Abstract
Live attenuated vaccines have been used for control of the disease caused by goose parvovirus (GPV), but the mechanism involved in attenuation of GPV remains elusive. This report presents the complete nucleotide sequences of two live attenuated strains of GPV (82-0321V and VG32/1) that were independently developed in Taiwan and Europe, together with the parental strain of 82-0321V and a field strain isolated in Taiwan in 2006. Sequence comparisons showed that 82-0321V and VG32/1 had multiple deletions and substitutions in the inverted terminal repeats region when compared with their parental strain or the field virus, but these changes did not affect the formation of the hairpin structure essential for viral replication. Moreover, 82-0321V and VG32/1 had five amino acid changes in the non-structural protein, but these changes were located at positions distant from known functional motifs in the non-structural protein. In contrast, 82-0321V had nine changes and VG32/1 had 11 changes in their capsid proteins (VP1), and the majority of these changes occurred at positions close to the putative receptor binding sites of VP1, as predicted using the structure of adeno-associated virus 2 as the model system. Taken together, the results suggest that changes in sequence near the receptor binding sites of VP1 might be responsible for attenuation of GPV. This is the first report of complete nucleotide sequences of GPV other than the virulent B strain, and suggests a possible mechanism for attenuation of GPV.
Introduction
Waterfowl parvovirus causes a disease of high mortality and morbidity in geese and ducks (Barnes, Citation1997; Gough, Citation2003). Studies using molecular analysis and virus neutralization tests have shown that waterfowl parvoviruses can be divided into two groups: the goose parvovirus (GPV)-related group, and the Muscovy duck parvovirus (MDPV)-related group (Le Gall-Reculé & Jestin, Citation1994; Zádori et al., Citation1994, Citation1995; Chang et al., Citation2000; Glávits et al., Citation2005; Poonia et al., Citation2006). To date only one strain of GPV, the virulent B strain, has been sequenced completely (Brown et al., Citation1995; Zádori et al., Citation1995). The genome of the virulent B strain is 5106 nucleotides in length and contains a long protein-encoding region bracketed by short inverted terminal repeats (ITRs) of 444 nucleotides. The distal 406 nucleotides of each repeat form a U-shaped hairpin structure consisting of a 181-base-pair (bp) double-stranded “stem” region and a 44-nucleotide “bubble” region (Zádori et al., Citation1995). The hairpin structure of the ITRs serves as the origin of virus replication. The protein-encoding region contains two major open reading frames (ORFs). The first ORF encodes the non-structural (NS) protein that is involved in viral replication, while the second ORF encodes three capsid proteins, VP1, VP2 and VP3. The three capsid proteins are derived from the same gene, and the entire amino acid sequences of VP2 and VP3 are contained within the C-terminal portion of VP1 (Zádori et al., Citation1995). Among the three capsid proteins, VP3 appears to be the most abundant in purified virions (Le Gall-Reculé & Jestin, Citation1994). Transcription analysis has revealed that the expression strategy of GPV exhibits features of both the Dependovirus and the Parvovirus genera (Qiu et al., Citation2005).
Little is known about the genetic mechanisms that determine the pathogenicity of waterfowl parvovirus. However, studies of other parvovirus systems—including canine and feline parvovirus, minute virus of mice and adeno-associated virus—have shown that changes of amino acids at the receptor binding site of the capsid protein play a key role in determining the host range and pathogenicity of parvoviruses (Chang et al., Citation1992; Govindasamy et al., Citation2003; López-Bueno et al., Citation2006; Palermo et al., Citation2006; Wu et al., Citation2006). Moreover, changes in non-coding nucleotide sequences close to the splice acceptor site of the NS2 gene are also involved in determination of the host range of minute virus of mice (D'Abramo et al., Citation2005). The receptor binding site at the capsid protein of waterfowl parvovirus has not yet been identified. However, previous studies have shown that the nucleotide sequences of GPV and MDPV are most closely related to that of adeno-associated virus 2 (AAV2). The VP1 protein of GPV and MDPV has 70.2% to 70.3% sequence similarity to that of AAV2 (Zádori et al., Citation1995). In AAV2, five amino acids of the VP1 protein (R484, R487, K532, R585 and R588) are directly involved in the binding of AAV2 to its receptor (Kern et al., Citation2003; Opie et al., Citation2003). These amino acids are located near the mounds surrounding the icosahedral three-fold axis of the virion (Wu et al., Citation2000; Xie et al., Citation2002; Opie et al., Citation2003). Because of the high degree of sequence similarity between waterfowl parvovirus and AAV2, it is possible to predict the receptor binding sites of GPV and MDPV using the structure of AAV2 as the model system.
In Taiwan, the disease caused by GPV is controlled using live attenuated vaccines. Two live vaccine strains of GPV have been used in Taiwan. One is 82-0321V, which was derived from the local field strain 82-0321; and the second is VG32/1, which was imported from Germany. Both strains have been found to be useful in the control of GPV, but the mechanism involved in attenuation of the two vaccine strains remains elusive. Tsai et al. (Citation2004) reported that strain 82-0321V had nine amino acid differences from 82-0321 in the VP1 sequence. However, the mechanism by which these differences might contribute to the attenuation of 82-0321V is unknown. Moreover, the sequence of strain VG32/1 was not included in the comparison, and differences outside the VP1 region were not identified. In this study, we obtained the complete nucleotide sequences of 82-0321V, 82-0321, VG32/1 and the field strain 06-0329, which was isolated in Taiwan in 2006. We identified all nucleotide differences between the complete genomes of these strains. We also mapped the changes in the attenuated vaccine strains at the predicted receptor binding site of the VP1 protein.
Materials and Methods
Viruses and DNA extraction
Two live vaccine strains (82-0321V and VG32/1) and two Taiwanese field strains (82-0321 and 06-0329) were sequenced in this study. The strain 82-0321V was derived from 82-0231 after 64 passages in Muscovy duck eggs, two in geese fibroblasts, and four in duck embryos. Strains 82-0321 and 06-0329 were isolated from geese in Taiwan in 1982 and 2006, respectively. Strains 82-0321 and 06-0329 produced 100% mortality when inoculated into 1-day-old Muscovy ducklings. In contrast, all ducklings inoculated with 82-0321V remained healthy. The strain VG32/1 was purchased from Impfstoffwerk Dessau-Tornau GmbH (Rodleben, Germany). VG32/1 possesses the K75N mutation in its VP1 sequence, which is a genetic marker of European vaccine strains (Tatár-Kis et al., Citation2004). DNA was extracted from 200 µl allantoic fluid or reconstituted vaccine using a DNeasy tissue kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions.
Polymerase chain reaction amplification of the complete genome of GPV
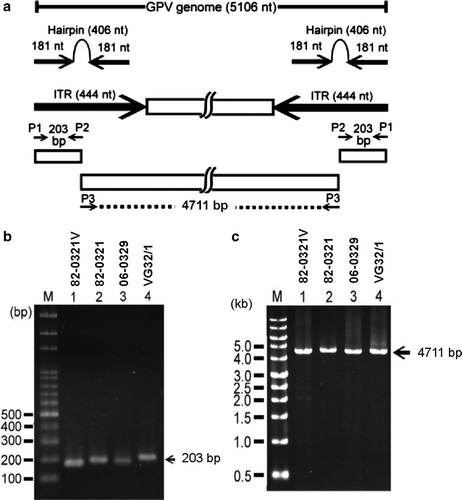
Three primers were used to amplify three DNA fragments covering the complete genome of GPV. The sequences of the three primers were: P1, 5′-CTC ATT GGA GGG TTC GTT CGT TCG AA-3′; P2, 5′-GCA TGC GCG TGG TCA ACC TAA CAG CCG GAA A-3′; and P3, 5′-GCA TGC CGC GCG GTC AGC CCA ATA-3′. The underlined sequences indicate the SphI cutting site at the “bubble” region of the hairpin structure. Because identical inverted terminal sequences were present at the two ends of the GPV genome, P1 was able to anneal to positions corresponding to nucleotides 1 to 25 and 5080 to 5105 of the GPV genome, whereas P2 annealed to nucleotides 173 to 203 and 4903 to 4933 (a). Therefore, polymerase chain reaction (PCR) amplification with primers P1 and P2 amplified two identical 203 bp DNA fragments located at the two ends of the GPV genome (a). In contrast, primer P3 annealed to nucleotides 198 to 221 and 4885 to 4908 and amplified a 4711 bp DNA fragment containing nucleotides 198 to 4908 of the GPV genome (a). All numbering used is consistent with the published sequences of the virulent B strain (Zádori et al., Citation1995). The PCR was performed in a 2720 Thermal Cycler (Applied Biosystems, Foster City, California, USA). The PCR mixture (25 µl) contained 16.5 µl water, 2.5 µl of 10x buffer, 2.5 µl dNTP blend (2.5 mM each of the four dNTPs), 2 µl primers (20 pmol each), 1 µl template DNA and 0.5 µl high-fidelity Taq XL DNA polymerase (Protech, Taipei, Taiwan). The PCR conditions were: one cycle of 94°C for 1 min, 35 cycles of 94°C for 15 sec, 55°C for 30 sec and 68°C for 30 sec (with primers P1 and P2) or for 5 min (with primer P3), and one cycle of 68°C for 5 min.
Figure 1. 1a: Strategy used for amplification of the complete genome of GPV. Primers P1, P2 and P3 are indicated by short horizontal arrows, and the PCR products (203 and 4711 bp) amplified are indicated by space bars. The ITRs (444 nucleotides (nt)) are shown by long arrows. The hairpin region (406 nt) is indicated by two inverted arrows connected by a loop. All numbering is consistent with the published sequence of the virulent B strain. 1b: PCR amplification using the primer pair P1 and P2. Lane M, size markers (100 bp ladder; PRO-tech, Taipei, Taiwan); lanes 1 to 4, amplification using DNA of 82-0321V, 82-0321, 06-0329 and VG32/1. 1c: PCR amplification using the primer P3. Lane M, size markers (1 kb ladder; PRO-tech); lanes 1 to 4, amplification using DNA of 82-0321V, 82-0321, 06-0329 and VG32/1.
Cloning and sequencing of PCR products
The amplified PCR products were purified using a QUAEX gel extraction kit (Qiagen), and were cloned into the vector pCR2.1-TOPO (Invitrogen, Carlsbad, California, USA) according to the protocol provided by the manufacturer. Recombinant plasmids from three clones of each PCR product were purified using a QIAprep Spin plasmid purification kit (Qiagen), and were sequenced using the Big Dye Terminator cycle sequencing kit (Applied Biosystems) in an automatic sequencer (ABI-3730XL DNA Analizer; Applied Biosystems). The plasmid containing the 203 bp fragment was sequenced with M13 forward and reverse primers. The plasmid containing the 4711 bp product was first sequenced with M13 forward and reverse primers and then with a total of five “walking” primers so that each nucleotide position of GPV was sequenced three to six times. In addition, the nucleotide sequence at the 5′-terminus of 82-0321V was confirmed by sequence analysis of a recombinant plasmid carrying the viral DNA purified directly from the virion without PCR amplification. In brief, 50 ml allantoic fluid containing 82-0321V was pelleted by centrifugation at 150 000×g in an SW41 rotor. The virus pellet was resuspended in 1 ml TE buffer (10 mM Tris, pH 8.0, 1 mM wthylenediamine tetraacetic acid). Viral DNA was extracted using a DNeasy tissue kit (Qiagen) and then digested with NcoI, which cut the genome of GPV at a single site to produce 1.2 kb and 3.8 kb fragments. The 1.2 kb fragment containing the 5′ terminus of the viral DNA was cloned into the EcoRV-NcoI site of the vector pGEM-5Zf(+) (Promega, Madison, Wisconsin, USA). The recombinant plasmid was sequenced using the M13 reverse primer to obtain sequences near the 5′ end of viral DNA.
Sequences were compiled using the Seqman program in the LASERGENE package (DNASTAR Inc., Madison, Wisconsin, USA). Prediction of ORFs and alignment of protein sequences were conducted using the GeneQuest and Megalign programs in the same package. Nucleotide and protein sequences were searched for homology in GenBank using the BLAST program (Altschul et al., Citation1990) provided by NCBI, USA.
Nucleotide sequence accession number
The nucleotide sequences determined in this study are available in GenBank under accession numbers EU583389 to EU583392.
Results
PCR amplification of the complete genome of GPV
Three primers (P1, P2 and P3) were used to amplify the complete genome of GPV. Primers P1 and P2 were expected to amplify two identical fragments of 203 bp in length, covering the 5′ and 3′ ends of the viral genome (a). Primer P3 was expected to amplify a fragment 4711 bp in length, covering the entire protein-encoding region of GPV (a). b shows that P1 and P2 amplified the expected 203 bp product from strain VG32/1. However, the products amplified from strains 82-0321V, 82-0321 and 96-0329 were only about 180 and 190 bp in length (b). This result indicated that the sizes of the ITRs of 82-0321V, 82-03211 and 96-0329 were shorter than that of VG32/1. Primer P3 amplified the expected 4711 bp product from all strains investigated in this study (c).
Nucleotide sequences of the complete genome
The PCR products amplified with primers P1, P2 and P3 were cloned and sequenced. The sequences obtained were used to compile the complete genomic sequences of GPV. The genomes of 82-0321V, 82-03211, 96-0329 and VG32/1 were found to be 4980, 5050, 5054 and 5104 nucleotides in length (), and were shorter than that of the virulent B strain (5106 nucleotides) (Zádori et al., Citation1995). The sizes of the full-length ITR, the “stem” and “bubble” regions in the ITR, and the NS and VP1 coding region of GPV are summarized in . It can be seen that 82-0321V has the shortest “stem” and “bubble” regions (152 and 39 nucleotides), followed by 82-0321 and 06-0329 (167 and 44 nucleotides), and by VG32/1 (181 and 43 nucleotides). In contrast, the lengths of the NS and VP1 coding regions were highly conserved; all strains had a NS coding region of 1884 nucleotides and a VP1 coding region of 2199 nucleotides ().
Table 1. Sizes of the genome and other genetic elements found in GPV sequenced in the present study
Overall comparison of the genomic sequences
To identify the nucleotide sequences responsible for attenuation of GPV, sequences of the attenuated strains were compared with those of their parental strains or field viruses. The comparison made in this report was primarily between 82-0321V and 82-0321, as well as between VG32/1 and the virulent B strain, but not between Taiwanese and European strains. This was to identify genetic changes associated with attenuation but not with geographical separation. Virulent strain B was chosen for comparison because a BLAST search using either the complete nucleotide sequence or the protein-encoding sequence showed that VG32/1 was most closely related to the virulent B strain. It is therefore possible that VG32/1 was derived from a strain similar to the virulent B strain, and sequence differences between VG32/1 and the virulent B strain might shed light on the mechanism involved in attenuation of VG32/1.
Comparison using the complete genome sequence showed that the nucleotide sequence of 82-0321V was 3.3% different from that of 82-0321, whereas that of VG32/1 was 1.4% different from that of the virulent B strain. The differences were found in both the ITRs and the protein-encoding regions. If sequences in the ITRs were excluded before comparison, 82-0321V was 2.9% different from 82-0321 whereas VG32/1 was 1.2% different from the virulent B strain. It is noteworthy that 06-0329, a virulent field strain isolated in 2006, had a nucleotide sequence that was only 0.8% different from that of 82-0321, which was isolated in 1982. This result indicates that the genomic sequence of GPV has been highly stable in the field for at least two decades.
Comparison of sequences in the ITRs
Alignment of sequences in the ITRs is shown in . All of the Taiwanese strains (82-0321V, 82-0321, 06-0329) had two deletions, designated the D1 and D5 regions, when compared with the European strains (VG32/1 and virulent B) (). Sequences in the D1 and D5 regions were complementary to each other; therefore, the presence of D1 and D5 did not affect the formation of the double-stranded “stem” region of the hairpin. Similarly, 82-0321V had three deletions, designated the D2, D3 and D4 regions, when compared with 82-0321 (). The D2 and D4 regions were complementary to each other, while D3 was located at the “bubble” region, so that the presence of D2, D3 and D4 also did not affect the formation of the double-stranded “stem” region of 82-0321V. VG32/1 had only a single nucleotide deletion when compared with the virulent B strain, but this deletion occurred within the “bubble region” (). In summary, deletions were found in the ITRs of both 82-0321V and VG32/1 when compared with 82-0321 or the virulent B strain, but no common deletion was found in 82-0321V and VG32/1, and no deletion affected the formation of the hairpin structure.
Figure 2. Alignment of nucleotide sequences in the inverted terminal repeat. The sequences aligned correspond to nucleotides 1 to 444 of the genome of the virulent B strain. The nucleotides in the “stem” region are indicated by filled circles whereas those in the “bubble” region are represented by open circles. The regions containing the deleted sequences (D1 to D5) are indicated by dashes. Nucleotide substitutions found between 82-0321V and 82-0321 and between VG32/1 and the virulent B strain are bold and underlined. The terminal resolution site (trs) is indicated by a vertical arrow, and the SphI cleavage site (GCATGC) is marked.
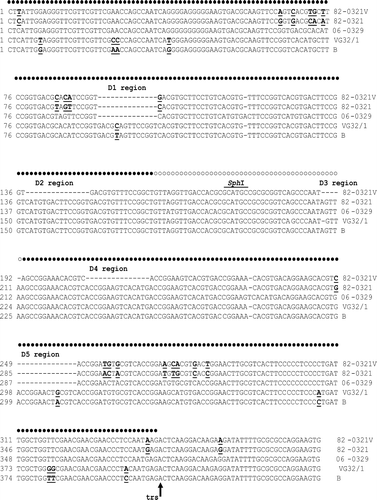
In addition to deletions, nucleotide substitutions were found between 82-0321V and 82-0321 and between VG32/1 and the virulent B strain. A total of 21 substitutions were found in 82-0321V and 10 substitutions in VG32/1 (). No common substitution was found in 82-0321V and VG32/1. Moreover, all substitutions occurred in pairs, and the nucleotide sequences in each pair were complementary to each other so that these substitutions did not affect the formation of the hairpin structure. For example, 82-0321V had a C to T substitution at nucleotide position 3 accompanied by a G to A substitution at position 403. The two substitutions were complementary to each other, and position 403 was three nucleotides upstream of the terminal resolution site (), so the two substitutions did not affect the formation of the double-stranded “stem” structure of the hairpin.
Comparison of amino acid sequences of the NS and VP1 proteins
Amino acid sequences of the NS and VP1 proteins were predicted from the nucleotide sequences. The NS protein of 82-0321V had five amino acid changes (H28D, E38G, H97R, S391F and N554K) when compared with 82-0321. The NS of VG32/1 also had five amino acid changes, but at different positions (S22P, Q27H, I123M, A223T and N609D), when compared with the virulent B strain. No common change was found in 82-0321V and VG32/1. Moreover, all changes were located at positions distant from the motif (residues 336 to 343) that is involved in NTP binding and the motif (residues 142 to 168) involved in DNA binding of the NS protein (Zádori et al., Citation1995).
The VP1 protein of 82-0321V had nine amino acid changes (G203E, V266G, K523E, A524R, L537I, G567V, K575R, D703N and S706N) when compared with 82-0321, whereas that of VG32/1 had 11 amino acid changes (K47R, K75N, V144I, Q479R, Y521H, L529I, E558D, T584I, D593E, G615W and D720K) when compared with the virulent B strain. Although no common change was found in 82-0321V and VG32/1, the majority of these changes were located in the vicinity of the putative receptor binding site of VP1, as predicted using the structure of AAV2 as the model system. In AAV2, five amino acids (R484, R487, K532, R585 and R588) of the VP1 are directly involved in binding of the virus to its receptor (Wu et al., Citation2000; Opie et al., 2003). shows that, of the nine amino acid changes found in 82-0321V, three changes (K523E, A524R and L537I) were close to K532 of AAV2, while two (G567V and K575R) were close to R585 of AAV2. Similarly, of the 11 amino acid changes found in VG32/1, two (Q479R and T584I) were next to R487 or R588 of AAV2, while four (Y521H, L529I, E558D and D593E) were close to either K532 or R588 of AAV2. Taken together, these results show that five of the nine changes found in 82-0321V and six of the 11 changes found in VG32/1 were located in close proximity to the putative receptor binding site of the VP1 protein of GPV.
Figure 3. Alignment of amino acid sequences in the putative receptor binding site of GPV. The sequences aligned correspond to residues 471 to 620 of the VP1 of GPV or residues 478 to 623 of the VP1 of AAV2. The sequences conserved in GPV and AAV2 are shaded. The residues (R484, R487, K532, K585 and K588) involved in receptor binding of AAV2 are indicated by asterisks. Amino acid changes occurring between 82-0321V and 82-0321 and between VG32/1 and the virulent B strain are bold and underlined.

Comparison of sequences at promoters and other regulatory regions
No change was found in 82-0321V and VG32/1 in regions containing the promoter sequences (P9 and P42), splice donor and acceptor sites, or polyadenylation sites (pAp and pAd) (Qiu et al., Citation2005). The nucleotide sequences of all these genetic elements were highly conserved in the virulent B strain and in all GPV strains sequenced in this study.
Discussion
The attenuated strains 82-0321V and VG32/1 contained multiple genetic changes in the ITRs and protein-encoding regions. Of these changes, deletions and substitutions occurred in the ITRs but they did not affect the formation of the hairpin essential for viral replication. Changes in the NS protein were located at positions distant from the NTP-binding and DNA-binding motifs, and thus might not play a key role in attenuation. In contrast, the majority of changes found in the VP1 protein clustered at the putative receptor binding site of GPV. Because 82-0321V and VG32/1 were developed independently in Taiwan and Europe, this clustering is not a mere coincidence but evidence strongly suggesting that changes in sequences at the receptor binding site are responsible for attenuation of GPV. This conclusion is consistent with previous studies on other parvoviruses, in which amino acid sequences at the receptor binding site were found to be critical for the pathogenicity and host range of the virus (Chang et al., Citation1992; Govindasamy et al., Citation2003; López-Bueno et al., Citation2006; Palermo et al., Citation2006; Wu et al., Citation2006). To our knowledge, this is the first report of prediction of receptor binding sites of GPV and the mechanism involved in attenuation of waterfowl parvovirus.
The receptor binding site of AAV2 contains five residues: R484, R487, K532, R585 and K588 (Xie et al., Citation2002; Kern et al., Citation2003; Opie et al., 2003). These five residues are all basic amino acids and constitute the basic patch capable of binding to the acidic heparin receptor on the cell surface. Of the five basic residues, only R487 is conserved in GPV. Moreover, the two residues (R585 and R588) that are critical for receptor binding in AAV2 are absent in GPV. This result suggests that GPV might use other basic amino acids for receptor binding. Possible candidates are K571 and K/R 575 of GPV. The two residues are located in a position only five residues away from R585 and R588, and may act as the substitutes for R585 and R588 in receptor binding. Site-directed mutagenesis and other studies are required to test this hypothesis.
Knowledge of the receptor binding site of waterfowl parvovirus is important in the development of live attenuated vaccine strains. In Taiwan, strain 82-0321V was derived from 82-0321 by passage in Muscovy duck embryos and 82-0321V has served as a good live attenuated vaccine strain of GPV (Tsai et al., Citation2004). However, a similar strategy failed to develop a safe attenuated vaccine strain of MDPV. To date, no live vaccine against MDPV has been used in the field, suggesting that the conventional method for developing live attenuated vaccine strains does not work for MDPV. A novel strategy for developing a live attenuated strain of MDPV is by site-directed mutagenesis of infectious DNA from MDPV. Infectious DNA of GPV has been successfully constructed (Qiu et al., Citation2005) and a similar strategy could be used for constructing infectious DNA of MDPV. This DNA could be used as the template for site-directed mutagenesis to develop an attenuated strain of MDPV. In the present study, changes in five residues in 82-0321V and six residues in VG32/1 were identified in the putative receptor binding site of VP1. These residues could be targets for site-directed mutagenesis to construct an attenuated strain. Experiments are underway in this laboratory to test this strategy. Successful development of a safe attenuated live vaccine for MDPV is useful for control of this disease.
To date only one strain of GPV and one strain of MDPV have been completely sequenced (Zádori et al., Citation1995). The lack of reports of complete sequences from GPV and MDPV strains may have resulted from the difficulty in sequencing the ITR region. Each repeat of the ITR contains palindromic sequences that could change between the “flip” and “flop” orientation when being cloned into a plasmid vector. The “flip” and “flop” orientation produces ambiguous sequencing results that are difficult to interpret (Zádori et al., Citation1995). Moreover, the palindromic sequences can fold up to form a hairpin structure, which resists PCR amplification of this region. In this report, we used primer pair P1 and P2 to amplify only one-half of the palindromic sequences in each ITR. Moreover, we used primer P3 to amplify the other half of the palindromic sequences. This methodology not only avoided formation of “flip” and “flop” orientation in the recombinant plasmid but also avoided formation of a hairpin structure during PCR amplification. With this method, we successfully amplified, cloned and sequenced the complete genome of four strains of GPV. The same methodology could be used for sequence analysis of other waterfowl parvoviruses.
Acknowledgements
This investigation was supported by grants 96-2313-B-005-014-MY2 from the National Science Council, Taiwan, Republic of China.
References
- Altschul , S.F. , Gish , W. , Miller , W. , Myers , E.W. and Lipman , D. J. 1990 . Basic local alignment search tool . Journal of Molecular Biology , 215 : 403 – 410 .
- Barnes , H.J. 1997 . “ Muscovy duck parvovirus ” . In Diseases of Poultry , 10th edn , Edited by: Calnek , B.W. 1032 – 1033 . Ames : Iowa State University Press .
- Brown , K.E. , Green , S.W. and Young , N.S. 1995 . Goose parvovirus—an autonomous member of the dependovirus genus? . Virology , 210 : 283 – 291 .
- Chang , P.C. , Shien , J.H. , Wang , M.S. and Shieh , H.K. 2000 . Phylogenetic analysis of parvoviruses isolated in Taiwan from ducks and geese . Avian Pathology , 29 : 45 – 49 .
- Chang , S.F. , Sgro , J.Y. and Parrish , C.R. 1992 . Multiple amino acids in the capsid structure of canine parvovirus coordinately determine the canine host range and specific antigenic and hemagglutination properties . Journal of Virology , 66 : 6858 – 6867 .
- D'Abramo , A.M. Jr , Ali , A.A. , Wang , F. , Cotmore , S.F. and Tattersall , P. 2005 . Host range mutants of Minute Virus of Mice with a single VP2 amino acid change require additional silent mutations that regulate NS2 accumulation . Virology , 340 : 143 – 154 .
- Glávits , R. , Zolnai , A. , Szabó , E. , Ivanics , E. , Zarka , P. , Mató , T. and Palya , V. 2005 . Comparative pathological studies on domestic geese (Anser anser domestica) and Muscovy ducks (Cairina moschata) experimentally infected with parvovirus strains of goose and Muscovy duck origin . Acta Veterinaria Hungarica , 53 : 73 – 89 .
- Gough , R.E. 2003 . “ Goose parvovirus infection ” . In Diseases of Poultry , 11th edn , Edited by: Saif , Y.M. 367 – 374 . Ames : Iowa State University Press .
- Govindasamy , L. , Hueffer , K. , Parrish , C.R. and Agbandje-McKenna , M. 2003 . Structures of host range-controlling regions of the capsids of canine and feline parvoviruses and mutants . Journal of Virology , 77 : 12211 – 12221 .
- Kern , A. , Schmidt , K. , Leder , C. , Müller , O.J. , Wobus , C.E. , Bettinger , K. , Von der Lieth , C.W. , King , J.A. and Kleinschmidt , J.A. 2003 . Identification of a heparin-binding motif on adeno-associated virus type 2 capsids . Journal of Virology , 77 : 11072 – 11081 .
- Le Gall-Reculé , G. and Jestin , V. 1994 . Biochemical and genomic characterization of muscovy duck parvovirus . Archives of Virology , 139 : 121 – 131 .
- López-Bueno , A. , Rubio , M.P. , Bryant , N. , McKenna , R. , Agbandje-McKenna , M. and Almendral , J.M. 2006 . Host-selected amino acid changes at the sialic acid binding pocket of the parvovirus capsid modulate cell binding affinity and determine virulence . Journal of Virology , 80 : 1563 – 1573 .
- Opie , S.R. , Warrington , K.H. Jr , Agbandje-McKenna , M. , Zolotukhin , S and Muzyczka , N. 2003 . Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding . Journal of Virology , 77 : 6995 – 7006 .
- Palermo , L.M. , Hafenstein , S.L. and Parrish , C.R. 2006 . Purified feline and canine transferrin receptors reveal complex interactions with the capsids of canine and feline parvoviruses that correspond to their host ranges . Journal of Virology , 80 : 8482 – 8492 .
- Poonia , B. , Dunn , P.A. , Lu , H. , Jarosinski , K.W. and Schat , K.A. 2006 . Isolation and molecular characterization of a new Muscovy duck parvovirus from Muscovy ducks in the USA . Avian Pathology , 35 : 435 – 441 .
- Qiu , J. , Cheng , F. , Yoto , Y. , Zádori , Z. and Pintel , D. 2005 . The expression strategy of goose parvovirus exhibits features of both the Dependovirus and Parvovirus genera . Journal of Virology , 79 : 11035 – 11044 .
- Tatár-Kis , T. , Mató , T. , Markos , B. and Palya , V. 2004 . Phylogenetic analysis of Hungarian goose parvovirus isolates and vaccine strains . Avian Pathology , 33 : 438 – 444 .
- Tsai , H.J. , Tseng , C.H. , Chang , P.C. , Mei , K. and Wang , S.C. 2004 . Genetic variation of viral protein 1 genes of field strains of waterfowl parvoviruses and their attenuated derivatives . Avian Diseases , 48 : 512 – 521 .
- Wu , P. , Xiao , W. , Conlon , T. , Hughes , J. , Agbandje-McKenna , M. , Ferkol , T. , Flotte , T. and Muzyczka , N. 2000 . Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism . Journal of Virology , 74 : 8635 – 8647 .
- Wu , Z. , Asokan , A. , Grieger , J.C. , Govindasamy , L. , Agbandje-McKenna , M. and Samulski , R.J. 2006 . Single amino acid changes can influence titer, heparin binding, and tissue tropism in different adeno-associated virus serotypes . Journal of Virology , 80 : 11393 – 11397 .
- Xie , Q. , Bu , W. , Bhatia , S. , Hare , J. , Somasundaram , T. , Azzi , A. and Chapman , M.S. 2002 . The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy . Proceedings of the National Academy of Sciences of the United States of America , 99 : 10405 – 10410 .
- Zádori , Z. , Erdei , J. , Nagy , J. and Kisary , J. 1994 . Characteristics of the genome of goose parvovirus . Avian Pathology , 23 : 359 – 364 .
- Zádori , Z. , Stefancsik , R. , Rauch , T. and Kisary , J. 1995 . Analysis of the complete nucleotide sequences of goose and muscovy duck parvoviruses indicates common ancestral origin with adeno-associated virus 2 . Virology , 212 : 562 – 573 .