
ABSTRACT
The most common strategy to assess the occupational exposure risk to hazardous drugs in cancer healthcare settings is by monitoring environmental contamination of work surfaces with cytotoxic drugs. Healthcare workers, especially pharmacy technicians and nursing, handle multiple hazardous drugs, so they really may be exposed to different cytotoxic drugs simultaneously. Therefore, simultaneous determination of several cytotoxic drugs provides a more accurate assessment of the real occupational exposure risk in healthcare settings. Our objective was to develop a method for surface wipe sampling and a quantification method for five cytotoxic drugs commonly used in chemotherapy treatments preparation: cyclophosphamide, 5-fluorouracil, epirubicin, gemcitabine and paclitaxel. Wiping procedure was performed using cellulose swabs moistened in a mixture solution containing acetonitrile, water, 2-propanol and methanol with 0.1% formic acid. The obtained extracts were simultaneously quantified by an LC-MS/MS method. Chromatographic separation was achieved using an Acquity UPLC® BEH C18 column by a gradient elution, and detection was carried out in multiple reaction monitoring mode with electrospray source operating in positive and negative ion mode. Wipe and extraction procedures developed were simple and easy to implement, being able to recover the cytotoxic target drugs from two type surfaces sampling, stainless steel surfaces and polyethylene bags surfaces. The analytical method was linear over the calibration range and the limits of quantification were 1 ng/mL for cyclophosphamide, gemcitabine and paclitaxel, 1.5 ng/mL for epirubicin and 5 ng/mL for 5-fluorouracil. Intra- and inter-day precision and accuracy for all drugs were always inferior to 15%. This work provides a suitable procedure to identify cytotoxic contamination on work surfaces, being a useful tool to monitor occupational exposure risk to hazardous drugs, as well as to evaluate the effectiveness of decontamination protocols applied and hazardous drug handling protocols in cancer healthcare settings.
1. Introduction
Handling hazardous drugs is associated with an occupational risk of residual exposure to cytotoxic drugs in cancer healthcare settings [Citation1,Citation2]. Both clinical and nonclinical workers may be exposed to hazardous drugs when they create aerosols, generate dust, clean up spills, or touch contaminated surfaces during the preparation, administration, or disposal of hazardous drugs. The most common routes of exposure are through inhalation, dermal absorption, accidental injection or oral via by the hand-to-mouth contact. At first, inhalation via droplets, particulates and vapours when creating aerosols was considered the main exposure route, but recent studies suggest that dermal contact with contaminated surfaces, during the preparation or handling of cytotoxic drugs, is actually the primary route of exposure [Citation3,Citation4]. The continuous and prolonged exposure to low doses of cytotoxic drugs can have mutagenic, embryotoxic, teratogenic and carcinogenic effects on healthcare workers. Although the long-term toxic effects of continued exposure have not been clearly established, and considering that latency period between exposure and the onset of a disease can be very long, the possible occupational risk that they represent and the serious consequences implied make it essential to adopt an adequate work system, with action measures that guarantee the safety of the healthcare workers, as well as the product and the environment.
Protection from hazardous drug exposures depends on safety programs established by employers and followed by workers. Factors that affect worker exposures include the following: drug handling circumstances (preparation, administration, or disposal), amount of drug prepared, frequency and duration of drug handling, potential for absorption, use of ventilated cabinets and work practices. The likelihood that a worker will experience adverse effects from hazardous drugs increases with the amount and frequency of exposure and the lack of proper work practices [Citation4,Citation5]. These programs should try to reduce occupational exposure to the lowest possible level and to reduce contamination generation by establishing safe cytotoxic drug handling protocols and adequate training of the personnel involved.
In order to assess the occupational exposure risk to hazardous drugs for healthcare workers and to be able to adopt the most suitable improvement actions, environmental contamination monitoring is required. Over last few years published safety guidelines for handling of hazardous drugs already recommend assessing environmental contamination on work surfaces and some of them also suggest the monitoring frequency [Citation6–13]. The most common practice to perform contamination monitoring is by work surfaces sampling, using wipes moistened in a cleaning solution, capable of recovering residual cytotoxic drugs from surface samples, on which an extraction method would be later applied. Thereafter, extracts obtained are analysed using a quantitative analytical technique in order to determine contamination level. Many authors have published methods of environmental sampling including a variety of cleaning materials, such as paper filters, swabs or tissues. A wide variety of solutions have also been described as wiping and extraction solutions, from sodium hydroxide or ammonium acetate solutions to aqueous solutions with different organic solvents [Citation6–16].
Currently, there is no standard for acceptable limits for cytotoxic drugs surface contamination. According to USP 797 (2004) data, levels higher than 1 ng/cm2 of cyclophosphamide can cause human absorption [Citation17]. In 2009, Schierl et al. published guidance values to facilitate interpretation of surface contamination monitoring. These guidance values were a statistical description of platinum and 5-fluorouracil surface monitoring data set from 102 German pharmacies. They proposed two threshold guidance values based on the 50th and 75th percentiles of all their results, so that contaminations below the 50th percentile were considered as good working practices and contaminations above 75th percentile were considered indicators of the need for optimising the procedures [Citation18]. Cyclophosphamide is frequently selected due to high toxicity, with high skin permeability, with possibility of vaporisation at room temperature and because large volumes of cyclophosphamide are prepared, which require to be previously dissolved. There are many studies using cyclophosphamide as marker drug have been published, so it available of sensitive analytical methods for its determination and these studies can be used as reference to compare results. In 2011, Sessink et al. analysed surface wiped samples and urine samples from healthcare workers, using cyclophosphamide as a marker drug and they concluded that an environmental contamination drug level lower than 0.1 ng/cm2 could be considered a safe reference value and values above 10 ng/cm2 could be considered not acceptable [Citation19]. In 2013, the MEWIP (Monitoring-Effect Study of Wipe Sampling in Pharmacies) project, a large-scale research study that included several cytotoxic drugs analysis, established a drug-independent guidance value of 0.1 ng/cm2 based on the 90th percentile of its results [Citation20]. At last USP published, USP 800 (2020), there is still no consensus and they reference again that more than 1 ng/cm2 of cyclophosphamide was found to cause human uptake [Citation21].
In order to quantify environmental contamination by monitoring samples, a highly sensitive analytical method with a low limit of quantification is needed. It has to be able to detect and quantify residual traces of cytotoxic drugs from surface samples [Citation22–28]. Different analytical methods are reported in the literature, ranging from liquid chromatography (LC) to gas chromatography (GC) coupled to different ultraviolet (UV) or mass spectrometry (MS) detection systems. Some of the published methods include a derivatisation step while others, to increase their sensitivity, incorporate a concentration step in the processing of the samples by liquid-liquid (LL) or solid phase (SPE) extraction. Mass spectrometry detection contributes with great sensitivity and specificity to the analytical methods and allows simultaneous analysis of different compounds. However, it requires complex equipment with a significant or an important associated economic cost, so they are not available in hospital pharmacy departments, in which chemotherapy treatments preparation is frequently centralised in Spain.
Some of the published methods perform quantification of two or three drugs [Citation8,Citation11,Citation15,Citation22,Citation23] including cyclophosphamide in combination with other drugs such as ifosfamide, both belonging to the same drug family [Citation6,Citation7,Citation12,Citation16,Citation24]. On publications that include a greater number of drugs, the analysis was performed under different chromatographic conditions or by separate analytical methods due to different physicochemical properties of drugs [Citation25,Citation26]. Consequently, there are few publications that perform the simultaneous determination of many drugs by using a single analytical method [Citation27,Citation28]. We consider simultaneous determination of several drugs to be a relevant improvement, as it gets a more complete and realistic assessment of the occupational exposure risk for healthcare workers, who handle multiple hazardous drugs.
The main objective of this work was, first, to develop a method for collecting surface samples in order to monitor environmental contamination from work surfaces. Second, to develop and validate an analytical method of liquid chromatography with detection by mass spectrometry in tandem (LC-MS/MS) which allowed to determine the presence of cytotoxic drugs. A wide variety of cytotoxic drugs are currently used for the preparation of chemotherapy treatments. However, five very highly used and toxic drugs were selected to be monitored as surrogate markers of contamination: cyclophosphamide (CP), 5-fluorouracil (5FU), epirubicin (EPI), gemcitabine (GEM) and paclitaxel (PAC).
2. Material and methods
2.1. Chemicals, reagents and materials
Certified reference standards of paclitaxel (853.9 g/mol; 99.2% pure), gemcitabine hydrochloride (299.66 g/mol; 99.9% pure) and epirubicin hydrochloride (579.98 g/mol; 96.4% pure) were purchased from European Pharmacopeia (European Directorate for the Quality of Medicines & HealthCare, Council of Europe, Strasbourg, France). Cyclophosphamide monohydrate (279.1 g/mol; ≥ 98% pure) and 5-fluorouracil (130.08 g/mol; ≥ 99% pure) were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Chemical structure and molecular formula are listed in .
Table 1. Chemical structure and molecular formula of the cytotoxic drugs selected for environmental contamination monitoring
LC-MS-grade water, methanol, acetonitrile and 2-propanol were obtained from Merck Millipore Group (Darmstadt, Germany). Formic acid, used as eluent additive for LC-MS, was supplied by Honeywell-Fluka (Seelze, Germany). Materials for wipe sampling were Texcel®-Pad, cellulose swabs pre-cut at 4 cm x 5 cm composed of 12 plies with high absorption, from Texpol (Barcelona, Spain).
2.2. Stock and working solutions, calibration standards and quality control solutions
From the certified reference standards, individual stock solutions of each target cytotoxic drug at a concentration of 1 mg/mL in methanol were prepared, distributed in aliquots and immediately stored at −80°C. Individual stock solutions were then freshly serial diluted to obtain three main working solutions containing all five drugs at 1, 10 and 100 µg/mL in a water and acetonitrile mixture (1:1, v/v). These main working solutions were further diluted in a mixture of acetonitrile, water, isopropanol and methanol (50:30:10:10, v/v) to obtain the calibrator standards and quality control solutions. Calibrator standards and quality control samples were freshly prepared before use. Concentrations of the calibration standards were 1, 2.5, 5, 15, 25 and 50 ng/mL for gemcitabine, cyclophosphamide and paclitaxel, 1.5, 2.5, 5, 15, 25 and 50 ng/mL for epirubicin and 5, 10, 15, 25, 50 and 100 ng/mL for 5-fluorouracil. Concentrations of the quality control samples were 5, 15 and 50 ng/mL for gemcitabine, cyclophosphamide, paclitaxel and epirubicin and 15, 25 and 100 ng/mL for 5-fluorouracil.
2.3. Equipment: LC-MS/MS conditions
Chromatographic analysis was carried out with an Acquity UPLC® integrated measurement system (Waters, Milford, MA, USA) which included a quaternary solvent manager, a sample manager-flow through needle and a thermostatic compartment with active column pre-heater. The chromatographic separation of the analytes was achieved on an Acquity UPLC® BEH C18 column (2.1 mm x 50 mm; packed with 1.7 µm particles) from Waters (Milford, MA, USA) with gradient elution. The mobile phases were: ultra-pure water with 0.01% formic acid (solution A) and acetonitrile (solution B), at the flow rate of 0.4 mL/min. Gradient elution scheme followed was: 100% A from 0 to 0.4 min, 70% B at 0.6 min, 100% B at 2 min, which was held for 1 min and finally the column was equilibrated for 2 min to the initial conditions. Total run time for each injection was 5 min and column temperature was kept at 40°C. The sample injection volume was 10 µL and the autosampler was held at a temperature of 10°C. After each injection cycle was done a washing of the needle with acetonitrile.
The chromatographic system was coupled to a triple quadrupole Xevo TQD® mass spectrometer (Waters, Milford, MA, USA) equipped with a Z-spray electrospray ionisation (ESI) source and controlled by Masslynx® version 4.1 software (Waters, Milford, MA, USA). Nitrogen was used as the nebulising and desolvation gas, and argon was used as collision gas. MS detector was operated in multiple reaction monitoring (MRM) mode and all the analytes were detected in positive ionisation, except 5-fluorouracil, which was analysed in negative ionisation mode. The optimised MS parameters were as follows: source temperature 110°C, desolvation temperature 400°C, desolvation gas flow rate 650 L/h, capillary potential 3.5 kV and 2.5 kV and cone voltage 32 V and 35 V for positive and negative ionisation, respectively. Acquisition, processing, integration and quantification of the chromatographic data was carried out using Masslynx® version 4.1 software (Waters, Milford, MA, USA).
2.4. Surface wipe samples preparation: wipe and extraction steps
To develop wipe and extraction procedures of surface samples, different solutions and cleaning materials were tested, evaluating which one results to have a greater recovery of the cytotoxic drugs on surface and that they were compatible with the subsequent step of chromatography analysis. These recovery tests were carried out on 10 cm x 10 cm areas of stainless-steel surface, which had previously been contaminated with a mixed standard solution that included all target cytotoxic drugs. Cleaning materials tested were cellulose swabs pre-cut at 4 cm x 5 cm and different sterile dry swabs composed of polystyrene or aluminium body with a cotton or viscose head. Wipe solutions tested were different aqueous mixtures with different proportions of one or more organic solvent (acetonitrile, methanol and 2-propanol), with or without eluent additives as formic or acetic acid. A single wiping or two consecutive wiping, following different directions of wipe movements and using one or more cleaning materials were also tested. Finally, different volumes of solutions, agitation times and speed in vortex and ultrasonic bath and centrifuged times were tested in order to optimise drugs recovery from cleaning material.
2.5. Analytical validation
A full validation procedure was performed evaluating linearity, limit of quantification (LQ), selectivity, carry-over, accuracy, precision, stability and recovery after sample preparation. According to the European Medicines Agency (EMA) guideline on bioanalytical method validation, the results obtained were evaluated [Citation29].
2.5.1. Linearity and limits of quantification
Six-level calibration standards in duplicate containing all target cytotoxic drugs were processed on different days. Calibration curves were generated by linear regression using a weighting factor of 1/X2 for gemcitabine and 1/X for all other drugs. Linearity was assessed by the average coefficient of determination (R2) of the calibration curves obtained on ten separate days. The limit of quantification is defined as the lowest concentration of the calibration curve, which can be measured with accuracy and precision within ± 20%. To estimate the limit of quantification, five replicates of lowest calibration standard were processed over one day.
2.5.2. Accuracy and precision
The accuracy and precision of the analytical method were evaluated by five independent analytical runs of quality controls at four-level concentrations: controls at low-, middle- and high-level concentration, and at the limit of quantification. All quality control samples were prepared in five replicates of each concentration per analytical run to determine the intra-day and inter-day accuracy and precision (repeatability and intermediate precision) of the assay. Accuracy and precision were estimated by calculating mean relative intra- and inter-daily biases and by coefficients of variation (CVs) on quality controls, respectively.
2.5.3. Stability
Stability studies included analysis of stock solutions stabilities for each of the five analytes and stabilities for samples. To evaluate stock solutions stability, individual stock solutions of each drug were conserved at −80°C for 6 months and were analysed at initial time and 1, 2 and 6 months. At initial time, when individual stock solutions were prepared, were used to obtain a mixed solution containing 100 ng/mL of each drug with which the peak area response for each drug was determined. After, at each stability time, a mixed solution was prepared from individual stock solutions and the peak area responses for each drug were compared with their response obtained at initial time. Mixed solutions were always prepared in triplicate, at initial time and in each evaluated stability times.
Samples stabilities studies included a short-term stability at refrigerated conditions and at in-autosampler. To evaluate samples short-term stability, quality control at low and high concentration were prepared and refrigerated at 2–8°C. These quality controls were analysed in triplicate at 0, 24 and 48 hours. To evaluate samples stability in autosampler, an analytical run of quality control at four-level concentration was reinjected at 24 hours, being conserved in autosampler at 10°C.
2.5.4. Recovery
Recovery of the target cytotoxic drugs in two different surfaces was analysed from surface control and quality control samples. Three independent analytical runs were analysed including three replicates of each type of control and at two-level concentrations. Low and high concentration of surface controls and quality controls were 5 ng/mL and 50 ng/mL for gemcitabine, epirubicin, cyclophosphamide and paclitaxel and 15 ng/mL and 100 ng/ml for 5-fluorouracil, respectively. To obtain the surface controls, firstly a volume of 50 µL of a mixed standard solution of the five drugs was spread using a micropipette over a 10 × 10 cm area of a stainless-steel surface and a polyethylene bag surface (NaCl 0.9%, 250 mL). Once the spread solutions had evaporated completely at room temperature, wipe and desorption procedures were applied on contaminated areas. For each evaluated surface, the recovery percentage for each drug was calculated from relationship between the response obtained from surface controls and quality controls, at two-level concentration. The resulting recovery rate was used as correction factor in order to quantify the level of target cytotoxic drugs on each type of surface.
3. Results
All preparations and dilutions were carried out following safe handling precautions, protocols for hazardous drugs and working in biological safety cabinets.
3.1. LC-MS/MS method
During LC-MS/MS conditions optimisation tests, the best compromise for all analytes was selected. For each target cytotoxic drug two transitions of mass-to-charge (m/z) were selected, the quantifier ion and the qualifier ion for confirmation, except 5-fluorouracil, which only the one m/z transition was selected as quantifier ion. MS/MS parameters configured and m/z transitions selected are summarised in . Typical MRM chromatograms of cytotoxic drugs are shown in .
Figure 1. Typical MRM chromatograms of cytotoxic drugs, obtained from a quality control with 100 ng/mL of 5-fluorouracil and 50 ng/mL of other analytes.
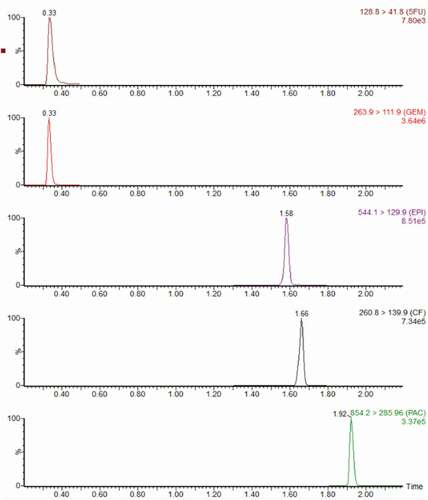
Table 2. MS/MS parameters and m/z transitions for cytotoxic drugs selected in study
3.2. Surface samples wipe and extraction procedure
Within wipe materials and solutions tested the best recovery results for all target cytotoxic drugs were provided by using cellulose swabs and a mixture solution of acetonitrile, water, 2-propanol and methanol (50:30:10:10, v/v) with 0.1% formic acid as a wipe solution. The surface wipe samples were collected by two consecutive wiping, using two cellulose swabs moistened with 0.1 mL of wipe solution each one of them and following two directions of wipe movements. The extraction procedure was then applied to the cellulose swab by vigorously agitation in vortex for 5 min and ultrasonication for 15 min following by a last centrifugation step.
3.3. Analytical validation
3.3.1. Linearity and limits of quantification
As shown in , analytical method was linear in the calibration ranges evaluated with R2 values greater than 0.99 for all target cytotoxic drugs. Calculated concentrations of the calibration standards were all within ± 20% of the nominal value for the limit of quantification and within ± 15% for all other calibration standards [Citation29], so calibration curves described adequately the concentration versus response relationship. The limits of quantification assessed were acceptable, with accuracy and precision values below 10% for all analytes.
Table 3. Linearity parameters and limits of quantification for the analytes
3.3.2. Selectivity and carry-over
All blank samples were free from any interference and no co-elution peaks were observed at the same mass transitions and retention times of the analytes. In carry-over assay results were considered correct and the peak area responses obtained in the three blank samples injected were not greater than 20% of limit of quantification of the analytes [Citation29]. Mean peak area responses observed in the blank samples after measurement of high-quality control were 0.57%, 0.26%, 2.46%, 0.45% and 0% for 5-fluorouracil, gemcitabine, epirubicin, cyclophosphamide and paclitaxel, respectively.
3.3.3. Accuracy and precision
Precision and accuracy values obtained of the analytical method are presented in , considered acceptable at all concentration levels of quality controls with values within 15% for quality controls and 20% for limit of quantification [Citation29]. Intra-day accuracy ranged between mean relative biases values of −8.4% and 3.5%, and repeatability ranged between coefficient of variation values of 0.7% and 9.0%. Inter-day accuracy ranged between −5.1% and 4.7%, and intermediate precision between 1.7% and 10.2%.
Table 4. Intra-day and inter-day accuracy and precision for the analytes
3.3.4. Stability
At each evaluated stability time individual stock solutions were considered to be stable, with a deviation percentage of peak area response of less than 15% [Citation29]. After solution storage at −80°C for 6 months, deviation percentage of peak area values were −13.1%, 0.5%, −0.4%, −12.6% and 3.2% for 5-fluorouracil, gemcitabine, epirubicin, cyclophosphamide and paclitaxel, respectively. Quality controls concluded that samples were stable while refrigerated at 48 hours, obtaining a mean concentration at each level concentration within ± 15% of the nominal concentration [Citation29]. Moreover, quality controls were stable in autosampler at 10°C for 24 h, with mean biases values within 15% for quality control and 20% for limit of quantification [Citation29]. In stability study results of quality controls are summarised.
Table 5. Short-term stability and in auto-sampler of quality control samples
3.3.5. Recovery
For all analytes, similar recovery percentages from low and high concentration surface controls were obtained, thus it was assumed that drug recovery is independent of concentration. The mean recovery of each drug was similar in both surfaces studied, except for paclitaxel which showed a slightly lower recovery on the polyethylene bag surfaces. Mean recoveries values of each target cytotoxic drug and by each surface are summarised in .
Table 6. Mean recoveries of the target cytotoxic drugs after their spreading on 100cm2 areas of stainless steel surfaces and polyethylene bag surfaces
4. Discussion
A simple and practical LC-MS/MS method was developed to be easily applied in the environmental cytotoxic contamination monitoring in occupational exposure risk assessment. Five cytotoxic drugs frequently used as chemotherapy treatments and associated with high toxicity in healthcare settings were selected to be monitored. The target cytotoxic drugs were 5-fluorouracil, gemcitabine, cyclophosphamide, epirubicin and paclitaxel.
During the LC-MS/MS method development, MS parameters and settings of each target cytotoxic drug were optimised manually by direct infusion into the MS/MS detector from mixed solution at concentration of 1 µg/mL of each analyte in a mixture of water and acetonitrile (50:50, v/v) with 0.1% formic acid. The MS responses of all analytes were tested under both ESI+ and ESI- ionisation modes and results obtained showed higher MS responses for the analytes under ESI+ mode, except for 5-fluorouracil, whose ESI- mode produced a higher MS signal intensity and more stable MS responses. Thus, initially polarity switching between ESI+ and ESI- modes was used in a single run. To avoid the loss of sensitivity during ESI positive-negative switching and to optimise conditions of each analyte, the MS detection process was divided into different times periods based on the LC retention times of the analytes.
After testing different elution gradients of the mobile phase and with or without addition of eluent additive to achieve a good resolution of peaks and to enhance MS sensitivity, under the chromatographic conditions selected, the total run time was 5 min, including the column equilibration time to ensure stable retention times. The retention times were 0.33 min, 0.34 min, 1.58 min, 1.66 min and 1.92 min for gemcitabine, 5-fluorouracil, epirubicin, cyclophosphamide and paclitaxel, respectively. Thus, three acquisition time periods were established: a first time period of 0.20 at 0.5 min, where 5-fluorouracil and gemcitabine were acquired with polarity switch, a second time period of 1.3 at 1.8 min, where cyclophosphamide and epirubicin were acquired in positive ionisation, and a third time period of 1.8 at 2.2 min, where only paclitaxel were acquired in positive ionisation.
Despite these acquisition time periods, because gemcitabine and 5-fluorouracil were chromatographic unresolved, the less dwell time involved in polarity switch resulted in low sensitivity, especially for 5-fluorouracil. Finally, in order to improve sensitivity and chromatographic reproducibility, ESI positive-negative switching in a single run was discarded and one injection in each ionisation mode per sample was carried out. At first injection, only 5-fluorouracil was acquired under ESI- and at second injection the other analytes were acquired under ESI+.
In method validation, different analytical parameters and stability conditions were evaluated. Method was linear over the calibration range and the limits of quantification were acceptable and compatible with the sensitivity required to determine of cytotoxic drug traces in environmental, considering the reference values recommended in the literature. Precision and accuracy, for all concentration levels of quality controls, were found within 15% of CV, so were better or at least similar to those of previous publications. No interfering peaks were present in any sample, nor was carry-over observed in any case. Based on the stability results obtained, all target cytotoxic drugs were stable at surface wipe samples for 48 hours at refrigeration conditions. Therefore, allowing collecting surface wipe samples in different centres and cooled shipping to a reference laboratory for analysis.
During the wipe and extraction steps development, different solutions and cleaning materials were tested. As cleaning materials were tested cellulose swabs and sterile dry swabs of different composition, with a polystyrene or aluminium body and cotton or viscose head. Sterile dry swabs were tested since they are easier to handle, with less risk of contamination, because only the head of swab is in direct contact with the surface to be wiped, but low recovery values for target cytotoxic drugs were obtained. Best recoveries were obtained using cellulose swabs. To avoid cross contamination during sample wiping, cellulose swabs were always handled with metal tweezers, which were cleaned with methanol before and after each use. As wipe solution several mixtures of water and one or more organic solvent were tested, with or without adding eluent additives as formic or acetic acid. Finally, the wipe solution selected was a mixture of acetonitrile, water, 2-propanol and methanol (50:30:10:10, v/v) with 0.1% formic acid.
All tests were carried out over 100 cm2 surface areas of stainless steel surfaces previously contaminated by spreading a mixed standard solution of all target cytotoxic drugs at known concentrations, which completely evaporated at room temperature. A single wiping or two consecutive wiping, using two cellulose swabs and following different directions of wipe movements, were also tested to improve the recovery rate. Best results were obtained when two consecutive surface wiping were performed, a first wipe movement from the top to the bottom and a second wipe movement from left to right. For each surface wiping, a cellulose swab cut at 2 × 2.5 cm and moistened with 0.1 mL of wipe solution was used. Both cellulose swabs collected per surface sample were placed into a single glass tube and then extraction procedure was applied.
Several conditions were tested to improve recovery of the analytes from cellulose swabs in extraction step: different mixture solutions and at different volumes were used, as well as different types and times agitation. Finally, the extraction procedure was performed in a final volume of 4 mL of same solution used at wipe step. Tubes were vigorously vortexed for 5 min and, to increase the recovery, were dispersed in an ultrasonic bath for 15 min. After that, tubes were centrifuged at 4000 rpm for 5 min to remove possible cellulose swab residues. Then, 200 µL of supernatant obtained were transferred into chromatographic vials.
The recovery of target cytotoxic drugs in two types of surfaces were also studied: stainless steel surface, the most common material used for work surfaces in the hazardous drug preparation units at hospital pharmacy services, and polyethylene bags surface (NaCl 0.9%, 250 mL), frequently used as containing bags for chemotherapy treatment preparations. Similar recovery rates were obtained for all drugs on both surfaces studied in the three independent series analysed with high and low concentration controls, being cyclophosphamide and paclitaxel the drugs with best recovery, while epirubicin recovery was close to 50%, 5-fluorouracil and gemcitabine presented lower recovery values, about 37%, probably due to adsorption or degradation issues. No differences were found in recovery values at high and low concentration controls, so it was assumed that recovery was independent of concentration. Thus, the recovery rate obtained for each drug was used as correcting factor to quantify the level of target cytotoxic drugs on each type of surface.
In conclusion, LC-MS/MS method described enables the rapid quantification of five cytotoxic drugs with different physicochemical properties (molecular size, polarity, etc.) in an aqueous solution with a high proportion of organic solvents. This type solution allows to sample surfaces and to analyse the presence of cytotoxic drugs traces on them, in order to monitor environmental contamination from surface wipe samples. The described method is suitable to identify cytotoxic environmental exposure at hospital pharmacy services, where chemotherapy treatment preparations are centralised. Moreover it can also be useful to evaluate the effectiveness of decontamination, cleaning and safe cytotoxic drug handling protocols, as well as their optimisation after improvement measures are applied.
Acknowledgements
The authors would like most sincerely thank pharmacy technicians of Pharmacy Department at Hospitalet centre of Catalan Institute of Oncology, who actively collaborated and facilitated study sample collection. Also, the authors thank Palex Medical S.A. (Company of Bidco Palex Group, Madrid, Spain) and Loccioni Company (Angeli di Rosora, Ancona, Italy) for support received and thank Waters Company (Milford, MA, USA) for technical assistance. Lastly, we also thank CERCA Programme/Generalitat de Catalunya for institutional support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- NIOSH Alert, Preventing Occupational Exposures to Antineoplastic and Other Hazardous Drugs in Healthcare Settings (NIOSH - National Institute for Occupational Safety and Health, Cincinnati, OH, USA, 2004).
- T.H. Connor and M.A. McDiarmid, CA Cancer J. Clin 56, 354 (2006). doi:https://doi.org/10.3322/canjclin.56.6.354.
- W. Fransman, R. Vermeulen and H. Kromhout, Int. Arch. Occup. Environ. Health 78, 403 (2005). doi:https://doi.org/10.1007/s00420-004-0595-1.
- L.A. Power and J.W. Coyne, Am. J. Health-Syst. Pharm 75, 1996 (2018). doi:https://doi.org/10.2146/ajhp180564.
- P.M. Lancharro, N. De Castro-Acuña Iglesias, F.J. González-Barcala and J.D. Moure González, Farm. Hosp 40, 604 (2016). doi:https://doi.org/10.7399/fh.2016.40.6.9103.
- A. Janes, C. Tanguay, N.J. Caron and J.F. Bussières, Can. J. Hosp. Pharm 68, 279 (2015). doi:https://doi.org/10.4212/cjhp.v68i4.1467.
- P.J. Sessink, T.H. Connor, J.A. Jorgenson and T.G. Tyler, J. Oncol. Pharm. Pract 17, 39 (2011). doi:https://doi.org/10.1177/1078155210361431.
- S. Viegas, M. Pádua, A.C. Veiga, E. Carolino and M. Gomes, Environ. Monit. Assess. 186, 7807 (2014). doi:https://doi.org/10.1007/s10661-014-3969-1.
- T.H. Connor, M.D. Zock and A.H. Snow, J. Occup. Environ. Hyg 13, 658 (2016). doi:https://doi.org/10.1080/15459624.2016.1165912.
- P. Marie, C. Christophe, R. Manon, M. Marc, B. Charleric and V. Patrice, Environ. Monit. Assess. 189, 2 (2017). doi:https://doi.org/10.1007/s10661-016-5762-9.
- A.W. Buning, T.H. Geersing and M. Crul, Int. J. Pharm. Pract 28, 66 (2020). doi:https://doi.org/10.1111/ijpp.12575.
- R.R. Larson, M.B. Khazaeli and H.K. Dillon, Am. J. Health-Syst. Pharm 59, 270 (2002). doi:https://doi.org/10.1093/ajhp/59.3.270.
- S. Valero-García, E. González-Haba, M.Q. Gorgas-Torner, J.M. Alonso-Herreros, A.C. Cercós Lletí, J.L. Poveda-Andrés, M.Á. Calleja-Hernandez and O. Delgado-Sánchez, Farm Hosp. 45, 2 (2021). doi:https://doi.org/10.7399/fh.11655.
- S. Nussbaumer, L. Geiser, F. Sadeghipour, D. Hochstrasser, P. Bonnabry, J.L. Veuthey and S. Fleury-Souverain, Anal. Bioanal. Chem 402, 2499 (2012). doi:https://doi.org/10.1007/s00216-011-5157-2.
- R. Schierl, C. Masini, S. Groeneveld, E. Fischer, A. Böhlandt, V. Rosini and D. Paolucci, J. Oncol. Pharm. Pract 22, 37 (2016). doi:https://doi.org/10.1177/1078155214551316.
- E. Ziegler, H.J. Mason and P.J. Baxter, Occup. Environ. Med 59, 608 (2002). doi:https://doi.org/10.1136/oem.59.9.608.
- USP <797> Pharmaceutical Compounding - Sterile Preparations. (2004).
- R. Schierl, A. Böhlandt and D. Nowak, Ann. Occup. Hyg. 53, 703 (2009). doi:https://doi.org/10.1093/annhyg/mep050.
- P.J. Sessink, 2011. Environmental Contamination with Cytostatic Drugs: Past, Present and Future.
- T.K. Kiffmeyer, J. Tuerk, M. Hahn, H. Stuetzer, C. Hadstein, A. Heinemann and U. Eixkmann, Ann. Occup. Hyg. 57, 444 (2013). doi:https://doi.org/10.1093/annhyg/mes081.
- USP General Chapter <800> Hazardous Drugs – Handling in Healthcare Settings. (2020) .
- G. Schmaus, R. Schierl and S. Funck, Am. J. Health Syst. Pharm 59, 956 (2002). doi:https://doi.org/10.1093/ajhp/59.10.956.
- L. Sabatini, A. Barbieri, M. Tosi and F.S. Violante, J. Mass. Spectrom 40, 669 (2005). doi:https://doi.org/10.1002/jms.840.
- C. Sottani, R. Turci, R. Schierl, R. Gaggeri, A. Barbieri, F.S. Violante and C. Minoia, Mass. Spectrom 21, 1289 (2007).
- J. Tuerk, T.K. Kiffmeyer, C. Hadtstein, A. Heinemann, H. Moritz, H. Stuetzer, H.M. Kuss and U. Eickmann, Intern. J. Environ. Anal. Chem 91, 1178 (2011). doi:https://doi.org/10.1080/03067319.2010.494769.
- A. Acramel, T. Chouquet, A. Plé, H. Sauvageon, S. Mourah, F. Jouenne and L. Goldwirt, Rapid Commun. Mass Spectrom. 34, e8594 (2020). doi:https://doi.org/10.1002/rcm.8594.
- S. Nussbaumer, S. Fleury-Souverain, P. Antinori, F. Sadeghipour, D.F. Hochstrasser, P. Bonnabry, J.L. Veuthey and L. Geiser, Anal. Bioanal. Chem 398, 3033 (2010). doi:https://doi.org/10.1007/s00216-010-4243-1.
- M. Jeronimo, M. Colombo, G. Astrakianakis and C.Y. Hon, Anal. Bioanal. Chem 407, 7083 (2015). doi:https://doi.org/10.1007/s00216-015-8868-y.
- EMA, European Medicines Agency. Guideline on Bioanalytical Method Validation. (2012).