
Abstract
Background
Our previous study has revealed that EphA7 was upregulated in patient-derived esophageal squamous cell carcinoma (ESCC) xenografts with hyper-activated STAT3, but its mechanism was still unclear.
Materials and methods
To assess the association between EphA7 and STAT3, western blotting, immunofluorescence, ChIP assay, and qRT-PCR were conducted. Truncated mutation and luciferase assay were performed to examine the promoter activity of EphA7. CCK-8 assay and colony formation were performed to assess the proliferation of ESCC. Cell-derived xenograft models were established to evaluate the effects of EphA7 on ESCC tumor growth. RNA-seq analyses were used to assess the effects of EphA7 on related signals.
Results
In this study, EphA7 was found upregulated in ESCC cell lines with high STAT3 activation, and immunofluorescence also showed that EphA7 was co-localized with phospho-STAT3 in ESCC cells. Interestingly, suppressing STAT3 activation by the STAT3 inhibitor Stattic markedly inhibited the protein expression of EphA7 in ESCC cells, in contrast, activation of STAT3 by IL-6 obviously upregulated the protein expression of EphA7. Moreover, the transcription of EphA7 was also mediated by the activation of STAT3 in ESCC cells, and the −2000∼−1500 region was identified as the key promoter of EphA7. Our results also indicated that EphA7 enhanced the cell proliferation of ESCC, and silence of EphA7 significantly suppressed ESCC tumor growth. Moreover, EphA7 silence markedly abolished STAT3 activation-derived cell proliferation of ESCC. Additionally, RNA-seq analyses indicated that several tumor-related signaling pathways were significantly changed after EphA7 downregulation in ESCC cells.
Conclusion
Our results showed that the transcriptional expression of EphA7 was increased by activated STAT3, and the STAT3 signaling may act through EphA7 to promote the development of ESCC.
Keywords:
Background
Esophageal cancer is one of the digestive tract tumors with the sixth mortality and the seventh incidence in the world [Citation1]. Esophageal cancer mainly includes esophageal adenocarcinoma (EAC) and esophageal squamous cell carcinoma (ESCC), and the majority of cases are ESCC in China [Citation2]. However, most ESCC patients are at an advanced stage upon diagnosis, and the 5-year survival rate is even less than 5% in most developing countries. Studies have found that chronic inflammation promoted the occurrence of ESCC, colorectal cancer, breast cancer, and liver cancer [Citation3–6]. Therefore, exploring the mechanism by which chronic inflammation promotes the occurrence and development of ESCC may provide novel insights into the therapy of ESCC.
Disruption of receptor tyrosine kinases (RTKs) function can cause a variety of diseases, such as vascular dysplasia, chronic inflammation, and cancer [Citation7,Citation8]. Eph receptors are a large group of RTKs. The human Eph family consists of 8 EphA proteins and 6 EphB proteins [Citation9]. Eph receptor activation occurs on binding to its ligand ephrin. Studies have shown that the Eph/ephrin axis exerted significant effects in the development of tumorigenesis [Citation10]. EphA7 was reported to be conserved in spinal animals [Citation11], and proven to be extensively expressed in the embryonic tissues [Citation12]. A number of studies have reported that EphA7 was dysregulated in tumors, such as non-small cell lung cancer [Citation13], osteosarcoma [Citation14], colorectal cancer [Citation15], prostate cancer [Citation16], and lymphoma [Citation17]. It may regulate tumor proliferation, metastasis, and apoptosis, and may also be inactivated in tumors. These studies suggest EphA7 exerts vital roles in the tumors.
It has been proven that JAK/STAT3 signaling plays vital roles in the biological processes, including proliferation, migration, angiogenesis, immune response, and inflammation [Citation18,Citation19]. Physiological STAT3 activation is short-lived and occurs only during the immune response; STAT3 quickly returns to its inactive form after being activated [Citation19]. However, in most cancers, including colon cancer, breast cancer, and prostate cancer, STAT3 is continuously activated [Citation20]. STAT3 overactivation leads to the overexpression of genes related to inflammation, proliferation, metastasis, and immunosuppression, which together promote the carcinogenesis [Citation21]. Our previous study found that constitutive activation of STAT3 played vital roles in the proliferation of ESCC, and RNA-seq results indicated that EphA7 was upregulated in patient-derived ESCC xenografts accompanied by a hyper-activated state of STAT3 [Citation22]. However, the relationship between EphA7 and STAT3 in ESCC has not been clarified yet.
In this study, we examined the relationship between constitutively phosphorylated STAT3 and EphA7, and explored EphA7 function in ESCC. We found that activation of STAT3 upregulated the transcriptional level of EphA7 in ESCC, and EphA7 enhanced the proliferation of ESCC cells. Our results revealed the important roles of STAT3-EphA7 axis in ESCC, and indicated that the STAT3 signaling may act through EphA7 to promote the development of ESCC, which suggested that targeting STAT3-EphA7 axis may be a novel strategy for the therapy of ESCC.
Materials and methods
Cells, cell culture, chemicals
Eca109, Kyse150, Kyse30, Kyse450, and Kyse510 cell lines were kindly provided by the Department of Pathophysiology, School of Basic Medicine, Zhengzhou University, Zhengzhou, China. The ESCC cells were cultured in Gibco RPMI 1640 medium with 10% FBS (Gibco) and 1% penicillin-streptomycin solution (Beyotime, Beijing, China). IL-6 was bought from PeproTech, USA. Stattic was received from Selleck Chemicals, Houston, USA.
Western blotting analysis
Cells or tissues were lysed with RIPA lysis (Beyotime, Beijing, China), and prepared for western blotting analysis. The denatured proteins were separated with BeyoGel™ SDS-PAGE Precast Gel (Beyotime, Beijing, China), and transferred to the PVDF membranes. The primary antibodies against p-STAT3, STAT3, GAPDH, and β-actin were purchased from Cell Signaling Technology Inc., Danvers, MA. The anti-EphA7 antibody was bought from Santa Cruz Biotechnology Inc., Dallas, TX.
Immunofluorescence staining
The cultured cells were fixed, permeabilized, blocked, and incubated with indicated primary antibodies overnight. Then, cells were incubated with FITC-labeled or cy3-labeled secondary antibodies (Thermo Fisher Scientific, Waltham, MA, USA) for 2 h in the dark in the next day. DAPI staining was performed for 10 min. After adding anti-fluorescence quencher, cells were examined using a laser confocal microscope.
Quantitative real-time PCR (qRT-PCR)
Total RNA from ESCC cells or tumors was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA), and reversely transcribed into cDNA, followed by qRT-PCR analysis with SYBR™ Select Master Mix (Thermo Fisher Scientific) following the manufacturer’s manual. The sequences of primers were as shown in Table S1 in the supplementary materials.
Luciferase assay
A series of human truncated EphA7 promoter fragments were subcloned into the pGL3 vector (Addgene), and the sequence of EphA7 promoter was shown in Figure S1 in the supplementary materials. The pRL-TK served as an internal control. The luciferase assay was conducted by using the dual-luciferase reporter kit (Promega, Madison, WI) following the manufacturer’s manual.
ChIP assay
The ChIP assay was carried out following the manufacturer’s manual (Millipore, Billerica, MA, USA). In brief, ESCC cells Eca109 were fixed with 1% formaldehyde, lysed and sonicated to shear genomic DNA. Then, supernatants were collected after centrifugation, and incubated with indicated antibodies overnight at 4 °C, followed by precipitation with protein A/G beads. The EphA7 promoter fragment −1670∼−1540 was identified by qRT-PCR analysis. The primers used were as shown in Table S1 in the supplementary materials.
shRNAs, plasmids, and lentivirus
The EphA7 shRNAs were synthesized by GeneChem, Shanghai, China. The target sequences for shEphA7s were as shown in Table S2 in the supplementary materials. The human overexpressing plasmid of STAT3 (OE-STAT3) was purchased from GeneChem (Shanghai, China). Plasmids were transfected into cells by using jetPRIME transfection reagent (Polyplus, Strasbourg, France) following the manufacturer’s protocol. The lentivirus overexpressing EphA7 was purchased from GeneChem, Shanghai, China. To establish stable cell lines, infected cells were selected with puromycin for 1 week, and positive cells were expanded.
Cell viability and proliferation
ESCC cells were cultured in the 96-well plates for different time points, and cell viability was measured by using the CCK-8 reagent (Meilun, Dalian, China) following the manufacturer’s manual. The absorbance value at 450 nm was measured.
Colony-formation assay
ESCC cells (2000 cells per well) were cultured in the 6-well plates. Ten days later, cells were fixed and prepared for crystal violet staining. Then, colonies were taken photos and counted.
Xenograft models
BALB/C nude mice were randomly assigned into two groups, and ESCC cells (Eca109 control cells and EphA7-knockdown cells) were subcutaneously injected. When tumors were palpable, the volume of tumors was measured with a vernier scale three times a week, and the volume was measured as follows: V = 1/2 (a × b × b). After an indicated amount of time, mice were sacrificed, and tumors were harvested for next analyses.
Immunohistochemistry
Tumor tissues from the xenografts were fixed, embedded in paraffin, sectioned, deparaffinized, and antigen retrieval. Sections were incubated with anti-EphA7 and anti-Ki67 antibodies (Santa Cruz Biotechnology) at 4 °C overnight. Sections were then incubated with the Ultra View Universal HRP multitimer (Ventana, Export, PA) at 37 °C for 30 min, and then developed by using the DAB reagent. Hematoxylin counterstaining was performed, and the tissue images were obtained using TissueFAXS Plus ST (Tissue Gnostics, Italy).
RNA-seq analysis
RNA-seq analyses were conducted to evaluate the expression of differential genes in the EphA7-knockdown cells compared with control cells. RNA-seq analyses were performed and analyzed by GENEWIZ (Suzhou, China). The differentially expressed genes were examined by KEGG and GO enrichment analysis.
Statistical analysis
Pictures were drawn by GraphPad Prism. Independent sample t test was used to compare the differences between two groups. One-way ANOVA was used to compare the differences among multiple groups. A p value less than 0.05 was considered to indicate significance.
Results
Activation of STAT3 upregulates the protein levels of EphA7 in ESCC cells
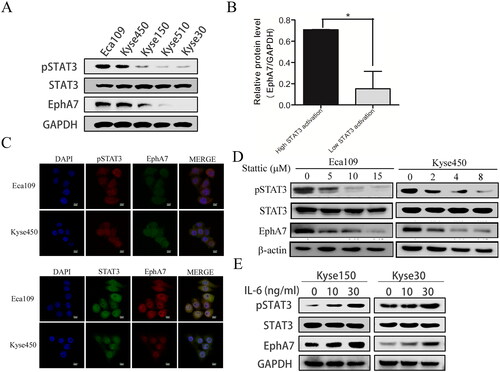
We previously discovered that EphA7 expression was correlated with constitutively phosphorylated STAT3 in patient-derived xenograft models [Citation22]. Here, we first explored the expression of EphA7 and phosphorylated STAT3 in ESCC different cell lines, and EphA7 was found upregulated in ESCC cell lines with high STAT3 activation (). EphA7 was also found co-localized with phospho-STAT3 in Eca109 and Kyse450 cells (). Additionally, suppressing STAT3 activation by the STAT3 inhibitor Stattic markedly down-regulated EphA7 protein levels in ESCC cells (), but in contrast, activation of STAT3 by the cytokine IL-6 obviously upregulated EphA7 protein levels (). Our current results indicated that STAT3 activation upregulated the expression levels of EphA7 in ESCC.
Figure 1. Activation of STAT3 promotes the protein expression of EphA7 in ESCC cells. (A and B) ESCC cell lines were lysed for western blotting against phospho-STAT3 (pSTAT3), STAT3, EphA7, and GAPDH (a), and the statistical analysis was conducted (B). (C) The co-localization of EphA7/pSTAT3 and EphA7/STAT3 in ESCC cells was analyzed by immunofluorescence staining (400×). (D) Eca109 and Kyse450 cells were incubated with indicated concentrations of Stattic for 6 h, followed by western blotting against phospho-STAT3 (pSTAT3), STAT3, EphA7, and β-actin. (E) Kyse150 and Kyse30 cells were incubated with indicated concentrations of IL-6 for 12 h, followed by western blotting against phospho-STAT3 (pSTAT3), STAT3, and EphA7. GAPDH was used as a loading control. *p < 0.05.
The transcription of EphA7 is increased by activated STAT3 in ESCC cells
As STAT3 is an important transcriptional factor, we then evaluated whether STAT3 regulated the transcription of EphA7. As shown in , the STAT3 inhibitor Stattic significantly inhibited the mRNA levels of EphA7 in ESCC cells. In contrast, activation of STAT3 by IL-6 induced the mRNA levels of EphA7 (). And overexpression of STAT3 also promoted the mRNA levels of EphA7 in ESCC cells (; Figure S2 in the supplementary materials). Then, we evaluated whether STAT3 regulated the promoter activity of EphA7 in ESCC cells. As shown in , the results from the luciferase assay indicated that STAT3 overexpression markedly increased EphA7 promoter activity (−2000∼−1). In addition, a series of truncated EphA7 promoter sequences were constructed, and the luciferase assay showed that the fragment of −2000∼−1500 retained a high level of promoter activity, but the luciferase activity was significantly decreased when it was driven by the fragments of −1500∼−1050, −1050∼−600, and −600∼−1, which suggested that the −2000∼−1500 region was essential for EphA7 transcription (). Moreover, the STAT3 inhibitor Stattic significantly down-regulated the luciferase activity of the EphA7 key promoter (−2000∼−1500) in ESCC cells (). But activation of STAT3 by IL-6 increased the luciferase activity of the EphA7 key promoter ().
Figure 2. STAT3 activation up-Regulates the transcription of EphA7 in ESCC cells. (A) Eca109 and Kyse450 cells were treated with indicated concentrations of Stattic for 6 h, followed by qRT-PCR against EphA7. GAPDH was used as an internal control. (B) Kyse150 and Kyse30 cells were incubated with indicated concentrations of IL-6 for 12 h, followed by qRT-PCR against EphA7 and GAPDH. (C) Kyse150 and Kyse30 cells were transfected with empty vector (EV) or STAT3-overexpressing plasmids (STAT3-OE) for 48 h, followed by qRT-PCR against EphA7 and GAPDH. (D) The truncated fragments of EphA7 promoter and STAT3-OE plasmids were co-transfected with pRL-TK into Eca109 cells. After 24 h of co-transfection, medium was replaced with fresh medium containing IL-6 for 12 h, and then cells were prepared for dual luciferase assay. (E) Eca109 and Kyse450 cells transfected with EphA7 promoter (−2000∼−1500) and pRL-TK plasmids were treated with indicated concentrations of Stattic for 6 h, followed by dual luciferase assay. (F) Kyse150 and Kyse30 cells transfected with EphA7 promoter (−2000∼−1500) and pRL-TK plasmids were incubated with indicated concentrations of IL-6 for 12 h, followed by dual luciferase assay. (G) Predicted binding sites of transcription factor STAT3 in EphA7 promoter −2000∼−1500 by JASPAR database. Black boxes indicate the predicted binding sites of STAT3. ‘+’ and ‘-’ means the sense and antisense stands. (H) Eca109 cells were collected and prepared for ChIP assay, followed by qRT-PCR analysis. *p < 0.05, **p < 0.01, ***p < 0.001.
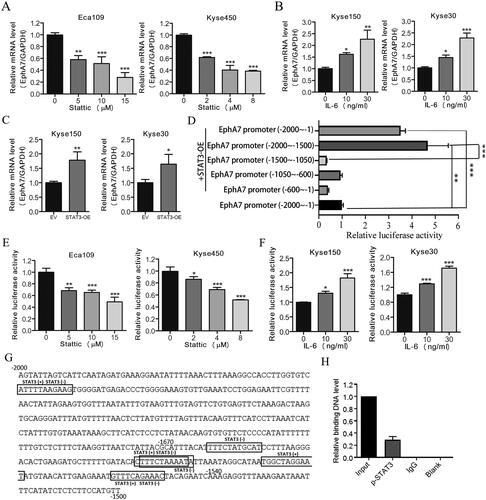
To further study the transcription mechanism of STAT3 regulating EphA7, the binding sites of STAT3 in EphA7 promoter −2000∼−1500 were predicted by JASPAR database. And several predicted binding sites of STAT3 were identified in EphA7 promoter −2000∼−1500 (; Table S3 in the supplementary materials). Moreover, the binding relationship of p-STAT3 as a transcription factor on the EphA7 promoter region was also verified through ChIP experiments (). These results above indicated that the transcription of EphA7 was positively regulated by STAT3 in ESCC cells.
EphA7 knockdown inhibits cell proliferation, and abolishes STAT3 activation-derived cell proliferation in ESCC
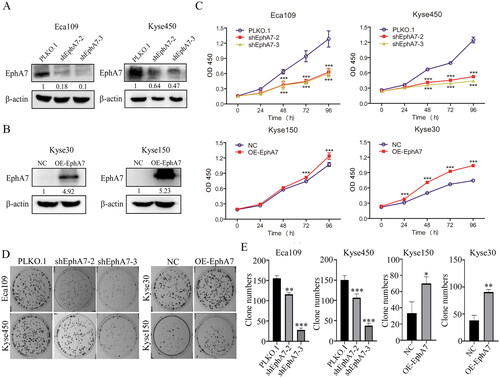
Then, EphA7 was knocked down by shRNAs in Eca109 and Kyse450 cells, and overexpressed in Kyse30 and Kyse150 cells (). To assess the effects of EphA7 on ESCC cell growth, CCK-8 assays were performed. As shown in , the growth ability was decreased when cells were knocked down of EphA7, while overexpression of EphA7 promoted cell growth in ESCC. Colony formation analysis also demonstrated that silence of EphA7 suppressed the colony formation ability of ESCC, but overexpression of EphA7 promoted the colony formation ability (). Additionally, EphA7 was also found mediate the cell migration, invasion and angiogenesis of ESCC (Figure S3 in the supplementary materials).
Figure 3. EphA7 promotes cell proliferation in ESCC cells. (A and B) Western blotting confirmed the Successful establishment of the EphA7-knockdown (A) and EphA7-overexpressing (B) stable cell lines. (C) Eca109 and Kyse450 cells with EphA7 knockdown and Kyse150 and Kyse30 cells with overexpressed EphA7 were prepared for CCK-8 assays as indicated. (D and E) Colony-formation assays of ESCC cells (D), and quantitative analysis was conducted (E). The overview of the colonies was taken photos by a camera. *p < 0.05, **p < 0.01, ***p < 0.001.
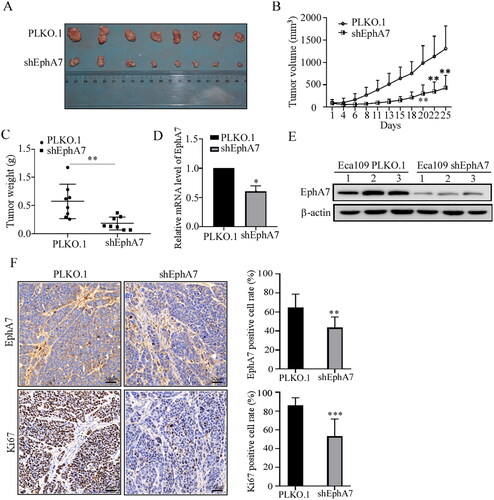
Our results above demonstrated the tumor-promoting effects of EphA7 on ESCC cells in vitro. Then, a cell-derived xenograft model was established to assess EphA7 function in vivo. As shown in , silence of EphA7 significantly suppressed the tumor growth of ESCC. Meanwhile, the results from the immunohistochemistry confirmed the down-regulation of EphA7 and the tumor cell proliferation marker Ki67 in the shEphA7 group compared with the control (). Collectively, our results confirmed that knockdown of EphA7 suppressed ESCC tumor growth in vivo.
Figure 4. Knockdown of EphA7 inhibits tumor growth of ESCC. (A) Photos of transplanted tumors of the indicated groups at the end of the experiment were taken by a camera. (B) Tumor growth curves. (C) Weight of the excised tumors. (D and E) Three tumors were randomly selected from the two groups, and the mRNA (D) and protein (E) levels of EphA7 were evaluated. (F) Representative images (20×) and quantitative analysis of the expression of EphA7 and Ki67 in tumor tissues detected by immunohistochemistry. *p < 0.05, **p < 0.01, ***p < 0.001.
Our previous study indicated that the activation factors of STAT3 signaling (such as IL-6) could induce the growth of ESCC cells [Citation22], we then evaluated whether EphA7 affected this effect. As shown in Figure S4 in the supplementary materials, we confirmed that IL-6 could significantly increase the cell growth of ESCC cells, but this effect could be significantly abolished by the knockdown of EphA7, which suggested that STAT3 signaling may act through EphA7 to promote the development of ESCC.
Knockdown of EphA7 modulates tumor-related signaling pathways in ESCC cells
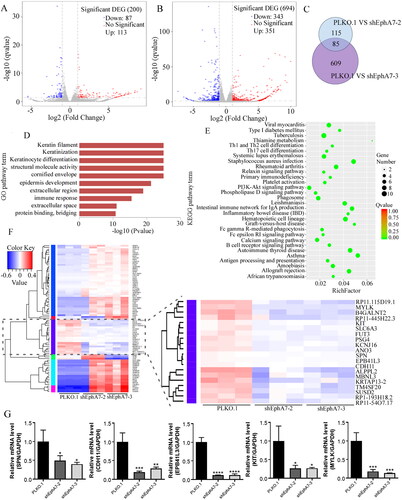
To explore the functional consequence of down-regulated EphA7 in ESCC cells, we performed RNA-seq on two sets of EphA7-knockdown cells (shEphA7-2 and shEphA7-3) and control cells (PLKO.1). In the shEphA7-2 group, 83 genes were found down-regulated and 113 genes were found up-regulated (); in the shEphA7-3 group, 343 genes were found down-regulated and 351 genes were found up-regulated (). In the two EphA7-knockdown groups, 85 overlapping genes were identified (). KEGG and GO enrichment analysis of the 85 overlapping genes revealed significant changes in tumor-related signaling pathways (). Through analysis of the 85 overlapping genes as well as verifying through qRT-PCR analyses, we found that SPN, CDH11, EPB41L3, KIT, and MYLK were significantly down-regulated (), which were closely related to tumor progression. Based on these results, we speculated that EphA7 may enhance the cell proliferation and metastasis of ESCC by regulating these genes, which would be further elucidated in our future work.
Figure 5. Knockdown of EphA7 modulates tumor-related signaling pathways in ESCC cells. (A) Volcano plot of gene changes in shEphA7-2-derived Eca109 cells. (B) Volcano plot of gene changes in shEphA7-3-derived Eca109 cells. (C) Venn diagram shows the intersection of differentially expressed genes in the two groups. (D) Cluster heat map of 85 overlapping differentially expressed genes. (E) Top 10 terms in the GO enrichment analysis results of 85 overlapping differentially expressed genes. (F) KEGG enrichment assay of 85 overlapping differentially expressed genes. (G) Eca109 cells with EphA7 knockdown were prepared for qRT-PCR against SPN, CDH11, EPB41L3, KIT, and MYLK. GAPDH was used as an internal control. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Discussion
Approximately 53% of ESCC cases worldwide occur in China [Citation23]. Because of the lack of early and effective clinical diagnosis methods for esophageal cancer, patients generally develop the resistance of cancer cells to radiotherapy or chemotherapy and the high recurrence rate of esophageal cancer after surgery [Citation24–26]. Thus, effective treatment of esophageal cancer is still a significant obstacle.
The Eph family proteins have been reported to participate in the signaling pathways of tissue formation and embryogenesis, and they are also involved in the development of tumors. They act as tumor suppressors or tumor promoters depending on the situation [Citation27–29]. The Eph family is also closely related to ESCC, and Miyazaki et al. found that elevated EphA2 was predicted as a negative index for ESCC patients [Citation30]. EphA3 was proven to inhibit the migration and invasion of ESCC cells by activating the mesenchymal-epithelial transition process and played an inhibitory role in ESCC [Citation31]. EphA5 was reported to be upregulated in ESCC [Citation32]. In our study, EphA7 was found enhance ESCC cell proliferation in vitro, and the xenograft models also showed that knockdown of EphA7 suppressed ESCC tumor growth in vivo, which suggested that EphA7 exerted tumor-promoting effect in ESCC.
The JAK-STAT3 signaling plays vital roles in human cancers. STAT3 is an oncogenic transcription factor involved in cell proliferation, growth, invasion, migration, inflammation, and immune function [Citation33]. For example, overexpression of some Toll-like receptors promotes the malignant activity in some tumor cells and immune cells through the JAK-STAT3 pathway, and some microRNAs that interact with the JAK-STAT3 pathway regulate the cancer-promoting inflammation and carcinogenesis [Citation34]. The results published previously by us indicated that STAT3 was constitutively activated in ESCC, and played important effects in the growth of inflammation-induced ESCC [Citation22]. We previously also observed EphA7 was upregulated in ESCC patient-derived xenograft models with constitutively phosphorylated STAT3 [Citation22]. In this study, the transcription of EphA7 was found upregulated by activated STAT3 in ESCC cells, which indicated that the STAT3 signaling may act through EphA7 to promote the development of ESCC. By nowadays, there still have been no potential therapeutic targets or strategies that could be derived from targeting the STAT3-EphA7 pathway, which will be studied in our future work.
Finally, transcriptome sequencing was performed in EphA7-knockdown cells. CDH11, EPB41L3, KIT, and MYLK genes were significantly down-regulated in ESCC cells with EphA7 knockdown, and they were closely related to cell adhesion, proliferation, metastasis, and inflammatory response. A previous study revealed that EphB6 mutation promoted cancer cell proliferation through EphA2 and downstream JNK/CDH11/RhoA/FAK signal transduction [Citation35], which suggested that CDH11, EPB41L3, KIT, and MYLK genes may be related to EphA7 function. In addition, KEGG and GO enrichment analysis showed that the signal pathways related to tumor progression were significantly changed in ESCC cells with EphA7 knockdown, which further suggested that EphA7 may play vital roles in the development of ESCC. Later, we will conduct a deeper study on the downstream molecules of EphA7 and its mechanism in the progression of ESCC based on the transcriptome results.
Collectively, this study suggested that the STAT3 signaling may act through EphA7 to promote the development of ESCC, which provided a novel theoretical basis for the therapy of ESCC.
Ethical approval
This study was approved by the Review Board and Ethical Committee of the First Affiliated Hospital of Zhengzhou University.
Author contributions
Li Wang: Methodology, Investigation, Data curation, Writing, Original draft preparation, Visualization. Qiao-Feng Zhao: Methodology, Investigation. Bing-Bing Yang: Methodology, Investigation. Hui-Jie Liang: Validation, Investigation. Xian-E Zhang: Validation, Investigation. Xiao-Yan Zhang: Validation, Investigation. Wan-Jing Yang: Formal analysis. Zhi-Yu Guo: Software. Xin Xu: Writing, Reviewing and Editing. Fang Tian: Conceptualization, Writing, Reviewing and Editing, Project administration, Funding acquisition. Qing-Hua Wu: Conceptualization, Supervision, Funding acquisition. All authors contributed to and approved the final manuscript.
| Abbreviations | ||
| ESCC | = | esophageal squamous cell carcinoma |
| EAC | = | esophageal adenocarcinoma |
| RTKs | = | receptor tyrosine kinases |
| FBS | = | fetal bovine serum |
| RT | = | room temperature |
| h | = | hour |
| qRT-PCR | = | quantitative real-time PCR |
| IL-6 | = | interleukin |
| CCK-8 | = | cell-counting kit-8 |
Supplemental Material
Download MS Word (3.6 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data generated or analyzed during this study are included in this published article. Data will be made available on request.
Additional information
Funding
References
- Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492.
- Chen J, Kwong DL, Cao T, et al. Esophageal squamous cell carcinoma (ESCC): advance in genomics and molecular genetics. Dis Esophagus. 2015;28(1):84–89. doi: 10.1111/dote.12088.
- Eyre R, Alferez DG, Santiago-Gomez A, et al. Microenvironmental IL1beta promotes breast cancer metastatic colonisation in the bone via activation of wnt signalling. Nat Commun. 2019;10(1):5016. doi: 10.1038/s41467-019-12807-0.
- Frohlich J, Mazza T, Sobolewski C, et al. GDF11 rapidly increases lipid accumulation in liver cancer cells through ALK5-dependent signaling. Biochim Biophys Acta Mol Cell Biol Lipids. 2021;1866(6):158920. doi: 10.1016/j.bbalip.2021.158920.
- Kuo CN, Pan JJ, Huang YW, et al. Association between nonsteroidal anti-inflammatory drugs and colorectal cancer: a population-based case-control study. Cancer Epidemiol Biomarkers Prev. 2018;27(7):737–745. doi: 10.1158/1055-9965.EPI-17-0876.
- Tulotta C, Lefley DV, Freeman K, et al. Endogenous production of IL1B by breast cancer cells drives metastasis and colonization of the bone microenvironment. Clin Cancer Res. 2019;25(9):2769–2782. doi: 10.1158/1078-0432.CCR-18-2202.
- Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–1134. doi: 10.1016/j.cell.2010.06.011.
- McDonell LM, Kernohan KD, Boycott KM, et al. Receptor tyrosine kinase mutations in developmental syndromes and cancer: two sides of the same coin. Hum Mol Genet. 2015;24(R1):R60–66. doi: 10.1093/hmg/ddv254.
- Taylor H, Campbell J, Nobes CD. Ephs and ephrins. Curr Biol. 2017;27(3):R90–R95. doi: 10.1016/j.cub.2017.01.003.
- Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133(1):38–52. doi: 10.1016/j.cell.2008.03.011.
- Taneja R, Thisse B, Rijli FM, et al. The expression pattern of the mouse receptor tyrosine kinase GeneMDK1Is conserved through evolution and requires Hoxa-2for rhombomere-specific expression in mouse embryos. Dev Biol. 1996;177(2):397–412. doi: 10.1006/dbio.1996.0173.
- Erturk E, Tuna G, Coskun D, et al. Investigation of anti-Cancer activity of newly synthesized 2,4-pentadien-1-one derivative containing benzofuran in human lung and colon cancer cells. Eurasian J Med Oncol. 2023;7(1):24–33. doi: 10.14744/ejmo.2023.61594.
- Li R, Sun Y, Jiang A, et al. Knockdown of ephrin receptor A7 suppresses the proliferation and metastasis of A549 human lung cancer cells. Mol Med Rep. 2016;13(4):3190–3196. doi: 10.3892/mmr.2016.4904.
- Tu Y, Cai Q, Zhu X, et al. Down-regulation of HCP5 inhibits cell proliferation, migration, and invasion through regulating EPHA7 by competitively binding miR-101 in osteosarcoma. Braz J Med Biol Res. 2021;54(2):e9161. doi: 10.1590/1414-431X20209161.
- Di W, Weinan X, Xin L, et al. Long noncoding RNA SNHG14 facilitates colorectal cancer metastasis through targeting EZH2-regulated EPHA7. Cell Death Dis. 2019;10(7):514. doi: 10.1038/s41419-019-1707-x.
- Li S, Wu Z, Ma P, et al. Ligand-dependent EphA7 signaling inhibits prostate tumor growth and progression. Cell Death Dis. 2017;8(10):e3122–e3122. doi: 10.1038/cddis.2017.507.
- Oricchio E, Wendel HG. Mining the cancer genome uncovers therapeutic activity of EphA7 against lymphoma. Cell Cycle. 2012;11(6):1076–1080. doi: 10.4161/cc.11.6.19451.
- Leonard WJ, O’Shea JJ. Jaks and STATs: biological implications. Annu Rev Immunol. 1998;16(1):293–322. doi: 10.1146/annurev.immunol.16.1.293.
- O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. 2013;368(2):161–170. doi: 10.1056/NEJMra1202117.
- Bowman T, Garcia R, Turkson J, et al. STATs in oncogenesis. Oncogene. 2000;19(21):2474–2488. doi: 10.1038/sj.onc.1203527.
- Carpenter RL, Lo HW. STAT3 target genes relevant to human cancers. Cancers (Basel). 2014;6(2):897–925. doi: 10.3390/cancers6020897.
- Tian F, Yang X, Liu Y, et al. Constitutive activated STAT3 is an essential regulator and therapeutic target in esophageal squamous cell carcinoma. Oncotarget. 2017;8(51):88719–88729. doi: 10.18632/oncotarget.20838.
- Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi: 10.3322/caac.21262.
- Chen L, Bi S, Hou J, et al. Targeting p21-activated kinase 1 inhibits growth and metastasis via Raf1/MEK1/ERK signaling in esophageal squamous cell carcinoma cells. Cell Commun Signal. 2019;17(1):31. doi: 10.1186/s12964-019-0343-5.
- Cheng X, Wei L, Huang X, et al. Solute carrier family 39 member 6 gene promotes aggressiveness of esophageal carcinoma cells by increasing intracellular levels of zinc, activating phosphatidylinositol 3-Kinase signaling, and up-regulating genes that regulate metastasis. Gastroenterology. 2017;152(8):1985–1997.e12. doi: 10.1053/j.gastro.2017.02.006.
- Lin DC, Hao JJ, Nagata Y, et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet. 2014;46(5):467–473. doi: 10.1038/ng.2935.
- Adams RH, Klein R. Eph receptors and ephrin ligands. Essential mediators of vascular development. Trends Cardiovasc Med. 2000;10(5):183–188. doi: 10.1016/s1050-1738(00)00046-3.
- Genander M, Frisen J. Ephrins and eph receptors in stem cells and cancer. Curr Opin Cell Biol. 2010;22(5):611–616. doi: 10.1016/j.ceb.2010.08.005.
- Kou CJ, Kandpal RP. Differential expression patterns of EPH receptors and ephrin ligands in human cancers. Biomed Res Int. 2018;2018:7390104–7390123. doi: 10.1155/2018/7390104.
- Miyazaki T, Kato H, Fukuchi M, et al. EphA2 overexpression correlates with poor prognosis in esophageal squamous cell carcinoma. Int J Cancer. 2003;103(5):657–663. doi: 10.1002/ijc.10860.
- Chen X, Lu B, Ma Q, et al. EphA3 inhibits migration and invasion of esophageal cancer cells by activating the mesenchymalepithelial transition process. Int J Oncol. 2019;54(2):722–732. doi: 10.3892/ijo.2018.4639.
- Zhang R, Liu J, Zhang W, et al. EphA5 knockdown enhances the invasion and migration ability of esophageal squamous cell carcinoma via epithelial-mesenchymal transition through activating WNT/beta-catenin pathway. Cancer Cell Int. 2020;20(1):20. doi: 10.1186/s12935-020-1101-x.
- Leeman RJ, Lui VW, Grandis JR. STAT3 as a therapeutic target in head and neck cancer. Expert Opin Biol Ther. 2006;6(3):231–241. doi: 10.1517/14712598.6.3.231.
- Yu H, Lee H, Herrmann A, et al. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736–746. doi: 10.1038/nrc3818.
- Yoon S, Choi JH, Kim SJ, et al. EPHB6 mutation induces cell adhesion-mediated paclitaxel resistance via EPHA2 and CDH11 expression. Exp Mol Med. 2019;51(6):1–12. doi: 10.1038/s12276-019-0261-z.