
ABSTRACT
Like other avian circovirus species, Pigeon circovirus (PiCV) is known to be genetically diverse with a relatively small circular single-stranded DNA genome of 2 kb that encodes for a capsid protein (Cap) and a replication initiator protein (Rep). Recent paleoviral evidence hints towards a probable Gondwanan origin of avian circoviruses, paralleling the evolution and dispersal of their hosts. Limited availability of PiCV genome sequence data in Australia has hindered phylogeographic studies in this species, so we screened clinically normal rock doves (Columba livia) in regional New South Wales, and demonstrated a high prevalence (76%) of PiCV infection by PCR. We also recovered 12 complete novel PiCV genomes and phylogenetic analyses revealed that PiCV circulating in Australian feral pigeons formed two strongly supported monophyletic clades. One clade resided with PiCV genomes from Poland, Australia, United Kingdom, Belgium, China, and Japan, and another basal clade was more closely related to PiCV genomes from Poland. A novel more distantly-related PiCV rep gene formed a solitary clade with weak posterior support. So we further analysed all selected partial rep gene sequences to demonstrate a likely naturally occurring spillover infection from a passerine circovirus candidate. The findings suggest that there is a high degree of genetic variation within PiCV in Columbiformes with potential greater admixture between avian circoviruses within Australia than previously known.
RESEARCH HIGHLIGHTS
Confirmed high prevalence rate of PiCV circulating in Australian wild pigeons.
Highlighted extensive recombination events within Australian PiCV.
Demonstrated a likely naturally occurring spillover infection from a passerine circovirus candidate.
Introduction
Pigeon circovirus (PiCV) belongs to the genus Circovirus (family Circoviridae, order unassigned) and has been identified as the likely aetiological agent of a multifactorial disease known as young pigeon disease syndrome or young bird sickness (Woods et al., Citation1993). Over the past two decades, PiCV infections have been documented in many parts of the world mainly in domesticated and feral rock doves, Columba livia (Woods et al., Citation1993; Woods et al., Citation1994; Abadie et al., Citation2001; Raue et al., Citation2005; Duchatel et al., Citation2006; Csagola et al., Citation2012; Stenzel et al., Citation2014; Stenzel & Koncicki, Citation2017). The virus infects pigeons of all ages; however, young birds are suggested to be more susceptible to PiCV infection (Tavernier et al., Citation2000). Affected birds with young pigeon disease syndrome exhibit clinical signs such as depression, lethargy, weight loss, anorexia, poor racing performance, regurgitation, polydipsia, diarrhoea and ruffled feathers; however, asymptomatic PiCV infection is also recognized (Abadie et al., Citation2001; Raue et al., Citation2005; Duchatel et al., Citation2006; Stenzel et al., Citation2014). Sometimes, the disease may be mild with birds showing non-specific signs such as poor racing performance, weight loss and / or respiratory difficulties (Raue et al., Citation2005). Due to the damage it causes to the bursa of Fabricius, PiCV infection likely causes some degree of immunosuppression, which makes infected individuals susceptible to secondary microbial infections (Abadie et al., Citation2001; Raue et al., Citation2005; Stenzel & Pestka, Citation2014; Stenzel & Koncicki, Citation2017)
The PiCV genome is relatively small, with two main genes that are bidirectionally transcribed from a double-stranded replicative intermediate (Todd et al., Citation2008). The virion sense gene rep encodes a replication-associated protein (Rep) and the complementary sense gene cap encodes a capsid protein (Cap) (Mankertz et al., Citation2000; Todd et al., Citation2001). Similar to other circoviruses, PiCV is highly genetically diverse and prone to genetic recombination (Stenzel et al., Citation2014; Wang et al., Citation2017). Recent paleoviral evidence hints towards a probable Gondwanan origin of avian circoviruses which parallels the evolution of their hosts (Das et al., Citation2016). Australasia is the biogeographical origin of a rich diversity of avian host species including the Passeriformes, Psittaciformes and Columbiformes all of which have their own avian circovirus species. In this study, we screened clinically normal birds in New South Wales to gain an insight into the epidemiology of PiCV infection in Australian feral pigeons as well as to better understand the genetic diversity of the virus in a biogeographic region with the highest degree of columbid diversity.
Materials and methods
Sampling and isolation of DNA
Samples used in the analyses described below were sourced from 17 clinically normal feral rock doves (C. livia) in New South Wales. Animal sampling was carried out in accordance with approved guidelines set by the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (1997) and approved by the Charles Sturt University Animal Ethics Committee (Research Authority permit 09/046). Total genomic DNA was isolated from the swab samples using a ReliaPrep gDNA Tissue Miniprep System (Promega, Madison, WI, USA). Virion enrichment was performed by centrifugation for 2 min at 800 × g to remove host cellular debris, and the supernatants were subsequently filtered through 5 μm centrifuge filters (Merck, Darmstadt, Germany). The filtrates were treated with nuclease to remove unprotected nucleic acids using 20 μl of RNase A (Promega), and incubated in microcentrifuges tubes at 56°C for 10 min. Viral nucleic acids were subsequently extracted using ReliaPrep binding columns (Promega) according to the manufacturer's guidelines.
PCR amplification, and sequencing of PiCV genome
Initially, extracted DNA was screened by an established nested PCR protocol using a set of degenerative primers for avian circovirus (Sarker et al., Citation2015; Sarker et al., Citation2016). Amplified PCR products, together with a standard molecular marker (Sigma, Saint Louis, MO, USA), were separated by electrophoresis in 0.8% agarose gel stained with GelRed (Biotium, Fremont, CA, USA) using 0.5×TBE buffer at 90 V for 1.5 h, and the appropriate bands were excised and purified using the Wizard® SV Gel and PCR Clean-Up System (Promega) according to the manufacturer’s instructions. Purified amplicons were sequenced with PCR primers by the Australian Genome Research Facility Ltd (Sydney, Australia) using an AB 3730xl unit (Applied Biosystems, AGRF, Sydney, Australia). For each amplicon, sequences were obtained at least twice in each direction for each isolate to ensure the validity of the data. The sequences were trimmed for primers, aligned to construct contigs using a minimum overlap of 35 bp and a minimum match percentage of 95%, and construction of partial sequences was carried out in Geneious Pro (version 10.2.2). Using partial sequence data generated in this study together with other selected circovirus genomes (Columbid circovirus AF252610, DQ915957, DQ915959, DQ915962, KF738867; PiCV LC035390; Canary circovirus (CaCV) AJ301633), a number of other primer sets () were designed to obtain full genome amplification and sequencing of circovirus from 13 positive feral pigeons. Reactions for different primer combinations were optimized, and the optimized reaction mixture contained 3 µl extracted genomic DNA, 2.5 µl of 10×High Fidelity PCR Buffer (Invitrogen, Carlsbad, CA, USA), 1 µl of 25 µM of each primer, 1 µl of 50 mM MgSO4, 4 µl of 1.25 mM dNTP’s, 1 U Platinum® Taq DNA Polymerase High Fidelity (Invitrogen, Carlsbad, CA, USA) and DEPC water added to a final volume of 25 µl. The optimized reaction was run as follows: 95°C for 3 min, followed by 40 cycles of 95°C for 30 s, 57°C for 45 s and 72°C for 2 min, and finally 72°C for 5 min. With the exception of primers sets C and D, the annealing temperature was 55°C for 45 s. In each set of reactions, PiCV positive genomic DNA and distilled H2O were included as positive and negative controls, respectively. The resulting PCR products were separated on a 0.8% agarose gel, and the appropriate bands were excised and purified using the Wizard® SV Gel and PCR Clean-Up System (Promega) according to the manufacturer’s instructions. The sequences were trimmed for primers, aligned to construct contigs using a minimum overlap of 35 bp and a minimum match percentage of 95%, and constructions of full genome sequence were carried out in Geneious Pro 10.2.2 (Biomatters, Auckland, New Zealand).
Table 1. Details of primers used in this study to amplify PiCV and CaCV genomes.
We also attempted to amplify a circovirus genome sequence sourced from a feral pigeon (#7), which was positive for CaCV using CaCV specific primers (, primer sets E and F) and the above-mentioned PCR conditions. Additionally, the isolated genomic DNA from pigeon #7 was further assessed by high-throughput next generation sequencing technology. A DNA library was prepared according to the published protocol adapted from the Illumina Nextera XT DNA Library Prep V3 Kit (Sarker et al., Citation2017; Sarker et al., Citation2018), and the library was sequenced on the Illumina MiSeq platform.
Phylogenetic analyses
All known PiCV sequence data were retrieved from the literature and NIH GenBank, and selected sequences (n = 119), representing all genotypic clades and geographical regions, were aligned in Geneious (version 10.2) using the MAFTT (version 7.309, algorithm; E-INS-I, scoring matrix; 100PAM / k = 2) (Katoh & Standley, Citation2013). To select the best construction method, a model testing was performed in CLC Genomics Workbench (version 9.5.4) using default parameters, and it favoured a general-time-reversible (GTR) substitution model for the maximum likelihood phylogeny. Bayesian phylogenetic tree was inferred using the program Beast v1.8.3 (Drummond, et al. Citation2012). In the MrBayes analysis, two independent Monte Carlo-Markov chains were implemented for 100 million generations each with trees sampled every 5000 generations. The Bayesian skyline coalescent demographic prior was chosen because it allows temporal changes in population size (Drummond et al., Citation2005). Each analysis was checked to ensure that a reasonable effective sample size (ESS > 200) had been reached for all parameters. Tracer version v1.6 was used to derive parameters and TreeAnnotator v1.8.1 was used to obtain the tree with the highest clade credibility and posterior probabilities for each node (Drummond et al., Citation2012), and FigTree v1.4.2 was used to generate the consensus tree (Andrew, Citation2009). Similarly, Bayesian phylogenetic tree was inferred using partial rep DNA sequences amplified in this study with other selected PiCV sequences.
Additionally, to identify the potential evolutionary history of a solitary clade generated by PiCV partial rep DNA sequence, a selection (n = 57) of other circoviruses were aligned in Geneious (version 10.2.3) using the MAFTT (version 7.309, algorithm; E-INS-I, scoring matrix; 100PAM / k = 2) (Katoh & Standley, Citation2013). Bayesian phylogenetic tree was inferred using similar software and parameter mentioned above. Furthermore, analyses of non-tree like evolutionary relationships amongst the selected different species of circoviruses were visualized using Neighbour-Net algorithm using default parameters implemented in SplitsTree4 (Huson & Bryant, Citation2006).
Recombination analyses
Evidence of recombination amongst the PiCV genomes sequenced in this study were screened using the RDP (Martin & Rybicki, Citation2000), GENECONV (Padidam et al., Citation1999), Bootscan (Martin et al., Citation2005), MaxChi (Smith, Citation1992), Chimaera (Posada & Crandall, Citation2001), Siscan (Gibbs et al., Citation2000) and 3Seq (Boni et al., Citation2007) methods contained in the RDP4 program (Martin et al., Citation2010). Recombination events that were detected by at least three of the aforementioned methods with significant P-values (<0.05) were considered plausible recombinant events. Sequences in the analysed dataset that most closely resembled the parental sequences of recombinants were defined as either “minor parents” or “major parents” based on the size of the genome fragments that these sequences had contributed to the detected recombinants (with the major parent contributing the larger fragment and the minor parent the smaller).
Results
Prevalence of PiCV in Australian feral pigeons
Using established PCR and primers targeting ∼350 bp of the rep gene, 13 of 17 (76.5%) Australian feral pigeon (C. livia) samples were positive for PiCV infection.
Analyses of PiCV genome sequences for patterns of relatedness
We recovered 12 complete PiCV genomes from 12 different pigeons. We obtained a partial genome sequence from one bird (pigeon #7) using a combination of Sanger and high-throughput next generation sequencing on samples from this bird. The 12 Australian PiCV genomes showed >85% genome-wide pairwise similarity to one another, and >81% similarity to the 107 other complete PiCV genomes available in GenBank which have been sampled from various other parts of the world, including Poland, Australia, United Kingdom, Belgium, China, Japan, North America and South Africa.
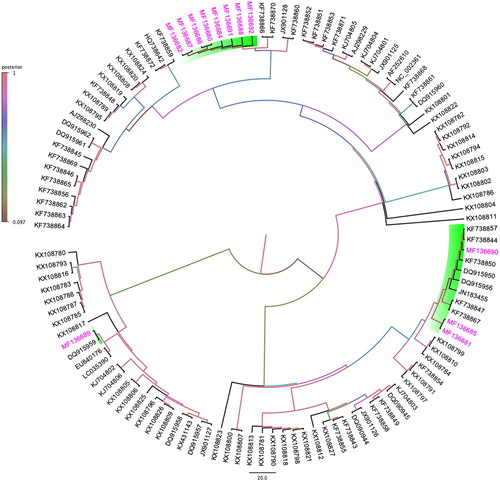
A Bayesian phylogenetic tree constructed using PiCV genomes sequenced (GenBank accession numbers: MF136680-82, and MF136684-92) in this study, along with 107 other selected PiCV genome sequences obtained from GenBank, demonstrated that the Australian PiCV circulating in wild feral pigeons formed three strongly supported monophyletic clades () (see Supplementary Figure S1 for original bootstraps support). Firstly, one clade comprised eight PiCV genomes isolated from Australian feral pigeon (MF136680, MF136682, MF136684, MF136686-88, MF136691-92), which demonstrated the strongest clade support (>98%) with spatially distant PiCV isolated from Poland (KF738860, KF738866 and KF738870) and Belgium (JX901128). Secondly, a strong geographic clustering of Australian PiCV genomes with other PiCV isolated globally indicated susceptibility to different genotypes (). Even though the PiCV genomes (MF136681, MF136685 and MF136690) from an Australian feral pigeon shared 91.1–92.5% sequence identity (Supplementary Table S1), Bayesian phylogenetic tree indicated that the Australian PiCV originated in this individual from a different ancestor (). For example, PiCV genomes (MF136681 and MF136685) from an Australian feral pigeon showed >97% clade support with other PiCV genomes isolated from China (KX108784, KX108799, KX108810) and Poland (KF738847, KF738867), whereas another Australian PiCV genome (MF136690) produced a separate and well-supported monophyletic clade with other PiCV genomes from Poland (KF738844, KF738857, KF738850). Thirdly, a PiCV genome obtained from an Australian feral pigeon (MF136689) had strong clade (>96%) support with the only other Australian PiCV genome (DQ915959) obtained from a feral Senegal dove (Streptopelia senegalensis) in Western Australia ().
Figure 1. Bayesian phylogenetic inference of evolutionary relationships amongst 12 PiCV full genome sequences isolated in this study from feral pigeon (C. livia) in Australia with 107 other selected PiCV genome sequences available in GenBank. A maximum clade credibility tree automatically rooted using an uncorrelated relaxed lognormal clock model in Beast v1.8.3, and FigTreev1.4.2 was used to generate the consensus tree. Labels at branch tips refer to GenBank accession number. Clade posterior probability values are highlighted by colours. Background shading clade and pink taxa highlight the PiCV genome sequences isolated from feral pigeons geographically located in Australia (colour online only).
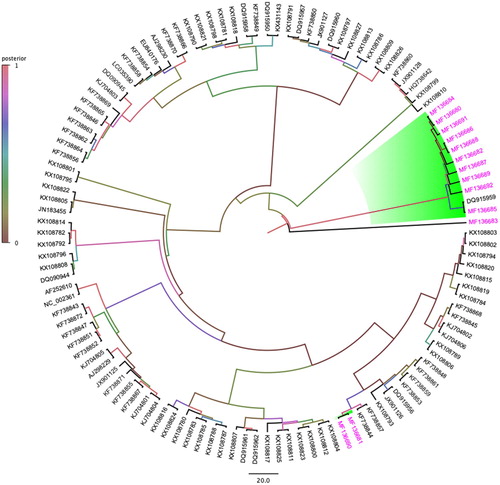
Tracking potential evolutionary relationships among partial PiCV rep gene
A Bayesian phylogenetic tree depicting the possible evolutionary relationship amongst 120 partial rep sequences, including 13 partial PiCV rep gene isolated in this study indicated that PiCV isolated from Australian feral pigeons fall into three different clades (). Interestingly, one of the novel PiCV rep genes isolated (MF136683) in this study made a solitary clade that has very strong posterior support (100%). The other two clades identified based on Australian PiCV partial rep gene sequences matched closely with full PiCV genome analysis, and highlighted a close evolutionary relationship amongst Australian and Polish PiCV sequences () (see Supplementary Figure S2 for original bootstraps support).
Figure 2. Bayesian phylogenetic inference of evolutionary relationships amongst 13 partial rep genes of PiCV sequences isolated in this study with 107 other selected partial rep gene sequences. A maximum clade credibility tree was automatically rooted in Beast v1.8.3 (GTR + I+G4 substitution model, an uncorrelated relaxed lognormal clock model, and coalescent Bayesian skyline prior). FigTreev1.4.2 was used to generate the consensus tree. Labels at branch tips refer to GenBank accession number. Clade posterior probability values are shown at tree nodes. Background shading clade and pink taxa highlight the PiCV sequences isolated from feral pigeons geographically located in Australia (colour online only).
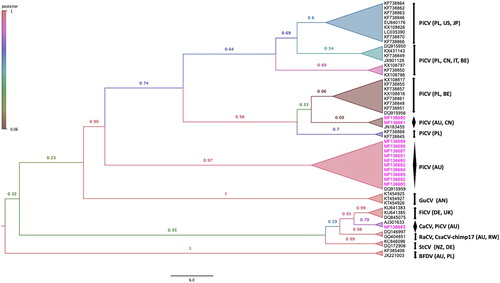
To investigate the relatedness of novel PiCV discovered in this study, we further analysed the partial rep gene sequenced in this study with partial rep gene sequences from other selected circoviruses, and demonstrated a likely spillover of a novel PiCV from a wild CaCV (), which was also supported by a non-tree like evolution as indicated by reticular circles (Supplementary Figure S3).
Figure 3. Bayesian phylogenetic inference of evolutionary relationships amongst 13 partial rep genes of PiCV sequences isolated in this study with 41 other selected partial rep gene sequences from other circoviruses. A maximum clade credibility tree was automatically rooted in Beast v1.8.3 (GTR + I+G4 substitution model, an uncorrelated relaxed lognormal clock model, and coalescent Bayesian skyline prior). FigTreev1.4.2 was used to generate the consensus tree. Labels at branch tips refer to GenBank accession number, and highlighted using different virus abbreviations (PiCV: Pigeon circovirus, GuCV: Gull circovirus, CaCV: Canary circovirus, FiCV: Finch circovirus, RaCV: Raven circovirus, CsaCV-chimp17: Chimpanzee stool avian-like circovirus-chimp17, StCV: Starling circovirus, BFDV: Beak and feather disease virus), followed by the country of origin code in parentheses (Australia: AU, Poland: PL, New Zealand: NZ, the Netherlands: AN, Germany: DE, Rwanda: RW, United Kingdom: UK, Belgium: BE, China: CN, Japan: JP, United States of America: US, Italy: IT). Clade posterior probability values are shown at tree nodes. Branch supports are collapsed and highlighted as different colours according to posterior probability. Pink taxa highlight the PiCV sequences isolated in this study from feral pigeons geographically located in Australia (colour online only).
Extensive recombination in Australian PiCV genomes
Viral recombination has played a significant role in the evolution of numerous ssDNA viruses and has been well presented for certain circoviruses (Cheung, Citation2009; Lefeuvre et al., Citation2009; Varsani et al., Citation2011; Sarker et al., Citation2014; Sarker et al., Citation2014; Stenzel et al., Citation2014). Accordingly, a large number of recombinations were detected in the PiCV genomes from Australian feral pigeons (). The strongest support for recombination was detected in the c-terminal portion of the capsid gene between PiCV isolated from Australian feral pigeons sourced from New South Wales and Polish pigeons originally isolated from fancy, racing, carrier pigeons (KF738844, KF738850, KF738857, respectively) (event 1). Importantly, a large segment (∼1350-bp) of an Australian PiCV (MF136690) was likely descended from a large number of ancestral PiCV genomes that were from geographically distant pigeon species (event 3).
Table 2. Details of recombination events (GenBank accession numbers) detected in Australian PiCV isolates.
Discussion
The results confirm a high prevalence of PiCV infection in clinically normal feral pigeons in regional Australia as well evidence of a high degree of genetic diversity that mirrors that for other avian circoviruses such as beak and feather disease virus (BFDV). The 12 Australian PiCV genomes we characterized showed >85% genome-wide pairwise similarity to one another, and >81% similarity to all the 107 other complete PiCV genomes available in GenBank. The findings suggest, that, like BFDV in Psittaciformes, there is a high degree of genetic variation within PiCV in Columbiformes with potential greater admixture between avian circoviruses within Australia than previously known. Phylogenetic analyses demonstrated that the novel PiCV genomes circulating in Australian feral pigeons formed two strongly supported monophyletic clades. One clade resided alongside PiCV genomes from various European countries, China, and Japan, and the other basal clade was more closely related to PiCV genomes from Poland. A novel more distantly-related PiCV rep gene formed a solitary clade with weak posterior support.
Previous research using BFDV (Das et al., Citation2016) has estimated avian circovirus speciation to have occurred in Australasia following the Triassic divergence of primordial Neoaves into passerine, columbid and larid host circoviruses deeply separated from those in waterfowl, consistent with a Triassic divergence of Galloanserae. The evolution of PiCV in pigeons and doves has probably taken a similar pathway to BFDV in parrots and cockatoos. Like the Psittaciformes, the order Columbiformes includes a widespread and diverse group of birds with 42 genera and 310 recognized species across the globe with the greatest diversity present in Australasia. The rock dove has the largest range of any columbid species with a natural distribution from Britain, throughout Europe, central Asia, India and China as well as a widespread urban range throughout the world as a beneficiary of human habitation. A greater range of PiCV genome sequence data is required from this region and the many native species of Columbiformes that inhabit Australia and New Guinea to better understand the phylogeographic niches of PiCV in pigeons.
Recent studies by Phan et al. (Citation2013) identified a wide range of known and unknown viral species belonging to the families Circoviridae, Parvoviridae, Picornaviridae, Reoviridae, Adenoviridae, Astroviridae, and Caliciviridae, as well as plant and insect viruses using droppings of wild urban pigeons (C. livia) from Hong Kong and Hungary. Therefore, it is not surprising that this study demonstrated a likely spillover of a novel passerine circovirus most closely related to CaCV ( and Supplementary Figure S3). As alluded to by Phan et al. (Citation2013), our study highlights that feral pigeons might act as the reservoirs of multiple viral pathogens and / or a likely example for cross-species transmission.
Viral recombination and reassortment are key evolutionary mechanisms driving pathogen diversity and host adaptation, as well as in the evolution of novel viral species (Awadalla, Citation2003; Martin et al., Citation2011). Similar to the study conducted by Stenzel et al. (Citation2014), this study has captured a large number of recombination events in Australian PiCV. Consistent with other circoviruses like BFDV (Sarker et al., Citation2014; Sarker et al., Citation2014), it seems likely that recombination is a key mechanism for the evolution of PiCV in Columbiformes but further research is required to analyse as yet undiscovered viruses in other pigeon and dove species.
This present work has demonstrated for the first time that PiCV from Australian feral pigeons had extensive recombination with other PiCV, and displayed a potential genetic admixture with other circoviruses. Such findings suggest that the high degree of genetic variation within the PiCV species as a whole is reflected in evolutionary dynamics within Australia, indicating the likely possible route of genetic admixture with other circoviruses. This work will underpin future studies and offers a better understanding of the role and significance of feral pigeons in the emergence of PiCV in racing and meat pigeons.
Supplemental Material
Download MS Word (11.1 MB)Acknowledgements
The authors are grateful to many of the VDL microbiologists for preserving original samples. S.S. and S.R.R. conceived the experiment; S.S. performed the experiments; S.S and S.D. analysed the sequencing data; S.S. and S.R.R. wrote the manuscript; all authors participated in reviewing and editing the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Subir Sarker http://orcid.org/0000-0002-2685-8377
Related Research Data
References
- Abadie, J., Nguyen, F., Groizeleau, C., Amenna, N., Fernandez, B., Guereaud, C., Guigand, L., Robart, P., Lefebvre, B. & Wyers, M. (2001). Pigeon circovirus infection: pathological observations and suggested pathogenesis. Avian Pathology, 30, 149–158. doi: 10.1080/03079450124811
- Andrew, R. (2009). FigTree v1.4 computer program and documentation distributed by the author.
- Awadalla, P. (2003). The evolutionary genomics of pathogen recombination. Nature Reviews Genetics, 4, 50–60. doi: 10.1038/nrg964
- Boni, M.F., Posada, D. & Feldman, M.W. (2007). An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics, 176, 1035–1047. doi: 10.1534/genetics.106.068874
- Cheung, A.K. (2009). Homologous recombination within the capsid gene of porcine circovirus type 2 subgroup viruses via natural co-infection. Archives of Virology, 154, 531–534. doi: 10.1007/s00705-009-0329-5
- Csagola, A., Lorincz, M., Tombacz, K., Wladar, Z., Kovacs, E. & Tuboly, T. (2012). Genetic diversity of pigeon circovirus in Hungary. Virus Genes, 44, 75–79. doi: 10.1007/s11262-011-0669-6
- Das, S., Sarker, S., Peters, A., Ghorashi, S.A., Phalen, D., Forwood, J.K. & Raidal, S.R. (2016). Evolution of circoviruses in lorikeets lags behind its hosts. Molecular Phylogenetics and Evolution, 100, 281–291. doi: 10.1016/j.ympev.2016.04.024
- Drummond, A.J., Rambaut, A., Shapiro, B. & Pybus, O.G. (2005). Bayesian coalescent inference of past population dynamics from molecular sequences. Molecular Biology and Evolution, 22, 1185–1192. doi: 10.1093/molbev/msi103
- Drummond, A.J., Suchard, M.A., Xie, D. & Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, 29, 1969–1973. doi: 10.1093/molbev/mss075
- Duchatel, J.P., Todd, D., Smyth, J.A., Bustin, J.C. & Vindevogel, H. (2006). Observations on detection, excretion and transmission of pigeon circovirus in adult, young and embryonic pigeons. Avian Pathology, 35, 30–34. doi: 10.1080/03079450500465692
- Gibbs, M.J., Armstrong, J.S. & Gibbs, A.J. (2000). Sister-Scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics (Oxford, England), 16, 573–582. doi: 10.1093/bioinformatics/16.7.573
- Huson, D.H. & Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution, 23, 254–267. doi: 10.1093/molbev/msj030
- Katoh, K. & Standley, D.M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: improvements in performance and usability. Molecular Biology and Evolution, 30, 772–780. doi: 10.1093/molbev/mst010
- Lefeuvre, P., Lett, J.-M., Varsani, A. & Martin, D.P. (2009). Widely conserved recombination patterns among single-stranded DNA viruses. Journal of Virology, 83, 2697–2707. doi: 10.1128/JVI.02152-08
- Mankertz, A., Hattermann, K., Ehlers, B. & Soike, D. (2000). Cloning and sequencing of columbid circovirus (coCV), a new circovirus from pigeons. Archives of Virology, 145, 2469–2479. doi: 10.1007/s007050070002
- Martin, D. & Rybicki, E. (2000). RDP: detection of recombination amongst aligned sequences. Bioinformatics (Oxford, England), 16, 562–563. doi: 10.1093/bioinformatics/16.6.562
- Martin, D.P., Biagini, P., Lefeuvre, P., Golden, M., Roumagnac, P. & Varsani, A. (2011). Recombination in eukaryotic single stranded DNA viruses. Viruses, 3, 1699–1738. doi: 10.3390/v3091699
- Martin, D.P., Lemey, P., Lott, M., Moulton, V., Posada, D. & Lefeuvre, P. (2010). RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics (Oxford, England), 26, 2462–2463. doi: 10.1093/bioinformatics/btq467
- Martin, D.P., Posada, D., Crandall, K.A. & Williamson, C. (2005). A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Research and Human Retroviruses, 21, 98–102. doi: 10.1089/aid.2005.21.98
- Padidam, M., Sawyer, S. & Fauquet, C.M. (1999). Possible emergence of new geminiviruses by frequent recombination. Virology, 265, 218–225. doi: 10.1006/viro.1999.0056
- Phan, T.G., Vo, N.P., Boros, Á, Pankovics, P., Reuter, G., Li, O.T.W., Wang, C., Deng, X., Poon, L.L.M. & Delwart, E. (2013). The viruses of wild pigeon droppings. PLoS One, 8, e72787–e72787. doi: 10.1371/journal.pone.0072787
- Posada, D. & Crandall, K.A. (2001). Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proceedings of the National Academy of Sciences of the United States of America, 98, 13757–13762. doi: 10.1073/pnas.241370698
- Raue, R., Schmidt, V., Freick, M., Reinhardt, B., Johne, R., Kamphausen, L., Kaleta, E.F., Müller, H. & Krautwald-Junghanns, M.-E. (2005). A disease complex associated with pigeon circovirus infection, young pigeon disease syndrome. Avian Pathology, 34, 418–425. doi: 10.1080/03079450500267825
- Sarker, S., Ghorashi, S.A., Forwood, J.K., Bent, J.S., Peters, A. & Raidal, S.R. (2014). Phylogeny of beak and feather disease virus in cockatoos demonstrates host generalism and multiple-variant infections within Psittaciformes. Virology, 460–464, 72–82. doi: 10.1016/j.virol.2014.04.021
- Sarker, S., Lloyd, C., Forwood, J. & Raidal, S.R. (2016). Forensic genetic evidence of beak and feather disease virus infection in a Powerful Owl, Ninox strenua. Emu, 116, 71–74. doi: 10.1071/MU15063
- Sarker, S., Moylan, K.G., Ghorashi, S.A., Forwood, J.K., Peters, A. & Raidal, S.R. (2015). Evidence of a deep viral host switch event with beak and feather disease virus infection in rainbow bee-eaters (Merops ornatus). Scientific Reports, 5, 14511. doi: 10.1038/srep14511
- Sarker, S., Patterson, E.I., Peters, A., Baker, B.G., Forwood, J.K., Ghorashi, S.A., Holdsworth, M., Baker, R., Murray, N. & Raidal, S.R. (2014). Mutability dynamics of an emergent single stranded DNA virus in a naïve host. PLoS One, 9, e85370.
- Sarker, S., Roberts, H.K., Tidd, N., Ault, S., Ladmore, G., Peters, A., Forwood, J.K., Helbig, K. & Raidal, S.R. (2017). Molecular and microscopic characterization of a novel Eastern grey kangaroopox virus genome directly from a clinical sample. Scientific Reports, 7, 16472.
- Sarker, S., Isberg, S.R., Milic, N.L., Lock, P. & Helbig, K.J. (2018). Molecular characterization of the first saltwater crocodilepox virus genome sequences from the world’s largest living member of the Crocodylia. Scientific Reports, 8, 5623.
- Smith, J. (1992). Analyzing the mosaic structure of genes. Journal of Molecular Evolution, 34, 126–129.
- Stenzel, T. & Koncicki, A. (2017). The epidemiology, molecular characterization and clinical pathology of circovirus infections in pigeons - current knowledge. Veterinary Quarterly, 37, 166–174. doi: 10.1080/01652176.2017.1325972
- Stenzel, T. & Pestka, D. (2014). Occurrence and genetic diversity of pigeon circovirus strains in Poland. Acta Veterinaria Hungarica, 62, 274–283. doi: 10.1556/AVet.2014.004
- Stenzel, T., Piasecki, T., Chrzastek, K., Julian, L., Muhire, B.M., Golden, M., Martin, D.P. & Varsani, A. (2014). Pigeon circoviruses display patterns of recombination, genomic secondary structure and selection similar to those of beak and feather disease viruses. Journal of General Virology, 95, 1338–1351. doi: 10.1099/vir.0.063917-0
- Tavernier, P., Herdt, P.D., Thoonen, H. & Ducatelle, R. (2000). Prevalence and pathogenic significance of circovirus-like infections in racing pigeons (Columba livia). Vlaams Diergeneeskundig Tijdschrift, 69, 338–341.
- Todd, D., Fringuelli, E., Scott, A.N.J., Borghmans, B.J., Duchatel, J.P., Shivaprasad, H.L., Raidal, S.R., Abadie, J.X., Franciosini, M.P. & Smyth, J.A. (2008). Sequence comparison of pigeon circoviruses. Research in Veterinary Science, 84, 311–319. doi: 10.1016/j.rvsc.2007.03.007
- Todd, D., Weston, J.H., Soike, D. & Smyth, J.A. (2001). Genome sequence determinations and analyses of novel circoviruses from goose and pigeon. Virology, 286, 354–362. doi: 10.1006/viro.2001.0985
- Varsani, A., Regnard, G.L., Bragg, R., Hitzeroth, I.I. & Rybicki, E.P. (2011). Global genetic diversity and geographical and host-species distribution of beak and feather disease virus isolates. Journal of General Virology, 92, 752–767. doi: 10.1099/vir.0.028126-0
- Wang, K.C., Zhuang, Q.Y., Qiu, Y., Wang, T. & Chen, J.M. (2017). Genome sequence characterization of pigeon circoviruses in China. Virus Research, 233, 1–7. doi: 10.1016/j.virusres.2017.03.007
- Woods, L.W., Latimer, K.S., Barr, B.C., Niagro, F.D., Campagnoli, R.P., Nordhausen, R.W. & Castro, A.E. (1993). Circovirus-like infection in a pigeon. Journal of Veterinary Diagnostic Investigation, 5, 609–612. doi: 10.1177/104063879300500417
- Woods, L.W., Latimer, K.S., Niagro, F.D., Riddell, C., Crowley, A.M., Anderson, M.L., Daft, B.M., Moore, J.D., Campagnoli, R.P. & Nordhausen, R.W. (1994). A retrospective study of circovirus infection in pigeons: nine cases (1986-1993). Journal of Veterinary Diagnostic Investigation, 6, 156–164. doi: 10.1177/104063879400600205