Abstract
Molecular analysis was conducted on 36 Mycoplasma gallisepticum DNA extracts from tracheal swab samples of commercial poultry in seven South African provinces between 2009 and 2012. Twelve unique M. gallisepticum genotypes were identified by polymerase chain reaction and sequence analysis of the 16S-23S rRNA intergenic spacer region (IGSR), M. gallisepticum cytadhesin 2 (mgc2), MGA_0319 and gapA genetic regions. The DNA sequences of these genotypes were distinct from those of M. gallisepticum isolates in a database composed of sequences from other countries, vaccine and reference strains. The most prevalent genotype (SA-WT#7) was detected in samples from commercial broilers, broiler breeders and layers in five provinces. South African M. gallisepticum sequences were more similar to those of the live vaccines commercially available in South Africa, but were distinct from that of F strain vaccine, which is not registered for use in South Africa. The IGSR, mgc2 or MGA_0319 sequences of three South African genotypes were identical to those of the ts-11 vaccine strain, necessitating a combination of mgc2 and IGSR targeted sequencing to differentiate South African wild-type genotypes from ts-11 vaccine. To identify and differentiate all 12 wild-types, mgc2, IGSR and MGA_0319 sequencing was required. Sequencing of gapA was least effective at strain differentiation. This research serves as a model for the development of an M. gallisepticum sequence database, and illustrates its application to characterize M. gallisepticum genotypes, select diagnostic tests and better understand the epidemiology of M. gallisepticum.
Introduction
Mycoplasma gallisepticum, the aetiologic agent of chronic respiratory disease in chickens and infectious sinusitis in turkeys, is the most pathogenic mycoplasmal pathogen of poultry (Ley, Citation2008). M. gallisepticum has a considerable economic impact on the poultry industry worldwide, through production losses, airsacculitis condemnations and mortality, and the associated costs of control and surveillance testing. In South Africa, M. gallisepticum has been implicated as the cause of severe respiratory disease. Farmers depend on vaccination (with live and/or killed oil emulsion vaccines) and antibiotic treatment to control this disease in infected flocks. Of the live vaccines available for M. gallisepticum control, only strains ts-11 (Whithear et al., Citation1990a, Citation1990b) and 6/85 (Evans & Hafez, Citation1992) are commercially available in South Africa; F strain vaccines (Adler et al., Citation1960; Luginbuhl et al., Citation1967) are not currently registered for use.
The genome of M. gallisepticum evolves extremely rapidly (Delaney et al., Citation2012), resulting in genetic variants (strains) with marked differences in antigenicity, pathogenicity and transmissibility (Rodriguez & Kleven, Citation1980; Lin & Kleven, Citation1984; Levisohn et al., Citation1986; Yoder, Citation1986; Kleven et al., Citation1988; Garcia et al., Citation1994; Kleven, Citation1998; Ley, Citation2008). These strain differences potentially have implications for both the success of vaccination and M. gallisepticum detection by diagnostic assays (Kleven et al., Citation1988; Garcia et al., Citation1994; Rosengarten & Yogev, Citation1996; Kleven, Citation1998; Ley, Citation2008). With the worldwide increase in live vaccine usage to control M. gallisepticum, techniques that allow differentiation of wild-type (field strain) M. gallisepticum from the vaccine strains are particularly important. In addition, strain differentiation techniques may be utilized as valuable tools for the control of M. gallisepticum, facilitating rapid recognition of outbreaks, epidemiological traceability and targeted control (Ferguson et al., Citation2005).
A number of molecular techniques have been described for M. gallisepticum strain differentiation, including restriction fragment length polymorphism (Kleven et al., Citation1988), amplified fragment length polymorphism (Hong et al., Citation2005), random amplified polymorphic DNA (RAPD) (Geary et al., Citation1994; Fan et al., Citation1995), and, recently, polymerase chain reaction (PCR) and high-resolution melting curve analysis (Ghorashi et al., Citation2010). While RAPD has been widely and successfully used for M. gallisepticum strain differentiation, it has intrinsic problems with reproducibility, hampering inter-laboratory comparisons and long-term epidemiological studies (Tyler et al., Citation1997; Ferguson et al., Citation2005).
Targeted sequencing of selected M. gallisepticum genomic regions has been demonstrated to facilitate very good strain differentiation, with sequencing of multiple genetic targets allowing better discriminatory power than RAPD analysis (Ferguson et al., Citation2005). Despite the extraordinary point substitution rate of M. gallisepticum (Delaney et al., Citation2012), selected genomic targets have proven sequence stability (Ferguson et al., Citation2005; Raviv et al., Citation2007). Targeted genetic sequencing is also a highly reproducible strain differentiation method, allowing the development of a reference database and global comparisons between laboratories (Ferguson et al., Citation2005). A significant advantage of targeted sequencing over RAPD is that is can be performed directly from clinical samples, without the requirement for mycoplasma isolation in pure culture (Ferguson et al., Citation2005; Raviv et al., Citation2007).
The first objective of this study was to develop an M. gallisepticum sequence database by targeted genetic sequencing of extracted M. gallisepticum DNA from a range of commercial poultry types and geographic locations within South Africa. The next objective was to use this sequence data to characterize South African M. gallisepticum wild-type genotypes (hereafter referred to as “wild-types”), determine their distribution within the country and their identity to M. gallisepticum vaccines and wild-types from other countries, and finally to assess the ability of the selected targets to differentiate South African wild-types. Four previously characterized genetic regions were selected for analysis: two genes encoding surface proteins involved in cytadhesion (M. gallisepticum cytadhesin 2 [mgc2] and gapA), one gene encoding a predicted conserved surface lipoprotein (MGA_0319), and the 16S-23S rRNA intergenic spacer region (IGSR) (Ferguson et al., Citation2005; Raviv et al., Citation2007). This research demonstrates the value of an M. gallisepticum sequence database, and is the first report on the diversity and distribution of M. gallisepticum wild-types in South Africa.
Materials and Methods
M. gallisepticum DNA
Tracheal swab samples were obtained from commercial broiler, broiler breeder and layer flocks from seven of the nine South African provinces between 2009 and 2012. An opportunity sampling approach was employed, which depended on voluntary sample submission from flocks known or suspected of being infected with M. gallisepticum. In addition, M. gallisepticum DNA extracts contributed by five South African diagnostic laboratories were incorporated into the study. All submitters were requested to complete a questionnaire detailing the province of origin, flock M. gallisepticum vaccination history, and poultry type from which samples were collected. The M. gallisepticum vaccination history of the 36 flocks from which samples were collected was as follows: non-vaccinated (15 flocks); ts-11 vaccinated (three flocks); recombinant fowlpox-M. gallisepticum vaccinated (two flocks); M. gallisepticum bacterin vaccinated (three flocks); and vaccination history unknown or not reported (13 flocks). DNA extraction from swab samples was performed at Allerton Provincial Veterinary Laboratory in South Africa (KwaZulu-Natal Department of Agriculture and Environmental Affairs, Pietermaritzburg, KwaZulu-Natal) using the Qiagen DNeasy® Blood and Tissue Kit (QIAGEN, Hilden, Germany) according to the manufacturer's instructions. Extracted DNA was submitted to the Poultry Diagnostic and Research Center (PDRC, University of Georgia, Athens, GA, USA) in 1.5 ml Eppendorf Safe-Lock tubes™ (Eppendorf AG, Hamburg, Germany) or on Whatman™ FTA™ cards (Whatman, Piscataway, NJ, USA). DNA was extracted from FTA™ cards using the Qiagen QIAmp® DNA Mini Kit (QIAGEN, Valencia, CA, USA). Then 200 µl Whatman™ FTA™ Purification Reagent (Whatman) was added to five 3-mm disks from each card, followed by vortexing for 15 sec and incubation on ice for 10 min with intermittent vortexing. The rest of the extraction procedure was performed according to the manufacturer's instructions.
PCR amplification and sequencing of targeted genes
The four genetic regions selected for analysis, with expected PCR product sizes and discrimination indices for targeted DNA sequencing, are presented in . Primer sequences, thermocycler programmes and PCR amplification methods used were as previously described (Ferguson et al., Citation2005; Raviv et al., Citation2007). Amplified DNA was purified using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA), and submitted to the Georgia Genomics Facility (University of Georgia) for sequencing. Purified amplicons were sequenced twice in both directions with forward and reverse amplification primers, using the ABI 3730xl DNA Analyzer capillary sequencer (Applied Biosystems). Raw sequence data were analysed with the EditSeq™ program (in Lasergene®; DNASTAR, Inc., Madison, WI, USA), and the complete overlapping of complementary sequences, editing and consensus construction were produced with the SeqMan™ program (in Lasergene®; DNASTAR, Inc.).
Table 1. Genetic regions selected for analysis, with expected PCR product sizes and discrimination (D) indices for targeted genetic sequencing.
Sequence analysis
DNA sequences of genome targets for the live M. gallisepticum vaccine strains (F-strain, ts-11 and 6/85) and for M. gallisepticum isolates from different countries were obtained by GenBank Internet search (http://www.ncbi.nlm.nih.gov/genbank) and from the database of the Mycoplasma laboratory at the PDRC. The PDRC database contains sequences of the IGSR and a 236 to 302 base pair (bp) fragment of the mgc2 gene (Garcia et al., Citation2005), with a total of 1380 sequences from 25 countries worldwide. South African wild-type mgc2 sequences were edited to start and end at equivalent sequence positions to the mgc2 gene fragment to facilitate comparison with all IGSR and mgc2 (fragment) sequences in the database. M. gallisepticum vaccine and reference strains and 25 M. gallisepticum isolates from 12 countries, for which both IGSR and mgc2 (fragment) sequences were available, were selected for more detailed comparison with the South African wild-types (). To conduct targeted sequence analysis, all consensus sequences were edited to start and end at equivalent sequence coding positions using the EditSeq™ program (in Lasergene®; DNASTAR, Inc.). Alignments of the sequences of individual and multiple genetic regions were constructed by the Clustal-W method using the MegAlign™ program (Lasergene®; DNASTAR, Inc.). Dendrograms were constructed from Clustal-W alignments by the neighbour-joining method with 1000-bootstrap replicate analysis using Molecular Evolutionary Genetics Analysis (MEGA) version 5.05 software (http://www.megasoftware.net). Amino acid translations and protein sequence alignments were performed using the EditSeq™ and MegAlign™ programs (Lasergene®; DNASTAR, Inc.), respectively.
Table 2. M. gallisepticum reference and vaccine strains and DNA samples from other countries used to generate the IGSR/mgc2 dendrogram for comparison with South African wild-type genotypes.
Designation of sequence types and wild-types
Sequences were named with a letter to designate the genetic region analysed (IGSR = “R”, mgc2=“C”, MGA_0319 = “P” and gapA=“G”), followed by a number, depending on their sequence similarity to the live M. gallisepticum vaccine strains. Sequences identical to those of the live vaccine strains were designated as follows: ts-11, “0”; 6/85, “1”; F strain, “2”. Sequences not identical to the vaccine sequences were assigned numbers greater than 2. If the sequence of one or more genetic regions was not identical to that of the live vaccine sequences, the sample was considered to be wild-type M. gallisepticum DNA, and was assigned a wild-type number (e.g. SA-WT#1).
Discrimination index of targeted genetic sequence analysis
The discriminatory power of a typing method depends on the number and relative frequency of the genotypes distinguished (Hunter & Gaston, Citation1988). Discriminatory power can be calculated using Simpson's index of diversity or the discrimination index (D), which is a measure of the probability that two unrelated strains will be placed into different typing groups (Hunter & Gaston, Citation1988). A D index > 0.90 is considered adequate, and a D index > 0.95 is considered good typing discrimination (Hunter & Gaston, Citation1988; Ferguson et al., Citation2005). Discrimination indices were calculated for targeted sequence analysis of individual and multiple genetic regions.
GenBank accession numbers
Accession numbers for IGSR and mgc2 (fragment) sequences of M. gallisepticum reference and vaccine strains and isolates from other countries selected for comparison with the South African wild-types are shown in . Accession numbers for the IGSR, mgc2, MGA_0319 and gapA sequences of South African M. gallisepticum genotypes and the ts-11 genotype are shown in . mgc2, MGA_0319 and gapA sequences of the vaccine strains 6/85 and F were previously submitted to GenBank under the following accession numbers: AY556231, AY556075 and AY556153 (mgc2, MGA_0319 and gapA, respectively, of strain 6/85) and AY556230, AY556074 and AY556152 (mgc2, MGA_0319 and gapA, respectively, of F strain) (Ferguson et al., Citation2005).
Table 3. South African M. gallisepticum genotypes identified, with corresponding sequence types and genotype prevalence.
Results
Genotyping of South African M. gallisepticum samples
Sequence data of adequate quality were obtained from 36 M. gallisepticum DNA samples. Thirty-five of the 36 M. gallisepticum positive samples were identified as wild-types, because the sequences of one or more genetic targets were not identical to those of the live vaccine strains ( and Supplementary Figures 1 to 5). One sample, which originated from a commercial layer flock vaccinated with ts-11, was identical to ts-11 vaccine strain in all four genetic regions, and was therefore designated “ts-11-like”. Twelve distinct wild-types (SA-WT#1 to #12) were identified based on combined sequence analysis ( and Supplementary Figures 1 to 5). Complete sequences of the IGSR or mgc2 targets could not be obtained for SA-WT#5, SA-WT#11 and SA-WT#12; nonetheless, these samples could be identified as unique wild-types based on sequencing of the other three genetic targets.
Eight different sequence types could be distinguished by mgc2 sequencing, four types by IGSR or MGA_0319 sequencing, and only two types by gapA sequencing ( and Supplementary Figures 1 to 5). The gapA sequences of ts-11 (type 0) and 6/85 (type 1) were identical, and were thus designated G0/1. SA-WT#7 was the most prevalent wild-type (42.9% of samples); the prevalence of the other wild-types ranged from 2.9 to 8.6%. SA-WT#1 and SA-WT#9 were identical to ts-11 in the mgc2 or IGSR regions respectively, while SA-WT#4 was identical to ts-11 in the IGSR, MGA_0319 and gapA regions. The mgc2 sequence of SA-WT#4 was 99.8% similar to that of ts-11, differing only by a single-nucleotide polymorphism (SNP) at position 345. This change represents a non-synonymous polymorphism, since the resulting codon encodes lysine (AAA) in SA-WT#4 and asparagine (AAC) in ts-11. Repeated amplification and sequence analysis of the mgc2 targets of both samples identified as SA-WT#4 yielded inconclusive results, with sequences either 99.8% or 100.0% similar to ts-11 (A or C at position 345, respectively). SA-WT#4 was isolated from broilers and layers in KwaZulu-Natal, which were not vaccinated with live M. gallisepticum vaccines. Respiratory disease was reported in both cases, although it is unclear whether other causes of respiratory infection were excluded. None of the South African sequences matched the F strain (type 2) sequence in any genetic region analysed.
Discrimination indices of genetic targets
To evaluate and compare the capability of targeted genetic sequencing to discriminate between M. gallisepticum isolates, D index values were estimated for individual genetic targets and for multiple targets (). The D index values of 0.745 (IGSR), 0.894 (mgc2), 0.615 (MGA_0319) and 0.295 (gapA) were estimated for sequencing of individual targets. Sequencing of more than one genetic target increased discriminatory power, with discrimination indices of 1.000 (IGSR/mgc2), 0.955 (mgc2/MGA_0319) and 0.927 (IGSR/MGA_0319) achieved by sequencing of multiple targets.
Table 4. Comparison of targeted genetic sequencing methods for discrimination of South African M. gallisepticum genotypes.
Origin of South African M. gallisepticum wild-types
The majority (40.0%) of samples submitted originated from KwaZulu-Natal, while 42.9% of samples originated from the six other provinces sampled (17.1% from the Eastern Cape, 11.4% from the Western Cape, 5.7% from the Free State and 2.9% each from the North-West Province, Gauteng and Mpumalanga), and 17.1% of samples were of unknown provincial origin. Seven of the 12 wild-types identified were detected in samples from KwaZulu-Natal, four from the Eastern Cape, three from the Western Cape, and one each from North-West Province, Gauteng, Mpumalanga and the Free State. SA-WT#7 was the most widespread M. gallisepticum wild-type, and was detected in five of the seven provinces sampled. The other wild-types were detected in one or at most two provinces. SA-WT#7 was detected in samples from broilers, broiler breeders and layers, while SA-WT#1, SA-WT#4, SA-WT#6 and SA-WT#8 were detected in two bird types, and SA-WT#2, SA-WT#3, SA-WT#5, SA-WT#9, SA-WT#10 and SA-WT#11 were detected in only one bird type.
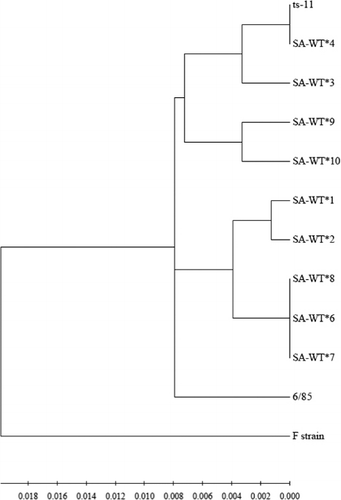
IGSR/mgc2/MGA_0319/gapA dendrogram
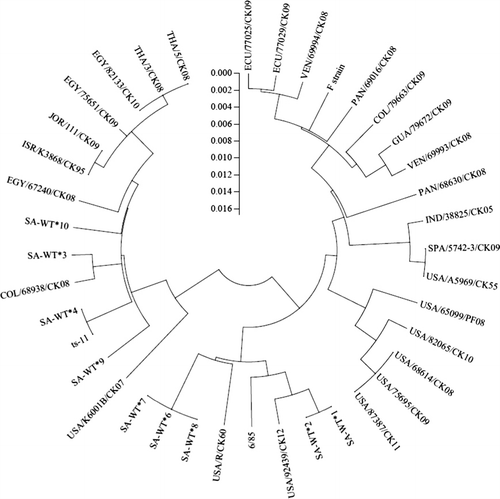
Dendrograms were constructed of multiple genetic sequence alignments to allow sample comparison based on two or more genetic regions. The dendrogram of the IGSR/mgc2/MGA_0319/gapA alignment was used to compare South African wild-types with each other and the vaccine strains (). In general, South African wild-type sequences were highly similar to each other, with sequence similarity ranging from 97.6 to 100%. (Note that although sequence similarity for SA-WT#6, SA-WT#7 and SA-WT#8 and SA-WT#4 and ts-11 was estimated to be 100% by the alignment software, these sequences were not identical.) The mgc2 sequence of SA-WT#6 differed from that of SA-WT#7 by a SNP, while SA-WT#8 differed from SA-WT#7 by a single nucleotide deletion in the IGSR region. A SNP of the mgc2 gene differentiated SA-WT#4 and ts-11, as previously discussed. The South African wild-type sequences were more similar to ts-11 (sequence distance range: 97.8 to 100%) and 6/85 (sequence distance range: 97.9 to 98.6%), but grouped distinctly from the F strain (sequence distance range: 96.0 to 96.5%).
Individual IGSR and mgc2 (fragment) alignments
To facilitate comparison of South African wild-types with all M. gallisepticum isolates in the PDRC database (including those isolates for which the sequence of only one genetic target was available), individual IGSR and mgc2 (fragment) alignments were also constructed. The South African wild-type IGSR sequences were unique, differing from all other IGSR sequences in our database. SA-WT#6, SA-WT#7, SA-WT#8, SA-WT#9, SA-WT#10 and SA-WT#11 had three characteristic deletions in the mgc2 gene fragment (49 to 51 bp, 100 to 123 bp and 163 to 165 bp), resulting in these sequences being 30 bp shorter than the other South African wild-type sequences. These deletions were absent in the vaccine strains and in all other mgc2 (fragment) sequences examined, except for one 2008 isolate from a peafowl in the USA (USA/65099/PF08), whose mgc2 (fragment) sequence was identical to those of SA-WT#7, SA-WT#8, SA-WT#9 and SA-WT#10. The mgc2 (fragment) sequence of SA-WT#3 was identical to those of two US isolates (USA/74235/CK09 and USA/94473/CK12) and one Australian isolate (AUS/019/CK97; GenBank accession number AY556302). The SA-WT#5 mgc2 (fragment) sequence was identical to that of the reference strain USA/A5969/CK55, and to several wild-types from the USA, Colombia, Spain, Malaysia and Thailand.
IGSR/mgc2 (fragment) dendrogram
The construction of an IGSR/mgc2 (fragment) dendrogram allowed South African wild-types to be compared with 25 wild-type sequences from 12 countries based on both the IGSR and mgc2 (fragment) sequences (; origins of foreign M. gallisepticum DNA samples are listed in ; sequence alignments in Supplementary Figures 1 and 2). The South African wild-type IGSR/mgc2 (fragment) sequences were unique and different from all other wild-types analysed. They were more similar to sequences from Egypt, Jordan, Israel, Thailand, and certain sequences from the United States and Colombia, with sequence distances ranging from 96.4 to 99.8%. In particular, a high degree of similarity was found between SA-WT#1 and SA-WT#2 and USA/92439/CK12 (sequence distances: 99.8%), and between SA-WT#3 and COL/68938/CK08 (sequence distance: 99.4%). In contrast, South African wild-types were most different from wild-type sequences from Ecuador, Venezuela, Panama, Guatemala, India, Spain, and other US and Colombian sequences, with sequence distances ranging from 96.0 to 98.5%. In general, IGSR/mgc2 (fragment) sequences formed distinct geographical clusters, represented by sequences from Asia and the Middle East, South Africa, the USA and Central and South America.
Discussion
Of the four targets analysed, mgc2 sequencing had the best discriminatory power for differentiation of South African genotypes, followed by IGSR and MGA_0319, with gapA having poor discriminatory power. The D index calculated for mgc2 in this study was 0.894, which is similar to the D index of 0.915 previously described for targeted genetic sequencing of isolates from the USA, Israel and Australia (Ferguson et al., Citation2005). However, D indices for MGA_0319, gapA and IGSR were considerably lower in this study than those previously reported (Ferguson et al., Citation2005; Raviv et al., Citation2007). In previous studies, D indices were estimated based on typing of larger numbers of M. gallisepticum isolates and laboratory strains from the USA (Raviv et al., Citation2007), as well as from other countries (Ferguson et al., Citation2005). Strain diversity in a restricted geographical area such as South Africa would be expected to be significantly lower than diversity across broader geographical regions, including multiple countries. Our estimates of discriminatory power are therefore conservative, and indicate the ability of sequencing of these targets to distinguish South African genotypes only.
Raviv et al. estimated the discriminatory power of IGSR sequencing to be higher than that of previous estimates for mgc2, and reported that strains with ts-11-like mgc2 sequences were correctly identified as wild-types by IGSR sequencing (Ferguson et al., Citation2005; Raviv et al., Citation2007). In this study, mgc2 sequencing gave better discriminatory power (D = 0.894) than IGSR sequencing (D = 0.745). This superior discriminatory power may be as a result of increased variability of the South African isolates in the mgc2 region. Some of this variability was attributed to three deletions of the mgc2 gene, resulting in the loss of a total of 30 nucleotides in certain South African wild-types (SA-WT#6, SA-WT#7, SA-WT#8, SA-WT#9, SA-WT#10 and SA-WT#11). These deletions were only observed in one other isolate in our sequence database (USA/65099/PF08). Size variation of the mgc2 gene was first described by Ferguson et al. (Citation2005), who concluded that this characteristic was not a reliable strain differentiation tool due to the relatively short insertions/deletions observed and the limited number of strains that could be differentiated. This study supports this conclusion, as only two genotypes could be differentiated based on mgc2 size polymorphism, and the difference in length was not sufficient to enable differentiation on ethidium bromide-stained agarose gels.
As previously reported (Ferguson et al., Citation2005), we found that sequencing more genetic targets increased the overall discrimination index, with maximum discriminatory power achieved by sequencing both IGSR and mgc2. In addition, sequencing of both of these targets was necessary to differentiate three wild-types (SA-WT#1, SA-WT#4 and SA-WT#9) from ts-11. In one case, MGA_0319 sequencing allowed the differentiation of an additional wild-type (SA-WT#12) for which the mgc2 sequence could not be obtained, probably as a result of sub-optimal DNA quality. Sequencing of MGA_0319 (a shorter region than mgc2 and IGSR) may thus be a useful adjunct to strain differentiation in cases where complete IGSR or mgc2 sequences cannot be obtained.
SA-WT#6 and SA-WT#7, and SA-WT#4 and ts-11 were only distinguishable by SNPs of the mgc2 gene, while SA-WT#7 and SA-WT#8 differed only by a single nucleotide deletion of the IGSR. The use of single nucleotide changes to differentiate strains is supported by the correlation of these differences with RAPD results, and by the demonstrated sequence stability of the genetic targets (Ferguson et al., Citation2005; Raviv et al., Citation2007; Gharaibeh et al., Citation2011). It is possible, however, that certain single nucleotide changes of particular targets do not result in strain differences. We recently sequenced the IGSR and mgc2 (fragment) genetic regions of seven US M. gallisepticum samples from ts-11 vaccinated broiler breeders and layers, and found that these sequences could only be distinguished from those of ts-11 by a single nucleotide change in the mgc2 (fragment) sequence at position 18 (corresponding to position 345 in the mgc2 sequence) (data not shown). Since the SNP in the mgc2 sequence that differentiated SA-WT#4 from ts-11 occurred at the same position, we suspect that SA-WT#4 may actually be derived from ts-11. This is an interesting observation, considering that SA-WT#4 was isolated from broilers and layers that were not vaccinated with ts-11. Alternatively, one could speculate that SA-WT#4 may be a field strain that closely resembles ts-11 in all of the particular genomic regions sequenced. Since targeted sequencing cannot differentiate mixed infections of M. gallisepticum strains, it is also theoretically possible that these non-vaccinated flocks were co-infected with ts-11 and a field strain. Complete genome sequencing of SA-WT#4 may be useful in determining the relationship of this genotype to ts-11.
Three of the 12 South African wild-types were identical to ts-11 in two or three of the four genetic regions analysed, and wild-types clustered consistently with ts-11 or 6/85 in the analysis of the combined data. In contrast, South African wild-types demonstrated low sequence similarity to the F strain. As both ts-11 and 6/85 vaccines have been used in South Africa for more than a decade while F strain vaccines are still not available, it may be theorized that vaccine usage has affected the wild-type sequences through horizontal gene transfer or homologous recombination with existing field strains. To further investigate this possibility, it would be necessary to evaluate M. gallisepticum DNA pre-dating the introduction of ts-11 and 6/85. Unfortunately, we did not have access to historical M. gallisepticum isolates for this study. Our results indicate that South African wild-types are readily differentiated from F strain by targeted sequencing; a fact that would facilitate differentiation of vaccinated and infected flocks should F strain vaccines be introduced into the country in the future. In contrast, the sequence similarity of South African M. gallisepticum wild-types to the commercially available live vaccines, particularly ts-11, highlights the importance of sequencing both the IGSR and mgc2 targets to allow differentiation of wild-type M. gallisepticum from the vaccine strains. Other genetic targets (e.g. pvpA or vlhA) could also be investigated for their ability to facilitate this strain differentiation (Pillai et al., Citation2003; Ferguson et al., Citation2005; Ghorashi et al., Citation2010). pvpA analysis was performed in a pilot study of South African M. gallisepticum wild-types (data not shown), but due to difficulty in amplification and alignment this target was not included in the present study.
The South African wild-type combined IGSR/mgc2 (fragment) sequences were unique, differing from those of wild-types from other countries in our database. Clustering of isolates according to geographical origin in the IGSR/mgc2 (fragment) dendrogram is consistent with the correlation of epidemiologically linked isolates previously reported for sequencing of multiple genetic targets (Ferguson et al., Citation2005). Individual analysis of IGSR and mgc2 (fragment) targets revealed that while South African wild-type IGSR sequences were unique, three different wild-type mgc2 (fragment) sequences were identical to certain sequences from other countries. One match of particular interest was that of the mgc2 (fragment) sequences of SA-WT#7, SA-WT#8, SA-WT#9 and SA-WT#10 and a peacock isolate from the USA (USA/65099/PF08). USA/65099/PF08 was the only isolate outside South Africa to have the characteristic 30 bp deletion of the mgc2 gene fragment observed in SA-WT#7, SA-WT#8, SA-WT#9 and SA-WT#10. This phenomenon could theoretically result from a horizontal gene transfer or homologous recombination event, although there is no known epidemiological link between USA/65099/PF08 and these South African wild-types. It is also possible that the mgc2 deletions in USA/65099/PF08 and SA-WT#7, SA-WT#8, SA-WT#9 and SA-WT#10 represent random mutational events, which occurred independently in unrelated genotypes.
Wild-type prevalence could not be determined in this study because an opportunity sampling approach was used, in which sample numbers, location and bird type sampled were not dictated, but were dependent on voluntary sample submission. However, our results do indicate that one wild-type (SA-WT#7) was the most widespread M. gallisepticum wild-type in South Africa between 2009 and 2012, since it was present in 42.9% of samples analysed, and was detected in samples from broilers, broiler breeders and layers in five of the seven provinces sampled. The majority of wild-types (7 of 12) were detected in samples from KwaZulu-Natal, with fewer wild-types from other provinces. It should be noted that 40.0% of all samples tested originated from KwaZulu-Natal, and it is likely that more wild-types would have been detected in other provinces had more samples from these provinces been tested.
In conclusion, targeted sequencing of South African M. gallisepticum DNA extracts from samples collected between 2009 and 2012 has been used to develop an M. gallisepticum sequence database. This database was applied to characterize 12 novel M. gallisepticum genotypes and select genetic targets to optimize differentiation of South African wild-types from each other and from the live vaccine strains. We are currently using the sequence data to develop cost-effective and broadly applicable molecular techniques, which will facilitate the differentiation of South African M. gallisepticum wild-types from live M. gallisepticum vaccine strains. In addition, this study has improved our understanding of the diversity and distribution of M. gallisepticum in South Africa, and has provided a foundation for future epidemiological investigation. Analysis of M. gallisepticum samples from South Africa in the event of the introduction or removal of live vaccines will allow further study of the effects of vaccination on the genetic variability in field strains. Internationally, this model could be used to guide the development of other M. gallisepticum sequence databases, which would have application in elucidating the epidemiology of M. gallisepticum and in refining diagnostic techniques for different countries.
cavp_a_819486_sm3005.pdf
Download PDF (321.8 KB)Acknowledgements
The authors are grateful to Ms Tracy Schmidt of Allerton Provincial Veterinary Laboratory in South Africa for performing the DNA extraction. They would also like to acknowledge all those who kindly submitted samples for this project, and thank Dr Stanley H. Kleven for reviewing the manuscript.
References
- Adler, H.E., McMartin, D. & Shifrine, M. (1960). Immunization against Mycoplasma infections of poultry. The American Journal of Veterinary Research, 21, 482–485.
- Delaney, N.F., Balenger, S., Bonneaud, C., Marx, C.J., Hill, G.E., Ferguson-Noel, N., Tsai, P., Rodrigo, A. & Edwards, S.V. (2012). Ultrafast evolution and loss of CRISPRs following a host shift in a novel wildlife pathogen, Mycoplasma gallisepticum. PLoS Genetics, 8, e1002511. 10.1371/journal.pgen.1002511
- Evans, R.D. & Hafez, Y.S. (1992). Evaluation of a Mycoplasma gallisepticum strain exhibiting reduced virulence for prevention and control of poultry mycoplasmosis. Avian Diseases, 36, 197–201. 10.2307/1591490
- Fan, H.H., Kleven, S.H. & Jackwood, M.W. (1995). Application of polymerase chain reaction with arbitrary primers to strain identification of Mycoplasma gallisepticum. Avian Diseases, 39, 729–735. 10.2307/1592409
- Ferguson, N.M., Hepp, D., Sun, S., Ikuta, N., Levisohn, S., Kleven, S.H. & Garcia, M. (2005). Use of molecular diversity of Mycoplasma gallisepticum by gene-targeted sequencing (GTS) and random amplified polymorphic DNA (RAPD) analysis for epidemiological studies. Microbiology, 151, 1883–1893. 10.1099/mic.0.27642-0
- Garcia, M., Elfaki, M.G. & Kleven, S.H. (1994). Analysis of the variability in expression of Mycoplasma gallisepticum surface antigens. Veterinary Microbiology, 42, 147–158. 10.1016/0378-1135(94)90014-0
- Garcia, M., Ikuta, N., Levisohn, S. & Kleven, S.H. (2005). Evaluation and comparison of various PCR methods for detection of Mycoplasma gallisepticum infection in chickens. Avian Diseases, 49, 125–132.
- Geary, S.J., Forsyth, M.H., Aboul Saoud, S., Wang, G., Berg, D.E. & Berg, C. M. (1994). Mycoplasma gallisepticum strain differentiation by arbitrary primer PCR (RAPD) fingerprinting. Molecular and Cellular Probes, 8, 311–316. 10.1006/mcpr.1994.1042
- Gharaibeh, S., Laibinis, V., Wooten, R., Stabler, L. & Ferguson-Noel, N. (2011). Molecular characterization of Mycoplasma gallisepticum isolates from Jordan. Avian Diseases, 55, 212–216. 10.1637/9526-091510-Reg.1
- Ghorashi, S.A., Noormohammadi, A.H. & Markham, P.F. (2010). Differentiation of Mycoplasma gallisepticum strains using PCR and high-resolution melting curve analysis. Microbiology, 156, 1019–1029. 10.1099/mic.0.031351-0
- Hong, Y., Garcia, M., Levisohn, S., Savelkoul, P., Leiting, V., Lysnyansky, I., Ley, D.H. & Kleven, S.H. (2005). Differentiation of Mycoplasma gallisepticum strains using amplified fragment length polymorphism and other DNA-based typing methods. Avian Diseases, 49, 43–49. 10.1637/7254-080504R
- Hunter, P.R. & Gaston, M.A. (1988). Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. Journal of Clinical Microbiology, 26, 2465–2466.
- Kleven, S.H. (1998). Mycoplasmas in the etiology of multifactorial respiratory disease. Poultry Science, 77, 1146–1149.
- Kleven, S.H., Morrow, C.J. & Whithear, K.G. (1988). Comparison of Mycoplasma gallisepticum strains by hemagglutination-inhibition and restriction endonuclease analysis. Avian Diseases, 32, 731–741. 10.2307/1590992
- Levisohn, S., Dykstra, M.J., Lin, M.Y. & Kleven, S.H. (1986). Comparison of in vivo and in vitro methods for pathogenicity evaluation for Mycoplasma gallisepticum in respiratory infection. Avian Pathology, 15, 233–246. 10.1080/03079458608436284
- Ley, D.H. (2008). Mycoplasma gallisepticum infection. In Y.M. Saif, A.M. Fadly, J.R. Glisson, L.R. McDougald, L.K. Nolan, & D.E. Swayne (Eds.). Diseases of Poultry 12th edn (pp. 807–834). Ames, IA.: Blackwell Publishing.
- Lin, M.Y. & Kleven, S.H. (1984). Evaluation of attenuated strains of Mycoplasma gallisepticum as vaccines in young chickens. Avian Diseases, 28, 88–99. 10.2307/1590131
- Luginbuhl, R.E., Tourtellotte, M.E. & Frazier, M.N. (1967). Mycoplasma gallisepticum – control by immunization. Annals of the New York Academy of Sciences, 143, 234–238. 10.1111/j.1749-6632.1967.tb27662.x
- Pillai, S.R., Mays, H.L., Jr., Ley, D.H., Luttrell, P., Panangala, V.S., Farmer, K.L. & Roberts, S.R. (2003). Molecular variability of house finch Mycoplasma gallisepticum isolates as revealed by sequencing and restriction fragment length polymorphism analysis of the pvpA gene. Avian Diseases, 47, 640–648. 10.1637/6095
- Raviv, Z., Callison, S., Ferguson-Noel, N., Laibinis, V., Wooten, R. & Kleven, S.H. (2007). The Mycoplasma gallisepticum 16S-23S rRNA intergenic spacer region sequence as a novel tool for epizootiological studies. Avian Diseases, 51, 555–560. 10.1637/0005-2086(2007)51[555:TMGSRI]2.0.CO;2
- Rodriguez, R. & Kleven, S.H. (1980). Pathogenicity of two strains of Mycoplasma gallisepticum in broilers. Avian Diseases, 24, 800–807. 10.2307/1589957
- Rosengarten, R. & Yogev, D. (1996). Variant colony surface antigenic phenotypes within mycoplasma strain populations: implications for species identification and strain standardization. Journal of Clinical Microbiology, 34, 149–158.
- Tyler, K.D., Wang, G., Tyler, S.D. & Johnson, W.M. (1997). Factors affecting reliability and reproducibility of amplification-based DNA fingerprinting of representative bacterial pathogens. Journal of Clinical Microbiology, 35, 339–346.
- Whithear, K.G., Soeripto, Harrigan, K.E. & Ghiocas, E. (1990a). Immunogenicity of a temperature sensitive mutant Mycoplasma gallisepticum vaccine. Australian Veterinary Journal, 67, 168–174. 10.1111/j.1751-0813.1990.tb07748.x
- Whithear, K.G., Soeripto, Harringan, K.E. & Ghiocas, E. (1990b). Safety of temperature sensitive mutant Mycoplasma gallisepticum vaccine. Australian Veterinary Journal, 67, 159–165. 10.1111/j.1751-0813.1990.tb07745.x
- Yoder, H.W., Jr. (1986). A historical account of the diagnosis and characterization of strains of Mycoplasma gallisepticum of low virulence. Avian Diseases, 30, 510–518. 10.2307/1590415