinical data.
All clinical data were retrospectively collected from electronic medical records.
Statistical analysis
Data were analyzed using R Studio version 1.2.5001 [Citation21] and SPSS Statistics version 25 [Citation22]. Continuous variables were summarized using mean, standard deviation, median, minimum, and maximum as appropriate. Categorical variables were summarized using number and percentage.
Results
Patient characteristics are presented in . The majority of patients had colorectal cancer (CRC; n = 113, 60.1%), non-small cell lung cancer (NSCLC; n = 21, 11.2%), or pancreatic cancer (n = 20, 10.6%).
Table 1. Patient characteristics at date of referral to the Phase 1 Unit.
KRAS subtypes
Codon 12 and 13 mutations accounted for 93.7% of all KRAS mutations (80.9% at codon 12 and 12.8% at codon 13). Across tumor types, G12D and G12V were the most frequent mutations (27.1% and 26.6%, respectively), followed by G12C (11.7%) and G13D (11.2%). The frequencies of KRAS mutation subtypes in CRC, NSCLC, pancreatic cancer, and all other cancers are outlined in , and the distribution of all diagnoses and subtypes are listed in Supplementary Table 1.
Figure 1. Pie charts of relative frequencies of different KRAS mutation subtypes among cohort. (a) All cancers. (b) Colorectal cancer (CRC). (c) Non-small cell lung cancer (NSCLC). (d) Pancreatic cancer. (e) All other cancers.
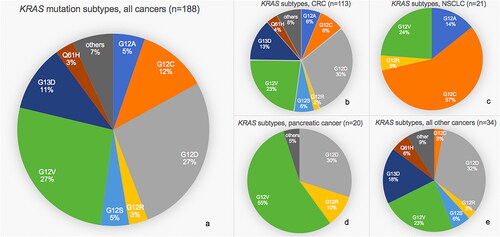
Among the 188 patients, 64 (34.0%) had no known KRAS mutation upon referral. The majority of these tumors (n = 56, 87.5%) had not previously been analyzed for KRAS (all 113 CRCs had prior KRAS analysis). The remaining eight tumors (12.5%) were initially KRAS wild-type (wt) (CRC n = 7, neuroendocrine carcinoma n = 1). Of the seven KRAS wt CRC patients, six had received anti-EGFR therapy prior to referral to the Phase 1 Unit. The initial GP of another patient showed a KRAS wt, BRAF V600E mutated breast cancer and the patient received 64 weeks of BRAF targeted therapy (dabrafenib + trametinib). Upon progression, a new GP revealed presence of KRAS mutations (G12V and G12A).
Of the 124 patients with known KRAS mutation upon referral, 119 (96.0%) had the same KRAS mutation subtype in the GP as the one previously found (or a mutation in the same codon if not specified further). Five tumors (4.0%) had a different subtype in the GP (Supplementary Table 2).
Co-occurring genomic alterations
In total, 175 different cancer-related co-occurring genomic alterations were recorded, with a median of four co-alterations per tumor. There were 164 (87.2%) tumors with at least one cancer-associated pathogenic co-mutation, and 138 tumors (73.4%) had high RNA expression of CEACAM5. HRD was found in 37 tumors (19.7%) and 13 tumors (6.9%) were TMB high (Supplementary Table 3).
The co-alterations present in >5% of CRCs, NSCLCs, pancreatic cancers, or all other cancers pooled together (across KRAS subtypes) included alterations in the genes APC, ATM, BRCA1/2, CDKN2A, CCND1, ERBB2, FBXW7, GNAS, PIK3CA, RB1, SMAD4, STK11, TP53, KRAS, MDM2, MYC, and PTEN (). The co-alterations present in >5% of tumors with KRAS mutations G12D, G12V, G12C, G13D, or all other subtypes pooled together (across cancers), included alterations in the genes APC, ATM, BRCA2, CDKN2A/B, ERBB2, FBXW7, PIK3CA, SMAD4, STK11, TP53, KRAS, MYC, and PTEN ().
Figure 2. Bar chart of relative frequencies of co-occurring genomic alterations in different tumors. (a) In colorectal cancer (CRC), non-small cell lung cancer (NSCLC), pancreatic cancer, or all other cancers across all KRAS subtypes. (b) In tumors with KRAS mutation G12D, G12V, G12C, G13D, or all other subtypes across all cancers. Only alterations present in >5% of tumors are shown. Pathogenic: pathogenic mutation; vus: variant of unknown significance; HRD: homologous recombination deficiency; TMB: tumor mutational burden. PTEN loss include PTEN biallelic deletions and PTEN pathogenic mutation+PTEN deletion and/or loss of heterozygosity.
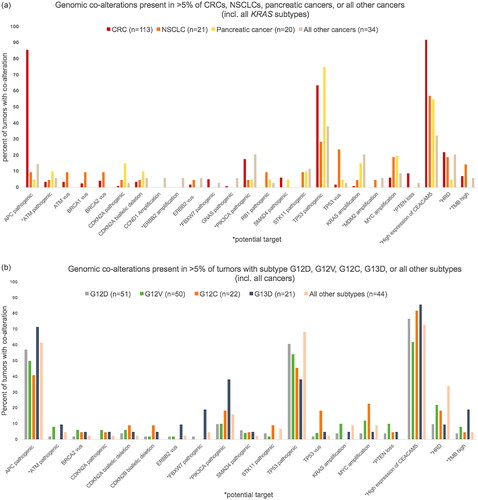
A complete overview of the distribution and frequency of all the different co-alterations recorded are presented in Supplementary Tables 3 and 4, stratified by tumor type including all KRAS subtypes (Supplementary Table 3) and by KRAS subtype including all tumor types (Supplementary Table 4).
Potential targets and treatment
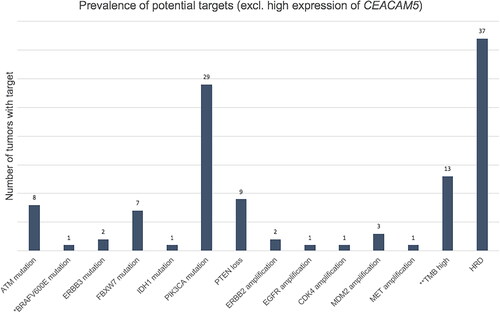
Potentially actionable targets were identified in 157 tumors (83.5%), of which 138 (73.4%) had high expression of CEACAM5, and 85 (45.2%) had other potential targets (). ESMO Scale for Clinical Actionability of molecular Targets (ESCAT) levels for all potential targets have been assessed and presented in Supplementary Table 5.
Figure 3. Prevalence of potential targets in 85 tumors with targets other than high expression of CEACAM5. *The patient with breast cancer who received BRAF targeted therapy. **With/without pathogenic mutations in mismatch repair genes. 62 tumors had one target, 17 tumors had two, five tumors had three and one tumor had four targets. HRD: homologous recombination deficiency; TMB: tumor mutational burden. PTEN loss include PTEN biallelic deletions and PTEN pathogenic mutation+PTEN deletion and/or loss of heterozygosity.
A total of 41 patients (21.8%) received treatment in the Phase 1 Unit (). Of the 41 patients, 15 received targeted treatment matched to their GP, of whom one patient had objective partial response (PR) according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1.
Figure 4. Patients treated according to genomic profile. *The patient received pembrolizumab based on high tumor mutation burden (TMB high). **Considered an actionable target at time of treatment. BR: best response according to RECIST 1.1; SD: stable disease; PR: partial response; PD: progressive disease; EOT: reason for end of treatment.
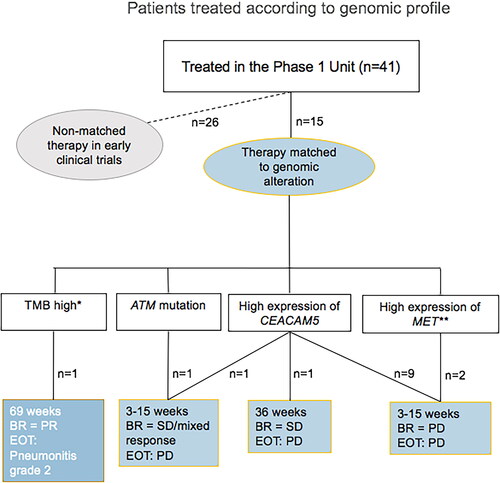
The remaining 26 patients received non-matched therapy in early clinical trials. Notably, 145 patients (77.1%) did not receive treatment in the Phase 1 Unit, 120 of whom despite potentially actionable targets.
There were 44 patients (23.4%) who subsequently received standard systemic treatment at their local hospital, but most received palliative care. The median survival time from referral to the Phase 1 Unit was 5.7 months (Supplementary Table 6).
Discussion
In this descriptive study of 188 patients with KRAS-mutated solid tumors representing 16 cancer types, we investigated the distribution and frequency of KRAS subtypes, genomic co-alterations, and potential targets.
The majority (93.7%) was KRAS-mutated in codon 12 or 13, and subtype G12D and G12V were the most frequent across tumor types, consistent with other studies [Citation2,Citation7,Citation23].
In NSCLC and pancreatic cancer, only codon 12 mutations were observed. Mutations outside codon 12 or 13 were only found in CRC. In concordance with this study, mutations in codon 13, 117, and 146 have been described as more common in CRC relative to NSCLC and pancreatic cancer. Furthermore, mutations in codon 117 and 146 have been described as nearly selective for CRC compared to all cancers [Citation8].
Our study population was highly selected, comprising patients with advanced disease, who had received several treatment regimens prior to referral to the Phase 1 Unit, and who were still in good general performance. This may explain the relatively small NSCLC and pancreatic cancer subgroups (21 and 20 patients, respectively), and the KRAS mutation subtype frequencies found in this study may therefore not represent the KRAS mutation frequencies in the general cancer population. Nonetheless, we found cancer-specific distribution of the various KRAS mutation subtypes, similar to other studies [Citation2,Citation4,Citation23].
We recorded 175 different cancer-related co-alterations. The most frequent were pathogenic mutations in the APC and TP53 gene and high expression of CEACAM5. Pathogenic APC mutations and high expression of CEACAM5 were most frequent in CRC (85.8% and 92%, respectively). Consistent with these findings, other studies have found co-occurrence of APC and KRAS mutations in 80% of CRCs [Citation23], and high expression of CEACAM5 in 90% of CRCs (KRAS status unknown) [Citation24]. In our study, co-occurring TP53 mutations appeared more common in CRC (63.7%) and pancreatic cancer (75%) compared to NSCLC (23.8%). Similarly, another study showed that KRAS mutations often occur together with TP53 in CRC, pancreatic cancer, and NSCLC, although at frequencies of 40%, 80%, and 40%, respectively [Citation23].
This study did not distinguish between left- and right-sided colon cancers, which have previously been shown to differ in regards to mutations and epigenetic changes [Citation25,Citation26]. Thus, as the distribution of the colon cancers was not known and which could potentially impact the co-alterations observed, we decided to pool colon and rectal cancers together. However, as the majority of patients had CRC (60.1%) it would have been relevant to compare diagnostic subgroups as the importance of KRAS subtypes and concurrent genomic alterations might be different in different diagnostic subgroups.
Mutations in the tumor suppressor genes APC and TP53 are frequent alterations in human cancers [Citation27,Citation28], but cannot yet be targeted with therapy. High expression of CEACAM5 was considered an actionable target as several trials with bispecific antibodies targeting CEACAM5 were accruing patients with high CEACAM5 expression at our facility during the study period. HRD, TMB high, loss of PTEN, pathogenic mutations in FBXW7, PIK3CA, ATM, BRAF (V600E), ERBB3, and IDH1, and amplifications of ERBB2, EGFR, CDK4, MDM2, and MET were also considered actionable, all of which were found in this study, with ESCAT levels ranging from I-A to IV (Supplementary Table 5).
Potentially actionable targets were identified in 83.5% of the patients. This is numerically higher than the 76.5% (352 of 460 GPs) previously reported by our facility (CoPPO) [Citation15], which could be explained by dynamics in the definition of targets and advances in precision oncology. However, a considerable smaller proportion of patients received targeted treatment matched to their GP in this study (7.4%) compared to 22% in the previous study by CoPPO, which included all patients with solid tumors referred to the Phase 1 Unit 2013–2017 [Citation15]. The response rate was also lower in this study: 0.5% vs. 3.3% (1 PR of 188 GPs vs. 15 PRs of 460 GPs), and 6.7% vs. 14.9% (1 PR of 15 treated matched to GP vs. 15 PRs of 101 treated matched to GP). This could be due to the fact that targeting alterations upstream or downstream of the KRAS signaling pathway are without significant success and the absence of other actionable driver mutations in the study population [Citation29].
In our study, 26 patients (13.8%) received non-matched therapy in early clinical trials. The majority (77.1%) did not enroll in any trials in Phase 1, most likely due to performance decline or non-eligibility in clinical trials.
Despite the high number of potentially actionable targets, few patients with KRAS-mutated tumors received targeted therapy matched to their genomic profile. Although KRAS mutations do not per se exclude other actionable driver mutations, these co-existing driver mutations are rare and often not actionable due to concomitant KRAS mutation (e.g., ERBB2 amplification or BRAF mutations). The co-occurring targets at DNA or chromosomal level are mainly characterized by being loss-of-function alterations (e.g., ATM and HRD) constituting a potential target for synthetic lethality drugs (PARP and ATR inhibitors) or TMB-high status constituting a potential target for immune checkpoint inhibitors.
Many genomic alterations co-occur with KRAS, and although some may not be currently targetable, the genomic landscape is changing. Genomic profiling of KRAS-mutated tumors contributes to this research and may uncover new treatable targets, especially loss-of-function mutations inferring targets of synthetic lethality and/or increasing our knowledge of resistance mechanisms, ultimately benefitting these patients. An example of the latter, is the breast cancer patient who received BRAF targeted therapy due to an activating BRAF mutation (V600E) found in the initial GP, and whose tumor developed KRAS mutations upon progression, indicating well-known resistance mechanisms [Citation30,Citation31].
Another potential benefit of performing extensive genomic analysis in KRAS-mutated solid tumors is the prognostic and treatment-predictive information which may be derived from KRAS subtypes and co-occurring alterations [Citation32–35]. Recent data from KRYSTAL-1, showed an even greater response to the KRAS G12C inhibitor adagrasib in patients with KRAS-mutated NSCLC, whose tumor also had a concurrent STK11 mutation [Citation36]. In our study, two patients could potentially have had increased benefit from receiving adagrasib. These patients had KRAS G12C mutated NSCLCs and STK11 co-mutations, one of which was pathogenic, and one was a VUS.
As G12C-selective inhibitors are being introduced, with observed objective response in NSCLC, CRC and other KRAS-mutated solid tumors [Citation37–39] and with the recent FDA approval of sotorasib in NSCLC, the possibility of combined targeted therapy arises. At present time, however, the outcome of combining KRAS G12C inhibitors with other treatments based on molecular tumor profiling is unknown. Double-targeted therapy is currently being investigated in clinical trials such as combining anti-EGFR therapy with a G12C inhibitor for KRAS G12C mutant CRC (NCT04793958), and combining immune checkpoint inhibitors with a G12C inhibitor for KRAS mutant solid tumors (NCT04699188).
A definite strength of this study is the single-center design with uniformity and continuity in interpretation and decision making of each GP. In 15 cases, genomic analysis was presumably performed on an assay with less coverage of the genome, no RNA expression and less possibility of detecting chromosomal aberrations (14 ctDNA and one FFPE) as biopsy was not possible or contained normal tissue. These GPs may therefore have presented with fewer co-alterations.
There is no clear definition of what constitutes a potential target, and far from all target-drug combinations have the same therapeutic potential [Citation40]. We included several potential targets with varying evidence of targetability, but the list of potential targets is not complete and will differ from site to site.
In conclusion, we found different patterns of KRAS mutation subtypes in different cancers, and varying frequencies of known genomic alterations co-occurring with KRAS in different cancers across subtypes and in different subtypes across cancers. The majority of patients had one or more potential targets but only a small percentage of the patients received targeted treatment matched to their genomic profile indicating limited clinical benefit from the analyses at present time.
The genomic landscape is continuously evolving and molecular characterization of concurrent alterations and potential targets as provided by our study will provide insight and add knowledge to this field. Furthermore, as KRAS G12C inhibitors are being introduced and an increasing number of loss-of-function alterations emerge as potential druggable targets in the synthetic lethality or functional restoration setting, the benefits of performing extensive genomic analysis in KRAS-mutated solid tumors may increase in the future.
Authors’ contributions
Guarantor of integrity of the entire study: KSR, UL. Study concepts and design: ICJ, IS, MH, UL, KSR. Literature research: ICJ, IS, KSR, MH, CQ. Clinical studies: IS, MH, CQ, KSR, UL. Experimental studies/data analysis: ICJ, LB. Statistical analysis: ICJ, IS, KSR. Manuscript preparation: All authors. Manuscript editing: All authors.
Supplemental Material
Download PDF (527.1 KB)Supplemental Material
Download MS Word (228.3 KB)Acknowledgements
The authors thank all the patients and their relatives for the participation. Conduction of this study would not have been possible without the work of the Kennedy Center and its laboratory staff at Glostrup, Denmark.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data that underlie the results reported in this article can be made available upon reasonable request to the corresponding author, and will require the completion of a data processing agreement.
Additional information
Funding
References
- Martin P, Leighl NB, Tsao MS, et al. KRAS mutations as prognostic and predictive markers in non-small cell lung cancer. J Thorac Oncol. 2013;8(5):530–542.
- Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129(7):1287–1292.
- Aredo JV, Padda SK, Kunder CA, et al. Impact of KRAS mutation subtype and concurrent pathogenic mutations on non-small cell lung cancer outcomes. Lung Cancer. 2019;133:144–150.
- Scheffler M, Ihle MA, Hein R, et al. K-ras mutation subtypes in NSCLC and associated co-occuring mutations in other oncogenic pathways. J Thorac Oncol. 2019;14(4):606–616.
- Chiorean EG, Coveler AL. Pancreatic cancer: optimizing treatment options, new, and emerging targeted therapies. Drug Des Devel Ther. 2015;9:3529–3545.
- Zenonos K, Kyprianou K. RAS signaling pathways, mutations and their role in colorectal cancer. World J Gastrointest Oncol. 2013;5(5):97–101.
- Oikonomou E, Koustas E, Goulielmaki M, et al. BRAF vs RAS oncogenes: are mutations of the same pathway equal? Differential signalling and therapeutic implications. Oncotarget. 2014;5(23):11752–11777.
- Haigis KM. KRAS alleles: the devil is in the detail. Trends Cancer. 2017;3(10):686–697.
- Dunnett-Kane V, Nicola P, Blackhall F, et al. Mechanisms of resistance to KRASG12C inhibitors. Cancers. 2021;13(1):151.
- Mustachio LM, Chelariu-Raicu A, Szekvolgyi L, et al. Targeting KRAS in cancer: promising therapeutic strategies. Cancers. 2021;13(6):1204.
- Sahin IH, Garrett CR. Predicting cetuximab efficacy in patients with advanced colorectal cancer. Curr Biomark Find. 2014;4:61–68.
- Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2(1):e17.
- Meric-Bernstam F, Hainsworth J, Bose R, et al. MyPathway HER2 basket study: pertuzumab (P)+trastuzumab (H) treatment of a large, tissue-agnostic cohort of patients with HER2-positive advanced solid tumors. J Clin Oncol. 2021;39(15_Suppl):3004–3004.
- Tuxen IV, Jønson L, Santoni-Rugiu E, et al. Personalized oncology: genomic screening in phase 1. APMIS. 2014;122(8):723–733.
- Tuxen IV, Rohrberg KS, Oestrup O, et al. Copenhagen prospective personalized oncology (CoPPO)—clinical utility of using molecular profiling to select patients to phase I trials. Clin Cancer Res. 2019;25(4):1239–1247.
- Harris PA, Taylor R, Thielke R, et al. Research electronic data capture (REDCap) – a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381.
- Harris PA, Taylor R, Minor BL, et al. The REDCap consortium: building an international community of software partners. J Biomed Inform. 2019;95:103208.
- Tuxen IV, Ahlborn LB, Mau-Soerensen M, et al. Plasma total cell-free DNA is a prognostic biomarker of overall survival in metastatic solid tumour patients. Br J Cancer. 2019;121(2):125–130.
- Szustakowski J, Green G, Geese WJ, et al. Evaluation of tumor mutational burden as a biomarker for immune checkpoint inhibitor efficacy: a calibration study of whole exome sequencing with FoundationOne. Cancer Res. 2018;78(13_Suppl):5528–5528.
- Marquard AM, Eklund AC, Joshi T, et al. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark Res. 2015;3:9.
- RStudio Team. RStudio: integrated development for R. Boston (MA): RStudio, PBC; 2020.
- IBM Corp. IBM SPSS statistics for windows, version 25.0. Armonk (NY): IBM Corp; 2017.
- Timar J, Kashofer K. Molecular epidemiology and diagnostics of KRAS mutations in human cancer. Cancer Metastasis Rev. 2020;39(4):1029–1038.
- Zhou J, Fan X, Chen N, et al. Identification of CEACAM5 as a biomarker for prewarning and prognosis in gastric cancer. J Histochem Cytochem. 2015;63(12):922–930.
- Slattery ML, Curtin K, Wolff RK, et al. A comparison of colon and rectal somatic DNA alterations. Dis Colon Rectum. 2009;52(7):1304–1311.
- Salem ME, Weinberg BA, Xiu J, et al. Comparative molecular analyses of left-sided colon, right-sided colon, and rectal cancers. Oncotarget. 2017;8(49):86356–86368.
- Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2(1):a001008.
- AACR Project GENIE Consortium. AACR project GENIE: powering precision medicine through an international consortium. Cancer Discov. 2017;7(8):818–831.
- Del Re M, Rofi E, Restante G, et al. Implications of KRAS mutations in acquired resistance to treatment in NSCLC. Oncotarget. 2018;9(5):6630–6643.
- Johnson DB, Menzies AM, Zimmer L, et al. Acquired BRAF inhibitor resistance: a multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer. 2015;51(18):2792–2799.
- Niemantsverdriet M, Schuuring E, ter Elst A, et al. KRAS mutation as a resistance mechanism to BRAF/MEK inhibition in NSCLC. J Thorac Oncol. 2018;13(12):249–251.
- Jones RP, Sutton PA, Evans JP, et al. Specific mutations in KRAS codon 12 are associated with worse overall survival in patients with advanced and recurrent colorectal cancer. Br J Cancer. 2017;116(7):923–929.
- Kulemann B, Rösch S, Seifert S, et al. Pancreatic cancer: circulating tumor cells and primary tumors show heterogeneous KRAS mutations. Sci Rep. 2017;7(1):4510.
- La Fleur L, Falk-Sörqvist E, Smeds P, et al. Mutation patterns in a population-based non-small cell lung cancer cohort and prognostic impact of concomitant mutations in KRAS and TP53 or STK11. Lung Cancer. 2019;130:50–58.
- Skoulidis F, Goldberg ME, Greenawalt DM, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8(7):822–835.
- Riely G, Ou SI, Rybkin I, et al. KRYSTAL-1: activity and preliminary pharmacodynamic (PD) analysis of adagrasib (MRTX849) in patients (pts) with advanced non-small-cell lung cancer (NSCLC) harboring KRASG12C mutation. J Thorac Oncol. 2021;16:S748–S802.
- Johnson ML, Ou SHI, Barve M, et al. KRYSTAL-1: activity and safety of adagrasib (MRTX849) in patients with colorectal cancer and other solid tumors harboring a KRAS G12C mutation. 2020 EORTC-NCI-AACR Symposium (abstr LBA4).
- Janne PA, Rybkin II, Spira AI, et al. KRYSTAL-1: activity and safety of adagrasib (MRTX849) in advanced/metastatic non-small-cell lung cancer harboring KRAS G12C mutation. 2020 EORTC-NCI-AACR Symposium (abstr LBA3).
- Hong DS, Fakih MG, Strickler JH, et al. KRASG12C inhibition with sotorasib in advanced solid tumors. N Engl J Med. 2020;383(13):1207–1217.
- Gyawali B, Kesselheim AS. The promise of ESCAT: a new system for evaluating cancer drug-target pairs. Nat Rev Clin Oncol. 2019;16(3):147–148.