Abstract
Type C botulinum neurotoxin (BoNT/C)-producing Clostridium botulinum causes animal botulism worldwide and has become a serious problem in poultry flocks and waterfowl in Sweden. The objectives of the present study were to isolate, characterize and subtype C. botulinum type C avian isolates in order to increase the knowledge of the genetic diversity. Isolates from 13 birds were identified by 16S rRNA sequencing and BoNT/C gene detection by real-time polymerase chain reaction (PCR). Conventional PCR was used to distinguish a chimeric BoNTC/D gene, often associated with avian botulism, from the BoNT/C gene. The isolates analysed all contained the gene coding for a chimeric toxin type C/D. Two fingerprinting techniques, pulsed-field gel electrophoresis (PFGE) and randomly amplified polymorphic DNA analysis (RAPD), were optimized and used to investigate the epidemiological relatedness among the strains. The isolates were divided into three different pulsotypes based upon their restriction profiles for SmaI and SalI. The RAPD system proved to be as discriminative as PFGE. This study reveals a small genetic diversity among Swedish type C strains, with a high similarity between strains from broilers and herring gulls.
Introduction
Clostridium botulinum is an anaerobic spore-forming Gram-positive bacterium that is distributed ubiquitously in soil and aquatic environments (Ward et al., Citation1967). The species C. botulinum is divided into four physiological groups (I to IV), which produce botulinum neurotoxins (BoNTs) of seven different serotypes (A to G). Most cases of animal botulism are caused by BoNT types C and D, belonging to the physiological group III (Hatheway, Citation1990). Type C botulism, the paralytic disease caused by BoNT/C, has become an emerging and serious problem in poultry flocks in Sweden (Blomqvist et al., Citation2007). During 2008, botulism outbreaks were confirmed in 13 different broiler farms in Sweden. This is the highest number since 2003, when the first outbreak was reported (Blomqvist et al., Citation2009). Type C botulism has also been reported in waterfowl in Sweden (Neimanis et al., Citation2007). Little is known about the epidemiology of the disease, and the factors behind the outbreaks are not identified.
The approximately 3.8 kb genes encoding BoNT/C and BoNT/D are carried by circular plasmid prophages that express unstable lysogeny in the bacteria (Eklund & Poysky, Citation1974; Sakaguchi et al., Citation2005). Takeda et al. (Citation2005) showed that the neurotoxin genes in Japanese avian isolates comprise chimeric C/D sequences, consisting of an amino terminal part from the BoNT/C gene and a carboxyl terminal domain from the BoNT/D gene. Toxin from most cases of botulism in poultry and wild birds in Sweden can be neutralized with both type C and D antitoxins, indicating that a chimeric form is dominating among Swedish avian cases.
Little has been published on genomic analysis of C. botulinum type C, probably due to difficulties in isolation of clinical strains. Obtaining C. botulinum type C strains in pure culture is difficult, partly because of the obligatory anaerobic growth, but also since there are no known traits suitable to use for selective cultivation (Franciosa et al., Citation1996). At present, the organism is isolated through repeated culturing in broth and on agar plates.
Fingerprinting methods such as pulsed-field gel electrophoresis (PFGE) and randomly amplified polymorphic DNA analysis (RAPD) have been applied to study the relatedness of types A, B and E (Lin & Johnson, Citation1995; Hielm et al., Citation1998; Hyytiä et al., Citation1999), but none of these methods has been thoroughly adapted for type C. PFGE is commonly used to subtype many different species of bacteria and is both reproducible and discriminatory. Limitations of the method typically encountered when analysing clostridia species are the production of endogenous DNases causing DNA degradation, and the difficulties in efficiently lysing the bacteria (Kristjansson et al., Citation1994; Hielm et al., Citation1998). The problem with DNases has previously been solved by fixing cells in formaldehyde before cell lysis (Gibson et al., Citation1994), but the difficulties in obtaining a high quantity of clostridia DNA remain. Furthermore, DNA from several Gram-positive bacteria has been reported to be degraded by Tris radicals formed during electrophoresis (Klaassen et al., Citation2002), something that can be prevented by replacing Tris by thiourea or HEPES buffer (Ray et al., Citation1992). RAPD is a fast and simple polymerase chain reaction (PCR)-based DNA fingerprinting method, first described by Williams et al. (Citation1990). A limitation of this technique is the unspecific binding of the primer, which can cause problems in reproducibility (Bidet et al., Citation2000; Samore et al., Citation1997). However, RAPD can be used as a complement to PFGE for fast characterization, and for strains untypable by PFGE due to DNA degradation.
The objectives of the present study were to isolate and characterize C. botulinum type C avian strains and to increase the knowledge of genetic diversity of Swedish strains by using optimized protocols of PFGE and RAPD.
Materials and Methods
Bacterial strains
The strains used in this study are presented in . The study includes nine clinical Swedish C. botulinum type C isolates from chickens, of which seven were isolated from organs collected during botulism outbreaks in 2008 and two from botulism outbreaks in 2007. The material used for isolation was collected throughout the southern part of Sweden from broilers housed in conventional systems. The study also contains one Norwegian isolate from a botulism outbreak on a broiler farm in 2007 and three from Swedish herring gulls collected in 2007 from an island in the south of Sweden. Strain C-Stockholm and CIP 104983 (Collection of Institute Pasteur, France) were included as reference strains. The botulism outbreaks were confirmed by the mouse bioassay performed according to the Nordic Committee on Food Analysis (Citation1991) at the Swedish National Veterinary Institute (SVA), also described by Lindberg et al. (Citation2010).
Table 1. C. botulinum strains characterized by PCR and 16S rRNA sequencing.
Isolation of C. botulinum strains
One gram of organ material, caecum or liver, was inoculated into 9 ml pre-reduced (boiled for 15 min) tryptose–peptone–glucose–yeast extract (TPGY) broth—5% Tryptone (Difco), 0.5% Proteose Peptone (Difco), 0.4% glucose, 2% yeast extract (Oxoid) and 0.1% starch (Merck)—supplemented with 0.1% l-cysteine–HCl and 0.14% NaHCO3, and then heated for 15 min at 70°C. The sample was incubated overnight at 37°C followed by subculturing of 0.5 ml from the lower part of the culture into fresh, pre-reduced TPGY. The subculture was heated and incubated overnight under the same conditions. An inoculum of the overnight culture was streaked onto a McClung Toabe Agar plate—4% Proteose Peptone (Difco), 0.2% NaCl, 0.2% glucose, 0.5% NaHPO4, 0.1% KH2PO4, 0.02% MgSO4, 0.5% yeast extract (Difco) and 2.5% Bacto agar (Difco)—supplemented with 0.1% l-cysteine–HCl and 0.14% NaHCO3, and incubated at 37°C for 2 days. On McClung Agar containing egg yolk, lipase breaks down free fats in the egg yolk, causing an iridescent sheen on the surface of the colonies. Lecithinase degrades lecithin in the egg yolk, producing an opaque precipitate surrounding the colony. One colony, positive for lipase and lecithinase, was cultured overnight at 37°C in 9 ml pre-reduced TPGY broth. Cells were harvested by centrifugation at 3000 x g for 15 min prior to DNA extraction. All microbial plating was performed in an anaerobic workstation (Electrotek, Shipley, UK) and all incubations were carried out in anaerobic jars (Merck, Darmstadt, Germany).
DNA extraction and detection of the BoNT gene by real-time PCR
The cell pellet was resuspended in 190 µl G2 buffer supplemented with 10 µl proteinase K (components of EZ1 DNA Tissue kit; Qiagen, Hilden, Germany), and then incubated for 30 min at 56°C. The sample was chilled on ice, then treated with 0.4 mg RNase A (Qiagen) and incubated for 5 min. DNA was automatically extracted in the BioRobot® EZ1 workstation (Qiagen) with an elution volume of 50 µl. The eluated DNA was analysed on a 1% (w/v) agarose gel (Agarose NA; GE Healthcare, Little Chalfont, UK) to determine the quality of the DNA. The presence of the BoNT gene was investigated by real-time PCR targeting the amino terminal part of the BoNT/C gene, according to Lindberg et al. (Citation2010). The primers and probe were from Applied Biosystems (Foster City, California, USA) and reactions were performed in a 7500 real-time PCR system (Applied Biosystems).
Storage of strains
Strains showing positive for the BoNT gene were frozen in TPGY broth with 15% glycerol (Sigma, St Louis, Missouri, USA) and stored at −70°C until use. Initiation of a culture from frozen stock had to be performed in liquid culture, after which an inoculum was streaked on a fastidious anaerobe agar plate (LabM, Bury, UK) with 5% defibrinated horse blood (SVA), to confirm an absence of contamination.
BoNT gene characterization
To distinguish the BoNT/C gene and the C/D chimeric neurotoxin gene, the BoNT gene of the isolates was characterized by conventional PCR using three primer pairs (C1Fw and CDRv, CDFw and C2Rv, and CDFw and D2Rv) as described by Takeda et al. (Citation2005). The 50 µl PCR mix for each reaction comprised 50 mM KCl, 10 mM Tris–HCl (pH 8.3), 2.5 mM MgCl2, 0.2 mM each deoxynucleotide, 1 U AmpliTaq Gold polymerase (Applied Biosystems), 1 µM each primer and 50 ng template. Amplification was performed in a PTC-100 thermal cycler (MJ Research, Watertown, Massachusetts, USA), with denaturation at 94°C for 1 min, annealing at 55°C for 1 min and extension at 72°C for 1 min for 40 cycles, followed by 72°C for 7 min. The products were analysed by electrophoresis on a 1.5% (w/v) agarose gel (Agarose NA; GE Healthcare).
Sequence analysis of the 16S rRNA gene
The 16S rRNA gene of the 13 clinical isolates and of the reference strain C-Stockholm was amplified with primers suitable for members of the phylum Firmicutes according to Johansson et al. (Citation1998). The products were used for cycle sequencing with labelled terminators (Big Dye; Applied Biosystems) and with the sequencing primers described by Johansson et al. (Citation2006). The sequencing products were analysed with an ABI Prism 3100 genetic analyser (Applied Biosystems), and contig sequences were generated and edited in the BioNumerics version 5.5 software (Applied Maths, Sint-Martens-Latem, Belgium) and compared in GenBank (Benson et al., Citation2008) using the BLASTn search program (Altschul et al., Citation1990). The sequences were deposited in the European Molecular Biology Laboratory Nucleotide Sequence Database (EMBL, Heidelberg, Germany).
PFGE analysis
The PFGE procedure was performed according to Hielm et al. (Citation1998) but with some modifications as described below. A test panel of five C. botulinum type C strains of different origin—three from Swedish broilers (07-BKT002873 or 07-BKT028387 or 08-BKT015925), one from Norwegian broiler (07-C6N), and one from herring gull (07-V891)—were chosen for evaluation of the different steps of the protocol. An 8 ml volume of overnight TPGY cultures was centrifuged at 3000 x g for 15 min at 10°C. Pellets were resuspended in 1 ml PIV buffer (10 mM Tris, 1 M NaCl, pH 7.5) with or without 4% or 10% formaldehyde and incubated for 1 h on ice. The cells were collected by 5 min centrifugation (3000 x g), washed twice in PIV buffer and resuspended in 330 µl double-strength lysis solution (12 mM Tris, pH 7.5, 2 M NaCl, 200 mM ethylenediamine tetraacetic acid [EDTA], pH 8.0, 1% Brij 58 (Sigma), 0.4% sodium deoxycholate, 1% sodium lauroyl sarcosine). During testing for increased lysis efficiency, 5 U/ml lysostaphin (Sigma) and 20 U/ml mutanolysin (Sigma) were added to each sample. PIV buffer or TE buffer (10 mM Tris and 1 mM EDTA, pH 7.6) were also used for plug casting in comparison with double-strength lysis solution. An equal volume of 2% low-melting point agarose (Agarose Prep; GE Healthcare) was mixed with the cell solution, which was left to solidify for 10 min in agarose plug moulds. Plugs were incubated for 4 h with gentle shaking at 37°C in 5 ml of 1x lysis solution supplemented with lysozyme (Roche, Basel, Switzerland) to a final concentration of 4 mg/ml. The buffer was exchanged for 2.5 ml ESP buffer (0.5 M EDTA and 10% sodium lauryl sarcosine, pH 8.0) containing 1.5 to 2 mg proteinase K (Roche) and incubation was performed for 2 days at 56°C with gentle shaking. To remove proteinase K, plugs were washed twice in double-distilled water for 15 min at 56°C, and four times for 20 min in 56°C TE buffer, and were stored at 4°C until use. Digestion by restriction endonucleases was performed according to the recommendations of the manufacturer with 40 U of the enzymes SmaI (New England BioLabs, Ipswich, Massachusetts, USA), SalI, MluI, ApaI, XbaI or XhoI (GE Healthcare). After digestion, gel slices were inserted into a 1% agarose gel (Agarose NA; GE Healthcare) and electrophoresis was performed at 14°C in 0.5x TBE buffer (45 mM Tris–borate, 1 mM EDTA) or HEPES buffer (16 mM HEPES–NaOH, 16 mM sodium acetate, 0.8 mM EDTA, pH 7.5). The settings were 6 or 4 V/cm for 20 to 30 h at a pulse switch time ramped from 1 to 40 s in a CHEF DRII apparatus (BioRad, Hercules, California, USA). A low-range PFG marker (New England BioLabs) was used as the size standard. DNA fragments were stained in distilled water with 0.5 µg ethidium bromide/ml for 30 min and destained in distilled water for 60 min prior to photography in the Gel Logic 200 Imaging System (Eastman Kodak Company, Rochester, New York, USA). The established protocol was used to analyse all of the strains by PFGE.
RAPD analysis
RAPD analysis was performed on all strains using Illustra Ready-To-Go RAPD Analysis beads (GE Healthcare) as described by the manufacturer, using 50 ng genomic DNA and 25 pmol of a single arbitrary primer from the same manufacturer. All six primers were used on each strain. To confirm consistency, the most discriminating primer was tested running two identical amplifications and one using a different set of template of the same isolates. The samples were denatured at 95°C for 5 min, amplified for 45 cycles of 1 min at 95°C, 1 min at 36°C and 2 min at 72°C, and subjected to a final extension step at 72°C for 5 min, with a PTC-100 thermal cycler. The amplified products were separated by electrophoresis on 1.5% (w/v) agarose gels (Agarose NA; GE Healthcare) in 1x TBE buffer at 120 V for 2.5 h in a Horizontal Electrophoresis Gel Box (Continental Lab Products, San Diego, California, USA). The 100 bp ladder (GE Healthcare) was used as a size marker. Gels were stained and photographed as for the PFGE gels.
Fingerprint analysis
Comparisons of PFGE and RAPD patterns and cluster analysis were performed with the BioNumerics version 5.5 software (Applied Maths). The level of similarity among the DNA profiles was estimated using the Dice coefficient correlation, and cluster analysis was performed with the unweighted pair group method using arithmetic averages. The position tolerance was set at 1.5% and the optimization value at 0.5%.
Results
Isolation of C. botulinum type C strains
Initially, TPGY broth supplemented with sodium thioglycollate was used, but this was exchanged for TPGY broth with l-cysteine–HCl and NaHCO3, which provided better growth of the clinical isolates. It was easier to isolate C. botulinum type C from liver than from caecum. However, the bacterium was not found in all liver samples that had a corresponding positive caecum sample, so sometimes isolation had to be performed from the caecum. Subcultivation by re-inoculation into fresh broth and subsequent heating also proved to be a helpful step to obtain a pure culture (see Supplementary ). Furthermore some isolates that were initially positive in the BoNT/C gene real-time PCR turned negative during the isolation process, which implies that they lost the phage during subcultivation. One of these isolates was included in the fingerprinting analyses (07-BKT028387). Strains were cultivable on McClung Toabe agar plates during the isolation process, but not after freezing in −70°C or in liquid nitrogen. Thus, after freezing, strains had to be cultivated in liquid media.
BoNT gene characterization and 16S rRNA sequencing
DNA from 11 of the strains could be amplified in the conventional BoNT gene-characterization PCR assays. Of those, all clinical strains were of C/D mosaic neurotoxin type, while the two reference strains accordingly were type C (). The three toxigenic strains that did not yield conventional PCR products generated high cycle threshold values in the BoNT/C gene real-time PCR, which thus appears to be more sensitive than the conventional PCR. The results of the sequencing of the 16S rRNA gene are presented in . The 16 S rRNA sequence of the reference strain C-Stockholm shared the highest similarity with Clostridium haemolyticum ATCC 9650 (GenBank accession number NR_024749) and Clostridium novyi ATCC 25758 (GenBank accession number AB035087). The contig sequences of 12 of the clinical strains were identical and differed on seven positions compared with the 16S rRNA sequence of the reference strain C-Stockholm, and by five compared with C. novyi ATCC 25758. Strain 07-BKT002873 differed by one additional nucleotide compared with C-Stockholm and also with C. novyi ATCC 25758. The clinical strains shared the highest similarity (differed by only one gap) to C. botulinum type C strain ATCC 17782 (GenBank accession number L37590).
Nucleotide sequence accession numbers
The three different 16S rRNA sequences have been deposited in the EMBL Nucleotide Sequence Database under accession numbers FN552456 (08-BKT008340), FN552457 (strain 07-BKT002873) and FN552458 (strain C-Stockholm), as presented in .
PFGE optimization
The PFGE protocol described by Hielm et al. (Citation1998) was essentially applicable for C. botulinum type C. The use of mutanolysin was removed from the protocol since it did not improve lysis. The other changes tested (addition of lysostaphin to the lysis buffer and casting plugs in different buffers) did not improve lysis (data not shown). Incubation with lysozyme resulted in slightly sharper bands, even though the incubation time was decreased to 4 h. Formaldehyde fixation of the cells clearly decreased endogenous DNase activity, which resulted in less smearing of the DNA (). However, increasing the percentage of formaldehyde in the PIV solution to 10%, which is recommended for C. botulinum group I (Nevas et al., Citation2005), did not improve the quality of the banding pattern. Using a Tris-containing buffer instead of HEPES buffer for electrophoresis resulted in completely degraded DNA, except for strain 07-BKT002873, even after pre-treatment with formaldehyde (). Electrophoresis in HEPES buffer therefore was incorporated into the PFGE protocol.
Figure 1. PFGE patterns of SmaI digests of four avian strains of C. botulinum type C. Lane 1, strain 08-BKT015925; lane 2, 07-BKT002873; lane 3, 07-C6N; and lane 4, 07-V891. The four left lanes in each gel represent samples fixed with formaldehyde, and the four right lines represent samples without. The outermost lanes contain the Lambda Ladder PFG marker (M). 1a: Gel electrophoresed in 0.5x TBE buffer. 1b: Gel electrophoresed in HEPES buffer. The pulse time was ramped from 1 to 40 s for 24 h at (1a) 6 V/cm and (1b) 4 V/cm. Note that the best result appears from fixing cells in formaldehyde and performing electrophoresis in HEPES buffer.
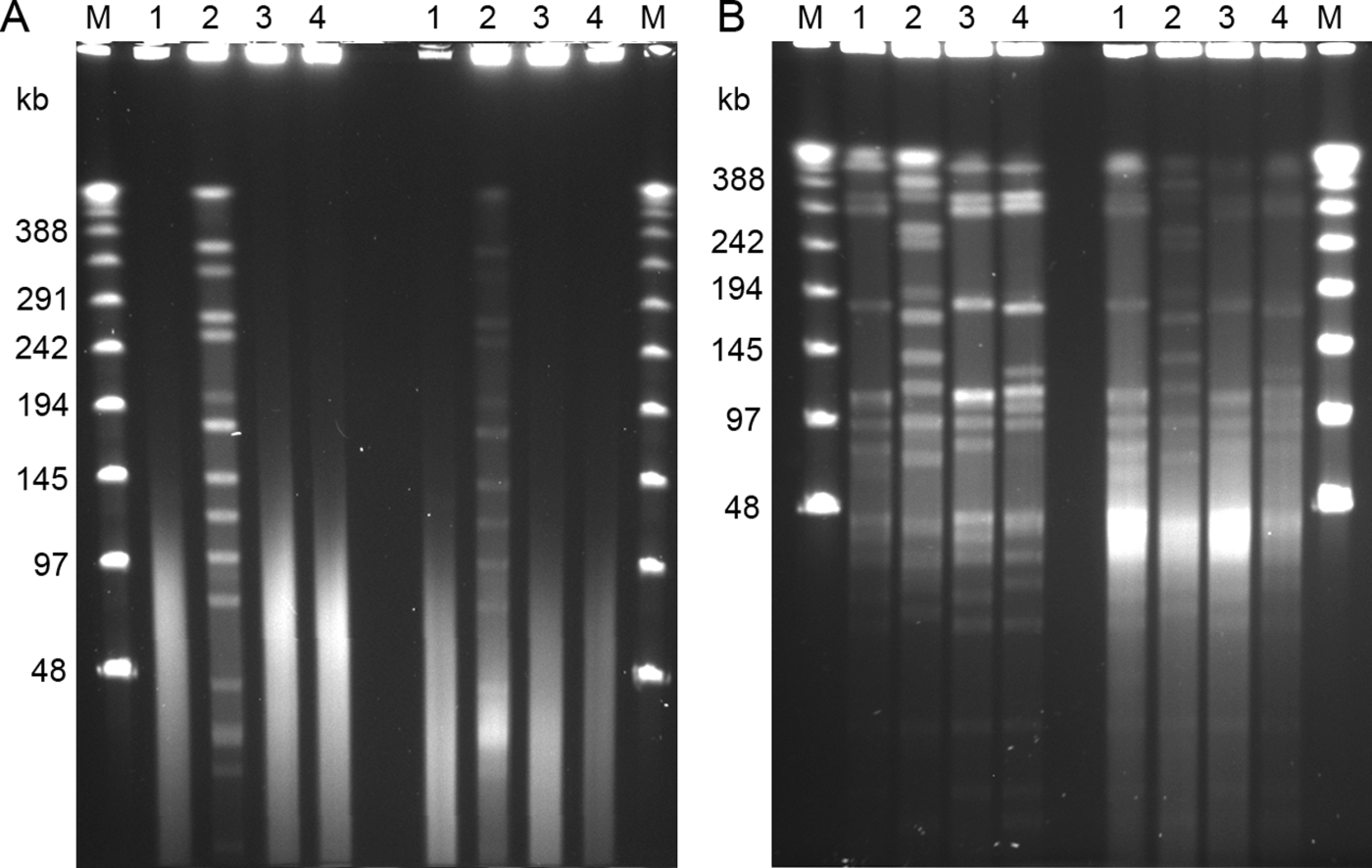
PFGE running conditions
Digestion with XbaI and XhoI resulted in smears of degraded DNA, while the remaining four enzymes resulted in clear fingerprint patterns (). On the basis of the number of bands produced, SmaI and SalI were chosen for epidemiological analysis. The fragments were between 23 and 400 kbp, and to resolve these fragments PFGE runs were ramped from 1 to 40 s. Since HEPES buffer has a higher ionic strength than TBE buffer, the voltage had to be decreased to 4 V/cm and the electrophoresis time increased to 30 h from 20 h. The smallest fragments included in the analysis were 45 kbp for both enzymes.
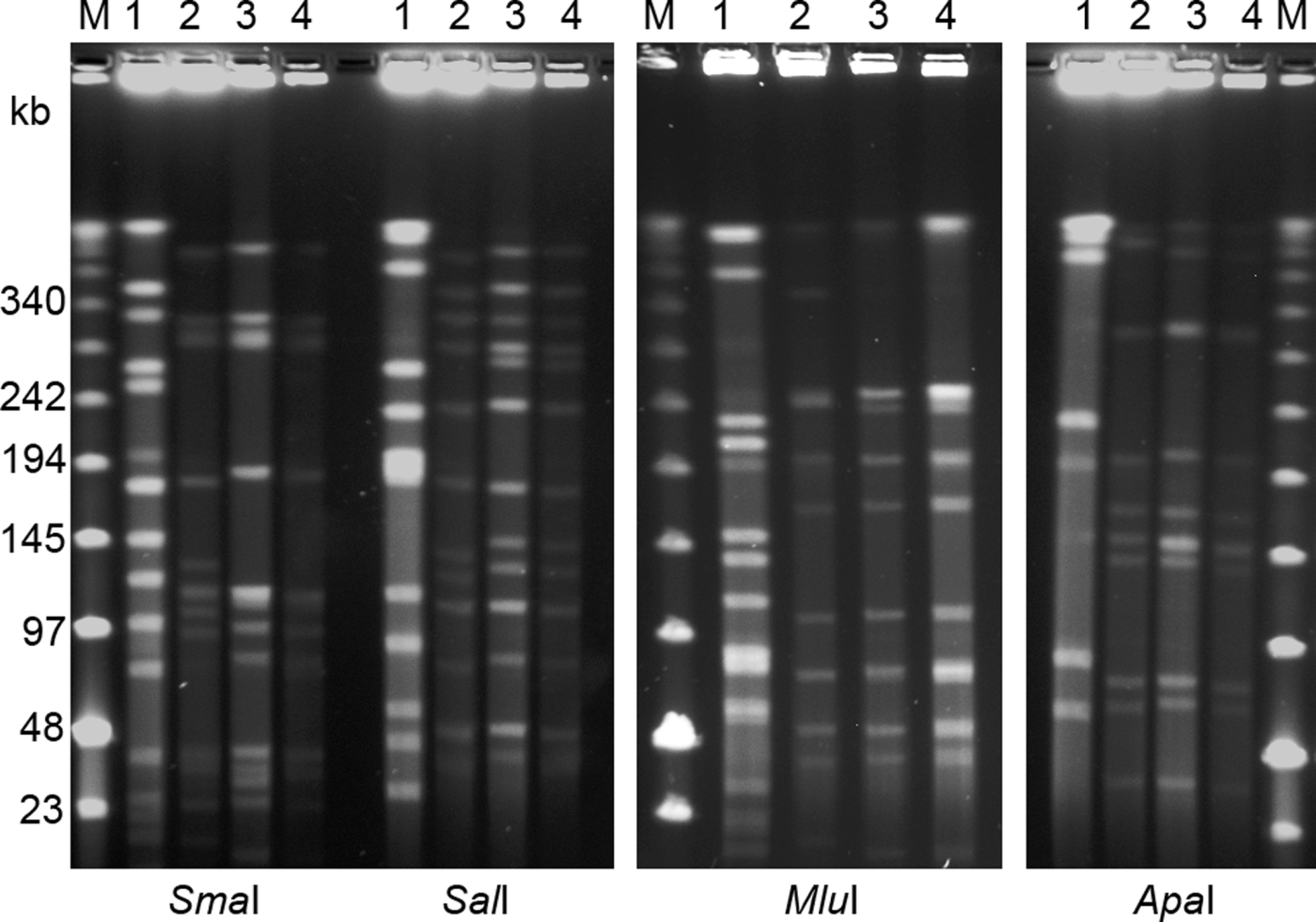
Figure 2. PFGE separation of macrorestriction digests of four avian C. botulinum type C strains using restriction enzymes SmaI, SalI, ApaI and MluI. Lane 1, strain 07-BKT002873; lane 2, 07-V891; lane 3, 07-BKT028387; and lane 4, 08-BKT015925. The outermost lanes contain a low range PFG marker (M). The pulse time was ramped from 1 to 40 s for 30 h at 4 V/cm.
Subtyping of C. botulinum type C strains by PFGE
The C. botulinum type C strains analysed in this study were divided into five different pulsotypes based upon the restriction profiles generated by SmaI and SalI. The two reference strains (strain C-Stockholm and CIP 104983) represented two different pulsotypes. The third pulsotype corresponded to one the Swedish broiler isolates from 2007 (07-BKT002873). The fourth pulsotype consisted of a cluster of the remaining broiler isolates, which generated identical SmaI restriction profiles, but with one strain (08-BKT008340) forming a subgroup among the SalI restriction profiles. However, the subgroup clustered very closely (96% similarity), with only one shifted-band difference. The last pulsotype consisted of the strains from gulls, which all gave rise to identical restriction profiles, sharing 89% similarity with the fourth pulsotype based upon the SalI restriction patterns (93% similarity using SmaI). Thus, the restriction enzyme SalI turned out to be the most discriminative choice in this study. Adding SmaI profiles to the same analysis did not improve differentiation. A dendrogram based on PFGE restriction profiles of SalI is presented in .
Figure 3. Dendrogram of 15 C. botulinum type C strains based on PFGE digestion patterns obtained by the SalI restriction enzyme. The percentage of similarity of the strains was calculated with the Dice coefficient, and the clustering was performed by the unweighted pair group method using arithmetic averages.
Subtyping of C. botulinum type C strains by RAPD
All the strains generated fragment patterns with all six primers, but primer two was the most discriminative. Primer two generated three to four stronger bands between 200 and 900 bp and several weaker bands (), but only the strong bands were included in the analysis. The reference strains (C-Stockholm and CIP 108943) created two individual unique patterns, and so did the strain from a Swedish broiler outbreak in 2007 (07-BKT002873). The three strains from gulls differed from the rest of the broiler strains by lacking one fragment of about 230 bp. Thus, the strains were separated into five profiles, corresponding to the pulsotypes determined by PFGE. These results were consistent during repeated amplifications, even when using DNA frozen for up to 6 months, or applying a different set of templates from the same strans. The other five primers also generated unique patterns for the two reference strains and for strain 07-BKT002873, but could not discriminate between the remaining strains from broilers and those from gulls.
Figure 4. Random-amplified polymorphic-DNA analysis patterns of 15 strains of C. botulinum type C after amplification by primer two (GE Healthcare, Little Chalfont, UK). Lanes 1 to 7, Swedish broiler isolates from 2008; lanes 8 and 10, Swedish broiler isolates from 2007; lane 9, a Norwegian broiler isolate from 2007; lanes 11 to 13, isolates from herring gulls; and lanes 14 and 15, reference strains C-Stockholm and CIP 108943. The outermost lanes are the 100 kb marker (M).
Discussion
Avian botulism is a serious and emerging problem. Molecular characterization of isolates from avian botulism cases has so far been performed to a small extent, and therefore little is known about the genetic diversity of the species. The present study demonstrates that the avian C. botulinum strains analysed contained the gene coding for a chimeric toxin type C/D. Comparison of the strains using PFGE and RAPD revealed a high genetic similarity between strains from broilers and herring gulls, but a high diversity compared with the type C reference strains.
Isolation of C. botulinum type C is considered difficult, mainly because of its poor growth in mixed culture and on agar plates and because there are no known traits suitable for selective cultivation. Another problem isolating toxigenic strains is the unstable lysogeny of the phage (Sakaguchi et al., Citation2005). In this study some strains lost the phage during subcultivation. This caused problems in the isolation process because the BoNT/C gene real-time PCR was used to confirm the presence of the bacteria. The current cultivation method for isolation is time-consuming, and a simpler and more efficient isolation method needs to be developed to enable larger molecular epidemiological studies. In this study, it was easier to isolate C. botulinum type C from liver samples than from caecum material. This has also been described by Myllykoski et al. (Citation2009) and is probably due to fewer competing microbes in the liver sample.
The 13 C. botulinum type C strains isolated in the present study were obtained from avian material collected in Sweden and Norway during 2007 and 2008. The neurotoxin gene of the nine strains that were characterized with conventional PCR assays were of type C/D, which is in agreement with previous characterization of Japanese avian strains (Takeda et al., Citation2005). It has been shown that type C/D toxin is more toxic to avian species than either type C or D (Miyazaki & Sakaguchi, Citation1978; Takeda et al., Citation2005), but the toxigenic properties of avian strains have not been satisfactorily explained. Given that chicken litter often is the suspected source of C. botulinum-contamination leading to cattle botulism (Appleyard & Mollison, Citation1985; McLoughlin et al., Citation1988; Neill et al., Citation1989), it would be interesting to know how susceptible cattle are to type C/D toxin.
Until recently when Nakamura et al. (Citation2009) published two type C strains analysed by PFGE, there were no reports about subtyping of C. botulinum type C with either PFGE or RAPD. Since type C botulism is an emerging disease, there is clearly a need for application and evaluation of such methods for epidemiological studies. Amplified fragment length polymorphism (AFLP) has been applied to C. botulinum type C (Hill et al., Citation2007) and the technique is useful in epidemiological studies where numerous strains are being compared. However, standardization of the technique is necessary before interlaboratory comparisons can be performed.
The PFGE procedure in the present study was based on a protocol developed for C. botulinum group II (Hielm et al., Citation1998) with the major exception that electrophoresis in TBE buffer resulted in smears of completely degraded DNA. The use of HEPES buffer has been recommended for prevention of DNA degradation during electrophoresis (Ray et al., Citation1992). In our study, band profiles emerged for all 15 strains using HEPES buffer, compared with only one strain when using TBE buffer. The addition of thiourea to the TBE buffer to prevent DNA degradation has been the more frequently used alternative (Zhang et al., Citation2004; Leclair et al., Citation2006), but since thiourea is a carcinogenic agent we chose not to use it. Endogenous DNases were efficiently inactivated by the formaldehyde treatment described for C. botulinum group II. Although lysis was not optimal, the adapted protocol generated sufficiently strong band profiles for subtyping. SmaI and XhoI have been used previously to type C. botulinum isolates (Hielm et al., Citation1998; Nevas et al., Citation2005), but the restriction enzyme SalI turned out to be more discriminative of the three. One strain that lost the phage during subculturing was included in the study and its SmaI and SalI profiles were identical to the toxigenic strain patterns within the same pulsotype. This indicated that there are no restriction sites in the phage and it allowed us to include a non-toxigenic strain in the comparison.
All strains could be typed by RAPD with the six primers tested, although only primer two was as discriminative as the PFGE system. Hyytiä et al. (1999), analysed C. botulinum group I and II strains with primers one to six from the same kit, and found primer five to be most discriminative of them. However, when primer five was applied in this study, it could not differentiate between strains from broilers and gulls. Like Hyytiä et al., we found the system with Ready-To-Go Analysis RAPD beads highly reproducible when only including strong bands in the clustering analysis. Furthermore, we did not find that frozen storage of the DNA (up to 6 months) affected the results. The disadvantage with the RAPD system is that it was more difficult to interpret the banding patterns than with the PFGE system. However, RAPD can be used as a complement to PFGE for pulsed-field untypable strains and also for primary screening because the results can be obtained much faster.
The cluster analysis based upon the PFGE results of digestion with SalI, and the RAPD analysis with primer two, revealed the strains from outbreaks in geographically separated Swedish broiler farms in late 2007 and 2008 to be almost identical. This could be due to contamination from a common source. However, little is known about the overall genetic diversity of type C strains, and in our study we show that these strains were also identical to one Norwegian strain from 2007. One strain from an outbreak in a Swedish broiler farm in early 2007 generated fingerprinting profiles that differed considerably from the rest of the clinical isolates. This strain also had one nucleotide difference in the 16S rRNA sequence compared with the other clinical strains that shared identical sequences. The clustering analysis also revealed how different the avian type C/D isolates were from the two type C reference strains. The nomenclature of C. botulinum is based upon the BoNT genes, which means that all bacteria with a BoNT/C gene are classified as a C. botulinum type C. Although the 16S rRNA sequence of the C-Stockholm reference strain was more similar to the corresponding genes in C. novyi and C. haemolyticum than to another C. botulinum type C strain (ATCC 17782), it is still considered a C. botulinum strain since it contains the gene for BoNT/C. A problem when comparing C. botulinum type C and also type D strains is the possibility that the phage with the BoNT gene may be harboured by other species of bacteria. It has been demonstrated that C. botulinum type C also can be infected by phages from type D and C. novyi type A (Eklund & Poysky, Citation1974; Eklund et al., Citation1974). However, the identification of C. botulinum type C will hopefully be simpler once the genome is assembled and chromosomal markers can be found and used for confirmation.
The results of the present study indicate that there is a small genetic diversity among Swedish type C strains, with a high similarity between strains from broilers and gulls. However, more strains from other wild birds need to be characterized for a better understanding of the genetic variation, since the three strains in this study were from the same geographic location. In a study by Hill et al. (Citation2007) where 19 C. botulinum type C strains were subtyped by AFLP , they clustered according to toxin type (C or C/D), the source of isolation, and to some extent also geographic location. In conclusion, the protocols in this study for subtyping of C. botulinum type C by PFGE and RAPD will be useful in further research in the so-far modestly explored area of type C botulism.
Supplementary Data
Download ()Acknowledgements
The present work was supported by grants from the Swedish Farmer's Foundation for Agricultural Research and the Swedish Environmental Protection Agency. Part of the work on the subtyping was financially supported by the National Veterinary Institute Research Facility for small-scale research and pilot projects. The authors are also grateful to Marianne Persson for her technical assistance in 16S rRNA sequencing, and to Henrik Uhlhorn and Bruce David at The National Veterinary Institute, Norway, for kindly providing isolation material.
References
- Altschul , S.F. , Gish , W. , Miller , W. , Myers , E.W. and Lipman , D.J. 1990 . Basic local alignment search tool . Journal of Molecular Biology , 215 : 403 – 410 .
- Appleyard , W.T. and Mollison , A. 1985 . Suspected bovine botulism associated with broiler litter waste . The Veterinary Record , 116 : 522
- Benson , D. A. , Karsch-Mizrachi , I. , Lipman , D. J. , Ostell , J. and Wheeler , D.L. 2008 . GenBank . Nucleic Acids Research , 36 : D25 – D30 .
- Bidet , P. , Lalande , V. , Salauze , B. , Burghoffer , B. , Avesani , V. Delmee , M. 2000 . Comparison of PCR-ribotyping, arbitrarily primed PCR, and pulsed-field gel electrophoresis for typing Clostridium difficile . Journal of Clinical Microbiology , 38 : 2484 – 2487 .
- Blomqvist G. Berndtson E. Jansson D.S. Båverud V. Engström B. 2007 Botulism on a broiler breeder farm in Sweden In Proceedings of the 15th World Veterinary Poultry Congress 553 Beijing , , China
- Blomqvist G. Skarin H. Lindberg A. Båverud V. Engström B. 2009 Surveillance for Clostridium botulinum type C or C/D in Swedish broilers In Proceedings of the 16th World Veterinary Poultry Congress 415 Marrakesh, Morocco
- Eklund , M.W. and Poysky , F.T. 1974 . Interconversion of type C and D strains of Clostridium botulinum by specific bacteriophages . Applied Microbiology , 27 : 251 – 258 .
- Eklund , M.W. , Poysky , F.T. , Meyers , J.A. and Pelroy , G.A. 1974 . Interspecies conversion of Clostridium botulinum type C to Clostridium novyi type A by bacteriophage . Science , 186 : 456 – 458 .
- Franciosa , G. , Fenicia , L. , Caldiani , C. and Aureli , P. 1996 . PCR for detection of Clostridium botulinum type C in avian and environmental samples . Journal of Clinical Microbiology , 34 : 882 – 885 .
- Gibson , J.R. , Sutherland , K. and Owen , R.J. 1994 . Inhibition of DNAse activity in PFGE analysis of DNA from Campylobacter jejuni . Letters in Applied Microbiology , 19 : 357 – 358 .
- Hatheway , C.L. 1990 . Toxigenic clostridia . Clinical Microbiological Reviews , 3 : 66 – 98 .
- Hielm , S. , Björkroth , J. , Hyytiä , E. and Korkeala , H. 1998 . Genomic analysis of Clostridium botulinum group II by pulsed-field gel electrophoresis . Applied and Environmental Microbiology , 64 : 703 – 708 .
- Hill , K.K. , Smith , T.J. , Helma , C.H. , Ticknor , L.O. , Foley , B.T. Svensson , R.T. 2007 . Genetic diversity among Botulinum neurotoxin-producing clostridial strains . Journal of Bacteriology , 189 : 818 – 832 .
- Hyyti , E. , Bjorkroth , J. , Hielm , S. and Korkeala , H. 1999 . Characterisation of Clostridium botulinum groups I and II by randomly amplified polymorphic DNA analysis and repetitive element sequence-based PCR . International Journal of Food Microbiology , 48 : 179 – 189 .
- Johansson , A. , Aspan , A. , Bågge , E. , Baverud , V. , Engström , B.E. and Johansson , K.E. 2006 . Genetic diversity of Clostridium perfringens type A isolates from animals, food poisoning outbreaks and sludge . BMC Microbiology , 6 : 47
- Johansson K.-E. Heldtander M. U. Pettersson B. Characterization of mycoplasmas by PCR and sequence analysis with universal 16S rDNA primers In R. Miles R. Nicholas Mycoplasma Protocols. Methods in Mdecular Biology 145 165 Totowa , NJ Humana Press
- Klaassen , C.H. , van Haren , H.A. and Horrevorts , A.M. 2002 . Molecular fingerprinting of Clostridium difficile isolates: pulsed-field gel electrophoresis versus amplified fragment length polymorphism . Journal of Clinical Microbiology , 40 : 101 – 104 .
- Kristjansson , M. , Samore , M.H. , Gerding , D.N. , DeGirolami , P.C. , Bettin , K.M. , Karchmer , A.W. and Arbeit , R.D. 1994 . Comparison of restriction endonuclease analysis, ribotyping, and pulsed-field gel electrophoresis for molecular differentiation of Clostridium difficile strains . Journal of Clinical Microbiology , 32 : 1963 – 1969 .
- Leclair , D. , Pagotto , F. , Farber , J.M. , Cadieux , B. and Austin , J.W. 2006 . Comparison of DNA fingerprinting methods for use in investigation of type E botulism outbreaks in the Canadian Arctic . Applied and Environmental Microbiology , 65 : 703 – 708 .
- Lin , W.J. and Johnson , E.A. 1995 . Genome analysis of Clostridium botulinum type A by pulsed-field gel electrophoresis . Applied and Environmental Microbiology , 61 : 4441 – 4447 .
- Lindberg A. Skarin H. Knutsson R. Blomqvist G. Baverud V. 2010 Real-time PCR for Clostridium botulinum type C neurotoxin (BoNTC) gene, also covering a chimeric C/D sequence—application on outbreaks of botulism in poultry Veterinary Microbiology in press . doi: 10.1016/j.vetmic.2010.04.030
- McLoughlin , M.F. , McLlroy , S.G. and Neill , S.D. 1988 . A major outbreak of botulism in cattle being fed ensiled poultry litter . The VeterinaryRecord , 122 : 579 – 581 .
- Miyazaki , S. and Sakaguchi , G. 1978 . Experimental botulism in chickens: the cecum as the site of production and absorption of botulinum toxin . Japanese Journal of Medical Science & Biology , 31 : 1 – 15 .
- Myllykoski , J. , Lindström , M. , Keto-Timonen , R. , Söderholm , H. , Jakala , J. Kallio , H. 2009 . Type C bovine botulism outbreak due to carcass contaminated non-acidified silage . Epidemiology and Infection , 137 : 284 – 293 .
- Nakamura , K. , Kohda , T. , Umeda , K. , Yamamoto , H. , Mukamoto , M. and Kozaki , S. 2009 . Characterization of the D/C mosaic neurotoxin produced by Clostridium botulinum associated with bovine botulism in Japan . Veterinary Microbiology , 140 : 147 – 154 .
- Neill , S.D. , McLoughlin , M.F. and McIlroy , S.G. 1989 . Type C botulism in cattle being fed ensiled poultry litter . The Veterinary Record , 124 : 558 – 560 .
- Neimanis , A. , Gavier-Widen , D. , Leighton , F. , Bollinger , T. , Rocke , T. and Morner , T. 2007 . An outbreak of type C botulism in herring gulls (Larus argentatus) in southeastern Sweden . Journal of Wildlife Diseases , 43 : 327 – 336 .
- Nevas , M. , Lindstrom , M. , Hielm , S. , Bjorkroth , K.J. , Peck , M.W. and Korkeala , H. 2005 . Diversity of proteolytic Clostridium botulinum strains, determined by a pulsed-field gel electrophoresis approach . Applied and Environmental Microbiology , 71 : 1311 – 1317 .
- Nordic Committee on Food Analysis 1991 Botulinum Toxin, Detection in Foods, Blood and Other Test Materials (Vol. 79) , 2nd edn Oslo Nordic Committee on Food Analysis.
- Ray , T. , Weaden , J. and Dyson , P. 1992 . Tris-dependent site-specific cleavage of Streptomyces lividans DNA . FEMS Microbiology Letters , 75 : 247 – 252 .
- Sakaguchi , Y. , Hayashi , T. , Kurokawa , K. , Nakayama , K. , Oshima , K. Fujinaga , Y. 2005 . The genome sequence of Clostridium botulinum type C neurotoxin-converting phage and the molecular mechanisms of unstable lysogeny . Proceedings of the National Academy of Sciences of the USA , 102 : 17472 – 17477 .
- Samore , M. , Killgore , G. , Johnson , S. , Goodman , R. , Shim , J. Venkataraman , L. 1997 . Multicenter typing comparison of sporadic and outbreak Clostridium difficile isolates from geographically diverse hospitals . Journal of Infectious Diseases , 176 : 1233 – 1238 .
- Takeda , M. , Tsukamoto , K. , Kohda , T. , Matsui , M. , Mukamoto , M. and Kozaki , S. 2005 . Characterization of the neurotoxin produced by isolates associated with avian botulism . Avian Diseases , 49 : 376 – 381 .
- Ward , B.Q. , Carroll , B.J. , Garrett , E.S. and Reese , G.B. 1967 . Survey of the U.S. Atlantic coast and estuaries from Key Largo to Staten Island for the presence of Clostridium botulinum . Applied Microbiology , 15 : 964 – 965 .
- Williams , J.G. , Kubelik , A.R. , Livak , K.J. , Rafalski , J.A. and Tingey , S.V. 1990 . DNA polymorphisms amplified by arbitrary primers are useful as genetic markers . Nucleic Acids Research , 18 : 6531 – 6535 .
- Zhang , Y. , Yakrus , M.A. , Graviss , E.A. , Williams-Bouyer , N. , Turenne , C. , Kabani , A. and Wallace , R.J. Jr . 2004 . Pulsed-field gel electrophoresis study of Mycobacterium abscessus isolates previously affected by DNA degradation . Journal of Clinical Microbiology , 42 : 5582 – 5587 .
Supplementary material
Figure 5. Clostridium botulinum type C, strain 07-BKT028387, in pure culture on McClung Toabe agar.