Abstract
The development of a reverse transcriptase-polymerase chain reaction (RT-PCR) test for detecting avian nephritis virus (ANV) is described. Primers, which amplified a fragment of 182 base pairs (bp), were located in the conserved 3′ untranslated region (UTR) of the genome. The limit of detection of the test was estimated to be approximately 18 viral copies using a 10-fold dilution series of in vitro transcribed RNA. Positive signals were produced with representative ANV samples, some of which were not detected by previously described RT-PCR tests for detecting ANV, but other avian astroviruses including chicken astrovirus isolates and duck hepatitis virus types 2 and 3 tested negative. When applied to gut content samples from UK, German and US broiler flocks with enteritis/growth problems, ANVs were detected by RT-PCR in 82/82 (100%) samples. ANVs were also detected in 80/96 (83%) pooled gut content samples from longitudinal surveys of four broiler flocks displaying below-average performance. Whereas all samples collected on day 0 from the surveys were negative for ANV, all samples collected at days 4/5, 7, 10, 14, 21 and 28 tested positive. Sequence determinations performed with amplicons produced with 14 field samples confirmed the ANV specificity of the test, while comparative and phylogenetic analyses based on 109-nucleotide 3′-UTR sequences demonstrated that the majority of ANVs investigated were more closely related to the serotype 2 ANV (accession number AB 046864) than to the serotype 1 ANV (accession number NC 003790).
Introduction
Avian nephritis virus (ANV) was first isolated from the rectal contents of a 1-week-old, apparently normal, broiler chicken (Yamaguchi et al., Citation1979). Experimental inoculation of 1-day-old specific pathogen free (SPF) chicks with the isolated virus (G-4260 strain) resulted in diarrhoea, weight loss, mortality and interstitial nephritis (Imada et al., Citation1979). On the basis of size, morphology and physicochemical properties, ANV was originally regarded as a picornavirus (Maeda et al., Citation1979). However, following genome sequence determination and analysis, it is now classified as a member of the genus Avastrovirus of the family Astroviridae (Imada et al., Citation2000).
Serological testing has shown that ANV infections are widespread in commercial chickens in Japan and in several European countries, and ANV-specific antibody has also been detected in turkeys and in SPF flocks (Imada et al. Citation1980; Connor et al., Citation1987). ANVs and viruses originally described as avian enteroviruses or enterovirus-like viruses (ELVs), which are antigenically related to ANV and that are hereafter called ANVs, have been detected on a number of occasions in broiler flocks with growth problems, known variously as uneven growth, infectious stunting and runting-stunting (McNulty et al., Citation1984; Decaesstecker et al., Citation1989; Shirai et al., Citation1992). Studies undertaken in Japan have shown that there are at least two different ANV serotypes, which, using indirect immunofluorescence tests, display relatively low levels of cross-reactive antibody (Shirai et al., Citation1992). ANV isolates can also vary with regard to pathogenicity (Shirai et al., Citation1991, Citation1992) and possibly tropism (Decaesstecker et al., Citation1989). The detection of ANVs in growth-retarded broilers and the ability of some ANVs to cause substantial degrees of growth retardation following experimental infection of SPF and broiler chickens have led to speculation that ANV may be important in causing growth problems in the field (McNulty et al., Citation1984; Decaesstecker et al., Citation1986; Shirai et al., Citation1992). However, the clinical significance of ANV infections is unknown, due largely to the lack of convenient diagnostic tests. These viruses are difficult to isolate in cell culture due to their comparatively poor growth in primary chicken cells and the common occurrence in enteric samples of other viruses such as reoviruses or adenoviruses, which are more readily isolated. In addition, antisera specific to antigenically different ANVs for use in the detection of virus antigen by immunostaining methods are not widely available.
Knowledge of the ANV genomic sequence has allowed the development of reverse transcription-polymerase chain reaction (RT-PCR) tests for the detection of virus RNA. However, the detection of any RNA virus by RT-PCR is often limited by both the low availability of sequence information and the high degree of sequence diversity. Two RT-PCR tests for detecting ANV have been reported. In one study, Mandoki et al. (Citation2006b) confirmed the presence of ANV in 36/56 (64%) kidney samples from clinical outbreaks of nephritis and gout in Hungary, in which diarrhoea and growth retardation were also observed. Their RT-PCR test was based on primers located within open reading frame (ORF) 1a, which encodes the non-structural viral protease. On the basis of partial ORF 1a sequences of 11 ANV amplicons, which showed 76 to 86% nucleotide identity, it was concluded that the ANVs were highly diverse (Mandoki et al., Citation2006a). An alternative RT-PCR test based on the use of degenerate primers located within ORF 1b (RNA polymerase) was described in a US study by Day et al. (Citation2007), who reported the detection of ANVs in enteric samples from sick and healthy chickens (61/109; 56%) and turkeys (7/31; 23%) originating in the USA. We have recently determined partial nucleotide sequences of 20 ANVs, the majority of which were collected from growth-retarded flocks originating in the UK, Belgium and Germany from the 1980s to 2008. Whereas regions of the capsid protein ORF 2 sequences displayed as little as 58% nucleotide identity, high levels (~90% identity) of conserved sequence were demonstrated when the untranslated regions at the 3′terminus (3′ untranslated region [UTR]) of the ANV genome were compared (Todd et al., unpublished results). This has enabled us to identify conserved sequences on which to base the design of RT-PCR primers. In this paper we report the development of a sensitive RT-PCR test for detecting ANV RNA and its application for the detection of ANV in gut contents and kidney samples from broiler flocks.
Methods and Materials
Viruses and virus growth
The G-4260 serotype 1 isolate of ANV (ANV-1), obtained from Dr S. Yamaguchi, National Institute of Animal Health, Gifu, Japan, was propagated in chick kidney cells, with the virus pool used in this investigation having an infectious titre of 105 median tissue culture infective doses (TCID50)/ml. Information relating to the source, propagation history and production of virus pools of the duck hepatitis virus type 2 (DHV-2; isolate M52), DHV-3 (isolate X1222A Calnek) and the 11672 and 612 isolates of chicken astrovirus (CAstV) was described by Todd et al. (Citation2009). An ELV, designated Northern Ireland ELV 1, produced in this laboratory and present as an extract of intestinal contents (pool 276; ELV276), was derived from broiler chickens with runting-stunting syndrome (McNulty et al., Citation1984). A second ELV, designated Belgian ELV 1, was supplied by Dr G. Meulemans, Brussels, Belgium, and was prepared as described previously (McNulty et al., Citation1991). Cell culture lysates, clarified 10% whole embryo or embryo liver homogenates, or clarified 10% faeces/intestinal content suspensions were used for RNA extraction.
Sample origin and processing
Intestinal contents or cloacal swabs were sampled from chickens collected from problem flocks in the UK and Germany, and were stored at –80°C. Samples from the UK were kindly provided by A. Fernandez-Gutierrez (Aviagen Ltd, Midlothian, UK), M. Alcorn (St David's Poultry Team Ltd, Exmouth, UK), B.H. Thorp (St David's Poultry Team at the Dick Vet, Easter Bush Veterinary Centre, Midlothian, UK), C. Prins, G. Hayes and A. Walker (Slate Hall Veterinary Practice Ltd, Cambridge, UK), P.W. Cargill (Wyatt Poultry Veterinary Services, Hereford, UK) and D. Pearson (VION UK Veterinary and Food Diagnostic Service, Grampian Country Chickens (Rearing) Ltd, West Lothian, UK).
Swabs were immersed in 0.5 ml chilled phosphate-buffered saline and vortexed thoroughly. The swab suspensions underwent one freeze/thaw cycle at –80°C before the swab was removed. Intestinal contents were diluted 1:10 in chilled phosphate-buffered saline and then shaken by hand briefly with glass beads to disrupt the solid material. Both suspensions were centrifuged at 3000 x g at 4°C for 30 min and the supernatants were transferred to fresh tubes for storage at –80°C until required. presents the samples investigated.
Table 1. Field samples from UK broiler flocks displaying growth problems.
Dr P. Otto (Friedrich Loeffler Institute, Federal Institute for Animal Health, Jena, Germany) provided samples 03V0358, 05V0096, 05V0298, 05V0105, 05V0377, 05V0089, 05V0321, 05V0339, 06V0084, 06V0040, 06V0062, 06V0082, 06V0540, 06V0567, 06V0574. Twelve samples (799 MO/2005, 802 AR/2005, 812 DE/2005, 836 NC/2005, 840 AR/2005, 866 GA/2006, 883 MO/2006, 916 CA/2006, 1254 GA/2008, 1255 GA/2008, 1335 GA/2009 and 1340 GA/2009), prepared from diagnostic submissions from US broiler flocks, were provided as clarified 10% faecal extracts by Dr Michael Day, Southeast Poultry Research Laboratory (USDA, Agricultural Research Service, Athens, Georgia, USA).
Longitudinal surveys
Four flocks that, based on recent performances, were predicted to exhibit average and below-average performances were sampled longitudinally. The flocks belonged to the same UK poultry organization, but were located on different sites. Gut contents from approximately 12 birds were sampled from each flock at days 0, 4 or 5, 7, 10, 14, 21 and 28. The samples were grouped into four pools and processed by homogenization as described above. In total, approximately 84 gut content samples were collected from each surveyed flock, from which 28 pooled samples were processed for RNA extraction. The performance of each flock was estimated after slaughter by calculating European production efficiency factor (EPEF) values, which represent standard measures of overall flock performance as determined by the equation:
RNA extraction
Viral RNA was extracted from 140 µl each supernatant using the QIAamp Viral RNA or RNeasy Mini Kits (Qiagen, Crawley, UK) according to the manufacturer's instructions. Each RNA was eluted in 30 µl RNase-free water.
RT-PCR assays
Primers (ANV forward, 5′-ACG GCG AGT ACC ATC GAG-3′; and ANV reverse, 5′-AAT GAA AAG CCC ACT TTC GG-3′) were designed to amplify a 182 base pair (bp) fragment (nucleotides 6715 to 6896 in sequence of the ANV-1 genome) from within the 3′-UTR of ANV genomes. One-step RT-PCR was performed on the RNAs using the above primers and the SuperScript III One-Step RT-PCR System with Platinum® Taq DNA Polymerase kit (Invitrogen, Paisley, UK). Each reaction contained 1 x reaction buffer, 1 µM each primer, 1 µl enzyme mix, 2.5 µl RNA and diethyl pyrocarbonate-treated H2O to 25 µl. Amplification occurred in a Veriti thermocycler (Applied Biosystems, Warrington, UK) starting with a reverse transcription step of 45°C for 30 min, then an initial denaturing step of 94°C for 2 min, followed by 40 cycles of denaturation at 94°C for 30 sec, annealing at 57°C for 30 sec and extension at 68°C for 30 sec. There was a final extension step at 68°C for 7 min. The PCR products were electrophoresed at 50 to 125 V for 40 to 60 min on a 2% agarose gel in Tris-acetate ethylenediamine tetraacetic acid buffer and were visualized by ethidium bromide staining and ultraviolet (UV) transillumination.
Previously described avian astrovirus degenerate primers that amplify 434 bp fragments from within ORF 1b (RNA polymerase gene) regions of a wide range of avian astroviruses were used in a one-step RT-PCR to verify the presence of specific astroviruses in CAstV, DHV-2 and DHV-3 isolates as previously described (Todd et al., Citation2009). Two previously described RT-PCR tests for detecting ANV RNA were carried out as reported (Mandoki et al., Citation2006b; Day et al., Citation2007).
Cloning and sequencing
Fragments of approximately 2.5 kbp were amplified from RNAs that had been extracted from a range of clinical samples including VF04-01/2, VF07-13/7, VF08-03, VF08-18/5 and VF08-29 using the forward primer 5′-ACC TTG AAT CCC TGT GGG GCA-3′ and the reverse primer 5′-AAA AGT TAG CCA ATT CAA AAT TAA TTC-3′ in a single-tube RT-PCR format. Amplification conditions involved a RT step of 45°C for 30 min, an initial denaturing step of 94°C for 2 min, followed by 35 to 40 cycles each comprising denaturation at 94°C for 30 sec, annealing at 57°C for 30 sec and extension at 68°C for 2.5 min. The amplified 2.5 kbp fragments, which corresponded in location to nucleotides 4379 to 6927 of the ANV-1 genomic sequence, were cloned using TOPO TA Cloning® Kit (Invitrogen). Two representative recombinant plasmids, each containing one of the 2.5 kbp ANV-specific fragments, were sequenced using M13 forward and reverse primers followed by primer-walking strategies. When the sequences of the two cloned 2.5 kbp fragments differed by more than 2%, the sequences were labelled alphabetically; for example, sequences VF08-03a and VF08-03b were obtained from 2.5 kbp fragments amplified from sample VF08-03. The sequences of the 3′ UTRs (approximately 307 nucleotides) of the cloned ANV fragments were aligned with corresponding regions of the ANV-1 and ANV-2 (the serotype 2 ANV isolate present in the sequence databases) genome sequences using the ClustalW algorithm programme.
Amplicons of 182bp were excised from the agarose gels and purified using the Wizard SV Gel and PCR Clean-Up System (Promega, Southampton, UK). Fourteen of the purified PCR products were sequenced in both directions using the same forward and reverse primers used for amplification and the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). The products were sequenced commercially. Sequencing data were analysed using the Vector NTI suite (Invitrogen). Phylogenetic analysis was conducted on trimmed amplicons of 109 nucleotides using MEGA version 4 (Tamura et al., Citation2007).
In vitro transcription
A 394 bp fragment of ANV-specific cDNA, which encompassed the 3′ UTR of the ANV genome (nucleotides 6530 to 6924 of the ANV-1 genome), was amplified from a recombinant pCR®2.1-TOPO plasmid containing a 2.5 kb ANV cDNA fragment amplified from sample VF04-01/2, and cloned into the pCR®II-TOPO® vector (Invitrogen). PCR used the forward primer 5′-GAT GAG TTT GAT TGT GTG GAT AAC-3′ and reverse primer 5′-AGT AGC AAT CAA ATT ATT C-3′, with amplification being performed over 40 cycles of denaturation at 95°C for 30 sec, annealing at 40°C for 30 sec, and extension at 68°C for 30 sec. The recombinant pCR®II-TOPO plasmid containing the 394 bp fragment was linearized by Xho1 digestion and in vitro transcribed from the SP6 promoter using the MEGAscript® SP6 kit (Ambion (Europe) Ltd, Huntingdon, UK), according to the manufacturer's instructions, but with an overnight incubation period at 37°C. Following transcription, the in vitro transcribed RNA was treated with Turbo DNase (Ambion), then further purified from template contamination by extraction with TRIzol (Invitrogen, Paisley, UK), and then High Pure RNA Tissue Kit (Roche, Welwyn Garden City, Herts, UK). The quality of the in vitro transcribed RNA was assessed by RT-PCR using the ANV forward and reverse primers, and its purity assessed by PCR using the same primers and cycling conditions, but excluding the RT step. The concentration of the in vitro transcribed RNA was determined using a Genova spectrophotometer (Jenway, Dunmow, UK).
Sequence accession numbers
The GenBank accession numbers of the 3′-UTR sequences, which were determined from the cloned 2.5 kbp fragments, were GU222330 (VF04-01/2), GU222331 (VF07-13/7), GU222332 (VF08-03a), GU222333 (VF08-18/5), and GU222334 (VF08-29a). The GenBank accession numbers of the 109-nucleotide sequences, which were determined from the 182 bp RT-PCR test amplicons, were GU222335 (VF04-01/2), GU222336 (VF05-01/1), GU222337 (VF05-01/5), GU222338 (VF05-01/7), GU222339 (VF05-01/8), GU222340 (VF05-01/10), GU222341 (VF05-01/12), GU222342 (VF06-01/4), GU222343 (VF06-02/8), GU222344 (VF07-13/1), GU222345 (VF07-13/3), GU222346 (VF07-13/7), GU222347 (VF08-07/1) and GU222348 (VF08-07/3). The accession number for the ANV-1 (G-4260 serotype 1) genome sequence is NC 003790 and for the ANV-2 (serotype 2) partial genome sequence is AB 046864.
Results
Comparison of 3′-UTR sequences
The 3′-UTR sequences (approximately 307 nucleotides) of 20 ANVs that were present in gut content samples from broiler flocks with severe growth problems were determined by sequencing cloned 2.5 kb ANV-specific cDNA fragments, which corresponded in location to nucleotides 4379 to 6927 of the ANV-1 genomic sequence. Alignment of seven representative ANV 3′-UTR sequences, including those of ANV-1 and ANV-2 isolates, is shown in . Sequence comparison showed that these sequences shared >92% nucleotide identity and allowed the identification of a number of conserved regions that could be used for the design of RT-PCR primers. The same sequences were substantially conserved when the corresponding regions from 15 additional ANV sequences were compared (data not shown). The locations of the ANV forward and ANV reverse primers, which flank a region of 182 bp, used for the diagnostic RT-PCR test are shown.
Figure 1. ClustalW alignment of 3′-UTR genomic regions of representative ANVs, including the G-4260 isolate of the serotype 1 ANV strain (ANV-1) a serotype 2 ANV strain (ANV-2), and five ANVs detected in field samples (VF04-01/2, VF07-13/7, VF08-03a, VF08-18/5 and VF08-29a).

Optimization of test and determination of the limit of detection
The performance of the RT-PCR test was initially evaluated using dilutions of RNA that had been extracted from a pool of ANV-1 grown in cell culture. An annealing temperature of 57oC was selected following consideration of the amount of PCR product detected and the absence of non-specific amplicon bands, which were observed with some samples when lower annealing temperatures, such as 50°C, were used.
The limit of detection of the RT-PCR assay was determined using RNA that had been in vitro transcribed from the recombinant pCR®II-TOPO plasmid containing the 394 bp ANV 3′-UTR insert plasmid. The last 10-fold serial dilution with which a visible RT-PCR product was amplified corresponded to approximately 18 viral RNA target molecules (data not shown). Application of the RT-PCR test to 10-fold dilutions of RNA that had been extracted from the ANV-1 pool, which had an infectivity titre of approximately 105 TCID50/ml, showed that amplicons were detected with dilutions up to and including 10–6. Dilutions of 10–5 of the virus RNA extract were included for positive control purposes in the majority of RT-PCR tests undertaken.
No RT-PCR amplicons were produced with RNAs that had been extracted from samples of DHV-2, DHV-3, and the 11672 and 612 isolates of CAstV, which were previously shown to be positive using the pan-avian astrovirus degenerate primer-based RT-PCR test (Todd et al., Citation2009), thereby indicating that the RT-PCR test was specific for ANV and not for other avian astroviruses.
Application of RT-PCR tests to field samples
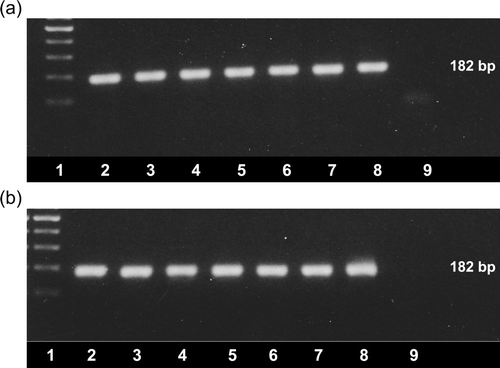
Gut content or faeces samples that had been collected from broiler chicken flocks experiencing enteritis and growth retardation problems were tested by the RT-PCR test. Fifty-five samples were received from October 2004 to May 2008 as part of 10 submissions obtained from six different UK poultry organizations (). Additional samples were obtained from affected broiler flocks in Germany (n = 15) and the USA (n = 12). Of 82 samples tested, 82 (100%) were positive by RT-PCR, the majority producing single DNA bands, sized 182 bp, after agarose gel electrophoresis, ethidium bromide staining and UV transillumination (). In addition, positive RT-PCR results were obtained with five pooled swab samples that were collected from broiler chickens affected by wet litter problems and five pooled samples collected from chicken flocks that were unaffected by wet litter problems. Additional amplicon bands were observed with some samples, especially those prepared from swabs that had been extracted with the RNeasy extraction kit.
Figure 2. Detection of ANV by the newly-developed RT-PCR. Amplicons (182 bp) were visualized using ethidium bromide staining and UV transillumination following agarose gel electrophoresis. 2a: Lane 1, 100 bp ladder; lanes 2 to 9, amplicons produced from RNA extracted from VF06-01/1 (lane 2), VF06-01/4 (lane 3), VF07-13/1 (lane 4), VF07-13/3 (lane 5), VF07-13/5 (lane 6), VF07-13/7 (lane 7) and VF08-07/1 (lane 8) and negative water control (lane 9). 2b: Lane 1, 100 bp ladder; lanes 2 to 9, amplicons produced from RNA extracted from VF08-07/3 (lane 2), VF06-02/2 (lane 3), VF06-02/6 (lane 4), VF06-02/8 (lane 5), VF05-01/1 (lane 6), VF05-01/3 (lane 7), VF05-01/5 (lane 8) and negative water control (lane 9).
Application of RT-PCR tests to longitudinal surveys samples
Of 96 pooled gut content samples, collected in longitudinal surveys of four broiler flocks from day 0 to day 28, 80 (83%) tested positive by the RT-PCR test. The 16 negative samples were those collected at day 0, when the chicks were introduced to the broiler house, but all pooled samples collected at later time points tested positive. Below-average EPEF values of 327, 315 and 308 were estimated for the three male broiler flocks, and an EPEF value of 238 was estimated for the female broiler flock that was surveyed.
Comparison of RT-PCR tests
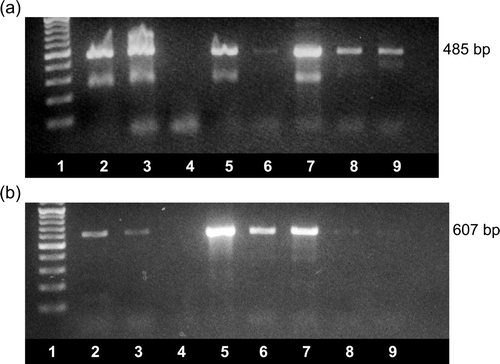
The RNAs extracted from 12 representative field samples (VF04-01/2, VF04-01/6, VF04-01/10, VF05-01/5, VF05-01/8, VF06-01/1, VF06-02/2, VF06-02/8, VF07-13/7, VF08-03, VF08-18/1 and VF08-18/5), which tested positive by our newly-developed RT-PCR test, were tested by two previously described RT-PCR tests. Using the test reported by Day et al. (Citation2007), 10 samples were positive, with one sample (VF05-01/5) of the 10 producing a faint amplicon band (a). Nine of the 12 samples were positive by the RT-PCR test described by Mandoki et al. (Citation2006b) (b). The nine samples that tested positive by this test included two samples (VF04-01/2 and VF04-01/6) that produced faint amplicon bands (b, lanes 8 and 9). Two samples (VF06-02/2 and VF08-18/5) tested negative by each of the previously described tests. It was noted that the RT-PCR products generated by the test described by Day et al. (Citation2007) very often contained extra DNA bands with mobilities corresponding to approximately 285 bp, in addition to the 485 bp expected amplicon, whereas single bands of 607 bp were obtained when the PCR products generated by the test described by Mandoki et al. (Citation2006b) were analysed.
Figure 3. Detection of ANV by (3a) the RT-PCR test described by Day et al. (Citation2007) and (3b) the RT-PCR test described by Mandoki et al. (Citation2006b). Amplicons were visualized using ethidium bromide staining and UV transillumination following agarose gel electrophoresis. Lane 1, 100 bp ladder; lanes 2 to 9, amplicons produced from RNA extracted from VF06-01/1 (lane 2), VF07-13/7 (lane 3), VF06-02/2 (lane 4), VF06-02/8 (lane 5), VF05-01/5 (lane 6), VF05-01/8 (lane 7), VF04-01/2 (lane 8) and VF04-01/6 (lane 9). The sizes of the amplicons are shown.
Sequence characterization of RT-PCR amplicons
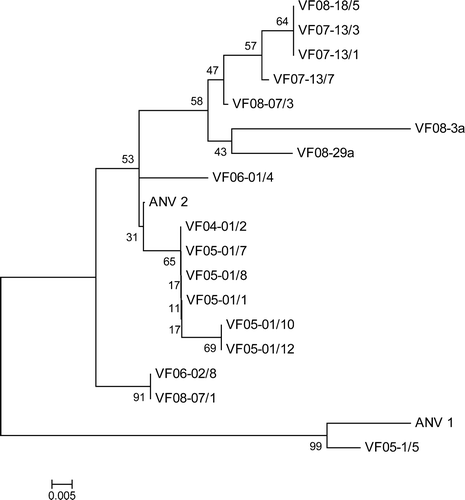
Nucleotide sequences, approximately 109 nucleotides in size, were determined from the amplicons produced with 14 field samples. The region for which sequence was determined corresponded to residues 6753 to 6861 in the genome of ANV-1. Comparisons confirmed that the 14 amplicon sequences shared between 79% and 97% nucleotide identity with the ANV-1 sequence and between 87% and 99% with the ANV-2 sequence, so confirming their ANV specificity. A phylogenetic tree constructed with these 14 sequences and corresponding sequences of five different ANVs, shown in , revealed the existence of two major groupings (). One grouping contained the most distantly related ANVs, namely ANV-1 and VF05-1/5. The second major grouping comprised two subgroups, including one containing VF06-02/8 and VF08-07/1, and a second comprising the sequences of 15 ANVs, which were more closely related to ANV-2.
Figure 4. Phylogenetic tree of ANVs based on 109-nucleotide 3′-UTR sequences. The ANV sequences include 14 determined from 182 bp amplicons and five different sequences selected from those shown in . The tree was constructed with Mega 4 (Tamura et al., Citation2007) using the neighbour-joining method and 1000 bootstrap replicates (bootstrap values are shown on tree). Isolate details are given in and accession numbers are given in Materials and Methods.
Discussion
This paper describes the development of a sensitive RT-PCR test for detecting ANV RNA and its application, which demonstrated the presence of ANV in all 82 gut content samples that had been collected from broiler flocks affected by growth retardation problems. The samples tested included those from 10 submissions collected from six UK poultry organizations between 2004 and 2008, and samples collected from affected broiler chickens in Germany and the USA. In addition, all 80 pooled gut content samples that had been collected in longitudinal surveys of four broiler flocks with below-average performances from day 4 to day 28 were also positive for ANV.
The primers used in the test are located in the 3′-UTR region of the ANV genome, which was found to be highly conserved when relevant sequences from 20 different ANVs were compared (Todd et al., unpublished results). The untranslated or non-coding regions located at the 5′ or 3′ termini of RNA virus genomes have been used previously for the development of RT-PCR tests for detecting a number of viruses, including infectious bronchitis virus (Callison et al., Citation2006) and the bovine pestivirus, bovine diarrhoea disease virus (La Rocca & Sandvik, Citation2009). Terminal untranslated regions are known to have roles in the replication of virus RNA and often contain sequences with substantial secondary and tertiary structure. The high levels of sequence conservation required to preserve functionality can be exploited for the design of RT-PCR primers. In contrast, due to codon degeneracy, it is often very difficult to identify completely conserved sequences of suitable length within protein-coding regions of the genome. The two previously described RT-PCR tests for detecting ANV RNA are both based in coding regions. Despite the use of degenerate primers located within the ORF 1b (polymerase gene), in our hands the test described by Day et al. (Citation2007) failed to amplify product from two of 12 RNAs extracted from representative field samples. The test employed by Mandoki et al. (Citation2006b), which used non-degenerate primers located within the ORF 1a (protease) region, also failed to amplify any RT-PCR product from three of 12 RNAs extracted from the field samples tested. The failure of these previously described RT-PCR tests to detect ANV RNA in some samples is probably due to mismatches between the primer and the target virus RNA sequences. Differences in test sensitivities may also account for the higher detection rates found in the present investigation when compared with the detection rate (56% with chicken samples) reported in the USA study (Day et al., Citation2007). Although the sequence determinations confirmed that the amplicons were ANV specific and the phylogenetic analysis demonstrated that the majority of ANVs compared were more closely related to ANV-2 than ANV-1, work by Strain et al. (Citation2008) has indicated that, due to the occurrence of recombination between astrovirus variants, sequences from several genomic regions should be considered when determining the phylogenetic relationships.
The availability of an RT-PCR test that can sensitively detect ANVs provides a useful diagnostic tool with which investigations into ANV epidemiology and pathogenicity can be initiated. In the four broiler flocks surveyed, the non-detection of ANV in day 0 samples and its subsequent detection at all time points thereafter suggested that most broiler chicks were infected with horizontally-acquired virus that was carried over in the broiler house or that was shed by small numbers of vertically-infected chicks. In this connection, given that ANVs can be difficult to isolate in cell cultures or in embryonated eggs, the RT-PCR test provides a very suitable method for investigating the occurrence of vertically-transmitted ANV in eggs.
In the absence of data from flocks displaying above-average performance, the exceptionally high ANV detection rates observed in this study are of uncertain significance in relation to pathogenic effects that ANV may be causing. Studies with turkey poults have shown that, in addition to being detected in approximately 80% of birds with enteritis and/or growth problems, turkey astroviruses were also detected in some 30% of birds from “normal” flocks of similar age (Reynolds et al., Citation1987). Although more work is needed to determine the prevalence of ANV infections in “normal” flocks showing above-average performances, it is likely that ANV infections are ubiquitous in broiler chickens and that many are subclinical. The highly sensitive RT-PCR that we have developed is unable to discriminate between subclinical infections and those infections that are causing disease problems for the broilers. The severity of the pathogenic effects caused by ANV infections may be dependent on a number of virus-related and host-related factors, including the age and route (vertical or horizontal) of infection, maternal antibody levels, virus strain, size of the infecting dose, and the presence of co-infecting pathogens. We have recently reported that CAstV was detected in 50/52 (96%) of gut content samples from problem broiler flocks that tested positive for ANV in the present investigation (Smyth et al., Citation2009). It remains to be determined whether the pathogenic effects of these two different astroviruses are additive or synergistic. It is possible that the ANV pathogenicity will be reflected by or will be a consequence of the extent of virus replication in the intestine and in other tissues such as the kidney and pancreas that are known to be affected by ANV (Decaesstecker et al., Citation1986; Shirai et al., Citation1992). The development of a real time RT-PCR test with the capacity to quantify virus load is underway to investigate this possibility.
Acknowledgements
The present work was funded in part by the Biotechnology and Biological Sciences Research Council, Department of the Environment, Food and Rural Affairs, and the Department of Agriculture and Rural Development for Northern Ireland.
References
- Callison , S.A. , Hilt , D.A. , Boynton , T.O. , Sample , B.F. , Robison , R. , Swayne , D.E. and Jackwood , M.W. 2006 . Development and evaluation of a real time Taqman RT-PCR assay for the detection of infectious bronchitis virus from infected chickens . Journal of Virological Methods , 138 : 60 – 65 .
- Connor , T.J. , McNeilly , F. , McFerran , J.B. and McNulty , M.S. 1987 . A survey of avian sera from Northern Ireland for antibody to avian nephritis virus . Avian Pathology , 16 : 15 – 20 .
- Day , J.M. , Spackman , E. and Pantin-Jackwood , M. 2007 . A multiplex RT-PCR test for differential identification of turkey astrovirus type 1, turkey astrovirus type 2, chicken astrovirus, avian nephritis virus and avian rotavirus . Avian Diseases , 51 : 681 – 684 .
- Decaesstecker , M. , Charlier , G. and Meulemans , G. 1986 . Significance of parvoviruses, entero-like viruses and reoviruses in the aetiology of the chicken malabsorption syndrome . Avian Pathology , 15 : 769 – 782 .
- Decaesstecker , M. , Charlier , G. , Peeters , J. and Meulemans , G. 1989 . Pathogenicity of fowl enteroviruses . Avian Pathology , 18 : 697 – 713 .
- Imada , T. , Yamaguchi , S. and Kawamura , H. 1979 . Pathogenicity for baby chicks of the G4260 strain of the picornavirus “avian nephritis virus” . Avian Diseases , 23 : 582 – 588 .
- Imada , T. , Yamaguchi , S. , Mase , M. , Tsukamoto , K. , Kubo , M. and Morooka , A. 2000 . Avian nephritis virus (ANV) as a new member of the family Astroviridae and construction of infectious ANV cDNA . Journal of Virology , 74 : 8487 – 8493 .
- Imada , T. , Yamaguchi , S. , Mura , N. and Kawamura , H. 1980 . Antibody survey against avian nephritis virus among chickens in Japan . National Institute Animal Health Quarterly , 20 : 79 – 80 .
- La Rocca , S.A. and Sandvik , T. 2009 . A short target real-time RT-PCR assay for detection of pestiviruses infecting cattle . Journal of Virological Methods , 161 : 122 – 127 .
- Maeda , M. , Imada , T. , Taniguchi , T. and Horiuchi , T. 1979 . Pathological changes in chicks inoculated with the picornavirus “avian nephritis virus” . Avian Diseases , 23 : 588 – 596 .
- Mandoki , M. , Bakonyi , T. , Ivanics , E. , Nemes , C. , Dobos-Kovacs , M. and Rusvai , M. 2006a . Phylogenetic diversity of avian nephritis virus in Hungarian chicken flocks . Avian Pathology , 35 : 224 – 229 .
- Mandoki , M. , Dobos-Kovacs , M. , Bakonyi , T. and Rusvai , M. 2006b . Molecular diagnosis of avian nephritis: preliminary report . Acta Veterinaria Hungarica , 54 : 51 – 60 .
- McNulty , M.S. , Allan , G.M. , Connor , T.J. , McFerran , J.B. and McCracken , R.M. 1984 . An entero-like virus associated with runting stunting syndrome in broiler chickens . Avian Pathology , 13 : 429 – 439 .
- McNulty , M.S. , Connor , T.J. , McNeilly , F. and McFerran , J.B. 1991 . Biological characterisation of avian enteroviruses and enterovirus-like viruses . Avian Pathology , 19 : 75 – 87 .
- Reynolds , D.L. , Saif , Y.M. and Thiel , K.W. 1987 . A survey of enteric viruses in turkey poults . Avian Diseases , 31 : 89 – 98 .
- Shirai , J. , Nakamura , K. , Shinohara , K. and Kawamura , H. 1991 . Pathogenicity and antigenicity of avian nephritis isolates . Avian Diseases , 35 : 49 – 54 .
- Shirai , J. , Tanimura , N. , Uramoto , K. , Narita , M. , Nakamura , K. and Kawamura , H. 1992 . Pathologically and serologically different avian nephritis virus isolates implicated in the etiology of baby chick nephropathy . Avian Diseases , 36 : 369 – 377 .
- Smyth , V.J. , Jewhurst , H.L. , Adair , B.M. and Todd , D. 2009 . Detection of chicken astrovirus by reverse transcription polymerase chain reaction . Avian Pathology , 38 : 293 – 301 .
- Strain , E. , Kelley , L.A. , Schulz-Cherry , S. , Muse , S.V. and Koci , M.D. 2008 . Genomic analysis of closely related astroviruses . Journal of Virology , 82 : 5099 – 5103 .
- Tamura , K. , Dudley , J. , Nei , M. & Kumar , S. 2007 MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0 . Molecular Biology and Evolution , 24 , 1596 1599 Available online at http://www.kumarlab.net/publications (accessed October 2009) .
- Todd , D. , Smyth , V.J. , Ball , N.W. , Donnelly , B.M. , Wylie , M. , Knowles , N.J. and Adair , B. 2009 . Characterisation of chicken enterovirus-like viruses, duck hepatitis virus (DHV) type 2 and DHV type 3 as astroviruses . Avian Pathology , 38 : 21 – 29 .
- Yamaguchi , S. , Imada , T. and Kawamura , H. 1979 . Characterization of a picornavirus from broiler chicks . Avian Diseases , 23 : 571 – 581 .