Abstract
The nucleotide sequence of the HN gene was determined for 21 isolates of avian paramyxovirus type 2 virus and compared with the published HN gene of APMV-2/chicken/California/Yucaipa/56. The HN gene of the 22 viruses had five different lengths in the range of 1737 to 1755 nucleotides coding for 579 to 585 amino acids. Phylogenetic analysis of a corresponding 1734-nucleotide sequence from the HN gene of each virus established five genetic groups (I to V), two of which (II and IV) could be divided into two sub-groups (IIa and IIb; and IVa and IVb). Although there were some exceptions, generally isolates placed in the same genetic group had >80% similarity in nucleotide sequence and <80% with the other isolates; while those in the same sub-group had >90% nucleotide sequence similarity.
Introduction
The genus Avulavirus, sub-family Paramyxovirinae, family Paramyxoviridae, order Mononegavirales (Lamb et al., Citation2005) consists of viruses that have been isolated primarily from birds and placed, on the basis of antigenic relatedness, into nine avian paramyxovirus (APMV) types (APMV-1 to APMV-9). Of these nine serotypes, APMV-2 viruses were the second to be recognized after APMV-1 viruses, which are more commonly known as Newcastle disease viruses. The first isolation of an APMV-2 virus was by Bankowski and co-workers in 1956 in Yucaipa, California, USA from chickens with laryngotracheitis (Bankowski et al., Citation1960). This virus has been named APMV-2/chicken/California/Yucaipa/56 (ck/Yucaipa/56). Since then, numerous viruses antigenically grouped with ck/Yucaipa/56 have been isolated worldwide from domestic poultry, captive caged birds and wild birds (Alexander & Senne, Citation2008). Since the early 1970s, testing of wild captured caged birds in quarantine has frequently resulted in the isolation of APMV-2 viruses, primarily from passerine species but also from psittacine species (Senne et al., Citation1983; Alexander, Citation1993, Citation2000), and these results plus isolations from wild passerine species sampled at the time of trapping (Fleury & Alexander, Citation1979; Tumova et al., Citation1979, Lipkind et al., Citation1982, Citation1995; Goodman & Hanson, Citation1988; Racnik et al., Citation2008) suggest these birds may represent reservoir species for APMV-2 viruses. APMV-2 infections have been reported in chickens in the USA, Canada, Russia, Japan, Israel, India, Saudi Arabia and Costa Rica, and turkeys in the USA, Canada, Israel, France and Italy (Alexander, Citation2000). However, the distribution in poultry is likely to be far more widespread and in recent years APMV-2 viruses have been reported to be common in chickens in the People's Republic of China (Zhang et al., Citation2006, Citation2007), and isolates were obtained from laying hens in Scotland in 2002 and 2006 (Wood et al., Citation2008) and pheasants in England in 2009.
Despite the widespread distribution of APMV-2 viruses and the various hosts from which they have been isolated, there have been few attempts to compare and group different isolates. Ozdemir et al. (Citation1990) prepared three mouse monoclonal antibodies with haemagglutination inhibition (HI) activity against the APMV-2-type species (ck/Yucaipa/56) and were able to place the 53 isolates tested with them into four groups. However, as these authors point out, many of the isolates examined were obtained from quarantine birds, often of undetermined origin, making it difficult to define host, temporal or geographical relationships between viruses placed in the same group.
Although the whole genome of ck/Yucaipa/56 has been sequenced (Subbiah et al., Citation2008), to date there has only been one report of phylogenetic analysis of APMV-2 isolates in which the haemagglutinin-neuraminidase (HN) and fusion (F) genes of four isolates were compared with ck/yucaipa/56. Two isolates were from Gouldian fuches (Erythrura gouldiae) in the same flock and two were from separate broiler chicken flocks in China. All isolates were closely related to ck/Yucaipa/56, with 99.5 to 100% nucleotide identity in the HN gene and 99.6 to 99.9% in the F gene (Zhang et al., Citation2006). In the present study we selected 21 APMV-2 isolates from different hosts and geographical locations and designed primers to allow reverse transcription (RT)-polymerase chain reaction (PCR) and nucleotide sequencing of the HN gene, which the earlier phenotypic study by Ozdemir et al. (Citation1990) had indicated was likely to be variable; phylogenetic trees were then constructed to allow comparisons of genetic relatedness of these and ck/Yucaipa/56.
Materials and Methods
Viruses
All APMV-2 viruses used in the present study () were obtained from the reference collection kept at the Veterinary Laboratories Agency, Weybridge. The viruses were grown in the allantoic cavities of 9-day-old to 11-day-old embryonated specific pathogen free domestic fowls’ (Gallus gallus domesticus) eggs (EFEs), from which the infective allantoic fluid was harvested and stored at –70°C until use. The viruses originated from birds in Europe, Asia and Africa and were isolated from wild birds, poultry and captive caged birds.
Table 1. APMV-2 isolates used in the present study.
Reverse transcription polymerase chain reactions
Viral RNA was extracted from the harvested allantoic fluids of each isolate using the Qiagen QIAamp viral RNA kit by the spin-column method, following the manufacturer's protocol (Qiagen, Crawley, UK). One-step PCR was carried out using the Qiagen OneStep RT-PCR kit in 50 µl final volumes as described in the kit instructions, with the reaction including 5 µl extracted RNA and RNasin® (8U per reaction; Promega, Southampton, UK). RT-PCRs were conducted with a variety of primer combinations designed to amplify within the APMV-2 HN genes; the final concentration of each primer was 50 µM. Thermocycling was conducted on a GS1 instrument (GRI Ltd, Essex, UK), the conditions being: (1) 50°C for 30 min; (2) 95°C for 15 min; and (3) 40 cycles of 94°C for 45 sec, 50°C for 45 sec, 72oC for 2 min.
The PCR products were separated by electrophoresis through a 2% w/v agarose gel in Tris-acetate ethylenediamine tetraacetic acid buffer, stained with ethidium bromide and visualized under ultraviolet light using standard protocols (Sambrook & Russell, Citation2006). All PCR products were purified after being excised from the gel using the Qiagen QIAquick gel extraction kit as described by the manufacturer.
Primer design
Initial RT-PCRs were conducted with the HNP3A/HNP6A primer pair based on ck/Yucaipa/56 (Zhang et al., Citation2006), but this produced the predicted approximately 400 bp amplicon with only 10 of the 21 APMV-2 isolates. Sequencing within this region affirmed clear diversity within the HN genes of the APMV-2 isolates. Consequently, RT-PCR primers were designed for the HN genes by the following cyclical approach. Published (Chang et al., Citation2001; Subbiah et al., Citation2008) HN full-length peptide sequences for ck/Yucaipa/56 (accession number D14030), APMV-1 strain La Sota (AF077761) and APMV-6 (NC_003043) were aligned. Conserved peptide sequences of identical and chemically similar amino acids were identified, and their corresponding nucleotide sequences were aligned and consensus sequences generated using the Clustal W algorithm in the Megalign programme of the Lasergene (DNastar, Madison, Wisconsin, USA) version 7.0 bioinformatics package. These were analysed by the PrimerSelect programme in the Lasergene software package, and probable PCR primer pairs identified that spanned between two conserved regions. The primer sequences were compared with the corresponding region in the APMV-1, APMV-6 and ck/Yucaipa/56 HN genes, as well as any new HN sequences from other APMV-2 isolates generated by amplification with the HNP3A/HNP6A primers. Among the non-APMV-2 HN gene sequences available, APMV-1 and APMV-6 HN genes were chosen because these displayed the greatest conservation with the published ck/Yucaipa/56 sequence. This examination guided introduction of degenerate nucleotides at variable positions within the 3′ half of the primers in order to maximize the chances of successful annealing and amplification of the various APMV-2 isolates. Introduction of degenerate nucleotides at the 3′ terminal end of the primer was avoided. The use of such generic APMV-2 primers resulted in the generation of new APMV-2 HN sequence data for more isolates used in the present study, and this was added to the alignment to similarly guide more primer design by a repeat of the above process. These generic primers were located to produce overlapping amplicons, which ranged in length from 400 to 1100 bp. This cyclic process of RT-PCR generic primer design, sequencing, increasing the amount of new APMV-2 HN sequence data in the alignment, followed by more primer design, continued until almost full-length HN open reading frames had been sequenced for all 21 APMV-2 isolates.
On occasion, primers were designed specifically for APMV-2 isolates that belonged to one monoclonal antibody (mAb) group where the guiding alignment consisted of only newly-generated sequences from the same group. This was successfully employed in, for example, the case of some mAb group 4 isolates in which amplification with generic APMV-2 primers in one region in the HN gene had consistently failed.
Conserved regions of the about 100 carboxy-terminal amino acids of the 5′ neighbouring F gene of the APMV-1, APMV-2 and APMV-6 isolates were aligned to identify regions of nucleotide sequence conservation for design of a conserved forward primer. These were analysed in PrimerSelect together with the about 400 bp APMV-2 HN consensus sequence adjacent to the 5′ end of this gene to identify primer pairs. Amplification across the junction of the HN and F genes facilitated completion of the sequencing to include the 5′ ends of the APMV-2 HN gene. Sequencing of the 3′ ends of the APMV-2 HN gene was similarly completed after amplification of a region that spanned from the HN gene and into the L gene to its 3′ side. Conserved peptides in the amino-terminal region of the L gene from APMV-1, APMV-2 and APMV-6 isolates served to design the reverse primer.
Nucleotide sequencing
Purified amplicons were sequenced using the BigDye DNA sequencing kit (Applied Biosystems (AB), Warrington, UK) and the Applied Biosystems 3130 16 capillary genetic analyser according to the company's instructions. The sequencing primers used were the same as the corresponding PCR primers for that gene region. For larger amplicons (>800 bp), internal sequencing primers were occasionally employed to ensure high-quality sequencing data. All amplicons were sequenced in both orientations, and HN sequence data for all APMV-2 isolates were assembled using the SeqmanII algorithm in the Lasergene bioinformatics package.
The sequences generated in the present study were submitted to the Genbank database. The accession numbers are presented in .
Bioinformatics analysis of sequenced HN genes
Alignment of all 21 new APMV-2 HN gene sequences and the re-sequenced ck/Yucaipa/56 HN was carried out by the Clustal W algorithm (Megalign; Lasergene). This alignment was analysed phylogenetically using the Molecular Evolutionary Genetics Analysis (MEGA version 4.0; Tamura et al., Citation2007). Phylogenetic trees were constructed using the method of minimum evolution (Tamura et al., Citation2007). Evolutionary distances were measured by the number of nucleotide substitutions occurring between isolates, which was done using the Maximum Composite Likelihood model (Tamura et al., Citation2007). Confidence of the estimated relationships was calculated using the bootstrapping procedure; by pseudo-repeating data collecting, the reliability of the tree was estimated by examining the frequency of its occurrence in a large number of trees. A bootstrap repetition value of 1000 was used for this test. The confidence levels for correctly constructing the corresponding clades were shown to be reliable. Predicted properties of the 22 HN amino acid sequences were investigated using the Protean algorithm (Lasergene), while putative signal peptide sequences plus O-glycosylation and N-glycosylation sites were identified using the Net glycosylation software available on the Danish Technical University website (www.dtu.dk/English.aspx).
Monoclonal antibodies
mAb sub-typing of EFE-grown APMV-2 isolates was as described by Ozdemir et al. (Citation1990). In brief, these authors utilized three mAbs (mAb20, mAb79, mAb87) raised against the APMV-2 prototype strain ck/Yucaipa/56 in HI tests (Alexander, Citation2008) and were able to place the viruses they studied into four groups.
Results
Monoclonal antibody sub-typing of isolates
Results of HI tests with the mAbs used by Ozdemir et al. (Citation1990) enabled the 22 viruses to be placed into four groups on the basis of the reaction of the isolates with the mAbs (); these groups corresponded to those reported in the earlier study.
Table 2. Haemagglutination inhibition titres with APMV-2 isolates and mouse monoclonal antibodies raised against ck/Yucaipa/56.
Sequencing results
The HN gene of the 22 APMV-2 viruses had five different lengths, ranging from 1737 nucleotides, coding for 579 amino acids, to 1755 nucleotides, coding for 585 amino acids (). To be able to carry out phylogenetic analysis, the sequences of the HN genes of the 22 viruses were trimmed from the 3′ carboxy terminal to 1734 nucleotides coding for the 578 amino acids from the amino terminus. This length of the HN gene was used for structural analysis using software on the Danish Technical University website. The signal peptide region of the HN gene for all 22 viruses was predicted to be the first 37 amino acids at the amino end of the protein ().
Figure 1. Structural analysis of HN gene. Bold and broken arrows show predicted N-glycosylation and O-glycosylation sites, bold lines with “C” refer to conserved cysteine residues, and shaded regions represent predicted antigenic regions. aa, amino acid.
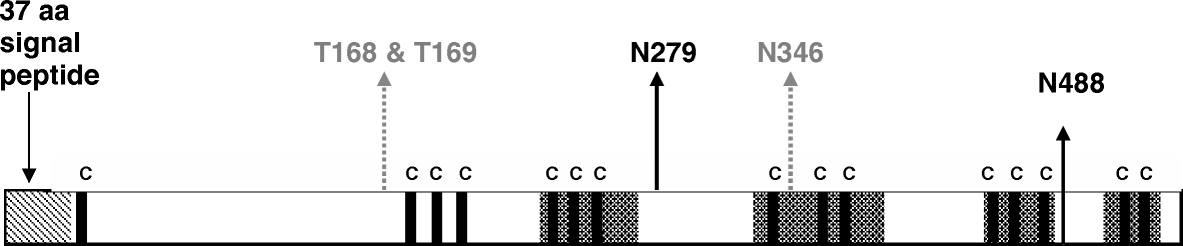
The sialic acid binding site, thought to be the hexapeptide motif NRKSCS, is located at positions 235 to 240 for all isolates, consistent with previous reports for ck/Yucaipa/56 (Mirza et al., Citation1994; Subbiah et al., Citation2008). The six amino acids of the neuraminidase active site identified previously as R175, E400, R415, R505, Y533, E554 (Subbiah et al., Citation2008) were also conserved.
The finding in the earlier report by Subbiah et al. (Citation2008) that the HN protein of strain ck/Yucaipa/56 has 11 conserved cysteine residues in the region corresponding to the globular head was confirmed for each of the other 21 isolates. One extra group of three cysteine residues was observed for all isolates between positions C173, C187 and C197. The cysteine residues, seen in four groups of three residues along the mature peptide, and in one group of two residues at the carboxy terminal of the protein, are shown in .
Sites of possible antigenic significance were determined by Protean (Version 7.0, Lasergene), which takes into account surface probability plots and regions scoring negative for hydrophobicity. As shown in , these regions are found in or adjacent to the conserved cysteine groups.
Of the five potential N-linked glycosylation sites previously identified at N120, N279, N346, N391, N488 (Subbiah et al., Citation2008), only N279 and N488 are conserved in all isolates examined. As predicted by the Net glycosylation software, there is a high likelihood that these two positions are N-glycosylated. A third asparagine residue at position 346 was commonly found in the APMV-2 isolates, but not in all, and was most common in the genetic group II viruses. Differences can be seen in terms of possible N-glycosylation sites of isolates in genetic groups III and IV, but there is not enough data to determine whether these differences are significant—more group III isolates would be needed to evaluate this. There are many sites throughout the HN sequence where O-glycosylation is predicted to occur due to the high abundance of serine and threonine in the protein. The two threonine residues where the predicted O-glycosylation potential is highest, at amino acid positions 168 and 169, are conserved in all 22 viruses and are shown in .
Phylogenetic analysis of isolates
The phylogenetic tree obtained by analysing the 1734 nucleotide sequence of the HN genes of the 22 viruses is shown in . The 22 viruses could be placed into five genetic groups, although groups I and III are represented by only one virus. Despite being the prototype virus, ck/Yucaipa/56 showed only between 67.4 and 70.1% similarity with the other 21 isolates in the sequence analysed and was placed on its own in group I. Eleven of the isolates (isolates 3, 4, 5, 7, 9, 12, 13 19, 20, 21 and 22) showing between 91.4 and 99.9% similarity with each other were placed in group IIa. Isolate 2, gadwall/Kenya/80, had between 82.0 and 83.7% similarity with the IIa viruses and was placed on its own in sub-group IIb. Isolate 8, c-b/Singapore/93, was also placed in a clade on its own (genetic group III); this isolate showed between 67.4 and 77.6% similarity with the other isolates, except isolate 6, robin/Q-Eng/88, with which 83% similarity was recorded. Genetic group IVa was formed from five isolates (6, 10, 14, 15 and 16) that had 90.3 to 97.9% similarity with each other, except that isolates 6 and 14 had only 83.4% similarity. Isolate 11, eagle/Africa/00, had 79.6 to 83.6% similarity with the group IVa viruses and was placed in sub-group IVb. The two remaining isolates 17 and 18, both isolated from passerine species in Slovenia in 2004 and 2005, showed 93.8% nucleotide similarity but only 68.3 to 72.4% with 19 of the other 20 viruses. Isolate 14 had 77.6% similarity with isolate 17 and 77.9% similarity with isolate 18. The Slovenian isolates were placed in genetic group V.
Figure 2. Phylogenetic tree based on data from 22 APMV-2 viruses. Presented as a minimum evolution tree, based on the nucleotide sequences of the HN gene (1734 nucleotides of the open reading frame). Branch lengths represent the predicted number of substitutions and are proportional to the differences between the isolates. The numbers represent confidence levels (%) for having inferred the particular clade correctly. Roman numerals show the genetic groups, the extents of which are shown by the arrows. For full names of isolates, see .
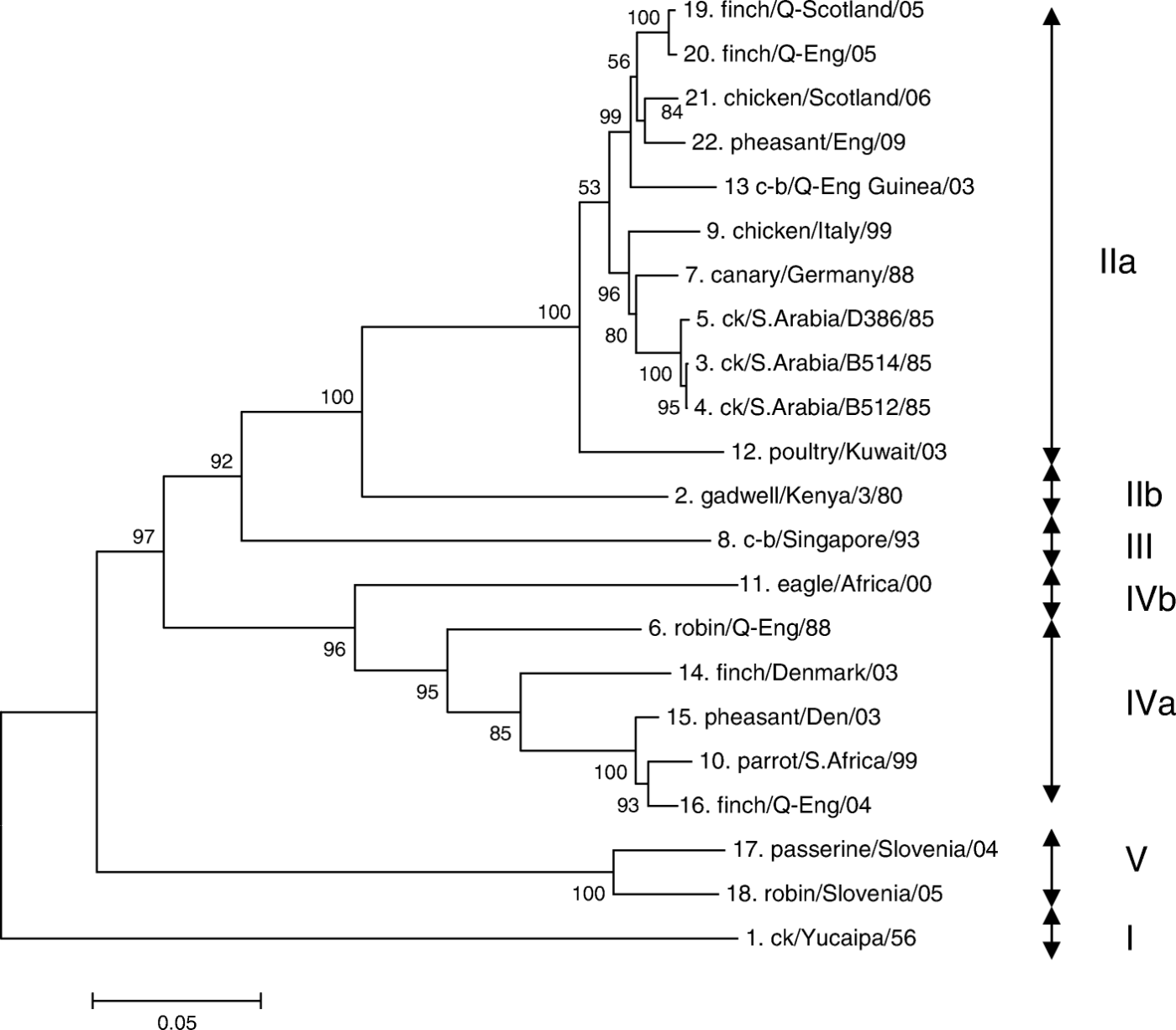
Analysis using amino acid sequences produced essentially the same phylogenetic tree with no important differences (tree not shown). Similarities using amino acid sequences were generally closer, between genetic groups; for example, ck/Yucaipa/56 showed between 73.0 and 77.3% amino acid sequence similarity with the other 21 viruses. The range within groups was very similar to nucleotide sequences; for IIa this was 90.3 to 99.1% amino acid similarity, and for IVa was 89.2 to 97.6%.
Discussion
Of the nine recognized avian paramyxovirus serotypes (APMV-1 to APMV-9), full genome sequences are available for a number of APMV-1 strains and at least one representative of the other serotypes: APMV-2 (Subbiah et al., Citation2008); APMV-3 (Kumar et al., Citation2008); APMV-4 (Nayak et al., Citation2008); APMV-5 (Samuel et al., Citation2010); APMV-6 (Chang et al., Citation2001); APMV-7 (Xiao et al., Citation2009); APMV-8 (Paldurai et al., Citation2009); APMV-9 (Samuel et al., Citation2009). However, although there have been many studies comparing multiple isolates of APMV-1 viruses (for example, Lomniczi et al., Citation1998; Aldous et al., Citation2003) by phylogenetic analysis, usually of partial genome sequences, which have led to a greater understanding of the evolution and epidemiology of these viruses, we believe the present study is the first genetic assessment of a significant number of isolates from any of the other APMV serotypes.
The HN proteins of the 22 APMV-2 viruses have deduced amino acid residues of five different lengths; each of these involves extension of the C-terminus of the open reading frame due to a change in position of the stop codon. This is similar to APMV-1, in which at least six different lengths have been observed for the HN0 protein (Gould et al., Citation2003). It remains to be determined whether this variation in protein length has any impact on the biological activity of the APMV-2 HN protein. In the case of APMV-1, post-translational cleavage of the HN0 precursor protein is required for the larger 616 amino acid protein but not for the smaller 571 amino acid or 577 amino acid lengths (Alexander & Senne, Citation2008). It has been shown for APMV-1 viruses that this variation in length is not linked to pathogenicity (Römer-Oberdörfer et al., Citation2003), but it remains to be seen whether there is an effect on virus replication and host range.
In genetic studies on APMV-1 viruses to date, no rigorous criteria have been established for placing viruses in the same lineage, sub-lineage or clade. In the present study our groupings were to some extent arbitrary, but this resulted in viruses with 80% or greater similarity in nucleotide sequence being placed in the same genetic group and those with more than 90% similarity in the same sub-group, although there were some exceptions. These are not dissimilar values to the relationships between viruses grouped or separated in studies with APMV-1 viruses (for example, Lomniczi et al., Citation1998; Aldous et al., Citation2003). In addition, each identified “grouping” was supported by a bootstrap value in excess of 90% at the relevant node ().
There was broad agreement between the groupings obtained by reactions in HI tests with the three mAbs and the genetic groupings. Ck/Yucaipa/56 (group I) and all genetic group II isolates were placed in mAb group 1, with the exception of isolates, 3, 4 and 5, the closely related APMV-2 viruses from chickens in Saudi Arabia, which were the only three representatives of mAb group 2. C-b/Singapore/93 was the only representative of both mAb group 3 and genetic group III. The isolates placed in genetic groups IV and V all showed group 4 reactions (i.e. HI titres <10) with the three mAbs.
The dissimilarity between ck/Yucaipa/56, which was the only virus isolated in America and by far the oldest isolate, and the other 21 Afro-Eurasian isolates suggested that genetic differences might correlate with temporal or geographical differences. However, it was difficult to establish trends and relationships between the isolates placed in the different genetic groups. As would be expected, the three 1985 isolates from chickens in Saudi Arabia (isolates 3 to 5) were closely related in sub-group IIa, but these were most closely related to an isolate from a canary made in Germany in 1988 (isolate 7) and one from a chicken in Italy (isolate 9) isolated in 1999 (). Four isolates from Great Britain obtained during 2005 to 2009 (isolates 19 to 22) also formed a close group in IIa, but two of these were obtained in 2005 from birds held in quarantine in Scotland and England with unknown countries of origin and the other obtained from layer hens in Scotland in 2006 and pheasants in England in 2009. Although the presence of two similar viruses from different sources in the same laboratory in Scotland always raises concerns of cross-contamination, infection of the layers was confirmed by serological tests showing antibodies to APMV-2 virus in serum samples taken from the birds (Wood et al., Citation2008) and the close similarity with pheasant/Eng/09 gives added reassurance. Three closely related viruses placed in sub-group IVa also showed geographical and temporal diversity, being isolated from pheasants in Denmark in 2003 (isolate 15), a parrot in South Africa in 1999 (isolate 10) and a finch in quarantine in England in 2004 (isolate 16).
Our study has demonstrated that there is considerable genetic diversity between Afro-Eurasian isolates of APMV-2 viruses, which is in contrast to the finding by Zhang et al. (Citation2006) who reported near identity of four isolates from the People's Republic of China with the prototype strain ck/Yucaipa/56. Although our genetic analysis showed clear similarities and differences between the isolates tested, it was difficult to infer significant ecological or epidemiological trends due to the diverse hosts and their uncertain origins, especially for captive caged birds. However, now that the technology for sequencing these viruses has been developed, sequences of additional past and future isolates will be added to the dataset and such trends may become apparent.
References
- Aldous , E.W. , Mynn , J.K. , Banks , J. and Alexander , D.J. 2003 . A molecular epidemiological study of avian paramyxovirus type 1 (Newcastle disease virus) isolates by phylogenetic analysis of a partial nucleotide sequence of the fusion protein gene . Avian Pathology , 32 : 239 – 357 .
- Alexander D.J. 1993 Paramyxovirus infections In J.B. McFerran M.S. McNulty M.C. Horzinek (Series Ed.) . Viral Infections of Vertebrates Volume 3: Viral Infections of Birds 321 340 Amsterdam Elsevier Science
- Alexander , D.J. 2000 . Newcastle disease and other avian paramyxoviruses . OIE Scientific and Technical Review , 19 : 443 – 462 .
- Alexander D.J. 2008 Newcastle disease World Organisation for Animal Health (OIE) Manual of Diagnostic Tests and Vaccines for Terrestrial Animals , 6th edn Chapter 2.3.14 576 589 Paris OIE
- Alexander , D.J. and Senne , D.A. 2008 . “ Newcastle disease, other avian paramyxoviruses and pneumovirus infections: Newcastle disease ” . In Diseases of Poultry , 12th edn , Edited by: Saif , Y.M. , Fadly , A.M. , Glisson , J.R. , McDougald , L.R. , Nolan , L.K. and Swayne , D.E. 75 – 100 . Ames : Blackwell Publishing .
- Bankowski , R.A. , Corstvet , R.E. and Clark , G.T. 1960 . Isolation of an unidentified agent from the respiratory tract of chickens . Science , 132 : 292 – 293 .
- Chang , P.C. , Hsieh , M.L. , Shien , J.H. , Graham , D.A. , Lee , M.S. and Shieh , H.K. 2001 . Complete nucleotide sequence of avian paramyxovirus type 6 isolated from ducks . Journal of General Virology , 82 : 2157 – 2168 .
- Fleury , H.J.A. and Alexander , D.J. 1979 . Isolation of twenty three Yucaipa-like viruses from 616 wild birds in Senegal, West Africa . Avian Diseases , 23 : 742 – 744 .
- Goodman , B.B. and Hanson , R.P. 1988 . Isolation of avian paramyxovirus-2 from domestic and wild birds in Costa Rica . Avian Diseases , 32 : 713 – 717 .
- Gould , A.R. , Hansson , E. , Selleck , K. , Kattenbelt , J.A. , Mackenzie , M. and Della-Porta , A.J. 2003 . Newcastle disease virus fusion and haemagglutinin-neuraminidase gene motifs as markers for viral lineage . Avian Pathology , 32 : 361 – 373 .
- Kumar , S. , Nayak , B. , Collins , P.L. and Samal , S.K. 2008 . Complete genome sequence of avian paramyxovirus type 3 reveals an unusually long trailer region . Virus Research , 137 : 189 – 197 .
- Lamb , R.A. , Collins , P.L. , Kolakofsky , D. , Melero , J.A. , Nagai , Y. Oldstone , M.B.A. 2005 . “ Family paramyxoviridae ” . In Virus Taxonomy, Eighth Report of the International Committee on Taxonomy of Viruses , Edited by: Fauquet , C.M. , Mayo , M.A. , Maniloff , J. , Desselberger , U. and Ball , L.A. 655 – 668 . San Diego : Elsevier Academic Press .
- Lipkind , M. , Alexander , D. , Shihmanter , E. , Weisman , Y. and Collins , M. 1995 . Antigenic heterogeneity of avian paramyxoviruses of serotype 2 (Yucaipa-like) isolated from domestic and wild birds in Israel . Comparative Immunology Microbiology and Infectious Diseases , 18 : 189 – 207 .
- Lipkind , M. , Shihmanter , E. , Weisman , Y. , Aronovici , A. and Shoham , D. 1982 . Characterization of Yucaipa-like avian paramyxoviruses isolated in Israel from domesticated and wild birds . Annales de l'Institut Pasteur, Virologie , 133 : 157 – 161 .
- Lomniczi , B. , Wehmann , E. , Herczeg , J. , Ballagi-Pordany , A. , Kaleta , E.F. Werner , O. 1998 . Newcastle disease outbreaks in recent years in Western Europe were caused by an old (VI) and a novel genotype (VII) . Archives of Virology , 143 : 49 – 64 .
- Mirza , A.M. , Deng , R. and Iorio , R.M. 1994 . Site-directed mutagenesis of a conserved hexapeptide in the paramyxovirus hemagglutinin-neuraminidase glycoprotein: effects on antigenic structure and function . Journal of Virology , 68 : 5093 – 5099 .
- Nayak , B. , Kumar , S. , Collins , P.L. and Samal , S.K. 2008 . Molecular characterization and complete genome sequence of avian paramyxovirus type 4 prototype strain duck/Hong Kong/D3/75 . Virology Journal , 5 : 124
- Ozdemir , I. , Russell , P.H. , Collier , J. , Alexander , D.J. and Manvell , R.J. 1990 . Monoclonal antibodies to avian paramyxovirus type 2 . Avian Pathology , 19 : 395 – 400 .
- Paldurai , A. , Subbiah , M. , Kumar , S. , Collins , P.L. and Samal , S.K. 2009 . Complete genome sequences of avian paramyxovirus type 8 strains goose/Delaware/1053/76 and pintail/Wakuya/20/78 . Virus Research , 142 : 144 – 153 .
- Racnik , J. , Slavec , B. , Trilar , T. , Zadravec , M. , Dovc , A. Krapez , U. 2008 . Evidence of avian influenza virus and paramyxovirus subtype 2 in wild-living passerine birds in Slovenia . European Journal of Wildlife Research , 54 : 529 – 532 .
- Römer-Oberdörfer , A. , Werner , O. , Veits , J. , Mebatsion , T. and Mettenleiter , T.C. 2003 . Contribution of the length of the HN protein and the sequence of the F protein cleavage site to Newcastle disease virus pathogenicity . Journal of General Virology , 84 : 3121 – 3129 .
- Sambrook , J. and Russell , D. 2006 . The Condensed Protocols from Molecular Cloning: A Laboratory Manual , Cold Spring Harbor , NY : Cold Spring Harbor Laboratory Press .
- Samuel , A.S. , Kumar , S. , Madhuri , S. , Collins , P.L. and Samal , S.K. 2009 . Complete sequence of the genome of avian paramyxovirus type 9 and comparison with other paramyxoviruses . Virus Research , 142 : 10 – 18 .
- Samuel A.S. Palduri A. Kumar S. Collins P.L. Samal S.K. 2010 Complete genome sequence of avian paramyxovirus (APMV) serotype 5 completes the analysis of nine PMV serotypes and reveals the longest APMV genome PLoS one, 5, e9269 doi: 10.371/journal.pone.0009269
- Senne , D.A. , Pearson , J.E. , Miller , L.D. and Gustafson , G.A. 1983 . Virus isolations from pet birds submitted for importation into the United States . Avian Diseases , 27 : 731 – 744 .
- Subbiah , M. , Xioa , S. , Collins , P.L. and Samal , S.K. 2008 . Complete sequence of the genome of avian paramyxovirus type 2 (strain Yucaipa) and comparison with other paramyxoviruses . Virus Research , 137 : 40 – 48 .
- Tamura , K. , Dudley , J. , Nei , M. and Kumar , S. 2007 . MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0 . Molecular Biology and Evolution , 24 : 1596 – 1599 .
- Tumova , B. , Stumpa , A. , Janout , V. , Uvizl , M. and Chmela , J. 1979 . A further member of the Yucaipa group isolated from the common wren (Troglodytes troglodytes) . Acta Virologica , 23 : 504 – 507 .
- Wood , A.M. , Dagless , M.D. , Pirie , J.O. , Garcia-Rueda , M.C. , Manvell , R.J. Cox , W.J. 2008 . Isolations of avian paramyxovirus type 2 (APMV-2) viruses from domestic fowl in Scotland in 2002 and 2006 . The Veterinary Record , 162 : 788 – 789 .
- Xiao , S. , Paldurai , A. , Nayak , B. , Subbiah , M. , Collins , P.L. and Samal , S.K. 2009 . Complete genome sequence of avian paramyxovirus type 7 (strain Tennessee) and comparison with other paramyxoviruses . Virus Research , 145 : 80 – 91 .
- Zhang , G. , Zhao , J. , Wang , H. , Yang , A. , Bu , C. and Wang , M. 2006 . Isolation, identification and comparison of four isolates of avian paramyxovirus serotype 2 in China . Avian Diseases , 50 : 386 – 390 .
- Zhang , G. , Zhao , J. and Wang , M. 2007 . Serological survey on the prevalence of antibodies to avian paramyxovirus serotype 2 in China . Avian Diseases , 51 : 137 – 139 .