Abstract
In recent years polymerase chain reaction (PCR) assays have become widely used as methods to confirm the presence of Mycoplasma gallisepticum and Mycoplasma synoviae in poultry flocks, but there has been limited standardization of the protocols used. Thirteen laboratories from five different countries participated in an interlaboratory comparison of detection of M. gallisepticum and M. synoviae DNA by PCR in samples that contained 10-fold dilutions of these bacteria. The concentration of bacteria ranged from 105 to 102 genome copies/100 µl sample, as quantified by real-time PCR, and the samples were supplied on dry cotton swabs. Each laboratory was asked to use its standard method for PCR testing of these pathogens. A questionnaire was supplied with the samples to obtain details of the methods that were used in testing. One-half of the laboratories used a commercially available test kit, while the others used an in-house protocol. The protocols used for DNA extraction varied greatly, even among those using commercially available test kits. Two laboratories had developed the primers for nucleic acid amplification themselves, and one of these used real-time PCR for amplification. While the majority of the laboratories detected M. synoviae down to the 100 copy limit of the comparison, the detection limit for M. gallisepticum was somewhat higher. Furthermore, different results were obtained from laboratories that used the same commercial test kit. To the best of our knowledge this is the first investigation of its kind in the field of avian diseases.
Essai interlaboratoire pour la détection de l'acide nucléique de Mycoplasma gallisepticum et de Mycoplasma synoviae par PCR
Depuis quelques années les réactions de polymérisation en chaîne sont devenues des méthodes largement utilisées pour confirmer la présence de Mycoplasma gallisepticum et de Mycoplasma synoviae dans les troupeaux de volailles, mais il y a eu très peu de standardisation des protocoles utilisés. Treize laboratoires appartement à 5 pays différents ont participé à un essai interlaboratoire pour la détection de l'ADN de M. gallisepticum et de M. synoviae par PCR dans des échantillons qui contenaient des dilutions de 10 en 10 de ces bactéries. La concentration des bactéries allait de 105 à 102 copies de génome/100 µl d’échantillon, quantifiée par PCR en temps réel, et les échantillons étaient fournis sous forme d’écouvillon en coton sec. Il a été demandé à chaque laboratoire d'utiliser sa méthode standard pour la détection par PCR de ces agents pathogènes. Un questionnaire a été fourni avec les échantillons pour obtenir les détails des méthodes employées. La moitié des laboratoires ont utilisé un kit disponible dans le commerce, alors que les autres ont utilisé un protocole maison. Les protocoles utilisés pour extraire l'ADN étaient très différents, même parmi ceux qui utilisaient les kits du commerce. Deux laboratoires ont développé eux-mêmes les amorces pour l'amplification de l'acide nucléique, et l'un d'eux a utilisé la PCR en temps réel pour l'amplification. Alors que la majorité des laboratoires a détecté M. synoviae jusqu’à la limite inférieure de 100 copies, la limite de détection de M. gallisepticum a été quelque peu supérieure. De plus, des résultats différents ont été obtenus par des laboratoires qui utilisaient le même kit commercial. Autant que l'on sache, c'est la première étude de cette sorte dans le cadre des maladies aviaires.
Ringtest zum Vergleich Qualifikation des Nukleinsäurenachweises von Mycoplasma gallisepticum und Mycoplasma synoviae mittels PCR
In den letzten Jahren werden in zunehmenden Maße Tests basierend auf der Polymerasekettenreaktion (PCR) für den Nachweis von Mycoplasma gallisepticum und Mycoplasma synoviae in Geflügelherden verwendet, wobei die verwendeten Protokolle allerdings nur wenig standardisiert sind. Dreizehn Laboratorien aus fünf verschiedenen Ländern beteiligten sich an einem Ringtest, zum Nachweis der DNS von M. gallisepticum und M. synoviae mittels PCR, wobei Proben zur Verfügung gestellt wurden, welche die entsprechenden Bakterien in 10-facher Verdünnung enthielten. Die Bakterienkonzentration, welche mittels Realtime PCR quantifiziert wurde, reichte von 105 bis 102 Genomkopien/100µl Probe. Die Proben wurden auf trockenen Tupfern zur Verfügung gestellt und jedes Labor wurde angehalten, seine Standardmethode für den Nachweis dieser Pathogene durchzuführen. Weiters wurde jedem Labor ein Fragebogen übersandt, in welchem Details bezüglich der verwendeten Methode angegeben werden sollten. Die Hälfte der Labors verwendete kommerziell erhältliche Test-Kits, während andere wiederum ein hausinternes PCR-Protokoll benutzten. Große Unterschiede wurden vor allem in der DNS-Extraktionsmethode festgestellt, welche sogar unter jenen Labors variierte, die dieselben kommerziellen Test-Kits verwendeten. Zwei Labore arbeiteten mit selbst entwickelten Primern und eines mit Realtime PCR für die DNS-Amplifikation. Während der Großteil der Laboratorien M. synoviae in Konzentrationen bis zu 100 Genomkopien nachweisen konnte, lag das Detektionslimit für M. gallisepticum etwas höher. Des weiteren wurden unterschiedliche Ergebnisse auch von jenen Labors erzielt, die die gleichen kommerziellen Test Kits verwendeten. Die vorliegende Untersuchung ist die erste Ihrer Art auf dem Gebiet der Geflügelkrankheiten.
Comparación interlaboratorial de la capacidad para detectar los ácidos nucleicos de Mycoplasma gallisepticum y Mycoplasma synoviae mediante PCR
En los últimos años se ha extendido el uso de la reacción en cadena de la polimerasa (PCR) como método para confirmar la presencia de Mycoplasma gallisepticum y Mycoplasma synoviae en lotes de aves. Trece laboratorios de 5 países distintos participaron en una comparación interlaboratorial de la detección del DNA de Mycoplasma gallisepticum y Mycoplasma synoviae mediante PCR en muestras que contenían diluciones seriadas de logaritmo 10 de estas bacterias. La concentración de bacterias variaba entre 105 a 102 copias de genoma/100 µl de muestra, cuantificado a través de PCR a tiempo real, y las muestras se suministraron en hisopos de algodón secos. Se pidió a cada laboratorio que utilizaran su método estándar para el análisis mediante PCR de estos patógenos. Se suministró un cuestionario con las muestras para obtener detalles de los métodos que se usaron en el análisis. La mitad de los laboratorios utilizó kits comerciales, mientras que el resto utilizó protocolos propios. Hubo mucha diversidad entre los protocolos de extracción del DNA, incluso entre los que utilizaban kits comerciales. Dos laboratorios habían desarrollado cebadores propios para la amplificación de los ácidos nucleicos, y uno de ellos utilizaba PCR a tiempo real para esta amplificación. Mientras que la mayoría de los laboratorios detectó M. synoviae por debajo del límite de 100 copias de la comparación, el límite de detección de M. gallisepticum fue algo mayor. Además, se obtuvieron distintos resultados entre laboratorios que usaban los mismos kits comerciales. Que sepamos, este es el primer estudio de este tipo en el campo de las enfermedades aviares.
Introduction
Infections in poultry with Mycoplasma gallisepticum or Mycoplasma synoviae are a major concern for the poultry industry (Kleven, Citation2003; Ley, Citation2003). Horizontal transmission occurs within a flock and the progeny of breeder birds can be infected by vertical transmission (Bradbury, Citation2005). Prevention of infection and precise diagnosis of breeder flocks is therefore an important element in control strategies.
Various diagnostic methods have been described for confirmation of the presence of a mycoplasma infection (Kleven, Citation1998). Serological monitoring systems for detection of antibody against M. gallisepticum and M. synoviae are widely used in routine diagnosis, even though there are problems with sensitivity and specificity (Levisohn & Kleven, Citation2000). Isolation of the organism is laborious and time-consuming and therefore far from a routine procedure (Zain & Bradbury, Citation1996). Furthermore, culture takes several days and sometimes is confounded by overgrowth by other bacteria or suppression of growth by antimicrobial treatments administered to the birds. As a consequence, the polymerase chain reaction (PCR) has been regarded as a valuable tool under practical diagnostic conditions and may offer similar, or even higher, sensitivity than isolation (Kempf et al., Citation1993; Ewing et al., Citation1996; Salisch et al. Citation1998; Mekkes & Feberwee, Citation2005). Consequently, nucleic acid detection methods are preferred as an adjunct to serological investigations (Kempf, Citation1997; Feberwee et al., Citation2005). In general, PCR tests for diagnostic purposes in the poultry industry are being increasingly used because of the limitations of conventional methods.
Most veterinarians, producers and even laboratory scientists have little experience using molecular tests, especially in the diagnostic laboratory, which is very different from the research environment (Apfalter et al., Citation2005), but PCR tests to detect Mycoplasma species are on the list of assays of nearly every laboratory working in the field of avian diseases. In recent years several different commercial test kits have been introduced to supply the market with appropriate tools for routine testing.
This investigation was set up to compare PCR assays of different laboratories for detection of M. gallisepticum and M. synoviae. Protocols that were used in the individual laboratories for DNA extraction and PCR were compared, as were the results obtained when testing identical samples in the different laboratories. In order to supply identical samples to all participating laboratories, a DNA standard was developed to determine the number of copies of DNA extracted from cultured bacteria.
Materials and Methods
Participating laboratories
Thirteen laboratories from five different countries participated. Eleven of these laboratories used a single method to process the swabs—either a commercially available kit or an in-house system. After extraction of DNA, two laboratories used two completely different methods in parallel to process the samples. One of these laboratories obtained identical results for both methods, whereas the other one did not. As a consequence the two differing results obtained by the second laboratory were regarded as coming from two different laboratories (Laboratories 4 and 5). Consequently, 14 different results are presented in the tables.
M. gallisepticum and M. synoviae strains
The reference strains M. gallisepticum ATCC 19610 and M. synoviae ATCC 25204 were obtained from the American Type Culture Collection (ATCC). Each reference strain was suspended in 2 ml Mycoplasma liquid medium (Mycoplasma Experience, Reigate, UK). Following inoculation, the broth cultures were incubated at 37°C, and were examined daily for acidity, as indicated by a colour change from red to orange or yellow.
Calculation of rRNA gene copy numbers
DNA was extracted from 200 µl cultures of each reference strain using Chelex® 100 Resin (BioRad, Munich, Germany) according to the manufacturer's instructions. For amplification of the 16S rRNA genes from extracted DNA from M. gallisepticum the forward primer was MG14F (5′-gagctaatctgtaaagttggtc-3′) and the reverse primer was MG13R (5′-gcttccttgcggttagcaac-3′) (Lauerman, Citation1998), while the rRNA genes of M. synoviae were amplified using the forward primer MS-1 (5′-gaagcaaaatagtgatatca-3′) and the reverse primer MS-2 (5′-gtcgtctccgaagttaacaa-3′) (Lauerman et al., Citation1993).
Following amplification the PCR fragments were cloned using the TA Topocloning® Kit for Sequencing (Invitrogen, Lofer, Austria) according to the manufacturer's instructions. Plasmid concentrations were measured by spectrophotometry (Smartspec, BioRad, Munich, Germany) and number of copies calculated as described by Whelan et al. (Citation2003).
Real-time PCRs were performed with the Brilliant® Sybr® Green QPCR Master Mix (Stratagene, Amsterdam, The Netherlands) in 25 µl PCR mixture containing a primer concentration of 1 pmol. For both species the PCRs were performed in a Stratagene MX 3000 with an initial denaturation step of 95°C for 10 min followed by 40 cycles of denaturation at 95°C for 30 sec, annealing at 56°C for 1 min and extension at 72°C for 1 min. Standard curves were created using a 10-fold dilution series of each plasmid, with 2 µl each dilution used in the real-time PCRs.
Preparation of samples
The number of copies of the rRNA genes in the cultures of each of the mycoplasma strains was quantified and each culture was diluted to contain concentrations of 105 to 102 copies of rRNA gene/100 µl. The swabs of the four dilutions of M. gallisepticum ATCC 19610 were assigned sample numbers 1, 8, 12 and 4, respectively, and the swabs of the four dilutions of M. synoviae ATCC 25204 were assigned the numbers 11, 13, 6 and 3, respectively. In addition, a series of four swabs (samples 5, 9, 10 and 7) were supplied that contained a 50 µl volume of the dilutions of both reference strains. The total volume of 100 µl of each of the dilutions of M. gallisepticum ATCC 19610 or M. synoviae ATCC 25204, as well as the mixtures of both (2×50 µl), were absorbed into sterile cotton swabs (Sarstedt, Nürmbrecht, Germany), which then dried at room temperature. One negative swab (sample 2) that contained 100 µl broth media was also included. Each swab was supplied as an individual sample in a separate tube.
Results
Quantification of samples by real-time PCR
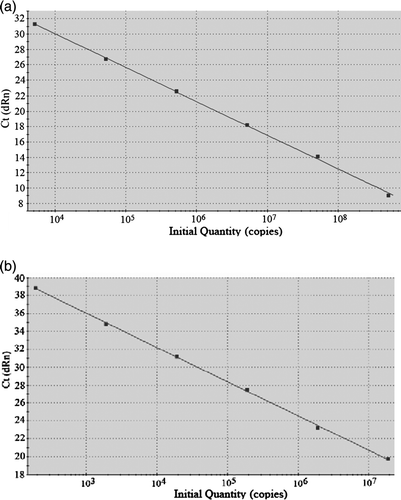
Standard curves derived from the 10-fold dilution series of M. gallisepticum ATCC 19610 and M. synoviae ATCC 25204 are shown in . Each reaction was performed in triplicate and the manufacturer's software was used to calculate the threshold cycle. Only reactions with an efficiency close to 100% were included in the calculation.
Figure 1. Standard curves derived from the 10-fold dilution series for (1a) M. gallisepticum and (1b) M. synoviae.
DNA extraction protocols
A summary of the extraction protocols used by the laboratories is presented in . There was considerable variation between the buffers used in the different protocols. Only six laboratories (Laboratories 1, 2, 7, 8, 12 and 13) processed the entire initial sample for DNA extraction, the others processing only a portion. For example, Laboratory 14 used only 2.5% of the material retrieved from the swabs. The extracted DNA was resuspended in different volumes of buffer, ranging from 25 µl to 250 µl. With the exception of laboratory 10, which used 15 µl extracted material, all other laboratories used 2 to 5 µl as template DNA in the PCR assay ().
Table 1. Methods used for DNA extraction and PCR according to the information supplied by each laboratory
PCR variables
One-half of the laboratories (Laboratories 1 to 7) used a commercial kit while the other seven laboratories used an in-house system (). Laboratories 1 and 2 used the same commercial test kit. A different commercial kit was used by Laboratories 3 and 4, and a third detection kit was used by three laboratories (Laboratories 5, 6 and 7). There were some variations in the protocol between laboratories using the same commercial test kit.
PCRs were performed in volumes of between 20 and 50 µl. With the exception of Laboratory 12, all used primers that hybridized within the 16S rRNA genes of M. gallisepticum or M. synoviae. Information on the target sequence was not supplied for the commercial kits as this information is not publicly available for some of the kits. In all laboratories the detection of M. gallisepticum and M. synoviae was done by separate PCR tests. Laboratory 14 used real-time PCR for detection. Some diversity was noticed with regard to the volume taken from the PCRs to confirm successful amplification of the nucleic acid. The processed volume ranged from 6% to 100%. One-half of the laboratories used at least 40% to 50% of the volume of the PCR to perform the final step of the protocol, the detection of amplified PCR products.
PCR results
The results of the PCR testing are presented in . All laboratories supplied the correct result for the negative control (sample 2). A false-positive result was only reported once. Laboratory 12 detected both M. gallisepticum and M. synoviae in sample 1, which contained only 105 copy numbers of the M. gallisepticum rRNA gene. Laboratory 8 provided a graded result for positive samples, ranging from + + + to +, which was in accordance with the concentrations on the swabs. Some laboratories (Laboratories 2, 3, 6 and 14) described samples that were closest to the detection limit as ‘weak positive’.
Table 2. Results from the different laboratories
Nearly all of the PCR results corresponded with the declining concentrations of target organisms supplied on the swabs. The exception was Laboratory 4, which reported a negative result for sample 6, which contained 103 copies of the M. synoviae rRNA gene, but a positive result for sample 3, which contained fewer bacteria. This also occurred at Laboratory 10 with the samples containing both M. gallisepticum and M. synoviae. Sample 9, with 104 copies, was reported as negative, while samples containing fewer bacteria were found to be positive.
The majority of laboratories detected the lowest concentrations of M. synoviae supplied (102 rRNA gene copies), whereas the detection limit for M. gallisepticum was no lower than 103 copies. Three laboratories (Laboratories 5, 7 and 9) detected M. gallisepticum only in samples with the highest concentrations supplied (105 copies).
Discussion
This investigation was set up to compare the results and methods of several laboratories performing PCR tests to diagnose the presence of M. gallisepticum and M. synoviae. Dried swabs were supplied to standardize the samples, even though wet swabs are sometimes used in field diagnostic investigations in order to allow isolation by culture (Zain & Bradbury, Citation1996).
A questionnaire enabled identification of variations in the DNA extraction methods. The dilution factor was calculated at two different stages of the procedure in order to investigate the possible influence of this on results. However, the extraction procedure had very little influence on the outcome of the PCR. Laboratories 12 and 13, and Laboratories 3 and 4, obtained different results and had different detection limits even though they transferred identical quantities of the samples through the steps of the extraction procedure. The extraction procedure also had little influence on the results obtained by the laboratory that analysed the samples by real-time PCR and achieved very high analytical sensitivities, even though a relatively small proportion of the extracted DNA was used in the assay. This might be expected given the high sensitivity reported for the detection of M. gallisepticum by real-time PCR (Carli & Eyigor, Citation2003) and confirmed by comparing this PCR technique with isolation (Mekkes & Feberwee, Citation2005).
All of the laboratories that commented about the intensity of the PCR signal obtained detected the weakest signal from the samples with the lowest concentrations of bacteria, whether the swabs contained one or both species. This confirmed the validity of the test and the samples that were supplied, despite the differing detection limits of the assays performed by different laboratories. As every laboratory processed each sample for M. gallisepticum and M. synoviae, a total of 364 (14×26) PCRs were performed by the laboratories and only one false-positive result (Laboratory 12) was obtained. One possible reason for this may be that the primers used by this laboratory may have been cross-reactive when high concentrations of DNA are present in the samples, as none of the other laboratories used the same primers to detect M. gallisepticum. To some extent this demonstrates the high standard of the participating laboratories, keeping in mind that processing a large number of samples at the same time increases the risk of cross-contamination of samples, potentially creating false positives (Kwok & Higuchi, Citation1989). This has been reported in a previous ring test in which field samples were used for investigation (Hess et al., Citation2004).
Only two laboratories obtained false negative results. However, to address this issue more fully a second testing method would need to be included, preferably isolation by culture, but this would not reflect the usual diagnostic situation. The influence of the medium on the results could be excluded as all samples were set up in an identical way using the same volume of medium. Two of the PCR kits used include an internal positive control, which is co-amplified in every sample and demonstrates the absence of inhibitors of the PCR. The PCR products obtained from the internal positive control can be easily distinguished from those obtained from the target sequences as they have different amplicon lengths. Therefore, false negative results due to inhibition of the PCR can be easily identified when using PCR in diagnostic investigations (Moalic et al., Citation1998; Carli & Eyigor, Citation2003). However, the presence of an internal positive control may interfere with amplification of the target DNA, resulting in false negative results (Sachadyn & Kur, Citation1998). In our investigation there was no evidence of an influence of internal positive controls on the likelihood of obtaining false negative results. Furthermore, the presence of both M. gallisepticum and M. synoviae in a sample had no influence on the likelihood of obtaining false negative results. In fact, some laboratories achieved a 10-fold higher sensitivity with mixed samples than with those containing a single species. This is difficult to explain, but it was noticed in the majority of laboratories. In swabs that contained both bacteria the samples used to inoculate the swabs were concentrated twofold in order to ensure that the volume of medium on the swab (100 µl) was identical to the volume on swabs containing only single species and the negative control.
The preparation of reference samples was based upon the determination of the number of copies of bacterial DNA using a standard curve established with a purified plasmid in which the gene for the 16S rRNA had been cloned (Whelan et al., Citation2003). This was considered more appropriate than enumeration of bacteria by culture for a comparison of PCR tests, as PCR is able to detect both viable and non-viable, or non-culturable, organisms. In addition, damaged and degraded DNA may serve as a template for PCR. This explains the numerous reports in which positive PCR results could not be confirmed by culture, as demonstrated recently for environmental samples and samples taken from experimentally infected birds (Marois et al., Citation2002). In our study none of the laboratories detected 100 copies of the M. gallisepticum rRNA gene. The decreased sensitivity of detection of M. gallisepticum could be a limitation of certain PCR protocols under practical conditions. In this context it needs to be considered that the calculation of the numbers of copies was performed with primers that hybridize to the 16S rRNA gene, and that there are two copies of this gene on the M. gallisepticum and M. synoviae genomes (Papazisi et al. Citation2003; Vasconcelos et al., Citation2005). This indicates that the detection limit is actually one-half of that presented in . However, Garcia et al. (Citation2005) found no differences in sensitivity between PCRs that targeted a single copy gene (mgc2) and those that targeted the 16S rRNA gene.
Test specificity was only assessed by examining the potential for cross-recognition of M. gallisepticum and M. synoviae, and other influences on specificity should be further examined in future investigations.
Most PCR diagnostic assays for mycoplasmas rely on detection of highly conserved regions of the 16S rRNA, and the low levels of genetic variation in this region reduce the likelihood that some strains will not be detected (Weisburg et al., Citation1989). In our investigation only one of the laboratories using an in-house system used oligonucleotides that did not hybridize with the 16S rRNA gene. It may well be that the relatively low sensitivity of the assays performed by this laboratory is compensated for by a somewhat increased specificity. Different levels of sensitivity between PCRs targeting four different genes of M. gallisepticum have been reported recently (Garcia et al., Citation2005). In this investigation the M. gallisepticum 16S rRNA gene PCR developed by Lauerman (Citation1998) had a higher analytical sensitivity than the other PCR methods tested. However, the 16S rRNA PCR method amplified DNA not only from M. gallisepticum but also from Mycoplasma imitans. These two phylogenetically related avian mycoplasmas have very similar 16S rRNA genes, and PCR primers that are targeted to this gene amplify both species (Boyle, Citation1993; Harasawa et al., Citation2004). M. imitans has been isolated from ducks, geese and partridges and therefore is considered of limited significance in diagnoses on samples from chickens (Abdul-Wahab et al., Citation1996).
Laboratory 7 achieved a much greater sensitivity of detection of M. synoviae than the other two laboratories (Laboratories 5 and 6) that used the same commercial kit. Comparison of the different test systems themselves is not appropriate for several reasons. For example, one test is only licensed for research purposes and one of the test kits used recommends the transport of the swabs in specific transport medium. This limits the comparison, as samples were not handled and the tests not used as the manufacturers recommend. However, the present investigation better reflects the situations where swabs are collected for PCR testing on several pathogens at the same time. In addition, the number of laboratories using the same test kit was too low to enable a full comparison. If more laboratories participate in future investigations, a scoring system could be established to enable the comparison of the outcomes of individual tests.
However, as more test kits become available on the market and are used by an increasing number of laboratories, the tendency will be to improve these test systems due to the increasing competition. This will further increase the standardization and q uality of the PCR diagnosis of mycoplasma infections in poultry. In this context, interlaboratory investigations are of some benefit for all the partners involved in order to establish a quality control system, and not only for in-house protocols, which are usually less well evaluated and therefore more prone to error under uncontrolled conditions (Apfalter et al., Citation2005).
Acknowledgement
The authors wish to thank the laboratories that participated in this investigation for their fruitful collaboration and their suggestions for further improvements.
References
- Abdul-Wahab , O.M.S. , Ross , G. and Bradbury , J.M. 1996 . Pathogenicity and cytadherence of Mycoplasma imitans in chicken and duck embryo tracheal organ cultures . Infection and Immunity , 64 : 563 – 568 .
- Apfalter , P. , Reischl , U. and Hammerschlag , M.R. 2005 . In-house nucleic amplification assays in research: How much quality control is needed before one can rely upon the results? . Journal of Clinical Microbiology , 43 : 5835 – 5841 .
- Boyle , J.S. ( 1993 ). Phylogeny and diagnosis of several avian mycoplasma species . Honours Thesis , The University of Melbourne , Australia .
- Bradbury , J.M. 2005 . Poultry mycoplasmas: Sophisticated pathogens in simple guise . British Poultry Science , 46 : 125 – 136 .
- Carli , K.T. and Eyigor , A. 2003 . Real-time polymerase chain reaction for Mycoplasma gallisepticum in chicken trachea . Avian Diseases , 47 : 712 – 717 .
- Ewing , M.L. , Lauerman , L.H. , Kleven , S.H. and Brown , M.B. 1996 . Evaluation of diagnostic procedures to detect Mycoplasma synoviae in commercial multiplier-breeder farms and commerical hatcheries in Florida . Avian Diseases , 40 : 798 – 806 .
- Feberwee , A. , Mekkes , D.R. , De Wit , J.J. , Hartman , E.G. and Pijpers , A. 2005 . Comparison of culture, PCR, and different serologic tests for detection of Mycplasma gallisepticum and Mycoplasma synoviae infections . Avian Diseases , 49 : 260 – 268 .
- Garcia , M. , Ikuta , N. , Levisohn , S. and Kleven , S.H. 2005 . Evaluation and comparison of various PCR methods for detection of Mycoplasma gallisepticum infection in chickens . Avian Diseases , 49 : 125 – 132 .
- Harasawa , R. , Pitcher , D.G. , Ramirez , A.S. and Bradbury , J.M. 2004 . A putative transposase gene in the 16S–23S rRNA intergenic spacer region of Mycoplasma imitans . Journal of Microbiology , 150 : 1023 – 1029 .
- Hess , M. , Neubauer , C. & Morrow , C. ( 2004 ). Durchführung eines PCR-Ringtestes zum Nachweis von Mycoplasma gallisepticum und Mycoplasma synoviae bei Mastelterntieren . Referatesammlung 66 (pp. 65 – 70 ). Hannover , , Germany : Fachgespräch Fachgruppe Geflügelkrankheiten .
- Kempf , I. 1997 . DNA amplification methods for diagnosis and epidemiological investigations of avian mycoplasmosis . Acta Veterinaria Hungaria , 45 : 373 – 386 .
- Kempf , I. , Blanchard , A. , Gesbert , F. , Guittet , M. and Bennejean , G. 1993 . The polymerase chain reaction for Mycoplasma gallisepticum detection . Avian Pathology , 22 : 739 – 750 .
- Kleven , S.H. 1998 . “ Mycoplasmosis ” . In A Laboratory Manual for the Isolation and Identification of Avian Pathogens , 4th edn , Edited by: Swayne , D.E. , John , R. , Jackwood , M.W. , Pearson , J.E. and Reed , W.M. 100 – 105 . Kennett Square , PA : University of Pennsylvania, New Bolton Center .
- Kleven , S.H. 2003 . “ Mycoplasma synoviae infection ” . In Diseases of Poultry , 11th edn , Edited by: Saif , Y.M. , Barnes , H.J. , Glisson , J.R. , Fadly , A.M. , McDougald , L.R. and Swayne , D.E. 756 – 766 . Ames , IA : Iowa State Press .
- Kwok , S. and Higuchi , R. 1989 . Avoiding false positives with PCR . Nature , 339 : 237 – 238 .
- Lauerman , L.H. 1998 . “ Mycoplasma PCR assays ” . In Nucleic Acid Amplification Assays for Diagnosis of Animal Diseases , Edited by: Lauerman , L.H. 41 – 52 . Davis , CA : American Association of Veterinary Laboratory Diagnosticians .
- Lauerman , L.H. , Hoerr , F.J. , Sharpton , A.R. , Shah , S.M. and van Santen , V.L. 1993 . Development and application of a polymerase chain reaction assay for Mycoplasma synoviae . Avian Diseases , 37 : 829 – 834 .
- Levisohn , S. and Kleven , S.H. 2000 . “ Avian mycoplasmosis (Mycoplasma gallisepticum) ” . In Diseases of Poultry: World Trade and Public Health Implications , Edited by: Beard , C.W. and McNulty , S. Vol. 19 , 425 – 442 . Paris , , France : Office International des Epizooties .
- Ley , D.H. 2003 . “ Mycoplasma gallisepticum infection ” . In Diseases of Poultry , 11th edn , Edited by: Saif , Y.M. , Barnes , H.J. , Glisson , J.R. , Fadly , A.M. , McDougald , L.R. and Swayne , D.E. 722 – 744 . Ames , IA : Iowa State Press .
- Marois , C. , Dufour-Gesbert , F. and Kempf , I. 2002 . Polymerase chain reaction for detection of Mycoplasma gallisepticum in environmental samples . Avian Pathology , 31 : 163 – 168 .
- Mekkes , D.R. and Feberwee , A. 2005 . Real-time polymerase chain reaction for qualitative and quantitative detection of Mycoplasma gallisepticum . Avian Pathology , 43 : 348 – 354 .
- Moalic , P.Y. , Gesbert , F. and Kempf , I. 1998 . Utility of an internal control for evaluation of a Mycoplasma meleagridis PCR test . Journal of Veterinary Microbiology , 61 : 41 – 49 .
- Papazisi , L. , Gorton , T.S. , Kutish , G. , Markham , P.F. , Browning , G.F. Nguyen , D.K. 2003 . The complete genome sequence of the avian pathogen Mycoplasma gallisepticum strain Rlow . Microbiology , 149 : 2307 – 2316 .
- Sachadyn , P. and Kur , J. 1998 . The construction and use of a PCR internal control . Molecular and Cellular Probes , 12 : 259 – 262 .
- Salisch , H. , Hinz , K.H. , Graack , H.D. and Ryll , M. 1998 . A comparison of a commercial PCR-based test to culture methods for detection of Mycoplasma gallisepticum and Mycoplasma synoviae in concurrently infected chickens . Avian Pathology , 27 : 142 – 147 .
- Vasconcelos , A.T. , Ferreira , H.B. , Bizarro , C.V. , Bonatto , S.L. , Carvalho , M.O. Pinto , P.M. 2005 . Swine and poultry pathogens: The complete genome sequences of two strains of Mycoplasma hyopneumoniae and a strain of Mycoplasma synoviae . Journal of Bacteriology , 187 : 5568 – 5577 .
- Weisburg , W.G. , Tully , J.G. , Rose , D.L. , Petzel , J.P. , Oyaizu , H. Yang , D. 1989 . A phylogenetic analysis of the mycoplasmas: Basis for their classification . Journal of Bacteriology , 12 : 6455 – 6467 .
- Whelan , J.A. , Russell , N.B. and Whelan , M.A. 2003 . A method for the absolute quantification of cDNA using real-time PCR . Journal of Immunological Methods , 278 : 261 – 269 .
- Zain , Z.M. and Bradbury , J.M. 1996 . Optimising the condition for isolation of Mycoplasma gallisepticum collected on applicator swabs . Journal of Veterinary Microbiology , 49 : 45 – 57 .