
Abstract
Objective: Fosamprenavir, a protease inhibitor (PI) to treat human immunodeficiency virus (HIV)-infected patients, has been approved in more than 40 countries and mainly used with nucleoside reverse transcriptase inhibitors. In Japan, Lexiva tablet (fosamprenavir calcium hydrate) has been marketed since January 2005 and used in clinical practice. The safety and effectiveness of fosamprenavir in HIV-infected Japanese patients were evaluated in an observational surveillance study (OTH112334).
Methods: A post-marketing surveillance study (PMS) of fosamprenavir usage in HIV-infected Japanese subjects evaluating drug safety was conducted under Good Post-marketing Study Practice from January 2005 to December 2014.
Results: Of 364 patients receiving fosamprenavir, 51% received emtricitabine/tenofovir disoproxil fumarate. Adverse events whose causal relationship could not be completely ruled out (adverse drug reactions; ADRs) were reported in 43.7%; the most common were diarrhoea (10.4%), hyperlipidaemia (8.5%) and hypertriglyceridaemia (6.9%). Serious ADRs were reported in 26 patients (32 events), including 1 death attributed to hepatic failure. Most ADRs occurred within 180 days after fosamprenavir was started. ADRs were more frequent in patients with the Centers for Disease Control and Prevention category B (AIDS or lipid disorders) or in those taking fosamprenavir combined with abacavir and lamivudine. Although spontaneous bleeding has been reported in hemophiliac patients taking other PIs, in this survey, only one muscle haemorrhage case was reported in 24 hemophiliac patients.
Conclusions: The results of this PMS analysis in Japan support its known safety profile and identified no new safety risks for people living with HIV/AIDS in Japan currently on, or beginning treatment with, fosamprenavir.
Introduction
The use of protease inhibitors (PIs) markedly improved the treatment options for a human immunodeficiency virus (HIV) infection.Citation1 This is because the PI-containing triple combination therapy can almost completely inhibit virus replication in vivo.Citation2 These triple combination therapies are currently referred to as combinational antiretroviral therapy (ART) or highly active ART (HAART).Citation3 With the advent of HAART, the number of patients who have died of AIDS has substantially decreased and HIV infection is now aggressively treated at the earlier stages of infection.Citation4
Fosamprenavir (Lexiva® Tablets 700, ViiV Healthcare, Tokyo), a prodrug of amprenavir (GlaxoSmithKline, United Kingdom), is one of the drugs available for treatment of HIV infection. The approved dosage of fosamprenavir is 1400 mg daily with ritonavir or 2800 mg daily without ritonavir.Citation5 The conditions for approval by the Japanese Ministry of Health, Labor and Welfare required that post-marketing surveillance of all patients who received fosamprenavir in Japan should be conducted to assess the actual use of the product at its initial approval. Post-marketing data were collected from 365 patients at 26 sites in Japan between January 2005 and December 2014 and analyzed for safety and effectiveness of fosamprenavir. When fosamprenavir was used as a component of ART, the incidence of adverse events whose causal relationship could not be completely ruled out (adverse drug reactions; ADRs) and serious adverse events were 43.7% and 21.7%, respectively.
Methods
Subjects
This prospective surveillance (trial registration number OTH112334) was conducted as part of a cooperative post-marketing survey (HIV-Related Drug [HRD] cooperative survey) by the pharmaceutical companies that market drugs for HIV infection treatment in Japan and used common survey forms. No statistical hypothesis testing was planned for both efficacy and safety. Since anti-retroviral drugs for HIV treatment are designated as orphan drugs and prescribed to a limited number of patients in Japan, and are generally administered with other drugs for treatment of HIV infection (including other anti-retrovirals and other drugs for treatment of HIV-related disease [such as opportunistic infection]), the HRD cooperative survey was conducted based on the agreement with Japanese regulatory authority (Pharmaceuticals and Medical Devices Agency, PMDA) in order to survey all of the patients who have received HIV infection treatments by the companies developing anti-HIV drugs and to reduce workload of healthcare personnel to fill out different forms for each drug per each patient. This surveillance was conducted by Nihon Ultmarc Inc. (CMIC-PMS Co., Ltd. since 1 April 2013) on behalf of the study sponsors and it included all fosamprenavir-treated patients who registered to participate in this survey between January 2005 and March 2013. The safety data continued to be collected until December 2014.
A second HRD cooperative survey was also conducted, involving pregnant women who received fosamprenavir during the survey period and whose deliveries were able to be followed.
Signed informed consent forms were not collected since these are not required to be collected according to Good Post-marketing Study Practice in Japan. As pre-specified in the study protocol, to ensure informed consent from all patients who would receive fosamprenavir, the use of fosamprenavir in treatment of HIV infection was explained to each potential subjects, and verbal agreement to use the product and study participation was obtained prior to taking the product.
Observed items
Over the course of the survey study the following demographic and safety information was collected: patient characteristics (sex, age, race, a history of HIV infection, medical history and complications); prior ART (drug name, dosage and duration of treatment); usage of other concomitant drugs; plasma HIV-RNA copies/mL; peripheral CD4+ cell counts; the Centers for Disease Control and Prevention (CDC) classification; presence/absence of abnormal changes in laboratory tests and these values if any abnormal values were observed; specific adverse events (AEs) as well as their dates of onset, course, treatment, seriousness, outcome and causal relationship to disease or drugs. The causal relationship of each AE to fosamprenavir was evaluated by the site physicians according to the four categories “definitely related,” “possibly related,” “not related,” “unknown.” Except for those categorized as “not related,” causal relationships could not be completely ruled out and so these AEs were defined as “ADRs.” Terminology of AEs in this report followed Medical Dictionary for Regulatory Activities (MedDRA) version 17.1. Furthermore, in hemophiliac patients, characteristic ADRs (e.g. spontaneous bleeding) were investigated as a specifically focused item. For HIV-infected pregnant women receiving ART, AEs associated with pregnancy and delivery were also collected and reported.
Statistical analysis
Relationship of patient characteristics and incidence of ADRs were examined by logistic regression. Univariate analysis for each patient’s characteristics was conducted. Variable selection was performed by forward, stepwise and backward method. Odds ratios with 95% confidence interval (CI) were estimated. All of the statistical analyses were conducted using SAS version 9.4.
Results
Surveyed patients
A total of 365 patients from 26 Japanese sites participating in this HRD cooperative survey were registered and completed the required survey forms. Of these, 364 patients were included in the safety analysis; one patient who had received fosamprenavir before the product was approved for use in Japan was excluded. The effectiveness analysis consisted of 336 patients, since 28 additional patients were excluded because CD4+ cell counts and HIV-RNA copies/mL at pre and post fosamprenavir treatment results were not available.
The median (min, max) duration of treatment period was 1138 (5, 2953) days with 1114 person-year for 362 patients (excluding 2 patients whose durations were unknown in the safety analysis), and 1239 (5, 2953) days with 1095 person-year for 334 patients in the effectiveness analysis.
Patient characteristics
The patient characteristics of the safety data set are shown in . Of 364 patients in the study, 94.2% (343 patients) were Japanese, and 92.9% (338 patients) were male. Ages at the initial treatment of fosamprenavir ranged from 19 to 76 years; the majority (95.6%) were between 15 and 64 years. The reason for use of fosamprenavir was HIV infection in all patients and 51% received emtricitabine/tenofovir disoproxil fumarate as concomitant medications. There were 24 HIV infected hemophiliac patients. The patient characteristics of the 336 patients included in the effectiveness analysis were similar to those of the 364 patients included in the safety analysis.
Table 1. Patient characteristics.
The numbers of patients categorized by dosage of fosamprenavir in 364 patients included in the safety analysis are shown in . Most patients (both ART naïve and ART-experienced) were treated with fosamprenavir 1400 mg + ritonavir. A total of 358 of 364 (98.4%) patients received fosamprenavir in combination with dual nucleoside reverse transcriptase inhibitors (NRTIs), consisting of 143 of 143 (100%) and 215 of 221 (97.3%) patients in the ART-naïve and ART-experienced groups, respectively.
Table 2. Numbers of patients categorized by dosage of fosamprenavir.
Adverse drug reactions
The listing of ADRs observed during this survey is shown in . ADRs were observed in 159 of 364 patients (43.7%; 276 events). The proportion of patients reporting ADRs by MedDRA system organ class in descending order were metabolism and nutrition disorders (18.7%; 68/364 patients), gastrointestinal disorders (14.6%; 53/364 patients), skin and subcutaneous tissue disorders (9.6%; 35/364 patients) and investigations (7.4%; 27/364 patients). The most common ADRs by MedDRA preferred terms were diarrhoea (10.4%; 38 events), hyperlipidaemia (8.5%; 31 events), hypertriglyceridaemia (6.9%; 25 events), rash (5.0%; 18 events) and nausea (3.6%; 13 events). All of these common ADRs had previously been listed in the precautions section of the Japanese package insert.Citation5
Table 3. Listing of adverse drug reactions.
Serious ADRs were reported in 26 patients (32 events) as shown in . The reported serious ADRs included immune reconstitution inflammatory syndrome, diabetes mellitus, hyperbilirubinaemia and lipodystrophy acquired (2 events in 2 patients each) while the other serious ADRs () were reported for 1 patient only. Of these, the outcome of hepatic failure reported in 1 patient (1 event) was death. The patient, a man in his sixties, was admitted to a hospital at 3.5 years after initiation of fosamprenavir treatment because of fall. He complained of lower back pain and underwent computed tomography, which revealed ascites. He died of hepatic cancer and hemorrhage on the next day. Outcomes of 12 events (11 patients) were reported as unresolved. For other serious ADRs, outcomes were reported as resolved or improved. Defining clearly the causal relationship between AEs and fosamprenavir was difficult because these patients were treated with other anti-HIV drugs in addition to fosamprenavir, and some ADRs were possibly related to their primary diseases or concomitant diseases, while some ADRs resolved during treatment with fosamprenavir.
Table 4. Listing of serious adverse drug reactions.
Duration of fosamprenavir treatment prior to onset of adverse drug reactions in anti-retroviral therapy naïve patients
The percentage of patients in whom ADRs occurred within 180 days, between 181 and 365 days (1 year), and on Day 366 and later after the start of fosamprenavir treatment was 68.9% (42/61 patients), 14.8% (9/61 patients) and 14.8% (9/61 patients), respectively, implying that most ADRs occurred within the first half a year after initiating fosamprenavir treatment. The most common ADRs which occurred within 180 days were diarrhoea (12 events), rash (8 events) and hyperlipidaemia (7 events), and those which occurred on Day 366 and later were diarrhoea and hyperlipidaemia (3 events each) (data not shown).
Adverse drug reactions by patient characteristics
In this survey, additional evaluation for ADRs was performed using univariate analysis with following factors that considered affecting the safety: age, sex, category of CDC classification at pretreatment, with or without complication diseases and combined backbone NRTI (). The results suggested that “category of CDC classification,” “with or without complications related to hyperlipidemia,” “combined backbone NRTI” were factors affecting safety, with 95% CI not including 1. Complications related with hyperlipidemia are defined as: increased blood triglycerides, abnormal lipid values, increased low density lipoprotein, hypercholesterolaemia, hypertriglyceridaemia, dyslipidaemia, lipid metabolism disorder, hyperlipidaemia and hypo-high-density lipoprotein cholesterolaemia.
Table 5. Univariate analysis of adverse drug reactions by patient characteristics.
A multivariate analysis was performed for these factors (). For CDC classification, the incidence of ADRs for patient in Category B was 64%, which indicated the impact on the occurrence of ADRs compared to category A (odds ratio, 2.59; 95% CI, 1.12–6.00), whereas there was no significant difference between category C and A (odds ratio, 0.67; 95% CI, 0.39–1.17). Since there were limited number of patients in category B (33 patients), these results need to be interpreted with caution. The incidence of ADRs in patients with complications related to hyperlipidemia was 59%, which was significantly higher than in patients without these complications (odds ratio, 2.08; 95% CI, 1.12–3.87). Results stratified by combined backbone NRTI indicated there was a lower incidence in patients treated with emtricitabine/tenofovir disoproxil fumarate (37%) than in those treated with lamivudine/abacavir sulfate (50%) (odds ratio, 0.59; 95% CI, 0.36–0.97).
Table 6. Multivariate analysis of adverse drug reactions by patient characteristics.
Since fosamprenavir can be used with ritonavir or without ritonavir at a higher dosage, we further explored difference in the incidence of ADRs between different doses of fosamprenavir. Data of ADRs by regimens based on 1400 mg or 2800 mg daily dose of fosamprenavir in ART naïve and experienced patients are shown in . The incidences of ADRs were 40.5% (109/269) in fosamprenavir 1400 mg + ritonavir group (44.0% [51/116] and 37.9% [58/153] in ART naïve and experienced patients, respectively) and 51.5% (34/66) in 2800 mg fosamprenavir treatment group (29.4% [5/17] and 59.2% [29/49], respectively). No prominent differences between groups were identified. In the 2800 mg fosamprenavir treatment group, difference in the incidence of ADRs between ART naïve and experienced patients was observed, but this needs to be interpreted with caution due to small number of patients.
Table 7. Incidence of adverse drug reactions in anti-retroviral therapy naïve and experienced patients.
Event of spontaneous bleeding in hemophiliac patients
When spontaneous bleeding was observed during or after fosamprenavir treatment, since this had been reported in hemophiliac patients who had received other PIs, additional investigation into product usage, concomitant medications, causal relationship to the product and presence/absence of abnormal changes in laboratory tests were performed. A total of 24 of 364 patients (6.6%) in the safety analysis set had haemophilia, including 13 patients (54.2%, 13/24 patients) who had 25 ADR events. Of these, only 1 event (in 1 patient) of muscle haemorrhage was observed.
Drug interactions
Amprenavir, the active metabolite of fosamprenavir, is primarily metabolized by cytochrome P450 (CYP) 3A4 and is also an inhibitor of CYP3A4. Therefore, coadministration of fosamprenavir with drugs having a narrow therapeutic window and metabolized by CYP3A4 may result in an increase in AEs of these drugs due to competitive inhibition of their metabolism.Citation6 There were 82 of the 364 patients (22.5%) who were receiving fosamprenavir in combination with drugs already listed in contraindications or precautions for coadministration in the Japanese package insert for fosamprenavir. The incidence of ADRs in these patients was 58.5% (48/82), which was significantly higher than that in patients taking fosamprenavir without coadministration of drugs listed in the contraindications or precautions (39.4% [111/282]) ().
Table 8. Incidence of adverse drug reactions in patients with or without combination with drugs listed in contraindications or precautions for coadministration in the package insert.
Pregnant cases
Only 1 pregnancy occurred during the survey period. In this case, induced abortion was chosen due to unexpected pregnancy, and the patient continued fosamprenavir treatment without any ADRs.
Additionally, 1 pregnancy occurred in 1 patient during fosamprenavir treatment in specified drug use surveillance, which was conducted at the same time as the PMS. This case was also terminated by induced abortion at the ninth week of gestation and did not lead to a delivery. There was no ADR in this case.
Effectiveness analysis in ART naïve patients
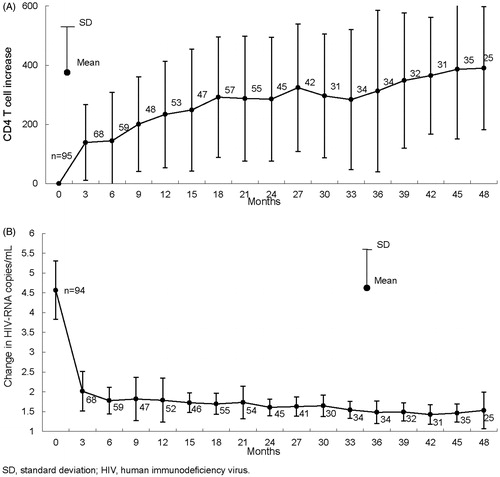
CD4+ cell counts and HIV-RNA copies/mL at pre and post fosamprenavir treatment were available for 137 ART naïve patients. Treatment effectiveness was evaluated in this population. Changes in CD4+ cell counts in 95 patients and HIV-RNA copies/mL in 94 patients are illustrated in . CD4+ cell counts rose after beginning treatment with fosamprenavir. A marked decrease in HIV-RNA copies/mL was observed.
Figure 1. Change in CD4+ cell counts and human immunodeficiency virus -RNA copies/mL after the first treatment of fosamprenavir in anti-retroviral therapy naïve patients. Changes in CD4+ cell counts (A) in 95 patients and in HIV-RNA copies/mL (B) in 94 patients are illustrated. CD4+ cell counts were increased after beginning treatment with fosamprenavir and marked decrease in HIV-RNA copies/mL was observed.
Discussion
In this survey, ADRs were observed in 43.7% (159/364) patients included in the safety analysis set. The common ADRs were all previously characterized and are already included in the precautions of the Japanese package insert. The majority of these ADRs occurred within 180 days after the initiation of fosamprenavir treatment. Since no previously uncharacterized safety signals were observed in this analysis, it provides additional confirmation that fosamprenavir can be used in ART in the HIV-positive Japanese population. No marked differences in the frequency of ADRs were noted between clinical trials conducted overseas and in Japan. No additional clinical concerns were detected from this post-marketing surveillance and the Japanese package insert revised in August 2016 incorporated the results from this analysis. However, this survey has certain limitations that the analyses were performed on the basis of anonymized data collected from 26 Japanese sites participating in this HRD cooperative survey. Moreover, in this survey, unlike in clinical trials, the characterizing of ADRs, their causal relationship, and treatment discontinuation were left to the discretion of physicians, and also, not all of the patients had whole laboratory data before or after the start of treatment, some items of which were obtained from only a few patients.
The evaluation of ADRs when stratified by patient characteristics suggested that several factors may impact ADR incidence, however since this was a relatively small retrospective study, further investigation would be warranted to confirm these observations. Nevertheless, it may be understandable that fosamprenavir, which is an HIV PI, deteriorates hyperlipidemia.Citation7 Likewise, patients with advanced diseases who generally are more vulnerable to ADRs, often receive lamivudine/abacavir sulfate as NRTI backbone to avoid renal impairment due to co-administration of emtricitabine/tenofovir disoproxil fumarate with opportunistic infection therapy.Citation8 The incidence of ADRs in patients receiving fosamprenavir in combination with medications listed in contraindications or precautions section of the fosamprenavir prescribing information was higher than for patients who did not receive such medications, confirming that appropriate use in accordance with the package insert is necessary when co-administering drugs metabolized by CYP3A4 in patients receiving fosamprenavir.
We also explored the difference in occurrence of ADRs between the two dosing options for fosamprenavir tablets since the level of exposure to amprenavir is known to be different between ritonavir-boosted fosamprenavir that is used at a 1400 mg daily dose compared with the non-boosted fosamprenavir which is dosed at 2800 mg daily.Citation5 No apparent difference in overall ADRs was observed, which suggests that non-boosted fosamprenavir could be used in patients for whom ritonavir use is not appropriate.
With regard to spontaneous bleeding in hemophiliac patients only 1 of 24 hemophiliac patients experienced muscle haemorrhage as an ADR. However due to the small sample size no conclusions can be drawn. Therefore healthcare providers should continue to exercise caution when using fosamprenavir for hemophiliac patients living with HIV/AIDS.
ViiV Healthcare K.K. and Glaxo SmithKline K.K. will continue to have strict pharmacovigilance to ensure safety of fosamprenavir tablet use in people living with HIV/AIDS.
Transparency
Declaration of funding
This work was supported by ViiV Healthcare K.K. However, the content is solely the responsibility of the authors and does not necessarily represent the views/opinions of any organization.
Declaration of financial/other relationships
AF, TN, TK, IK and TM are employees of ViiV Healthcare K.K. AK is an employee of GlaxoSmithKline K.K. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgements
We would like to express our gratitude to the physicians who cooperated in the present survey and provided valuable data.
Data availability statement
These data were submitted to the Pharmaceuticals and Medical Devices Agency (PMDA) and are disclosed on the following website: http://www.pmda.go.jp/drugs_reexam/2016/P20160630001/166272000_21600AMZ00652_A100_1.pdf.
References
- Palella FJ, Jr, Delaney KM, Moorman AC, et al. the HIV outpatient study investigators. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N Engl J Med. 1998;338(13):853–860.
- Bartlett JA, Fath MJ, Demasi R, et al. An updated systematic overview of triple combination therapy in antiretroviral-naive HIV-infected adults. Aids. 2006;20(16):2051–2064.
- Research group for therapy of HIV infection [Internet]. Guideline for anti-HIV treatment. Ver.14; 2010. [cited 2017 May 16]. http://www.hivjp.org/guidebook/hiv_14.pdf
- Hogg RS, Yip B, Chan KJ, et al. Rates of disease progression by baseline CD4 cell count and viral load after initiating triple-drug therapy. Jama. 2001;286(20):2568–2577.
- LEXIVA [PACKAGE INSERT]. Tokyo: ViiV Healthcare K.K.; 2016.
- Wire MB, Shelton MJ, Studenberg S. Fosamprenavir: clinical pharmacokinetics and drug interactions of the amprenavir prodrug. Clin Pharmacokinet. 2006;45(2):137–168.
- Dubé MP, Qian D, Edmondson-Melançon H, et al. Prospective, intensive study of metabolic changes associated with 48 weeks of amprenavir-based antiretroviral therapy. Clin Infect Dis. 2002;35(4):475–481.
- Nishijima T, Kawasaki Y, Mutoh Y, et al. Prevalence and factors associated with chronic kidney disease and end-stage renal disease in HIV-1-infected Asian patients in Tokyo. Sci Rep. 2017;7(1):14565.