
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Objective
Regulatory use of real-world evidence (RWE) has been recognized as a useful supplement to clinical trial evidence and could benefit patients by reducing time to treatment. However, commercial benefits have not been documented. The aim was to determine commercial impact of regulatory RWE, using ambrisentan as an illustrative example.
Methods
A Markovian-like transition model was constructed to simulate the drug development workflow across a simulation time of t = 20 years. RWE was assumed to be incorporated at pII–pIII and pII–pIII–pIV, and its multiplicative median transition rate was determined by biopharma expert opinion. Each model was subjected to “with” and “without” RWE rates. Commercial impact was estimated using potential decrease in time to launch. Time to first medicine adoption and potential lives saved were also estimated.
Results
Based on cumulative first prescriptions for ambrisentan among pulmonary arterial hypertension patients (N = 487), in comparison to standard drug development, RWE incorporation has the potential to expedite first medicine adoption by 10.4 weeks. The duration of market launch was estimated at 2.5–3.0 years earlier than standard, and approximately 9% of patients would benefit in survival. Potential earnings for an earlier launch would be GBP £43,597.86 per patient, with launch being brought forward from 2009 to 2007.
Conclusions
Regulatory RWE has the potential to increase overall survival rates and potential earnings by reducing time to launch. This study provides further support for industry efforts to generate RWE in time for regulatory approval.
Introduction
Real world evidence (RWE) has been defined in various ways by pharmaceutical industry stakeholders, although it is generally agreed to be scientific knowledge produced from the analysis of real-world observational data (RWD)Citation1–4. Stakeholders worldwide have been in the process of developing quality standards and guidelines regarding the generation and use of RWE for medicines development since the landmark Twenty-first Century Cures Act was legislated in the United States in 2016Citation1–5. PayorsCitation6 and regulatorsCitation7 have identified the use of RWD as a strategic priority to improve innovation and access to new treatments, although they have distinct quality criteria for RWE studiesCitation8. While the use of RWE for payor health technology assessment and evidence-based medicine for prescribers is widely accepted, regulatory use is still relatively uncertain and debatedCitation9,Citation10.
Limitations of RWE studies, such as the high number of potential confounders, have restricted regulatory RWE to post-approval use, although the landscape regarding regulatory RWE is evolvingCitation5. RWE has been recognized as a potential solution due to its ability to supplement randomized controlled trial evidence by including routinely excluded subgroups and providing additional evidence that cannot be generated by interventional clinical trials, for example that generated during routine care within the bounds of current medical and diagnostic practice. In addition, real-world data have led to the development of novel analytical tools, approaches, and techniques to address specific limitations in observational researchCitation11. This is evident in the use of medical records and patient registries used to support decision-making on the effectiveness of orphan drugs in rare diseases ()Citation5,Citation12. Regulatory RWE may be especially helpful in rare disease due to small sample sizes and the number of subgroups routinely excluded from randomized controlled trials (RCTs). However, its use is not yet widely accepted. Regulatory bodies have suggested a number of quality criteria necessary for RWE to be suitable for regulatory use; however, firm guidelines have yet to be establishedCitation1–3,Citation13.
Table 1. Regulatory use of RWE.
Endeavors by regulatory authorities to advance the use of RWE in regulatory decision-making have the potential to improve clinical outcomes and overall survival by reducing time to treatment. The inclusion of RWE at the regulatory stage may decrease the time to maximal medicine adoption by 22%Citation14. Avoiding treatment delay in rare disease may avoid irreversible clinical worsening, which may include hospitalization, disease progression and death for some patientsCitation15. Reduced time to effective treatment may reduce the substantial direct and indirect costs associated with hospitalization and healthcare utilization in some diseasesCitation16; however economic burdens related to unemployment and treatment costs may still impact patients, even in regions with health insurance coverageCitation17. It may be reasonable to assume that reduced time to treatment may improve clinical outcomes such that more patients may remain in work and able to contribute to the economy, although these costs would be difficult to estimate. Including RWE in regulatory decision-making may increase the depth of maximal medicine adoption by 31%Citation14. A possible explanation for this is that regulatory RWE may increase the breadth of populations considered for indication compared with RCT evidence alone, which may lead to a broader indication population at regulatory approvalCitation18. Competitive advantages of avoiding delay during drug development have also been recognized in an industry contextCitation19,Citation20.
In recent years, in addition to regulatory interest in the use of RWE, there has been growing interest from payors regarding the prospective benefits and relevance of RWE in HTA assessments, during the drug development pipeline and the associated marketing and reimbursement stagesCitation21,Citation22. Furthermore, real-world clinical information and data usually not collected in a trial setting may be amassed and analyzed allowing significant undetected patterns and trends to be observed; notably, patient population size and representativeness, the duration of follow-up data, comparisons of outcomes from multiple sites across the world over time, and assessments of the impact of risk-mitigation measures of medicinesCitation23,Citation24. Though the incorporation of RWE has been shown to be restricted predominantly to observational studies at clinical trial phase IV, current digital records on health, disease-onset and longevity may encourage the application of RWE at phase II–III–IV (pII–IV)Citation25, although specific benefits of inclusion at this stage are not well documented.
The aim of this study was to develop a simulation model to assess the potential time saving offered by the addition of RWE at phases pII to pIV by estimating time to early market access and time to earliest medicine adoption, using this to estimate potential number of lives saved and potential increased earnings.
Methods
Case selection
Ambrisentan is a non-sulfonamide, propanoic acid-class, endothelin receptor antagonist that is selective for the endothelin type-A (ET(A)) receptorCitation26 indicated for pulmonary arterial hypertension (PAH). PAH is considered a secondary disease resulting from persistent life-threatening diseases, such as cardiac complications. In comparison to other rare adult-onset genetic diseases, such as acromegaly, PAH has a high diagnosis detection rate, inclusive of patients presenting with mild symptoms. Ambrisentan has been granted orphan drug designation for the treatment of PAH in the United States and European Union. Ambrisentan was chosen as an appropriate case due to the author’s prior experience of bringing this drug to market and knowledge of its background with regards to the relevance of RWE to its medicine adoption.
Model framework
Model selection and assumptions
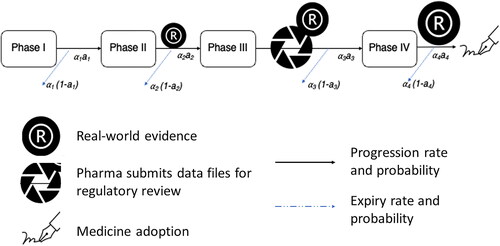
To determine cost savings as a result of earlier launch due to the addition of RWE at various phases of clinical development, it was necessary to construct a model capable of predicting progression rates of treatments through clinical development under the various RWE conditions. The drug development workflow is a state-based evolution of binary gatekeeping events with the possibility of exit or progression at the discrete stages of regulatory approval, payor recommendation and medicine adoptionCitation14. A Markov model was selected due to its greater suitability for discrete states than, for example, time series analysis. As Markov is a stochastic model, an ODE element was introduced to account for the effect of deterministic factors on progression through clinical development. Thus, a Markovian-like transition model was constructed to incorporate the progression rates and probabilities of success at each transition stage aka. clinical phase; Phase I (pI); Phase II (pII); Phase III (pIII) and Phase IV (pIV) under the various RWE conditions.
Support for use of Markovian models to simulate the various transition phases of clinical development can be found in existing medical literature. For example, Abbas et al.Citation27 estimated that the incorporation of RWE during orphan drug development could bring forward market access launch by 3 years. Additionally, multistate models based on Markov processes are a well-established method of estimating rates of transition between stages of diseaseCitation28. A semi-Markov model has been used to estimate patient progression through clinical trials for many patient classes, trial sites and start-timesCitation29. The model presented here is a first step toward a more sophisticated approach to progression rates and probabilities of success at each transition through the various stages of a clinical trial.
Drug development workflow
Potential time saving was determined by first modeling the drug development workflow, including regulatory approval, pricing and reimbursement and medicine adoption. A Markovian-like transition model was constructed to incorporate the progression rates and probabilities of success at each transition stage. Transition stages were defined as the clinical phase, Phase I (pI), Phase II (pII), Phase III (pIII) and Phase IV (pIV). This transition model is commonly known as an ordinary differential equation (ODE) or a Markovian model with a time entity determined by progression rate (). In drug development, each novel drug is subjected to a probability of failure and success at each phase, and upon successful review, the drug progresses to the next trial phase. Due to the non-interrupted and mandatory review process, this Markovian-like model was made feasible with the inclusion of the average time spent at each transition phase, that is, rate, a1,2,3,4.
Figure 1. ODE-Markovian model flow for clinical trial progression in drug development.
Simulation of RWE addition and model inputs
The model underwent a simulation time t = 20 years with the assumption that the transition rates and review stringency probabilities for successes were non-selective to all trials. The probability of success at each clinical phase was as follows: pI to pII: 70%; pII to pIII: 30%; pIII to pIV: 33%; with a success rate of 80% from pIV to first medicine adoption. At the beginning of the simulation cycle, t = 0, the hypothetical scenario was that N(t = 0) = 1000 novel drugs would enter pI trial, and workflow progression rate would be subject to a probability of expiry and success regarding transit to the next clinical phase. A phase-transition rate determined the duration of stay at each clinical phase (Supplementary Table S1). This information was dependent on previous reports by regulatory bodiesCitation30. RWE was assumed to be incorporated at pII–pIII and pII–pIII–pIV, and its multiplicative median transition rate was determined by biopharma expert opinion, as described in the source literature. Each model was subjected to “with” and “without” RWE rates. Model 1: Standard (no RWE); Model 2: RWE at pII–pIII; Model 3: RWE at pII–pIII–pIV to medicine adoption. Refer to Supplementary Materials Table S2, S3 and Figure S1 & S2 for Models 1 & 2.
Sensitivity analysis
A sensitivity analysis was conducted using Morris-Screening ODE with a preset minimum and maximum range for each parameter – for transition rates, probabilities and Morris-screening parameter range refer to Supplementary Table S1. The estimated time differences between “with” and “without” RWE would be the estimated time duration for earlier market access.
Model estimates
Decrease in time to medicine adoption
Time to first medicine adoption was defined as the duration from the ODE transition phase pIV to first prescription. The first clinical prescription at launch was estimated from the pIV to medicine adoption transition with its corresponding “time to crude” number equating to 1.00 (). For time to full medicine adoption, it was assumed that in a modern digital network from first adoption to 100% completion, the cumulative growth rate would meet the minimum requisite of an exponential characteristic,
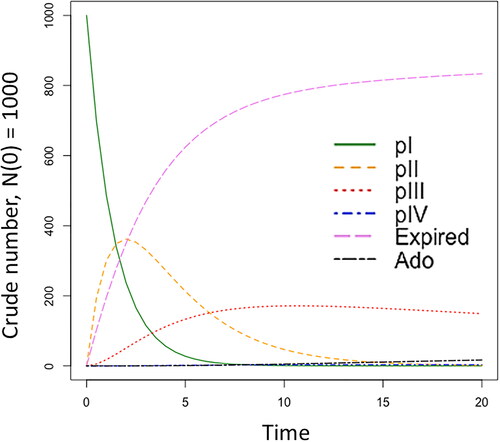
Figure 2. Simulated clinical trial regulatory-review dynamics using the ODE model with RWE. Abbreviations. Ado: medicine adoption; expired: discontinued trials; pI: phase I, pII: phase II, pIII: phase III, pIV: phase IV. Refer to Supplementary Material Table S4 for the numeric output.
Potential lives saved
Earlier launch of market products that benefit lifesaving is likely to result in a lower disease-specific mortality rate. To obtain an estimate for the potential number of lives saved, two empirical rates were derived from reports containing contemporary disease mortality and observational survival rates from PAH patients treated with ambrisentanCitation27,Citation28. In the literature, contemporary three-year mortality rates of PAH patients have been described as m(y1) = 5%, m(y2) = 16%, m(y3) = 21%. The first two-year survival rates of PAH patients treated with ambrisentan were S(y1) = 96% and S(y2) = 89%.
Software and data
All analyses were conducted using R version 4.2.0 and RStudio 2022.02.3 + 492 “Prairie Trillium” Release (1db809b8323ba0a87c148d16eb84efe39a8e7785, 2022-05-16) for macOS Mozilla/5.0 (Macintosh; Intel Mac OS X 12_3_1) AppleWebKit/537.36 (KHTML, like Gecko) QtWebEngine/5.12.10 Chrome/69.0.3497.128 Safari/537.36.
Results
RWE-ODE dynamics during drug development
presents RWE-ODE dynamics incorporated at clinical trial pII–pIV with increasing time in years based on the model described in . The four clinical trial phases pI-IV are represented as the transition phases in the model. Trials that discontinue before entering the progression phase enter the expired phase, while trials that progress to pIV are likely to enter medicine adoption, which begins at first clinical prescription.
Decrease in time to medicine adoption
Based on cumulative first prescriptions for ambrisentan among PAH patients (N = 487) and the assumption that an exponential characteristic were to occur, 100% medicine adoption for PAH patients treated with ambrisentan was estimated at approximately 30 weeks from date of launch in the standard model (“without RWE”; Supplementary Figure S5). The medicine adoption phase and the incorporation of RWE at clinical phases was determined with a rate value > 1.0. Otherwise, the model would return a standard ODE transition for clinical trials. The model estimated that the inclusion of RWE at pII–IV would result in medicine adoption occurring six months earlier than in the standard model (“without” RWE; ). Inclusion of RWE at pIV may encourage first medicine adoption to be brought forward by 73 days or 10.4 weeks.
Table 2. Estimated time in years to first medicine adoption. Numbers presented in pI to “ado” columns are transition trials in crude numbers; N(0) = 1000.
Potential lives saved
For the illustrative purpose of estimating potential lives saved from RWE incorporation in drug development, disease prevalence of PAH patients in the UK and Ireland is discussed in this study. Based on previous reportsCitation30,Citation31 and the re-estimated prevalence rate using the exact estimation approach, there were N = 487 PAH patients in 2001-2009. Though the previous article reported 482 diagnosed patients, it was within expectation that the exact estimation approach would capture patients that were yet-to be diagnosed, adding five to the reported rates. PAH has been considered a secondary disease resulting from persistent life-threatening diseases such as cardiac complications. In comparison to other rare adult-onset genetic diseases such as acromegaly, PAH has a high diagnosis detection rate including patients presenting mild symptoms. Therefore, the re-estimated disease prevalence is within an expectant spectrum; 4 per 10,000, and a total difference remaining of five cases that were to be diagnosed.
With the assumption that the age distribution of diagnosed PAH patients is a normal distribution for mean μ = 50.1 and standard deviation σ = 17.1, the person-years (Lx.PAH) for PAH patients in UK and Ireland can be obtained using EquationEquation (1)(1)
(1) . The prevalence rate must contain a denominator that describes the respective total general population, and it shares the same entity as its numerator, that is, in person-years Lx, EquationEquation (2)
(2)
(2) .
EquationEquation (1)(1)
(1) : Person-years for PAH patients with diagnosis age 35–70 in years 2001–2009.
(1)
(1)
EquationEquation (2)(2)
(2) : Exact prevalence rate for in the UK and Ireland from years 2001 to 2009 and reported diagnosed ages between 35 and 70. The denominator was Lx.Ireland = 30,106,559.00 and Lx.UK = 30,080,481.00.
(2)
(2)
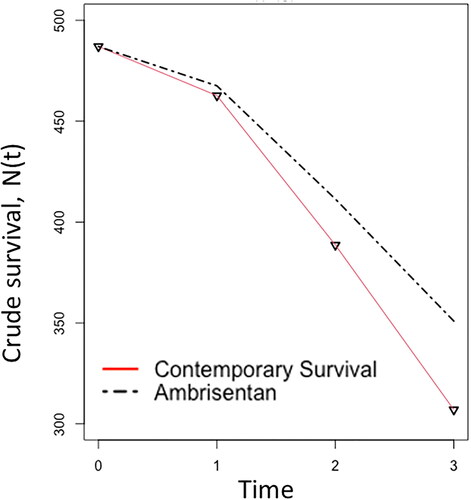
In conjunction with published mortality and survival rates in observational studies cited above, and the following equations: probability of death: q(t) = 1 − S(t) and mortality rate: m(t)= − log(1-q(t)), it is within mathematical approach to reconstruct a crude survival table (). Since the third-year survival rate for PAH-ambrisentan patients was not presented, a reasonable assumption has been made for PAH-ambrisentan mortality rate in year three at 70% proportional to contemporary mortality rate.
Table 3. Crude survival count for PAH patients: contemporary and ambrisentan-treated.
From the re-constructed empirical records presented in and , if RWE were to be considered as a supportive element during drug development and regulatory approval, approximately 9% of PAH patients would have benefited in survival from earlier market access to ambrisentan; that is, 350.94 − 307.01 = 44.
Figure 3. Crude survival count for PAH patients. N(0) can be a hypothetical or empirical number.
Increase in potential earnings
It is estimated that with the incorporation of RWE in orphan drug development, the most optimistic launch for market access can be brought forward by three years; refer to Supplementary Materials Table S2 and S3 for the total completion of clinical trials at t = 20 in the standard ODE Model 1, which corresponds to t = 17.0 ∼ 17.5 in Model 2 with RWE. The potential earnings for an earlier launch would be GBP £43,597.86 per patient, with the launch being brought forward from 2009 to 2007 (). Inflation rate was estimated at an average of 2% per annum, excepting years 2021 and 2022 at 3% and 4%, respectively.
Table 4. UK pricing for ambrisentan 5 mg or 10 mg.
Discussion
Real-world evidence has received widespread attention in recent years, and its use for regulatory decision-making has been actively promoted worldwide. Although regulatory bodies in different countries have established distinct strategies to develop RWE suitable to their specific needs, generation of RWE by pharmaceutical companies in time for regulatory approval is not widespread. Additionally, benefits to patients and pharmaceutical companies of early RWE generation have not been detailed in the literature. In this study we present a simulation model, where RWE has been incorporated at phases pII–pIV, evaluating the potential streamlining obtainable in the drug development cycle, leading to prompt approval and patient access.
Drug launch and market access
Novel drugs are required to undergo all clinical development phases (clinical trial phases 1–3) and those that are successfully approved subsequently enter the medicine adoption phase. This is often regarded as the first instance of prescription to a patient. Prescribers require the appropriate evidence to select a novel medicine. Often described as both a top-down and bottom-up process, decisions on the adoption of new drugs are frequently arrived at based on evidence in published literatureCitation9. As alluded to earlier, this impacts the patient negatively by delaying access to treatmentCitation14. This study aimed to estimate the amount of time by which a decision on the adoption of a medicine could be reduced as a result of the addition of RWE, particularly for orphan drugs and rare diseases.
Our simulation incorporates RWE as a multiplicative rate factor to the standard progression rate in regulatory review dynamics (). The model permits RWE to interact with pII, pIII, & pIV if required (RWEi=1,2,3 ≥1.0) (Table S1). With the incorporation of RWE, in orphan drug development, the model estimated that the most optimistic launch for market access could be brought forward by three years (Tables S2 & S2). In the case of ambrisentan, our model has predicted that with the inclusion of RWE, the launch could have been brought forward to 2007. Although this is a time saving of fewer than 12 months, the implications for patient access and prescriber adoption are numerous. In addition, the potential earnings per patient from earlier launch of ambrisentan, as a particular example, would have amounted to a total of GBP £43,597.86 between 2007 and 2009 alone (). Remarkably, our model has indicated that the inclusion of pIV-RWE would achieve 100% medicine adoption within 30 weeks after launch (Figure S5).
While there may be many benefits for patients of inclusion of RWE at the regulatory stage, there are ethical issues regarding early market access and exposure of patients to riskCitation32, as long-term safety evidence is often unavailable for years post-approval. However, early evidence is promising, with documented case studies showing that early phase RWE findings have been confirmed by phase 3 trialsCitation5 or have been comparable to global clinical trials and post-approval safety studiesCitation12. Mitigation of risk could be achieved by ensuring that RWE meets quality criteria and avoids common issues noted by regulatory bodies that restrict its use, such as poor data quality/reliability, low relevance of data and lack of pre-specification of study design and analysisCitation10.
Limitations
The model sought to estimate potential cost savings due to lives saved from reduced time to treatment of PAH patients with ambrisentan. While it was beyond the scope of the research question, further health economic considerations exist around issues such as healthcare utilization due to comorbidity and/or side effectsCitation16,Citation33 and possible reduced effectiveness due to low adherenceCitation17,Citation34. The use of epidemiological data has allowed a population-based perspective that accounts for variation in many of the real-world variables around treatment for PAH. It may be interesting to determine whether findings in this study are similar for patients treated with dual therapy in which ambrisentan is combined with other agents to target multiple pathways and improve real-world effectiveness. Additionally, early access pathways and regulatory/HTA legislation may impact time to market accessCitation35 and the probability of successful progression through the clinical development pathwayCitation36 as well as the inclusion of regulatory RWE. Overall, this study provides further support for industry efforts to generate RWE in time for regulatory approval.
Conclusion
The ODE-RWE model was constructed based on the fundamentals of clinical trial design, the stringency in review made by drug regulators and the likelihood for medicine adoption. With the exception of phase I, RWE can be incorporated to interact with the specified transition rate, and without penalizing on the stringency during drug review and approval. In comparison to standard drug development, RWE incorporation has the potential to expedite first medicine adoption by 10.4 weeks. The duration of market launch was estimated at 2.5 ∼ 3.0 years earlier than standard, and approximately 9% of patients would benefit in survival.
Transparency
Declaration of financial/other relationships
Dr Ravi Jandhyala is a visiting senior lecturer at the Centre for Pharmaceutical Medicine Research at King’s College London and is responsible for research into real-world evidence approaches. He is also the founder and CEO of Medialis Ltd, a medical affairs consultancy and contract research organization involved in the design and delivery of real-world evidence in the pharmaceutical industry. No conflict of interest has been registered from the author. The Jandhyala method was developed by Dr Jandhyala but is free of commercial licensing restrictions and while used as part of proprietary methodology, is not a direct means of commercial gain for the author.
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contributions
The author conducted the study and developed and approved the manuscript. The author affirms that the manuscript is an honest, accurate, and transparent account of the study being reported; that no important aspects of the study have been omitted, and any discrepancies from the study as planned (and, if relevant, registered) have been explained.
Ethical approval
This study was exempt from international regulations governing research ethics, as collection of data from human participants was not part of the study protocol.
Supplemental Material
Download Zip (658 KB)Acknowledgements
The author would like to thank Ashkay Patil and the Biostatistics Department at Medialis Ltd for their contribution to the study and manuscript as part of their role as employees of Medialis Ltd. Brendon Pearce and Lauri Naylor contributed to the manuscript by providing medical writing services as part of their roles as employees of Medialis Ltd.
Data availability statement
The author confirms that the data supporting the findings of this study are available within the article [and/or] its supplementary materials.
Additional information
Funding
References
- Medicines and Healthcare Products Regulatory Agency. MHRA guidance on the use of real-world data in clinical studies to support regulatory decisions. GOV.UK [accessed 2022 Nov 21]. Available from: https://www.gov.uk/government/publications/mhra-guidance-on-the-use-of-real-world-data-in-clinical-studies-to-support-regulatory-decisions/mhra-guidance-on-the-use-of-real-world-data-in-clinical-studies-to-support-regulatory-decisions
- US Food and Drug Administration. Framework for FDA’s real-world evidence program; 2018 [accessed 2022 Nov 21]. Available from: https://www.fda.gov/media/120060/download
- Cave A, Kurz X, Arlett P. Real-World data for regulatory decision making: challenges and possible solutions for Europe. Clin Pharmacol Ther. 2019;106(1):36–39.
- National Institute for Health and Care Excellence. NICE real-world evidence framework; corporate document [ECD9]; 2022 [accessed 2022 Nov 21]. Available from: https://www.nice.org.uk/corporate/ecd9/chapter/overview
- Baumfeld Andre E, Reynolds R, Caubel P, et al. Trial designs using real-world data: the changing landscape of the regulatory approval process. Pharmacoepidemiol Drug Saf. 2020;29(10):1201–1212.
- National Institute for Health and Care Excellence. The NICE strategy 2021 to 2026 [accessed 2022 Nov 22]. Available from: https://www.nice.org.uk/about/who-we-are/corporate-publications/the-nice-strategy-2021-to-2026
- European Medicines Agency. EMA regulatory science to 2025. 2020 [accessed 2022 Nov 21]. Available from: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/ema-regulatory-science-2025-strategic-reflection_en.pdf
- Jandhyala R. The multiple stakeholder approach to real-world evidence (RWE) generation: observing multidisciplinary expert consensus on quality indicators of rare disease patient registries (RDRs). Curr Med Res Opin. 2021;37(7):1249–1257.
- Khosla S, White R, Medina J, et al. Real world evidence (RWE)—a disruptive innovation or the quiet evolution of medical evidence generation? F1000Res. 2018;7:111.
- Mahendraratnam N, Mercon K, Gill M, et al. Understanding use of real-world data and real-world evidence to support regulatory decisions on medical product effectiveness. Clin Pharm Therapeut. 2022;111(1):150–154.
- Corrao G, Cantarutti A. Building reliable evidence from real-world data: needs, methods, cautiousness and recommendations. Pulm Pharmacol Ther. 2018;53:61–67.
- Storm NE, Chang W, Lin TC, et al. A novel case study of the use of Real-World evidence to support the registration of an osteoporosis product in China. Ther Innov Regul Sci. 2022;56(1):137–144.
- Breckenridge AM, Breckenridge RA, Peck CC. Report on the current status of the use of real-world data (RWD) and real-world evidence (RWE) in drug development and regulation. Br J Clin Pharmacol. 2019;85(9):1874–1877.
- Jandhyala R. A medicine adoption model for assessing the expected effects of additional real-world evidence (RWE) at product launch. Curr Med Res Opin. 2021;37(9):1645–1655.
- Oudiz RJ, Galiè N, Olschewski H, et al. Long-term ambrisentan therapy for the treatment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(21):1971–1981.
- Sikirica M, Iorga SR, Bancroft T, et al. The economic burden of pulmonary arterial hypertension (PAH) in the US on payers and patients. BMC Health Serv Res. 2014;14(1):676.
- Helgeson SA, Menon D, Helmi H, et al. Psychosocial and financial burden of therapy in USA patients with pulmonary arterial hypertension. Diseases. 2020;8(2):22.
- Jandhyala R. The effect of adding real-world evidence to regulatory submissions on the breadth of population indicated for rare disease medicine treatment by the European Medicines Agency. J Pharm Policy Pract. 2022;15(1):36.
- Chorniy A, Bailey J, Civan A, et al. Regulatory review time and pharmaceutical research and development. Health Econ. 2021;30(1):113–128.
- Krishna D, Rittié L, Tran H, et al. Short Time to market and forward planning will enable cell therapies to deliver R&D pipeline value. Hum Gene Ther. 2021;32(9-10):433–445.
- Sherman RE, Anderson SA, Dal Pan GJ, et al. Real-world evidence—what is it and what can it tell us. N Engl J Med. 2016;375(23):2293–2297.
- Sherman RE, Davies KM, Robb MA, et al. Accelerating development of scientific evidence for medical products within the existing US regulatory framework. Nat Rev Drug Discov. 2017;16(5):297–298.
- Ferreira-González I, Marsal JR, Mitjavila F, et al. Patient registries of acute coronary syndrome: assessing or biasing the clinical real world data? Circ Cardiovasc Qual Outcomes. 2009;2(6):540–547.
- Naidoo P, Bouharati C, Rambiritch V, et al. Real-world evidence and product development: opportunities, challenges and risk mitigation. Wien Klin Wochenschr. 2021;133(15-16):840–846.
- Kim HS, Lee S, Kim JH. Real-world evidence versus randomized controlled trial: clinical research based on electronic medical records. J Korean Med Sci. 2018;33(34):e213.
- Vatter H, Seifert V. Ambrisentan, a non-peptide endothelin receptor antagonist. Cardiovasc Drug Rev. 2006; 24(1):63–76.
- Abbas I, Rovira J, Casanovas J. Clinical trial optimization: Monte Carlo simulation Markov model for planning clinical trials recruitment. Contemp Clin Trials. 2007;28(3):220–231.
- Jackson CH, Sharples LD, Thompson SG, et al. Multistate Markov models for disease progression with classification error. J R Stat Soc Ser D Stat. 2003;52(2):193–209.
- Felli JC, Anderson WH, Kremidas JP, et al. A semi-Markov model for patient progression through clinical trials. Eur J Oper Res. 2007;176(1):542–549.
- Wijeratne DT, Lajkosz K, Brogly SB, et al. Increasing Incidence and prevalence of world health organization groups 1 to 4 pulmonary hypertension. Circ Cardiovasc Qual Outcomes. 2018;11(2):e003973.
- Strange G, Playford D, Stewart S, et al. Pulmonary hypertension: prevalence and mortality in the armadale echocardiography cohort. Heart. 2012;98(24):1805–1811.
- Eichler HG, Pignatti F, Flamion B, et al. Balancing early market access to new drugs with the need for benefit/risk data: a mounting dilemma. Nat Rev Drug Discov. 2008;7(10):818–826.
- Lang IM, Palazzini M. The burden of comorbidities in pulmonary arterial hypertension. Eur Heart J Suppl. 2019;21(Suppl K):K21–K28.
- Frantz RP, Hill JW, Lickert CA, et al. Medication adherence, hospitalization, and healthcare resource utilization and costs in patients with pulmonary arterial hypertension treated with endothelin receptor antagonists or phosphodiesterase type-5 inhibitors. Pulm Circ. 2020;10(1):2045894019880086.
- Tsekov I, Topova B, Doychev P, et al. PIN143 Analysis of the time to market access in Bulgaria of new daa medicines for chronic HCV infection authorized under the EMA specific procedures for EARLY access. Value Health. 2020;23:s567.
- Fontrier AM. Market access for medicines treating rare diseases: association between specialised processes for orphan medicines and funding recommendations. Soc Sci Med. 2022;306:115119.