
Abstract
Objective
To conduct a content analysis of IRB webpages of select universities (academic health centers) in the USA that describe post IRB- approval monitoring activities.
Method
This was a qualitative study. Thematic analysis was the method to review the webpage content of selected academic health centers (AHC) within the USA.
Results
Some US academic health “centers” IRB administrative or research compliance offices conduct post- approval monitoring (PAM) of human subjects’ research including clinical trials. The goals of this PAM programmes are to (a) ensure compliance to approved protocols, (b) preserve research integrity, (c) manage institutional risks, d) provide advisory/educational support to researchers, (e) recommend corrective actions for identified issues, and most importantly, (f) to protect the safety, rights, and well-being of research participants. Although not a requirement by law, the PAM program has legislative support in the US Code of Federal Regulations as part of the US Office for Human Research Protection’s (OHRP) Federal Wide Assurance (FWA). This is especially for institutions that conduct studies funded by the Federal government. PAM on-site checks reveal various incidents of protocol deviations and violations. This includes issues with recruitment processes, informed consent discrepancies, and incidents of non-compliance. When a study protocol is identified as non-compliant, the principal investigator works with the PAM monitor to develop a corrective action plan that would allow the study to become compliant and avoid sanctions from the IRB or the regulatory authority.
Conclusions
REC/IRB post-approval monitoring of clinical trials is a valuable mechanism of protection for research participants while giving educational and quality assurance support to researchers. The program enables early detection and resolution of non-compliance to approved protocols. The impact of the program in the USA requires further exploration.
1. Introduction
The balancing act between advancing scientific knowledge while protecting research participants has been challenging. In the modern era, many historical incidents of unethical conduct in research with humans include instances of exploitation, deceit, scientific misconduct, and cruelty. In response to these troubling and sometimes tragic events, research ethics has emerged as a means to define and articulate the ethical principles necessary to protect and establish as values the rights, well-being, and best interests of research participants. Along with ethics, human “subjects” research regulations have been created to promote and safeguard these values. The development of this field of ethics includes normative guidelines, enforcement institutions, and compliance measures to facilitate prior vetting of research to ensure integrity in research while protecting the most vulnerable from the passionate pursuits of scientistsCitation1. The Research Ethics Committee (REC) is one of the main developments during this period. Savulescu, Chalmers and Blunt notes that
Research Ethics Committees (RECs) are uniquely important institutions for at least two reasons: Firstly, they are the only regulatory point through which all proposed clinical research is likely to pass. Secondly, unlike other players who influence the research industry, they are unlikely to have strong vested interests in seeing particular results from research.Citation2
However, RECs are not without their critics. On the one hand, there are complaints of excess oversight of clinical research, particularly for the initial review and approval of the protocols by Ethics Committees. On the other hand, there are outcries regarding unethical practices and scientific misconduct. Successful clinical research outcomes contribute positively to public health. However, unethical research significantly impacts trust. Without trust, the entire research enterprise is compromised. Therefore the fundamental goal of clinical research is not merely to generate a safe product/intervention but to ensure the scientific and ethical integrity of the process. There are many approaches to achieving this goal, one of which is post-approval monitoring by Ethics CommitteesCitation3,Citation4.
Research ethics Committees (RECs) or Institutional Review Boards (IRBs) are integral to research oversight. The US National Institute of Health first established an Ethics Committee for the prior approval of research in the 1950s. This became a legal obligation in the US by the subsequent promulgation of the National Health Research Act of 1974Citation5. International and regional normative documents outline what ought to be the activities of ethics committees after the approval of clinical trial protocols in the EU and the USACitation6. A review of normative documents reveals that RECs are expected to (1) conduct continuing review, (2) receive notification of adverse events, (3) review and approve protocol amendments, (4) receive notifications of protocol deviations and violations, (5) suspend or terminate trials if necessary, and (6) withdraw favorable opinion or stop trialsCitation6. The authors highlight a difference in the legislative support for an active post-approval role for REC/IRBs in the USA compared to the European Union’s (EU) regulatory documents for clinical research. A study of the experiences of REC representatives in Europe indicates that post-approval activities are mainly limited to the review of protocol amendments and receipt of end-of-the-trial reports. Active or passive monitoring of research is considered the remit of the National Regulatory authorities or Medicine AgenciesCitation7. Although European REC representatives acknowledge the possible benefits of post-approval monitoring, they note challenges with a lack of legislative support, organizational structure, and financial/human resourcesCitation7. Brown et al. have also raised similar concerns regarding post approval monitoring in resource constrained countries conducting US initiated studies. Ethics post-approval monitoring seems to be an established practice within the USCitation8.
Anecdotal web and literature search on US IRB post-approval activities reveals that some research institutions, particularly academic health centers (AHCs), have implemented what is known as post-approval monitoring (PAM). These programs may be part of the institution’s wider human research protection program (HRPP), which encompasses audit and research integrity activities, or may be directly connected to the IRB administrative offices. IRB administrative office support includes the prior review and approval of research and compliance monitoring of IRB-approved researchFootnotei. Brown et al. and Melinda Young offers insight into the approaches to PAM. These include (1) administrative check-ins, (2) full on-site assessments, (3) self-assessments, (4) consent process review, (5) consent process observation, and (6) project team reviewCitation8–10. The program’s on-site or direct review assessment includes (1) observation of the research activity, (2) assistance to and education of the principal investigators (PI) in identifying deviations from the approved protocol, (3) implementation of any required changes, and (4) documentation of the findings of the PAM assessmentCitation9,Citation10. Young’s paper provides a basis for further exploration to identify the extent to which US institutions have implemented PAM and related programs and to assess its impact. This explorative study seeks to identify the post-approval activities of Institutional Review Boards (IRBs) within the US.Footnoteii However, it seeks only to provide a descriptive content analysis on select US academic health centers (AHC)Footnoteiii,Citation11 based on the publicly available content on their websites. The focus of the paper is on post-approval activities that are connected to the IRB offices and the IRB itself. It may not address all the elements of the more comprehensive aspects of HRPP programs.
2. Method
2.1. Research question and scope
This study was guided by the research question, “What are the activities of IRBs after the approval of clinical research on humans in the USA?” It is part of a project with the overarching theme: Ethics and compliance post clinical trial approval: the role of Research Ethics Committees. The project’s scope covers Europe and the USA, however, this paper only reflects on findings in the USA.
2.2. Data sources and search strategy
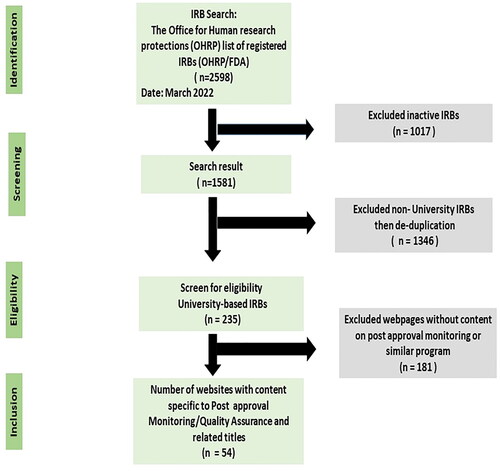
This study was a review of content on webpages of select AHCs in the USA. The Office for Human Research Protection (OHRP) database for Registered IORGs and IRBs was searched to identify the number of active IRBs in the USA. Of the 2598 registered university and hospital based IRBs, 1581 were active. To prevent duplication, only IRBs with designate IRB#1 for universities with multiple IRBs listed was selected. The final number for analysis was 235 (see ). Further screening of content is described under Section 2.4.
Figure 1. PRISMA flowchart of the selection process.
2.3. Eligibility criteria
Webpages of AHCs were eligible for inclusion if they broadly described post approval monitoring as a heading and/or note post-approval activities that were related to compliance checks of IRB approved protocols. Web pages that described quality assurance or quality improvement as an institutional audit function or risk management function but did not have content referring to IRB-approved protocols and compliance with these protocols were excluded. The focus on AHCs is based on an initial google search to identify which US IRBs mentioned PAM on their webpages. We then tailored our inclusion/exclusion criteria towards these institutions.
2.4. Title and content relevance screening
First author did a search of the selected webpages to identify post-approval activities listed on the webpages under the title of post approval “monitoring.” This search was limited to human subject research. Of the 235 IRB websites selected for analysis, 24 explicitly used the title Post approval monitoring (PAM) or noted the term in the general web content. Additionally, other programs under headings such as: quality improvement, quality assurance, routine monitoring, research congruency, audits, research or compliance monitoring program were identified as relevant to the study. After deliberation, the authors agreed to narrow the focus to webpages that outlined post approval monitoring activities despite variations in headings. After several readings of webpages designated for screening, a total of fifty-four (54) active University IRB webpages from 30 States were selected and the content extracted for analysis ().
Table 1. States (30) and names of selected institutions (54).
2.5. Data summary and synthesis
A spreadsheet was created of the active registered OHRP/FDA University-based IRBs and imported into Microsoft Excel 2016. A list of the webpages that met the eligibility criteria was generated. The relevant content for selected webpages was copied into Microsoft word and uploaded to NVIVO 12 Pro for analysis and coding.
2.6. Data analysis
Thematic analysis was used to identify themes according to Braun and Clarke’s six-step procedure for qualitative data analysisCitation12,Citation13. The extracted data from the IRB websites in the USA was read several times to identify patterns and themes. The phase of familiarization enabled initial code creation and subsequent organization of these codes into categories. After several discussions and revisions, the final themes are reported in the research findings below. Braun and Clarke’s method was appropriate because the inductive process and codes were not predefined.
2.7. Researcher characteristics and reflexivity
The researchers are a PhD research fellow, one attorney-at-law and two ethicists. The researchers have experience in pharmacy, research ethics, health law, and research governance/compliance. This study is part of a larger ongoing project on the topic of post-approval activities of research ethics committees. The preunderstanding of the research topic influenced the interpretation of data and subsequently the themes. However, the process was inductive. All themes were generated from the data.
2.8. Ethics approval and process
The Norwegian Centre for Research data reviewed and approved the research project Incorporation of ethics in Pharmaceutical Authorization Regulatory Procedures (REGULATORY ETHICS): Ethics and compliance post clinical trial approval- the role of Research Ethics Committees. Reference number 360856. According to Norwegian law governing research, the study is excluded from review by a research ethics committee as its focus is not considered health research.
3. Findings
3.1. Search results
A total of fifty-four (54) active AHC IRB webpages from 30 States were identified and the content extracted for analysis (see ).
3.2. AHC-IRB PAM “programs” thematic groupings and explanations
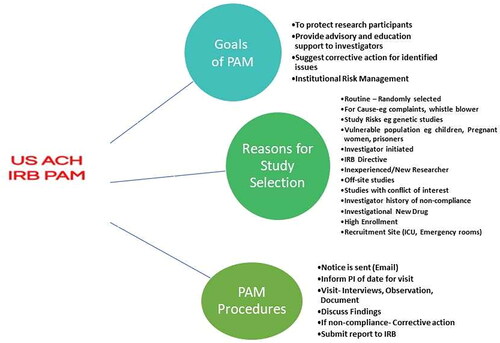
The main themes from content analysis of IRB websites in the USA regarding the role of the PAM programs are organized into three main thematic categories with sub-themes. The main categories are (1) Goals of PAM, (2) Reasons for study selection, and (3) PAM procedures. The sub-themes are discussed under the main thematic categories (see ).
Figure 2. Final thematic map showing main themes and explanations.
3.2.1. Goals of PAM
The reviewed AHCs indicate that the overarching goal of the PAM program is to ensure compliance with IRB-approved protocols and to preserve research integrity. Other goals include (1) to protect research participants, (2) to provide advisory and educational support to investigators, (3) to suggest corrective action for identified issues, and (4) institutional risk management. There is consistency across all institutions regarding the expectations of the program.
3.2.1.1. Theme 1: Ensuring compliance and preserving research integrity
All institutions indicate that the objective of the PAM program is to confirm compliance with IRB-approved protocols. However, as emphasized on the webpage of the University of Binghamton,
PAM visits are not designed to “catch” individuals. Rather, they are conducted to verify that research is being carried out as approved. The IRB recognizes that if noncompliance is detected, it may be a result of a lack of understanding or inadequate training (Binghamton University).
In addition to compliance, the verification of data integrity is integral to the validity of the results
The PAM program functions to maximize the safety of research participants and ensure data integrity by confirming that research is implemented in a manner consistent with the IRB approved protocol and in compliance with applicable regulations and institutional policies (University of Wisconsin).
Compliance checks are usually routine (not for cause). There are also for-cause reviews which may be prompted by complaints or IRB-directed reviews due to questionable observations during the annual review (continuing review). Examples of non-compliance include (1) modifications of protocols without IRB approval, (2) failure to report unanticipated serious adverse events, (3) deficient documentation for eligibility assessments, and (4) incomplete or missing informed consent forms (see ). Compliance checks may include document reviews, interviews, and observations. Document review may include informed consent forms, participant records, lab and dispensing records, and approved protocols. Interviews are usually done with research teams and/or participants. Observations are usually of the informed consent and research processes.
Table 2. Thematic examples of reasons for PAM.
3.2.1.2. Theme 2: Protection of research participants
The institutions explicitly state that the protection of research participants is the ultimate purpose of their programs. Therefore, it is emphasized that while balancing the role of educating researchers, the monitors after identifying non-compliance issues in the research records, they would notify the PI, IRB Chair, and any other relevant department for reporting and reconciliation purposes.
The compliance unit performs various post-approval monitoring and directed review activities primarily to ensure the rights and welfare of research participants are protected (Northwestern University)
The aim of the Program is to ensure maximum protection of human participants involved in research activities and promotion of best practices in the conduct of human research. (East Carolina University)
3.2.1.3. Theme 3: Educational and advisory support
Educational and advisory support is a significant part of PAM. As noted by Northwestern University
the program aims to ensure research staff have the educational resources and guidance necessary to successfully conduct research and provide the research community the study support tools, and other resources needed to perform compliant research (Northwestern University).
Educational support is distinguished from advisory support in that the former emphasize formal didactic courses/seminars/training while the latter is the provision of ongoing guidance on issues that may arise throughout the study. The didactic component may include training on Good Clinical practice, national and local regulations governing clinical research, and institutional best practices. Boston University notes the following:
reviews are intended to be educational and consultative in nature. The educational component involves providing the study staff with up-to-date information on best practices based in Good Clinical Practice (GCP). The consultative aspect is to find and help correct potential problems in study conduct, documentation, or process, including problems arising from IRB noncompliance (Boston University)
3.2.1.4. Theme 4: Recommend corrective actions for identified issues
Whenever the PAM monitor identifies non-compliance, a report is written and corrective actions recommended. Corrective actions enable researchers to avoid sanctions by regulatory authorities or unfavorable reports by sponsor monitors. However, if a researcher fails to comply or obey the directives of the PAM monitor, and continues to have clear protocol violations, then the matter is reported to the research compliance office director or a sub-committee and then, if necessary, reported to the IRB. Several of the AHCs note that it is the IRB that has the authority to suspend or terminate previously approved protocols. We do not interpret this to mean that it is only the IRB that may have this authority within an institution.
The IRB monitoring representative will observe the research activities, prepare reports, provide recommendations for maintaining compliance, provide training, if needed, and, if appropriate, assist in the execution of corrective and/or preventative actions. (Binghamton University)
The final visit report will list actionable findings, as well as areas in which deficiencies were identified. In cases where problems are noted, the investigator has the opportunity to respond to recommendations of the monitor or to provide clarifications or to develop a plan of corrective action to eliminate the potential for future problems. (John Hopkins University)
PAM staff will generate a draft report of findings outlining the concern/allegations that prompted the monitoring visit, the findings of the monitoring visit, any required corrective actions and the time frame within which the corrective actions should be addressed, and recommendations for best practices. (East Carolina University)
3.2.1.5. Theme 5: Institutional risk management
Several of the institutions indicate that PAM form part of their institutional risk management policy. Institutional risk management is an essential mechanism for Universities to achieve organizational objectives through strategic risk identification and mitigation. This is especially important for federal and externally funded projects and to protect the reputation of the institution. Some AHCs note the benefits of PAM as part of its overall institutional risk strategy:
A Post-Approval Monitoring (PAM) program functions as the most significant quality assurance and improvement component of the Human Research Protection Program (HRPP) (Charlotte University)
A quality assurance program provides many benefits to the University and its research program. It is imperative that research programs can assure adherence to federal research compliance mandates in order to protect the operation and reputation of the University, its human research program, and researchers (St Louis University)
3.2.2. Reasons for study selection
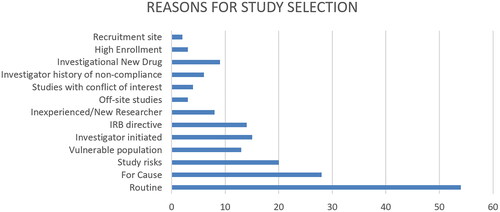
Thirteen descriptors were identified and coded as reasons for study selection (see ). Routine or periodic review and for cause visits were the most listed reasons on the web pages for a monitoring visit. Routine reviews are randomly selected IRB-approved protocols for monitoring. Routine monitoring may be annual or more frequently if non-compliance is identified. For cause reviews are the second highest noted reason for study selection. These may be initiated by a participant or employee complaint, IRB suspension/termination, allegation of non-compliance, or a whistleblower. Studies with Investigational new drugs, high risk and involving vulnerable participants are also regularly reviewed. A few AHCs note genetic studies may be selected for PAM, but many of the webpages only list but did not define or describe high risk. The IRB may also direct the PAM monitor to review a study that it is of the opinion requires more monitoring than annually. Some AHCs also note that PAM visits may be initiated by the PIs to solicit assistance in resolving concerns, prepare for external audits, or for educational purposes. This kind of assistance is also given if the principal investigator (PI) is new, a student, or has a history of non-compliance. Other reasons include studies with high enrollment of research participants, located off-site research, and studies with conflicts of interest. A few AHCs specifically highlighted recruitment site as important. Examples of flagged recruitment sites are emergency rooms and intensive care units.
Figure 3. Descriptors identified as reasons for study selection. The diagram is intended to provide insight into some of the listed reasons for study selection. It should not be interpreted to mean that some AHCs do not include these reasons in their programs. The data analyzed is from webpages and as such is limited. AHCs may have internal practice, policies and procedures that are not reflected on the webpages. This is a limitation of this type of study.
3.3.3. Pam procedures and findings
The process includes a notice, usually via electronic mail, informing the principal investigator of the date for a PAM monitor/compliance officer’s visit. Visits include interviews, observation of procedures, and document reviews. The monitor reviews the documents relevant to the IRB-approved protocol. At the end of the visit, the PAM monitor will discuss the findings with the principal investigators. If non-compliance is identified, a corrective action plan is discussed and implemented. gives an overview of some of the documents reviewed during a PAM visit and commonly detected non-compliance issues. The PAM monitor usually submits the report of the visit to the IRB or research compliance office director who may directly communicate with the IRB chair or a sub-committee established by the IRB.
Table 3. Documents reviewed by PAM monitors and commonly identified non-compliance issues.
4. Discussion
Ethicists across various jurisdictions have argued the benefits of active monitoring of research by Ethics CommitteesCitation3,Citation4,Citation14–16. Theoretically, the concept of REC/IRB monitoring seems very plausible. However, in some jurisdictions, such as the EU, there is skepticism about its practicality with suggestions regarding lack of legislative support, resource constraints, and negatively influencing trust between REC and researchersCitation7,Citation14,Citation17. An examination of normative documents, such as the Declaration of Helsinki, identified various activities that RECs/IRBs are expected to undertake following the initial review and approval of research protocols.
4.1. Organizational model
This review of US AHCs webpages indicates the feasibility and practicality of Ethics post-approval monitoring. The AHCs note that PAM aims to:
confirm that clinical research complies with the approved protocol,
educate researchers/investigators,
ensure the well-being of research participants, and
assist investigators in preparing for external audits.
These AHCs appear to operationalize an organizational model in line with one of several models proposed by Charles Weijer for Ethics Committee monitoring. He proposed that research misconduct and better compliance could be addressed if ethics monitoring is organized to conduct both passive and active monitoring of approved protocolsCitation3. Passive monitoring of health research includes document review/self-assessment. In contrast, active monitoring is an in-person review of adherence to the approved protocols, assessment of study records and participant files, evaluation of other research activities, and/or an observation of the consent process) or bothCitation4,Citation14. Passive monitoring would predominantly be the usual course of action of for the actual committee, while active monitoring is done by qualified administrative staff who submit reports to the committee.
4.2. Legislative support, OHRP Federal Wide Assurance and compliance oversight
The US OHRP outlines the legislative support (see ) and the Federal Wide Assurance (FWA) terms for institutions that receive federal funding. Pursuant to their FWAs, human subject research in AHCs must be reviewed, approved, and overseen by an IRB. Particular emphasis would be on terms 4 and 5 of the FWA. Succinctly put, terms 4 and 5 require IRBs to have written procedures and institutional support (staff and space) for conducting the review, identifying non-compliance, and prompt reporting when necessaryCitation18. The outlined policies described by the AHCs PAM programs appear to conform to the terms. Many AHCs note that PAM programs form part of their OHRP FWA policies and procedures. The AHCs also indicate that PAM programs were facilitated by the administrative staff of the IRB or research compliance offices.
Table 4. Legislative and policy support for HRPP in the USA.
The administrative involvement of AHCs in bolstering IRB's compliance with OHRP requirements was in response to pressure placed on federally funded institutions following several scandals due to lax institutional oversight and overworked IRBsCitation19. These occurred in the late 1990s to early 2000s. One case of notoriety is that of Gelsinger who participated in a clinical trial at University of Pennsylvania. Jesse should have been excluded based on the inclusion/exclusion criteria of the trial but due to lax oversight he was enrolled and diedCitation20,Citation21. Another relevant case is that of 24-year-old Ellen Roche, a healthy volunteer, who died a few days after inhaling hexamethoniumCitation22. Federal funding of research at John Hopkins was suspended by the OHRP. Subsequently, the AHC admitted fault and sought to address their shortcomings. The OHRP noted that the AHC's IRB failed in its responsibility to protect research participants in both the initial review and the monitoring of the research. A third relevant case is that of Hoiyan Wan, another healthy volunteer, who died while participating in a clinical trial at the University of Rochester. Wan died due to a deviation from usual procedure where a higher than usual dose of lidocaine was administered. These deaths emphasized the importance of independent verification by AHC IRBs to ensure they are fulfilling their mandateCitation19. The main strengths of the responses in the various cases were (1) the influence of the enforcement of the federal regulations via institutions such as OHRP, (2) the connection of FWA to funding of research, and (3) the public scrutiny and reputational damage to an institution when research participants are harmed. The financial sanctions and accompanying requirements forced many AHCs to implement stringent administrative measures to prevent fall out with the regulators.
Another very important institutional consideration is accreditationCitation19. The Association for the Accreditation of Human Research Protection Programs (AAHRPP) plays an integral role by including compliance and quality auditing and monitoring as an element of their accreditation process. Specifically, AAHRPP evaluates an AHC's HRRP policies and procedures to assure the quality of their programmeCitation19 AAHRPP is a private non-profit organization that is focused on setting standards sometimes considered even more rigorous than those of the regulators but without the punitive characteristic or consequence that the regulators may assertCitation19. Sociologist Sarah Babb describes this period in her book –Regulating Human Research as the period of hyper-compliance that was replete with bureaucracyCitation23.
A relevant legislative area is that the US Code of Federal Regulations (CFR) permits IRBs or authorized third parties to observe informed consent processesCitation24,Citation25. IRBs are also authorized to suspend or terminate research when deemed necessary. Several AHCs note that violations may be related to a breakdown in the informed consent processes. AHCs receiving federal funding/support must conform to the OHRP's Compliance Oversight Procedures for Evaluating Institutions. Similar to what obtains in the PAM programs, the AHCs are subject to OHRP's “for cause” and “routine” compliance evaluations. If an institution is non-compliant, the OHRP may either restrict or attach conditions to its FWA until full compliance is achieved or suspend the institution and prohibit further fundingCitation18 It may be for this reason that many AHCs, despite recent federal exemptions of some research from IRB continuing review, still require these studies to be reviewed under the PAM programs as part of their institutional risk strategy.
4.3. Compliance by means of cooperation, not coercion
A majority of the AHCs emphasize fostering an atmosphere of cooperation between the researcher and the PAM monitor/administrator. They note that PAM is not designed to “catch” bad researchers. The compelling strength of PAM is that the researcher considers it part of the institutional risk management strategy conducive to a pro-research environment and not external regulatory oversight rife with sanctions. The PAM program involves direct interactions between PIs and the IRB compliance or monitoring representative. It is important to distinguish between the roles of the IRB monitoring representative and the monitor mentioned in Good Clinical Practice (GCP) guidelines. The GCP monitor is usually a pharmaceutical industry sponsor or designated representative (an institution with overall responsibility for the trial) who checks whether a trial complies with laws and guidelines. The sponsor monitor reports directly to the sponsor. Despite the presence of sponsor monitors, non-compliance with protocols is still identified during and at the end of clinical trials by Regulatory Authority inspectorsCitation26–28. Shafiq et al. highlight this incongruity in their paper regarding a pilot IRB monitoring of research in India. They note that IRB monitors identified breaches at clinical trial sites even after the site was audited by sponsorsCitation4. They suggest that sponsor monitors might not have sufficient experience or clinical training to identify some violations. Although this observation is highly subjective, one could also argue that sponsor monitors have biased interests in the continuation of a study and may be less inclined to point out issues that an IRB may find relevant. The same argument could be advanced about GCP inspectors whose focus is more on the safety of the drug and less on ethical issues.
Sponsor and GCP representatives are important players in research compliance oversight, however, researchers may be intimidated by their presence and oversight due to the perceived risk to losing research funding or credibility. PI initiated PAM reviews was the 4th most reported reason for a study selection. These requests were in line with tentative audits or regulatory checks. When challenges occur, the researcher ought to be aware that he/she can receive the requisite support from the REC/IRBs. If researchers consider the PAM monitor as an advisor towards achieving their intended goals, they may be more willing to engage them for advice. A 1992 survey of Australian researchers on their views of monitoring by a REC revealed that researchers were supportive of an advisory/educational post-approval role. They also admitted to protocol deviations without REC approval and noted that monitoring by the REC ensured compliance with the approved protocolCitation16. The concern that RECs/IRBS have assumed the role of “ethics police” – stifling research and indirectly cultivating an environment for research misconduct may be changed if this approach to ethics monitoring is adopted globallyCitation29. Researchers and Ethics Committees should work together to achieve the common goal of scientifically and ethically sound research outcomes.
5. Limitations
This study was based on webpage content which is limited due to a lack of human validation. Our findings report only 54 AHCs with specific webpage content on PAM. However, we note that webpage content analysis has limitations as an AHC may have implemented PAM or a similar programme that is not published. Additionally, information on the extracted web pages may not be current. The primary objective of this study was to report what may be considered some post-approval activities of IRBs in the USA. Therefore, while it is our opinion that there are limits in terms of generalizability, content analysis of these AHC web pages is a valuable method for gaining insight into what organizations assert as part of their mandate. Although there is implicit positivity in our description of PAM, we acknowledge that inherent challenges may not be gleaned based on our selected method of analysis and data source. Further exploration using observation, in-depth interviews/surveys of relevant stakeholders such as IRB representatives, PAM monitors, researchers, participants, and evaluation of reports would provide greater insight into the programme.
6. Conclusions and recommendations
The surveyed US AHC-IRB PAM programs provide insight into the organizational structure, goals, and administrative models necessary to operationalize Ethics Committee’s passive and active monitoring of human subjects research. The US PAM model asserts a cooperative research environment between IRB administrative staff and researchers. It is our opinion that this cooperative model of research oversight could yield scientifically sound and ethically responsible research, thereby bolstering trust and facilitating scientific pursuits. Supplementary research is recommended to identify whether there is a difference in the number of identified protocol non-compliance between AHCs with PAM compared to programs without. Consideration of this type of a REC PAM model could be given in future reviews of the Declaration of Helsinki, ICH: GCP, and similar international normative guidelines. However, the established US programs would require supplementary investigation to aptly conclude that this model achieves the overarching goal of protecting research participants through education, cooperation, and quality assurance. For this type of research governance model to be operationalized in other jurisdictions, government funding, legislative support, organizational restructuring, and hiring and training of competent staff are important first steps.
Transparency
Author contributions
All the authors were involved in the writing of this manuscript. First author, a PhD research fellow, is the main author and did most of the core research. There were regular meetings with 2nd and 4th authors throughout the research process who were involved in conceptualization, design, interviews, analysis, and interpretation. Third author’s expertise in US Law and experience in research governance at a US AHC was important for the correct interpretation of regulatory terms and policy procedures. The manuscript was drafted by 1st author, while other authors were involved in the revision and final version to be published.
Acknowledgements
None.
Declaration of financial/other relationships
The authors are employees of the University of Oslo, Norway. The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
Notes
i The PAM monitor throughout this paper is not referring to the compliance monitoring by pharmaceutical sponsor agencies which is described within the ICH: Good Clinical Practice guidelines.
ii Throughout this paper, reference to REC/IRB is regarding REC/IRB or research compliance offices and administrative staff employed to the office and not the actual committee members. IRBs/RECs are usually supported by staff who carry out various functions on the Committee’s behalf. PAM monitors/administrators may report their findings directly to the IRB chair or to other relevant institutional managerial staff such as research integrity office based on the organizational structure of that institution. A Human Research Protection program (HRPP) is an institutional compliance program that encompasses a wide range of quality assurance and institutional risk management systems which may include auditing the IRB itself. This paper does not intend to go into the range of activities within this program. The focus is on activities of the PAM monitor/administrator regarding IRB approved protocols only. However, a majority of the reviewed webpages note these activities are part of their institutional HRPP.
iii Association of Academic Health Centers defines an academic health center as: “An academic health center encompasses all the health-related components of universities, including their health professions schools, patient care operations, and research enterprise” (29).
References
- Kim WO. Institutional Review Board (IRB) and ethical issues in clinical research historical views on human subject research. Korean J Anesthesiol. 2012;62(1):3–12.
- Savulescu J, Chalmers I, Blunt J. Are research ethics committees behaving unethically? Some suggestions for improving performance and accountability. BMJ. 1996;313(7069):1390–1393.
- Weijer C, Shapiro S, Fuks A, et al. Monitoring clinical research: an obligation unfulfilled. CMAJ. 1995;152(12):1973–1980.
- Shafiq N, Kumari S, Kumar V, et al. On-site monitoring of clinical trials by an ethics committee in India: a road less travelled. Res Ethics. 2021;17(1):45–54.
- Rice TW. The historical, ethical, and legal background of human-subjects research. Respir Care. 2008;53(10):1325–1329.
- Cox S, Solbakk JH, Bernabe RDLC. The role of research ethics committees after the approval of clinical trial protocols in the EU and the USA: a descriptive content analysis of international and regional normative documents. Curr Med Res Opin. 2021;37(6):1061–1069.
- Cox S, Solbakk JH, Bernabe RDLC. Research ethics committees and post-approval activities: a qualitative study on the perspectives of european research ethics committee representatives. Curr Med Res Opin. 2022;38(11):1897–1907.
- Brown B, Kinsler J, Folayan MO, et al. Post-approval monitoring and oversight of U.S.-initiated human subjects research in resource-constrained countries. J Bioeth Inq. 2014;11(2):119–123.
- Young M. In new common rule era, there is role for post-approval monitoring. Relias Media. 2019. [cited 2021 Sep 11]. https://www.reliasmedia.com/articles/145106-in-new-common-rule-era-there-is-role-for-post-approval-monitoring.
- Young M. Sample models of post-approval monitoring programs. Relias Media. 2019. [cited 2021 Sep 11]. https://www.reliasmedia.com/articles/145107-sample-models-of-post-approval-monitoring-programs.
- AAHCI. Academic health centers. Association of Academic Health Centers. 2022. [cited 2022 Sep 15]. https://www.aahcdc.org/About/Academic-Health-Centers.
- Braun V, Clarke V. Using thematic analysis in psychology. Qual Res Psychol. 2006;3(2):77–101.
- Braun V, Clarke V. One size fits all? What counts as quality practice in (reflexive) thematic analysis? Qual Res Psychol. 2020;18(3):328–352.
- Boateng O, Ndebele P, Mwesiga-Kayongo D. Research ethics in Africa: a resource for research ethics committees. Stellenbosch: SUN MeDIA; 2014.
- Berry J, Piwncess T, Hospital R, et al. Local research ethics committees can audit ethical standards in research. J Med Ethics. 1997;23(6):379–381.
- McNeill PM, Berglund CA, Webster IW. Do Australian researchers accept committee review and conduct ethical research. Soc Sci Med. 1992;35(3):317–322.
- Pickworth E. Should local research ethics committees monitor research they have approved. J Med Ethics. 2000;26(5):330–333. ?
- OHRP. Federalwide assurance instructions. HHS.gov. 2017. [cited 2021 Feb 09]. https://www.hhs.gov/ohrp/register-irbs-and-obtain-fwas/forms/fwa-instructions/index.html.
- Steinbrook R. Improving protection for research subjects. N Engl J Med. 2002;346(18):1425–1430.
- Rinde M. The death of Jesse Gelsinger, 20 years later [Online]. Science History Institute. Distillations. 2019. [cited 2022 Mar 18]. https://www.sciencehistory.org/distillations/the-death-of-jesse-gelsinger-20-years-later.
- Stephenson J. Gene therapy trials oversight. JAMA. 2000;283(15):1950–1950.
- Savulescu J, Spriggs M. The hexamethonium asthma study and the death of a normal volunteer in research. J Med Ethics. 2002;28(1):3–4.
- Babb S. Regulating human research: IRBs from peer review to compliance bureaucracy. Stanford: Stanford University Press; 2020.
- FDA. IRB continuing review after clinical investigation. Rockville (MD): US Food and Drug Administration; 2012. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/irb-continuing-review-after-clinical-investigation-approval.
- FDA. 21 CFR 56. 2019. https://www.ecfr.gov/cgi-bin/text-idx?SID=7681dfad0d169cfef7a609461bcf00b4&mc=true&node=pt21.1.56&rgn=div5.
- Bernabe R, van Thiel G, Breekveldt N, et al. Drug regulators and ethics: which GCP issues are also ethical issues. Drug Discov Today. 2016;21(2): 217–224.
- Bernabe R, van Thiel G, Breekveldt N, et al. Ethics in clinical trial regulation: ethically relevant issues from EMA inspection reports. Curr Med Res Opin. 2019;35(4):637–645.
- Seife C. Research misconduct identified by the US food and drug administration: out of sight, out of mind, out of the peer-reviewed literature. JAMA Intern Med. 2015;175(4):567–577.
- Klitzman R. The ethics police?: ‘IRBs’ views concerning their power. PLoS One. 2011;6(12):e28773.