
Abstract
Objective
The totality-of-evidence approach requires that similarity between a proposed biosimilar and a reference biologic is demonstrated across a range of analytical, preclinical, and clinical parameters to establish biosimilarity. We describe the totality of evidence for Sandoz biosimilar pegfilgrastim (LA-EP2006 [marketed as Ziextenzo]) that supported its regulatory approval in Europe and the United States.
Methods
Analytical similarity to the reference biologic [marketed by Amgen as Neulasta] was first investigated with regard to physiochemical quality attributes such as primary structure, pegylation, higher-order structures, variants and impurities, molecular size variants, and formulation (protein content, pH, excipients, etc.). In vitro biological activity studies were performed to examine the primary mechanism of action of pegfilgrastim. Bioequivalence (clinical pharmacokinetics [PK] and pharmacodynamics [PD]) of Sandoz biosimilar pegfilgrastim to the reference biologic was studied in healthy volunteers; efficacy, safety, and immunogenicity were assessed during confirmatory clinical efficacy studies in patients undergoing treatment for breast cancer.
Results
No meaningful or relevant differences were identified between Sandoz biosimilar pegfilgrastim and the reference biologic during analytical testing. Similar receptor binding and induction of cellular proliferation in vitro confirmed no functional differences between the biologics. Clinical studies in healthy adult participants demonstrated PK/PD biosimilarity and a similar safety profile between biosimilar and reference pegfilgrastim. Clinical studies in a sensitive patient population also demonstrated similar efficacy, safety, and immunogenicity between Sandoz biosimilar pegfilgrastim and the reference biologic.
Conclusions
The totality of evidence confirms that Sandoz biosimilar pegfilgrastim matches the reference biologic and will therefore provide equivalent efficacy and safety in all eligible indications.
Introduction
Biosimilars and the totality-of-evidence approach
A biosimilar is a biologic medicine that is fundamentally the same biologic entity as – and has clinically equivalent efficacy and safety to – an established reference medicineCitation1. The US Food and Drug Administration (FDA) defines a biosimilar as “a biological product that is highly similar to, and has no clinically meaningful differences from, an existing FDA-approved reference product”Citation2.
A biosimilar is developed to be commercialized once the patent of the reference medicine has expiredCitation3. Biologic medicines are produced using highly complex, specialized, and proprietary processes in living cells; this means that it is not possible to produce an identical version of the reference biologic, either in further batches of the reference medicine or in biosimilarsCitation4,Citation5.
The development of biosimilars follows a distinct regulatory pathway that is essentially a stepwise process designed to demonstrate equivalence to the reference medicine, based on the totality of evidence concept. This process establishes similarity to the reference medicine across a range of measures (analytical, preclinical, and clinical) which, if assessed separately in isolation from the others, would not be sufficient to establish biosimilarity. Establishing biosimilarity therefore involves considerable structural and functional characterization, which aims to establish fundamental biosimilarity at the molecular and mechanistic level, in addition to preclinical and clinical assessments via pharmacokinetic (PK), efficacy, and safety studies in patients to confirm clinical equivalenceCitation6. Following this concept, it is only possible to achieve biosimilarity after evaluation of the entire totality of evidence, unlike reference biologic medicines where the approval is mainly driven by pivotal studies.
Once totality of evidence has been demonstrated, the European Medicines Agency (EMA) and the FDA permit the use of a biosimilar to be extended to other eligible indications in which its reference medicine is approved, without conducting a clinical safety and efficacy study in such indications, provided this can be justified medically and scientificallyCitation7,Citation8. This concept is termed extrapolation and is based on the fact that similarity has been demonstrated at multiple levels (molecular, mechanistic, PK, immunogenicity, efficacy, and safety) between the reference and the biosimilar; thus there is no need for repeating the biosimilarity exercise in all other indications as long as the mechanism of action is the same in all the approved indicationsCitation3,Citation9. While the therapeutic response to biologics can vary between patients due to factors such as patient geneticsCitation10,Citation11, established biosimilarity implies that therapeutic response to an approved biosimilar should not vary in a clinically meaningful way from the response to the reference medicine.
The combination of a lower price and a similar efficacy and safety has raised interest in biosimilarsCitation12,Citation13. In the field of oncology, biosimilars are now available as treatment options for both supportive and therapeutic cancer careCitation12. Available data indicate that cost savings from adoption of biosimilars enables expanded access to therapeutic or supportive care treatments for the same overall budgetCitation12. The number of expiring patents for biologics is increasing, which in turn will lead to a significant increase in available biosimilarsCitation12,Citation14. Despite considerable efforts during the recent past, the use of biosimilars in medical practice is still limited due to a poor understanding of the biosimilar terminology, changing regulatory guidance, and a degree of uncertainty in their prescription and applicationCitation15.
Sandoz biosimilar pegfilgrastim
Pegfilgrastim is formed by the covalent conjugation of polyethylene glycol (PEG) to the N-terminal methionine of filgrastim. Filgrastim is a recombinant methionyl human granulocyte colony-stimulating factor (G-CSF)Citation16 that is produced in genetically modified Escherichia coli (E. coli) cells. Its amino acid sequence is identical to that of natural human G-CSF, except for the addition of the N-terminal methionine that is necessary for expression in E. coli. Recombinantly produced G-CSF is not glycosylated, unlike G-CSF isolated from human cells. By binding to and activating hematopoietic cells via the G-CSF receptor, filgrastim promotes the proliferation, differentiation, and activation of neutrophils, and helps manage neutropenia caused by cytotoxic chemotherapy, as well as other types of neutropenia associated with myeloablative therapy or advanced human immunodeficiency virus infection. PEG conjugated to filgrastim decreases the renal clearance and so increases the elimination half-life, resulting in an increased duration of activity. These pharmacological properties offer the advantage of only one dose of pegfilgrastim being required per cycle of chemotherapy as opposed to the multiple daily doses required with filgrastim.
Sandoz biosimilar pegfilgrastim (LA-EP2006Footnotei), as with the reference biologicFootnoteii, consists of a filgrastim protein moiety with 175 amino acids and a single covalently-linked 20-KDa PEG at the N-terminusCitation17. Its protein primary structure, protein-PEG linker, conjugation site, occupancy, and PEG moiety are all indistinguishable from the reference pegfilgrastim. Sandoz biosimilar pegfilgrastim was developed for the same indication, dosage, and administration route as the reference pegfilgrastim. It was approved in the EU in 2018 for reduction in the duration of neutropenia and the incidence of febrile neutropenia in adult patients treated with cytotoxic chemotherapy for malignancy (with the exception of chronic myeloid leukemia and myelodysplastic syndrome)Citation17. Approval was granted in the US in 2019Citation18 and was followed by approval in several other highly regulated markets; submissions for approval in other territories globally are ongoing.
This article reviews the step-by-step development of Sandoz biosimilar pegfilgrastim that generated the totality of evidence and resulted in its approval as a biosimilar version of the reference biologic.
Biosimilar development – building a scientific bridge
Whereas small-molecule generics are identical to their reference medicines, biosimilars can only be similar to their reference biologicsCitation19, albeit with no clinically meaningful differences in terms of quality attributes, efficacy, safety, and immunogenicityCitation6,Citation20. The fact that biosimilar and reference biologics cannot be identical is due, at least in part, to the usage of different cell lines and purification processesCitation20. Guidelines of the FDA and the EMA list particular considerations for the non-clinical and clinical development of a biosimilarCitation15. In both cases, a step-by-step totality-of-evidence approach is proposed. This starts with identification and assessment of critical quality attributes of the biosimilar and reference molecules, followed by analytical characterization to confirm that any differences between them have a low risk of resulting in functional consequences. After this, further testing in preclinical and clinical studies (pharmacology, efficacy, safety, and immunogenicity) is intended to address any residual uncertaintiesCitation21. Unlike clinical studies for reference medicines, the purpose of clinical evaluation of a biosimilar is to demonstrate that the proposed biosimilar is neither less nor more effective than the originator; furthermore, the two medicines also need to be similar in terms of immunogenicity and safetyCitation22. Importantly, clinical assessments of efficacy, safety, and immunogenicity should be conducted in a population that is sensitive enough to detect any meaningful differences (if these exist) between the biosimilar and reference medicineCitation23.
It is important to note that to establish a scientific bridge in legal terms, a reference medicine is considered as a specific entity approved by a local jurisdictionCitation22. Consequently, a reference medicine approved by the EU is classified as a foreign product in the US and vice versaCitation24. Due to this condition, similarity in terms of physiochemical/analytical properties and clinical pharmacology (PK and pharmacodynamics [PD]) must be demonstrated between a proposed biosimilar and the reference biologic sourced in the US and in the EUCitation7,Citation8. Furthermore, similarity in these properties must be verified between the US- and EU-reference medicines. This approach provides a scientific bridge between both biologics, which is necessary for development and authorization of a biosimilarCitation7,Citation8 for both markets while preventing redundant work packages. Based on this scientific rationale, national regulators accept the use of a foreign-sourced comparator in subsequent comparative clinical studiesCitation7,Citation8. For Sandoz biosimilar pegfilgrastim, the scientific bridge was established by demonstrating its similarity to the pegfilgrastim reference biologics from the US and EU in terms of PK, PD, safety, and immunogenicityCitation25. Furthermore, similarity was proven between the pegfilgrastim reference medicines sourced in the US and EUCitation25.
Unlike in Europe or the US, usage of a foreign-approved reference medicine is suitable in clinical evaluations for the Japanese authorities as long as Japanese patients participate in either a comparative PK or comparative clinical efficacy studyCitation26.
Analytical characterization and criticality/risk assessment of quality attributes
Analytical characterization (both physiochemical and biological) and similarity are the foundation of the stepwise process used to develop a biosimilar. The structure, physiochemical properties, and biological functionalities of Sandoz biosimilar pegfilgrastim were characterized using an array of highly sensitive orthogonal analytical methods () in line with regulatory authorities’ current recommendations about demonstration of analytical similarity.
Table 1. Orthogonal analytical methods used to characterize Sandoz biosimilar pegfilgrastim and for comparisons with the reference biologic.
A criticality assessment of quality attributes was carried out during the development by ranking their impacts on the PK/PD, efficacy, safety, and immunogenicity, based on knowledge from published literature, functional characterization, and clinical experience. Quality attributes with the highest impact on potential clinical outcome are defined as critical quality attributes, which must be kept within appropriate limits to ensure the desired biosimilar quality. Quality attributes which are involved in the mechanism of action received the most attention during analytical and process development. This ensured a risk-based biosimilar development by focusing on selected quality attributes.
Similarity in structural and functional attributes
As mentioned above, the most critical quality attributes potentially influencing PK, PD, immunogenicity, safety, and efficacy were identified. Based on this, a comprehensive analytical structural and functional characterization of Sandoz biosimilar pegfilgrastim and the reference biologics from the EU and US was carried out using a set of orthogonal methods as listed in Citation17.
Identical primary structure, with identical protein-PEG linker structure and pegylation site, was confirmed between Sandoz biosimilar pegfilgrastim and the reference biologics, in addition to indistinguishable higher-order structure. Sandoz biosimilar pegfilgrastim also showed similar variant and impurity (with minor differences) profiles, with no meaningful differences from the reference medicines. The purity of Sandoz biosimilar pegfilgrastim is either within or higher than the purity range of reference pegfilgrastim, including at the end of shelf life. Furthermore, similar biological activities were demonstrated between Sandoz pegfilgrastim and EU- and US-reference pegfilgrastim, including potency (using an in vitro NFS-60 cell proliferation assay) and binding affinity to the G-CSF receptor. Typical comparison of some quantitative quality attributes of Sandoz biosimilar pegfilgrastim with the quality ranges of the reference biologic is given in .
Figure 1. Comparison of some quantitative quality attributes of Sandoz biosimilar pegfilgrastim with the quality ranges of EU-licensed and US-licensed reference pegfilgrastim. Potency assessed by NFS-60 cell proliferation; content by ultraviolet spectroscopy; aggregates (high molecular weight variants [HMWV]) by size-exclusion chromatography (SEC), and other variants/impurities such as acidic variants in cation-exchange chromatography (CEX), di-pegylated filgrastim, deamidation, etc. in reverse-phase chromatography as post-peaks.
![Figure 1. Comparison of some quantitative quality attributes of Sandoz biosimilar pegfilgrastim with the quality ranges of EU-licensed and US-licensed reference pegfilgrastim. Potency assessed by NFS-60 cell proliferation; content by ultraviolet spectroscopy; aggregates (high molecular weight variants [HMWV]) by size-exclusion chromatography (SEC), and other variants/impurities such as acidic variants in cation-exchange chromatography (CEX), di-pegylated filgrastim, deamidation, etc. in reverse-phase chromatography as post-peaks.](/cms/asset/c7026670-aa04-4755-829f-917f5f8c6e2d/icmo_a_2061707_f0001_c.jpg)
To provide a solid base for the analytical similarity assessment and an analytical bridge between the biosimilar medications used in different development phases and in the various (non-)clinical studies, in accordance with ICH Q5ECitation27, several head-to-head process comparability studies were also performed. The studies also included routine release and in-process control (IPC) tests; additional physiochemical characterization and biological activity methods; and stability studies under intended storage conditions as well as accelerated, stressed, and forced degradation conditions. In various stability studies of the final drug product, the stability profiles of Sandoz biosimilar pegfilgrastim and reference biologic were shown to be indistinguishable.
Furthermore, the analytical similarity assessment was concluded with a more comprehensive statistical evaluation of quantitative quality attributes based on broader batch ranges and data for Sandoz biosimilar and reference biologics acquired throughout the development period. The final analytical similarity assessment included a risk-based, tiered statistical evaluation of similarity on critical quality attributes based on comparison with quality ranges of EU, US, and combined EU/US-reference pegfilgrastim global ranges. The results further support similarity between Sandoz biosimilar pegfilgrastim and reference pegfilgrastim, removing any residual uncertainty. Head-to-head studies of EU/US-reference pegfilgrastim with reference pegfilgrastim sourced from other highly regulated markets also support the conclusion that Sandoz biosimilar pegfilgrastim matches the reference pegfilgrastim globally.
Overall, the broad range of quality attributes investigated in the physiochemical and in vitro biological characterization and similarity studies confirmed that Sandoz biosimilar pegfilgrastim and the reference biologics are highly similar in terms of structural and functional attributesCitation17,Citation18. This forms a foundation for the totality of evidence for Sandoz biosimilar pegfilgrastim compared to US- and EU-reference pegfilgrastim.
Preclinical in vivo studies
According to current regulatory guidelines for the development of biosimilars, comparative animal studies are only necessary when there is remaining uncertainty concerning the comparative in vivo biological effects after the analytical and in vitro comparisonsCitation7. For Sandoz biosimilar pegfilgrastim, both comparative G-CSF receptor-binding and G-CSF receptor-expressing NFS-60 cell proliferation assays demonstrated similar bioactivity. Therefore, in the context of overall analytical similarity, there was no residual uncertainty to be addressed by animal studies.
However, at the time of early development of Sandoz biosimilar pegfilgrastim, the regulatory pathway for pegfilgrastim biosimilars was not fully defined. Therefore, some comparative animal studies were required by regulatory agencies for the demonstration of biosimilarity. Consequently, several animal studies were performed to assess PK/PD, general toxicity, and toxicokinetics of subcutaneously administered Sandoz biosimilar pegfilgrastim in comparison with EU-reference pegfilgrastim.
PK/PD parameters were assessed in sensitive settings in naïve, non-rodent and rodent species, as well as neutropenic settings resulting from myelosuppressive chemotherapy, which more closely resemble the clinical use setting. No significant differences in the neutrophil responses were found in either normal or neutropenic circumstances.
A comparative toxicity study including immunogenicity and local tolerance assessment was performed in rats using the intended subcutaneous route of administration. Both Sandoz biosimilar pegfilgrastim and EU-reference pegfilgrastim were well tolerated, and treatment-related changes were limited to the adverse effects expected after G-CSF administration. The type and severity of the observed changes were considered similar for both Sandoz biosimilar pegfilgrastim and reference pegfilgrastim, as was the local subcutaneous tolerability.
In summary, although current biosimilar development guidelines require animal studies only if the comparability exercise identifies issues that would block direct entrance into human clinical studies, an extensive program of animal studies for Sandoz biosimilar pegfilgrastim confirmed a similar biological response in terms of efficacy and safety to the reference pegfilgrastim.
Clinical studies
Clinical pharmacology
The PK and PD of Sandoz biosimilar pegfilgrastim were investigated in three studies in healthy male and female participants (). A further PK sub-study provided supportive PK data in patients with breast cancer. Absolute neutrophil count (ANC) was a surrogate efficacy endpoint in these trials.
Table 2. Overview of Sandoz biosimilar pegfilgrastim phase I PK/PD and phase III confirmatory clinical studies.
The primary PK endpoints of these studies included pegfilgrastim serum concentration, evaluated by area under the serum concentration–time curve (AUC) measured from time of dosing and extrapolated to infinity (AUC0–inf) or to the last measurable concentration (AUC0-last), and maximum observed serum concentration (Cmax). The primary PD endpoints were ANC area under the effect curve measured from time of dosing to last measurable concentration (AUEC0–last) and maximum effect (Emax). Secondary endpoints included safety and immunogenicity of the analyzed biologicsCitation17,Citation25,Citation28.
Study LA-EP06-101 showed similarity with respect to the PD endpoints AUEC0–last and EmaxCitation17, but was found to be under-powered for demonstration of PK biosimilarity as a result of high inter-subject variability for pegfilgrastim PK, a finding that is consistent with data from an earlier publicationCitation29. Since LA-EP06-101 was conducted in a parallel design, the high inter-subject variability for pegfilgrastim PK had a high impact on the resultsCitation17. Therefore, this issue was addressed in subsequent studies by using a crossover design in which each participant served as their own control to prevent inter-individual variabilityCitation25,Citation28.
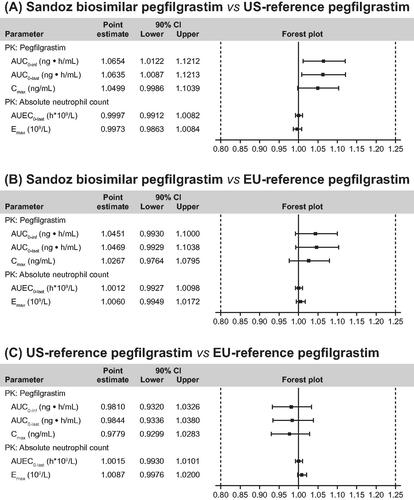
Study LA-EP06-103 was conducted using a two-arm crossover design and other measures to further reduce sample heterogeneityCitation28. This study demonstrated PK/PD biosimilarity to 6 mg of the EU-reference pegfilgrastim. The key study to demonstrate clinical PK/PD biosimilarity between Sandoz biosimilar pegfilgrastim and EU- and US-reference biologics was a phase I bridging study (LA-EP06-104) using a three-way crossover six-sequence designCitation25. The study was powered at 90% to achieve confidence intervals (CIs) within the predefined biosimilarity margins 0.8–1.25 in pairwise comparisons (biosimilar vs US reference, biosimilar vs EU reference, and US reference vs EU reference). shows the data from this study for mean pegfilgrastim serum concentration and ANC profile. For each pairwise comparison, although some point estimates were slightly more than 1.00, the 90% CIs for the geometric mean ratios for secondary PK/PD parameters were all contained within the predefined similarity margins (). This difference did not translate to any clinically meaningful differences in safety or efficacy of the biosimilar. The safety profile (Supplementary Table 1) and incidence of anti-drug antibodies were also comparable between Sandoz biosimilar pegfilgrastim and the reference medicines.
Figure 2. (A) Mean pegfilgrastim serum concentration and (B) absolute neutrophil count (ANC) profiles following a fixed single subcutaneous injection of 6 mg of the Sandoz pegfilgrastim biosimilar or the pegfilgrastim reference biologics in healthy volunteers (study LA-EP06-104). Reproduced from Bellon et al.Citation25 under the CC-BY-NC license; https://creativecommons.org/licenses/by-nc/4.0/legalcode. Abbreviation. SD, standard deviation.
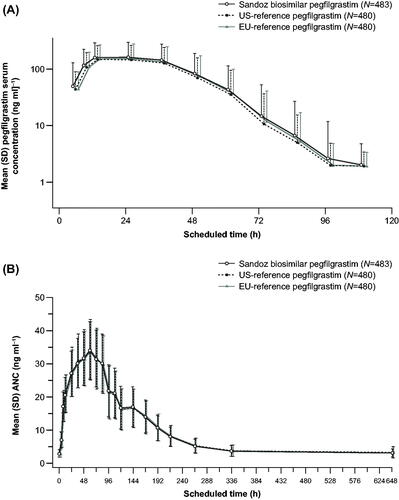
Figure 3. Similarity for primary PK and PD parameters between Sandoz biosimilar pegfilgrastim, US-licensed pegfilgrastim, and EU-licensed pegfilgrastim (study LA-EP06-104). (Reproduced from Bellon et al.Citation25, with minor modifications to layout, under the CC-BY-NC license; https://creativecommons.org/licenses/by-nc/4.0/legalcode.) For each pairwise comparison, the 90% CIs for the geometric mean ratios for secondary PK/PD parameters were all contained within the predefined similarity margins. Due to an intrinsic variability that is inherent in all biological medicines as well as the complex manufacturing of these products, it is not possible to produce identical products. Minor differences can be found in different batches as in products produced in different countriesCitation4,Citation5. Most important, these differences must be within predefined margins without being clinically meaningfulCitation7,Citation8. Abbreviations. AUC, area under the serum concentration–time curve; AUC0–inf, AUC measured from time of dosing and extrapolated to infinity; AUC0–last, AUC measured from time of dosing to last measurable concentration; AUEC0–last, area under the effect curve measured from time of dosing to last measurable concentration; CI, confidence interval; Cmax, maximum observed serum concentration; Emax, maximum effect attributable to the investigational medicinal product; PD, pharmacodynamic; PK, pharmacokinetic.
A recent meta-analysisCitation30 evaluated the PK and PD of Sandoz biosimilar pegfilgrastim versus US- and EU-reference pegfilgrastim using data from three phase I studies: LA-EP06-101Citation17; LA-EP06-103Citation28; and a six-sequence, three-way, crossover studyCitation25. For each treatment comparison, the 90% CIs for the geometric mean ratios for PK/PD parameters were within the equivalence margins of 0.80–1.25 (Supplementary Figure 1), supporting PK/PD biosimilarity of Sandoz biosimilar pegfilgrastim to US- and EU-reference pegfilgrastimCitation30.
Together, these studies demonstrate PK and PD biosimilarity between Sandoz biosimilar pegfilgrastim and both EU- and US-reference biologics, thereby establishing the PK and PD similarity component of the totality of evidence.
Phase III confirmatory clinical studies
The PROTECT-1 and PROTECT-2 studies () were designed to hierarchically assess equivalence and then non-inferiority of Sandoz biosimilar pegfilgrastim and EU-reference pegfilgrastim, in terms of the duration of severe neutropenia (DSN) and the safety and immunogenicity of both medicines, in female patients with breast cancer receiving established myelosuppressive chemotherapyCitation31,Citation32. The studies were essentially of identical design; double-blind, randomized, parallel group, multicenter clinical trialsCitation31,Citation32. Both were powered at 90% to evaluate the primary efficacy parameter, the mean DSN during cycle 1 of chemotherapy with docetaxel, doxorubicin, and cyclophosphamide (TAC). The DSN was calculated as the number of consecutive days from the first day when a patient’s ANC was <0.5 × 109/L until the patient reached an ANC ≥0.5 × 109/L in cycle 1. Women with breast cancer receiving TAC chemotherapy are known to represent a sensitive population in which differences between a proposed biosimilar and reference G-CSF can be identifiedCitation33.
Baseline characteristics of patients were balanced across the different groupsCitation33. After a screening period of up to 21 days, patients were randomized 1:1 to receive either Sandoz biosimilar pegfilgrastim or the reference medicine. All participants received pegfilgrastim (biosimilar or reference biologic) at a dose of 6 mg subcutaneously, at least 24 h after the end of chemotherapyCitation32,Citation33.
A pooled analysis of data from PROTECT-1 and PROTECT-2 was conductedCitation33. The primary endpoint in both studies, mean DSN during the first chemotherapy cycle, was comparable in the Sandoz biosimilar pegfilgrastim and reference medicine (1.056 ± 1.055 vs 1.016 ± 0.958 days) groups. Since the difference between both groups was only 0.04 days [95% CI: −0.19 to 0.11], the equivalence criteria were met (the defined margin was ±1 day)Citation33. The nadir of ANC and the incidence of febrile neutropenia (both secondary endpoints) were also equivalent between the biosimilar and reference medicine (). Finally, the safety profile of Sandoz biosimilar pegfilgrastim and that of the reference medicine showed no clinically meaningful differences (). No treatment-related binding and neutralizing antibodies were detected during PROTECT-1 or PROTECT-2, including in the 6-month safety follow-up of PROTECT-1 (). This supports that Sandoz biosimilar pegfilgrastim has no increased immunogenic potential compared with reference pegfilgrastim.
Table 3. Primary and secondary efficacy parameters with Sandoz biosimilar pegfilgrastim and reference pegfilgrastim in patients with breast cancer – pooled data from LA-EP06-301 and LA-EP06-302.
Table 4. Clinical safety of Sandoz biosimilar pegfilgrastim and reference biologic in patients with breast cancer.
Table 5. Number of patients with confirmed positive ADA results – studies LA-EP06-103, LA-EP06-104, LA-EP06-301, and LA-EP06-302 (safety analysis set).
These results confirm, as a part of the totality-of-evidence approach, that Sandoz biosimilar pegfilgrastim matches the reference medicine in terms of efficacy, safety, and immunogenicityCitation33.
Summary
The totality of evidence across its development program confirms the comprehensive biosimilarity between Sandoz biosimilar pegfilgrastim and reference pegfilgrastim. The analytical and functional similarity of Sandoz biosimilar pegfilgrastim and reference pegfilgrastim have been demonstrated through comparison of physicochemical quality attributes, as well as binding assays and cell-based in vitro assays that assess the functionality of the molecule. Data from extensive preclinical pharmacology and toxicity studies provided further support for the biosimilarity of Sandoz biosimilar pegfilgrastim and the reference biologic.
Extensive clinical data further contributed to the assessment of biosimilarity between Sandoz biosimilar pegfilgrastim and reference pegfilgrastim. Phase I studies have demonstrated biosimilarity of Sandoz biosimilar pegfilgrastim to the EU- and US-reference biologics from a PK and PD perspectiveCitation25,Citation28; a meta-analysis of the three PK/PD studies provided further confirmationCitation30. Similar efficacy, safety, and immunogenicity for Sandoz biosimilar pegfilgrastim and EU-reference biologic were also demonstrated by two confirmatory studies in patients with cancer after up to 6 months of follow-upCitation31,Citation32. The availability of clinical efficacy and safety data for Sandoz biosimilar pegfilgrastim may be helpful to physicians, who are used to looking at such data when making treatment decisionsCitation34.
In conclusion, the biosimilarity of Sandoz biosimilar pegfilgrastim to the reference biologic has been demonstrated from an analytical, functional, preclinical, and clinical PK/PD perspective. In addition, phase III confirmatory studies conducted in patients with breast cancer under chemotherapy showed that Sandoz biosimilar pegfilgrastim matched the reference pegfilgrastim in terms of efficacy, safety, and immunogenicity. Clinicians can be confident that Sandoz biosimilar pegfilgrastim may be used safely and effectively in approved indications and will provide equivalent efficacy and safety as the reference medicine at a favorable price.
Transparency
Declaration of funding
The conducted studies, which are the basis of this manuscript, were funded by Sandoz.
Declaration of financial/other relationships
Ulrich Nagl, Xinghua Guo, Jens Heyn, Yu-Ming Shen, Gregor Schaffar, Martin Humphrey, Natalia Koptelova, and Sreekanth Gattu are employees of Sandoz. Anne Bellon, Miryana Dimova-Dobreva, and Nicola Mathieson are former employees of Sandoz. Sanjiv S. Agarwala has no relationships to declare. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contributions
Conception and planning of the work that led to the manuscript, analysis and interpretation of the data, critical revision of the manuscript for important intellectual content, and approval of the final submitted version of the manuscript were done by all authors.
Supp_Fig_1.eps
Download EPS Image (2 MB)Totality_of_evidence_review_ms_Suppl_17Nov21.docx
Download MS Word (142.1 KB)Acknowledgements
Medical writing support was provided by Caroline McGown and Tony Reardon of Aura, a division of Spirit Medical Communications Ltd, and funded by Sandoz.
Notes
i Marketed as Ziextenzo.
ii Marketed by Amgen as Neulasta.
References
- Bui LA, Hurst S, Finch GL, et al. Key considerations in the preclinical development of biosimilars. Drug Discov Today. 2015;20(Suppl 1):3–15.
- US Food and Drug Administration. Biosimilar and interchangeable products. 2020. [cited 2021 Aug 16]. Available from: https://www.fda.gov/drugs/biosimilars/biosimilar-and-interchangeable-products.
- Weise M, Kurki P, Wolff-Holz E, et al. Biosimilars: the science of extrapolation. Blood. 2014;124(22):3191–3196.
- Schiestl M, Stangler T, Torella C, et al. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29(4):310–312.
- Planinc A, Dejaegher B, Vander Heyden Y, et al. Batch-to-batch N-glycosylation study of infliximab, trastuzumab and bevacizumab, and stability study of bevacizumab. Eur J Hosp Pharm. 2017;24(5):286–292.
- Windisch J. Biosimilars versus originators: similarities and differences from development to approval. Int J Clin Rheumatol. 2015;10(6):501–510.
- European Medicines Agency Committee for Medicinal Products for Human Use. Guideline on similar biological medicinal products containing monoclonal antibodies—non-clinical and clinical issues. 2014. [cited 2021 Aug 16]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-2.pdf.
- US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry. 2015. [cited 2021 Aug 16]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/scientific-considerations-demonstrating-biosimilarity-reference-product.
- McCamish M, Pakulski J, Sattler C, et al. Toward interchangeable biologics. Clin Pharmacol Ther. 2015;97(3):215–217.
- Murdaca G, Gulli R, Spano F, et al. TNF-α gene polymorphisms: association with disease susceptibility and response to anti-TNF-α treatment in psoriatic arthritis. J Invest Dermatol. 2014;134(10):2503–2509.
- Murdaca G, Spano F, Contatore M, et al. Pharmacogenetics of etanercept: role of TNF-α gene polymorphisms in improving its efficacy. Expert Opin Drug Metab Toxicol. 2014;10(12):1703–1710.
- McBride A, Balu S, Campbell K, et al. Expanded access to cancer treatments from conversion to neutropenia prophylaxis with biosimilar filgrastim-sndz. Future Oncol. 2017;13(25):2285–2295.
- Yoo DH, Choe JY, Shim SC, et al. Switching to biosimilars in the treatment of rheumatic diseases. Expert Rev Clin Immunol. 2018;14(7):557–571.
- Busse A, Lüftner D. What does the pipeline promise about upcoming biosimilar antibodies in oncology? Breast Care. 2019;14(1):10–16.
- Kolberg HC, Colleoni M, Santi P, et al. Totality of scientific evidence in the development of ABP 980, a biosimilar to trastuzumab. Target Oncol. 2019;14(6):647–656.
- Molineux G. The design and development of pegfilgrastim (PEG-rmetHuG-CSF, Neulasta)®). Curr Pharm Des. 2004;10(11):1235–1244.
- European Medicines Agency Committee for Medicinal Products for Human Use. Assessment report – Ziextenzo. 2018. [cited 2021 Aug 16]. Available from: https://www.ema.europa.eu/en/documents/assessment-report/ziextenzo-epar-public-assessment-report_en.pdf.
- US Food and Drug Administartion. Clinical pharmacology review(s) – Ziextenzo. 2019. [cited 2021 Aug 16]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761045Orig1s000ClinPharmR.pdf.
- Kabir ER, Moreino SS, Sharif Siam MK. The breakthrough of biosimilars: a twist in the narrative of biological therapy. Biomolecules. 2019;9(9):410.
- Declerck P, Danesi R, Petersel D, et al. The language of biosimilars: clarification, definitions, and regulatory aspects. Drugs. 2017;77(6):671–677.
- Markus R, Liu J, Ramchandani M, et al. Developing the totality of evidence for biosimilars: regulatory considerations and building confidence for the healthcare community. BioDrugs. 2017;31(3):175–187.
- Thomas M, Thatcher N, Goldschmidt J, et al. Totality of evidence in the development of ABP 215, an approved bevacizumab biosimilar. Immunotherapy. 2019;11(15):1337–1351.
- Goel N, Chance K. Biosimilars in rheumatology: understanding the rigor of their development. Rheumatology. 2017;56(2):187–197.
- Strand V, Cronstein B. Biosimilars: how similar? Intern Med J. 2014;44(3):218–223.
- Bellon A, Wang J, Skerjanec A, et al. A large multicentre, randomized, double-blind, cross-over study in healthy volunteers to compare pharmacokinetics, pharmacodynamics and safety of a pegfilgrastim biosimilar with its US- and EU-reference biologics. Br J Clin Pharmacol. 2020;86(6):1139–1149.
- Kuribayashi R, Sawanobori K. Current Japanese regulatory systems for generics and biosimilars. J Pharm Sci. 2018;107(3):785–787.
- European Medicines Agency. ICH Topic Q 5 E: comparability of biotechnological/biological products - step 5. 2005. [cited 2021 Aug 16]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-5-e-comparability-biotechnological/biological-products-step-5_en.pdf.
- Nakov R, Gattu S, Wang J, et al. Proposed biosimilar pegfilgrastim shows similarity in pharmacokinetics and pharmacodynamics to reference pegfilgrastim in healthy subjects. Br J Clin Pharmacol. 2018;84(12):2790–2801.
- Yang BB, Morrow PK, Wu X, et al. Comparison of pharmacokinetics and safety of pegfilgrastim administered by two delivery methods: on-body injector and manual injection with a prefilled syringe. Cancer Chemother Pharmacol. 2015;75(6):1199–1206.
- Gattu S, Wang J, Bellon A, et al. Meta-analysis of pharmacokinetic/pharmacodynamic results of 3 phase 1 studies with biosimilar pegfilgrastim. Clin Pharmacol Drug Dev. 2021;10(10):1130–1141.
- Blackwell K, Donskih R, Jones CM, et al. A comparison of proposed biosimilar LA-EP2006 and reference pegfilgrastim for the prevention of neutropenia in patients with early-stage breast cancer receiving myelosuppressive adjuvant or neoadjuvant chemotherapy: pegfilgrastim randomized oncology (supportive care) trial to evaluate comparative treatment (PROTECT-2), a phase III, randomized, double-blind trial. Oncologist. 2016;21(7):789–794.
- Harbeck N, Lipatov O, Frolova M, et al. Randomized, double-blind study comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Future Oncol. 2016;12(11):1359–1367.
- Blackwell K, Gascon P, Jones CM, et al. Pooled analysis of two randomized, double-blind trials comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Ann Oncol. 2017;28(9):2272–2277.
- Markus R, McBride HJ, Ramchandani M, et al. A review of the totality of evidence supporting the development of the first adalimumab biosimilar ABP 501. Adv Ther. 2019;36(8):1833–1850.