Abstract
Intensive effort has been invested in the search for new effective vaccines that can be conveniently administered, with minimal handling of birds. Heat-labile enterotoxin (LT) of Escherichia coli H10407 is known as a powerful adjuvant for injection and oral administration. However, to use the toxin as an immunostimulator in animals, its toxicity must be neutralized. To this aim, we modified the LT gene by changing two amino acids (threonine 50 and valine 53) of the A subunit to proline. The modified non-toxic LT gene (nLT) was cloned and expressed in E. coli as a soluble protein. The protein was efficiently purified, reaching levels of about 20 mg active hexameric nLT per litre of induced culture. The mutated protein, nLT, and the wild-type protein (wtLT) were administered orally to chickens. Those treated with the wtLT exhibited diarrhoea, whereas chickens treated with the nLT showed no signs of disease compared with untreated birds. The new non-toxic, purified nLT stimulated antibody production in birds treated by injection or by oral administration. A field trial with layers that included a series of injections of Bovine Serum Albumin mixed with nLT showed this modified LT's ability to act as an adjuvant for the antigen mixed with it. This study demonstrates the efficient expression and purification of LT, in which toxicity was neutralized by genetic modification. Such an approach will enable the use of a non-toxic LT molecule with a modified A subunit by the poultry industry, to enhance immune responses against antigens co-vaccinated with it.
Détoxification génétique et conservation de l'activité adjuvante de l'entérotoxine LT d'Escherichia coli
De gros efforts ont été investis dans la recherche de nouveaux vaccins efficaces qui peuvent être administrés d'une manière commode, avec une manipulation minimale des animaux. L'entérotoxine labile à la chaleur d'Escherichia coli H10407 est connue comme étant un adjuvant puissant pour l'injection et l'administration orale. Cependant, pour utiliser la toxine comme un immunostimulant chez l'animal, sa toxicité doit être neutralisée. Dans ce but, nous avons modifié le gène LT en changeant en proline deux acides aminés (thréonine 50 et valine 53) de la sous unité A. Le gène LT modifié non toxique (nTL) a été cloné et exprimé dans E. coli comme une protéine soluble. La protéine a été efficacement purifiée, atteignant des niveaux de d'environ 20 mg de nLT héxamérique par litre de culture induite. La protéine mutée, nLT, et la protéine de type sauvage (wtLT) ont été administrées par voie orale à des poulets. Ceux traités avec la wtLT ont présenté de la diarrhée, alors que les poulets traités avec la nLT n'ont présenté aucun signe de la maladie, comparés aux poulets non traités. La nouvelle nLT purifiée non toxique a stimulé la production d'anticorps chez les poulets traités par injection ou par administration orale. Un essai terrain avec des pondeuses qui incluait des séries d'injections de BSA mélangée à la nLT a montré la capacité de cette LT modifiée à jouer le rôle d'adjuvant pour l'antigène mélangé avec elle. Cette étude montre l'efficacité de l'expression et de la purification de la LT dont la toxicité a été neutralisée par modification génétique. Une telle approche doit permettre l'utilisation, par l'industrie avicole, d'une molécule LT non toxique avec une sous unité A modifiée, pour améliorer les réponses immunitaires contre les antigènes qui entrent avec elle dans la composition des vaccins.
Genetische Entgiftung des Escherichia coli-Enterotoxins LT unter Aufrechterhaltung der Adjuvansaktivität
In die Suche nach neuen effektiven Vakzinen, die bequem und mit minimaler Handhabung der Vögel verabreicht werden können, wurden intensive Bemühungen gesteckt. Das hitzelabile Enterotoxin (LT) des Escherichia coli-Stamms H10407 ist als ein starkes Adjuvans der Injektion und der oralen Verabreichung bekannt. Für die Verwendung des Toxins als Immunstimulator in Tieren muss seine Toxizität jedoch neutralisiert werden. Zu diesem Zweck modifizierten wir das LT-Gen durch den Austausch von zwei Aminosäuren (Threonin 50 und Valin 53) der Untereinheit A gegen Prolin. Das veränderte nicht-toxische LT-Gen (nLT) wurde kloniert und in E. coli als lösliches Protein exprimiert. Das Protein wurde hochgereinigt, wobei in der induzierten Kultur Mengen von 20 mg des aktiven hexameren nLT pro Liter erreicht wurden. Das mutierte (nLT) und das Wildtyp-Protein (wtLT) wurden oral an Hühner verabreicht. Diejenigen, die das wtLT erhalten hatten, bekamen Diarrhoe, während die Hühner, die mit dem nLT behandelt worden waren, im Vergleich zu den unbehandelten Tieren keine Krankheitssymptome zeigten. Das neue nicht-toxische, gereinigte nLT stimulierte nach Injektion oder oraler Gabe in den Tieren eine Antikörperproduktion. In einem Feldversuch mit Legehennen konnte nach einer Serie von Injektionen von BSA gemischt mit nLT die Fähigkeit dieses modifizierten LTs nachgewiesen werden, in einer Mischung mit diesem Antigen als Adjuvans zu wirken. Diese Studie zeigt die effiziente Exprimierung und Reinigung von LT, dessen Toxizität durch genetische Modifikation neutralisiert wurde. Diese Methode ermöglicht in der Geflügelindustrie die Verwendung eines durch die Modifizierung der Untereinheit A nicht toxischen LT-Moleküls, um als Bestandteil der Vakzine die Immunantworten gegen Antigene zu verstärken.
Detoxificación genética y retención de la actividad adyuvante de la enterotoxina LT de Escherichia coli
Se han invertido grandes esfuerzos en la búsqueda de nuevas vacunas efectivas que puedan ser administradas convenientemente, con el mínimo manejo de las aves. La enterotoxina termolábil (LT) de Escherichia coli H10407 es conocida como un potente adyuvante para la administración oral y parenteral. Sin embargo, para usar esta toxina como inmunoestimulador en animales, su toxicidad debe ser neutralizada. Con este objetivo, modificamos el gen LT mediante la sustitución de dos aminoácidos (treonina 50 y valina 53) de la subunidad A por prolina. Se clonó el gen modificado LT no-tóxico (nLT) y se expresó en E. coli como una proteína soluble. La proteína se purificó eficientemente, llegando a niveles de 20 mg de hexámero nLT activo por litro de cultivo inducido. La proteína mutada, nlT, y la proteína de tipo salvaje se administraron oralmente a pollos. Aquéllos tratados con la wtLT mostraron diarrea, mientras que los pollos tratados con la nLT no mostraron signos de enfermedad, en comparación con las aves no tratadas. La nueva nLT no tóxica purificada estimuló la producción de anticuerpos en las aves tratadas mediante vía oral o parenteral. Un ensayo de campo en ponedoras que incluyó una serie de inyecciones de BSA en combinación con nLT mostraron esta capacidad de la LT modificada para actuar como adyuvante para el antígeno en combinación con éste. Este estudio demuestra la expresión y purificación eficientes de LT, en la cual se había neutralizado su toxicidad mediante modificación genética. Este tipo de enfoque permitirá el uso de la molécula de LT no-tóxica con la subunidad A modificada en la industria avícola, para mejorar la respuesta inmune frente a los antígenos combinados en la vacuna.
Introduction
The poultry industry is currently experiencing an increasingly pressing need for a new generation of effective vaccines that require minimal handling of birds during administration (Tripathy, Citation2004). The development of new vaccines includes approaches that involve pathogen modification and immune-system manipulation. Genetically engineered vaccines have many advantages, such as the prevention of disease outbreaks, the use of pure and well-characterized material, more efficient and less costly production, their use in multivalent vaccines, and the elimination of the hazards of pathogenicity intrinsic to inactivated or attenuated vaccines (Pitcovski et al., Citation2003). However, one of the problems of vaccination with recombinant antigenic subunit vaccines is low immunogenicity as compared with vaccination with the whole pathogen. Therefore, the development of effective adjuvants in parallel to new vaccines is a central task (Singh & O'Hagan, Citation1999; Gasparini et al., Citation2001).
Oral administration of vaccines is more convenient and economic for the poultry industry. Moreover, it stimulates a mucosal immune response and IgA production, which are important barriers for mucosal pathogens (McGhee et al., Citation1992; Boyaka et al., Citation1999; Ernst et al., Citation1999). To stimulate the immune response by oral vaccination, the antigen of interest has to be stable under digestive-tract conditions. Heat-labile enterotoxin of enteropathic Escherichia coli (LT) and cholera toxin of Vibrio cholera (CT) have both been found to be effective systemic and mucosal adjuvants, and are able to transfer antigens through the digestive tract (De Haan et al., Citation1999; Rappuoli et al., Citation1999; Hagiwar et al., Citation2001). In addition, LT has been found to enhance both cellular and humoral responses (Nicollier-Jamot et al., Citation2004). Enhancement of both branches of the immune system is known to yield more efficient vaccination (Hilleman, Citation1966; Gupta et al., Citation1993).
LT and CT exhibit a high similarity in structure and activity (Spangler, Citation1992). E. coli H10407 produces an LT that is pathogenic to humans, being the primary causative agent of traveller's diarrhoea. This LT is identical to one that is pathogenic to chickens, at the amino-acid level as well as biologically, immunologically and physiochemically (Sugii & Tsuji, Citation1989; Tsuji et al., Citation1990; Inoue et al., Citation1993). LT consists of two subunits: the 27-kDa catalytic A domain (LTA) anchored in a ring of five identical 11.6-kDa B subunits (LTBs). The A and B subunits are synthesized separately with a leader peptide, and transferred to the cell periplasm, where the leader peptides are removed and a toxin unit is assembled by non-covalent linkage between one LTA and five LTBs (Yamamoto & Yokota, Citation1983; Sixma et al., Citation1991; Spangler, Citation1992; Cheng et al., Citation1999). The B pentamers of LT bind to ganglioside receptors on the surface of eukaryotic cells, mainly GM1 (Holmgren et al., Citation1985; Fukuta et al., Citation1988; Sugii & Tsuji, Citation1989; Spangler, Citation1992). Following this binding, the toxin uses the host protein's retention and degradation pathways to reach the cytoplasm (Hazes & Read, Citation1997). In the cytosol, LTA is activated by digestive cleavage of the disulfide bond at amino acids 187 and 199, and proteolytic digestion at amino acids 192 and 195, to produce fragments A1 and A2 (Feil et al., Citation1996). The A1 fragment catalyses the transfer of an ADP-ribose from NAD to stimulatory α-subunits of G proteins (Gsα). After ADP-ribosylation, Gsα binds to adenylate cyclase and constitutively activates it, leading to a sustained increase in intracellular cyclic AMP concentration (Feil et al., Citation1996; Cassel & Selinger, Citation1977).
Although LT is highly immunogenic and stimulates both systemic and mucosal responses in the immune system (Hagiwar et al., Citation2001), its use as an adjuvant is limited, partially due to its toxicity. Moreover, studies have shown the importance of ADP-ribosyl transferase in LT's adjuvant activity (Lucke et al., Citation1992; Feil et al., Citation1996). LT detoxification has been achieved by changing the amino acids at a specific region on the A subunit while retaining adjuvant activity (Pizza et al., Citation1994; Douce et al., Citation1995 Citation1997; Feil et al., Citation1996; Giuliani et al., Citation1998; Cheng et al., Citation1999; Rappuoli et al., Citation1999). The use of non-toxic LT as an adjuvant may be safer and less costly than using other commercial adjuvants (Lebens & Holmgren, Citation1994; Partidos et al., Citation1996).
Another problem that limits the use of LT as an adjuvant is the difficulty involved in expressing large amounts of it and purifying it efficiently (Amin & Hirst, Citation1994; De Mattos Areas et al., Citation2002). The purpose of this research was to construct, express, purify and characterize the adjuvant activity of a mutant non-toxic LT molecule for its future use as an adjuvant.
Materials and Methods
Design and cloning of the wild-type protein and modified non-toxic LT genes
E. coli H10407 was kindly supplied by the American Tissue Culture Center. The plasmid carrying the LT was isolated with a Wizard Plus Minipreps kit (GibcoBRL, Paisley, UK). All primers were produced by Biotechnology General (Rehovot, Israel) according to the LT sequence (GenBank accession number AB011677). The primer homologous to the 5′ end of LT (A start), 5′-ggatccttaaacaaaacaagtggcg-3′, and the primer homologous to the 3′ end of LT (B stop), 5′-ctgcagctagttttccatactgattgccgc-3′, were used. Underlined letters indicate the position of BamHI (A start) and PstI (B stop) restriction sites. The restriction sites were added to the 5′ end of each primer to allow cloning of the resultant polymerase chain reaction (PCR) fragment into pQE30 plasmid (Qiagen, Valencia, California, USA). The gene coding for the wild-type LT protein (wtLT) was amplified by PCR, as described previously (Pitcovski et al., Citation1996). Briefly, the PCR solution contained 1 u Taq polymerase (Promega, Madison, Wisconsin, USA), 5 µl Taq buffer (20 mM Tris–HCl, pH 8.3, 50 mM KCl, 2 mM MgCl2) and 20 pmol each primer, in a final volume of 50 µl. The PCR scheme was as follows: 5 min at 94°C, 2 min at 42°C, 60 sec at 74°C (60 sec at 94°C, 2 min at 50°C, 3 min at 74°C) for 28 cycles, 15 min at 74°C. The full-length LT gene was used as a template to produce the modified non-toxic mutant (nLT), by two-step PCR amplification under the described conditions, using primers for the 5′ and 3′ ends of LT and two primers carrying mutations: change start 5′-cgcgagaggaacacaaccgggctttccgagatatgatgacgg-3′ and its reverse complementary primer, change stop 5′-ccgtcatcatatctcggaaagcccggttgtgttcctctcgcg-3′. The underlined letters are the substitutions of threonine and valine to proline at positions 50 and 53 in the A1 domain of LTA.
The first step of the PCR was carried out using the A start and Change stop primers, producing a 300-base-pair fragment of 5′ nLT, and another reaction using B stop and Change start primers, resulting in a 900-base-pair fragment of 3′ nLT. The second step used the two fragments produced in the first step as primers to each other, since they have complementary regions. This PCR step yielded the entire mutated code for nLT. The amplified segments (wtLT and nLT) were separated on a 0.8% agarose gel, and the appropriate bands were excised and purified with a PCR Products Purification System kit (GibcoBRL). The DNA was digested by restriction enzymes BamHI and PstI, incorporated into the primers. After additional purification, the fragments were ligated into multiple cloning sites of the pQE30 plasmid (Qiagen), which was digested by the same enzymes. The recombinant plasmids were transformed into E. coli JM109 (Invitrogen, San Diego, California, US) by heat-shock transformation according to the manufacturer's instructions. Bacterial colonies carrying the wtLT and nLT genes were detected by PCR, using the same primers as for cloning as earlier, and by analysis with restriction enzymes. The DNA sequences of these colonies were determined from both sides of the gene (Hebrew University, Biotechnology Services, Jerusalem, Israel) using primers of the plasmid and internal primers of the genes.
Expression of wtLT and nLT in E. coli
Bacteria carrying the wtLT-containing and nLT-containing plasmids with the correct sequences were grown on LB medium (10 g/l yeast extract, 5 g/l Bacto tryptone, and 10 g/l NaCl) supplemented with 100 µg/ml ampicillin from OD 0.5 to 0.7. Production of recombinant proteins was induced by adding 1 mM IPTG (isopropyl (β)-d-thiogalactoside) to the medium and growing the bacteria for 3 h at 37°C. The cells were centrifuged, the pellet was dissolved in disruption buffer (50 mM Tris–HCl, 50 mM NaCl, 1 mM EDTA, and pH 8.0), and 400 µg/ml lysozyme was added to the suspension, which was then incubated for 30 min at 4°C with gentle shaking. This was followed by the addition of 400 µg/ml DNase, 1 mg/ml deoxycholic acid, and 10 mM MgCl2, and incubation at room temperature for 30 min. The disrupted bacteria were centrifuged (30 min, 4°C, 17 000×g). The wtLT and the nLT proteins were found solubilized in the disruption buffer medium. Screening for colonies expressing the proteins was performed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting. The positive colonies were grown in 1 l LB medium containing ampicillin for large-scale expression of the recombinant proteins.
Purification of wtLT and nLT
The wtLT and nLT proteins were purified by affinity chromatography (Clements & Finkelstein, Citation1979; Guidry et al., Citation1997) and by size-exclusion chromatography (Takeda et al., Citation1981). The wtLT and nLT proteins were further purified by cation-exchange chromatography using 20 mM phosphate buffer, pH 7.2. The bacterial disruption medium (30 ml containing approximately 1 mg/ml recombinant proteins) was applied to 20 ml strong cation-exchange resin (S Sepharose fast-flow resin; Amersham-Pharmacia Biotech, Uppsala, Sweden) equilibrated in 20 mM buffered phosphate, pH 7.2, packed in an XK column (2.6×20 cm; Amersham-Pharmacia Biotech). The column was integrated with the AKTA prime system (Amersham-Pharmacia Biotech). After sample loading at 3 ml/min, the column was washed with five column volumes of equilibration buffer. Thereafter, the recombinant proteins were eluted in a gradient of 10 column volumes of 1 M NaCl in phosphate buffer. Fractions containing the wtLT and nLT, as determined by 12% SDS-PAGE, were pooled and stored for further analyses. The eluted fractions were analysed by 12% SDS-PAGE, western blotting and enzyme-linked immunosorbent assay (ELISA). The concentration of the purified wtLT and nLT was calculated by Bradford test (BioRad, Hercules, California, USA). Bacteria carrying the wild-type plasmid (pQE30) were processed in the same way as wtLT and nLT, for use as negative and positive controls.
Mass spectrometry analysis
Purified wtLT and nLT proteins were cut from 12% SDS-PAGE gel and sent to the Smoler Proteomics Center (Technion, Israel) for further analysis.
In-gel proteolysis
The proteins in the gel were reduced with 10 mM dithiothreitol (30 min, 60°C) and modified with 100 mM iodoacetamide in 10 mM ammonium bicarbonate (30 min, 24°C). The gel pieces were dehydrated with acetonitrile and rehydrated with 10% acetonitrile in 10 mM ammonium bicabonate containing modified trypsin (Promega) at a 1:100 enzyme-to-substrate ratio. The gel pieces were incubated overnight at 37°C and the resulting peptides were recovered and analysed.
Mass spectrometry analysis
The tryptic peptides were resolved by reverse-phase chromatography on 0.1×200-mm fused silica capillaries packed with reversed-phase material (Grace Vydac, California, USA). The peptides were eluted with linear 50 min gradients of 5 to 95% of acetonitrile with 0.1% formic acid in water at flow rates of 0.4 µl/min. Mass spectrometry was performed by an ion-trap mass spectrometer (LCQ-DecaXP; Finnigan, San Jose, California, USA) in a positive mode.
The mass spectrometry data were clustered and analysed using the Sequest software (J. Eng and J. Yates, University of Washington and Finnigan, San Jose), searching the NR-NCBI database or a specific database.
Protein and antibody assays
The assays were performed as described previously (Fingerut et al., Citation2005).
Western blotting
Supernatant of the disrupted bacteria was analysed by 12% SDS-PAGE and western blotting. A nitrocellulose filter (Hybond C; Amersham International, Little Chalfont, UK), after protein transfer from the polyacrylamide gel, was blocked in a milk buffer and the membrane was incubated for 1 h at 37°C with rabbit anti-CT antibody (Sigma, St Louis, Missouri, USA) diluted 1:1000 in milk buffer. Filters were washed twice in phosphate-buffered saline (PBS) and incubated with goat anti-rabbit IgG-peroxidase conjugate (Sigma) diluted 1:1000, followed by incubation with the substrate solution 3,3′-diaminobenzidine (Sigma).
Enzyme-linked immunosorbent assay
The ability of the wtLT and nLT proteins to bind to GM1 receptor was tested as described previously (Sanchez et al., Citation1990). ELISA plates were incubated for 2 h at 37°C or overnight at 4°C with GM1 receptor (monosialoganglioside-GM1; Sigma) diluted in PBS to a final concentration of 15 µg/ml. Each of the steps was followed by three washes in PBS buffer with 0.05% Tween 20 (v/v) and drying on a paper towel. Skim milk in PBS (1:1, v/v) was added for 1 h at 37°C as a blocking step. The tested protein was incubated for 1 h at 37°C and rabbit anti-CT antibody diluted 1:1000 in PBS buffer was used to detect the protein. This was followed by incubation with a secondary antibody—goat anti-rabbit IgG conjugated to horseradish peroxidase diluted 1:1000 in PBS buffer. A substrate solution, o-phenylenediamine dihydrochloride (Sigma), was added and the OD450 was determined by an ELISA reader (Lumitron). To test the recognition of CT by anti-wtLT or anti-nLT, ELISA plates were covered with GM1 receptor (monosialoganglioside-GM1; Sigma), and incubated with commercial CT as the antigen. The tested serum was diluted 1:250 in PBS buffer and added to the washed plates. Antibodies were detected by a secondary antibody, rabbit anti-chicken IgG conjugated to horseradish peroxidase and diluted 1:1000 in PBS buffer. OD450 was determined by an ELISA reader. To test the anti-Bovine Serum Albumin (BSA) antibodies, ELISA plates were covered with BSA diluted in carbonate-coating buffer (pH 9.6) to a final concentration of 1 mg/ml as the antigen. The sera were tested as previously.
In-vivo trials
The safety of nLT relative to the toxic wtLT protein was tested on three groups of laying hens, six birds per group, treated orally with: wtLT, 3 mg per bird; nLT, 3 mg per bird; or no treatment. The clinical symptoms and egg yield were followed for 10 days. Chicken excrement was collected twice before the treatment and 24 h after it. The same amount (15 g) of excrement was collected per bird, samples were pooled for each treatment group, and the percentage of water was calculated by weighing the excrement before and after 48 h desiccation at 65°C.
Immunogenicity of purified nLT protein was examined in six groups of laying hens, six birds per group. The birds were treated with: intramuscular injections of CT, of wtLT, or of nLT; or oral administrations of CT, of wtLT, or of nLT. Each bird was treated with 50 µg protein, twice with a 2-week interval. A purified fraction of the wild-type plasmid supernatant was used as a negative control in parallel to the LT protein fractions. No adjuvant was added to the proteins. Blood was drawn 3 weeks after the second vaccination and sera were kept at −20°C until use. The presence of antibodies in the sera was tested by western blotting and ELISA using CT as the antigen, as already described.
The adjuvant effect of purified nLT was examined. Two-week-old broilers were divided into five groups, six birds per group and treated by intramuscular injection twice, at a 2-week interval. The treatments were (per bird in a group): 100 µg BSA (Sigma); 50 µg nLT mixed with 100 µg BSA; 100 µg BSA with Freund's incomplete adjuvant (Sigma); 100 µg BSA mixed with a purified fraction of the wild-type plasmid supernatant, used as a negative control; or no treatment. Blood was drawn 3 weeks after the second vaccination and sera were kept at −20°C until use. The presence of antibodies in the sera was tested by western blotting and ELISA using CT and BSA as the antigens, as already described.
Statistical analyses
Data were evaluated by single-factor analysis of variance (ANOVA) for statistical significance.
Results
Design and cloning of wtLT and nLT genes
The E. coli H10407 plasmids were extracted and used as a template for wtLT and nLT synthesis. The wtLT (1.2 kb) was amplified by PCR using the A start and B stop primers. The nLT gene was constructed by two-step PCR. First 5′ nLT (0.3 kb) and 3′ nLT (0.9 kb) were amplified. In the second step, the two parts of the nLT were used as primers to each other and elongation resulted in the complete nLT gene (1.2 kb). The DNA fragments were purified, ligated into the multiple cloning site of the pQE30 plasmid and transformed into the E. coli bacteria. The colonies were screened and the sequences were determined. Colonies with the correct sequence of wtLT or nLT genes were used.
Expression of wtLT and nLT in E. coli
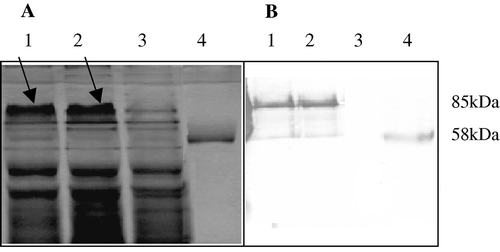
Following 3 h of stimulation with IPTG, the bacteria were disrupted and centrifuged. The supernatant was analysed by Coomassie Blue staining (a) and western blotting (b). The hexameric forms of wtLT and nLT were seen in the supernatant fraction of the disrupted bacteria (a, lanes 1 and 2, respectively) and identified by specific antibodies against CT at 85 kDa (b, lanes 1 and 2, respectively). No such band was seen in the negative control (a, b, lane 3). The CTB protein was used as a positive control (a, b, lane 4). wtLT and nLT proteins undergo partial disassembly, which is detected by anti-CT antibodies as B-subunit pentamers at 58 kDa (b, lanes 1 and 2, respectively).
Figure 1. Expression of wtLT and nLT on an SDS-polyacrylamide gel (1a) stained with Coomassie blue and (1b) on western blot. Lanes 1, 2 and 3, supernatant of bacteria expressing wtLT, nLT or wild-type plasmid, respectively; lane 4, commercial LTB pentamer of CT. Samples were loaded onto a 12% denaturing SDS-polyacrylamide gel without boiling to avoid separation of the pentameric structure into monomers. Anti-CT antibodies were used in the western blot.
Purification of wtLT and nLT
The eluted fractions from the affinity purification performed according to Clements & Finkelstein (Citation1979), as well as from the method based on size-exclusion chromatography (Takeda et al., Citation1981), contained the recombinant LT mixed with many other proteins (data not shown). Affinity chromatography performed according to Guidry et al. (Citation1997) yielded non-functional LT that could not bind to the GM1 receptor, as observed by ELISA using GM1 as the coating for the ELISA plates and anti-CT antibodies (purified nLT, OD450 nm 0.46±0.1; non-purified nLT, OD450 nm 1.81±0.04; negative control, OD450 nm 0.49±0.16). The supernatant of the disrupted bacteria containing the nLT protein was purified by cation-exchange chromatography. The proteins were bound to the column under neutral conditions and were eluted by a continuous NaCl gradient (). The nLT protein was eluted with over 90% purity (a, lane 3), identified by the commercial anti-CT (b, lane 3) and quantified by the Bradford test. Hexamers of LT were disassembled after boiling both unpurified and purified fractions for 3 min (a,b, lanes 2 and 4, respectively). Monomers of A (27 kDa) and B (11.6 kDa) were recognized in the boiled purified fraction stained with Coomassie blue (a, lane 4). The commercial anti-CT was able to recognize B monomers only (b, lanes 2 and 4). Hexamers of LT (a, lane 3), monomers of A and monomers of B (a, lane 4) were cut from gel and identified by mass spectrometry analysis. The total yield of nLT expressed and purified as described was 20 mg/l of induction culture at a concentration of 1.2 mg/ml elution buffer. Bacteria carrying the wtLT were processed in the same way as those carrying nLT (data not shown).
Figure 2. Purification of nLT protein by cation-exchange chromatography. 2a: Separation of the supernatant of disrupted bacteria containing wild-type plasmid (negative control). 2b: Separation of the supernatant of disrupted bacteria containing the nLT protein. The elution began at 45 min and the nLT was eluted at 55 min with 42% NaCl.
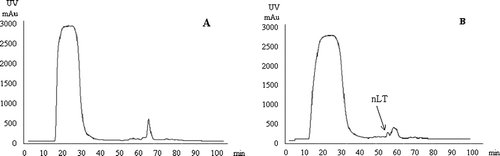
Figure 3. Cation-exchange-chromatography-purified nLT on an SDS-polyacrylamide gel (3a) stained with Coomassie blue and (3b) on western blot. Lanes 1 and 2, supernatant of bacteria expressing nLT before and after 3 min boiling, respectively; lanes 3 and 4, elution fraction of nLT (42% NaCl) before and after 3 min boiling, respectively. Samples were loaded onto a 12% denaturing SDS-polyacrylamide gel. Anti-CT antibodies were used in the western blot.
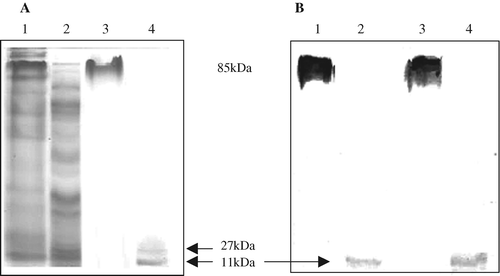
The purified proteins were detected only as hexamers and retained the ability to bind to the GM1 receptor, as observed by ELISA using GM1 as the coating for the ELISA plates and anti-CT antibodies (wtLT, OD450 nm 1.71±0.12; nLT, OD450 nm 1.76±0.9; CT, OD450 nm 1.79±0.04; negative control, OD450 nm 0.39±0.12). However, this ability partially disappeared following boiling, indicating that wtLT and nLT are assembled properly (wtLT, OD450 nm 0.49±0.02; nLT, OD450 nm 0.53±0.9; CT, OD450 nm 0.48±0.13; negative control, OD450 nm 0.36±0.5).
The safety of nLT
Each bird was orally administered 3 mg purified wtLT or nLT. Within approximately 3 h of the treatment, birds from the wtLT treatment group developed severe diarrhoea, which lasted for 48 h. Birds treated with nLT and untreated birds showed no signs of disease.
Chicken excrement was collected from the birds and its water content was calculated. The water percentage in the pooled chicken excrement was calculated by weighing the sample before and after desiccation. The water content in the chicken excrement prior to the treatment was 9.3 to 9.5% in all groups. More than threefold the percentage of water was found in the pooled excrement of birds treated with wtLT 24 h after the treatment (31.6%), as compared with excrement from the group treated with nLT and the untreated group (9.3 and 9.7% water, respectively). Furthermore, in comparison with the nLT-treated and untreated birds, the egg yield of birds from the wtLT-treated group decreased to one egg per bird every 2 days; after 10 days, yield returned to normal (approximately one egg per bird per day).
Immunogenicity of nLT protein
The purified nLT protein was intramuscularly injected or orally administered at 50 µg per bird. In response to vaccination with nLT, regardless of the administration method, antibodies were produced and detected by ELISA at levels similar to those produced by birds vaccinated with wtLT or CT. A significantly higher antibody titre was found in the experimental groups as compared with the negative control treatment (consisting of a fraction extracted in parallel from bacteria carrying the wild-type plasmid), which did not induce development of anti-CT antibodies (one-way ANOVA test; ).
Table 1. Anti-CT antibodies developed in response to vaccination with nLT, as tested by ELISA
The adjuvant effect of nLT
Broilers were treated by intramuscular injection of 50 µg nLT mixed with 100 µg BSA per bird. The ability of nLT to enhance anti-BSA antibody secretion was tested by ELISA, using BSA as the antigen. The birds in the nLT + BSA treatment group showed significantly higher levels of antibody against BSA, similar to the group treated with BSA mixed with a commercial adjuvant, relative to birds from the negative control treatment (the fraction extracted from bacteria carrying the wild-type plasmid, mixed with BSA) or those injected with pure BSA (one-way ANOVA test; ).
Table 2. Adjuvant effect of nLT, as tested by ELISA
Discussion
In this study, the A subunit of LT was genetically modified in order to neutralize its toxicity (Feil et al., Citation1996). The wtLT and nLT genes were efficiently expressed in bacterial cells and recognized by commercial anti-CT antibodies. The E. coli expression system, combining the pQE30 plasmid with bacterial strain JM109, was found to be a simple and efficient way of producing this toxin. Expression of the toxin in E. coli may enhance the probability of obtaining the protein in its correct form, which is essential for its adjuvant activity. In most studies, LT and CT have only been produced in relatively small quantities in the original organisms (E. coli H10407 or Vibrio cholerae, respectively), with the danger of reversible mutations that might induce the production of toxic LT (Sugii & Tsuji, Citation1988; Amin & Hirst, Citation1994; De Mattos Areas et al., Citation2002), or in complicated, inconvenient expression systems, such as bacteriophages or yeast (Feil et al., Citation1996). In the original bacteria, only part of the LT is secreted from the cell (Clements & Finkelstein, Citation1979). In contrast to other reports, we found a high concentration of nLT in the supernatant of the disrupted bacteria, and not in the inclusion bodies (De Mattos Areas et al., Citation2002). Soluble wtLT and nLT were found at a rate of 10% of the total soluble bacterial proteins. Several purification methods were tested before establishing the method detailed in this report. One aspect that must be taken into account when purifying LT is that following its purification, it must retain its ability to bind GM1 receptor, a requirement that is crucial for its immunostimulatory functions (Dickinson & Clements, Citation1995; Douce et al., Citation1995 Citation1997). A method based on the affinity of LTB pentamers to agar (Clements & Finkelstein, Citation1979) yielded the elution of many other soluble proteins together with the recombinant LT. Apparently, the method was designed for LT, which had been partially purified by crystallization (Cheng et al., Citation1999) or which had been secreted from the original bacteria (Tsuji et al., Citation1991). Affinity chromatography (Guidry et al., Citation1997), based on the recognition of LT by specific antibody followed by the former's elution, yielded non-functional LT that could not bind to the GM1 receptor. A method of separating the LT protein by size (Takeda et al., Citation1981) was not sensitive enough and elution fractions of LT were loaded with a large amount of irrelevant, similar-sized proteins. The most successful method proved to be ion-exchange chromatography, taking into consideration the physiochemical parameters of the protein. wtLT and nLT proteins were purified at pH 7.2, and the ability to bind GM1 receptor was retained. The purification conditions were adjusted to obtain only hexameric forms in the elution fraction. Since the pI of the whole toxin and each possible structure (e.g. subunit A, subunit B, or B pentamer) are different, only the hexamers are able to bind to the resin under such conditions. In this method, the protein was efficiently purified and relatively high yields (20 mg purified protein per litre) were obtained. In comparison, LT or CT and their subunits are usually obtained by different methods with similar purity at maximal yields of hundreds of micrograms or a few milligrams per litre (Tsuji et al., Citation1991; Amin & Hirst, Citation1994; Feil et al., Citation1996; De Mattos Areas et al., Citation2002).
LT was mutated to eliminate its toxic activity. Mutations at amino acid 50 (threonine to proline) or amino acid 53 (valine to proline) were tested separately by Feil et al. (Citation1996), and each was found to be considerably less toxic than the wild type. In this study, our goal was to produce a safe molecule, which could be employed in the future as a vaccine carrier or adjuvant in the poultry industry. Thus, in order to reduce the probability of reversal to the original toxic form, two mutations were inserted simultaneously. In order to prove the safety of nLT for use as an adjuvant, wtLT and nLT proteins were administered orally to birds. Birds treated with nLT showed no signs of disease, whereas birds treated with the toxic protein developed severe diarrhoea and exhibited decreased egg-laying. This result confirmed that the mutations in the LTA subunit had neutralized its toxic activity; in fact, even at a 10-fold higher concentration than that used in vaccines, nLT does not cause disease (Khoury & Meinersmann, Citation1995; Girard et al., Citation1999). On the other hand, the mutations did not influence nLT's capacity as a systemic and mucosal immunostimulator when administered by injection or orally. Moreover, the newly designed protein exhibited adjuvant activity when mixed with BSA as the antigen. The level of anti-BSA antibodies was significantly higher in birds vaccinated by injection with BSA + nLT than in those injected with BSA alone, and was similar to the antibody titre of birds vaccinated with BSA and a commercial adjuvant. In a study in our laboratory, a mixture of nLT with inactivated virus induced antibody production similar to the commercial vaccine, and significantly higher than the virus in PBS (data to be published).
In conclusion, this work describes the production of a genetically modified LT with neutralized toxicity. The methods established for the production and purification of the modified LT were found to be very efficient, yielding large amounts of highly purified protein. The modified LT protein was found to be safe and immunogenic in birds via both injection and oral administration and showed the ability to enhance immune responses against an antigen co-administered with it. nLT, produced and purified by the methods described in this study, may be used by the poultry industry as an efficient and inexpensive adjuvant for systemic and mucosal vaccinations.
Acknowledgments
The authors thank the Smoler Proteomics Center at the Technion, Israel, for the Mass spectrometry analysis.
Related Research Data
References
- Amin , T. and Hirst , T.R. 1994 . Purification of the B-subunit oligomer of Escherichia coli heat-labile enterotoxin by heterologous expression and secretion in a marine Vibrio . Protein Expression and Purification , 2 : 198 – 204 .
- Boyaka , P.N. , Marinaro , M. , Vancott , J.L. , Takahashi , I. , Fujihash , K. , Yamamoto , M. , van Ginkel , F.W. , Jackson , R.J. , Kiyono , H. and McGhee , J.R. 1999 . Strategies for mucosal vaccine development . The American Journal of Tropical Medicine and Hygiene , 60 : 35 – 45 .
- Cassel , D. and Selinger , Z. 1977 . Mechanism of adenylate cyclase activation by cholera toxin: inhibition of GTP hydrolysis at the regulatory site . Proceedings of the National Academy of Sciences of the USA , 74 : 3307 – 3311 .
- Cheng , E. , Cardenas-Freytag , L. and Clements , J.D. 1999 . The role of cAMP in mucosal adjuvanticity of Escherichia coli heat-labile enterotoxin (LT) . Vaccine , 18 : 38 – 49 .
- Clements , J.D. and Finkelstein , R.A. 1979 . Isolation and characterization of homogeneous heat-labile enterotoxins with high specific activity from Escherichia coli cultures . Infection and Immunity , 24 : 760 – 769 .
- De Haan , L. , Holtrop , M. , Verweij , W.R. , Agsteribbe , E. and Wilschut , J. 1999 . Mucosal immunogenicity and adjuvant activity of the recombinant A subunit of the Escherichia coli heat-labile enterotoxin . Immunology , 97 : 706 – 713 .
- De Mattos Areas , A.P. , Sarno de Oliveira , M.L. , Ramos , C.R. , Sbrogio-Almeida , M.E. , Raw , I. and Ho , P.L. 2002 . Synthesis of cholera toxin B subunit gene: cloning and expression of a functional 6Xhis-tagged protein in Escherichia coli . Protein Expression and Purification , 25 : 481 – 487 .
- Dickinson , B.L. and Clements , J.D. 1995 . Dissociation of Escherichia coli heat-labile enterotoxin adjuvanticity from ADP-ribosyltransferase activity . Infection and Immunity , 63 : 1617 – 1623 .
- Douce , G. , Turcotte , I. , Cropley , M. , Roberts , M. , Pizza , M. , Domenghini , R. , Rappuoli , R. and Dougan , G. 1995 . Mutant of Escherichia coli heat-labile toxin lacking ADP-ribosyltransferase activity act as nontoxic, mucosal adjuvants . Proceedings of the National Academy of Sciences of the USA , 92 : 1644 – 1648 .
- Douce , G. , Fontana , M. , Pizza , M. , Rappuoli , R. and Dougan , G. 1997 . Intranasal immunogenicity and adjuvanticity of site-direct mutant derivatives of cholera toxin . Infection and Immunity , 65 : 2821 – 2828 .
- Ernst , P.B. , Song , F. , Klimpel , G.R. , Haeberle , H. , Bamford , K.B. , Crowe , S.E. , Ye , G. and Reyes , V.E. 1999 . Regulation of the mucosal immune response . The American Journal of Tropical Medicine and Hygiene , 60 : 2 – 9 .
- Feil , I.K. , Reddy , R. , de Haan , L. , Merritt , E.A. , van den Akker , F. , Storm , D.R. and Hol , W.G.J. 1996 . Protein engineering studies of A-chain loop 47–56 of Escherichia coli heat-labile enterotoxin point to a prominent role of this loop for cytotoxicity . Molecular Microbiology , 20 : 823 – 832 .
- Fingerut , E. , Gutter , B. , Meir , R. , Eliahoo , D. & Pitcovski , J. (2005) . Vaccine and adjuvant activity of recombinant subunit B of E. coli enterotoxin produced in yeast . Vaccine , 23 , 4685 – 4696 .
- Fukuta , S. , Magnani , J.L. , Twiddy , E.M. , Holmes , R.K. and Ginsburg , V. 1988 . Comparison of the carbohydrate-binding specificities of cholera toxin and Escherichia coli heat-labile enterotoxins LTh-I, LT-IIa, and LT-Iib . Infection and Immunity , 56 : 1748 – 1753 .
- Gasparini , R. , Pozzi , T. , Montomoli , E. , Fragapane , E. , Senatore , F. , Minutello , M. and Podda , A. 2001 . Increased immunogenicity of the MF59-adjuvanted influenza vaccine compared to a conventional subunit vaccine in elderly subjects . European Journal of Epidemiology , 17 : 135 – 140 .
- Girard , F. , Pery , P. , Naciri , M. and Quere , P. 1999 . Adjuvant effect of cholera toxin on systemic and mucosal immune responses in chickens infected with E. tenella or given recombinant parasitic antigen per os . Vaccine , 17 : 1516 – 1524 .
- Giuliani , M.M. , Del Giudice , G. , Giannelli , V. , Dougan , G. , Douce , G. , Rappuoli , R. and Pizza , M. 1998 . Mucosal adjuvanticity and immunogenicity of LTR72, a novel mutant of Escherichia coli heat-labile enterotoxin with partial knockout of ADP-ribosyltransferase activity . The Journal of Experimental Medicine , 187 : 1123 – 1132 .
- Guidry , J.J. , Cardenas , L. , Cheng , E. and Clements , J.D. 1997 . Role of receptor binding in toxicity, immunogenicity, and adjuvanticity of Escherichia coli heat-labile enterotoxin . Infection and Immunity , 65 : 4943 – 4950 .
- Gupta , R.K. , Relyveld , E.H. , Lindblad , E.B. , Bizzini , B. , Ben-Efraim , S. and Gupta , C.K. 1993 . Adjuvants—a balance between toxicity and adjuvanticity . Vaccine , 11 : 294 – 306 .
- Hazes , B. and Read , R.J. 1997 . Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells . Biochemistry , 36 : 11051 – 11054 .
- Hagiwar , Y. , Tsuji , T. , Iwasaki , T. , Kadowaki , S. , Asanuma , H. , Chen , Z. , Komase , K. , Suzuki , Y. , Aizawa , C. , Kurata , T. and Tamura , S. 2001 . Effectiveness and safety of mutant Escherichia coli heat-labile enterotoxin (LT H44A) as an adjuvant for nasal influenza vaccine . Vaccine , 19 : 2071 – 2079 .
- Hilleman , M.R. 1966 . Critical appraisal of emulsified oil adjuvants applied to viral vaccine . Progress in Medical Virology , 8 : 131 – 132 .
- Holmgren , J. , Lindblad , M. , Fredman , P. , Svennerholm , L. and Myrvold , H. 1985 . Comparison of receptors for cholera and Escherichia coli enterotoxins in human intestine . Gastroenterology , 89 : 27 – 35 .
- Inoue , T. , Tsuji , T. , Koto , M. , Imamura , S. and Miyama , A. 1993 . Amino acid sequence of heat-labile enterotoxin from chicken enterotoxigenic Escherichia coli is identical to that of human strain H 10407 . FEMS Microbiology Letters , 108 : 157 – 161 .
- Khoury , C.A. and Meinersmann , R.J. 1995 . A genetic hybrid of the Campylobacter jejuni flaA gene with LT-B of Escherichia coli and assessment of the efficacy of the hybrid protein as an oral chicken vaccine . Avian Diseases , 39 : 812 – 820 .
- Lebens , M. and Holmgren , J. 1994 . Mucosal vaccines based on the use of cholera toxin B subunit as immunogen and antigen carrier . Developments in Biological Standardization , 82 : 215 – 227 .
- Lucke , N. , Tsuji , T. and Holmgren , J. 1992 . The adjuvant effect of Vibrio cholerae and Escherichia coli heat-labile enterotoxin is linked to their ADP-ribosyltransferase activity . European Journal of Immunology , 22 : 2277 – 2281 .
- McGhee , J.R. , Mestecky , J. , Derzbaugh , M.T. , Eldridge , J.H. , Hirasawa , M. and Kiyono , H. 1992 . The mucosal immune system: from fundamental concepts to vaccine development . Vaccine , 10 : 75 – 88 .
- Nicollier-Jamot , B. , Ogier , A. , Piroth , L. , Pothier , P. and Kohli , E. 2004 . Recombinant virus-like particles of a norovirus (genogroup II strain) administered intranasally and orally with mucosal adjuvants LT and LT(R192G) in BALB/c mice induce specific humoral and cellular Th1/Th2-like immune responses . Vaccine , 22 : 1079 – 1086 .
- Partidos , C.D. , Pizza , M. , Rappuoli , R. and Steward , M.W. 1996 . The adjuvant effect of a non-toxic mutant of heat-labile enterotoxin of Escherichia coli for the induction of measles virus-specific CTL responses after intranasal co-immunization with a synthetic peptide . Immunology , 9 : 483 – 487 .
- Pitcovski , J. , Di-Castro , D. , Shaaltiel , Y. , Azriel , A. , Gutter , B. , Yarkoni , E. , Michael , A. , Krispel , S. and Levi , B.Z. 1996 . Insect cell-derived VP2 of infectious bursal disease virus confers protection against the disease in chickens . Avian Diseases , 40 : 753 – 761 .
- Pitcovski , J. , Gutter , B. , Gallili , G. , Goldway , M. , Perelman , B. , Gross , G. , Krispel , S. , Barbakov , M. and Michael , A. 2003 . Development and large-scale use of recombinant VP2 vaccine for the prevention of infectious bursal disease of chickens . Vaccine , 21 : 4736 – 4743 .
- Pizza , M. , Fontana , M.R. , Giuliani , M.M. , Domenighini , M. , Magagnoli , C. , Giannelli , V. , Nucci , D. , Hol , W. , Manetti , R. and Rappuoli , R. 1994 . A genetically detoxified derivative of heat-labile Escherichia coli enterotoxin induces neutralizing antibodies against the A subunit . The Journal of Experimental Medicine , 180 : 2147 – 2153 .
- Rappuoli , R. , Pizza , M. , Douce , G. and Dougan , G. 1999 . Structure and mucosal adjuvanticity of cholera and Escherichia coli heat-labile enterotoxins . Immunology Today , 20 : 493 – 500 .
- Sanchez , J. , Holmgren , J. and Svennerholm , A. 1990 . Recombinant fusion protein for simple detection of Escherichia coli heat-stable enterotoxin by GMI enzyme-linked immunosorbent assay . Journal of Clinical Microbiology , 28 : 2175 – 2177 .
- Singh , M. and O'Hagan , D. 1999 . Advances in vaccine adjuvants . Nature Biotechnology , 17 : 1075 – 1081 .
- Sixma , T.K. , Pronk , S.E. , Kalk , K.H. , Wartna , E.S. , van Zanten , G.A.M. , Witholt , B. and Hol , W.G.J. 1991 . Crystal structure of a cholera toxin-related heat-labile enterotoxin from E. coli . Nature , 351 : 371 – 377 .
- Spangler , B.D. 1992 . Structure and function of cholera toxin and the related Escherichia coli heat-labile enterotoxin . Microbiological Reviews , 56 : 622 – 647 .
- Sugii , S. and Tsuji , T. 1988 . Hemagglutinating activity of the B subunits of the heat-labile enterotoxins isolated from chicken enterotoxigenic Escherichia coli . FEMS Microbiology Letters , 57 : 105 – 108 .
- Sugii , S. and Tsuji , T. 1989 . Binding specificities of heat-labile enterotoxins isolated from porcine and human enterotoxigenic Escherichia coli for different gangliosides . Canadian Journal of Microbiology , 35 : 670 – 673 .
- Takeda , Y. , Honda , T. , Taga , S. and Miwatani , T. 1981 . In vitro formation of hybrid toxins between subunits of Escherichia coli heat-labile enterotoxin and those of cholera enterotoxin . Infection and Immunity , 2 : 341 – 346 .
- Tripathy , D.N. 2004 . The impact of vaccines and the future of genetically modified poxvirus vaccines for poultry . Animal Health Research Reviews , 5 : 263 – 266 .
- Tsuji , T. , Joya , J.E. , Honda , T. and Miwatani , T. 1990 . A heat-labile enterotoxin (LT) purified from chicken enterotoxigenic Escherichia coli is identical to porcine LT . FEMS Microbiology Letters , 55 : 329 – 332 .
- Tsuji , T. , Honda , T. , Miwatani , T. and Miyama , A. 1991 . Detection and purification of the free A subunit of heat-labile enterotoxin produced by enterotoxigenic Escherichia coli . FEMS Microbiology Letters , 61 : 277 – 282 .
- Yamamoto , T. and Yokota , T. 1983 . Sequence of heat-labile enterotoxin of Escherichia coli pathogenic for humans . Journal of Bacteriology , 155 : 728 – 733 .