
Lysosomal acid Lipase A (LIPA), also known as Lysosomal acid lipase (LAL) or cholesterol ester hydrolase, is located on the long arm of human chromosome 10Citation1. This enzyme is of fundamental importance for the hydrolysis of cholesteryl ester and triglyceride in lysosomes (). LAL deficiency (LAL-D) is an inherited autosomal recessive disorder, with an estimated prevalence between 1:40,000–1:300,000Citation2. While reduced LAL activities cause cholesteryl ester storage disease (CESD), the lack or production of an enzyme with no residual activity causes the more severe early-onset form known as Wolman disease (WD).
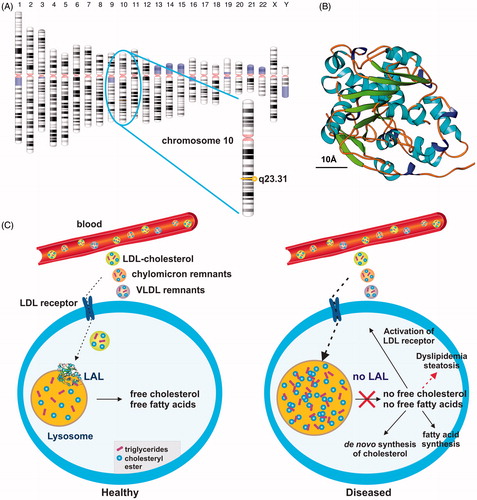
Figure 1. Simplified scheme of lysosomal acid lipase activity in cellular fat and cholesterol metabolism. (a) Lysosomal acid lipase is located on human chromosome 10q23.31. (b) Human LAL bears the characteristic structural features of the α/β hydrolase fold family consisting of typical α-helixes and β-sheetsCitation17. The predicted model of human LAL was generated using the fully automated protein structure homology-modeling server SWISS MODELCitation18, human LAL protein sequence deposited in the NCBI Protein data baseCitation19 (access. no. GenBank: AAB60328.1), and the crystal structure coordinates of dog gastric lipaseCitation20, sharing an overall sequence identity of 60.22% with human LAL. The estimated global model quality (GMQE) and global and local model quality (QMEAN) of the model are 0.77 and –2.99, respectively. A size marker (10 Å) is given. According to a Prosite scanCitation21, the active serine site is located at position 174 within the human LAL protein, consisting of a total of 399 amino acids. (c) In healthy subjects, LAL is located in lysosomes where it plays an important role in breaking down cholesteryl esters and triglycerides imported via the LDL/LDL receptor route from the blood into the cell. (d) The lack of LAL prevents breakdown of fatty material and trafficking of lipids from the lysosome into the cytoplasm. As a consequence, the control circuit for regulation of the intracellular cholesterol concentration is interrupted. In turn, the lower cellular cholesterol level provokes increased LDL receptor activity and stimulates cholesterol synthesisCitation22,Citation23. As a consequence, the cell is overloaded with cholesterol. Some of the extra cholesterol is a source for bile acid synthesis, while another portion becomes esterified and incorporated into VLDL, which ultimately can become trapped in the lysosomal compartment.
Although the phenotypic spectrum of LAL-D is highly variable, clinical features of LAL-D typically include marked dyslipidemia, elevated liver enzymes, hepatomegaly, steatosis, and splenomegaly. If left untreated, the progressive hepatic dysfunction associated with LAL-D leads to liver failure, making liver transplantation necessaryCitation3. While the treatment of LAL-D was in the past limited to control of cholesterol levels and prevention of premature atherosclerosisCitation4, the introduction of enzyme replacement strategies enabling correction of respective metabolic defects offers hope to patients suffering from this disorderCitation5–7.
Sebelipase alfa is a recombinant human LAL produced by molecular farming in the egg white of transgenic hensCitation8. The enzyme is N-glycosylated at five glycosylation sites, predominantly N-acetylglucosamine and mannose-terminated N-linked sugars targeting the enzyme to cells expressing the mannose-6-phosphate receptor and further conferring ability to traffic via endosomes to the lysosomeCitation9. The replacement therapy with Sebelipase alfa was approved in December 2015 under the trade name Kanuma by the Food and Drug Administration in the US. It is applied as an intravenous infusion drug once weekly in patients with rapidly progressive LAL deficiency occurring in the first 6 months of life, and once every other week in all other patientsCitation10. Although the biochemical and clinical consequences of LAL-D in the pathogenesis of hepatic disease are well characterized, the clinical course of late onset of LAL-D is still unknown.
In an article in this issue of Current Medical Research and Opinion, Burton et al.Citation11 summarized the results of an observational study conducted in pediatric and adult LAL-D patients. In particular, the study was designed to get information about the time to progression from first clinical manifestation to first documentation of hepatic disease. In their study, the authors analyzed data from 31 liver biopsy patients and one patient without biopsy data who had undergone liver transplantation. As expected, the patients of this collective were characterized by a high frequency of hepatomegaly, splenomegaly, and elevated levels of alanine aminotransferase and LDL cholesterol. In addition, most subjects had documented liver insults (fibrosis, cirrhosis, liver transplant). Importantly, first clinical manifestations occurred at a mean age of 9.2 years, while the median time to an event was 3.1 years. Unfortunately, the authors provided no separate correlation of genotype and time to an event for pediatric and adult patients. However, compared to other liver disorders associated with accumulation of excess fat, the data of this study highlight the fact that LAL-D is a rapidly progressing disease associated with a high risk for liver failure and transplant.
Of note, independent studies suggested lowered LAL activities as an under-recognized cause of dyslipidemia and a causal link to the pathogenesis of non-alcoholic fatty liver disease (NAFLD) in pediatric and adult patientsCitation12–14. In this context, it is thoroughly reasonable to implement LAL-D screening programs to detect patients at risk. Actually, there are more than 40 loss-of-function mutations known, giving rise to lowered LAL activitiesCitation2. Both genetic testing and direct quantitation of LAL concentrations by immunogenic tests are suitable to identify respective patients with LAL alterations. A further enrichment of diagnostic strategies was the implementation of the LAL/Lalistat 2 testCitation15. This test comparatively measures total lipase activity and lipase activity in the presence of the highly competent LAL inhibitor Lalistat 2 (a thiadiazole carbamate). In the respective assay that can be conducted in 96 well plates, LAL deficient samples show significantly reduced activity, with results close to zero activity, while LAL mutant allele carriers show activity grouped nearly between healthy and LAL deficient cases, with all results below the reference rangeCitation15. The evaluation of this method in normal controls (n = 140), carriers (n = 15), and CESD patients (n = 11) showed that this novel method for the measurement of LAL in dried blood spots offers an alternative, cheap, and reliable diagnostic, possibility to discriminate between unaffected, carriers, and affected subjectsCitation15. This method will allow straightforward screening for CESD and WD in high risk populations. In addition, this Lalistat 2 test was also successfully applied to demonstrate a strong association between impaired LAL activity and fatty liver diseaseCitation13.
Besides these improvements in diagnostics, there is an urgent need for programs that provide information about disease progression time in LAL-D and subjects at higher risk of LAL-associated liver failures. In this regard, Alexion Pharmaceuticals (New Haven, CT, USA) has recently launched a screening program (NCT02926872) in which children from 2–16 years will be recruited to provide more focused guidance to the medical community as to which pediatric patients should be tested for LAL-DCitation16.
To sum up, the study by Burton et al.Citation11 once more demonstrates that LAL-D is a life-threatening disorder with short progression times. In addition, concomitant work demonstrated low LAL activity in both children and adults suffering from NAFLD. Thus, screening programs for LAL-D mutations or reduced enzymatic activities might not only be relevant for patients with suspected LAL-D, but also for subjects with a documented family history of idiopathic steatosis or steatohepatitis.
Ralf Weiskirchen Institute of Molecular Pathobiochemistry, Experimental Gene Therapy and Clinical Chemistry (IFMPEGKC), RWTH University Hospital Aachen, Aachen, Germany [email protected]
Transparency
Declaration of funding
No funding information was provided for this manuscript.
Declaration of financial/other interests
The work of RW is financially supported by the German Research Foundation (SFB/TRR 57, projects P13 and Q3) and the Interdisciplinary Center for Clinical Research (IZKF) at the University Hospital Aachen (Project E7-6). The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. CMRO peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
RW acknowledges the help of Sabine Weiskirchen, who prepared the figure for this editorial.
References
- Wild PS, Zeller T, Schillert A, et al. A genome-wide association study identifies LIPA as a susceptibility gene for coronary artery disease. Circ Cardiovasc Genet 2011;4:403-12
- Desai NK, Wilson DP. Lysosomal acid lipase deficiency. In: De Groot LJ, Chrousos G, Dungan K, et al., editors. Endotext [Internet]. South Dartmouth, MA: MDText.com, Inc.; 2000 [updated 2016 Jun 22]. https://www.ncbi.nlm.nih.gov/books/NBK395569/. Accessed May 8, 2017
- Burton BK, Deegan PB, Enns GM, et al. Clinical features of lysosomal acid lipase deficiency. J Pediatr Gastroenterol Nutr 2015;61:619-25
- Porto AF. Lysosomal acid lipase deficiency: diagnosis and treatment of Wolman and cholesteryl ester storage diseases. Pediatr Endocrinol Rev 2014;12(S1):125-32
- Su K, Donaldson E, Sharma R. Novel treatment options for lysosomal acid lipase deficiency: critical appraisal of sebelipase alfa. Appl Clin Genet 2016;9:157-67
- Frampton JE. Sebelipase Alfa: A review in lysosomal acid lipase deficiency. Am J Cardiovasc Drugs 2016;16:461-8
- Valayannopoulos V, Malinova V, Honzík T, et al. Sebelipase alfa over 52 weeks reduces serum transaminases, liver volume and improves serum lipids in patients with lysosomal acid lipase deficiency. J Hepatol 2014;61:1135-42
- Sheridan C. FDA approves ‘farmaceutical’ drug from transgenic chickens. Nat Biotechnol 2016;34:117-9
- Balwani M, Breen C, Enns GM, et al. Clinical effect and safety profile of recombinant human lysosomal acid lipase in patients with cholesteryl ester storage disease. Hepatology 2013;58:950-7
- FDA U.S. Food & drug Administration. FDA News Release: FDA approves first drug to treat a rare enzyme disorder in pediatric and adult patients. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm476013.htm. Accessed May 8, 2017
- Burton BK, Silliman N, Marulkar S. Progression of liver disease in children and adults with lysosomal acid lipase deficiency. Curr Med Res Opin 2017; in press doi: 10.1080/03007995.2017.1309371
- Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency—an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 2014;235:21-30
- Baratta F, Pastori D, Del Ben M, et al. Reduced lysosomal acid lipase activity in adult patients with Non-alcoholic fatty liver disease. EBioMedicine 2015;2:750-4
- Selvakumar PK, Kabbany MN, Lopez R, et al. Reduced lysosomal acid lipase activity - A potential role in the pathogenesis of non alcoholic fatty liver disease in pediatric patients. Dig Liver Dis 2016;48:909-13
- Hamilton J, Jones I, Srivastava R, et al. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta 2012;413:1207-10
- Carr PD, Ollis DL. Alpha/beta hydrolase fold: an update. Protein Pept Lett 2009;16:1137-48
- ClinicalTrials.gov [Internet]. A service of the U.S. National Institutes of Health. 2016 https://clinicaltrials.gov/ct2/show/NCT02926872. Accessed May 8, 2017
- SWISS MODEL [Internet]. Universität Basel, The Center for Molecular Life Sciences. 2014 https://swissmodel.expasy.org/. Accessed March 31, 2017
- NCBI (National Center for Biotechnology Information) [Internet]. U.S. National Library of Medicine. 1992 https://www.ncbi.nlm.nih.gov/. Accessed May 8, 2017
- RCSB Protein Data Bank [Internet]. Research Collaboratory for Structural Bioinformatics. Accession number: 1K8Q. 2002 http://www.rcsb.org/pdb/explore/explore.do?structureId=1k8q. Accessed May 8, 2017
- Prosite. [Internet]. ExPASy Bioinformatics Resource Portal. Database of protein domains, families and functional sites. http://prosite.expasy.org/. 2011 Accessed May 8, 2017
- Ginsberg HN, Le NA, Short MP, et al. Suppression of apolipoprotein B production during treatment of cholesteryl ester storage disease with lovastatin. Implications for regulation of apolipoprotein B synthesis. J Clin Invest 1987;80:1692-7
- Cummings MH, Watts GF. Increased hepatic secretion of very-low-density lipoprotein apolipoprotein B-100 in cholesteryl ester storage disease. Clin Chem 1995;41:111-4