
ABSTRACT
To better understand the prevalence of Gallibacterium anatis in different poultry species, a rapid and accurate method was developed to detect G. anatis using a TaqMan fluorescent quantitative polymerase chain reaction (qPCR). Specific primers and a TaqMan probe were designed based on the reference gtxA gene sequence. The qPCR standard curve showed a good linear relationship, and the method showed good reproducibility, sensitivity, and specificity, indicating its suitability for G. anatis identification and quantitative analysis. A comparison of the detection results in 160 clinical swab samples showed that the detection rate (54.4%) of the qPCR for G. anatis was better than that of two conventional methods: gyrB gene-based qPCR for G. anatis (51.9%) and culture-based identification (34.4%). G. anatis was detected in layer chicken (77.3%), Silkie chicken (72.7%), and duck (27.1%) with relatively high detection rates, whereas dove (8.8%) and quail (3.0%) showed lower detection rates, indicating the different prevalence of G. anatis in different fowl species.
Research highlights
G. anatis infection induces serious economic losses to the poultry industry.
Identification of bacteria by culture-based methods is a cumbersome process.
A TaqMan fluorescent qPCR method was developed for detecting G. anatis.
Introduction
Gallibacterium is a genus in the Pasteurellaceae family of Gram-negative bacteria, which was classified by Christensen et al. (Citation2003) to include avian [Pasteurella haemolytica]–“Actinobacillus salpingitidis”–Pasteurella anatis complex. Gallibacterium anatis is the type species of the genus Gallibacterium (Bisgaard et al., Citation2009). Recent studies (Jordan et al., Citation2005; Vazquez et al., Citation2006; Paudel et al., Citation2013) have shown that G. anatis is closely linked to salpingitis, peritonitis, and other infections, and may result in decreased egg quantity and quality, leading to an increased mortality rate and thus inducing serious economic losses to the poultry industry. Members of Gallibacterium show a broad spectrum of infection in avian species, including chicken, duck, goose, turkey, pheasant, egret, partridge, and pigeon (Mushin et al., Citation1980; Bisgaard, Citation1993; Bojesen, Christensen et al., Citation2007; Yang et al., Citation2014). Gallibacterium infection is often found in combination with infection by other pathogens such as Escherichia coli or infectious laryngotracheitis virus (Shaw et al., Citation1990; Neubauer et al., Citation2009); in the case of simultaneous injection with other pathogens, the test chickens showed more severe clinical signs (Zheng, Citation2010; Huangfu, Zhao, Yang, Li et al., Citation2012). G. anatis is primarily transmitted horizontally, and the respiratory tract has been confirmed as a primary transmission route (Zepeda et al., Citation2010). In addition, in 2014, the recovery of G. anatis from yolk at 10 days post-infection indicated the potential of vertical transmission (Paudel et al., Citation2014).
Identifying bacteria through traditional culture-based methods can be a cumbersome process. Therefore, the development of a rapid, accurate detection method for diagnosing G. anatis infection would be of great significance to the poultry industry. Several methods have been developed for detecting the pathogen to date (Bojesen, Vazquez et al., Citation2007; Huangfu, Zhao, Yang, Chen et al., Citation2012; Wang et al., Citation2016), including Gallibacterium-specific polymerase chain reaction (GS-PCR) and SYBR Green dye-based fluorescent quantitative PCR (qPCR), but no TaqMan fluorescent qPCR detection method based on the gtxA gene has yet been reported. Kristensen et al. (Citation2010) first discovered the gtxA gene encoding the GtxA toxin, which has leukotoxic and haemolytic activities. In the present study, PCR primers and a TaqMan probe were designed based on the G. anatis gtxA gene sequence to develop a TaqMan fluorescent qPCR method for detecting G. anatis. Our aim was to develop a method with high specificity, high sensitivity, and a rapid reaction rate that would be suitable for the portable, rapid identification and detection of G. anatis in different birds, and to better understand the prevalence of G. anatis in poultry.
Materials and methods
Bacterial strains and clinical samples
A total of 47 bacterial strains () were used in this study, including 32 strains of G. anatis, 20 of which were identified by the PCR amplification of the 16S rRNA gene and sequence comparison of the database (Weisburg et al., Citation1991; Drancourt et al., Citation2000), and the other 12 strains, including strain F 149, were provided as a gift from the Poultry Diseases Institute of Henan Agricultural University. The remaining 15 strains were stocks maintained in the laboratory, representing other poultry-associated bacteria from the families Pasteurellaceae, Enterobacteriaceae, and Staphylococcaceae for comparison (). To evaluate the specificity of the method, 80 chickens in one farm of Shanxi Province, which was found to be positive for Gallibacterium in a preliminary survey, were investigated by the newly developed method as well as with two conventional methods: gyrB gene-based qPCR and culture-based identification. Swab samples were collected from the upper cleft palate and the cloaca of each of the 80 chickens, for a total of 160 samples. In addition, to determine the prevalence of G. anatis in different bird species, 458 birds were randomly sampled from six different species in China for G. anatis detection.
Table 1. Basic information of bacterial strains used in this study.
Primers and probe
For qPCR of the gtxA gene, fluorescent qPCR primers (Primer-F, Primer-R) and a probe (Probe_IN) were designed based on the gtxA gene sequence of G. anatis 12656-12 (FJ917356) publicly available in the GenBank database, and were synthesized by Sangon Biotech (Shanghai, China). For GS-PCR, two pairs of primers were synthesized as reported by Bojesen, Vazquez et al. (Citation2007): Rpob-L and Rpob-R are located in the rpoB gene, and 1133fgal and 114r are located in the 16S rRNA and 23S rRNA genes, respectively. For gyrB gene-based qPCR, the primer and probe were synthesized as reported by Wang et al. (Citation2016). The sequences of all primers and the probes are shown in .
Table 2. Primer and probe sequences.
Sample DNA preparation
To extract bacterial DNA, the bacteria were inoculated in Luria–Bertani medium (composition: 10 g tryptone, 5 g yeast extract, 10 g NaCl, 1000 ml H2O, pH adjusted to 7.2 with 5 M NaOH) containing 10% calf serum and incubated at 37°C for 18 h. One millilitre of the cultured bacterial broth was placed in a 1.5-ml centrifuge tube and centrifuged at 7378×g for 5 min. The supernatant was discarded, and 600 µl of Tris-ethylenediaminetetraacetic acid (TE) buffer (10 mM Tris-HCl, 1 mM EDTA, pH: 8) was added and thoroughly mixed. The bacterial mixture was placed in boiling water for 10 min, immediately transferred to an ice bath for 2 min, and centrifuged at 10,625×g for 10 min. The supernatant was collected and stored at −20°C until use.
To extract DNA from the clinical samples, cotton swabs were placed in a 1.5-ml Eppendorf tube and immersed in 0.8 ml of TE buffer for 10 min. After vortexing for 30 s, the cotton swab was discarded (squeezing out as much liquid as possible) and the mixture was centrifuged at 10,625×g for 5 min. The supernatant was discarded, the pellet was re-suspended in 100 µl of TE buffer, and the mixture was centrifuged at 10,625×g for 5 min. The supernatant was discarded, and the same volume of TE buffer was employed to re-suspend the pellet. The mixture was placed in boiling water for 10 min, immediately transferred to an ice bath for 2 min, and centrifuged at 10,625×g for 10 min. The supernatant was collected and stored at −20°C until use.
Standard positive recombinant plasmid preparation
A fragment of the gtxA gene was synthesized by Sangon Biotech (Shanghai, China) with the following sequence: 5′-ATCCAACTCCATTATTGATATGGGACATTCTATCGCAAACCTAATTCAATCGGATGTTAATACAAGTAGCGGGAAAGCGGCTATTGCAGAAGGATCAATTAAATTAATTGGCAATATTAACGGATTAGTCAGCGATGTGTTAAGCCTTTCTAATGCCTCTACCGCCGTT-3′.
The gtxA gene fragment was cloned into the pUC57 plasmid via the SmaI cloning site, creating a recombinant plasmid. The plasmid was serially diluted 10-fold to reach 2.53 × 100 copies/µl. Five serial dilutions from 2.53 × 106 to 2.53 × 102 copies/µl were used as standard templates for optimizing the fluorescent qPCR conditions and to construct a standard curve.
TaqMan fluorescent qPCR standard curve construction
The five 10-fold dilutions of recombinant plasmid DNA (2.53 × 106–2.53 × 102 copies/µl) were applied as templates for fluorescent qPCR amplification, using Ace-Q qPCR Probe Master Mix (Vazyme Biotech, Nanjing, China). The total volume of the reaction mixture was 25 µl, including 1 µl of 10 µM Primer-F, 1 µl of 10 µM Primer-R, 0.5 µl of 10 µM Probe_IN, 1 µl of ROX reference dye, 1 µl of template DNA, and 20.5 µl of sterile Milli-Q ultrapure water (Millipore, Billerica, MA, USA). PCR was run on the ABI7500 FAST fluorescent qPCR system (Applied Biosystems, Foster City, CA, USA) with the following reaction programme: 95°C for 5 min; 95°C for 10 s and 60°C for 34 s for 40 cycles. The results were used for analysis and standard curve construction.
Specificity, sensitivity, and repeatability testing
The specificity of the qPCR method for G. anatis detection was tested with genomic DNA collected from each of the 47 bacterial strains listed in , using the recombinant plasmid as the positive control and ultrapure water as the negative control. Sensitivity was tested using nine serial dilutions of recombinant plasmid DNA from 2.53 × 108 to 2.53 × 100 copies/µl, and the results were compared with those of conventional PCR detection using the same primers. Intra-group repeatability of the method was assessed by performing qPCR in triplicate with the plasmid template at concentrations of 2.53 × 108, 2.53 × 106, and 2.53 × 104 copies/µl. To evaluate the inter-assay variability, these plasmids were detected by the present qPCR method, and then tested again four and eight days later.
Clinical sample detection
A total of 160 cotton swabs of the 80 chickens collected from a single farm were tested by the present qPCR, gyrB gene-based qPCR, and culture-based identification. Swabs were inoculated on blood agar plates under sterile conditions, and incubated at 37°C for 24 h. Subsequently, individual haemolytic colonies were utilized to inoculate Luria–Bertani liquid medium containing 5% calf serum for purification. GS-PCR was employed for isolate identification. Genomic DNA was collected from the cotton swab samples of 458 poultry birds, including the 80 chickens mentioned above, for the present qPCR detection.
Statistical analysis
The detection data of the swabs were statistically analysed using the chi-squared test; differences were considered statistically significant when P < 0.05.
Results
Construction of the qPCR standard curve
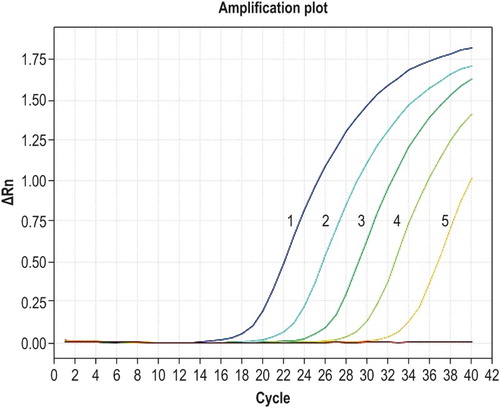
The five concentrations of recombinant plasmid DNA from 2.53 × 106 to 2.53 × 102 copies/µl were used as templates for fluorescent qPCR amplification. Dynamic amplification curves are shown in . The slope of the standard curve was −3.333, the intercept was 41.727, and the correlation coefficient (R2) was 0.998; the amplification efficiency was 99.5%.
Figure 1. Amplification curves for qPCR of known standards. Curves 1–5 correspond to the concentrations of 2.53 × 106, 2.53 × 105, 2.53 × 104, 2.53 × 103, and 2.53 × 102 copies/µl of recombinant plasmid DNA, respectively.
Specificity, sensitivity, and repeatability of qPCR detection
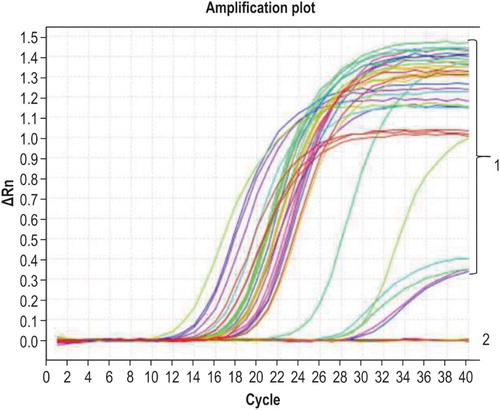
The qPCR amplification results showed that only G. anatis was successfully amplified, and none of the remaining 15 bacterial strains exhibited a specific amplification signal ().
Figure 2. Specificity of qPCR. Curve 1 corresponds to G. anatis and the positive control sample; curve 2 corresponds to other bacteria and the negative control.
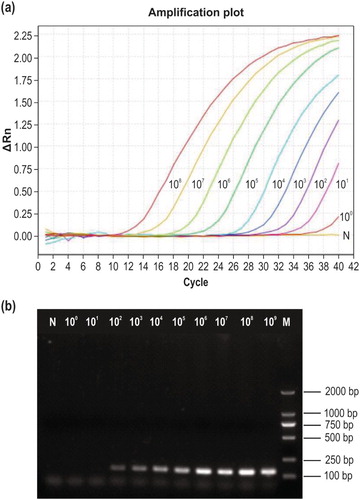
The nine serial dilutions of recombinant plasmid DNA (2.53 × 108–2.53 × 100 copies/µl) were used as templates for qPCR detection, and the amplification curves are shown in (a). The lowest template concentration at which a fluorescent signal appeared with a cycle threshold (Ct) value less than 35 was 2.53 × 101 copies/µl (Ct = 33.6). The lower limit of detection by using conventional PCR amplification was 2.53 × 102 copies/µl ((b)), demonstrating that the new qPCR method is 10 times more sensitive.
Figure 3. Sensitivity testing. (a) Sensitivity of the newly developed qPCR method. Curves 108–100 correspond to 2.53 × 108–2.53 × 100 copies/µl of recombinant plasmid DNA, respectively; N, negative control. (b) Sensitivity of conventional PCR detection using the same primers. M, DL2000 DNA marker; 109–100, 2.53 × 109–2.53 × 100 copies/µl of recombinant plasmid DNA; N, negative control.
Three sample concentrations (2.53 × 108, 2.53 × 106, and 2.53 × 104 copies/µl) in triplicate were selected as templates for quantitative amplification. There was no clear difference in the Ct values of the S-shaped amplification curves, and the coefficients of variation for both inter- and intra-group repeatability were less than 2%, demonstrating that the method had good reproducibility. Intra- and inter-group repeatability results are shown in .
Table 3. Intra- and inter-group qPCR repeatability results.
Clinical sample detection
The detection rates using the present qPCR, gyrB gene-based qPCR, and culture-based identification of the 160 swabs were 54.4%, 51.9%, and 34.4%, respectively. There was no significant difference in the detection rates of the two fluorescent qPCR methods (P > 0.05), whereas the detection rate of the culture-based identification was significantly lower than those of the two qPCR methods (P < 0.01). The detection rates of the newly developed fluorescent qPCR method in the seven bird species were, from highest to lowest, 77.3% (layer chicken), 72.7% (Silkie chicken), 27.1% (duck), 8.8% (dove), 3.0% (quail), and 0 (goose) ().
Table 4. Detection of G. anatis in several poultry species.
Discussion
In this study, we developed a TaqMan fluorescent qPCR method for detecting G. anatis, showing a correlation coefficient (R2) of 0.998 and an amplification efficiency of 99.5%, which meet the requirements of fluorescent qPCR. Moreover, the method showed good specificity, with the 32 known G. anatis strains successfully detected among 47 samples from different genera of bacteria, and the method was 10 times more sensitive than the traditional PCR (detection limit of 2.53 × 102 copies/ml). In addition, the coefficients of variation for both inter- and intra-group repeatability were less than 2%, demonstrating the good reproducibility of this method.
The detection rates in 160 swab samples were highest using the newly developed qPCR, followed by gyrB gene-based qPCR, with culture-based identification showing a significantly lower detection rate.
G. anatis was detected in five out of six fowl species examined, indicating that this bacterium is present in many different fowl species. The different detection rates also provide a clue as to the different prevalence of G. anatis in different fowl species: layer chickens, Silkie chickens, and ducks all had high prevalence, whereas doves and quails had comparatively lower prevalence. However, as G. anatis prevalence is associated with the production system and biosecurity in chicken flocks (Bojesen et al., Citation2003), this deduction requires further experimental validation. Although no goose samples were found to be positive for G. anatis in this study, this does not rule out the possibility of geese carrying this pathogen, and could have also been due to the small sample size.
Compared with the qPCR method based on SYBR Green dye we developed in 2012 (Huangfu, Zhao, Yang, Chen et al., Citation2012), the present TaqMan-based method is more sensitive and suitable for use with the ABI7500 FAST fluorescent qPCR system. This can help avoid the use of the glass capillary on the Roche fluorescence qPCR system so as to further reduce the detection costs.
Recently, a real-time qPCR method for the culture-independent identification and quantification of G. anatis was reported (Wang et al., Citation2016), which allows for the species-specific identification of G. anatis. However, we speculate that the diagnostic method we established herein has a wider detection range since this qPCR is established based on detection of the gtxA gene, which can theoretically be applicable for amplification of any Gallibacterium species with the gtxA gene, such as G. anatis as well as Gallibacterium genomospecies 1 and 2 (Kristensen et al., Citation2011). However, only G. anatis was identified with the new qPCR method in this study, and thus we could not determine whether the method is also suitable for amplification of Gallibacterium genomospecies 1 and 2, given their unavailability for testing. Thus, further studies are needed to verify this hypothesis.
Overall, these results showed that the constructed standard curve meets the needs of the TaqMan fluorescent qPCR method and is suitable for the portable gtxA gene-based identification of G. anatis. G. anatis prevalence was observed to different degrees in layer chickens, Silkie chickens, ducks, quails, and doves. Given the close relationship between G. anatis and salpingitis, oviductal cysts, oophoritis, peritonitis, and respiratory disease, the qPCR method developed in this study should prove to be a valuable rapid diagnostic tool for G. anatis infection and help to prevent and control the infection.
Acknowledgements
We acknowledge the help of senior experimentalist Chuanbi Zhao, retired staff member of Henan University of Animal Husbandry and Economy, for collecting the clinical samples.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bisgaard, M. (1993). Ecology and significance of Pasteurellaceae in animals. Zentralblatt für Bakteriologie, 279, 7–26. doi: 10.1016/S0934-8840(11)80487-1
- Bisgaard, M., Korczak, B.M., Busse, H.J., Kuhnert, P., Bojesen, A.M. & Christensen, H. (2009). Classification of the taxon 2 and taxon 3 complex of Bisgaard within Gallibacterium and description of Gallibacterium melopsittaci sp. nov., Gallibacterium trehalosifermentans sp. nov. and Gallibacterium salpingitidis sp. nov. International Journal of Systematic and Evolutionary Microbiology, 59, 735–744. doi: 10.1099/ijs.0.005694-0
- Bojesen, A.M., Christensen, H., Nielsen, S.S. & Bisgaard, M. (2007). Host-specific bacterial lineages in the taxon 2 and 3 complex of Pasteurellaceae. Systematic and Applied Microbiology, 30, 119–127. doi: 10.1016/j.syapm.2006.04.002
- Bojesen, A.M., Nielsen, S.S. & Bisgaard, M. (2003). Prevalence and transmission of haemolytic Gallibacterium species in chicken production systems with different biosecurity levels. Avian Pathology, 32, 503–510. doi: 10.1080/0307945031000154107
- Bojesen, A. M., Vazquez, M.E., Robles, F., Gonzalez, C., Soriano, E.V., Olsen, J.E. & Christensen, H. (2007). Specific identification of Gallibacterium by a PCR using primers targeting the 16S rRNA and 23S rRNA genes. Veterinary Microbiology, 123, 262–268. doi: 10.1016/j.vetmic.2007.02.013
- Christensen, H., Bisgaard, M., Bojesen, A.M., Mutters, R. & Olsen, J.E. (2003). Genetic relationships among avian isolates classified as Pasteurella haemolytica, “Actinobacillus salpingitidis” or Pasteurella anatis with proposal of Gallibacterium anatis gen. nov., comb. nov. and description of additional genomospecies within Gallibacterium gen. nov. International Journal of Systematic and Evolutionary Microbiology, 53, 275–287. doi: 10.1099/ijs.0.02330-0
- Drancourt, M., Bollet, C., Carlioz, A., Martelin, R., Gayral, J.P. & Raoult, D. (2000). 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. Journal of Clinical Microbiology, 38, 3623–3630.
- Huangfu, H., Zhao, J., Yang, X., Chen, L., Chang, H., Wang, X., Yao, H. & Wang, C. (2012). Development and preliminary application of a quantitative PCR assay for detecting gtxA-containing Gallibacterium species in chickens. Avian Diseases, 56, 315–320. doi: 10.1637/9907-082511-Reg.1
- Huangfu, H., Zhao, J., Yang, X., Li, Q., Wang, C., Chen, L., Chang, H., Wang, X., Liu, H. & Yao, H. (2012). Tissue distribution of Gallibacterium anatis in chickens co-infected with infectious bronchitis virus. Acta Veterinaria et Zootechnica Sinica, 43, 1623–1629.
- Jordan, F.T., Williams, N.J., Wattret, A. & Jones, T. (2005). Observations on salpingitis, peritonitis and salpingoperitonitis in a layer breeder flock. Veterinary Record, 157, 573–577. doi: 10.1136/vr.157.19.573
- Kristensen, B.M., Frees, D. & Bojesen, A.M. (2010). Gtxa from Gallibacterium anatis, a cytolytic RTX-toxin with a novel domain organisation. Veterinary Research, 41, 25. doi: 10.1051/vetres/2009073
- Kristensen, B.M., Frees, D. & Bojesen, A.M. (2011). Expression and secretion of the RTX-toxin GtxA among members of the genus Gallibacterium. Veterinary Microbiology, 153, 116–123. doi: 10.1016/j.vetmic.2011.05.019
- Mushin, R., Weisman, Y. & Singer, N. (1980). Pasteurella haemolytica found in respiratory tract of fowl. Avian Diseases, 24, 162–168. doi: 10.2307/1589775
- Neubauer, C., De Souza-Pilz1, M., Bojesen, A.M., Bisgaard, M. & Hess, M. (2009). Tissue distribution of haemolytic Gallibacterium anatis isolates in laying birds with reproductive disorders. Avian Pathology, 38, 1–7. doi: 10.1080/03079450802577848
- Paudel, S., Alispahic, M., Liebhart, D., Hess, M. & Hess, C. (2013). Assessing pathogenicity of Gallibacterium anatis in a natural infection model: the respiratory and reproductive tracts of chickens are targets for bacterial colonization. Avian Pathology, 42, 527–535. doi: 10.1080/03079457.2013.843160
- Paudel, S., Liebhart, D., Hess, M. & Hess, C. (2014). Pathogenesis of Gallibacterium anatis in a natural infection model fulfils Koch’s postulates: 1. Folliculitis and drop in egg production are the predominant effects in specific pathogen free layers. Avian Pathology, 43, 443–449. doi: 10.1080/03079457.2014.955782
- Shaw, D.P., Cook, D.B., Maheswaran, S.K., Lindeman C.J. & Halvorson, D.A. (1990). Pasteurella haemolytica as a co-pathogen in pullets and laying hens. Avian Diseases, 34, 1005–1008. doi: 10.2307/1591397
- Vazquez, M.E., Gonzalez, C.H., de la Mora, R. & Bojesen, A.M. (2006). Prevalence of Gallibacterium anatis in Mexico and their effect in laying hens. Proceedings of the 4th International Veterinary and Diagnostics Conference. Oslo, Norway.
- Wang, C., Robles, F., Ramirez, S., Riber, A.B. & Bojesen, A.M. (2016). Culture-independent identification and quantification of Gallibacterium anatis (G. anatis) by real-time quantitative PCR. Avian Pathology, 45, 538–544. doi: 10.1080/03079457.2016.1184743
- Weisburg, W.G., Barns, S.M., Pelletier, D.A. & Lane, D.J. (1991). 16S ribosomal DNA amplification for phylogenetic study. Journal of Bacteriology, 173, 697–703. doi: 10.1128/jb.173.2.697-703.1991
- Yang, X., Chen, Y., Yu, S., Zou, N., Wu, R., Xia, J., Niu, T., Wang, F. & Huang, Y. (2014). Isolation and identification of one pigeon-origin Gallibacterium genomo sp. 3. Journal of Zhejiang University (Agricultural & Life Sciences), 40, 249–256.
- Zepeda, V.A., Calderón-Apodaca, N.L., Paasch, M.L., Martin, P.G., Paredes, D.A., Ramirez-Apolinar, S. & Soriano-Vargas, E. (2010). Histopathologic findings in chickens experimentally infected with Gallibacterium anatis by nasal instillation. Avian Diseases, 54, 1306–1309. doi: 10.1637/9423-061410-ResNote.1
- Zheng, L. (2010). The first isolation and identification of Gallibacterium anatis from fowls in China and preliminary research on its biological characteristics (pp. 34–37). Zhengzhou: Henan Agricultural University.