
Abstract
Aim
Our goal was to describe a precision medicine program in a regional academic hospital, characterize features of included patients and present early data on clinical impact.
Materials and methods
We prospectively included 163 eligible patients with late-stage cancer of any diagnosis from June 2020 to May 2022 in the Proseq Cancer trial. Molecular profiling of new or fresh frozen tumor biopsies was done by WES and RNAseq with parallel sequencing of non-tumoral DNA as individual reference. Cases were presented at a National Molecular Tumor Board (NMTB) for discussion of targeted treatment. Subsequently, patients were followed for at least 7 months.
Results
80% (N = 131) of patients had a successful analysis done, disclosing at least one pathogenic or likely pathogenic variant in 96%. A strongly or potentially druggable variant was found in 19% and 73% of patients, respectively. A germline variant was identified in 2.5%. Median time from trial inclusion to NMTB decision was one month. One third (N = 44) of patients who underwent molecularly profiling were matched with a targeted treatment, however, only 16% were either treated (N = 16) or are waiting for treatment (N = 5), deteriorating performance status being the primary cause of failure. A history of cancer among 1st degree relatives, and a diagnosis of lung or prostate cancer correlated with greater chance of targeted treatment being available. The response rate of targeted treatments was 40%, the clinical benefit rate 53%, and the median time on treatment was 3.8 months. 23% of patients presented at NMTB were recommended clinical trial participation, not dependent on biomarkers.
Conclusions
Precision medicine in end-stage cancer patients is feasible in a regional academic hospital but should continue within the frame of clinical protocols as few patients benefit. Close collaboration with comprehensive cancer centers ensures expert evaluations and equality in access to early clinical trials and modern treatment.
Introduction
Treatment guidance based on genomic analysis has the potential to improve the outcome of cancer patients with limited treatment options. By molecular profiling, mutations or mutational signatures may be detected in tumor cells, in the tumor microenvironment, or in normal tissue, that are biomarkers of the effect of or resistance to specific treatments [Citation1]. This concept is usually named precision or personalized medicine [Citation2,Citation3]. With an increasing number of druggable variants of low frequency, new predictive markers, lower costs, and higher speed of processing, extensive genomic tumor analysis is becoming cost-efficient and may in the near future become part of the routine work-up of most cancer patients [Citation4].
Genomic analysis is sparing valuable tissue compared to multiple sequential assays, enables the design of a personalized plan for treatment, improves recruitment of patients to trials, discloses codrivers and predictors of resistance, identifies germline variants, and may contribute to the diagnosis [Citation4,Citation5]. The analyses can furthermore lead to the disclosure of rare, but potentially druggable variants across many tumor types [Citation1]. The tissue agnostic approach has recently been approved for drugs targeting neurotrophic tyrosine receptor kinase (NTRK) fusions, and for the treatment of microsatellite instability (MSI)-high or tumor mutational burden (TMB)-high tumors [Citation6]. However, the principle is not universally applicable [Citation7]. Therefore, most trials in precision medicine investigate drug efficacy in diagnosis-specific cohorts, demanding multicenter and international collaboration to obtain a sufficient sample size [Citation8].
The feasibility of precision medicine for patients’ refractory to standard oncological treatment has been demonstrated in several studies [Citation9]. Although unambiguous benefit has been observed casuistically [Citation10,Citation11], high-level scientific evidence is lacking [Citation12,Citation13], as well as new molecular predictive factors and new targeted drugs are constantly evolving making previous reports obsolete. More patients on targeted treatment have longer progression-free survival (PFS) as compared to prior, non-targeted treatment [Citation14,Citation15], they achieve higher response rates (RR) [Citation16], and a much larger difference in median overall survival (OS) has been observed for responders compared to non-responders [Citation17]. Additionally, meta-analyses of early-phase clinical studies showed that patients who received targeted treatment had better outcomes than those who received unspecific treatment, and the treatment-related mortality was lower [Citation9, Citation18].
Many comprehensive cancer centers offer patients with incurable cancer extensive genomic sequencing in search for druggable variants [Citation19]. The implementation of precision medicine in daily routine is, however, not trivial and rarely described in less exclusive environments [Citation20]. The disparity in the outcome of cancer patients may result from the delayed introduction of new treatments with higher efficacy at smaller hospital units [Citation21]. From a national health perspective, it is important to ensure equality in access to new diagnostics and modern treatment, independently of geography, ethnicity, and socioeconomical factors [Citation3, Citation22].
The aims of the current study are to describe the interdisciplinary workflow of a precision medicine program established in the setting of a regional public academic hospital, to characterize the genomic, pathological, and clinical features of included patients, as well as to present early data on the clinical impact in the first two years of the program.
Materials and methods
Patients were recruited from the North Denmark Region with 590,000 inhabitants, constituting 1/10 of the Danish population [Citation23]. Cancer patients from the region, who are candidates for oncological treatment, are all referred to the Department of Oncology at Aalborg University Hospital. Exceptions include patients with uncommon cancer diagnoses and patients in demand of specific treatment of high complexity. The number of new patients referred annually to the Department is approximately 3300. All costs for Danish patients are covered by the public health care system.
In 2020 a precision medicine program based on in-house whole exome sequencing (WES) and RNA sequencing (RNAseq) was initiated. All eligible patients are offered enrollment in the Proseq Cancer trial (NCT05695638), which allows for biobanking, registration of clinical and laboratory data, and sharing of genomic data with the purpose of research while fulfilling the Danish General Data Protection Regulation (GDPR) requirements. The trial was approved by the Ethics Committee of Northern Jutland, Denmark (N-20200018).
Inclusion criteria and study endpoints
Patients included are at least 18 years of age, eligible for treatment, and have incurable, progressing, and/or life-threatening cancer (of any diagnosis) with an expected residual survival of at least 3 months and no efficient remaining standard treatment options. All candidate patients are identified by specialist oncologists and are reviewed by the primary investigator (Professor Morten Ladekarl) for eligibility prior to inclusion. Patients with hematological malignancies are included as well but not assessed in the current analysis. All patients signed informed consent.
The primary endpoint per protocol is the fraction of patients, for whom a molecular variant can be identified that potentially can be matched with a targeted drug, labeled for use in cancer, i.e., a ‘druggable’ variant [Citation24], among patients with no further efficient standard treatment options who had a tumor molecular profiling done. A positive outcome is defined as 10% the first year linearly rising to 25% after 10 years.
Clinical-pathological data
After informed consent, clinical-pathological baseline data is registered in REDCap [Citation25] by oncological investigators. Results of new pathological and molecular assessments are prospectively registered as well as decisions made at the National Molecular Tumor Board (NMTB) (Supplementary Table A.1). For the present analysis, the outcome of all patients was registered at the time of data cutoff (January 3rd, 2023) and no patients were lost to follow-up.
Biological samples
New 1.2 mm core needle biopsies are taken from the most easily accessible, preferably progressing tumor lesion that has not been irradiated, usually guided by ultrasound. Biopsies from centrally located lung lesions or other sites with increased risk of complications, or procedures requiring general anesthesia are generally avoided. In each case, one biopsy is fixed in neutral-buffered formalin and allocated to histopathological assessment. Another, adjacent 1–2 biopsies are kept in RNAlater until processing for molecular analysis within 5 d. Patients with intracranial tumors are biopsied at the time of diagnostic surgery or if surgery is otherwise required, and tissue is kept frozen at −80 °C until the clinical course indicates that patients are eligible for the protocol. In parallel, a 10 ml EDTA blood sample is drawn for analysis of non-tumoral DNA.
Unfixed or fresh frozen tissue material is preferred. However, in selected patients for whom a new biopsy is unobtainable and no fresh frozen tissue is available, DNA is extracted from the most recent FFPE archival sample of tumor tissue. Cell-free tumor-DNA (ctDNA) from peripheral blood samples is used in cases with insufficient tissue for analysis. Samples for ctDNA analysis are collected in cell-free DNA blood collection tubes (Streck).
Pathological analysis
The FFPE tumor biopsies are cut into four sections of 4 µm and one of 1 µm. One 4 µm section is stained with hematoxylin and eosin and another with HER2-antibody. Cancer and histological subtype are diagnosed by senior consultant pathologists. Additional pathological analyses, including immunohistochemistry or targeted NGS, are done on FFPE tissue at the initiative of the pathologist, or if requested by investigators or the tumor board.
Molecular profiling
The molecular profiling is performed in-house at the Department of Molecular Diagnostics. Tissue biopsies in RNAlater or fresh frozen tissue are homogenized, and RNA and DNA is extracted using Qiagen AllPrep®. Non-tumoral DNA for profiling is extracted from peripheral blood leukocytes using the QiaSymphony DSP DNA midi kit (Qiagen). WES library preparation is performed using the Sureselect XT HS Library Prep Kits (Agilent). Exome capture is performed using the SureSelect XT HS Clinical Research Exome V2 (Agilent), while RNAseq library preparation is done using the TruSeq Stranded mRNA Library Prep Kit (Illumina). Sequencing is carried out as 2 × 150 bp paired-end on a NovaSeq 6000 (Illumina), producing a minimum 26 Gb, 18 Gb, or 33 million reads of raw sequence data for tumor DNA, non-tumoral DNA, or tumor RNA samples, respectively.
For molecular profiling, the tumor DNA is subjected to somatic short variant detection using tumor/normal WES analysis, detection of copy number alterations, TMB and MSI status, and mutational signature analysis. Tumor RNA is used for the generation of an expression profile used as input for tissue classification [Citation26]. Moreover, RNAseq enables the detection of fusion transcripts for the identification of larger chromosomal abbreviations.
ctDNA is extracted from 10 ml blood samples or, in a few cases, from peritoneal fluid in cell-free DNA blood collection tubes (Streck) using QiaSymphony Virus/pathogen Midi Kit (Qiagen). TruSight Oncology 500 assay (Illumina) is used for library preparation. 2 × 150 bp paired-end sequencing is performed on a NovaSeq 6000 (Illumina) producing minimum 1500× coverage of the targeted regions.
Using the non-tumoral DNA, clinically relevant pathogenic variants are detected in a small set of genes (BRCA1, BRCA2, ATM, MLH1, MSH2, MSH3, PMS1, MLH3, MSH6, PMS2, PALB2, RAD51C, RAD51D, MBD4, ANKRD26, CEBPA, DDX41, ETV6, GATA3, RUNX1, TERC, TERT, and TP53). Germline variants in other genes are not analyzed unless specifically requested by clinical geneticists and consented by the patient.
Raw sequencing data are processed and stored under the regional IT-system. Sequencing data and supplementary metadata are submitted to the Danish National Genome Center.
Bioinformatics
The bioinformatic procedure used in this study has previously been described [Citation27]. Using the Genome Analysis Tool Kit (GATK) the process largely follows the GATK-recommendations [Citation28]. The filtered variant file (VCF) is uploaded together with information on sex, age, diagnosis, and detected fusion transcripts, gene losses or gains, to Qiagen Clinical Insight Interpret (QCI) for automatic classification of variants [Citation29]. All variants of clinical significance are manually verified by visual inspection of the DNA and RNAseq data. Variants are classified as pathogenic, likely pathogenic, variants of unknown significance (VUS), or benign/likely benign [Citation30]. QCI is also used prospectively to link variants and approved treatments or clinical trials into tiers [Citation31]: Tier 1 – variants of strong clinical significance and tier 2 – variants of potential clinical significance. Tier 3 – variants of unknown significance – is in the present analysis merged with variants with no QCI-annotation.
Non-tumoral variants are classified according to the 2015 ACMG/AMP guidelines [Citation32] and are, together with the history of patients, reviewed by an MD specialized in clinical genetics. If a germline variant with a likely consequence for the health of the patient or a relative is identified, patients who had requested this information at protocol consent are offered referral to the Department of Clinical Genetics.
Reports are generated using a dedicated platform (PrOnco) from clinical data stored in REDCap and variants data. The platform follows the ‘FAIR’ principles in data stewardship [Citation33] and enables a tumor board report to be generated on demand or made accessible through an interactive interface. PrOnco also includes custom reporting tools to monitor the inclusion of patients live, integrates a variant search functionality, and can be used to implement validation tools and utilities to ensure the quality of the data collected [Citation34].
Tumor board
Cases are presented at a weekly web-based multidisciplinary NMTB, directed by the phase I unit at Rigshospitalet, Copenhagen. The NMTB has participants from all eight Danish oncological centers engaged in clinical precision cancer medicine. Besides medical oncologists, the participation of pathologists, clinical genetics, and molecular biologists is mandatory. Based on patients’ history and characteristics, disclosed molecular variants, gene signatures, or other tumor characteristics, and available trials or drugs, matched treatments, and early-phase clinical trials are suggested. Treatment suggestions are not influenced by geography, as patients can be referred to any center.
Targeted treatment
Treatment can be offered on-site if a targeted drug of a nationally approved indication is suggested by the NMTB. If not, the patient may be treated in an available clinical protocol. This especially includes the ProTarget trial – a nation-wide, investigator-initiated basket trial of targeted treatment, studying the efficacy of approved drugs used ‘off label’ in cohorts of patients with a similar target, similar drug, and similar diagnosis (NCT04341181) [Citation35]. The trial was initiated at the local site January 2022.
If no approved drug or relevant protocol is available or feasible, treatment with a targeted drug used outside a clinical protocol is pursued. This includes treatment with a drug that is labeled but not approved by the Danish authorities, however, may be used after individual permission. It also includes ‘off-label’ treatment after individual permission, or ‘compassionate use’ with an unapproved drug in a ‘named user program’. Supplementary advises for individual cases can be sought from the Danish National Board of Health Committee for Experimental Treatment. Patients treated with matched targeted drugs outside a clinical drug trial are evaluated by CT- or MR-scans and biomarkers at baseline and every 8th or 9th week until progression. Blood samples for research are drawn and stored in Bio-and Genome Bank Denmark, while treatment data is registered prospectively as shown in Supplementary Table A.2.
Results
The total number of patients included in the protocol from June 1st 2020 to May 31st 2022 was 164, corresponding to an average monthly inclusion rate of 6.8 patients. One patient withdrew consent. The protocol did not allow for the registration of eligible patients that were not asked or declined trial participation. The patients included in the study were not selected at random. Baseline clinical characteristics for the ‘intention-to-treat’ (ITT) population, the subpopulation with tumor WES performed and the subpopulation recommended a matched targeted treatment at the NMTB are shown in . Most patients were heavily pretreated, two-thirds having received at least 3 prior lines of systemic treatment, and most had metastatic disease. A high fraction of patients had a history of asbestos exposure, which was expected from demography [Citation31], whereas 17% had a prior or concurrent other malignancy diagnosed. The most common primary tumor sites were breast, ovary, and lung, totaling 44%. Those, who had a history of 1st-degree relatives with cancer or a diagnosis of lung or prostate cancer, were overrepresented in the group of patients with the recommendation of a matched targeted treatment.
Table 1. Baseline characteristics of all ‘intention-to-treat’ (ITT) patients, patients with a tumor WES performed and patients with a tumor WES performed and a matched targeted treatment recommended at the National Molecular Tumor Board.
The most frequent sites of new biopsies were the liver (23%), abdominal cavity (17%), lung (10%), and brain (9%). In 28 patients more than one biopsy procedure was required. Excluding procedures performed on archived samples or ctDNA, the median time from protocol consent to the presentation of results at NMTB was 31 d (lower quartile (LQ): 25 d; upper quartile (UQ): 42 d), including a median of 12 d (LQ: 7 d; UQ: 18 d) spend from consent to decisive biopsy, 15 d (LQ: 14 d; UQ: 20 d) spend from biopsy to molecular analysis done, and 2 d (LQ: 1 d; UQ: 2 d) spend from molecular analysis done to NMTB presentation. Archived samples including fresh frozen tumor tissue was used as a source of molecular tumor profiling in 21 patients and ctDNA in seven patients. In all cases where tumor sequencing was attempted leucocyte DNA could be analyzed for non-tumoral variants.
At this interim analysis, the fraction of patients with molecular tumor profiling, who were recommended a matched targeted treatment, was 34% (27% in ITT-analysis). A chart demonstrating the flow of patients is shown in . 20% of patients included had no tumor sequencing performed due to biopsy not possible, failed or refused (N = 22), or deteriorating general condition (N = 10). The QCI tier-level of actionability was highly correlated with having a matched targeted treatment recommended at NMTB vs no targeted treatment recommended; 60% vs 40% for tier 1 (in 40 patients), 24% vs 76% for tier 2 (in 82 patients), and 0% vs 100% for tiers >2 (in 9 patients) (p < .001 by a Chi-squared test). 44 patients had a total of 62 druggable targets as determined by the NMTB. The pathological and molecular characteristics of these patients and the corresponding recommended targeted treatments are shown in Supplementary Table B. Most of the alterations (42%) were matched with immune check point inhibitors, while 18% were ERBB2-alterations, and 11% HRD-associated alterations. More rarely, druggable alterations were found in PIK3CA, BRAFV600E, FGFR, KRASG12C, MET, EGFR, CDKN2A, NTRK3, AKT1, and KDR.
Figure 1. (a) A chart demonstrating the flow of patients from trial inclusion to treatment recommendation at the National Molecular Tumor Board. If several QCI tier variants were found in samples from a patient, the highest tier was used. QCI tier >2 includes tier 3 and non-annotated pathogenic or likely pathogenic variants. ITT: ‘intention-to-treat’; WES: whole exome sequencing; QCI: Qiagen Clinical Insight Interpret [Citation29]; NA: not assessed; NMTB: National Molecular Tumor Board. (b) Outcome of National Molecular Tumor Board recommendations of a matched targeted treatment in 44 patients. (c) Outcome of National Molecular Tumor Board recommendations of a non-targeted trial in 33 patients.
![Figure 1. (a) A chart demonstrating the flow of patients from trial inclusion to treatment recommendation at the National Molecular Tumor Board. If several QCI tier variants were found in samples from a patient, the highest tier was used. QCI tier >2 includes tier 3 and non-annotated pathogenic or likely pathogenic variants. ITT: ‘intention-to-treat’; WES: whole exome sequencing; QCI: Qiagen Clinical Insight Interpret [Citation29]; NA: not assessed; NMTB: National Molecular Tumor Board. (b) Outcome of National Molecular Tumor Board recommendations of a matched targeted treatment in 44 patients. (c) Outcome of National Molecular Tumor Board recommendations of a non-targeted trial in 33 patients.](/cms/asset/d6789596-3bd6-4e8e-99ba-c86c1acb7a9e/ionc_a_2185542_f0001_c.jpg)
The outcomes of NMTB recommendations of a matched targeted treatment are illustrated in . At the time of data cutoff, 21 patients were eligible of the 44 patients, who had been recommended a matched targeted treatment, and 16 of these had started the suggested treatment indicating that benefit rate is low. Approximately half of the patients were ineligible, most frequently due to deteriorating performance status. 33 patients (23% of the ITT population) were recommended a non-targeted clinical trial (). Of these, five have started treatment in trials, while four patients are on a waiting list. Overall, the median OS from study inclusion in the ITT-cohort was 7.3 months (95% Confidence lnterval (CI): 6.1–9.4 months) and the estimated 1-year survival was 33% (CI: 26–42%).
The pathogenic or likely pathogenic variants found and their QCI-annotated ‘actionability’ are shown in . A total of 42 QCI-tier 1 and 305 QCI-tier 2 variants were disclosed, distributed in 120 patients (92% of patients having a tumor WES successfully done). In only 3.8% of molecularly profiled tumors, no pathogenic or likely pathogenic variants were disclosed. Within the limited panel of genes screened, pathogenic germline variants were found in 4 patients (2.5% of the ITT-population), in BRCA1, BRCA2, DDX41, and PALB2.
Figure 2. Genes with clinically relevant alterations and mutational signatures distributed according to diagnoses. Each row represents a patient and is grouped by primary tumor site. Columns represent genes with relevant short nucleotide variants, fusions, and CNV’s and these are sorted by frequency. Signatures of MSI-high (cut-point 3.5%) and TMB-high (cut-point 10 mut./Mb) [Citation27] are shown in the right columns. Variants were merged for five patients who had their analysis repeated. The color of the squares indicates the actionability of the variant classified prospectively according to Qiagen Clinical Insight Interpret (QCI) [Citation29]. QCI tier 1 A includes variants with an FDA-approved therapy and can be found in professional guidelines, tier 1B includes variants with strong evidence for sensitivity to therapy from well-powered studies supported by consensus from experts, tier 2C includes variants with FDA-approved therapies for a different tumor type, and tier 2D includes variants with a plausible sensitivity to therapy indicated by a few case reports or preclinical trials [Citation30]. mut.: mutations; Mb: megabases; HC: head and neck cancer; CRC: colorectal cancer; MM: malignant melanoma.
![Figure 2. Genes with clinically relevant alterations and mutational signatures distributed according to diagnoses. Each row represents a patient and is grouped by primary tumor site. Columns represent genes with relevant short nucleotide variants, fusions, and CNV’s and these are sorted by frequency. Signatures of MSI-high (cut-point 3.5%) and TMB-high (cut-point 10 mut./Mb) [Citation27] are shown in the right columns. Variants were merged for five patients who had their analysis repeated. The color of the squares indicates the actionability of the variant classified prospectively according to Qiagen Clinical Insight Interpret (QCI) [Citation29]. QCI tier 1 A includes variants with an FDA-approved therapy and can be found in professional guidelines, tier 1B includes variants with strong evidence for sensitivity to therapy from well-powered studies supported by consensus from experts, tier 2C includes variants with FDA-approved therapies for a different tumor type, and tier 2D includes variants with a plausible sensitivity to therapy indicated by a few case reports or preclinical trials [Citation30]. mut.: mutations; Mb: megabases; HC: head and neck cancer; CRC: colorectal cancer; MM: malignant melanoma.](/cms/asset/4ba7a356-1127-4e8e-961f-866b0b73a0bb/ionc_a_2185542_f0002_c.jpg)
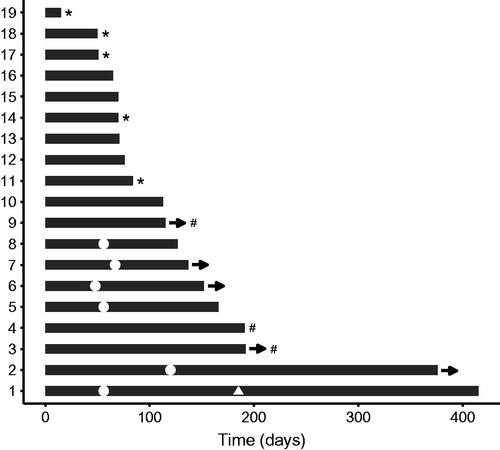
16 patients started on a matched targeted treatment; one patient was treated twice against the same target and another twice against the same and once against a different target. A plot of treatment duration and efficacy of these 19 targeted treatments is shown in . 15 targeted treatments were given in 13 patients with measurable disease resulting in five partial responses (PR) and one complete response (CR), accounting to an overall RR of 40%. The clinical benefit rate (CBR) [Citation8] of 17 treatments was 53% in 15 patients with evaluable disease. Five patients are still on treatment. The median treatment duration was 3.8 months.
Figure 3. Treatment duration and response for 19 molecularly matched targeted treatments in 16 patients. Each treatment is shown as bars. The time from treatments start to end is indicated on the x-axis. Complete response (triangle) or partial response (dots) is indicated at the time of response for patients evaluable by RECIST. # Stable disease lasting >16 weeks as the best response (for patients evaluable for Clinical Benefit Rate). * Clinically progressive disease. All other treatments aborted were caused by progression according to RECIST. -> Treatment continues.
Discussion
In this interim analysis, we found that one-third of patients was recommended a matched targeted treatment based on tumor molecular profiling. A huge variation in rates of profiled patients matched to treatment in clinical trials has been reported, ranging from 4% to 36% [Citation36], mainly caused by different definitions of variants being ‘druggable’ [Citation37,Citation38]. In the current study, we defined ‘druggable’ in a pragmatic way by registering treatment recommendations at the NMTB, reflecting an expert consensus on available targeted treatments, guidelines, and clinical trials in the country [Citation39]. The ESCAT classifier may be used as a manual tool for a listing of molecular variants based on the evidence available and based on consensus [Citation40]. However, one study found that ESCAT tier levels were not well correlated with PFS in patients treated [Citation41]. Several tools providing automatic annotations of ‘druggability’ are available [Citation42,Citation43], but large inconsistencies among different classifiers have been reported [Citation37, Citation44]. The QCI classifier, used to generate the molecular reports in our study but not used for clinical decisions, is indeed not systematically pertinent. For example, it does not consider coalterations, annotates some alterations typically regarded as non-actionable as ‘druggable’ and match various chemotherapy to alterations [Citation38]. Despite these limitations, we found a strong association between QCI tier-levels and the chance of having a matched treatment recommended. The development of artificial intelligence models for the allocation of targeted agents to patients, integrating molecular profiling information as well as extensive clinical data including detailed patient trajectory, exposures, and comorbidities could be of great interest [Citation45]. A ‘patients like me’ tool may also be developed to facilitate decision-making by clinicians and communication to patients [Citation46].
Another explanation to the variation in rates of patients matched to treatment may be different access to relevant drugs. Sometimes even marketed drugs may not be available to the treating physician due to reimbursement issues, or – in public health care funded systems – lack of national approval [Citation47]. In our setup, patients could be referred to treatment at any center in the country and marketed drugs were available inhouse through a multidrug basket trial or by permission from local authorities. However, the basket trial was first initiated locally on January 2022 and clinical trials of targeted drugs were before that restricted mainly to those offered at a distant phase I trial unit. Logistics might have reduced the referral of patients to such trials [Citation48].
Finally, heterogeneous inclusion criteria and biological differences among populations may contribute to variations in the fraction of patients eligible [Citation49,Citation50]. For example, it is known that the prevalence of ‘druggable’ variants is dependent on the histology and anatomical site of the primary tumor [Citation51,Citation52]. Moreover, many patients in studies of late-stage cancer have been treated by targeted agents earlier in their disease, rendering some targets clinically un-druggable [Citation53].
In agreement with others [Citation54], our study points to improvements in patient selection, methodology, and logistics that could increase the feasibility of precision medicine. The 20% of patients included in the ITT analysis that did not have a molecular tumor profiling done, mainly due to issues related to biopsies, is at least on par with similar reports [Citation54,Citation55], and substantiates the problems of preferred use of fresh histological biopsies [Citation56]. Fine needle aspiration is less risky, induces less discomfort, and may provide similar NGS-results [Citation57], however, generally exclusive of sufficient material for the histopathological and immunohistochemical analysis. ctDNA is increasingly used, either complimentary to biopsies or alone [Citation56,Citation58]. Liquid biopsies may be less sensitive to heterogeneity and may provide early information of progression and resistance [Citation58,Citation59]. The main limitations are small amounts of tumor DNA, lack of tissue for pathological diagnosis, and contamination by non-tumoral variants [Citation60]. The number of patients with immediately available tissue could be increased by storing fresh frozen tissue at diagnosis of the incurable disease. Indeed, we used archived fresh frozen tissue in patients with primary brain tumors and obtained a successful molecular analysis in all cases (data not shown). Using archival materials, temporal evolutions of molecular alterations are missed [Citation60], although a prior study of changes in druggable variants over time showed very limited clinical impact [Citation61].
Turnaround time is especially important for end-stage patients [Citation62]. In our study, the median time to the presentation at NMTB was approximately 1 month, but even so more than half of those recommended a matched treatment did not receive it, most often due to deteriorating performance status. In another recent, single-institution study, only 16% of 591 patients recommended a targeted treatment at the tumor board actually started on treatment at the institution [Citation14], and in patients with non-small-cell lung cancer, 65% of those who were potentially eligible did not benefit from precision therapy for a number of reasons [Citation54]. Our data indicates that using ctDNA or frozen archival tissue, a decision of treatment could be done within two weeks.
In this report with short follow-up, we found a high RR and CBR of targeted treatments of 40% and 53%, respectively, but a short median treatment duration of 3.8 months. The small number of patients and the heterogeneous populations preclude a formal comparison, however, with increasingly efficient drugs and more precise molecular predictors, improved outcome of precision medicine can be expected. For example, the CoPPO trial [Citation63] showed a RR of only 15%, whereas a more recent report of 215 patients in the DRUP trial found a CBR of 34% with a median treatment duration of 9 months [Citation8].
Cancers of late-stage patients are often therapy resistant, the tumor burden is high, and poor performance status or late effects may preclude further treatment. Results of precision medicine in late-stage cancer patients have proved the concept, but a significant impact on survival may require that targeted treatment is used in earlier lines. Except for highly druggable and truncal targets, it is debatable whether the one target – one drug approach over different cancer types will work [Citation53], and drug combinations might be a way forward [Citation64]. Furthermore, precision medicine could benefit further by knowledge in proteomics, cellomics, and metabolomics [Citation65] as well as by therapeutic drug monitoring to allow appropriate dosing of drugs selected [Citation66].
The discovery of pathogenic germline variants is an important element of precision medicine. In a previous report, pathogenic or likely pathogenic germline variants associated with cancer could be detected by WES in 18% of 636 patients with advanced cancer, and in 4% these variants had possible therapeutic consequences for the patient [Citation67]. We found pathogenic germline variants in only 2.5% of patients using a similar pipeline, however, germline variants reported in our trial were intentionally restricted to a specific panel of genes.
At our institution, experience in molecular tumor profiling was acquired through clinical research [Citation27]. It has been reported, however, that outside comprehensive cancer centers, oncologists may have sparse access to experts for interpretation of genomic test reports to guide decisions about the optimal use of targeted agents [Citation68], and access to trials and new targeted agents may also be limited. Through national collaboration, including discussion of patients at NMTB, inhouse initiation of a multidrug basket trial, and local access to labeled targeted drugs, we could allocate most eligible patients to treatment locally.
Conclusions
Genomic profiling of cancers and matched targeted treatment is feasible in a regional academic hospital setting. Although one-third of patients having molecular profiling done was matched with targeted treatment, the clinical benefit was limited as only 13% of the ITT-population started on treatment or are still eligible. Hence, precision medicine should move toward the 1st or 2nd line setting. Rapid evolutions in ‘omics’ require continuous awareness and adaption to ensure the most modern and optimal diagnostics at all facilities, not least the non-comprehensive cancer hospitals. In smaller hospital units, close collaboration with comprehensive cancer centers is necessary to ensure expert interpretation of results and equality in access to the most modern treatment.
Supplemental Material
Download MS Word (106.6 KB)Disclosure statement
Dr. Morten Ladekarl received a research grant from Scandion Oncology A/S, Denmark. Dr. Charlotte Aa. Haslund received honoraria for lectures from GSK and Bristol Myers Squibb. All other authors declare no conflicts of interest.
Data availability statement
The data that support the findings of this study are available on request from the corresponding author, ML. The data are not publicly available due to their containing information that could compromise the privacy of research participants.
Additional information
Funding
References
- Kamps R, Brandão R, Bosch B, et al. Next-Generation sequencing in oncology: genetic diagnosis, risk prediction and cancer classification. Int J Mol Sci. 2017;18(2):308.
- Yates LR, Seoane J, Le Tourneau C, et al. The european society for medical oncology (ESMO) precision medicine glossary. Ann Oncol. 2018;29(1):30–35.
- Morganti S, Tarantino P, Ferraro E, et al. Next generation sequencing (NGS): a revolutionary technology in pharmacogenomics and personalized medicine in cancer. Adv Exp Med Biol. 2019;1168:9–30.
- Malone ER, Oliva M, Sabatini PJB, et al. Molecular profiling for precision cancer therapies. Genome Med. 2020;12(1):8.
- Berger MF, Mardis ER. The emerging clinical relevance of genomics in cancer medicine. Nat Rev Clin Oncol. 2018;15(6):353–365.
- Duke ES, J. Fusco M, DeMoss P, et al. Highlights of FDA oncology approvals in 2022: tissue-agnostic indications, dosage optimization, and diversity in drug development. Cancer Discov. 2022;12(12):2739–2746.
- Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–103.
- van der Velden DL, Hoes LR, van der Wijngaart H, et al. The drug rediscovery protocol facilitates the expanded use of existing anticancer drugs. Nature. 2019;574(7776):127–131.
- Schwaederle M, Zhao M, Lee JJ, et al. Impact of precision medicine in diverse cancers: a meta-analysis of phase II clinical trials. J Clin Oncol. 2015;33(32):3817–3825.
- Wheeler DA, Takebe N, Hinoue T, et al. Molecular features of cancers exhibiting exceptional responses to treatment. Cancer Cell. 2021;39(1):38.e7–53.e7.
- Sharma MB, Carus A, Sunde L, et al. BRCA-associated pancreatico-biliary neoplasms: four cases illustrating the emerging clinical impact of genotyping. Acta Oncol. 2016;55(3):377–381.
- Zhang Q, Fu Q, Bai X, et al. Molecular profiling–based precision medicine in cancer: a review of current evidence and challenges. Front Oncol. 2020;10:532403.
- Le Tourneau C, Delord J-P, Gonçalves A, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16(13):1324–1334.
- Miller RW, Hutchcraft ML, Weiss HL, et al. Molecular tumor board–assisted care in an advanced cancer population: results of a phase II clinical trial. JCO Precis Oncol. 2022;(6):e2100524.
- Von Hoff DD, Stephenson JJ, Rosen P, et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol. 2010;28(33):4877–4883.
- Zimmer K, Kocher F, Spizzo G, et al. Treatment according to molecular profiling in relapsed/refractory cancer patients: a review focusing on latest profiling studies. Comput Struct Biotechnol J. 2019;17:447–453.
- Tsimberidou AM, Eggermont AMM, Schilsky RL. Precision cancer medicine: the future Is now, only better. Am Soc Clin Oncol Educ Book. 2014;(34):61–69.
- Nakamura Y, Fujisawa T, Taniguchi H, et al. SCRUM‐Japan GI‐SCREEN and MONSTAR‐SCREEN: path to the realization of biomarker‐guided precision oncology in advanced solid tumors. Cancer Sci. 2021;112(11):4425–4432.
- Meric-Bernstam F, Farhangfar C, Mendelsohn J, et al. Building a personalized medicine infrastructure at a major cancer center. J Clin Oncol. 2013;31(15):1849–1857.
- American Association for Cancer Research. Bringing precision medicine to community oncologists. Cancer Discov. 2017;7(1):6–7.
- Ladekarl M, Rasmussen LS, Kirkegård J, et al. Disparity in use of modern combination chemotherapy associated with facility type influences survival of 2655 patients with advanced pancreatic cancer. Acta Oncol. 2022;61(3):277–285.
- Bruno DS, Hess LM, Li X, et al. Disparities in biomarker testing and clinical trial enrollment among patients with lung, breast, or colorectal cancers in the United States. JCO Precis Oncol. 2022;6:e2100427.
- Danmarks Statistik. Befolkningstal. 2022. Available from: https://www.dst.dk/da/Statistik/emner/borgere/befolkning/befolkningstal
- Finan C, Gaulton A, Kruger FA, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med. 2017;9(383):eaag1166.
- Harris PA, Taylor R, Thielke R, et al. Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381.
- Johnson NT, Dhroso A, Hughes KJ, et al. Biological classification with RNA-seq data: can alternatively spliced transcript expression enhance machine learning classifiers? RNA. 2018;24(9):1119–1132.
- Bødker JS, Sønderkær M, Vesteghem C, et al. Development of a precision medicine workflow in hematological cancers, aalborg university hospital, Denmark. Cancers. 2020;12(2):312.
- McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303.
- QIAGEN. QIAGEN Digital Insights. Advancing molecular intelligence from bench to bedside. [cited 2023 Jan 1]. Available from: https://digitalinsights.qiagen.com/
- Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer. J Mol Diagn. 2017;19(1):4–23.
- Røe OD, Stella GM. Malignant pleural mesothelioma: history, controversy and future of a manmade epidemic. Eur Respir Rev. 2015;24(135):115–131.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424.
- Vesteghem C, Brøndum RF, Sønderkær M, et al. Implementing the FAIR data principles in precision oncology: review of supporting initiatives. Brief Bioinform. 2020;21(3):936–945.
- Vesteghem C, Dahl S, Brøndum R, et al. Implementing a data infrastructure for precision oncology projects leveraging REDCap. medRxiv. 2022. DOI:10.1101/2022.05.09.22274599
- Kringelbach T, Højgaard M, Rohrberg K, et al. ProTarget: a danish nationwide clinical trial on targeted cancer treatment based on genomic profiling – a national, phase 2, prospective, multi-drug, non-randomized, open-label basket trial. BMC Cancer. 2023;23(1):182.
- Yam C, Ma BBY, Yap TA. Global implementation of precision oncology. JCO Precis Oncol. 2021;(5):854–858.
- Owens J. Determining druggability. Nat Rev Drug Discov. 2007;6(3):187–187.
- Perakis SO, Weber S, Zhou Q, et al. Comparison of three commercial decision support platforms for matching of next-generation sequencing results with therapies in patients with cancer. ESMO Open. 2020;5(5):e000872.
- Koopman B, van der Wekken AJ, ter Elst A, et al. Relevance and effectiveness of molecular tumor board recommendations for patients with non–small-cell lung cancer with rare or complex mutational profiles. JCO Precis Oncol. 2020;4:393–410.
- Condorelli R, Mosele F, Verret B, et al. Genomic alterations in breast cancer: level of evidence for actionability according to ESMO scale for clinical actionability of molecular targets (ESCAT). Ann Oncol. 2019;30(3):365–373.
- Moreira A, Masliah-Planchon J, Callens C, et al. Efficacy of molecularly targeted agents given in the randomised trial SHIVA01 according to the ESMO scale for clinical actionability of molecular targets. Eur J Cancer. 2019;121:202–209.
- Fairley JA, Deans ZC, Treacy RJL, et al. An independent assessment of a commercial clinical interpretation software indicates that software can mitigate variation in human assessment. J Mol Pathol. 2022;3(3):125–139.
- Tamborero D, Dienstmann R, Rachid MH, et al. The molecular tumor board portal supports clinical decisions and automated reporting for precision oncology. Nat Cancer. 2022;3(2):251–261.
- Katsoulakis E, Duffy JE, Hintze B, et al. Comparison of annotation services for next-generation sequencing in a large-scale precision oncology program. JCO Precis Oncol. 2020;(4):212–221.
- Molica S, Seymour JF, Polliack AA. Perspective on prognostic models in chronic lymphocytic leukemia in the era of targeted agents. Hematol Oncol. 2021;39(5):595–604.
- Ortega-Martorell S, Riley P, Olier I, et al. Breast cancer patient characterisation and visualisation using deep learning and fisher information networks. Sci Rep. 2022;12(1):14004.
- Koleva-Kolarova R, Buchanan J, Vellekoop H, et al. Financing and reimbursement models for personalised medicine: a systematic review to identify current models and future options. Appl Health Econ Health Policy. 2022;20(4):501–524.
- Wercholuk AN, Parikh AA, Snyder RA. The road less traveled: transportation barriers to cancer care delivery in the rural patient population. JCO Oncol Pract. 2022;18(9):652–662.
- Lam M, Tran B, Beck S, et al. Precision oncology using a clinician-directed, tailored approach to molecular profiling. Asia Pac J Clin Oncol. 2018;14(1):84–90.
- Ma BB, Hui EP, Mok TS. Population-based differences in treatment outcome following anticancer drug therapies. Lancet Oncol. 2010;11(1):75–84.
- Andre F, Mardis E, Salm M, et al. Prioritizing targets for precision cancer medicine. Ann Oncol. 2014;25(12):2295–2303.
- Aaltonen LA, Abascal F, Abeshouse A, et al. Pan-cancer analysis of whole genomes. Nature. 2020;578(7793):82–93.
- Garraway LA, Verweij J, Ballman KV. Precision oncology: an overview. J Clin Oncol. 2013;31(15):1803–1805.
- Sadik H, Pritchard D, Keeling D-M, et al. Impact of clinical practice gaps on the implementation of personalized medicine in advanced non–small-cell lung cancer. JCO Precis Oncol. 2022;6:e2200246.
- Réda M, Richard C, Bertaut A, et al. Implementation and use of whole exome sequencing for metastatic solid cancer. EBioMedicine. 2020;51:102624.
- Bayle A, Peyraud F, Belcaid L, et al. Liquid versus tissue biopsy for detecting actionable alterations according to the ESMO scale for clinical actionability of molecular targets in patients with advanced cancer: a study from the french national center for precision medicine (PRISM). Ann Oncol. 2022;33(12):1328–1331.
- Dupain C, Masliah‐Planchon J, Gu C, et al. Fine‐needle aspiration as an alternative to core needle biopsy for tumour molecular profiling in precision oncology: prospective comparative study of next‐generation sequencing in cancer patients included in the SHIVA02 trial. Mol Oncol. 2021;15(1):104–115.
- Cheng F, Su L, Qian C. Circulating tumor DNA: a promising biomarker in the liquid biopsy of cancer. Oncotarget. 2016;7(30):48832–48841.
- Topham JT, O’Callaghan CJ, Feilotter H, et al. Circulating tumor DNA identifies diverse landscape of acquired resistance to anti–epidermal growth factor receptor therapy in metastatic colorectal cancer. J Clin Oncol. 2023;41(3):485–496.
- Chan HT, Chin YM, Low S-K. Circulating tumor DNA-Based genomic profiling assays in adult solid tumors for precision oncology: recent advancements and future challenges. Cancers. 2022;14(13):3275.
- van de Haar J, Hoes LR, Roepman P, et al. Limited evolution of the actionable metastatic cancer genome under therapeutic pressure. Nat Med. 2021;27(9):1553–1563.
- Vesteghem C, Brøndum RF, Mouritzen MT, et al. Thirty-day mortality following systemic anticancer therapy: evaluating risk factors without selection bias in a real-world, population-based cohort from 2009 to 2019. Clin Oncol. 2022;34(8):487–496.
- Tuxen IV, Rohrberg KS, Oestrup O, et al. Copenhagen prospective personalized oncology (CoPPO)—clinical utility of using molecular profiling to select patients to phase I trials. Clin Cancer Res. 2019;25(4):1239–1247.
- Plana D, Palmer AC, Sorger PK. Independent drug action in combination therapy: implications for precision oncology. Cancer Discov. 2022;12(3):606–624.
- Olivier M, Asmis R, Hawkins GA, et al. The need for multi-omics biomarker signatures in precision medicine. Int J Mol Sci. 2019;20(19):4781.
- Veal GJ, Amankwatia EB, Paludetto M-N, et al. Pharmacodynamic therapeutic drug monitoring for cancer: challenges, advances, and future opportunities. Ther Drug Monit. 2019;41(2):142–159.
- Bertelsen B, Tuxen IV, Yde CW, et al. High frequency of pathogenic germline variants within homologous recombination repair in patients with advanced cancer. NPJ Genom Med. 2019;4(1):13.
- Tsimberidou AM, Iskander NG, Hong DS, et al. Personalized medicine in a phase I clinical trials program: the MD anderson cancer center initiative. Clin Cancer Res. 2012;18(22):6373–6383.