
ABSTRACT
Infectious bursal disease virus (IBDV) of chickens is a birnavirus with a bi-segmented double-stranded RNA genome, the segments designated as A and B. We performed phylogenetic analysis using a 366-bp fragment of segment A (nt 785–1150) and a 508-bp fragment of segment B (nt 328–835) of IBDV. A total of 463 segment A and 434 segment B sequences from GenBank, including the sequences of eight recent Bangladeshi isolates, were used in the analysis. The analysis revealed eight genogroups of segment A under serotype 1, designated as A1 (classical), A2 (US antigenic variant), A3 (very virulent), A4 (dIBDV), A5 (atypical Mexican), A6 (atypical Italian), A7 (early Australian) and A8 (Australian variant), and a single genogroup under serotype 2, designated as A0. On the other hand, segment B could be categorized into five genogroups irrespective of serotype, these being B1 (classical-like), B2 (very virulent-like), B3 (early Australian-like), B4 (Polish & Tanzanian) and B5 (Nigerian). Segment B of serotype 2 strains clustered within genogroup B1. With the bi-segmented genome of IBDV, these differences would allow for a total of 45 possible assortments. Based on the combinations of segment A and segment B genogroups observed in 463 IBDV strains, a total of 15 genotypes could be recognized. Recent Bangladeshi IBDV strains, isolated in 2016, appeared to be segment reassortants having segment A of genogroup A3 (very virulent) and segment B of genogroup B3 (early Australian-like). An extended system of nomenclature of IBDV strains is proposed.
Introduction
Infectious bursal disease (IBD), also known as Gumboro disease, is an economically important viral disease of young chickens causing acute death or immunosuppression (Becht, Citation1980; Kibenge et al., Citation1988; Müller et al., Citation1992; van den Berg, Citation2000; Müller et al., Citation2003; Müller et al., Citation2012; Alkie & Rautenschlein, Citation2016; Eterradossi & Saif, Citation2019). The disease is characterized by lesions in the primary lymphoid organ bursa of Fabricius with the destruction of growing B-lymphocytes (Müller, Citation1986). Chickens between 3 and 6 weeks of age with a well-developed bursa are most commonly affected.
The aetiological agent, infectious bursal disease virus (IBDV) has a double-stranded bi-segmented RNA genome and belongs to the genus Avibirnavirus under the family Birnaviridae (Dobos et al., Citation1979; Leong et al., Citation2000; Delmas et al., Citation2019). IBDV is a non-enveloped isometric particle, approximately 60 nm in diameter, with an icosahedral capsid. The capsid is single-shelled consisting of 260 VP2 trimers arranged in T = 13 laevo lattices (Böttcher et al., Citation1997; Coulibaly et al., Citation2005). The IBDV genome comprises two segments of double-stranded RNA (Müller et al., Citation1979) – the larger segment A and the smaller segment B. Segment B (2.8 kbp) contains a single open reading frame (ORF) that encodes the viral RNA dependent RNA polymerase VP1. Segment A (3.2 kbp) contains two ORFs. The larger ORF encodes a polyprotein pVP2-VP4-VP3, which is autocatalytically and co-translationally cleaved into a precursor capsid protein pVP2, a viral protease VP4 – a non-canonical lon protease, and a capsid scaffolding protein VP3. Further serial cleavage at the C terminus of pVP2 (512 amino acids, aa) is catalyzed by VP4, which releases the mature capsid protein VP2 (441 aa) and four small peptides (Delmas, Citation2008). Another small overlapping ORF in segment A encodes a non-structural protein VP5, which is not essential for virus replication but plays an important role in the non-lytic egress of the virus particles from the infected cell (Mundt & Müller, Citation1995; Mundt et al., Citation1997; Lombardo et al., Citation2000).
The capsid subunits consist primarily of VP2 trimers. The monomeric VP2 has three distinct domains, designated as base (B), shell (S) and projection (P) (Coulibaly et al., Citation2005; Garriga et al., Citation2006; Lee et al., Citation2006). The central P domain contains a hypervariable region spanning aa positions 206–350 (Bayliss et al., Citation1990) and contains four exposed loops designated as PBC, PDE, PFG and PHI (Delmas, Citation2008). The nucleotide sequence of the VP2 hypervariable region is commonly used for molecular characterization of IBDV and a large number of sequence data is available in GenBank.
There are two serotypes of IBDV (McFerran et al., Citation1980); serotype 1 is pathogenic for chickens, while serotype 2 is nonpathogenic. The early strains of serotype 1 IBDV, commonly known as classical virulent strains (cvIBDV), were relatively less virulent causing low mortality; however, the recovered birds remained immunosuppressed and refractory to immunization against common poultry diseases. In the early 1980s, antigenic variant strains of IBDV (avIBDV) emerged in the USA, which were not fully controlled by antibodies to classical IBDV strains (Snyder et al., Citation1988). Since the late 1980s, a dramatic increase in specific mortality in chickens due to IBD was noticed, first in Europe (Chettle et al., Citation1989; van den Berg et al., Citation1991) and subsequently in other parts of the world. These new isolates are known as very virulent IBDV (vvIBDV). These vvIBDVs are now considered widely distributed all over the world except New Zealand and Australia. Interestingly, cvIBDV and vvIBDV appeared to be antigenically very similar (Öppling et al., Citation1991; van den Berg et al., Citation1996). Apart from these field strains, many attenuated vaccine strains also have been developed from cv and av IBDVs in different laboratories. Moreover, phylogenetically distinct IBDV (dIBDV) strains have been reported recently from South America (Hernandez et al., Citation2015), which were similar to the viruses detected earlier in retrospective samples from Eastern Europe (Domanska et al., Citation2004). Atypical IBDV strains also have been reported from Italy (Lupini et al., Citation2016) and from Mexico, the latter being considered to be natural recombinant strains (Jackwood, Citation2012; Michel & Jackwood, Citation2017).
Because of the bi-segmented nature of the genome, IBDV may undergo segment reassortment. It has been suggested that vvIBDV might have evolved through segment reassortment between the segment A of the circulating IBDV strains and segment B from an unknown IBDV strain (Islam, Zierenberg & Müller, Citation2001; Hon et al., Citation2006). In recent years, naturally occurring reassortant IBDV strains have been reported frequently from different parts of the world, which included reassortment between serotype 1 and serotype 2 (Jackwood et al., Citation2011; Soubies et al., Citation2017; Stoute et al., Citation2019) and reassortment between different pathotypes/genotypes of serotype 1 IBDVs (Sun et al., Citation2003; Le Nouen et al., Citation2006; Gao et al., Citation2007; Kasanga et al., Citation2007; Xia et al., Citation2008; Chen et al., Citation2012; Fernandes et al., Citation2012; He et al., Citation2014; Lu et al., Citation2015; Patel et al., Citation2016; Raja et al., Citation2016; Abed et al., Citation2018; Pikula et al., Citation2018; Stoute, et al., Citation2019; Wang et al., Citation2019; Mato et al., Citation2020; Pikula et al., Citation2020).
Traditionally, nucleotide sequencing of the VP2 hypervariable region is used for genotypic characterization of IBDV strains, though the OIE Manual (OIE, Citation2019) has been promoting for some years that the genetic characterization of both IBDV genome segments should be performed as the genetic variations in both genome segments may influence strain pathogenicity (Le Nouën et al., Citation2006; Le Nouën et al., Citation2012; Escaffre et al., Citation2013; Gao et al., Citation2018). Recently, Michel & Jackwood (Citation2017) and Jackwood et al. (Citation2018) proposed a classification of IBDV into seven genogroups based on the sequences of the VP2 hypervariable region of segment A. However, genotypic classification of IBDV based on segment B is not yet available. As both genome segments contribute to the virulence of IBDV and segment reassortment plays a role in virus evolution, it is now necessary to classify IBDVs based on both genome segments.
In the present paper, we propose a unified scheme of genotypic classification of IBDV based on the phylogenetic analysis of sequence datasets of both segment A and segment B. We also report the detection of reassortant IBDV strains for the first time in Bangladesh.
Materials and methods
The study utilized the sequence data of eight recent isolates of IBDV from Bangladesh, generated in the present study, and the sequence data that are publicly available in GenBank.
Detection and sequencing of IBDV from recent outbreaks in Bangladesh
Sample collection
A total of eight suspected IBD outbreaks in layer chickens in the Sakhipur and Bhaluka sub-districts of the Tangail and Mymensingh districts, respectively, were investigated during the period from March to October 2016. Affected flocks were small-scale layer farms having 1000–2000 replacement stocks of 20–53 days of age at the onset of the outbreaks. Mortality was variable ranging from 2% to 40%. Whitish diarrhoea, drowsiness and loss of appetite were consistently found in all sick birds. At necropsy, all birds showed marked gross lesions in their bursa of Fabricius, which was swollen, haemorrhagic or necrotic. Haemorrhages in the breast and thigh muscles were also found in some of the affected birds. Small pieces of tissues from the bursa of Fabricius were collected aseptically. The tissue samples were homogenized in PBS to make a 20% (w/v) suspension. The tissue suspension was centrifuged at 1500 × g for 10 min and the supernatant was collected in fresh sterile Falcon tubes, to which an antibiotic (Gentamycin, 500 µg/ml) was added and was stored at −80°C.
RT–PCR for IBDV and sequencing
Total RNA was extracted from the tissue homogenates with the KingFisher™ mL Purification System (Thermo Scientific, Waltham, MA, USA) using the MagMAX™ Viral RNA Isolation Kit (Applied Biosystems, Beverly, MA, USA). The RNA was used to amplify IBDV gene fragments from segment A and segment B. A previously reported primer set (VP2F or INCO-DC#3F: 5´-AAC AGC CAA CAT CAA CG-3´ and VP2R or INCO-DC#4R: 5´-GCT CGA AGT TGC TCA CCC-3´) (Zierenberg et al., Citation2001; Islam, Zierenberg, Eterradossi et al., Citation2001) was used to amplify a 677-bp fragment from the hypervariable region (nt 571–1247) of the VP2 gene in segment A. For segment B, the previously described primer set B-Univ-F: 5´-AAT GAG GAG TAT GAG ACC GA-3´and B-Univ-R: 5´-CCT TCT CTA GGT CAA TTG AGT ACC-3´ (Islam et al., Citation2012) was used to amplify a 1051-bp fragment (nucleotides 319–1369) from the VP1 gene. A one-tube RT–PCR reaction was performed on a thermocycler using the SuperScript™ III One-Step RT–PCR System with Platinum™ Taq High Fidelity DNA Polymerase (Invitrogen, Waltham, MA, USA). RT–PCR for both segments A and B was run simultaneously using a reaction profile as described earlier (Islam et al., Citation2012).
The RT–PCR products were purified using FavorPrep™ GEL/PCR Purification Kit (Favorgen Biotech Corp, Ping-Tung, Taiwan) and sequenced directly by a commercial sequencing laboratory (1st BASE, Selangor, Malaysia) using both forward and reverse RT–PCR primers. The raw sequence data were first checked for quality and then edited and assembled with the Bioedit and MEGA7 (www.megasoftware.net) software.
Sequence data sets from public domains
All nucleotide sequences of segment A and segment B of serotype 1 and serotype 2 strains IBDV available as of 11 June 2020 in GenBank (https://www.ncbi.nlm.nih.gov/nucleotide/) were downloaded. After a preliminary alignment, a total of 434 strains/isolates of IBDV were selected for phylogenetic analysis based on the following criteria: (a) both segment A and segment B sequences were available, and (b) the sequence data covered the targeted region. Deviating from the preset criteria, 29 more IBDV strains having sequence data of segment A only were also included in the phylogenetic analysis of segment A as their inclusion appeared to be important for the comprehensiveness of the analysis.
Phylogenetic analysis
Phylogenetic analysis was performed for both genome segments A and B of IBDV with the sequences of recent Bangladeshi isolates and those downloaded from GenBank. Based on the initial alignment, a 366-bp region of segment A and a 508-bp region of segment B were identified that would allow us to include the maximum number of sequence data sets in the analysis using equal sequence length. In the case of segment A, the selected region (nt 785–1150, aa 219–340) corresponded to the VP2 hypervariable region. On the other hand, for segment B the selected region (nt 328–835, aa 73–241) corresponded to a part of the N-terminal domain and a part of the finger subdomain of the VP1 polymerase. The region largely overlapped the “B marker region” (nt 439–867, aa 110–252), recently described to be phylogenetically representative (Alfonso-Morales et al., Citation2015). The sequence data were aligned with the ClustalW algorithm of MEGA7 software (www.megasoftware.net). Maximum likelihood (ML) phylogenetic trees were constructed using IQ-TREE programme (Nguyen et al., Citation2015). The ModelFinder programme embedded in the IQ-Tree (Kalyaanamoorthy et al., Citation2017) was used to select the best fit substitution model according to the Bayesian Information Criterion. An ultrafast bootstrapping of 1000 replicates was performed in IQ-TREE (Hoang et al., Citation2018). Finally, trees were visualized and edited in FigTree. In parallel, ML phylogenetic trees were also constructed with the respective best fit model (TN93+G for both segment A and segment B) using the MEGA7 programme. The stability of the nodes in the phylogenetic trees was tested by bootstrapping with 1000 replications. For quick reference, short phylogenetic trees were constructed using IQ-TREE programme for both segment A and segment B using selected sequences representing different phylogenetically distinct groups in the big trees.
For segment A, VP2 hypervariable region is commonly used for phylogenetic analysis; however, for segment B there is no universally accepted region for phylogenetic analysis. Therefore, the phylogenetic tree obtained with the selected region (nt 328–835) of segment B was compared with that obtained with the full-length sequence of segment B. To this end, we downloaded the complete segment B sequences of 135 strains of IBDV available in GenBank. Possible recombinant sequences were identified using Geneconv, RDP, MaxChi, Chimaera, BootScan and SiScan methods implemented in a recombination detection programme RDP4 (Martin et al., Citation2015). After removing recombinant sequences, 120 complete segment B sequences were available. Then we constructed phylogenetic trees with the complete segment B gene sequences and the corresponding 508-bp (nt 328–835) partial sequences of these 120 IBDV strains using the IQ-TREE programme and analysed the congruence between the two trees (Nguyen et al., Citation2015).
Identification of genogroups and genotypes
Based on this phylogenetic analysis, possible genogroups of IBDV were identified for segment A and segment B sequences separately. The estimates of average evolutionary distances between different genogroups of IBDV were calculated with MEGA7 (Kumar et al., Citation2016) using the Maximum Composite Likelihood model (Tamura et al., Citation2004). The rate variation among sites was modelled with a gamma distribution (shape parameter = 1).
Finally, based on the combination of the individual segment A and segment B genogroups, IBDV strains were assigned to different genotypes.
Results
Phylogenetic analysis and identification of segment-specific genogroups of IBDV
The phylogenetic analysis for segment A was done with 463 sequences including eight sequences of the new Bangladeshi isolates from the present study, while that for segment B was performed with 434 sequences including eight sequences of the new Bangladeshi isolates. The Maximum Likelihood phylogenetic trees obtained with IQ-TREE programme based on a 366-bp fragment in the hypervariable region of the VP2 gene in segment A (nt 785–1150) and a 508-bp fragment of segment B (nt 328–835) are presented in concise circular form (without isolate/strain names) in and , respectively. Detailed trees including taxa names are given in Supplementary Figure S1 and Supplementary Figure S2. Phylogenetic analysis with MEGA7 software produced similar trees (Supplementary Figures S3 and S4). For quick reference, short phylogenetic trees with selected sequences representing different clusters of segment A and segment B were also prepared with IQ-TREE programme and are presented in and , respectively.
Figure 1. Concise circular phylogenetic tree of segment A of 463 strains of IBDV based on VP2 hypervariable region sequences (nt 785–1150). The tree was generated by the use of a maximum likelihood (ML) tree method and ultrafast bootstrapping with 1000 replicates with the IQ-Tree software (Nguyen et al., Citation2015; Hoang et al., Citation2018). ModelFinder embedded in the IQ-Tree (Kalyaanamoorthy et al., Citation2017) was used to select the best fitted substitution model according to the Bayesian Information Criterion (SYM + I+G4). The tree was visualized in FigTree. The tree is drawn to scale and genotypic information is shown next to the tree. There were 366 positions in total in the final dataset. (Name and GenBank accession number of the strains is provided in the Supplementary Figure S1).
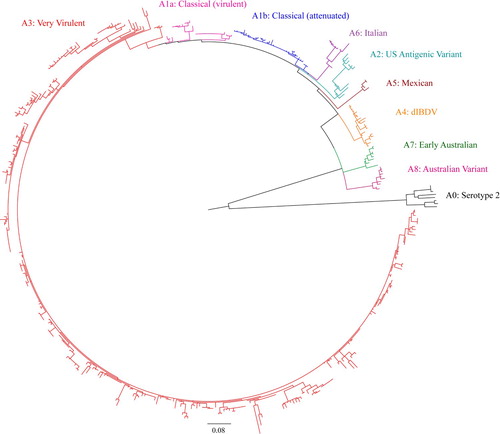
Figure 2. Concise circular phylogenetic tree of segment B of 434 strains of IBDV based on the 508-bp sequences corresponding to the VP1 N-terminal domain and the finger subdomain of the central polymerase (nt 328–835). The tree was generated by the use of a maximum likelihood (ML) tree method and ultrafast bootstrapping with 1000 replicates with the IQ-Tree software (Nguyen et al., Citation2015; Hoang et al., Citation2018). ModelFinder embedded in the IQ-Tree (Kalyaanamoorthy et al., Citation2017) was used to select the best fitted substitution model according to the Bayesian Information Criterion (GTR + F+G4). The tree was visualized in FigTree. The tree is drawn to scale and genotypic information is shown next to the tree. There were 508 positions in total in the final dataset. (Name and GenBank accession number of the strains is provided in the Supplementary Figure S2).
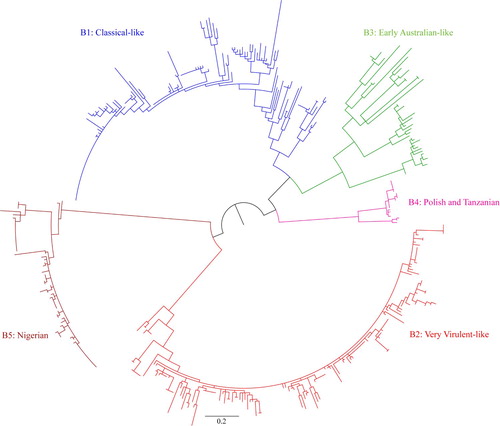
Figure 3. Phylogenetic tree of segment A of 96 selected representative strains of IBDV based on VP2 hypervariable region sequences (nt 785–1150). The tree was generated by the use of a maximum likelihood (ML) tree method and ultrafast bootstrapping with 1000 replicates with the IQ-Tree software (Nguyen et al., Citation2015; Hoang et al., Citation2018). ModelFinder embedded in the IQ-Tree (Kalyaanamoorthy et al., Citation2017) was used to select the best fitted substitution model according to the Bayesian Information Criterion (SYM + I+G4). The tree was visualized in FigTree. The tree is drawn to scale and genotypic information is shown next to the tree. There were 366 positions in total in the final dataset. Isolates under present study are marked with filled circles (●).
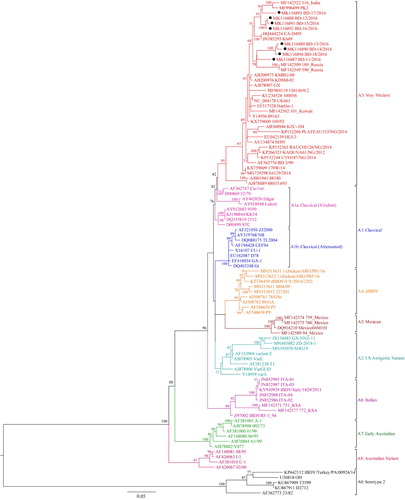
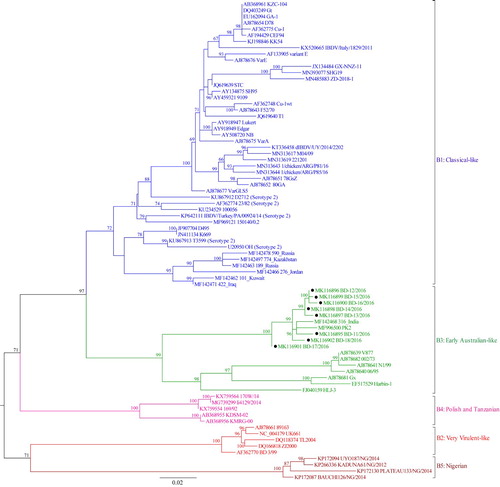
Figure 4. Phylogenetic tree of segment B of 77 selected representative strains of IBDV based on the 508-bp sequences corresponding to the VP1 N-terminal domain and the finger subdomain of the central polymerase (nt 328–835). The tree was generated by the use of a maximum likelihood (ML) tree method and ultrafast bootstrapping with 1000 replicates with the IQ-Tree software (Nguyen et al., Citation2015; Hoang et al., Citation2018). ModelFinder embedded in the IQ-Tree (Kalyaanamoorthy et al., Citation2017) was used to select the best fitted substitution model according to the Bayesian Information Criterion (GTR + F+G4). The tree was visualized in FigTree. The tree is drawn to scale and genotypic information is shown next to the tree. There were 508 in total positions in the final dataset. Isolates under present study are marked with filled circles (●).
On phylogenetic analysis of segment A, serotype 1 and serotype 2 IBDVs formed two major branches. Serotype 1 IBDVs can be further divided into eight genogroups, whereas serotype 2 IBDVs belong to a single genogroup (Figures 1, 3, Supplementary Figures S1, S3). In Serotype 1, segment A genogroups are A1 (classical), A2 (US antigenic variant), A3 (very virulent), A4 (early European and recent South American distinct IBDV, dIBDV), A5 (atypical or recombinant Mexican strains), A6 (atypical Italian), A7 (early Australian) and A8 (Australian variant). The single genogroup under serotype 2 is designated as A0. The average pairwise evolutionary distance between serotype 1 and serotype 2 strains was 0.658 base substitutions per site (i.e. 65.8%) and that between the eight genogroups of serotype 1 ranged from 0.90 to 0.224 base substitutions per site (i.e. 9% to 22.4%) (). It may be noted that genogroup A1 (classical IBDVs) comprises: (a) classical virulent strains and (b) classical attenuated strains adapted to cell culture. The classical virulent strains further segregate into four subclusters identified by a significant bootstrap and represented by strains: (i) F52/70 or Cu-1wt, (ii) Lukert or Edgar, (iii) Winterfield 2512 or its derivatives, and (iv) STC. In genogroup 3 (very virulent IBDVs), a small group of West African atypical vvIBDV formed a sub-cluster having an evolutionary divergence of 0.085 base substitutions per site (8.5%) from other vvIBDVs (data not shown).
Table 1. Estimates of evolutionary distances between two serotypes and between serotype 1 genogroups of IBDV based on VP2 hypervariable region of segment A.
Based on segment B sequences, IBDV strains can be divided into five distinct genogroups (Figure 2, 4, Supplementary Figure S2, S4): B1 (classical-like), B2 (very virulent-like), B3 (early Australian-like), B4 (Polish & Tanzanian), and B5 (Nigerian). Pairwise evolutionary distances between these five genogroups of segment B ranged from 0.129 to 0.179 base substitutions per site (12.9% to 17.9%) (). In general, genogroup B1 comprises sequences of classical strains (virulent and attenuated), US antigenic variant strains, dIBDVs and atypical Italian strains (A1, A2, A4 and A6 genogroups of segment A, respectively). Unlike segment A, phylogenetic analysis of segment B does not segregate serotype 1 and serotype 2 IBDV strains into separate groups. Segment B of serotype 2 strains, together with previously described interserotypic reassortant strains (segment A of serotype 1 and segment B similar to serotype 2), form a paraphyletic group within genogroup B1 (Figures 2, 4 and Supplementary Figure S2, S4). It may be noted that a group of isolates from the Middle East and Russia formed a distinct sub-cluster under genogroup B1. All of these isolates have segment A of very virulent genogroup A3.
Table 2. Estimates of evolutionary distances between IBDV segment B genogroups based on the 508-bp sequences corresponding to the VP1 N-terminal domain and the finger subdomain of the central polymerase (nt 328–835).
We compared the phylogenetic tree produced with the partial (508-bp) gene sequence of segment B with that derived from the full-length segment B sequence of 120 isolates. Both the trees had similar topology, and four genogroups (B1–B4) were clearly segregated (). Only a single isolate with full-length segment B sequence under genogroup 4 was available. However, no full-length sequence of any isolate under genogroup 5 was available for inclusion in this analysis. The analysis revealed a strong congruence between the phylogenetic trees obtained with the partial and full-length segment B sequences with a congruence index (Icong) 3.66 and P-value 1.83e-28.
Figure 5. Congruence analysis between two phylogenetic trees constructed based on the complete genome sequences and 508-bp sequences corresponding to the VP1 N-terminal domain and the finger subdomain of the polymerase (nt 328–835) of segment B of IBDV. The trees have 120 leaves. The Maximum Agreement SubTree (MAST) has 59 leaves. Icong = 3.66400736895919, P-value = 1.82572412756908e-28. The trees are more congruent than expected by chance. The analysis was conducted with the IQ-Tree software (Nguyen et al., Citation2015).
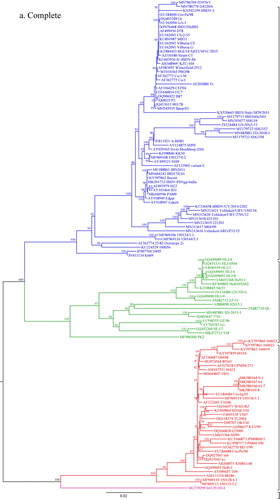
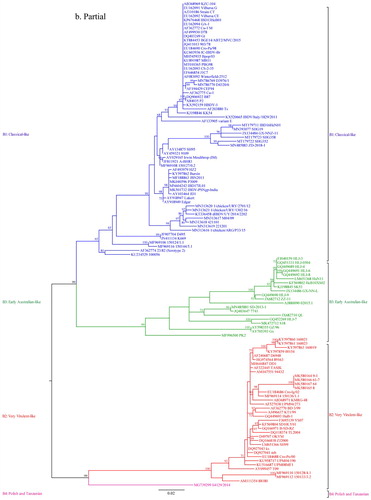
Sequence data of eight recent Bangladeshi isolates of IBDV (GenBank accession no. MK116887 to MK116902) were also analysed in the present study. Segment A of these isolates clustered with vvIBDV under genogroup A3, though interestingly distributed in two separate small branches indicating minor variations. On the other hand, segment B of all the eight new Bangladeshi isolates clustered with early Australian-like IBDVs under genogroup B3. This finding suggests that these viruses are segment reassortants having very virulent segment A and early Australian-like segment B.
Genotypic diversity of IBDV
Based on the observed combinations of segment A and segment B genogroups of 463 isolates/strains, a total of 15 genotypes of IBDV could be recognized (; Supplementary Table S1). We propose a binomial scheme for genotype designation using segment A and segment B genogroup numbers. It was interesting to note that several genogroups have emerged through apparent reassortments between segment A and segment B of previously circulating or newly emerged genogroups, as shown in .
Table 3. Genotypes of IBDV based on segment A and segment B genogroup combination.
Usually, viruses belonging to classical (A1), US antigenic variant (A2), dIBDV (A4) and atypical Italian (A6) genogroups of segment A have a classical-like segment B (genogroup B1) resulting in genotypic designations as A1B1, A2B1, A4B1 and A6B1, respectively. Serotype 2 strains (A0) also have classical-like (B1) segment B, leading to genotypic designation of A0B1. Viruses belonging to the very virulent (A3) genogroup of segment A usually have very virulent-like segment B (genogroup B2), and hence are designated as genotype A3B2. The early Australian (A7) and Australian variant (A8) genogroups of segment A have early Australian-like segment B (genogroup B3), resulting in genotypic designations of A7B3 and A8B3, respectively. The two recently described groups of IBDV isolates from Poland & Tanzania and Nigeria had very virulent (A3) segment A but distinct segment B (genogroup B4 and B5), giving rise to new genotypes of A3B4 and A3B5, respectively. No segment B sequence is yet available for the Mexican atypical or recombinant (A5) strains, so they are provisionally designated as A5Bx. Moreover, apart from these well-established genotypes, random reassortments between segment A and segment B of different genogroups are giving rise to new genotypes, such as A1B2, A1B3, A3B1 and A3B3 ( and Supplementary Table S1).
Discussion
Because of the bi-segmented nature of the genome, it may well be possible that segment reassortment has been playing a crucial role in the evolution of IBDV since its emergence. Both the segments contribute to virulence of IBDV (Le Nouën et al., Citation2006; Le Nouën et al., Citation2012; Escaffre et al., Citation2013; Gao et al., Citation2018). Hence molecular epidemiological studies on IBDV require characterization of both genome segments. In the present study, we have proposed a scheme of genotypic classification of IBDV based on comprehensive phylogenetic analyses of segments A and B of both serotype 1 and serotype 2 strains. We also report on the detection of segment reassortant IBDVs in Bangladesh for the first time.
For phylogenetic analyses, we relied on the sequence data publicly available in GenBank. Based on the initial alignment, a 366-bp region of segment A (nt 785–1150, aa 219–340) and a 508-bp region of segment B (nt 328–835, aa 73–241) were identified that would allow us to include the maximum number of sequence data sets in the analysis using all positions without deletion. In the case of segment A, the selected region corresponded to the hypervariable region of the VP2 gene, which is commonly used for phylogenetic analysis; for segment B our selected region (nt 328–835) largely overlapped the “B marker region” (nt 439–867), previously described to be phylogenetically representative (Alfonso-Morales et al., Citation2015). However, recently Pikula et al. (Citation2020) reported that the B-marker region was unable to reproduce the phylogenetic topology of the complete segment B genome. We compared the phylogenetic tree obtained with our selected region (nt 328–835) of segment B with that obtained for the full-length sequence of segment B of 120 selected isolates. A strong congruence between the two phylogenetic trees was observed (Icong = 3.66, P-value = 1.83e-28), indicating that the results of phylogenetic analysis with the selected region of segment B could be representative.
We propose the classification of serotype 1 segment A into eight genogroups (A1 through A8) and serotype 2 segment A into a single genogroup (A0). Recently, Michel & Jackwood (Citation2017) proposed seven genogroups for serotype 1 segment A, designated as G1 to G7. To avoid any future confusion we followed the numbering scheme of Jackwood et al. (Citation2018) and Michel & Jackwood (Citation2017) for designating segment A genogroups. However, as we proposed genogrouping for both segment A and segment B, we used prefix A and B before the genogroup number for segment A and segment B, respectively. Michel & Jackwood (Citation2017) proposed a single genogroup for Australian strains (G7) but we found that early Australian strains and Australian variant strains (Sapats & Ignjatovic, Citation2000) formed two distinct branches in the phylogenetic tree having as much as 16.2% evolutionary divergence. Hence, we proposed them as two separate genogroups, A7 and A8, respectively.
For segment B, we propose five genogroups (B1 through B5). Although for segment A the classical (virulent and attenuated), US antigenic variant, dIBDV and atypical Italian strains of IBDVs formed four different genogroups, for segment B they all belonged to genogroup B1. Segment B sequences derived from serotype 2 IBDVs (A0 segment A) also fell in genogroup B1. However, serotype 2 strains did not form a separate monophyletic cluster, nor did they cluster into any of the other B1 subclusters, instead they remained as a paraphyletic group. It may be noted that Jackwood et al. (Citation2011) and Soubies et al. (Citation2017) previously described “interserotypic reassortments” having a vvIBDV-like segment A (segment A genogroup A3), but a segment B exhibiting highest nucleotide similarity with that of serotype 2 strains. Consistent with previous findings, segment B sequences from these “interserotypic reassortants” are indeed positioned in our phylogenetic analyses together with segment B sequences of typical serotype 2 strains within the B1 genogroup. Abed et al. (Citation2018) described some Algerian very virulent strains (segment A genogroup A3) with segment B of unknown origin. In our analysis, segment B of these strains also clustered with the segment B of serotype 2 strains within genogroup B1. Interestingly, the segment Bs of a group of IBDVs from the Middle East and Russia form a phylogenetically significant cluster within genogroup B1. The segment As of these isolates belong to very virulent genogroup (A3). It remains to be observed if this group, due to continued B segment evolution, turns out as a new genogroup in the future.
The segment B of very virulent IBDVs was shown to be phylogenetically distinct from that of classical virulent, attenuated and US antigenic variant strains (Islam et al., Citation2001b). The segment Bs of very virulent IBDVs constitute the genogroup B2.
In all previous phylogenetic analyses, the early Australian strain 002/73 was always an outlier. From the present analysis it is evident that the segment B of early Australian strains (represented by 002/73, V877) forms a distinct genogroup (B3). A single segment B sequence of an Australian variant strain (05/5) also clusters in this genogroup. Interestingly, the segment B of some reassortant strains (e.g. Gx, Harbin-1, HLJ-3 and our recent Bangladeshi isolates) also clustered with this early Australian-like genogroup B3. However, the South Asian reassortant strains from Bangladesh, India and Pakistan formed a distinct subcluster within this genogroup.
Based on our analyses, we recognized two more distinct genogroups of segment B. These are genogroup B4 (comprising strains isolated in Poland and Tanzania) and genogroup B5 (for strains isolated in Nigeria). The segment A counterpart of these two genogroups cluster with segment A sequences of very virulent IBDVs and belong, therefore, to genogroup A3.
Based on the nucleotide divergence figures obtained in our analyses, we tentatively propose that, within an IBDV serotype, future segment A clusters (comprising at least three detected strains from two different outbreaks) identified with at least 9% nucleotide divergence from previous genetic lineages, should be considered as a new genogroup. Similarly, future segment B clusters (comprising at least three detected strains as above), identified with at least 12% nucleotide divergence should be considered as a new segment B genogroup. These figures compare well with those proposed to define new genotypes of Newcastle disease virus (10%) (Diel et al., Citation2012; Dimitrov et al., Citation2019), another important pathogenic RNA virus of domestic fowl.
In theory, the nine segment A genogroups, together with the five segment B genogroups, might allow a total of 45 possible genotypic combinations. Based on the presently known segment A and segment B genogroups, as shown in this study, we defined combinations leading to 15 genotypes of IBDV (). The classical virulent IBDV strains detected in the early 1960s and the classical attenuated strains generated in the laboratories or isolated from the field comprise the genotype A1B1. The US antigenic variant strains that emerged in early 1980s (Snyder et al., Citation1988) and contain a variant segment A but segment B similar to that of classical IBDVs constitute the genotype A2B1. The very virulent IBDVs that emerged in late 1980s (Chettle et al., Citation1989; van den Berg et al., Citation1991) and spread very quickly across the globe are designated as genotype A3B2. Two distinct lineages of IBDVs were detected in Australia and remained almost restricted in Australia. These are early Australian IBDVs detected in the early 1970s (Firth, Citation1974) and Australian variant strains detected in the 1990s (Sapats & Ignjatovic, Citation2000); these two groups of viruses are designated as genotype A7B3 and A8B3, respectively. So far, the segment B sequence of only a single isolate of genotype A8B3 is available, so the genotypic classification A8B3 needs to be confirmed with segment B sequences of more strains. Members of another lineage of IBDV, known as distinct IBDV (dIBDV), with phylogenetically distinct segment A have been reported from South America (Hernandez et al., Citation2015; Tomas et al., Citation2020), and were similar to the viruses detected earlier in retrospective samples from Eastern Europe (Domanska et al., Citation2004). Segment Bs of these viruses are similar to classical IBDVs. Hence, these distinct IBDVs are designated as genotype A4B1. Recently, atypical IBDV strains have been reported in Italy (Lupini et al., Citation2016; Lupini et al., Citation2020), which have phylogenetically distinct segment A. Viruses with atypical Italian type segment A have also been detected in Russia and the Kingdom of Saudi Arabia (Michel & Jackwood, Citation2017). Segment B sequence is available for one Italian strain only, which was similar to classical IBDVs. This genotype has been designated as A6B1, but needs to be confirmed with segment B sequences of more strains. Another group of atypical viruses appeared to have established in Mexico since early 2000s, having phylogenetically distinct segment A (Michel & Jackwood, Citation2017). Segment B sequences are not yet available. This group of viruses is designated provisionally as genotype A5Bx. Our present analysis also revealed two new groups of IBDVs having very virulent-like segment A, but phylogenetically distinct segment B. These two groups of viruses are designated as genotype A3B4 and A3B5. So far, the viruses of the genotype A3B4 have been detected from Poland between 1992 and 2015 (Pikula et al., Citation2020); similar viruses were detected in Tanzania in 2001 and 2004 (Kasanga et al., Citation2007; Kasanga, Citation2015). One virus of this genotype (li4129/2014) was also detected in Finland in 2014 (Tammiranta et al., Citation2018). The viruses of genotype A3B5 were isolated only in Nigeria between 2009 and 2014 (Nwagbo et al., Citation2016).
Apart from the genotypes mentioned above, a few more IBDV genotypes have emerged through random segment reassortment between the existing genotypes ( and Supplementary Table S1). Viruses of genotype A1B2 (segment A classical-like/segment B very virulent-like) have been detected sporadically in China (strain ZJ2000 and TL2004), Egypt (strain 160019) and Japan (strain CT). On the contrary, viruses of genotype A3B1 (segment A very virulent-like/segment B classical-like) have been isolated more frequently. As many as 50 such isolates have been recorded in the present analysis.
Viruses of another novel reassortant genotype A3B3 (segment A very virulent-like/segment B early Australian-like) have been frequently detected in recent years. Viruses belonging to this genotype were originally detected in Venezuela, such as 02015.1 and 02015.3 (Le Nouen et al., Citation2006), and China, such as Gx, Harbin-1 (Gao et al., Citation2007; Xia et al., Citation2008; Chen et al., Citation2012), but now are being detected in other Asian countries like India (Patel et al., Citation2016), Pakistan (Hussain et al., Citation2019), Bangladesh (isolates characterized in the present study), Thailand, SK53 (GenBank accession no. KJ198843 and KJ198845), Vietnam, 753_Vietnam (Michel & Jackwood, Citation2017) and USA, CAHFS7741 (Jackwood et al., Citation2012). In the present analysis 37 isolates of this genotype have been recorded.
The first IBDV serotype 2 isolates originated from turkeys, but they were later also isolated from chickens, and antibodies to serotype 2 IBDVs are common in both chickens and turkeys (Eterradossi & Saif, Citation2019). The serotype 2 strains have a distinct segment A, genogroup A0, but their B segments cluster within genogroup B1, i.e. classical-like. Hence the genotype designation of serotype 2 strains would be A0B1. By the generation of serotype 1/serotype2 reassortant IBDVs through reverse genetics and their investigation in vitro and in vivo, it was shown that genome segment A determines the bursa tropism of IBDV whereas segment B is involved in the efficiency of viral replication, particularly by an optimized interaction of the polymerase encoded by segment B with the structural protein VP3 encoded by segment A (Zierenberg et al., Citation2004). It was speculated that such reassortant IBDVs also exist in the field but remain undetected as they do not cause overt clinical signs. In fact, several “interserotypic reassortants” have been reported recently, which included viruses with very virulent-like segment A and serotype 2-like segment B (CA-K785, CA-D495, K669, 100056, 150124) (Jackwood et al., Citation2011; Soubies et al., Citation2017; Abed et al., Citation2018). These interserotypic reassortants are designated as genotype A3B1 together with other interpathotypic reassortants having very virulent-like segment A and classical-like segment B, as discussed above.
Taking together all this historical evidence, it is obvious that segment reassortments between the circulating or novel strains have contributed significantly to the expansion of genetic and pathogenic diversity of IBDV. The proposed system of genotypic classification of IBDV strains based on the sequence information of both genome segments will help for a better understanding of IBDV molecular epidemiology and evolution.
Comparative pathogenicity studies with the naturally occurring reassortant strains revealed that segment reassortments may influence the pathogenicity of IBDV. Jackwood et al. (Citation2016) compared the pathogenicity of six reassortant IBDV strains with that of very virulent, classical virulent and US antigenic variant strains and observed that the reassortant IBDV with a very virulent genome segment A and non-very virulent segment B was less pathogenic than the very virulent strain but more pathogenic than the classical virulent strain. Similarly, Lu et al. (Citation2015) reported that a reassortant strain having segment A from a very virulent strain but segment B from an attenuated strain was less pathogenic than a very virulent strain. Hence, it might be possible to relate the phenotypic properties of a virus to its genotypic characteristics based on both genome segments.
Jackwood et al. (Citation2018) proposed a system of nomenclature of IBDV strains following the scheme adopted for influenza viruses and coronaviruses. For example, the serotype 1 IBDV strain known commonly as STC was proposed to be designated as 1/chicken/ USA/STC/67(G1). We propose to further extend this nomenclature by adding genotypic identity including both segment A and segment B genogroup numbers, instead of only segment A genogroup number, at the end in parenthesis. Moreover, we propose to use S1 or S2 instead of 1 or 2 as serotypic designation at the beginning to avoid confusion with genotype numbers. The year of isolation could be in four digits to avoid any confusion in the future. Hence the updated nomenclature for classical STC strain would stand as S1/chicken/USA/STC/1967(A1B1), very virulent UK661 strain as S1/chicken/UK/UK661/1994(A3B2), dIBDV strain M04/09 as S1/chicken/Uruguay/M04/09/2009(A4B1), reassortant strain BD-11 as S1/chicken/Bangladesh/BD-11/2016(A3B3), serotype 2 strain 23/82 as S2/turkey/UK/23/82/1982(A0B1) and the “interserotypic reassortant” strain 100056 as S1/chicken/France/100056/2010(A3B1). It should be noted that, as the serotypic determinant lies in segment A, the serotypic designation (S1 or S2) of a strain should be based on segment A sequence irrespective of the nature of segment B.
Conclusion
The scheme of genotypic classification proposed in the present study will help reporting molecular characteristics of IBDV in a systematic and evolutionary way. A binomial way of genotypic designation of strains reflecting the segment A- and the segment B- genogroup is introduced here. It should be noted that point mutations in both the VP2 and VP1 genes may greatly affect antigenicity or pathogenicity (Eterradossi et al., Citation2004; Escaffre et al., Citation2013). As the VP2 hypervariable region contains the major antigenic domains, genogrouping based on segment A would broadly represent antigenic diversity. This binomial genotyping system of IBDV strains will additionally help in quickly describing the full genomic relatedness of any IBDV strain and, as a consequence, help for a better understanding of IBDV molecular epidemiology and evolution. However, the determination of antigenic diversity between the genogroups or genotypes, and thus effective IBD protection by vaccination, will still require serological studies and protection experiments using representative strains of different genogroups and/or genotypes. Similarly, pathogenicity of different genotypes should also be studied in controlled experiments.
Ethical approval
This study did not involve handling of any live animals or birds.
Supplemental Material
Download Zip (7.4 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Related Research Data
References
- Abed, M., Soubies, S., Courtillon, C., Briand, F.X., Allée, C., Amelot, M., De Boisseson, C., Lucas, P., Blanchard, Y., Belahouel, A., Kara, R., Essalhi, A., Temim, S., Khelef, D. & Eterradossi, N. (2018). Infectious bursal disease virus in Algeria: detection of highly pathogenic reassortant viruses. Infection Genetics and Evolution, 60, 48–57.
- Alfonso-Morales, A., Rios, L., Martinez-Perez, O., Dolz, R., Valle, R., Perera, C.L., Bertran, K., Frías, M.T., Ganges, L., Díaz de Arce, H., Majó, N., Núñez, J.I. & Pérez, L.J. (2015). Evaluation of a phylogenetic marker based on genomic segment B of infectious bursal disease virus: facilitating a feasible incorporation of this segment to the molecular epidemiology studies for this viral agent. PLoS One, 10, e0125853.
- Alkie, T.N. & Rautenschlein, S. (2016). Infectious bursal disease virus in poultry: current status and future prospects. Veterinary Medicine, 7, 9–18.
- Bayliss, C.D., Spies, U., Shaw, K., Peters, R.W., Papageorgiou, A., Müller, H. & Boursnell, M.E. (1990). A comparison of the sequences of segment A of four infectious bursal disease virus strains and identification of a variable region in VP2. Journal of General Virology, 71, 1303–1312.
- Becht, H. (1980). Infectious bursal disease virus. Current Topics in Microbiology and Immunology, 90, 107–121.
- Böttcher, B., Kiselev, N.A., Stel’Mashchuk, V.Y., Perevozchikova, N.A., Borisov, A.V. & Crowther, R.A. (1997). Three-dimensional structure of infectious bursal disease virus determined by electron cryomicroscopy. Journal of Virology, 71, 325–330.
- Chen, F., Liu, J., Yan, Z., Liu, D., Ji, J., Qin, J., Li, H., Ma, J., Bi, Y. & Xie, Q. (2012). Complete genome sequence analysis of a natural reassortant infectious bursal disease virus in China. Journal of Virology, 86, 11942–11943.
- Chettle, N., Stuart, J.C. & Wyeth, P.J. (1989). Outbreak of virulent infectious bursal disease in East Anglia. Veterinary Record, 125, 271–272.
- Coulibaly, F., Chevalier, C., Gutsche, I., Pous, J., Navaza, J., Bressanelli, S., Delmas, B. & Rey, F.A. (2005). The birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell, 120, 761–772.
- Delmas, B. (2008). Birnaviruses. In B.W.J. Mahy & M.H.V. van Regenmortel (Eds.), Encyclopedia of Virology 3rd edn. (Vol. 1, pp. 321–328). Oxford: Elsevier.
- Delmas, B., Attoui, H., Ghosh, S., Malik, Y.S., Mundt, E., Vakharia, V.N. & Report Consortium, I.C.T.V. (2019). ICTV virus taxonomy profile: Birnaviridae. Journal of General Virology, 100, 5–6.
- Diel, D.G., da Silva, L.H., Liu, H., Wang, Z., Miller, P.J. & Afonso, C.L. (2012). Genetic diversity of avian paramyxovirus type 1: proposal for a unified nomenclature and classification system of Newcastle disease virus genotypes. Infection Genetics and Evolution, 12, 1770–1779.
- Dimitrov, K.M., Abolnik, C., Afonso, C.L., Albina, E., Bahl, J., Berg, M., Briand, F.X., Brown, I.H., Choi, K.S., Chvala, I., Diel, D.G., Durr, P.A., Ferreira, H.L., Fusaro, A., Gil, P., Goujgoulova, G.V., Grund, C., Hicks, J.T., Joannis, T.M., Torchetti, M.K., Kolosov, S., Lambrecht, B., Lewis, N.S., Liu, H., Liu, H., McCullough, S., Miller, P.J., Monne, I., Muller, C.P., Munir, M., Reischak, D., Sabra, M., Samal, S.K., Servan de Almeida, R., Shittu, I., Snoeck, C.J., Suarez, D.L., Van Borm, S., Wang, Z. & Wong, F.Y.K. (2019). Updated unified phylogenetic classification system and revised nomenclature for Newcastle disease virus. Infection Genetics and Evolution, 74, 103917.
- Dobos, P., Hill, B.J., Hallett, R., Kells, D.T., Becht, H. & Teninges, D. (1979). Biophysical and biochemical characterization of five animal viruses with bisegmented double-stranded RNA genomes. Journal of Virology, 32, 593–605.
- Domanska, K., Mato, T., Rivallan, G., Smietanka, K., Minta, Z., de Boisseson, C., Toquin, D., Lomniczi, B., Palya, V. & Eterradossi, N. (2004). Antigenic and genetic diversity of early European isolates of infectious bursal disease virus prior to the emergence of the very virulent viruses: early European epidemiology of infectious bursal disease virus revisited? Archives of Virology, 149, 465–480.
- Escaffre, O., Le Nouën, C., Amelot, M., Ambroggio, X., Ogden, K.M., Guionie, O., Toquin, D., Müller, H., Islam, M.R. & Eterradossi, N. (2013). Both genome segments contribute to the pathogenicity of very virulent infectious bursal disease virus. Journal of Virology, 87, 2767–2780.
- Eterradossi, N., Gauthier, C., Reda, I., Comte, S., Rivallan, G., Toquin, D., De Boisseson, C., Lamande, J., Jestin, V., Morin, Y., Cazaban, C. & Borne, P.M. (2004). Extensive antigenic changes in an atypical isolate of infectious bursal disease virus and experimental clinical control of this virus with an antigenically classical live vaccine. Avian Pathology, 33, 423–431.
- Eterradossi, N. & Saif, Y.M. (2019). Infectious bursal disease. In D.E. Swayne, M. Boulianne, C.M. Logue, L.R. McDougald, V. Nair, D.L. Suarez, S. de Wit, T. Grimes, D. Johnson, M. Kromm, T.Y. Prajitno, I. Rubinoff & G. Zavala (Eds.), Diseases of Poultry 14th edn (pp. 257–283). NJ: John Wiley.
- Fernandes, M.J., Simoni, I.C., Harakava, R., Rivas, E.B. & Arns, C.W. (2012). Partial VP1 sequencing of Brazilian infectious bursal disease virus strains. Brazilian Journal of Microbiology, 43, 1015–1021.
- Firth, G.A. (1974). Letter: Occurrence of an infectious bursal syndrome within an Australian poultry flock. Australian Veterinary Journal, 50, 128–130.
- Gao, H.L., Wang, X.M., Gao, Y.L. & Fu, C.Y. (2007). Direct evidence of reassortment and mutant spectrum analysis of a very virulent infectious bursal disease virus. Avian Diseases, 51, 893–899.
- Gao, L., Li, K., Qi, X., Gao, Y., Wang, Y., Gao, H. & Wang, X. (2018). N-terminal domain of the RNA polymerase of very virulent infectious bursal disease virus contributes to viral replication and virulence. Science China. Life Sciences, 61, 1127–1129.
- Garriga, D., Querol-Audí, J., Abaitua, F., Saugar, I., Pous, J., Verdaguer, N., Castón, J.R. & Rodriguez, J.F. (2006). The 2.6-Angstrom structure of infectious bursal disease virus-derived T=1 particles reveals new stabilizing elements of the virus capsid. Journal of Virology, 80, 6895–6905.
- He, X., Xiong, Z., Yang, L., Guan, D., Yang, X. & Wei, P. (2014). Molecular epidemiology studies on partial sequences of both genome segments reveal that reassortant infectious bursal disease viruses were dominantly prevalent in southern China during 2000-2012. Archives of Virology, 159, 3279–3292.
- Hernandez, M., Tomás, G., Marandino, A., Iraola, G., Maya, L., Mattion, N., Hernández, D., Villegas, P., Banda, A., Panzera, Y. & Pérez, R. (2015). Genetic characterization of South American infectious bursal disease virus reveals the existence of a distinct worldwide-spread genetic lineage. Avian Pathology, 44, 212–221.
- Hoang, D.T., Chernomor, O., von Haeseler, A., Minh, B.Q. & Vinh, L.S. (2018). UFBoot2: improving the ultrafast bootstrap approximation. Molecular Biology and Evolution, 35, 518–522.
- Hon, C.C., Lam, T.Y., Drummond, A., Rambaut, A., Lee, Y.F., Yip, C.W., Zeng, F., Lam, P.Y., Ng, P.T. & Leung, F.C. (2006). Phylogenetic analysis reveals a correlation between the expansion of very virulent infectious bursal disease virus and reassortment of its genome segment B. Journal of Virology, 80, 8503–8509.
- Hussain, A., Wu, T., Li, H., Fan, L., Li, K., Gao, L., Wang, Y., Gao, Y., Liu, C., Cui, H., Pan, Q., Zhang, Y., Aslam, A., Muti-Ur-Rehman, K., Munir, M., Butt, S.L., Wang, X. & Qi, X. (2019). Pathogenic characterization and full length genome sequence of a reassortant infectious bursal disease virus newly isolated in Pakistan. Virologica Sinica, 34, 102–105.
- Islam, M.R., Rahman, S., Noor, M., Chowdhury, E.H. & Müller, H. (2012). Differentiation of infectious bursal disease virus (IBDV) genome segment B of very virulent and classical lineage by RT-PCR amplification and restriction enzyme analysis. Archives of Virology, 157, 333–336.
- Islam, M.R., Zierenberg, K., Eterradossi, N., Toquin, D., Rivallan, G. & Müller, H. (2001). Molecular and antigenic characterization of Bangladeshi isolates of infectious bursal disease virus demonstrate their similarities with recent European, Asian and African very virulent strains. Journal of Veterinary Medicine. B, Infectious Diseases and Veterinary Public Health, 48, 211–221.
- Islam, M., Zierenberg, K. & Müller, H. (2001). The genome segment B encoding the RNA-dependent RNA polymerase protein VP1 of very virulent infectious bursal disease virus (IBDV) is phylogenetically distinct from that of all other IBDV strains. Archives of Virology, 146, 2481–2492.
- Jackwood, D.J. (2012). Molecular epidemiologic evidence of homologous recombination in infectious bursal disease viruses. Avian Diseases, 56, 574–577.
- Jackwood, D.J., Crossley, B.M., Stoute, S.T., Sommer-Wagner, S., Woolcock, P.R. & Charlton, B.R. (2012). Diversity of genome segment B from infectious bursal disease viruses in the United States. Avian Diseases, 56, 165–172.
- Jackwood, D.J., Schat, K.A., Michel, L.O. & de Wit, S. (2018). A proposed nomenclature for infectious bursal disease virus isolates. Avian Pathology, 47, 576–584.
- Jackwood, D.J., Sommer-Wagner, S.E., Crossley, B.M., Stoute, S.T., Woolcock, P.R. & Charlton, B.R. (2011). Identification and pathogenicity of a natural reassortant between a very virulent serotype 1 infectious bursal disease virus (IBDV) and a serotype 2 IBDV. Virology, 420, 98–105.
- Jackwood, D.J., Stoute, S.T. & Crossley, B.M. (2016). Pathogenicity of genome reassortant infectious bursal disease viruses in chickens and turkeys. Avian Diseases, 60, 765–772.
- Kalyaanamoorthy, S., Minh, B.Q., Wong, T.K.F., von Haeseler, A. & Jermiin, L.S. (2017). Modelfinder: fast model selection for accurate phylogenetic estimates. Nature Methods, 14, 587–589.
- Kasanga, C.J. (2015). Partial molecular characterization of infectious bursal disease virus detected in Africa: potential evidence for virus recombination and genome segments A and B reassortment in nature. Research Opinions in Animal & Veterinary Sciences, 5, 468–475.
- Kasanga, C.J., Yamaguchi, T., Wambura, P.N., Maeda-Machang’u, A.D., Ohya, K. & Fukushi, H. (2007). Molecular characterization of infectious bursal disease virus (IBDV): diversity of very virulent IBDV in Tanzania. Archives of Virology, 152, 783–790.
- Kibenge, F.S., Dhillon, A.S. & Russell, R.G. (1988). Biochemistry and immunology of infectious bursal disease virus. Journal of General Virology, 69, 1757–1775.
- Kumar, S., Stecher, G. & Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33, 1870–1874.
- Le Nouen, C., Rivallan, G., Toquin, D., Darlu, P., Morin, Y., Beven, V., de Boisseson, C., Cazaban, C., Comte, S., Gardin, Y. & Eterradossi, N. (2006). Very virulent infectious bursal disease virus: reduced pathogenicity in a rare natural segment-B-reassorted isolate. Journal of General Virology, 87, 209–216.
- Le Nouën, C., Toquin, D., Müller, H., Raue, R., Kean, K.M., Langlois, P., Cherbonnel, M. & Eterradossi, N. (2012). Different domains of the RNA polymerase of infectious bursal disease virus contribute to virulence. PLoS One, 7, e28064.
- Lee, C.C., Ko, T.P., Chou, C.C., Yoshimura, M., Doong, S.R., Wang, M.Y. & Wang, A.H. (2006). Crystal structure of infectious bursal disease virus VP2 subviral particle at 2.6A resolution: implications in virion assembly and immunogenicity. Journal of Structural Biology, 155, 74–86.
- Leong, J.C., Brown, D., Dobos, P., Kibenge, F., Ludert, J.E., Müller, H., Mundt, E., & Nicholson, B. (2000). Family Birnaviridae. Virus taxonomy: classification and nomenclature of viruses. In M.H.V. van Regenmortel, C.M. Fauquet, D.H.L. Bishop, E.B. Carstens, M.K. Estes, S.M. Lemon, J. Maniloff, M.A. Mayo, D.J. McGeoch, C.R. Pringle & R.B. Wickner (Eds.), Seventh report of the International Committee on Taxonomy of viruses (pp. 481–490). San Diego: Academic Press.
- Lombardo, E., Maraver, A., Espinosa, I., Fernandez-Arias, A. & Rodriguez, J.F. (2000). VP5, the nonstructural polypeptide of infectious bursal disease virus, accumulates within the host plasma membrane and induces cell lysis. Virology, 277, 345–357.
- Lu, Z., Zhang, L., Wang, N., Chen, Y., Gao, L., Wang, Y., Gao, H., Gao, Y., Li, K., Qi, X. & Wang, X. (2015). Naturally occurring reassortant infectious bursal disease virus in northern China. Virus Research, 203, 92–95.
- Lupini, C., Felice, V., Silveira, F., Mescolini, G., Berto, G., Listorti, V., Cecchinato, M. & Catelli, E. (2020). Comparative in vivo pathogenicity study of an ITA genotype isolate (G6) of infectious bursal disease virus. Transboundary and Emerging Diseases, 67, 1025–1031.
- Lupini, C., Giovanardi, D., Pesente, P., Bonci, M., Felice, V., Rossi, G., Morandini, E., Cecchinato, M. & Catelli, E. (2016). A molecular epidemiology study based on VP2 gene sequences reveals that a new genotype of infectious bursal disease virus is dominantly prevalent in Italy. Avian Pathology, 45, 458–464.
- Martin, D.P., Murrell, B., Golden, M., Khoosal, A. & Muhire, B. (2015). RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evolution, 1, vev003.
- Mato, T., Tatár-Kis, T., Felföldi, B., Jansson, D.S., Homonnay, Z., Bányai, K. & Palya, V. (2020). Occurrence and spread of a reassortant very virulent genotype of infectious bursal disease virus with altered VP2 amino acid profile and pathogenicity in some European countries. Veterinary Microbiology, 245, 108663.
- McFerran, J.B., McNulty, M.S., McKillop, E.R., Connor, T.J., McCracken, R.M., Collins, D.S. & Allan, G.M. (1980). Isolation and serological studies with infectious bursal disease viruses from fowl, turkeys and ducks: demonstration of a second serotype. Avian Pathology, 9, 395–404.
- Michel, L.O. & Jackwood, D.J. (2017). Classification of infectious bursal disease virus into genogroups. Archives of Virology, 162, 3661–3670.
- Müller, H. (1986). Replication of infectious bursal disease virus in lymphoid cells. Archives of Virology, 87, 191–203.
- Müller, H., Islam, M.R. & Raue, R. (2003). Research on infectious bursal disease - the past, the present and the future. Veterinary Microbiology, 97, 153–165.
- Müller, H., Mundt, E., Eterradossi, N. & Islam, M.R. (2012). Current status of vaccines against infectious bursal disease. Avian Pathology, 41, 133–139.
- Müller, H., Schnitzler, D., Bernstein, F., Becht, H., Cornelissen, D. & Lutticken, D.H. (1992). Infectious bursal disease of poultry: antigenic structure of the virus and control. Veterinary Microbiology, 33, 175–183.
- Müller, H., Scholtissek, C. & Becht, H. (1979). The genome of infectious bursal disease virus consists of two segments of double-stranded RNA. Journal of Virology, 31, 584–589.
- Mundt, E., Kollner, B. & Kretzschmar, D. (1997). VP5 of infectious bursal disease virus is not essential for viral replication in cell culture. Journal of Virology, 71, 5647–5651.
- Mundt, E. & Müller, H. (1995). Complete nucleotide sequences of 5'- and 3'-noncoding regions of both genome segments of different strains of infectious bursal disease virus. Virology, 209, 10–18.
- Nguyen, L.T., Schmidt, H.A., von Haeseler, A. & Minh, B.Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular Biology and Evolution, 32, 268–274.
- Nwagbo, I.O., Shittu, I., Nwosuh, C.I., Ezeifeka, G.O., Odibo, F.J., Michel, L.O. & Jackwood, D.J. (2016). Molecular characterization of field infectious bursal disease virus isolates from Nigeria. Veterinary World, 9, 1420–1428.
- OIE. (2019). Infectious bursal disease (Gumboro disease). In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals 2019. Paris: World Organisation for Animal Health. Available at: https://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/3.03.12_IBD.pdf (accessed on 13 January 2021).
- Öppling, V., Müller, H. & Becht, H. (1991). Heterogeneity of the antigenic site responsible for the induction of neutralizing antibodies in infectious bursal disease virus. Archives of Virology, 119, 211–223.
- Patel, A.K., Pandey, V.C. & Pal, J.K. (2016). Evidence of genetic drift and reassortment in infectious bursal disease virus and emergence of outbreaks in poultry farms in India. Virusdisease, 27, 161–169.
- Pikula, A., Lisowska, A., Jasik, A. & Smietanka, K. (2018). Identification and assessment of virulence of a natural reassortant of infectious bursal disease virus. Veterinary Research, 49, 89.
- Pikula, A., Smietanka, K. & Perez, L.J. (2020). Emergence and expansion of novel pathogenic reassortant strains of infectious bursal disease virus causing acute outbreaks of the disease in Europe. Transboundary and Emerging Diseases, 67, 1739–1744.
- Raja, P., Senthilkumar, T.M., Parthiban, M., Thangavelu, A., Gowri, A.M., Palanisammi, A. & Kumanan, K. (2016). Complete genome sequence analysis of a naturally reassorted infectious bursal disease virus from India. Genome Announcements, 4, e00709-16. doi:10.1128/genomeA.00709-16
- Sapats, S.I. & Ignjatovic, J. (2000). Antigenic and sequence heterogeneity of infectious bursal disease virus strains isolated in Australia. Archives of Virology, 145, 773–785.
- Snyder, D.B., Lana, D.P., Cho, B.R. & Marquardt, W.W. (1988). Group and strain-specific neutralization sites of infectious bursal disease virus defined with monoclonal antibodies. Avian Diseases, 32, 527–534.
- Soubies, S.M., Courtillon, C., Briand, F.X., Queguiner-Leroux, M., Courtois, D., Amelot, M., Grousson, K., Morillon, P., Herin, J.B. & Eterradossi, N. (2017). Identification of a European interserotypic reassortant strain of infectious bursal disease virus. Avian Pathology, 46, 19–27.
- Stoute, S.T., Jackwood, D.J., Crossley, B.M., Michel, L.O. & Blakey, J.R. (2019). Molecular epidemiology of endemic and very virulent infectious bursal disease virus genogroups in backyard chickens in California, 2009-2017. Journal of Veterinary Diagnostic Investigation, 31, 371–377.
- Sun, J.H., Lu, P., Yan, Y.X., Hua, X.G., Jiang, J. & Zhao, Y. (2003). Sequence and analysis of genomic segment A and B of very virulent infectious bursal disease virus isolated from China. Journal of Veterinary Medicine. B, Infectious Diseases and Veterinary Public Health, 50, 148–154.
- Tammiranta, N., Ek-Kommonen, C., Rossow, L. & Huovilainen, A. (2018). Circulation of very virulent avian infectious bursal disease virus in Finland. Avian Pathology, 47, 520–525.
- Tamura, K., Nei, M. & Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proceedings of the National Academy of Sciences of the United States of America, 101, 11030–11035.
- Tomas, G., Marandino, A., Techera, C., Olivera, V., Perbolianachis, P., Fuques, E., Grecco, S., Hernández, M., Hernández, D., Calleros, L., Craig, M.I., Panzera, Y., Vagnozzi, A. & Pérez, R. (2020). Origin and global spreading of an ancestral lineage of the infectious bursal disease virus. Transboundary and Emerging Diseases, 67, 1198–1212.
- van den Berg, T.P. (2000). Acute infectious bursal disease in poultry: a review. Avian Pathology, 29, 175–194.
- van den Berg, T.P., Gonze, M. & Meulemans, G. (1991). Acute infectious bursal disease in poultry: isolation and characterisation of a highly virulent strain. Avian Pathology, 20, 133–143.
- van den Berg, T.P., Gonze, M., Morales, D. & Meulemans, G. (1996). Acute infectious bursal disease in poultry: immunological and molecular basis of antigenicity of a highly virulent strain. Avian Pathology, 25, 751–768.
- Wang, Q., Hu, H., Chen, G., Liu, H., Wang, S., Xia, D., Yu, Y., Zhang, Y., Jiang, J., Ma, J., Xu, Y., Xu, Z., Ou, C. & Liu, X. (2019). Identification and assessment of pathogenicity of a naturally reassorted infectious bursal disease virus from Henan, China. Poultry Science, 98, 6433–6444.
- Xia, R.X., Wang, H.Y., Huang, G.M. & Zhang, M.F. (2008). Sequence and phylogenetic analysis of a Chinese very virulent infectious bursal disease virus. Archives of Virology, 153, 1725–1729.
- Zierenberg, K., Raue, R. & Müller, H. (2001). Rapid identification of “very virulent” strains of infectious bursal disease virus by reverse transcription-polymerase chain reaction combined with restriction enzyme analysis. Avian Pathology, 30, 55–62.
- Zierenberg, K., Raue, R., Nieper, H., Islam, M.R., Eterradossi, N., Toquin, D. & Müller, H. (2004). Generation of serotype 1/serotype 2 reassortant viruses of the infectious bursal disease virus and their investigation in vitro and in vivo. Virus Research, 105, 23–34.