
ABSTRACT
The complete mitogenome of Haliotis iris, an economically important shellfish endemic to New Zealand, was sequenced for the first time. The mitogenome was 17,131 base pairs (bp) in length and contained 13 protein-coding genes (PCGs), 2 ribosomal RNA (rRNA) genes, 22 transfer RNA (tRNA) genes and a control region. All 13 genes were initiated by the start codon ATG, except for nad5 (ATA). Two typical stop codons, TAA and TAG, were present. All of the tRNAs could be folded into typical cloverleaf secondary structures except tRNASer1 and tRNALys, which lacked a DHU stem and complete amino acid acceptor stem, respectively. The control region was 1132 bp in length and contained six AT tandem repeats. According to the gene order of the mitogenome, the 30 analysed Vetigastropoda species could be classified into three types—type I: over half of the studied species were very similar to the gastropod ancestral gene order, and the rearrangements occurred in five tRNAs; type II: eight species were found to be missing several tRNA genes; type III: Fissurellidae, Lepetodrilidae showed a large inverted fragment.
Introduction
The black-footed abalone Haliotis iris is endemic to New Zealand, and inhabits intertidal and subtidal rocky reefs surrounding the North, South, Stewart and Chatham Islands (Will et al. Citation2011, Citation2015). Haliotis iris, also well-known as pāua by local Māori, is one of the biggest abalone species in the world and can reach 18 cm (Cunningham et al. Citation2016). As an economically important shellfish, it has become the third largest wild-caught abalone fishery in the world (Will et al. Citation2015). Research on the molecular biology of H. iris is scanty in recent years, with a focus on population genetics structure using two mitochondrial regions (cytochrome oxidase I and ATPase 8–ATPase 6), microsatellites (Will et al. Citation2011, Citation2015) and three actin genes (Bryant et al. Citation2006).
Mitochondrial DNA (mtDNA) has been widely used as a genetic marker for species identification and phylogenetic analyses for animals because the mitochondrial genome is small and highly conserved, has strictly orthologous protein-coding genes (PCG) and ribosomal RNAs (rRNAs), lacks recombination, possesses no introns and has very short intergenic regions (Xin et al. Citation2011; Wort et al. Citation2017; Zhu et al. Citation2017). Although many complete or near-complete animal mtDNAs are available in GenBank databases (> 55,000) (Ghiselli et al. Citation2017), among the 56 extant Haliotis species (Geiger Citation2000), only five of them, as shown in , have been deposited into GenBank, so there is a notable lack of mtDNA information for Haliotidae. To better understand the mtDNA dynamics within Haliotis species, it is necessary to adopt a broader approach to gathering and analysing data.
Table 1. GenBank accession number, composition and skew for different Vetigastropoda mitogenomes.
The complete mtDNA of H. iris is presented here for the first time. A comparative analysis of the mtDNA sequences of H. iris and other known Vetigastropoda species was performed to provide meaningful insights into the evolutionary pattern of mitogenomic features. The sequence will enrich the GenBank database, and provide more data for population genetic structure studies for H. iris.
Materials and methods
Sample collection and DNA extraction
One adult H. iris (body length 15.20 cm, body weight 530 g) was collected from the Christchurch coast, New Zealand. The foot muscle was extracted, transferred into 95% alcohol, and stored at –20°C until use. The total DNA was extracted with a TIANamp Marine Animals DNA Kit (TIANGEN Biotech Co., Ltd, Beijing, China) according to the manufacturer’s instructions.
Polymerase chain reaction amplification and sequencing
To amplify the entire mtDNA of H. iris, 12 pairs of primers were designed based on the conserved sequences of Haliotis discus hannai (GenBank Number: KF724723), Haliotis rubra (AY588938) and Haliotis laevigata (KJ472483) (). All amplifications were performed in 25 μL reaction volumes, including 0.2 μm of each primer, 5.0 μL of 10 × Taq Plus polymerase buffer, 0.2 mm of dNTPs, 2 units of Taq Plus DNA polymerase with proof-reading characteristic (TIANGEN Biotech) and 1 μL of DNA template. The amplification protocols were performed with the following cycling parameters: 95°C for 3 min, followed by 32 cycles of denaturation at 95°C for 30 s, annealing at 58°C for 45 s, extension at 72°C for 1–3 min (depending on the length of the fragments), and final extension at 72°C for 10 min. The polymerase chain reaction products were electrophoresed on 1% agarose gels, purified using a SanPrep Column DNA Gel Extraction Kit (Sangon Biotech Co., Ltd, Shanghai, China) and sequenced on an ABI 3730XL automated sequencer (Sangon Biotech Co., Ltd).
Table 2. Primers used to amplify the complete mitogenome of Haliotis iris.
Sequence assembly, annotation and analysis
To investigate the mitogenomic features between H. iris and other Vetigastropoda species, 30 Vetigastropoda mitogenome sequences were downloaded from GenBank (). The mitochondrial sequence was assembled with DNAMAN software (Feng et al. Citation2014), and sequence annotation was performed using NCBI BLAST. The sequence annotation, and the location of 22 transfer RNA (tRNA) genes and secondary structures were conducted using the online MITOS software (Bernt et al. Citation2013). The location of the PCGs and two rRNAs were identified using DOGMA software (Wyman et al. Citation2004). The boundaries of 13 PCGs and two rRNA genes were determined with BLAST searches in NCBI and confirmed by alignment with homologous genes from five Haliotis species available in GenBank. The base composition and the relative synonymous codon usage values were calculated using MEGA 6.0 (Tamura et al. Citation2013). A circular map of the H. iris mtDNA sequence was drawn with CGView (Grant and Stothard Citation2008). The skew composition was calculated using the following formula: AT skew = (A – T)/(A + T), GC skew = (G – C)/(G + C) (Basso et al. Citation2017). The A + T-rich repeat fragments of control region were identified using the Tandem Repeats Finder software (Benson Citation1999).
Results and discussion
Genome organisation and structure
The complete mitochondrial genome of H. iris was found to be a closed circular molecule of 17,131 base pairs (bp) (GenBank accession number: KU310895). Like the mitochondrial genome of other mollusc species, it included 13 PCGs (cox1–3, nad1–6, nad4 L, atp6/8 and cytb), 22 tRNA genes, 2 rRNA genes (12S and 16S rRNA) and a control region (, ). The genome was slightly larger than those of H. laevigata (16,545 bp, Robinson et al. Citation2016), Haliotis diversicolor (16,187 bp, Xin et al. Citation2011), H. rubra (16,907 bp, Maynard et al. Citation2005), Haliotis tuberculata (16,521 bp, Van Wormhoudt et al. Citation2009) and H. discus hannai (16,886 bp, Yang et al. Citation2015), as shown in . The sequence identity between H. iris and the other five abalone species exceeded 80% based on comparisons with BLAST software. The main differences were in the control region; the lengths of other regions of the genome were approximately equal among these species. Among the 37 genes, 15 genes were transcribed from the heavy strand, whereas the light strand encoded the remaining 22 genes.
Figure 1. Circular map of the mitochondrial genome of Haliotis iris. Genes on the outside of the circle were transcribed from the heavy strand in a counterclockwise direction, genes on the inside of the circle were transcribed from the light strand in a clockwise direction. Gene identities: nd1–6 and nd4L, NADH dehydrogenase subunits 1–6 and 4L (in yellow); cox1–3, cytochrome c oxidase subunits I–III (in pink); cytb, cytochrome b (in purple); atp6/8, ATP synthase subunit 6/8 (in light green); 12S/16S ribosomal RNA, small and large subunits of ribosomal RNA (in red). tRNA: Transfer RNA genes (in blue). The line between cox3 and tRNA-Glu indicates control region, the lines between other genes indicate intergenic spacer regions.
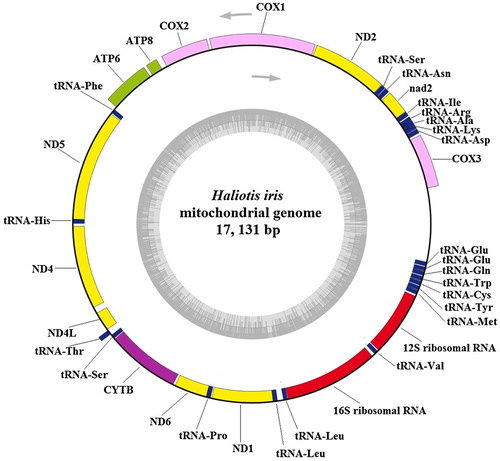
Table 3. Mitochondrial genome order and organisation of Haliotis iris.
One gene overlap and 32 intergenic spacers were found in the mitogenome of H. iris. The overlap, which was 3 bp in length, was located between tRNAPhe and nad5. Haliotis diversicolor also has one overlap region located between nad5 and tRNAHis, whereas H. rubra has seven overlap genes. The intergenic spacers ranged from 1 to 95 bp, with a total length of 758 bp. This phenomenon is commonly observed in mtDNA (Maynard et al. Citation2005; Ma et al. Citation2015; Lin et al. Citation2017). The nucleotide compositions and skews of H. iris and the 29 other Vetigastropoda species are shown in . The overall A + T content of H. iris was 59.8%. This A + T bias can also be seen in the Vetigastropoda mtDNAs already sequenced (), and it ranges from 57.8% in H. laevigata (Robinson et al. Citation2016) and Diodora graeca (Uribe et al. Citation2016) to 69.6% in Variegemarginula punctata (Lee et al. Citation2016). The A + T richness of mitochondrial genomes is common due to the presence of elements controlling the regulation of replication and transcription (Boore Citation1999). The AT skew (0.1999) for the H. iris mitogenome is slightly positive, indicating a higher occurrence of A than T, and its GC skew (–0.3343) is negative, indicating a higher content of C than G. These values are similar for the other abalone species, except H. laevigata, which has a negative AT skew (–0.1745) and a positive GC skew (0.2492). Astralium haematragum, Bolma rugosa, Chlorostoma argyrostomum, Tegula brunnea, Lepetodrilus nux and Lepetodrilus schrolli also had negative AT skews and negative GC skews.
Protein-coding genes
The total length of the 13 PCGs in H. iris was 11,263 bp, coding 3754 amino acids. The start and stop codons of the 13 PCGs are shown in . All 13 genes were initiated by the start codon ATG, except for nad5 (ATA), and two typical stop codons were present (TAA and TAG). Eight PCGs were terminated by a TAA codon; the genes in the nad4, nad4 L, cytb, nad6 and nad1 cluster were terminated by a TAG codon. It is notable that these five genes were all transcribed from the light strand, whereas all other genes were transcribed from the heavy strand, except for nad5, suggesting a strand-specific pattern of stop codon usage in H. iris mtDNA, which is similar to H. rubra mtDNA (Maynard et al. Citation2005) and different from H. laevigata and H. diversicolor mtDNA, in which there is no relationship between the strand and stop codon types.
The relative synonymous codon usage analysis for H. iris is shown in . The codons encoding Phe (8.36%), Leu (15.5%), Val (8.68%) and Gly (7.8%) were the most frequent, while those encoding Arg (1.71%) and Cys (1.6%) were rare, and the codon usage was biased, with a high frequency of AT compared with GC in the third codon position. The A + T content of the 13 PCGs was 56.8%, which is slightly less than that of the complete mitogenome, but both presented an A + T bias. The AT skew and GC skew values of the 29 Vetigastropoda species were negative and positive, respectively, except for the values in Lepetodrilus nux (AT skew: – 0.1063, GC skew: – 0.2011). These results suggest that, in most PCGs, more T than A and more C than G are present, which is consistent with the findings of Sun et al. (Citation2018), who classified 144 molluscan species into two groups: the first group included only 34 species (eight Gastropoda species and all Cephalopoda species) presenting positive AT skews and negative GC skews; the second one included 110 species (almost all Gastropoda species and all Bivalvia species) characterised by negative AT skews and positive GC skews, which revealed that these 144 species had strand asymmetry reversal on the entire plus strand. The nucleotides A and C appear more frequently in single-stranded DNA than in double-stranded DNA, resulting in strand asymmetry (Rocha et al. Citation2006; Sun et al. Citation2018).
Table 4. Codon usages of the protein-coding genes in Haliotis iris mitochondrial genome.
Transfer and ribosomal RNA genes
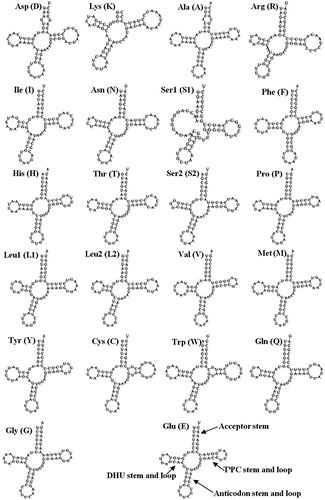
A total of 22 tRNAs, ranging in size from 63 bp (tRNALys) to 74 bp (tRNATrp), were identified in the H. iris mitogenome (). Eight tRNAs were encoded by the H-strand, and the remaining 14 tRNAs were encoded by the L-strand. All of the tRNAs could be folded into typical cloverleaf secondary structures except tRNASer1 and tRNALys, which lacked a DHU stem and complete amino acid acceptor stem, respectively. Yamazaki et al. (Citation1997) reported that Gastropoda the tRNA structures existed unique features, which lack either the DHU or TΨC stem. This has also been observed in H. rubra, H. diversicolor and Lophiotoma cerithiformis (Bandyopadhyay et al. Citation2006). A 7 bp amino acid acceptor stem was conserved in all tRNAs, except for tRNALys, in which it is just 3 bp. Sixty-one unmatched base pairs were detected in the H. iris tRNA genes, and most tRNA genes, except for tRNAAsp, tRNAArg and tRNAIle, had G-U mismatches in their secondary structures, which leads to a weak bond. A G-G mismatch in tRNAAsp and an A-C mismatch in tRNAAla were found in the amino acid acceptor stem, and two U-U mismatches were found in the TψC stem.
Figure 2. Putative secondary structure of the 22 transfer RNAs (tRNAs) identified in the mitochondrial genome of Haliotis iris. The tRNAs are labelled with the abbreviations of their corresponding amino acids.
The location of two rRNA genes was identified using DOGMA software. The boundaries of the 16S and 12S rRNA genes were determined with BLAST searches in NCBI and confirmed by alignment with homologous genes from five Haliotis species available in GenBank. The 16S rRNA gene was 1464 bp long and located between tRNALeu2 and tRNAVal; the 12S rRNA gene was 1008 bp long and located between tRNAVal and tRNAMet. The A + T contents of the 16S and 12S rRNA genes were 62.6% and 60.4%, respectively, higher than that of the overall mitogenome (59.8%). The pairwise distances between H. iris and the other 24 abalone species ranged from 0.05 to 0.13, indicating that 16S rRNA in Haliotidae had a high sequence identity (Guo et al. Citation2017).
Control region
The control region was 1132 bp in size and located between cox3 and tRNAGlu, with an A + T content of 66%. Six AT tandem repeats were found in the control region: (AT)23 (10 bp–55 bp), (AT)13 (16871 bp–16896 bp), (AT)10 (16917 bp–16936 bp), (AT)12 (16957 bp–16980 bp), (AT)14 (17000 bp–17027 bp) and (AT)23 (17048 bp–17093 bp). Tandem repeats are common within the control region of animal mtDNAs (Lunt et al. Citation1998; Sun et al. Citation2016; Feng et al. Citation2017). While H. rubra has two repeat tracts, (AT)80 and (AT)40, which are separated by a possible G + C-rich region, H. discus hannai has just one AT repeat of (AT)18. This indicates that interspecific differences in AT repeats exist. However, the mtDNA control regions of invertebrates are not well characterised, and their lack of, discrete conserved sequence blocks, makes internal primer design difficult (Maynard et al. Citation2005). There have been few studies on molluscan control regions to date.
Gene rearrangement
The gene arrangement of the 30 Vetigastropoda species is shown in . The gene orders of the six abalone species were identical, and similar to the hypothetical ancestral gene order of gastropods (Osca et al. Citation2014; Uribe et al. Citation2016), except for the location of trnD and trnN. According to the mitogenome gene order, the 30 Vetigastropoda species can be classified into three types. The first type: the families Haliotidae, Turbinidae, Trochidae, Tegulidae, Phasianellidae and Chilodontidae had almost the same genome organisation, which was very similar to the gastropod ancestral gene order. Rearrangements occurred in five tRNAs (trnN, trnD, trnE, trnG, trnT). The second type: Astralium haematragum, Stomatella planulata, Chlorostoma argyrostomum, Omphalius nigerrimus, Tegula brunnea and Tegula lividomaculata were found to be missing trnE. Tegula lividomaculata also missed trnG. Tectus virgatus and Margarites vorticiferus both had a partial genome, lacking the trnM-trnY-trnC-trnW-trnQ-trnG-trnE cluster. The third type: Fissurella volcano and Diodora graeca showed a large inverted fragment that included the cob–nad6–trnP–nad1–trnL1–trnL2–rrnL–trnV–rrnS–trnM–trnY–trnC–trnW–trnQ–trnG–trnE genes, whereas the inverted fragment including the trnV–rrnS–trnM–trnY–trnC–trnW–trnQ–trnG–trnE genes existed in Variegemarginula punctata. Lepetodrilus nux and Lepetodrilus schrolli also showed one inversion event affecting a large fragment that included the trnD–atp8–atp6–trnF–nad5–trnH–nad4–trnT–trnS–cob–nad6–trnP–nad1–trnL1–trnL2 genes. Chrysomallon squamiferum showed two inverted fragments that included the trnF–atp6–atp8–trnD genes and the trnQ–trnW–trnC–trnY genes. These results are similar to those of Lee et al. (Citation2016), Nakajima et al. (Citation2016), Uribe et al. (Citation2016, Citation2017) and Williams et al. (Citation2014). The 30 Vetigastropoda species shared the same PCG order for the 13 PCGs, except for species in the families Fissurelloidea, Lepetodriloidea and Peltospiridae. Rearrangements of tRNA are generally considered to be a consequence of the tandem duplication of part of the mitogenome (Lavrov et al. Citation2002; Tang et al. Citation2017; Zhu et al. Citation2017). The particularly high number of rearrangements in the Fissurelloidea and Lepetodriloidea mitogenomes correlates with the high evolutionary rates exhibited by species in these families (Williams et al. Citation2014; Lee et al. Citation2016; Uribe et al. Citation2016).
Figure 3. Gene arrangements of 30 Vetigastropoda mitogenomes. Genes without colour correspond to the hypothetical ancestral gastropod arrangement. Genes translocated are coloured in yellow; inversions are in light blue; grey-coloured boxes represent missing genes.

Acknowledgements
We would like to thank staff of Southern Rainbow Limited for supplying the black-footed abalone.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bandyopadhyay PK, Stevenson BJ, Cady MT, Olivera BM, Wolstenholme DR. 2006. Complete mitochondrial DNA sequence of a Conoidean gastropod, Lophiotoma (Xenuroturris) cerithiformis: Gene order and gastropod phylogeny. Toxicon. 48:29–43. doi: 10.1016/j.toxicon.2006.04.013
- Basso A, Babbucci M, Pauletto M, Riginella E, Patarnello T, Negrisolo E. 2017. The highly rearranged mitochondrial genomes of the crabs Maja crispata and Maja squinado (Majidae) and gene order evolution in Brachyura. Scientific Reports. 7:4096. doi: 10.1038/s41598-017-04168-9
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research. 27(2):573–580. doi: 10.1093/nar/27.2.573
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Molecular Phylogenetics and Evolution. 69(2):313–319. doi: 10.1016/j.ympev.2012.08.023
- Boore JL. 1999. Animal mitochondrial genomes. Nucleic Acids Research. 27(8):1767–1780. doi: 10.1093/nar/27.8.1767
- Bryant MJ, Flint HJ, Sin FYT. 2006. Isolation, characterization, and expression analysis of three actin genes in the New Zealand black-footed abalone, Haliotis iris. Marine Biotechnology. 8(2):110–119. doi: 10.1007/s10126-005-5139-5
- Cunningham SC, Smith AM, Lamare MD. 2016. The effects of elevated pCO2 on growth, shell production and metabolism of cultured juvenile abalone, Haliotis iris. Aquaculture Research. 47:2375–2392. doi: 10.1111/are.12684
- Feng S, Powell SM, Wilson R, Bowman JP. 2014. Extensive gene acquisition in the extremely psychrophilic bacterial species Psychroflexus torquis and the link to sea-ice ecosystem specialism. Genome Biology and Evolution. 6:133–148. doi: 10.1093/gbe/evt209
- Feng YW, Li Q, Yu H, Kong LF. 2017. Complete mitochondrial genome sequence of Cucullaea labiata (Arcoida: Cucullaeidae) and phylogenetic implications. Genes & Genomics. 39:867–875. doi: 10.1007/s13258-017-0548-1
- Geiger DL. 2000. Distribution and biogeography of the Haliotidae (Gastropoda: Vetigastropoda) world-wide. Bollettino Malacologico. 35:57–120.
- Ghiselli F, Milani L, Iannello M, Procopio E, Chang PL, Nuzhdin SV, Passamonti M. 2017. The complete mitochondrial genome of the grooved carpet shell, Ruditapes decussatus (Bivalvia, Veneridae). PeerJ. 5:e3692. doi: 10.7717/peerj.3692
- Grant JR, Stothard P. 2008. The CGViewServer: A comparative genomics tool for circular genomes. Nucleic Acids Research. 36:W181–W184. doi: 10.1093/nar/gkn179
- Guo ZS, Zhang JW, Li L, Hou XG, Zhang HT, Zhang XJ. 2017. Sequence analysis of mitochondrial 16S rRNA gene and molecular phylogeny of three species of abalones. Journal of Shandong University (Natural Science. 52:1–8. (In Chinese with English abstract).
- Lavrov DV, Boore JL, Brown WM. 2002. Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: duplication and non-random loss. Molecular Biology and Evolution. 19:163–169. doi: 10.1093/oxfordjournals.molbev.a004068
- Lee H, Samadi S, Puillandre N, Tsai MH, Dai CF, Chen WJ. 2016. Eight new mitogenomes for exploring the phylogeny and classification of Vetigastropoda. Journal of Molluscan Studies. 82:534–541. doi: 10.1093/mollus/eyw027
- Lin LL, Li XJ, Zhang HL, Zheng ZM. 2017. Mitochondrial genomes of three Tetrigoidea species and phylogeny of Tetrigoidea. PeerJ. 5:e4002. doi: 10.7717/peerj.4002
- Lunt DH, Whipple LE, Hyman BC. 1998. Mitochondrial DNA variable number tandem repeats (VNTRs): utility and problems in molecular ecology. Molecular Ecology. 7:1441–1455. doi: 10.1046/j.1365-294x.1998.00495.x
- Ma HY, Ma CY, Li CH, Lu JX, Zou X, Gong YY, Wang W, Chen W, Ma LB, Xia LJ. 2015. First mitochondrial genome for the red crab (Charybdis feriata) with implication of phylogenomics and population genetics. Scientific Reports. 5:11524. doi: 10.1038/srep11524
- Maynard BT, Kerr LJ, McKiernan JM, Jansen ES, Hanna PJ. 2005. Mitochondrial DNA sequence and gene organization in the Australian blacklip abalone Haliotis rubra (Leach). Marine Biotechnology. 7:645–658. doi: 10.1007/s10126-005-0013-z
- Nakajima Y, Shinzato C, Khalturina M, Nakamura M, Watanabe H, Satoh N, Mitarai S. 2016. The mitochondrial genome sequence of a deep-sea, hydrothermal vent limpet, Lepetodrilus nux, presents a novel vetigastropod gene arrangement. Marine Genomics. 28:121–126. doi: 10.1016/j.margen.2016.04.005
- Osca D, Irisarri I, Todt C, Grande C, Zardoya R. 2014. The complete mitochondrial genome of Scutopus ventrolineatus (Mollusca: Chaetodermomorpha) supports the Aculifera hypothesis. BMC Evolutionary Biology. 14:1–10. doi: 10.1186/1471-2148-14-1
- Robinson NA, Hall NE, Ross EM, Cooke IR, Shiel BP, Robinson AJ, Strugnell JM. 2016. The complete mitochondrial genome of Haliotis laevigata (Gastropoda: Haliotidae) using MiSeq and HiSeq sequencing. Mitochondrial DNA. 27(1):437–438. doi: 10.3109/19401736.2014.900611
- Rocha EPC, Touchon M, Feil EJ. 2006. Similar compositional biases are caused by very different mutational effects. Genome Research. 16:1537–1547. doi: 10.1101/gr.5525106
- Sun SE, Li Q, Kong LF, Yu H. 2018. Multiple reversals of strand asymmetry in molluscs mitochondrial genomes, and consequences for phylogenetic inferences. Molecular Phylogenetics and Evolution. 118:222–231. doi: 10.1016/j.ympev.2017.10.009
- Sun YX, Wang L, Wei GQ, Qian C, Dai LS, Sun Y, Abbas MN, Zhu BJ, Liu CL. 2016. Characterization of the complete mitochondrial genome of Leucoma salicis (Lepidoptera: Lymantriidae) and comparison with other Lepidopteran insects. Scientific Reports. 6:39153. doi: 10.1038/srep39153
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA 6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution. 30(12):2725–2729. doi: 10.1093/molbev/mst197
- Tang BP, Xin ZZ, Liu Y, Zhang DZ, Wang ZF, Zhang HB, Chai XY, Zhou CL, Liu QN, Yang WX. 2017. The complete mitochondrial genome of Sesarmops sinensis reveals gene rearrangements and phylogenetic relationships in Brachyura. PLoS ONE. 12(6):e0179800. doi: 10.1371/journal.pone.0179800
- Uribe JE, Kano Y, Templado J, Zardoya R. 2016. Mitogenomics of Vetigastropoda: insights into the evolution of pallial symmetry. Zoologica Scripta. 45(2):145–159. doi: 10.1111/zsc.12146
- Uribe JE, Williams ST, Templado J, Abalde S, Zardoya R. 2017. Denser mitogenomic sampling improves resolution of the phylogeny of the superfamily Trochoidea (Gastropoda: Vetigastropoda). Journal of Molluscan Studies. 83:111–118. doi: 10.1093/mollus/eyw049
- Van Wormhoudt A, Le Bras Y, Huchette S. 2009. Haliotis marmorata from Senegal; a sister species of Haliotis tuberculata: morphological and molecular evidence. Biochemical Systematics and Ecology. 37:747–755. doi: 10.1016/j.bse.2009.12.020
- Will M, Hale ML, Schiel DR, Gemmell NJ. 2011. Low to moderate levels of genetic differentiation detected across the distribution of the New Zealand abalone, Haliotis iris. Marine Biology. 158:1417–1429. doi: 10.1007/s00227-011-1659-x
- Will M, McCowan T, Gemmell NJ. 2015. Broad-scale genetic patterns of New Zealand abalone, Haliotis iris, across a distribution spanning 13° latitude and major oceanic water masses. Genetica. 143:487–500. doi: 10.1007/s10709-015-9847-0
- Williams ST, Foster PG, Littlewood DTJ. 2014. The complete mitochondrial genome of a turbinid vetigastropod from MiSeq Illumina sequencing of genomic DNA and steps towards a resolved gastropod phylogeny. Gene. 533:38–47. doi: 10.1016/j.gene.2013.10.005
- Wort EJG, Fenberg PB, Williams ST. 2017. Testing the contribution of individual genes in mitochondrial genomes for assessing phylogenetic relationships in Vetigastropoda. Journal of Molluscan Studies. 83:123–128. doi: 10.1093/mollus/eyw044
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255. doi: 10.1093/bioinformatics/bth352
- Xin Y, Ren JF, Liu X. 2011. Mitogenome of the small abalone Haliotis diversicolor Reeve and phylogenetic analysis within Gastropoda. Marine Genomics. 4:253–262. doi: 10.1016/j.margen.2011.06.005
- Yamazaki N, Ueshima R, Terrett JA, Yokobori S, Kaifu M, Segawa R, Kobayashi T, Numachi K, Ueda T, Nishikawa K, et al. 1997. Evolution of pulmonate gastropod mitochondrial genomes: comparisons of gene organizations of Euhadra, Cepaea and Albinaria and implications of unusual tRNA secondary structures. Genetics. 145:749–758.
- Yang EC, Nam BH, Noh SJ, Kim YO, Kim DG, Jee YJ, Park JH, Noh JH, Yoon HS. 2015. Complete mitochondrial genome of Pacific abalone (Haliotis discus hannai) from Korea. Mitochondrial DNA. 26(6):917–918. doi: 10.3109/19401736.2013.863289
- Zhu KC, Wu N, Sun XX, Guo HY, Zhang N, Jiang SG, Zhang DC. 2017. Characterization of complete mitochondrial genome of five stripe wrasse (Thalassoma quinquevittatum, Lay & Bennett, 1839) and phylogenetic analysis. Gene. 598:71–78. doi: 10.1016/j.gene.2016.10.042