
ABSTRACT
Latency is an important feature of infectious laryngotracheitis virus (ILTV) yet is poorly understood. This study aimed to compare latency characteristics of vaccine (SA2) and field (CL9) strains of ILTV, establish an in vitro reactivation system and examine ILTV infection in peripheral blood mononuclear cells (PBMC) in specific pathogen-free chickens. Birds were inoculated with SA2 or CL9 ILTV and then bled and culled at 21 or 35 days post-inoculation (dpi). Swabs (conjunctiva, palatine cleft, trachea) and trigeminal ganglia (TG) were examined for ILTV DNA using PCR. Half of the TG, trachea and PBMC were co-cultivated with cell monolayers to assess in vitro reactivation of ILTV infection. ILTV DNA was detected in the trachea of approximately 50% of ILTV‐inoculated birds at both timepoints. At 21 dpi, ILTV was detected in the TG only in 29% and 17% of CL9- and SA2-infected birds, respectively. At 35 dpi, ILTV was detected in the TG only in 30% and 10% of CL9- and SA2-infected birds, respectively. Tracheal organ co-cultures from 30% and 70% of CL9- and SA2-infected birds, respectively, were negative for ILTV DNA at cull but yielded quantifiable DNA within 6 days post-explant (dpe). TG co-cultivation from 30% and 40% of CL9-and SA2-infected birds, respectively, had detectable ILTV DNA within 6 dpe. Latency characteristics did not substantially vary based on the strain of virus inoculated or between sampling timepoints. These results advance our understanding of ILTV latency and reactivation.
RESEARCH HIGHLIGHTS
Following inoculation, latent ILTV infection was detected in a large proportion of chickens, irrespective of whether a field or vaccine strain was inoculated.
In vitro reactivation of latent ILTV was readily detected in tracheal and trigeminal ganglia co-cultures using PCR.
ILTV latency observed in SPF chickens at 21 days post-infection was not substantially different to 35 days post-infection.
Introduction
Herpesviruses are among the most successful pathogens affecting both humans and animals (Szpara et al., Citation2014; Sehrawat et al., Citation2018). This success is largely due to their capacity to establish latent infections following primary infection (Nicoll et al., Citation2012). Based on host range, genetic organization and replication strategies, the Herpesviridae family of viruses is divided into three subfamilies: Alpha-, Beta-, and Gamma herpesvirinae (Sehrawat et al., Citation2018). The vast majority of known human and animal pathogens belong to the Alphaherpesvirinae subfamily and they typically establish latency in sensory ganglia (Szpara et al., Citation2010).
Gallid alphaherpesvirus 1 or infectious laryngotracheitis virus (ILTV) is an avian alphaherpesvirus that causes infectious laryngotracheitis (ILT), an acute respiratory tract infection of chickens, which may cause mild to severe disease (Kirkpatrick, Mahmoudian, Colson, et al., Citation2006). Mortality in infected flocks ranges between 5% and 70% depending on the virulence of the strain involved (Saif et al., Citation2011). Similar to other herpesviruses, ILTV is capable of establishing latency. The principal sites of ILTV latent infection are the trigeminal ganglia (TG) (Williams et al., Citation1992) and the trachea (Bagust, Citation1986). ILTV DNA has been detected in TG during latent infection by several researchers (Hughes et al., Citation1991; Williams et al., Citation1992; Han & Kim, Citation2003; Chacón et al., Citation2015; Parra et al., Citation2015). Although reactivation of latent ILTV from trachea has been demonstrated using a culture-based approach (Bagust, Citation1986) the location of the latent ILTV in the trachea has not been determined. Stressors leading to immune suppression are known to reactivate latent ILTV infections (Hughes et al., Citation1989), and even attenuated vaccine strains are capable of establishing latent infections and reactivating later in life (Hughes et al., Citation1991).
Attenuated strains of ILTV are widely used as vaccines and they play an important role in protecting poultry flocks against ILT (García et al., Citation2013). SA2 was the first attenuated ILTV vaccine developed in Australia by serial passage of a low pathogenic ILTV field isolate in chicken embryos (Kirkpatrick, Mahmoudian, Colson, et al., Citation2006; Lee et al., Citation2011). Since then, it has been widely used to control ILT in Australia; however, the high level of residual virulence of this vaccine has made it unsuitable for use in young chickens (Kirkpatrick, Mahmoudian, Colson, et al., Citation2006; Lee et al., Citation2011). The contemporaneous use of Australian-origin vaccine strains (SA2 and A20) and the European-origin Serva vaccine strain in commercial chickens in Australia led to the emergence of natural recombinant strains that caused widespread outbreaks of disease (Lee et al., Citation2012). One of these recombinants, belonging to genotype class 9, according to the Australian genotyping system, has replaced the previously predominant field ILTV strains in many Australian poultry producing areas (Lee et al., Citation2015) and it is currently the most prevalent ILTV field strain in the Australian state of Victoria (Agnew-Crumpton et al., Citation2016). However, the latency characteristics of this field strain have not been investigated.
The capacity to establish latent infections, with subsequent reactivation later in life, is a known limitation associated with attenuated ILTV vaccine strains (Bagust, Citation1986; Hughes et al., Citation1991; Han & Kim, Citation2003) which may facilitate recombination events. Latency and reactivation of SA2 have been previously investigated in vivo as well as in vitro by performing TG or tracheal explant cultures (Bagust, Citation1986); however, these culture techniques have not been replicated in recent times. The other member of the Alphaherpesvirinae that infects chickens, Gallid alphaherpesvirus 2 (Marek’s disease virus (MDV)) establishes latency in peripheral blood mononuclear cells (PBMC) (Parcells et al., Citation2003). Similarly, equine alphaherpesvirus 1 (another alphaherpesvirus) infects leukocytes, which allow for systemic viral dissemination (Poelaert et al., Citation2019) and also serve as a site of latency (Chesters et al., Citation1997). However, the capacity of ILTV to infect and establish latency in PBMC is unknown. It has also been suggested that PBMC, in particular macrophages, could potentially play a role in the systemic dissemination of ILTV during acute infection (Calnek et al., Citation1986; Rodríguez-Avila et al., Citation2007).
The current study aimed to establish a latency infection model and to better understand the latency and reactivation characteristics of SA2 and CL9 ILTV in the natural host. The SA2 vaccine strain and the CL9 field strain of ILTV were used in this study to allow a comparison between a vaccine strain and a virulent field strain. Understanding the latency characteristics of CL9 is important as this is a prevalent recombinant strain of ILTV that has shown improved fitness and a high level of virulence (Lee et al., Citation2015; Agnew-Crumpton et al., Citation2016).
Materials and methods
ILTV strains
The SA2 and CL9 ILTV strains were used in this study. Poulvac® Laryngo SA2 (Zoetis, Rhodes, Australia) was obtained from the manufacturer and reconstituted according to the manufacturer’s instructions. CL9 ILTV was propagated in the Leghorn male hepatoma (LMH) cells (Kawaguchi et al., Citation1987) and growth medium consisting of Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 10% v/v foetal bovine serum (FBS), 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, pH 7.7) and 50 µg/ml ampicillin. Both strains were titrated using plaque assays on LMH cells as previously described (Devlin et al., Citation2006) before eye-drop or intra‐tracheal inoculation.
In vivo ILTV inoculation study
The in vivo study was conducted with the approval from the Animal Ethics Committee, Faculty of Veterinary and Agricultural Sciences, The University of Melbourne (ethics identification number 1714129.1). One hundred one-day-old White Leghorn type specific-pathogen-free (SPF) chickens were randomly divided into three groups (40 birds in groups 1 and 2, and 20 birds in group 3) and wing-tagged for identification. The three groups were separately housed in negatively pressured Horsfall-Bauer-type isolator units at the Asia-Pacific Centre for Animal Health animal facility, and provided with irradiated feed and sterilized water ad libitum.
Medium consisting of DMEM supplemented with 10% v/v FBS was used to dilute the ILTV strains to reach the desired viral titre. At 3 weeks of age, birds in group 1 were inoculated with SA2 ILTV strain at a dose of 103 plaque forming units (PFU) per bird. Similarly, birds in group 2 were inoculated with CL9 ILTV at a dose of 103 PFU/bird. All birds in group 3 were mock-inoculated with sterile medium (DMEM supplemented with 10% v/v FBS). Half of the dose was inoculated via eye-drop in 40 µl to each eye and the other half (80 µl) was inoculated directly into the trachea in each inoculated birds.
Birds were monitored for clinical disease after inoculation. Any birds showing signs of severe disease were euthanased to prevent suffering. At 21 days post-inoculation (dpi), six birds from group 1, seven birds from group 2 and 10 birds from group 3 were randomly selected, bled from the brachial vein and culled by exposure to halothane. At 35 dpi all remaining birds were bled and euthanased in a similar manner.
Sample collection
Approximately 1 ml of blood was collected from each bird into heparinized BD Vacutainer tubes (BD Biosciences, Franklin Lakes, NJ) and stored on ice until processing for flow cytometry. At post mortem examination, swab samples were collected from the conjunctiva of both eyes, the palatine cleft, and the trachea of each bird. All the swabs collected were placed in 1 ml viral transport medium (VTM; DMEM supplemented with 3% v/v FBS, 0.02 M HEPES (pH 7.7) and 0.25 mg/ml of both gentamicin and ampicillin) and stored at −80°C until DNA extraction.
Tracheas and TG, for co-culture with cultured cells, were collected only at 35 dpi from 25 randomly selected birds (n = 10 from each of groups 1 and 2, and n = 5 from group 3). The trachea and larynx were aseptically excised from the neck of each bird and placed in 50 ml centrifuge tubes (Falcon BD, Franklin Lakes, NJ) containing 20 ml of VTM and stored on ice until further processing. Each bird was decapitated, and TG were aseptically removed. A section of brain tissue (from the olfactory lobe) immediately next to TG was collected from each bird to be used as a dissection contamination control of the TG tissue. No ILTV DNA was expected in the control brain tissue and its presence would indicate potential contamination with viral DNA during dissection. TG for co-culture were stored on ice until further processing. The TG collected from the remaining birds were snap-frozen in dry ice and stored at –80°C until processing for molecular detection of ILTV DNA.
Peripheral blood mononuclear cell isolation
Whole blood from each bird was processed separately. One ml of whole blood was overlaid onto 1 ml of Ficoll-Paque PLUS (GE Healthcare, Chicago, IL) at room temperature. This was centrifuged (Allegra X-12R; Beckman, Radnor, PA) at 400 × g for 30 min at 18°C without a brake. Using a sterile transfer pipette, the white interphase containing mononuclear cells was aspirated and resuspended in 10 ml of chilled PBMC buffer (1 mM EDTA, 25 mM HEPES pH 7.7, 1 % v/v FBS in phosphate-buffered saline (PBS)) before centrifugation at 800 × g for 5 min at 4°C. The resulting cell pellet was resuspended in PBMC buffer and centrifuged as above. The cell pellet was then gently resuspended in 200 μl PBMC buffer. Visual inspection of PBMC was done by preparing a Diff‐Quik™ (Microptic S.L., Barcelona, Spain) stained smear. Viable cells were counted using Trypan blue exclusion (0.4 % w/v Trypan blue; Sigma-Aldrich, St. Louis, MO) in a haemocytometer (Brightline; Hausser Scientific, Horsham, PA).
Co-culture of PBMC, TG or trachea with LMH cells
Co-culturing with LMH cell monolayers was performed in 12-well tissue culture trays. The growth medium of the pre-established LMH cell monolayers was replaced with 1 ml of a maintenance medium (MM; DMEM supplemented with 5% v/v FBS, 2 μM L-glutamine, 2.5 μg/ml amphotericin B and 0.2 mg/ml of both ampicillin and gentamicin). PBMC, TG and tracheas from each bird were individually processed within a class II biological safety cabinet.
When co-culturing PBMC, 100 μl (approximately 2 × 106 cells) of the isolated PBMC suspension from each bird was added to a single well in a tissue culture tray.
During TG co-culture, TG from each bird were transferred aseptically into a sterile Petri dish and were minced using a sterile scalpel blade. TG pieces from each bird were then transferred using a 200 µl pipette tip into a single tissue culture well containing an LMH cell monolayer.
Tracheal organ co-culture (TOC) was followed by transferring each trachea to a plastic Petri dish and removing the adipose and connective tissue surrounding the trachea. The trachea was then divided into three equal size segments: upper, middle and lower trachea. Four tracheal ring slices (2–3 mm thick) were aseptically transected from each segment to be used for tracheal organ co-culture with LMH cells in 12 well tissue culture trays. The rings from each segment were cut from similar regions of each trachea; just below the larynx from the upper tracheal segment, the centre of the middle tracheal segment, and at the proximal end of the lower tracheal segment. Separate wells were used to culture the tracheal rings from each segment.
Maintenance and sample collection from cultures
Tissue culture trays with PBMC, TG or tracheal co-cultures were incubated at 37°C in a humidified atmosphere of 5% v/v CO2 in air for up to 12 days. From all cultures, 500 μl of culture supernatants were collected at 3-day intervals and stored at −80°C and a similar volume was replaced with fresh MM. In consideration of the integrity and viability of the LMH cell monolayers, TG and tracheal tissues were transferred to a new sub-confluent monolayer after 6 days of co-culture. When transferring to a fresh LMH cell monolayer, a 200 µl pipette tip or small forceps were used to pick up the pieces of TG or the tracheal rings, respectively. After transferring the tissues, all medium and the old LMH cell monolayer in each well were collected. Culture supernatants collected during co-culture were stored at –80°C until further use. These supernatants were used for viral DNA detection by PCR and/or detection of viable viruses infecting a secondary monolayer.
DNA extraction and polymerase chain reactions
DNA was extracted from the VTM from swabs, co-culture supernatants, snap-frozen TG and the control brain tissue samples. DNA extractions were performed using 200 μl of swab suspension or culture supernatants, the whole TG or 0.5 cm3 of the brain tissue as the starting material. MagMAX™ CORE Nucleic Acid Purification Kit (Applied Biosystems™, Waltham, MA) together with the KingFisher™ Flex Purification System (Thermo Scientific™, Waltham, MA) were used for automated DNA extraction according to the manufacturers’ instructions. Extracted DNA was eluted in 90 μl of elution buffer and subjected to the UL15 gene targeted quantitative PCR (UL15qPCR) and/or the UL15 gene targeted nested PCR (UL15NPCR). The approach was to quantify the viral genome copy numbers (GCN) when possible, or, alternatively, to qualitatively detect ILTV DNA using the UL15NPCR. Trigeminal ganglia, control brain tissue, PBMC co-culture supernatants and TG culture supernatants were subjected to UL15NPCR because low viral genome copy numbers (<100) were expected from these samples and the UL15NPCR is more sensitive than UL15qPCR (Thilakarathne et al., Citation2019). Viral transport medium from swabs and tracheal co-culture supernatants was subjected to UL15qPCR as quantifiable numbers of viral copies were expected.
UL15 gene targeted quantitative PCR (UL15qPCR)
Primers for this qPCR have been previously described (Mahmoudian et al., Citation2011). The modified protocol used in this study included the use of GoTaq® system (Promega, Madison, WI, USA) with the conditions previously described (Thilakarathne et al., Citation2019).
UL15 gene targeted nested PCR (UL15NPCR)
This nested PCR was developed to overcome the limitations associated with the widely used universal herpesvirus nested PCR (VanDevanter et al., Citation1996) including a lower sensitivity, low specificity and prolonged cycling time. Extracted DNA was subjected to UL15NPCR using the primers and conditions described at previously (Thilakarathne et al., Citation2019).
Testing culture supernatant for viable viruses
Pre-established LMH cell monolayers with ∼70% confluency were used. Growth medium (GM-DMEM supplemented with 10% v/v FBS, 10 mM HEPES (pH 7.5) and 50 µg/ml ampicillin) on LMH cell monolayers was removed and the monolayers were washed twice with PBS. Two hundred μl of each UL15qPCR-positive culture supernatant were added to a single well on 12-well tissue culture trays. The culture plate was gently swirled to cover the monolayer with the culture supernatant.
The plates were incubated for 1 h in a humidified atmosphere of 5% v/v CO2 in air at 37°C to allow viral adsorption, and this was further facilitated by gentle swirling of the plates/flasks at 15-min intervals. After 1 h incubation, GM was added and re-incubated in same conditions for up to 72 h. The capacity of reactivated viruses to infect cells was assessed by observing cytopathic effects in LMH cell monolayers.
Data analysis
Microsoft® Excel (Version 16.16.2) was used for the log transformation of GCN and Prism 5 for Windows version 5.03 (GraphPad Software, Inc., San Diego, CA) was used to generate figures. Statistical analyses were performed using Minitab (Version 17) statistical software (Minitab Pty Ltd, Sydney, Australia). Fisher’s exact test was used to compare the proportions of ILTV DNA-positive birds between groups. One-way analysis of variance (ANOVA), in conjunction with Tukey’s multiple comparison test, was used to compare mean GCN between groups. During viral quantification, samples that returned GCN values at or below the cut-off limit of the UL15qPCR (100 GCN) were considered negative. All qPCR-negative samples were assigned the cut-off value for subsequent statistical analyses. In each statistical test a P-value ≤0.05 was considered statistically significant.
Results
Mortalities
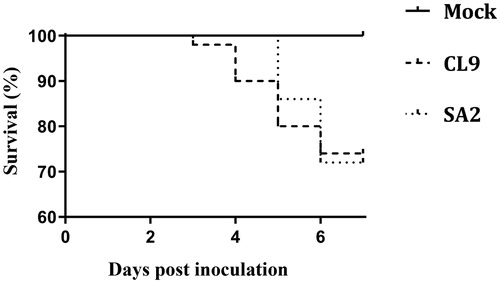
Survival percentage in each group is shown in . Mortalities (sudden deaths or birds euthanased due to severe clinical disease) occurred up to 6 dpi in both CL9, and SA2-infected groups. The cumulative mortality percentages until 7 dpi were 32.5% and 35% in CL9- and SA2‐infected groups, respectively. These mortality rates were not significantly different between infected groups. Post mortem examinations revealed severe haemorrhagic tracheitis in birds that died due to CL9 infection, whereas those that died due to infection with SA2 had diphtheritic plaques and muco-caseous plugs on the upper tracheal mucosal surface.
Figure 1. Survival rates in specific pathogen-free birds up to 7 days post-inoculation with SA2 or CL9 ILTV, or those birds that remained uninoculated (Mock). There were no statistically significant differences between infected groups.
Viral genome copy numbers in conjunctival, palatine cleft or tracheal swabs at the time of culling
The percentage of UL15qPCR-positive swabs observed with each group and their mean log10 viral genome copy numbers at 21 and 35 dpi are shown in Supplementary Table 1. All the conjunctival and palatine cleft swabs collected from SPF chickens at 21 dpi with CL9 or SA2 ILTV were negative. qPCR-positive tracheal swab samples were detected in 43% and 67% of the birds inoculated with either CL9 or SA2 ILTV, respectively, at this same timepoint. Neither the percentage of UL15qPCR-positive tracheal swab samples nor the mean log10 GCN were significantly different between infected groups at this timepoint. Only birds inoculated with SA2 ILTV had U15qPCR-positive percentage and log10 GCN significantly higher than those of the mock-inoculated group.
At 35 dpi, all the conjunctival swabs and most of the palatine cleft swabs (96%) collected from both of the ILTV-inoculated groups were UL15qPCR-negative. At this timepoint, 50% and 40% of the tracheal swabs collected from chickens inoculated with CL9 or SA2, respectively, were UL15qPCR-positive. These percentages of UL15qPCR‐positive tracheal swab samples in both groups of birds were significantly higher than that of the mock-inoculated group of birds, which had no positive results. The log10 GCN detected in ILTV-inoculated groups were not significantly different to each other or the mock-inoculated birds.
No significant differences in the percentage of UL15qPCR-positive tracheal swab samples were observed between 21 and 35 dpi within each inoculation group.
Molecular detection of ILTV DNA in TG
shows the individual UL15NPCR data for each TG and control brain tissue samples that were immediately snap-frozen after collection at 21 or 35 dpi. The percentages of UL15NPCR-positive TG collected from SPF chickens at 21 dpi with CL9 or SA2 ILTV were 29% and 83%, respectively. At 35 dpi with CL9 or SA2 ILTV, the percentages of UL15NPCR-positive TG were 70% and 50%, respectively. No statistically significant differences were observed between inoculation groups at each sampling timepoint or within each inoculation group between timepoints. None of the birds with UL15NPCR-positive TG had detectable ILTV DNA in the corresponding control brain tissue (). Interestingly, 37% of the ILTV-inoculated birds that had detectable ILTV DNA in their TG did not have quantifiable ILTV DNA in their palatine cleft and tracheal swabs. In contrast, 18% of ILTV-inoculated birds that had quantifiable ILTV DNA in their palatine cleft and tracheal swabs had no detectable ILTV DNA in TG tested using the UL15NPCR ().
Table 1. UL15qPCR results for swab samples and UL15 nested PCR for control brain tissue and trigeminal ganglia collected from SPF chickens at 21 or 35 days post-inoculation with CL9 or SA2 ILTV via the intra-tracheal route.
Detection of viral genomes from TG co-culture supernatants
Supernatants collected from the TG co-culture of 30% and 40% of the birds inoculated with CL9 or SA2 ILTV, respectively, were UL15NPCR-positive (). These proportions were not significantly different. The majority (71%) of the UL15NPCR-positive samples were first detected at 6 days post-explant (dpe). Additionally, 71% of the ILTV DNA-positive TG co-culture supernatants were from birds that had no quantifiable viral DNA in palatine cleft and tracheal swabs at the time of culling at 35 dpi (birds 135, 145, 177, 180 and 189; ). In addition, 25% of the ILTV-inoculated birds that had quantifiable ILTV DNA in their palatine cleft and tracheal swab samples at the time of euthanasia had no UL15NPCR-positive results in their culture supernatant samples at any of the sampling timepoints post-explant (birds 136, 138, 140, 182 and 193; ).
Table 2. UL15qPCR results for swab samples and UL15 nested PCR results for control brain tissue collected during post-mortem examination at 35 days post-inoculation with SA2 or CL9 ILTV, or mock inoculated with sterile medium (mock), and UL15 nested PCR results for culture supernatant collected during trigeminal ganglia co-cultivation with LMH cell monolayers at the indicated time points post-explant.
Detection of viral genomes from tracheal organ co-culture supernatants
Upper and/or middle tracheal co-culture supernatants from 50% of the birds inoculated with CL9 ILTV and all 10 of those inoculated with SA2 were UL15qPCR-positive at one or both sampling timepoints post explant (). The majority (93%) of the UL15qPCR-positive co-culture supernatants were first detected positive at 3 dpe and most (92.8%) of those co-cultures remained positive at 6 dpe. Sixty per cent and 70% of those inoculated with CL9 or SA2 ILTV, respectively, which yielded UL15qPCR-positive co‐culture supernatants at one or both sampling timepoints post explant, had UL15qPCR-negative tracheal swabs at the time of euthanasia at 35 dpi (highlighted in ).
Table 3. UL15qPCR results of tracheal swab samples collected from SPF chickens at 35 dpi with CL9 or SA2 ILTV, or mock inoculated with sterile medium, and UL15qPCR results of tracheal organ co-culture supernatant samples collected at days 3 and 6 post-explant.
Supernatant samples collected at 3 dpe from upper tracheal co-cultures prepared from those inoculated with SA2 ILTV which produced PCR-detectable ILTV DNA, had significantly higher (P = 0.005) mean log10 GCN (3.22) compared to those observed in supernatant samples from similar co-cultures obtained from birds inoculated with CL9 ILTV (2.42) or the mock-inoculated birds (2.00). Similarly, at 6 dpe, ILTV PCR-positive supernatant samples collected from upper tracheal co-cultures obtained from birds inoculated with SA2 ILTV had significantly higher (P = 0.006) mean log10 GCN (2.95) compared to those observed in supernatant samples from similar co-cultures obtained from birds inoculated with CL9 ILTV (2.09) or the mock-inoculated group (2.00). In both ILTV-inoculated groups of chickens, the mean log10 GCN observed in upper tracheal co-culture supernatants at 3 dpe were slightly higher than at 6 dpe, but the difference was not significant. None of the lower tracheal co-culture supernatant samples from ILTV-inoculated chickens were UL15qPCR-positive at any sampling timepoint. Similarly, none of the supernatant samples collected from tracheal co‐cultures prepared from the mock-inoculated group of chickens were UL15qPCR-positive at any timepoint.
Detection of cytopathic effects in LMH cells inoculated with supernatants collected from TG or tracheal co-cultures
Cytopathic effects on the LMH cell monolayer were not observed when UL15NPCR-positive TG co‐culture supernatants or UL15qPCR-positive tracheal co-culture supernatants were sub-cultured on secondary LMH cell monolayers.
Of the birds that had no qPCR detectable viruses in their conjunctival, palatine cleft and tracheal swab samples by 35 dpi, only one bird had ILTV DNA detected after co-culture from TG only (bird 135), whereas six birds had ILTV DNA detected after co-culture of trachea only (birds 142, 153, 175, 176, 185, and 186) and an additional four birds had ILTV DNA positive results from both sites after co-culture (birds 145, 177, 180, 189) ( and ).
None of the PBMC co-culture supernatants were positive for UL15NPCR at any of the sampling points tested.
Discussion
The experiment described here developed a TG co-culture system and reproduced a TOC system to reactivate latent ILTV in vitro. Further, it compared the characteristics of latency established by a virulent field ILTV strain (CL9) and a moderately attenuated ILTV vaccine strain (SA2).
The dose of the inoculum was chosen based on previous ILTV infection studies (Kirkpatrick, Mahmoudian, Colson, et al., Citation2006; Lee et al., Citation2015). Under these conditions we hypothesized that less severe disease and lower mortality would be observed in SA2-inoculated chickens compared to those inoculated with CL9. However, no significant differences were observed in mortality rates between birds inoculated with CL9 (32.5%) or SA2 (35%). Inoculating the virus at two primary replication sites (both conjunctiva and trachea) may have resulted in more severe clinical disease in those inoculated with the SA2 vaccine strain, consistent with previous findings (Kirkpatrick, Mahmoudian, O’Rourke, et al., Citation2006). Alternatively, host factors, including genetic background, the weight and size of the eggs or chicks, or incubation conditions may have also had an influence in the severity of clinical presentation. The relationship between host genetics and susceptibility to ILTV infection is poorly understood (Coppo et al., Citation2013). It is possible that this relationship is similar to that observed during infection with MDV. Previous studies have shown that the genetic background of the chickens can affect their susceptibility to MDV infection and disease (Hepkema et al., Citation1993; Kreager, Citation1998; Yonash et al., Citation1999; Emara et al., Citation2001).
Approximately 50% of the inoculated birds had quantifiable viruses in trachea at both 21 or 35 dpi. According to the literature, acute ILTV infection in trachea subsides by 14 dpi (Rodríguez-Avila et al., Citation2007). Therefore, quantifiable ILTV DNA observed after 21 dpi could be explained by the presence of residual ILTV DNA after cessation of primary replication, or horizontal transmission of infection from another bird, or from reactivation of latent ILTV infection. The observation of a similar pattern of detection at 35 dpi favours the latter alternative, as it seems unlikely that residual DNA would not be cleared by the host immune system this long after inoculation. Previous studies with SA2 have shown a similar pattern of reactivation at approximately 6 weeks post-inoculation (Coppo et al., Citation2012). Further support for this hypothesis is the relatively high levels of ILTV DNA detected in the tracheal swabs collected from SA2-infected birds at 35 dpi. This long-term detection of ILTV in these groups of birds suggests that latency may be established in a relatively high proportion of chickens. Consistent with findings in commercial layers (Thilakarathne et al., Citation2019), at any one time, long after infection, approximately half of the population has reactivated vaccine virus in their trachea without any clinical signs of infection. This is likely to have a significant impact on the control of ILTV in commercial poultry, and the emergence of recombinant ILTV.
After three decades from the initial report (Bagust, Citation1986), the current study was able to reproduce the results obtained by Bagust using TOC, and demonstrated the presence of ILTV DNA mainly in the upper segments of the trachea of infected birds after explant and co-culture. In this study, tracheal swabs were used to quantify viral GCN in the tracheal lumen or on the tracheal epithelium. Two thirds of the birds did not have detectable ILTV DNA in their trachea, as assessed by qPCR, at the time of euthanasia. These birds may potentially represent latently infected birds. Therefore, it is possible that these tracheal segments released ILTV from sites of latency in tracheal tissues. The exact histologic location of latency sites in the trachea has not been determined, but may include neuronal cell body aggregations (tracheal peripheral ganglia), tracheal epithelial cells or both. The glossopharyngeal and vagus cranial nerves innervate the pharynx, larynx and cervical trachea. The proximal and distal ganglia of the glossopharyngeal, and vagus cranial nerves have been identified as sites of cell bodies of the neurons that innervate the trachea (Williams et al., Citation1992). Even though alphaherpesviruses typically establish latency in sensory ganglia (Szpara et al., Citation2010) there have been records of their capacity to establish latency in non-neuronal sites, for example corneal latency observed in HSV-1 (Fukuda et al., Citation2003; Polcicova et al., Citation2005; Kaye & Choudhary, Citation2006). Since qPCR-positive tracheal co-culture supernatants did not induce any cytopathic effects on secondary LMH cell monolayers, it is hard to interpret whether the detection of increasing genome copy numbers in subsequently collected supernatants indicates a persistent non-productive type of infection or latent infection. Similarly, further investigations are required to better understand the role of monocytes/macrophages in the biology of ILTV. It is currently unknown if these leukocyte populations are susceptible to ILTV infection in vivo. If they are, results from the current study indicate that PBMC are not actively engaged in release of ILTV DNA during latent stages of infection under experimental in vitro culture conditions.
Consistent with Bagust (Citation1986) and our previous ILTV vaccine latency study (Thilakarathne et al., Citation2019), the current study was unable to detect infectious ILTV virions in TG co-culture supernatants. This is possibly associated with a low number of neurons being infected or being capable of in vitro reactivation. Previous studies in mice with HSV-1 have determined that only a fraction of latently infected neurons will undergo reactivation under in vitro co‐cultivation conditions (Bloom, Citation2016).
In the current study, a limited number of TG co-culture supernatants had detectable ILTV DNA as assessed by the UL15NPCR. It is likely that these UL15NPCR-positives represent birds with latent ILTV in TG tissue because the majority (71%) of them did not have quantifiable ILTV DNA in their trachea at the time of euthanasia. The observation that these UL15NPCR-positive TG co-culture supernatants did not produce cytopathic effects when cultured on secondary LMH cell monolayers, raises the possibility that host cells were not releasing infectious virus, or the culture conditions were not appropriate for virus propagation. Further studies to optimize experimental culture conditions to favour in vitro reactivation are warranted.
Previous reports by Williams et al. (Citation1992), indicating that TG is the main site of latency for ILTV, used molecular methods of detection, rather than viral cultivation. The absence of ILTV DNA in the control brain tissue of individual birds used in the current study helped to validate the detection of ILTV DNA in the TG from the same bird, providing evidence that ILTV DNA detected in TG is unlikely to be the result of contamination from the conjunctival, palatine cleft or tracheal mucosae during post mortem dissection and examination. Viral DNA detection by UL15NPCR in TG revealed no significant differences between the proportion of positives in inoculated groups at any sampling timepoint (21 or 35 dpi) or over the whole time period. Thus, this study provides evidence that latency observed in TG after 21 dpi is not substantially different from what is observed at 35 dpi under experimental conditions. Further, these results show that latent infections can be readily identified by detecting ILTV DNA in sites of latency using molecular methods, which is consistent with previous findings by Williams et al. (Citation1992).
A proportion of birds that had no quantifiable ILTV DNA in their conjunctival, palatine cleft or tracheal swab samples at the time of euthanasia, had detectable ILTV DNA in the co-culture supernatant of either one or both sites of latency (i.e. trachea and TG). These data suggest that both the trachea and the TG may be sites of latency and reactivation of ILTV. Whether latency and reactivation characteristics at each of these sites are similar remains to be elucidated.
The pathology observed in the trachea varied with the inoculated virus, with diphtheritic plaques and plugs observed in SA2-infected birds and haemorrhagic tracheitis observed in CL9-infected birds. These differences could be associated with the higher levels of virus amplification in TOC from SA2-infected birds compared to CL9-infected birds, with sloughing of the tracheal epithelium to expose the blood vessels in the CL9-infected birds (Thilakarathne et al., Citation2020) resulting in less epithelium for virus replication post-explant. In contrast, the diphtheritic pathology associated with SA2 infection (Purcell & Surman, Citation1974) may facilitate retention and replication of virus in the comparatively intact epithelium.
The advantages of molecular methods and the limitations of in vitro culture techniques to detect latent infections identified in the current study provide useful insights that can help in the design of future experimental studies aiming to investigate ILTV latency in chickens.
Supplemental Material
Download MS Word (55.5 KB)Acknowledgements
We acknowledge Paola K. Vaz, Adepeju Esther Onasanya, Pollob Shil, June Daly and Jenece Wheeler for their help in conducting the in vivo experiment.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Agnew-Crumpton, R., Vaz, P.K., Devlin, J.M., O’Rourke, D., Blacker-Smith, H.P., Konsak-Ilievski, B., Hartley, C.A. & Noormohammadi, A.H. (2016). Spread of the newly emerging infectious laryngotracheitis viruses in Australia. Infection, Genetics and Evolution, 43, 67–73. doi: 10.1016/j.meegid.2016.05.023
- Bagust, T. (1986). Laryngotracheitis (gallid-1) herpesvirus infection in the chicken. 4. Latency establishment by wild and vaccine strains of ILT virus. Avian Pathology, 15, 581–595. doi: 10.1080/03079458608436317
- Bloom, D.C. (2016). Alphaherpesvirus latency: a dynamic state of transcription and reactivation. Advances in Virus Research, 94, 53–80. doi: 10.1016/bs.aivir.2015.10.001
- Calnek, B., Fahey, K. & Bagust, T. (1986). In vitro infection studies with infectious laryngotracheitis virus. Avian Diseases, 30(2), 327–336. doi: 10.2307/1590536
- Chacón, J.L., Núñez, L.F.N., Vejarano, M.P., Parra, S.H.S., Astolfi-Ferreira, C.S. & Ferreira, A.J.P. (2015). Persistence and spreading of field and vaccine strains of infectious laryngotracheitis virus (ILTV) in vaccinated and unvaccinated geographic regions, in Brazil. Tropical Animal Health and Production, 47, 1101–1108. doi: 10.1007/s11250-015-0834-3
- Chesters, P., Allsop, R., Purewal, A. & Edington, N. (1997). Detection of latency-associated transcripts of equid herpesvirus 1 in equine leukocytes but not in trigeminal ganglia. Journal of Virology, 71, 3437–3443. doi: 10.1128/JVI.71.5.3437-3443.1997
- Coppo, M.J., Devlin, J.M. & Noormohammadi, A.H. (2012). Comparison of the replication and transmissibility of an infectious laryngotracheitis virus vaccine delivered via eye-drop or drinking-water. Avian Pathology, 41, 99–106. doi: 10.1080/03079457.2011.643222
- Coppo, M.J., Hartley, C.A. & Devlin, J.M. (2013). Immune responses to infectious laryngotracheitis virus. Developmental & Comparative Immunology, 41, 454–462. doi: 10.1016/j.dci.2013.03.022
- Devlin, J., Browning, G. & Gilkerson, J. (2006). A glycoprotein I- and glycoprotein E-deficient mutant of infectious laryngotracheitis virus exhibits impaired cell-to-cell spread in cultured cells. Archives of Virology, 7, 1281–1289. doi: 10.1007/s00705-005-0721-8
- Emara, M.G., Abdellatif, M.A., Pollock, D.L., Sadjadi, M., Cloud, S.S., Pope, C.R., Rosenberger, J.K. & Kim, H. (2001). Genetic variation in susceptibility to Marek's disease in a commercial broiler population. Avian Diseases, 45(2), 400–409. doi: 10.2307/1592979
- Fukuda, M., Deai, T., Hibino, T., Higaki, S., Hayashi, K. & Shimomura, Y. (2003). Quantitative analysis of herpes simplex virus genome in tears from patients with herpetic keratitis. Cornea, 22, S55–S60. doi: 10.1097/00003226-200310001-00008
- García, M., Volkening, J., Riblet, S. & Spatz, S. (2013). Genomic sequence analysis of the United States infectious laryngotracheitis vaccine strains chicken embryo origin (CEO) and tissue culture origin (TCO). Virology, 440, 64–74. doi: 10.1016/j.virol.2013.02.007
- Han, M.G. & Kim, S.J. (2003). Efficacy of live virus vaccines against infectious laryngotracheitis assessed by polymerase chain reaction-restriction fragment length polymorphism. Avian Diseases, 47, 261–271. doi: 10.1637/0005-2086(2003)047[0261:EOLVVA]2.0.CO;2
- Hepkema, B.G., Hensen, E.J., Blankert, J.J., Zijpp, A.J., Albers, G.A.A., Tilanus, M.G.J. & Egberts, E. (1993). Mapping of susceptibility to Marek's disease within the major histocompatibility (B) complex by refined typing of White Leghorn chickens. Animal Genetics, 24, 283–287. doi: 10.1111/j.1365-2052.1993.tb00312.x
- Hughes, C., Gaskell, R., Jones, R., Bradbury, J. & Jordan, F. (1989). Effects of certain stress factors on the re-excretion of infectious laryngotracheitis virus from latently infected carrier birds. Research in Veterinary Science, 46, 274–276. doi: 10.1016/S0034-5288(18)31158-5
- Hughes, C.S., Williams, R.A., Gaskell, R.M., Jordan, F.T.W., Bradbury, J.M., Bennett, M. & Jones, R.C. (1991). Latency and reactivation of infectious laryngotracheitis vaccine virus. Archives of Virology, 121, 213–218. doi: 10.1007/BF01316755
- Kawaguchi, T., Nomura, K., Hirayama, Y. & Kitagawa, T. (1987). Establishment and characterization of a chicken hepatocellular carcinoma cell line, LMH. Cancer Research, 47, 4460–4464.
- Kaye, S. & Choudhary, A. (2006). Herpes simplex keratitis. Progress in Retinal and Eye Research, 25, 355–380. doi: 10.1016/j.preteyeres.2006.05.001
- Kirkpatrick, N.C., Mahmoudian, A., Colson, C.A., Devlin, J.M. & Noormohammadi, A.H. (2006). Relationship between mortality, clinical signs and tracheal pathology in infectious laryngotracheitis. Avian Pathology, 35, 449–453. doi: 10.1080/03079450601028803
- Kirkpatrick, N.C., Mahmoudian, A., O'Rourke, D. & Noormohammadi, A.H. (2006). Differentiation of infectious laryngotracheitis virus isolates by restriction fragment length polymorphic analysis of polymerase chain reaction products amplified from multiple genes. Avian Diseases, 50, 28–33. doi: 10.1637/7414-072205R.1
- Kreager, K.S. (1998). Chicken industry strategies for control of tumor virus infections. Poultry Science, 77, 1213–1216. doi: 10.1093/ps/77.8.1213
- Lee, S.-W., Devlin, J.M., Markham, J.F., Noormohammadi, A.H., Browning, G.F., Ficorilli, N.P., Hartley, C.A. & Markham, P.F. (2011). Comparative analysis of the complete genome sequences of two Australian origin live attenuated vaccines of infectious laryngotracheitis virus. Vaccine, 29, 9583–9587. doi: 10.1016/j.vaccine.2011.10.055
- Lee, S.-W., Hartley, C.A., Coppo, M.J., Vaz, P.K., Legione, A.R., Quinteros, J.A., Noormohammadi, A.H., Markham, P.F., Browning, G.F. & Devlin, J.M. (2015). Growth kinetics and transmission potential of existing and emerging field strains of infectious laryngotracheitis virus. PloS One, 10, e0120282. doi: 10.1371/journal.pone.0120282
- Lee, S.-W., Markham, P.F., Coppo, M.J.C., Legione, A.R., Markham, J.F., Noormohammadi, A.H., Browning, G.F., Ficorilli, N., Hartley, C.A. & Devlin, J.M. (2012). Attenuated vaccines can recombine to form virulent field viruses. Science, 337, 188–188. doi: 10.1126/science.1217134
- Mahmoudian, A., Kirkpatrick, N.C., Coppo, M., Lee, S.-W., Devlin, J.M., Markham, P.F., Browning, G.F. & Noormohammadi, A.H. (2011). Development of a SYBR Green quantitative polymerase chain reaction assay for rapid detection and quantification of infectious laryngotracheitis virus. Avian Pathology, 40, 237–242. doi: 10.1080/03079457.2011.553582
- Nicoll, M.P., Proença, J.T. & Efstathiou, S. (2012). The molecular basis of herpes simplex virus latency. FEMS Microbiology Reviews, 36, 684–705. doi: 10.1111/j.1574-6976.2011.00320.x
- Parcells, M., Arumugaswami, V., Prigge, J., Pandya, K. & Dienglewicz, R. (2003). Marek's disease virus reactivation from latency: changes in gene expression at the origin of replication. Poultry Science, 82, 893–898. doi: 10.1093/ps/82.6.893
- Parra, S., Nuñez, L., Astolfi-Ferreira, C. & Ferreira, J. (2015). Occurrence of infectious laryngotracheitis virus (ILTV) in 2009–2013 in the State of São Paulo-Brazil. Revista Brasileira de Ciência Avícola, 17, 117–120. doi: 10.1590/1516-635x1701117-120
- Poelaert, K.C.K., Van Cleemput, J., Laval, K., Favoreel, H.W., Couck, L., Van den Broeck, W., Azab, W., Nauwynck, H.J. & Longnecker, R.M. (2019). Equine herpesvirus 1 bridles T lymphocytes to reach its target organs. Journal of Virology, 93, e02098–e02018. doi: 10.1128/JVI.02098-18
- Polcicova, K., Biswas, P.S., Banerjee, K., Wisner, T.W., Rouse, B.T. & Johnson, D.C. (2005). Herpes keratitis in the absence of anterograde transport of virus from sensory ganglia to the cornea. Proceedings of the National Academy of Sciences, 102, 11462–11467. doi: 10.1073/pnas.0503230102
- Purcell, D. & Surman, P. (1974). Aerosol administration of the SA-2 vaccine strain of infectious leryngotracheitis virus. Australian Veterinary Journal, 50, 419–420. doi: 10.1111/j.1751-0813.1974.tb05357.x
- Rodríguez-Avila, A., Oldoni, I., Riblet, S. & García, M. (2007). Replication and transmission of live attenuated infectious laryngotracheitis virus (ILTV) vaccines. Avian Diseases, 51, 905–911. doi: 10.1637/8011-041907-REGR.1
- Saif, Y., Fadly, A.M., Glisson, J.R., McDougald, L.R., Nolan, L.K. & Swayne, D.E. (2011). Diseases of Poultry. Wiley-Blackwell: Wiley.
- Sehrawat, S., Kumar, D. & Rouse, B.T. (2018). Herpesviruses: harmonious pathogens but relevant cofactors in other diseases? Frontiers in Cellular and Infection Microbiology, 8, 177. doi: 10.3389/fcimb.2018.00177
- Szpara, M.L., Gatherer, D., Ochoa, A., Greenbaum, B., Dolan, A., Bowden, R.J., Enquist, L.W., Legendre, M. & Davison, A.J. (2014). Evolution and diversity in human herpes simplex virus genomes. Journal of Virology, 88, 1209–1227. doi: 10.1128/JVI.01987-13
- Szpara, M.L., Kobiler, O. & Enquist, L.W. (2010). A common neuronal response to alphaherpesvirus infection. Journal of Neuroimmune Pharmacology, 5, 418–427. doi: 10.1007/s11481-010-9212-0
- Thilakarathne, D.S., Hartley, C.A., Diaz-Méndez, A., Coppo, M.J. & Devlin, J.M. (2019). Development and application of a combined molecular and tissue culture-based approach to detect latent infectious laryngotracheitis virus (ILTV) in chickens. Journal of Virological Methods, 277, 113797.
- Thilakarathne, D.S., Noormohammadi, A.H., Browning, G.F., Quinteros, J.A., Underwood, G.J., Hartley, C.A., Coppo, M.J., Devlin, J.M. & Diaz-Méndez, A. (2020). Pathogenesis and tissue tropism of natural field recombinants of infectious laryngotracheitis virus. Veterinary Microbiology, 243, 108635. doi: 10.1016/j.vetmic.2020.108635
- VanDevanter, D.R., Warrener, P., Bennett, L., Schultz, E.R., Coulter, S., Garber, R.L. & Rose, T.M. (1996). Detection and analysis of diverse herpesviral species by consensus primer PCR. Journal of Clinical Microbiology, 34, 1666–1671. doi: 10.1128/JCM.34.7.1666-1671.1996
- Williams, R., Bennett, M., Bradbury, J., Gaskell, R., Jones, R. & Jordan, F. (1992). Demonstration of sites of latency of infectious laryngotracheitis virus using the polymerase chain reaction. Journal of General Virology, 73, 2415–2420. doi: 10.1099/0022-1317-73-9-2415
- Yonash, N., Bacon, L., Witter, R. & Cheng, H. (1999). High resolution mapping and identification of new quantitative trait loci (QTL) affecting susceptibility to Marek’s disease. Animal Genetics, 30, 126–135. doi: 10.1046/j.1365-2052.1999.00457.x