
ABSTRACT
Mycoplasma synoviae is an economically important avian pathogen worldwide, causing respiratory disease, infectious synovitis, airsacculitis and eggshell apex abnormalities in commercial chickens. Despite the widespread use of MS-H as a live attenuated vaccine over the past two decades, the precise molecular basis for loss of virulence in this vaccine is not yet fully understood. To address this, the whole genome sequence of the vaccine parent strain, 86079/7NS, was obtained and compared to that of the MS-H vaccine. Except for the vlhA expressed region, both genomes were nearly identical. Thirty-two single nucleotide polymorphisms (SNPs) were identified in MS-H, including 11 non-synonymous mutations that were predicted, by bioinformatics analysis, to have changed the secondary structure of the deduced proteins. One of these mutations caused truncation of the oppF-1 gene, which encodes the ATP-binding protein of an oligopeptide permease transporter. Overall, the attenuation of MS-H strain may be caused by the cumulative and complex effects of several mutations. The SNPs identified in MS-H were further analyzed by comparing the MS-H and 86079/7NS sequences with the strains WVU-1853 and MS53. In the genomic regions conserved between all strains, 30 SNPs were found to be unique to MS-H lineage. These results have provided a foundation for developing novel biomarkers for the detection of virulence in M. synoviae and also for designing new genotyping assays for discrimination of MS-H from field strains.
Introduction
Mycoplasmas are a group of bacteria that are distinguished phenotypically from other bacteria by their very small size and the lack of a cell wall. The organisms belong to the class Mollicutes, order Mycoplasmatales, family Mycoplasmataceae, and genus Mycoplasma (Johansson & Pettersson, Citation2002). Mycoplasma synoviae (MS) is an important avian pathogen worldwide. Infection with MS was first described in 1954 (Olson et al., Citation1954), before isolation the causative agent in 1956 (Olson, Citation1956). The first isolation of MS in Australia was reported in 1974 (Gilchrist & Cottew, Citation1974).
Mycoplasma synoviae can cause subclinical upper respiratory infection, infectious synovitis (Ferguson-Noel & Noormohammadi, Citation2013), airsacculitis (Kleven et al., Citation1972), and eggshell apex abnormalities (EAA) (Feberwee et al., Citation2009) in poultry. The severity of the clinical signs may vary between different strains and routes of infection (Lockaby et al., Citation1998; Kang et al., Citation2002). The organism is transmitted horizontally through direct contact, or vertically via fertile eggs (Kleven, Citation1998). Although the disease is rarely associated with bird mortality, it can cause a significant economic impact in the poultry industry due to condemnation of carcasses, decreased weight gain, reduced egg quality and production.
Vaccination is a common strategy used to control infection with MS in commercial flocks. The MS-H live attenuated vaccine (Vaxsafe MS®; Bioproperties Pty. Ltd., Ringwood, Victoria, Australia) has been widely used in a large number of countries since its registration in Australia in 1996. The MS-H strain is a temperature-sensitive (ts+) mutant obtained by N-nitro-N’-methyl-N-nitrosoguanidine (NTG) mutagenesis of an Australian field strain 86079/7NS (Morrow et al., Citation1998), which was originally isolated from the choanal cleft of a commercial layer hen with respiratory disease (Morrow, Citation1990). NTG mainly causes mutations by methylation of nucleotides, especially guanine, which can then pair with thymine or cytosine (Lucchesi et al., Citation1986) and give rise to GC-to-AT conversions. Treatment of 86079/7NS with NTG produced a panel of 15 ts+ mutants, from which MS-H was selected as a vaccine on the basis of its consistent colonization and temperature-sensitivity (Morrow et al., Citation1998). MS-H grows well at 33°C but its growth is significantly reduced at 39°C (the core body temperature of birds). It is postulated that this temperature sensitivity phenotype is the reason that MS-H colonizes only the upper respiratory tract of its host. MS-H has been shown to induce long-term protective mucosal immunity (Jones et al., Citation2006a; b). Despite the widespread use of the MS-H vaccine, the genetic basis for its attenuation is still poorly understood, owing to the random nature of NTG mutagenesis. The first study that attempted to identify mutations linked to attenuation described an alteration, G123R, in Obg (a GTP binding protein), which could be a potential factor contributing to the ts+ phenotype of MS-H (Shahid et al., Citation2013). As the ts+ MS-H strain can revert to a non-temperature-sensitive (ts-) phenotype in rare cases without resulting in reversion to the full virulent phenotype of the parent strain 86079/7NS, it is believed that more factors are involved in the attenuation of virulence seen in the vaccine (Noormohammadi et al., Citation2003).
Recently, the whole genome sequence of MS-H was sequenced in our laboratory using a combination of Illumina sequence data and long reads generated by Oxford Nanopore Technology (Zhu et al., Citation2018). A major step towards defining the molecular basis of attenuation in MS-H was the completion of the genome sequence of the parent strain 86079/7NS. In this study, the same sequencing strategies were applied to 86079/7NS, followed by a comparative genomic analysis of these two strains. The vaccine and the parent strains had almost identical genomic sequences, but a large number of single nucleotide polymorphisms (SNPs) were detected in MS-H. A few of these SNPs caused nonsynonymous or frameshift mutations. These specific genomic differences may provide insights into determinants responsible for the avirulent nature of MS-H and the genetic basis of virulence in MS.
Materials and Methods
Strain, culture conditions and DNA extraction
M. synoviae strain 86079/7NS (the 4th passage of the original clinical isolate, which is in the order of the passaging of the previously published MS-H vaccine) was grown in mycoplasma culture medium supplemented with 10% swine serum and 0.01% (w/v) of nicotinamide adenine dinucleotide (NAD) (Whithear, Citation1993) at 37°C in a 50 ml final volume until late logarithmic phase (approximately pH 6.8). Cells were collected by centrifugation at 10,000 × g for 30 min. Genomic DNA was prepared by proteinase K digestion of the pellet, phenol–chloroform extraction and ethanol precipitation (Green & Sambrook, Citation2016, Citation2017). The DNA concentration was determined by measuring the optical density at 260 nm (OD260), and the purity was determined by calculating the OD260/280 ratio using a NanoDrop™ 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Sequencing and assembly
An Illumina MatePaired (3-kbp insert size) genomic DNA library was prepared and sequenced by the Micromon DNA Sequencing Facility (Monash University, Australia) using a Genome Analyzer IIx system (Illumina, San Diego, CA, USA). Two Oxford Nanopore long read genomic DNA libraries were independently prepared using the sequencing kit 2D SQK-LSK208 (Oxford Nanopore Technologies, Oxford, UK) according to the manufacturer’s instructions using 1.6 or 2.3 µg of DNA sheared with a Covaris-g Tube on an Eppendorf centrifuge 5424 at 6000 rpm (3381 × g) as per the specifications provided by the manufacturer. Sequencing data was generated on a MinION MK-I device fitted with a FLO-MIN106 flowcell (R9.4 chemistry) and processed using the ONT Albacore Sequencing Pipeline (version 1.2.1). To confirm the accuracy of de novo assembly, 86079/7NS genomic DNA was digested with the restriction endonuclease AflII using the OpGen enzyme kit and the MapCard kit and subjected to Argus™ optical mapping (BGI, Hong Kong). Optical mapping data were analyzed using MapSolver (version 3.1). Oligonucleotides (Geneworks, Australia) were designed using Primer3 (version 2.3.4) to perform PCR amplifications to close gaps in the assembly, and PCR amplicons were sequenced using the Sanger method at the Micromon DNA Sequencing Facility (Monash University, Australia).
De novo sequence assembly, gene prediction and annotation
MatePaired Illumina reads were filtered using Nesoni 0.122 to trim bases below Phred quality 20, and remove the adapter sequences and reads with a length less than 91 bp (https://pypi.python.org/pypi/nesoni/0.122). The quality of the filtered reads was confirmed using FastQC (Andrews, Citation2010). The Illumina reads were then de novo assembled using Velvet (Zerbino & Birney, Citation2008) (version 1.2.10) using k-mer values of 81 and 91 and coverage cut-off values of 5, 10, 20, 50, 100 and 200.
The Nanopore 2D reads with a minimal length of 1000 bp were extracted from a set of fast5 files returned by the Albacore basecaller and converted into “.fasta” format using the Poretools programme (Loman & Quinian, Citation2014). The Nanopore reads were then de novo assembled using Canu 1.4 (Berlin et al., Citation2015) with the parameter genomesize = 0.9 m and errorRate = 0.1. The Illumina short reads were mapped against the Canu contig using the Geneious® v.7.0.6 (Kearse et al., Citation2012) sequence manipulation suite to generate the consensus sequence. Automatic annotation of the 86079/7NS sequence was performed on the RAST webserver with default parameters for mycoplasmas (Aziz et al., Citation2008). The whole-genome sequence of 86079/7NS has been deposited in GenBank under accession number CP029258.
Genome comparison and analysis
Whole genome alignment of MS-H (GenBank accession CP021129) and 86079/7NS was performed using MAUVE (Darling et al., Citation2004). General nucleotide sequence alignments were conducted with MAFFT v.7.017. Screening for synonymous and non-synonymous SNPs was conducted with Find Variations/SNPs in Geneious®. Secondary structures of the deduced protein sequences were predicted using EMBOSS 6.5.7 tool garnier (Garnier et al., Citation1978; Rice et al., Citation2000). The genome sequences of MS strains MS53 and WVU-1853 (GenBank accession numbers AE017245 and CP011096) were also included as references for analysis of SNPs.
Results and Discussion
Illumina and Nanopore sequence reads for assembling the complete 86079/7NS genome, including the highly repetitive vlhA pseudogene locus
A set of 12,817,968 Illumina MatePaired reads from strain 86079/7NS were de novo assembled using Velvet with a series of K-mer and coverage cutoff settings. Using the best K-mer and cutoff values of 91 and 10, respectively, a set of 55 contigs was produced, with sizes ranging from 181 bp to 569,106 bp. The contig sequences were converted into in silico AflII restriction endonuclease cleavage maps and aligned with the 86079/7NS optical map to determine their locations and orientations during assembly. However, the high similarity and repetitive nature of insertion sequences (IS), prevented assembly of many of the IS-carrying genomic regions solely from the Illumina datasets. Sanger sequencing of 38 PCR products combined with scaffolding of the Velvet data generated a 766,256 bp contig, representing an almost complete genome, but failed to assemble the vlhA pseudogene region, a 50 kb locus containing a large number of highly repetitive sequences. This procedure was successfully used to complete the genome sequencing of MS vaccine strain MS-H, which carries an equivalent, difficult to assemble, locus (Zhu et al., Citation2018). To resolve the vlhA pseudogene region and circularize the 86079/7NS genome, a set of 12,991 Nanopore reads, with a minimal length of 1000 bp, was independently assembled. After trimming two short overlapping regions at both ends, the assembly was circularized into a single 809,647 bp contig. Due to the high error rate of Nanopore sequences, the Illumina reads were mapped against the contig, revealing an average depth of 1776 ± 305. The extraction of the consensus sequence resulted in a fully assembled genome encompassing the entire vlhA cluster and all the other regions that couldn’t be assembled solely from Illumina reads. The whole genome sequence result was further verified by aligning the previous Velvet contigs with the new Illumina/Nanopore hybrid assembly. Most discrepancies were found in the highly repeated transposase genes, to which the misassembly of the Velvet contigs was attributable. All nucleotide discrepancies between these two datasets were resolved manually by inspecting the alignments of Illumina reads or Sanger sequences of PCR products to produce a finished genome assembly. Single molecule restriction endonuclease cleavage fragments generated using AflII were converted into a circular optical map using the OpGen optical map assembler, with a total size of 0.803 Mb, covering 97.69% of the genome. Using OpGen MapSolver (v.3.1), the complete 86079/7NS genome sequence was converted in silico into an AflII restriction map and aligned to its optical map (). The genomic sequence of 86079/7NS was in complete agreement with the optical map, confirming the accuracy of the assembly.
Figure 1. Mapping of complete genomic sequence of 86079/NS against its optical map.

MS-H and 86079/7NS have almost identical genomic sequences
The genome of the vaccine parent strain, 86079/7NS, was 818,795 bp in length and highly similar to that of MS-H, with no multinucleotide insertions or deletions, or genomic rearrangements (except for the vlhA expressible region) (). In our previous study, a 55 kb genomic inversion delimited by a tRNAGly and an IS30 was identified in MS-H (Zhu et al., Citation2018). The same inverted region was also noted in 86079/7NS, which confirmed that the genomic inversion was not introduced by chemical mutagenesis, but occurred naturally at an earlier stage. A number of genes thought to be unique to MS-H (Zhu et al., Citation2018), including a deoxyribose-phosphate aldolase (deoC) fragment, an ATP-dependent DNA helicase, and an N-acetylneuraminate lyase variant (nanA), were also found in 86079/7NS, suggesting that these genes were horizontally acquired by the vaccine parental strain or its ancestor. Whether the presence of these genes is a common feature of Australian MS strains remains to be explored. The most distinctive region in the MS-H and 86079/7NS genomes was located in the expressed vlhA gene (Narat et al., Citation1998; Bencina, Citation2002). The vlhA gene from MS-H shared 81% nucleotide and 75.4% amino acid sequence identity with 86079/7NS, which further highlights the variable nature of this gene (Noormohammadi et al., Citation2000). The predicted VlhA protein sequence diverged from residue 224 to the carboxyl terminus, similar to those previously described in a variant vlhA gene of strain WVU-1853 (Noormohammadi et al., Citation2000). Clonal variations of the VlhA protein are common in MS and may influence cytadherence in the host and therefore contribute to disease (May et al., Citation2014). In MS, vlhA pseudogenes are used as a repertoire to generate sequence diversity in the expressed vlhA gene (Noormohammadi et al., Citation2000). The vlhA pseudogenes are highly conserved between MS-H and its parent, except for a “G→A” mutation at nucleotide 255672 and a deletion of two bases (TT) before nucleotide 258581 in MS-H. In addition, a total of 32 mutations were distributed across the remainder of the genome of MS-H. The majority of the mutations (85%) were caused by transitions, with a strong preference for “G→A” substitutions (). The extreme bias in single base substitutions confirmed one of the properties of the mutagen, NTG, which mainly leads to transition mutations, as observed in previous studies (Gordon et al., Citation1988; Ohnishi et al., Citation2008).
Figure 2. MAUE alignment of the complete genomic sequence of M. synoviae strains 86079/NS and MS-H. The expressed vlhA gene in highlighted with a black box.
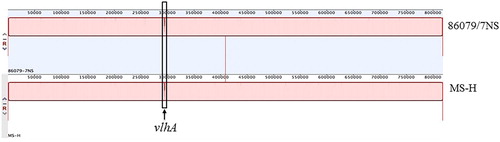
Table 1. Nucleotide and coding differences identified between the genomes of MS-H and 86079/7NS*.
A few genetic mutations found in MS-H may have functional significance
Of the 32 SNPs identified between virulent strain 86079/7NS and MS-H, 26 were located in putative coding regions, and six were found in intergenic spaces where a coding sequence with an unknown function was located downstream. No coding sequence had a SNP resulting in more than one amino acid change. While synonymous SNPs (sSNPs) may have functional consequences in rare cases (Kimchi-Sarfaty et al., Citation2007), nonsynonymous SNPs (nsSNPs), frameshifts, or intergenic SNPs that affect gene expression or protein structure are the most likely causes of phenotypic variation. An apparent bias in the numbers of nsSNPs was noted, in favour of those that introduced amino acid substitutions or frameshifts that might cause differences in protein functions between MS-H and 86079/7NS (). These affected genes were grouped into categories involved in amino acid metabolism, environmental sensing, central metabolism, transport, tRNA metabolism and nucleic acid metabolism. A total of 11 substitutions were predicted to have disrupted the secondary structures of the deduced proteins and were therefore subjected to further sequence analysis to predict the potential effects of the 11 substitutions on the deduced proteins described ().
In MS, the vlhA locus is flanked by identical homologues of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which were putatively transferred from Mycoplasma gallisepticum (Vasconcelos et al., Citation2005). These sequences share 100% amino acid similarity amongst MS strains 86079/7NS, WVU-1853 and MS53. However, each of the GAPDH coding genes in MS-H was found to carry a mutation that introduced an amino acid change in the product. GAPDH is a NAD+ dependent glycolytic enzyme that catalyses the conversion of glyceraldehyde 3-phosphate to 1, 3-diphosphoglycerate, and can be located in the cytoplasm as well as on the cell surface (Tunio et al., Citation2010). In addition to their metabolic role, surface-localised GAPDHs from several bacterial species, including Streptococcus pneumoniae (Ling et al., Citation2004), Staphylococcus aureus, Staphylococcus epidermidis (Modun & Williams, Citation1999), Listeria monocytogenes (Schaumburg et al., Citation2004) and Escherichia coli (Egea et al., Citation2007), have also been demonstrated to have a role in pathogenesis, including facilitating host immune evasion (Terao et al., Citation2006) and colonization and invasion of host tissues (Alvarez et al., Citation2003). It is worth noting that the conservative substitutions, A185 V and R306K, caused by the two nsSNPs, each in one of the two copies of the GAPDH gene (nt 242451 and nt 296526, respectively), were located in the catalytic domain of the enzyme, which encompasses residues 149–333 (Walker et al., Citation1980; Branlant & Branlant, Citation1985). One substitution was found located in the S loop (residues 178–201), which is believed to be associated with the thermal stability of the enzyme (Walker et al., Citation1980). However, as the biological structure of GAPDH has not been well characterized in MS, whether such amino acid changes can disrupt the core binding motifs, and therefore affect function, still needs to be explored.
In MS-H, a single base pair substitution within a gene coding for a protein homologous to haemolysin C (HlyC) resulted in a change from alanine to valine compared to the corresponding protein in 86079/7NS. Haemolysins are potential virulence factors in mollicutes (Chambaud et al., Citation2001). Membrane-associated haemolysin activity is usually dependent on the conversion of a nontoxic prohaemolysin, proHlyA, to toxic haemolysin, HlyA, by an internal protein acyltransferase, HlyC, using the acyl-acyl carrier protein (ACP) as the essential acyl donor (Minion & Jarvill-Taylor, Citation1994; Trent et al., Citation1999). These proteins are known to be present in Mycoplasma pulmonis (Chambaud et al., Citation2001), Ureaplasma urealyticum (Glass et al., Citation2000) and Mycoplasma penetrans (Sasaki et al., Citation2002). However, although orthologues of HlyC and ACP synthase were found in MS, a CDS for HlyA was not identified and thus a catalyst performing the function of HlyC remains elusive.
Mycoplasma synoviae encodes a VACB-like ribonuclease II (RNase R, the gene product of vacB), the orthologues of which have been implicated in virulence in several pathogens (Tobe et al., Citation1992; Cheng et al., Citation1998; Erova et al., Citation2008; Tsao et al., Citation2009). In MS-H, a nsSNP (nt 352952) was found in the vacB gene. BLAST-P analysis indicated that the vacB mutation in MS-H occurred at a serine residue that is highly conserved across the orthologues from other mycoplasmal species. Even though a role for this protein in virulence in MS is yet to be established, RNase R has been shown to be essential for the growth of Mycoplasma genitalium (Hutchison et al., Citation1999; Lalonde et al., Citation2007).
Non-conservative amino acid substitutions were also identified in three other putative proteins associated with nucleic acid metabolism, including uvrB (G26E), rpoC (R1270I) and dnaE (S103F), which are involved in DNA repair, transcription of RNA, and replication/repair, respectively. Mutations in the uvrB gene of Mycobacterium tuberculosis have been shown to increase sensitivity to acidified nitrite, a reactive nitrogen intermediate in the phagosome that helps protect the host during infection (Darwin et al., Citation2003) and an insertion mutation in this gene attenuated virulence in mice (Darwin & Nathan, Citation2005). A mutation (G to A) in the rpoC gene of E. coli leading to the substitution D1033G in the RNA polymerase beta subunit causes a temperature-dependent increase in chromosome copy number (Rasmussen et al., Citation1983) through effects on chromosomal replication control by altering the expression of the dnaA gene (Petersen & Hansen, Citation1991). DnaE is the principal chromosomal replication enzyme in many bacteria, although it is also involved in DNA repair in some mycoplasmas (Koonin & Bork, Citation1996; Hutchison et al., Citation1999). In E. coli the N-terminal region of DnaE has been shown to be responsible for its catalytic activity (Kim et al., Citation1997). A single amino acid substitution within the N-terminal region of DnaE results in a ts+ phenotype in both E. coli (Flett et al., Citation1999) and Caulobacter crescentus, possibly by affecting the initiation of DNA replication (Lo et al., Citation2004). As very little is known about the mechanism of chromosomal replication and its regulation in MS, it would be interesting to find out whether the mutations detected in nucleotide metabolism related genes in MS-H had an influence on its chromosomal replication and therefore its growth rate under permissive/non-permissive temperatures.
A non-conservative H275Y substitution was identified within the MS-H gene encoding KtrB, the membrane-spanning subunit of the Na+/K+ symporter KtrAB which mediates Na+-dependent high affinity K+ uptake in many prokaryotes (Nakamura et al., Citation1998; Durell et al., Citation1999; Johnson et al., Citation2009). K+ transport is especially important for cytoplasmic pH homeostasis for bacteria exposed to an alkaline external pH (Nakamura et al., Citation1984). A substitution in the P-loop selectivity filter in KtrB, resulting in a decrease in affinity for K+, has been seen in Vibrio alginolyticus (Lü & Miller, Citation1995; Tholema et al., Citation1999). Alignment with the consensus sequence of KtrB of Bacillus subtilis (Albright et al., Citation2007) revealed that the H275Y mutation of MS-H was not located within the TM-P-loop-TM domain, indicating that this mutation might be tolerated in MS and was less likely to be linked with the attenuation of virulence seen in MS-H.
A frameshift mutation in oppF-1 may result in the loss of function of its product
M. synoviae possesses two Opp systems that are located distantly from each other within the chromosome, hereafter named opp-I and opp-II. The deletion of one nucleotide (T) at position 397721 led to a codon frameshift in the oppF gene of opp-I in MS-H, compared to the parent strain 86079/7NS, while the second system was unchanged. OppF is proposed to act as the ATP-binding component of an ABC transporter involved in uptake of oligopeptides. The frameshift is likely to have inactivated protein function by interrupting the coding sequence, allowing translation of only the amino-terminal 156 amino acids. Among the molecular factors associated with the virulence of pathogenic bacteria, the importance of oligopeptide permease (Opp) transporters has been highlighted in both Gram-negative and Gram-positive pathogens (Eitinger et al., Citation2011; Jones et al., Citation2014; Moraes et al., Citation2014). These multi-subunit protein complexes belong to the ABC transporter family, and are typically composed of an extracellular substrate binding lipoprotein, OppA, two transmembrane proteins, OppB and OppC (forming the pore) and two ATP-binding proteins, OppD and OppF (Garai et al., Citation2017). Studies on several pathogenic bacteria have indicated that Opp proteins are involved not only in importing peptides as nutrients, but also in adhesion to the host, acting as colonization and virulence factors (Cundell et al., Citation1995; Podbielski et al., Citation1996; Samen et al., Citation2004; Moraes et al., Citation2014; Tseng et al., Citation2017). In the case of M. tuberculosis, the Opp system was shown to be required for chronic infection and for expression of virulence-associated surface lipid (Flores-Valdez et al., Citation2009) and opp mutants exhibited in vivo survival defects, indicating diminished virulence of the organism (Sassetti & Rubin, Citation2003; Flores-Valdez et al., Citation2009). Partial impairment of the OppD1 function in M. gallisepticum strain Ap3AS reduced its colonization ability and virulence. A mutant that could express the N-terminal 34% of OppD1 was less pathogenic than a mutant that could express only the N-terminal 7.9% of OppD1 (Tseng et al., Citation2017). The phylogenic relationships between opp operons within the same species are complex, although it has been suggested that the opp operons in the hominis group underwent duplication and divergence. Homologous proteins encoded by the two gene clusters share low sequence similarity, possibly indicating that they have different biological functions. It is possible that the pseudogenization of oppF in opp-I in MS-H is not fully compensated for by the single oppF gene copy present in opp-II, which could result in attenuation of virulence. Alternatively, as has been hypothesized for OppD1 in M. gallisepticum (Tseng et al., Citation2017), it is tempting to speculate that, as OppF proteins form dimers in the transport complex, the product of the partial copy of oppF-I leads to the formation of heterodimers with the product of the full-length copy from oppF-II, resulting in less efficient substrate transport and thus a decrease in virulence.
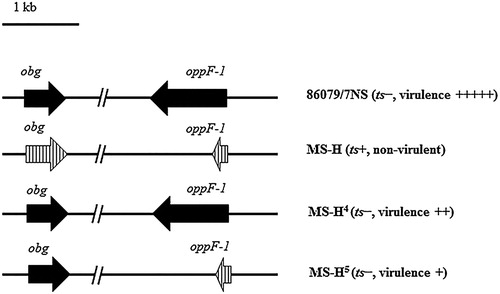
A substitution (G123R) within Obg has been predicted to be one of the causes of the ts+ phenotype of MS-H (Shahid et al., Citation2013). However, the ts+ MS-H strain can occasionally revert to a ts− phenotype, possibly due to the selection pressure in the field (Markham et al., Citation1998). Although such loss of the ts+ phenotype was not associated with a full reversion to the level of virulence of the original parent strain 86079/7NS, partial virulence was observed in other MS-H clones reisolated from vaccinated flocks and displaying a ts− phenotype (Noormohammadi et al., Citation2003). For example, the ts− reisolate MS-H4 caused upper tracheal mucosal thicknesses that were significantly greater than those caused by ts− MS-H5, although they were both less pathogenic than the parental strain (Markham et al., Citation1998; Noormohammadi et al., Citation2003). Comparative sequence analyses of the oppF-1 gene revealed that the frameshift mutation within oppF-1 was present in MS-H5, but not found in MS-H4 (). It is tempting to speculate that the mutation in oppF-1 could have resulted in a greater reduction of virulence observed in MS-H5, indicating that the mutation in oppF-1 could be another factor that might slow down growth without affecting survival, and therefore contribute to the attenuation of the vaccine strain.
Figure 3. Phenotypes of appF-I and obg genes in 86079/NS, MS-H re-isolates. Solid arrows represent wild-type genes. Vertically hatched arrows represent vaccine-type genes.
The mutations reported in this study should be confirmed in conjunction with whole genome sequences of a large number of MS-H reisolates such as those reported previously (Shahid et al., Citation2013). This would determine if any of the mutations are reversible under in vivo or in vitro conditions and which mutation(s) should be further characterized in future studies focusing on the attenuation of MS-H.
SNPs for discriminating infected from vaccinated animals (DIVA assays)
As MS-H has been used widely to prevent disease caused by MS, differentiation of the vaccine strain from field isolates would be useful during vaccination and eradication programmes. Comparative genomic sequence analyses of strains MS-H, 86079/7NS, combined with two other field strains, WVU-1853 and MS53 (for which full genome sequences are available in GenBank), revealed 30 SNPs unique to the MS-H vaccine strain, which were located in regions conserved between the strains (). It was noted that most SNPs (except for the SNPs in obg and oppF-1) (Shahid et al., Citation2013; Zhu et al., Citation2017) reported here haven’t been targeted in previous MS typing assays, including those based on a multi-locus sequence typing scheme (Cizelj et al., Citation2015; Dijkman et al., Citation2016; El-Gazzar et al., Citation2017). Therefore, these SNPs could be used as novel vaccine markers to develop diagnostic assays, such as polymerase chain reaction based high-resolution melting curve analysis (PCR-HRM), for discriminating MS-H from wild-type MS strains. As the MS-H vaccine is now widely used in a large number of countries, and back mutation within MS-H may occasionally occur under selective pressure in the field, it would be ideal to perform complete genome sequencing of MS-H reisolates from different geographical locations to confirm the stability of the SNPs unique to MS-H. Such studies will facilitate selecting the most reliable marker(s) that can be used for a MS-H specific molecular assay. Several molecular techniques, based on sequence analyses of the conserved region of the vlhA gene, are currently available for the discrimination of MS strains (Hong et al., Citation2004; Jeffery et al., Citation2007; Hammond et al., Citation2009; Harada et al., Citation2009; Wetzel et al., Citation2010; El-Gazzar et al., Citation2012; Bayatzadeh et al., Citation2014; Dijkman et al., Citation2014). However, given that several Australian, and possibly two European, isolates share the same vlhA sequence type as MS-H (Dijkman et al., Citation2014; Catania et al., Citation2016), the discriminatory power of vlhA sequence typing for molecular epidemiological studies of MS isolates has limitations. A number of studies have therefore used the mutations found in the obg gene for discriminating MS-H from field strains (Shahid et al., Citation2014; Dijkman et al., Citation2017). As there are Australian field strains that are identical to the MS-H vaccine, as well as rare cases where ts− MS-H re-isolates are identical in the targeted region of the obg gene, the differentiation of the MS-H vaccine from field strains using these techniques may not be totally reliable, especially in Australia. Therefore, development of a typing technique with improved discriminatory power, based on novel genetic markers of the MS-H vaccine, is desirable.
Table 2. Unique single nucleotide variations detected in M. synoviae strains MS-H, 86079/7NS, WVU-1853 and MS53.
Conclusion
Few virulence determinants or virulence-associated determinants have been identified in MS. Elucidation of the genetic basis of attenuation of the MS-H vaccine will contribute to understanding the pathogenic mechanisms employed by MS, and to the development of improved vaccines and better strategies for control of the disease it causes. The genomic comparison of MS-H and its wild-type progenitor 86079/7NS was a first step in the identification of potential MS virulence factors. The SNPs with putative functional effects, identified by comparison of the two strains, have confirmed that the attenuation observed in MS-H is likely to be the result of the cumulative and complex effects of several mutations. As predicting the impact of intergenic SNPs or nsSNPs is problematic without experimental validation, we have adopted a conservative approach in the interpretation of the SNP data, and instead see them as the basis for facilitating future focused studies on the basis of attenuation of MS-H. A total of 30 SNPs were found to be unique to MS-H. These results have provided a rich source of novel biomarkers that can be used to develop new genotyping assays for discrimination of MS-H from field strains.
Acknowledgements
The authors would like to acknowledge the assistance from the staff of Asia-Pacific Centre for Animal Health (APCAH), Faculty of Veterinary and Agricultural Sciences, the University of Melbourne. Funding for this study was provided by APCAH.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Ling Zhu http://orcid.org/0000-0002-1570-8584
References
- Albright, R.A., Joh, K. & Morais-Cabral, J.H. (2007). Probing the structure of the dimeric KtrB membrane protein. Journal of Biological Chemistry, 282, 35046–35055. doi: 10.1074/jbc.M704260200
- Alvarez, R.A., Blaylock, M.W. & Baseman, J.B. (2003). Surface localized glyceraldehyde-3-phosphate dehydrogenase of Mycoplasma genitalium binds mucin. Molecular Microbiology, 48, 1417–1425. doi: 10.1046/j.1365-2958.2003.03518.x
- Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. Retrieved from http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- Aziz, R.K., Bartels, D., Best, A.A., DeJongh, M., Disz, T., Edwards, R.A., Formsma, K., Gerdes, S., Glass, E.M., Kubal, M., Meyer, F., Olsen, G.J., Olson, R., Osterman, A.L., Overbeek, R.A., McNeil, L.K., Paarmann, D., Paczian, T., Parrello, B., Pusch, G.D., Reich, C., Stevens, R., Vassieva, O., Vonstein, V., Wilke, A. & Zagnitko, O. (2008). The RAST Server: Rapid annotations using subsystems technology. BMC Genomics, 9, 75. doi: 10.1186/1471-2164-9-75
- Bayatzadeh, M.A., Pourbakhsh, S.A., Ashtari, A., Abtin, A.R. & Abdoshah, M. (2014). Molecular typing of Iranian field isolates Mycoplasma synoviae and their differentiation from the live commercial vaccine strain MS-H using vlhA gene. British Poultry Science, 55, 148–156. doi: 10.1080/00071668.2013.878781
- Bencina, D. (2002). Haemagglutinins of pathogenic avian mycoplasmas. Avian Pathology, 31, 535–547. doi: 10.1080/0307945021000024526
- Berlin, K., Koren, S., Chin, C.S., Drake, J.P., Landolin, J.M. & Phillippy, A.M. (2015). Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nature Biotechnology, 33, 623–630. doi: 10.1038/nbt.3238
- Branlant, G. & Branlant, C. (1985). Nucleotide sequence of the Escherichia coli gap gene. FEBS Journal, 150, 61–66.
- Catania, S., Gobbo, F., Ramirez, A.S., Guadagnini, D., Baldasso, E., Moronato, M.L. & Nicholas, R.A. (2016). Laboratory investigations into the origin of Mycoplasma synoviae isolated from a lesser flamingo (Phoeniconaias minor). BMC Veterinary Research, 12, 52. doi: 10.1186/s12917-016-0680-1
- Chambaud, I., Heilig, R., Ferris, S., Barbe, V., Samson, D., Galisson, F., Moszer, I., Dybvig, K., Wróblewski, H., Viari, A. & Rocha, E.P. (2001). The complete genome sequence of the murine respiratory pathogen Mycoplasma pulmonis. Nucleic Acids Research, 29, 2145–2153. doi: 10.1093/nar/29.10.2145
- Cheng, Z.F., Zuo, Y., Li, Z., Rudd, K.E. & Deutscher, M.P. (1998). The vacB gene required for virulence in Shigella flexneri and Escherichia coli encodes the exoribonuclease RNase R. Journal of Biological Chemistry, 273, 14077–14080. doi: 10.1074/jbc.273.23.14077
- Cizelj, I., Berčič, R.L., Slavec, B., Narat, M., Dovč, P. & Benčina, D. (2015). Multilocus sequence analysis for Mycoplasma synoviae molecular genotyping. British Poultry Science, 56, 658–665. doi: 10.1080/00071668.2015.1113506
- Cundell, D.R., Pearce, B.J., Sandros, J., Naughton, A.M. & Masure, H.R. (1995). Peptide permeases from Streptococcus pneumoniae affect adherence to eucaryotic cells. Infection and Immunity, 63, 2493–2498.
- Darling, A.C.E., Mau, B., Blattner, F.R. & Perna, N.T. (2004). Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Research, 14, 1394–1403. doi: 10.1101/gr.2289704
- Darwin, K.H., Ehrt, S., Gutierrez-Ramos, J.C., Weich, N. & Nathan, C.F. (2003). The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science, 302, 1963–1966. doi: 10.1126/science.1091176
- Darwin, K.H. & Nathan, C.F. (2005). Role for nucleotide excision repair in virulence of Mycobacterium tuberculosis. Infection and Immunity, 73, 4581–4587. doi: 10.1128/IAI.73.8.4581-4587.2005
- Dijkman, R., Feberwee, A. & Landman, W.J. (2017). Development, validation and field evaluation of a quantitative real-time PCR able to differentiate between field Mycoplasma synoviae and the MS-H live vaccine strain. Avian Pathology, 46, 403–415. doi: 10.1080/03079457.2017.1296105
- Dijkman, R., Feberwee, A. & Landman, W.J.M. (2014). Variable lipoprotein haemagglutinin (vlhA) gene sequence typing of mainly Dutch Mycoplasma synoviae isolates: Comparison with vlhA sequences from GenBank and with amplified fragment length polymorphism analysis. Avian Pathology, 43, 465–472. doi: 10.1080/03079457.2014.958980
- Dijkman, R., Feberwee, A. & Landman, W.J.M. (2016). Development and evaluation of a multi-locus sequence typing scheme for Mycoplasma synoviae. Avian Pathology, 45, 426–442. doi: 10.1080/03079457.2016.1154135
- Durell, S.R., Hao, Y., Nakamura, T., Bakker, E.P. & Guy, H.R. (1999). Evolutionary relationship between K+ channels and symporters. Biophysical Journal, 77, 775–788. doi: 10.1016/S0006-3495(99)76931-6
- Egea, L., Aguilera, L., Gimenez, R., Sorolla, M.A., Aguilar, J., Badia, J. & Baldoma, L. (2007). Role of secreted glyceraldehyde-3-phosphate dehydrogenase in the infection mechanism of enterohemorrhagic and enteropathogenic Escherichia coli: Interaction of the extracellular enzyme with human plasminogen and fibrinogen. International Journal of Biochemistry & Cell Biology, 39, 1190–1203. doi: 10.1016/j.biocel.2007.03.008
- Eitinger, T., Rodionov, D.A., Grote, M. & Schneider, E. (2011). Canonical and ECF-type ATP-binding cassette importers in prokaryotes: Diversity in modular organization and cellular functions. FEMS Microbiology Reviews, 35, 3–67. doi: 10.1111/j.1574-6976.2010.00230.x
- El-Gazzar, M., Ghanem, M., McDonald, K., Ferguson-Noel, N., Raviv, Z. & Slemons, R.D. (2017). Development of multilocus sequence typing (MLST) for Mycoplasma synoviae. Avian Diseases, 61, 25–32. doi: 10.1637/11417-040516-Reg
- El-Gazzar, M.M., Wetzel, A.N. & Raviv, Z. (2012). The genotyping potential of the Mycoplasma synoviae vlhA gene. Avian Diseases, 56, 711–719. doi: 10.1637/10200-041212-Reg.1
- Erova, T.E., Kosykh, V.G., Fadl, A.A., Sha, J., Horneman, A.J. & Chopra, A.K. (2008). Cold shock exoribonuclease R (VacB) is involved in Aeromonas hydrophila pathogenesis. Journal of Bacteriology, 190, 3467–3474. doi: 10.1128/JB.00075-08
- Feberwee, A., De Wit, J.J. & Landman, W.J.M. (2009). Induction of eggshell apex abnormalities by Mycoplasma synoviae: Field and experimental studies. Avian Pathology, 38, 77–85. doi: 10.1080/03079450802662772
- Ferguson-Noel, N. & Noormohammadi, A.H. (2013). Mycoplasma synoviae infection. In D.E. Swayne, J.R. Glisson, L.R. McDougald, L.K. Nolan, D.L. Suarez & V.L. Nair (Eds.), Diseases of poultry 13th edn (pp. 875). Hoboken, NJ: Wiley-Blackwell.
- Flett, F., De Mello Jungmann-Campello, D., Mersinias, V., Koh, S.L.M., Godden, R. & Smith, C.P. (1999). A ‘gram-negative-type’ DNA polymerase III is essential for replication of the linear chromosome of Streptomyces coelicolor A3 (2). Molecular Microbiology, 31, 949–958. doi: 10.1046/j.1365-2958.1999.01237.x
- Flores-Valdez, M.A., Morris, R.P., Laval, F., Daffé, M. & Schoolnik, G.K. (2009). Mycobacterium tuberculosis modulates its cell surface via an oligopeptide permease (Opp) transport system. FASEB Journal, 23, 4091–4104. doi: 10.1096/fj.09-132407
- Garai, P., Chandra, K. & Chakravortty, D. (2017). Bacterial peptide transporters: Messengers of nutrition to virulence. Virulence, 8, 297–309. doi: 10.1080/21505594.2016.1221025
- Garnier, J., Osguthorpe, D.J. & Robson, B. (1978). Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. Journal of Molecular Biology, 120, 97–120. doi: 10.1016/0022-2836(78)90297-8
- Gilchrist, P.T. & Cottew, G.S. (1974). Isolation of Mycoplasma synoviae from respiratory diseases in chickens. Australian Veterinary Journal, 50, 81. doi: 10.1111/j.1751-0813.1974.tb05263.x
- Glass, J.I., Lefkowitz, E.J., Glass, J.S. & Heiner, C.R. (2000). The complete sequence of the mucosal pathogen Ureaplasma urealyticum. Nature, 407, 757–762. doi: 10.1038/35037619
- Gordon, A.J., Burns, P.A. & Glickman, B.W. (1988). N-Methyl-N′-nitro-N-nitrosoguanidine-induced mutation in a RecA strain of Escherichia coli. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 201, 219–228. doi: 10.1016/0027-5107(88)90129-7
- Green, M.R. & Sambrook, J. (2016). Precipitation of DNA with ethanol. Cold Spring Harbor Protocols, 12, 1116–1120, pdb-prot093377.
- Green, M.R. & Sambrook, J. (2017). Isolation of high-molecular-weight DNA using organic solvents. Cold Spring Harbor Protocols, 4, 356‐359, pdb-prot093450.
- Hammond, P.P., Ramírez, A.S., Morrow, C.J. & Bradbury, J.M. (2009). Development and evaluation of an improved diagnostic PCR for Mycoplasma synoviae using primers located in the haemagglutinin encodig gene vlhA and its value for strain typing. Veterinary Microbiology, 136, 61–68. doi: 10.1016/j.vetmic.2008.10.011
- Harada, K., Kijima-Tanaka, M., Uchiyama, M., Yamamoto, T., Oishi, K., Arao, M. & Takahashi, T. (2009). Molecular typing of Japanese field isolates and live commercial vaccine strain of Mycoplasma synoviae using improved pulsed-field gel electrophoresis and vlhA gene sequencing. Avian Diseases, 53, 538–543. doi: 10.1637/8934-052309-Reg.1
- Hong, Y., García, M., Leiting, V., Benčina, D., Dufour-Zavala, L., Zavala, G. & Kleven, S.H. (2004). Specific detection and typing of Mycoplasma synoviae strains in poultry with PCR and DNA sequence analysis targeting the hemagglutinin encoding gene vlhA. Avian Diseases, 48, 606–616. doi: 10.1637/7156-011504R
- Hutchison, C.A., Peterson, S.N., Gill, S.R., Cline, R.T., White, O., Fraser, C.M., Smith, H.O. & Venter, J.C. (1999). Global transposon mutagenesis and a minimal mycoplasma genome. Science, 286, 2165–2169. doi: 10.1126/science.286.5447.2165
- Jeffery, N., Gasser, R.B., Steer, P.A. & Noormohammadi, A.H. (2007). Classification of Mycoplasma synoviae strains using single-strand conformation polymorphism and high-resolution melting-curve analysis of the vlhA gene single region. Microbiology, 153, 2679–2688. doi: 10.1099/mic.0.2006/005140-0
- Johansson, K.E. & Pettersson, B. (2002). Taxonomy of mollicutes. In S. Razin & R. Herrmann (Eds.), Molecular Biology and Pathogenicity of mycoplasmas (pp. 1–32). Boston: Springer.
- Johnson, H.A., Hampton, E. & Lesley, S.A. (2009). The Thermotoga maritima Trk potassium transporter—from frameshift to function. Journal of Bacteriology, 191, 2276–2284. doi: 10.1128/JB.01367-08
- Jones, J.F., Whithear, K.G., Scott, P.C. & Noormohammadi, A.H. (2006a). Duration of immunity with Mycoplasma synoviae: Comparison of the live attenuated vaccine MS-H (Vaxsafe MS) with its wild-type parent strain, 86079/7NS. Avian Diseases, 50, 228–231. doi: 10.1637/7465-103005R.1
- Jones, J.F., Whithear, K.G., Scott, P.C. & Noormohammadi, A.H. (2006b). Onset of immunity with Mycoplasma synoviae: Comparison of the live attenuated vaccine MS-H (Vaxsafe MS) with its wild-type parent strain (86079/7NS). Avian Diseases, 50, 82–87. doi: 10.1637/7428-083005R.1
- Jones, M.M., Johnson, A., Koszelak-Rosenblum, M., Kirkham, C., Brauer, A.L., Malkowski, M.G., Murphy, T.F. (2014). Role of the oligopeptide permease ABC transporter of Moraxella catarrhalis in nutrient acquisition and persistence in the respiratory tract. Infection and Immunity, 82, 4758–4766. doi: 10.1128/IAI.02185-14
- Kang, M.S., Gazdzinski, P. & Kleven, S.H. (2002). Virulence of recent isolates of Mycoplasma synoviae in turkeys. Avian Diseases, 46, 102–110. doi: 10.1637/0005-2086(2002)046[0102:VORIOM]2.0.CO;2
- Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., Buxton, S., Cooper, A., Markowitz, S., Duran, C., Thierer, T., Ashton, B., Meintjes, P. & Drummond, A. (2012). Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics (Oxford, England), 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
- Kim, D.R., Pritchard, A.E. & McHenry, C.S. (1997). Localization of the active site of the α subunit of the Escherichia coli DNA polymerase III holoenzyme. Journal of Bacteriology, 179, 6721–6728. doi: 10.1128/jb.179.21.6721-6728.1997
- Kimchi-Sarfaty, C., Oh, J.M., Kim, I.W., Sauna, Z.E., Calcagno, A.M., Ambudkar, S.V. & Gottesman, M.M. (2007). A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science, 315, 525–528. doi: 10.1126/science.1135308
- Kleven, S.H. (1998). Mycoplasmas in the etiology of multifactorial respiratory disease. Poultry Science, 77, 1146–1149. doi: 10.1093/ps/77.8.1146
- Kleven, S.H., King, D.D. & Anderson, D.P. (1972). Airsacculitis in broilers from Mycoplasma synoviae: Effect on air sac lesions of vaccinating with infectious bronchitis and Newcastle virus. Avian Diseases, 16, 915–924. doi: 10.2307/1588772
- Koonin, E.V. & Bork, P. (1996). Ancient duplication of DNA polymerase inferred from analysis of complete bacterial genomes. Trends in Biochemical Sciences, 21, 128–129. doi: 10.1016/S0968-0004(96)80165-4
- Lalonde, M.S., Zuo, Y., Zhang, J., Gong, X., Wu, S., Malhotra, A. & Li, Z. (2007). Exoribonuclease R in Mycoplasma genitalium can carry out both RNA processing and degradative functions and is sensitive to RNA ribose methylation. RNA, 13, 1957–1968. doi: 10.1261/rna.706207
- Ling, E., Feldman, G., Portnoi, M., Dagan, R., Overweg, K., Mulholland, F., Chalifa-Caspi, V., Wells, J. & Mizrachi-Nebenzahl, Y. (2004). Glycolytic enzymes associated with the cell surface of Streptococcus pneumoniae are antigenic in humans and elicit protective immune responses in the mouse. Clinical and Experimental Immunology, 138, 290–298. doi: 10.1111/j.1365-2249.2004.02628.x
- Lo, T., van Der Schalie, E., Werner, T., Brun, Y.V. & Din, N. (2004). A temperature-sensitive mutation in the dnaE gene of Caulobacter crescentus that prevents initiation of DNA replication but not ongoing elongation of DNA. Journal of Bacteriology, 186, 1205–1212. doi: 10.1128/JB.186.4.1205-1212.2004
- Lockaby, S.B., Hoerr, F.J., Lauerman, L.H. & Kleven, S.H. (1998). Pathogenicity of Mycoplasma synoviae in broiler chickens. Veterinary Pathology, 35, 178–190. doi: 10.1177/030098589803500303
- Loman, N.J. & Quinian, A.R. (2014). Poretools: A toolkit for analyzing nanopore sequence data. Bioinformatics (Oxford, England), 30, 3399–3401. doi: 10.1093/bioinformatics/btu555
- Lü, Q. & Miller, C. (1995). Silver as a probe of pore-forming residues in a potassium channel. Science, 268, 304–307. doi: 10.1126/science.7716526
- Lucchesi, P., Carraway, M. & Marinus, M.G. (1986). Analysis of forward mutations induced by N-methyl-N'-nitro-N-nitrosoguanidine in the bacteriophage P22 mnt repressor gene. Journal of Bacteriology, 166, 34–37. doi: 10.1128/jb.166.1.34-37.1986
- Markham, J.F., Scott, P.C. & Whithear, K.G. (1998). Field evaluation of the safety and efficacy of a temperature-sensitive Mycoplasma synoviae live vaccine. Avian Diseases, 42, 682–689. doi: 10.2307/1592703
- May, M., Dunne, D.W., Brown, D.R. (2014). A sialoreceptor binding motif in the Mycoplasma synoviae adhesin VlhA. PLoS One, 9, e110360. doi: 10.1371/journal.pone.0110360
- Minion, F.C. & Jarvill-Taylor, K. (1994). Membrane-associated hemolysin activities in mycoplasmas. FEMS Microbiology Letters, 116, 101–106. doi: 10.1111/j.1574-6968.1994.tb06682.x
- Modun, B. & Williams, P. (1999). The staphylococcal transferrin-binding protein is a cell wall glyceraldehyde-3-phosphate dehydrogenase. Infection and Immunity, 67, 1086–1092.
- Moraes, P.M., Seyffert, N., Silva, W.M., Castro, T.L., Silva, R.F., Lima, D.D., Hirata, R., Silva, A., Miyoshi, A. & Azevedo, V. (2014). Characterization of the Opp peptide transporter of Corynebacterium pseudotuberculosis and its role in virulence and pathogenicity. BioMed Research International, 2014, 1–7, Article ID 489782. doi: 10.1155/2014/489782
- Morrow, C.J., (1990). Mycoplasma synoviae (PhD thesis), the University of Melbourne.
- Morrow, C.J., Markham, J.F. & Whithear, K.G. (1998). Production of temperature-sensitive clones of Mycoplasma synoviae for evaluation as live vaccines. Avian Diseases, 42, 667–670. doi: 10.2307/1592700
- Nakamura, T., Tokuda, H. & Unemoto, T. (1984). K+/H+ antiporter functions as a regulator of cytoplasmic pH in a marine bacterium, Vibrio alginolyticus. Biochimica et Biophysica Acta (BBA) - Biomembranes, 776, 330–336. doi: 10.1016/0005-2736(84)90222-0
- Nakamura, T., Yuda, R., Unemoto, T. & Bakker, E.P. (1998). KtrAB, a new type of bacterial K+ uptake system from Vibrio alginolyticus. Journal of Bacteriology, 180, 3491–3494.
- Narat, M., Bencina, D., Kleven, S.H. & Habe, F. (1998). The hemagglutination-positive phenotype of Mycoplasma synoviae induces experimental infectious synovitis in chickens more frequently than does the hemagglutination-negative phenotype. Infection and Immunity, 66, 6004–6009.
- Noormohammadi, A.H., Jones, J.F., Harrigan, K.E. & Whithear, K.G. (2003). Evaluation of the non-temperature-sensitive field clonal isolates of the Mycoplasma synoviae vaccine strain MS-H. Avian Diseases, 47, 355–360. doi: 10.1637/0005-2086(2003)047[0355:EOTNFC]2.0.CO;2
- Noormohammadi, A.H., Markham, J.F., Kanci, A., Whithear, K.G. & Browning, G.F. (2000). A novel mechanism for control of antigenic variation in the haemagglutinin gene family of Mycoplasma synoviae. Molecular Microbiology, 35, 911–923. doi: 10.1046/j.1365-2958.2000.01766.x
- Ohnishi, J., Mizoguchi, H., Takeno, S. & Ikeda, M. (2008). Characterization of mutations induced by N-methyl-N′-nitro-N-nitrosoguanidine in an industrial Corynebacterium glutamicum strain. Mutation Research/Genetic Toxicology and Environmental Mutagenesis, 649, 239–244. doi: 10.1016/j.mrgentox.2007.10.003
- Olson, N.O. (1956). Studies of infectious synovitis in chickens. American Journal of Veterinary Research, 17, 747–754.
- Olson, N.O., Bletner, J.K., Shelton, D.C., Munro, D.A. & Anderson, G.C. (1954). Enlarged joint condition in poultry caused by infectious agent. Poultry Science, 33, 1075.
- Petersen, S.K. & Hansen, F.G. (1991). A missense mutation in the rpoC gene affects chromosomal replication control in Escherichia coli. Journal of Bacteriology, 173, 5200–5206. doi: 10.1128/jb.173.16.5200-5206.1991
- Podbielski, A., Pohl, B., Woischnik, M., Körner, C., Schmidt, K.H., Rozdzinski, E. & Leonard, B.A. (1996). Molecular characterization of group A streptococcal (GAS) oligopeptide permease (Opp) and its effect on cysteine protease production. Molecular Microbiology, 21, 1087–1099. doi: 10.1046/j.1365-2958.1996.661421.x
- Rasmussen, K.V., Atlung, T., Kerszman, G., Hansen, G.E. & Hansen, F.G. (1983). Conditional change of DNA replication control in an RNA polymerase mutant of Escherichia coli. Journal of Bacteriology, 154, 443–451.
- Rice, P., Longden, I. & Bleasby, A. (2000). EMBOSS: The European Molecular Biology Open Software Suite. Trends in Genetics, 16, 276–277. doi: 10.1016/S0168-9525(00)02024-2
- Samen, U., Gottschalk, B., Eikmanns, B.J. & Reinscheid, D.J. (2004). Relevance of peptide uptake systems to the physiology and virulence of Streptococcus agalactiae. Journal of Bacteriology, 186, 1398–1408. doi: 10.1128/JB.186.5.1398-1408.2004
- Sasaki, Y., Ishikawa, J., Yamashita, A., Oshima, K., Kenri, T., Furuya, K., Yoshino, C., Horino, A., Shiba, T., Sasaki, T. & Hattori, M. (2002). The complete genomic sequence of Mycoplasma penetrans, an intracellular bacterial pathogen in humans. Nucleic Acids Research, 30, 5293–5300. doi: 10.1093/nar/gkf667
- Sassetti, C.M. & Rubin, E.J. (2003). Genetic requirements for mycobacterial survival during infection. Proceedings of the National Academy of Sciences of the United States of America, 100, 12989–12994. doi: 10.1073/pnas.2134250100
- Schaumburg, J., Diekmann, O., Hagendorff, P., Bergmann, S., Rohde, M., Hammerschmidt, S., Jänsch, L., Wehland, J. & Kärst, U. (2004). The cell wall subproteome of Listeria monocytogenes. Proteomics, 4, 2991–3006. doi: 10.1002/pmic.200400928
- Shahid, M.A., Markham, P.F., Marenda, M.S., Agnew-Crumpton, R., Noormohammadi, A.H. (2014). High-resolution melting-curve analysis of obg gene to differentiate the temperature-sensitive Mycoplasma synoviae vaccine strain MS-H from non-temperature- sensitive strains. PLoS One, 9, e92215. doi: 10.1371/journal.pone.0092215
- Shahid, M.A., Markham, P.F., Markham, J.F., Marenda, M.S., Noormohammadi, A.H. (2013). Mutations in GTP binding protein Obg of Mycoplasma synoviae vaccine strain MS-H: Implications in temperature-sensitivity phenotype. PLoS One, 8, e73954. doi: 10.1371/journal.pone.0073954
- Terao, Y., Yamaguchi, M., Hamada, S. & Kawabata, S. (2006). Multifunctional glyceraldehyde-3-phosphate dehydrogenase of Streptococcus pyogenes is essential for evasion from neutrophils. Journal of Biological Chemistry, 281, 14215–14223. doi: 10.1074/jbc.M513408200
- Tholema, N., Bakker, E.P., Suzuki, A. & Nakamura, T. (1999). Change to alanine of one out of four selectivity filter glycines in KtrB causes a two orders of magnitude decrease in the affinities for both K+ and Na+ of the Na+ dependent K+ uptake system KtrAB from Vibrio alginolyticus. FEBS Letters, 450, 217–220. doi: 10.1016/S0014-5793(99)00504-9
- Tobe, T., Sasakawa, C., Okada, N., Honma, Y. & Yoshikawa, M. (1992). Vacb, a novel chromosomal gene required for expression of virulence genes on the large plasmid of Shigella flexneri. Journal of Bacteriology, 174, 6359–6367. doi: 10.1128/jb.174.20.6359-6367.1992
- Trent, M.S., Worsham, L.M. & Ernst-Fonberg, M.L. (1999). Hlyc, the internal protein acyltransferase that activates hemolysin toxin: Role of conserved histidine, serine, and cysteine residues in enzymatic activity as probed by chemical modification and site-directed mutagenesis. Biochemistry, 38, 3433–3439. doi: 10.1021/bi982491u
- Tsao, M.Y., Lin, T.L., Hsieh, P.F. & Wang, J.T. (2009). The 3′-to-5′ exoribonuclease (encoded by HP1248) of Helicobacter pylori regulates motility and apoptosis-inducing genes. Journal of Bacteriology, 191, 2691–2702. doi: 10.1128/JB.01182-08
- Tseng, C.W., Chiu, C.J., Kanci, A., Citti, C., Rosengarten, R., Browning, G.F. & Markham, P.F. (2017). The oppD gene and putative peptidase genes may be required for virulence in Mycoplasma gallisepticum. Infection and Immunity, 85, e00023–e00017. doi: 10.1128/IAI.00023-17
- Tunio, S.A., Oldfield, N.J., Ala'Aldeen, D.A., Wooldridge, K.G. & Turner, D.P. (2010). The role of glyceraldehyde 3-phosphate dehydrogenase (GapA-1) in Neisseria meningitidis adherence to human cells. BMC Microbiology, 10, 280. doi: 10.1186/1471-2180-10-280
- Vasconcelos, A.T., Ferreira, H.B., Bizarro, C.V., Bonatto, S.L., Carvalho, M.O., Pinto, P.M., Almeida, D.F., Almeida, R., Alves-Filho, L., Assuncao, E.N., Azevedo, V.A., Bogo, M.R., Brigido, M.M., Brocchi, M., Helio, A.B., Camargo, A.A., Camargo, S.S., Carepo, M.S., Carraro, D.C., de Mattos Cascardo, J.C., Castro, L.A., Cavalcanti, G., Chemale, G., Collevatti, R.G., Cunha, C.W., Dallagiovanna, B., Dambros, B.P., Dellagostin, O.A., Falcão, C., Fantinatti-Garboggini, F., Felipe, M.S.S., Fiorentin, L., Franco, G.R., Freitas, N.S.A., Frías, D., Grangeiro, T.B., Grisard, E.C., Guimarães, C.T., Hungria, M., Jardim, S.N., Krieger, M.A., Laurino, J.P., Lima, L.F.A., Lopes, M.I., Loreto, E.L.S., Madeira, H.M.F., Manfio, G.P., Maranhão, A.Q., Martinkovics, C.T., Medeiros, S.R.B., Moreira, M.A.M., Neiva, M., Ramalho-Neto, C.O., Nicolás, M.F., Oliveira, S.C., Paixão, R.F.C., Pedrosa, F.O., Pena, S.D.J., Pereira, M., PereiraFerrari, L., Piffer, I., Pinto, L.S., Potrich, D.P., Salim, A.C.M., Santos, F.R., Schmitt, R., Schneider, M.P.C., Schrank, A., Schrank, I.S., Schuck, A.F., Seuanez, H.N., Silva, D.W., Silva, R., Silva, S.C., Soares, C.M.A., Souza, K.R.L., Souza, R.C., Staats, C.C., Steffens, M.B.R., Teixeira, S.M.R., Urmenyi, T.P., Vainstein, M.H., Zuccherato, L.W., Simpson, A.J.H. & Zaha, A. (2005). Swine and poultry pathogens: The complete genome sequences of two strains of Mycoplasma hyopneumoniae and a strain of Mycoplasma synoviae. Journal of Bacteriology, 187, 5568–5577. doi: 10.1128/JB.187.16.5568-5577.2005
- Walker, J., Wonacott, A. & Harris, J.I. (1980). Heat stability of a tetrameric enzyme, D-glyceraldehyde-3-phosphate dehydrogenase. FEBS Journal, 108, 581–586.
- Wetzel, A.N., Lefevre, K.M. & Raviv, Z. (2010). Revised Mycoplasma synoviae vlhA PCRs. Avian Diseases, 54, 1292–1297. doi: 10.1637/9349-033010-ResNote.1
- Whithear, K.G. (1993). Avian mycoplasmosis. In T.J. Bagust & L.A. Corner (Eds.), Australian Standard Diagnositc Techniques for Animal Diseases, CSIRO for the Standing Committee on Agriculture and Resource Management (pp. 1–12). East Melbourne: CSIRO Publications.
- Zerbino, D.R. & Birney, E. (2008). Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Research, 18, 821–829. doi: 10.1101/gr.074492.107
- Zhu, L., Shahid, M.A., Markham, J., Browning, G.F., Noormohammadi, A.H. & Marenda, M.S. (2018). Genome analysis of Mycoplasma synoviae strain MS-H, the most common M. synoviae strain with a worldwide distribution. BMC Genomics, 19, 117. doi: 10.1186/s12864-018-4501-8
- Zhu, L., Konsak, B.M., Olaogun, O.M., Agnew-Crumptona, R., Kanci, A., Marenda, M.S., Browning, G.F. & Noormohammadi, A.H. (2017). Identification of a new genetic marker in Mycoplasma synoviae vaccine strain MS-H and development of a strategy using polymerase chain reaction and high-resolution melting curve analysis for differentiating MS-H from field strains. Veterinary Microbiology, 210, 49–55. doi: 10.1016/j.vetmic.2017.08.021