
ABSTRACT
Real-time reverse transcription PCR (rRT-PCR) is a widely used method for Newcastle disease virus (NDV) diagnosis and classification. The virus matrix (M) gene is usually used for general NDV identification and the fusion (F) gene for virulence differentiation. This method is rapid and sensitive, but the high mutation rate of the viral genome is a source of concern, as it may cause diagnostic failure. In late 2019, we encountered a problem classifying virulent NDV in samples from a few poultry flocks in which clinical signs were observed. Although the general NDV identification test was positive, the rRT-PCR virulence differentiation test gave negative results or higher cycle threshold values (Ct) than expected. Partial F gene sequencing for unclassified clinical NDV samples was used to investigate the cause of the rRT-PCR virulence differentiation test failure. The cause of the diagnostic failure was found to be a mismatch at the 3′ end of the reverse primer and adjustments were made to the diagnostic assay. The partial sequencing also allowed us to differentiate the virus as virulent according to the amino acid sequence of the F gene cleavage site and to genetically classify the virus as sub-genotype VII.2 NDV. After realizing that this is a resurgence event of sub-genotype VII.2 virus in an already endemic area of sub-genotype VII.1, we traced the spread of the new virus for several months. In addition, whole-genome sequencing of two 2020 sub-genotype VII.2 isolates was performed. GenBank search yielded related sequences from Jordan (2018) and Pakistan (2015–2016).
In 2019, there was a resurgence of NDV from sub-genotype VII.2 in Israel, in an already endemic area of sub-genotype VII.1.
A mismatch at the 3′ end of the reverse primer caused a diagnostic failure of the NDV virulence differentiation rRT-PCR assay.
The 2019 NDV sub-genotype VII.2 virus is genetically close to viruses from Jordan (2018) and Pakistan (2015–2016).
RESEARCH HIGHLIGHTS
Introduction
Newcastle disease virus (NDV), formally called Avian orthoavulavirus-1 (Afonso et al., Citation2016; Amarasinghe et al., Citation2017; ICTV, Citation2019), is a non-segmented, single-stranded negative sense RNA virus. The viral genome codes for six proteins: the nucleoprotein (NP), phosphoprotein (P), matrix (M), fusion (F), haemagglutinin-neuraminidase (HN), and RNA polymerase (L).
Since low virulence strains (lentogenic) are widely used as live vaccines, only strains with intermediate (mesogenic) and high (velogenic) virulence are considered by the OIE (Office of International Epizootics) to be the cause of Newcastle disease (ND) (OIE, Citation2018). The virulence classification is based on the amino acid composition at the cleavage site of the fusion protein, or the intracerebral pathogenicity index (ICPI) (OIE, Citation2018).
The primary methods for NDV diagnosis are virus isolation and real-time reverse transcription PCR (rRT-PCR). Detection by rRT-PCR usually has two steps. The first step is a general identification of any NDV by targeting conserved genes such as NP, M and L. The most widely used method targets the M gene (Wise et al., Citation2004). The second step is the classification of the virus virulence by targeting the cleavage site of the F gene with rRT-PCR specific probes or by sequencing this region and deducing the amino acid motif of the cleavage site (Fuller et al., Citation2009; Ferreira & Suarez, Citation2019). rRT-PCR is a rapid and sensitive diagnostic method, but it is prone to diagnostic failure due to the high mutation rate of the viral genome (Miller et al., , Citation2010).
The genetic classification of NDV isolates is based on the nucleotide sequence of the complete coding region of the fusion gene (F) (Diel et al., Citation2012; Dimitrov et al., Citation2019). The current field strains in Israel belong to class II genotype VII, sub-genotypes VII.1 (previously classified as VIIb and VIId) and sub-genotype VII.2 (previously classified as VIIi). Sub-genotype VII.2 was present in Israel during the years 2011–2013 alongside genotype VII.1 and was not detected during the years 2013-2017. It reappeared in 2018–2020 (Wiseman et al., Citation2018; Israel Veterinary Services and Animal Health, Citation2019, Citation2021).
ND is endemic to Israel and all commercial poultry are vaccinated. At the end of 2019, we encountered a problem classifying virulent NDV by rRT-PCR in a few poultry flocks from Avdon village in northern Israel, in which clinical signs were observed. The clinical signs included low mortality and drop in egg production followed by typical shape deformity of the eggs in layer flocks and moderate mortality with neurological signs (torticollis) in broiler flocks. Our general NDV identification that targets the virus matrix (M) gene was positive, but the virulence differentiation test, based on the fusion (F) gene, gave negative results or higher cycle threshold values (Ct) than expected. We used partial F gene sequencing to investigate the cause of the diagnostic failure.
Materials and methods
RNA extraction
Total RNA was extracted from allantoic fluid or suspension of tracheal and cloacal swabs using the QIAmp Viral RNA Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions.
Real-time reverse transcriptase PCR (rRT-PCR)
The rRT-PCR assay that targeted the matrix (M) gene was performed according to Wise et al. (Citation2004). The virulence differentiation test that targeted the fusion (F) gene was based on Fuller et al. (Citation2009) with modification (different reverse primer that produces a shorter DNA fragment of 94 instead of 160 nucleotides). After the cause of the diagnostic failure was found, adjustments were made to the primers and probes of the virulence differentiation test and the identification of lentogenic and velogenic viruses was separated into two different rRT-PCR assays. The former and updated primers and probes are listed in . The AgPath-ID One-Step RT-PCR kit was used (Ambion, Austin, TX, USA) with the following reaction conditions. For the former test: 12.5 µl of kit-supplied buffer, 0.5 µl of each primer, 0.4 µl of probe, 1 µl of kit-supplied enzyme, 5.1 µl nuclease-free water to a total individual reaction volume of 25 µl containing 5 µl of RNA. For the updated velogenic test: 12.5 µl of kit-supplied buffer, 1.25 µl of each primer, 1.25 µl of probe, 1 µl of kit-supplied enzyme, 2.75 µl nuclease-free water to a total individual reaction volume of 25 µl containing 5 µl of RNA. Primers and probes were used at concentrations of 10 µM, and 5 µM respectively, in all reactions. Using the 7500 RT-PCR system or Step One Plus (Applied Biosystems), the cycling conditions were: after an initial reverse transcription step at 45°C for 10 min and an initial denaturation step at 95°C for 10 min, 40 cycles (95°C for 15 sec, 50°C for 30 sec, and 60°C for 32 sec) were performed, with fluorescence detection at the end of the annealing-extension step. Cycle threshold values (Ct) were calculated using the Applied Biosystems software. Reaction curve with Ct ≤40 was considered positive.
Table 1. List of primers and probes targeting NDV F gene for virulence differentiation rRT-PCR test.
Analytic sensitivity and specificity of the updated rRT-PCR test
Two representative NDV isolates from sub-genotypes VII.1 (sample number 344194) and VII.2 (sample number 352768) were selected. Tenfold serial dilutions of extracted RNA were used to evaluate the detection limit of the test. The titres of each isolate were determined in embryonated chicken eggs, calculated using the Reed-Muench method and expressed as log 50% egg-infective doses (log EID50) per microlitre of viral suspension. Comparisons were made between the former and new tests. The efficiency [efficiency % = (10(−1/slope) −1) × 100], slope, and a coefficient of determination (R2) of the rRT-PCR assay were determined using the Applied Biosystems 7500 real-time PCR or Step One Plus SDS software. Specificity testing was performed using lentogenic NDV, APMV-2, APMV-3 and other viruses and bacteria diagnosed by rRT-PCR in our laboratories.
Virology
Virus isolation was performed by injection of a suspension of tracheal and cloacal swabs into the allantoic cavity of 9- to 11-day-old-specific pathogen-free (SPF) embryonated chicken eggs (OIE, Citation2018). The swabs were tested in pools of five swabs (one to two pools for every source), and each pool was injected into five eggs. The allantoic fluid of eggs showing embryo mortality was collected and tested by haemagglutination assay (HA) and rRT-PCR in order to confirm the diagnosis.
Nucleotide sequencing of the F gene rRT-PCR primers and probe binding sites
Total RNA was extracted as described above. A ∼300 bp fragment of the F gene (positions 461-157), covering the region of the rRT-PCR primers and probe binding sites, was amplified with the primers NOH-For (5′-TACACCTCATCCCAGACAGG-3′) and NOH-Rev (5′-AGTCGGAGGATGTTGGCAGC-3′) as described by Cattoli et al. (Citation2010). After the amplification, the cDNA fragment was sequenced by the Sanger method (HyLabs, Israel).
Whole-genome sequencing (WGS)
We performed WGS of two isolates (sample numbers: 348118, 352768). Extraction of RNA was completed from 100 μl of the isolate in allantoic fluid using the Viral RNA Mini kit (Qiagen). Ribosomal RNA (rRNA) depletion was performed by using the Terminator 5′-Phosphate-Dependent Exonuclease (Epicentre Biotechnologies, Madison, WI, USA), followed by removal of deoxyribonucleic acid (DNA) using the DNase I (RNase-free) kit (New England BioLabs, Ipswich, MA, USA), according to the manufacturer’s instructions. After stopping the reaction by adding ethylenediaminetetraacetic acid (EDTA) to a concentration of 5 mM, the sample was cleaned up using a Viral RNA Mini kit (Qiagen). The elution volume was 60 μl (final RNA concentration was measured by Nanodrop: 134 ng/μl for sample 348118, and 117 ng/μl for sample 352768). Library preparation and WGS was done in the Technion Genome Center, Haifa, Israel. The cDNA libraries were prepared using the TruSeq RNA sample preparation kit. Paired-end sequencing (2X150) was performed using 300 cycles Illumina MiSeq Nano run. Consensus sequences were assembled by alignment of the contigs to a reference sequence using Geneious assembler (Geneious Prime version 2021.0.3). 384,818 of 1,666,066 reads and 280,523 of 1,546,422 reads were used to assemble samples 348118 and 352768, respectively. The sequence coverage was 99.3% (base coverage mean:3490) for sample 348118 and 99.5% (base coverage mean:2590) for sample 352768.
Accession numbers
The sequences of the NDV isolates included in the study were submitted to GenBank and received the accession numbers MZ712001 and MZ712002.
Outbreak data
Data on diagnosed ND cases were collected from the database of the Poultry Health Laboratories of Israel Egg and Poultry Board (Berman & Samberg, Citation1991). This database records veterinary data on all commercial flocks. A ND outbreak was defined according to the OIE guidelines (OIE, Citation2018).
Geographical data
The maps presented in this article were created with ArcMap™ 10.4. The geographical location (longitude and latitude) of all commercial poultry farms was obtained from the Israel Ministry of Agriculture GIS database.
Phylogenetic tree construction and estimates of evolutionary divergence between sequences
Phylogenetic analysis and estimates of evolutionary divergence between sequences of complete genome was performed with MEGA7 software (Tamura et al., Citation2004; Kumar et al., Citation2016). The evolutionary history was inferred using the Neighbor-Joining method. A phylogenetic tree was constructed based on 500 bootstrap replicates.
Results
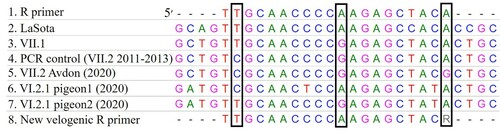
In order to investigate the cause of the rRT-PCR virulence differentiation test failure, partial sequencing of the F gene was performed for unsettled samples (when general NDV identification was positive, but the virulence differentiation test gave negative results or higher Ct values than expected). The amino acid sequence of the F gene cleavage site with basic amino acids at positions 115 and 116 and phenylalanine at 117 (112RRQKRF117) confirmed that the isolates were virulent (velogenic). The partial sequencing also allowed us to classify the virus genotype and revealed the resurgence of sub-genotype VII.2 in Israel. In the analysis of the nucleotide sequencing of the F gene rRT-PCR primers and probe binding sites, we found two mismatches in the forward primer, one mismatch in the probe and three mismatches in the reverse primer. Only one mismatch, at the 3′ end of the reverse primer (F gene position 372), was different between the new VII.2 virus and the VII.1 viruses or older VII.2 viruses (). We decided to separate the virulence differentiation test into two sets of probes and primers, one for lentogenic viruses and another for velogenic viruses. The velogenic probe and primers were modified to better fit the field strains in Israel (). The new rRT-PCR test is also suitable for the diagnosis of pigeon NDV strains (genotype VI) that are occasionally isolated in Israel from pigeons and wild birds.
Figure 1. The nucleotide sequence of the new VII.2 virus (5) differs from the R primer (1) at three positions (black frames), but only at the first 3′ position is there a difference which is not found in other field viruses (3, 4, 6, 7) (sub-genotype VII.1, older sub-genotype VII.2 and sub-genotype VI.2.1). In the new primer, this position was replaced with R (mix of A and G).
The sensitivity and the limit of detection (LOD) of the updated rRT-PCR test were assessed with two representative NDV isolates from sub-genotypes VII.1 (sample number 344194) and VII.2 (sample number 352768) at determined initial titres of 109.7 and 1010.4 EID50/ml, respectively. The endpoints of detection were 102.7 and 103.4 EID50/ml, respectively. According to the linear segment of the reaction slope, the limit of quantification was defined between 107.7 and 102.7 for sub-genotypes VII.1 (Y = −3.36x + 45.11, R2 = 0.99, efficiency = 98%) and between 1010.4 and 103.4 for sub-genotypes VII.2 (Y = −3.29x + 47.65, R2 = 0.99, efficiency = 101%).
A comparison was made between the former and the new velogenic rRT-PCR tests with nine sub-genotype VII.1 samples and seven newly introduced 2019–2020 sub-genotype VII.2 samples. The sample type and Ct values obtained are shown in .
Table 2. Comparison of Ct values of general NDV identification and the former and updated velogenic rRT-PCR tests for sub-genotype VII.1 and newly introduced 2019-2020 VII.2 samples.
The specificity for velogenic NDV was tested using viruses and bacteria diagnosed by rRT-PCR in our laboratories: lentogenic NDV, APMV-2, APMV-3, IBV, avian influenza (H5N1, H7N3, H9N2), aMPV, TMEV, reovirus, Salmonella Enteritidis, Salmonella Typhimurium, Mycoplasma gallisepticum, Mycoplasma synoviae, Mycoplasma meleagridis and Avibacterium paragallinarum. The results were negative for all tested pathogens with the velogenic NDV probes and primers (data not shown). Positive controls of each strain of pathogen, in an rRT-PCR assay with a primer-probe set designed for its detection, yielded a positive result in all cases.
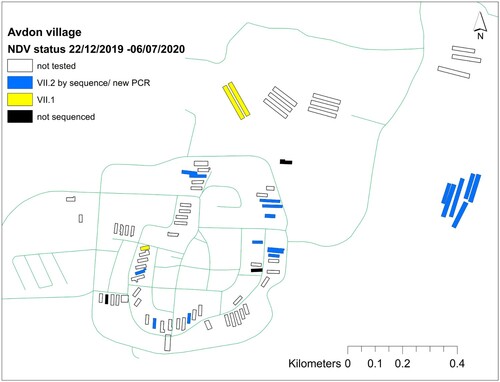
Following the identification of the new sub-genotype VII.2 introduction event, we further traced the virus spread by sampling additional viruses for partial F gene sequencing. A total of 51 samples isolated from 21 villages between April 2019 and December 2020 were sequenced (supplementary material). The phylogenetic analysis of the 51 short sequences is shown in Figure S1. In total, the VII.2 sub-genotype was detected in five villages (). In three of the villages (Avdon, Goren, Yaara), we found cocirculation of viruses from both sub-genotypes. is an example from Avdon village of the cocirculation of viruses in different farms in the same village.
Figure 2. Villages with ND outbreaks between April 2019 and December 2020. Viruses from sub-genotype VII.2 were detected in five villages. In three of the villages (Avdon, Goren, Yaara), we found cocirculation of viruses from both sub-genotypes.
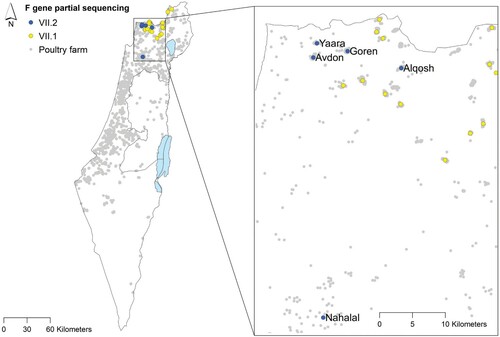
Figure 3. Cocirculation of viruses from sub-genotypes VII.1 and VII.2 in different farms in Avdon village. This is a typical example of a village with poultry farms in northern Israel.
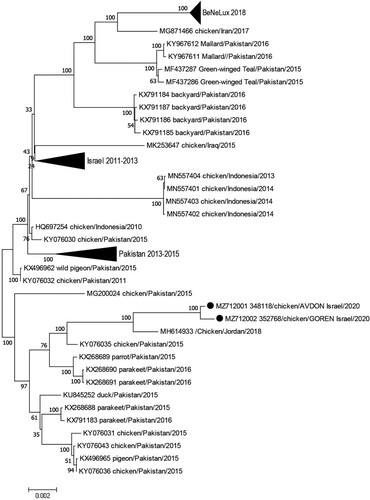
A whole-genome sequencing was performed for two of the 2020 sub-genotype VII.2 isolates. GenBank blast found high nucleotide similarity to a virus isolated in Jordan in 2018 (99.2%) and to viruses isolated in Pakistan in 2015–2016 (98.1–98.8%). There is about 2% difference between the new virus and sub-genotype VII.2 viruses isolated in Israel in 2011–2013. A phylogenetic tree of sub-genotype VII.2 viruses with up to 97% similarity is shown in .
Figure 4. Phylogenetic tree based on the complete sequence of sub-genotype VII.2 viruses with up to 97% similarity to the sub-genotype VII.2 isolated in Israel in 2020 (marked by a black circle). The evolutionary history was inferred using the Neighbor-Joining method (Saitou & Nei, Citation1987). The optimal tree with the sum of branch length = 0.15091515 is shown. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown next to the branches (Felsenstein, Citation1985). The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Maximum Composite Likelihood method (Tamura et al., Citation2004) and are in the units of the number of base substitutions per site. The analysis involved 85 nucleotide sequences. All positions with less than 80% site coverage were eliminated. That is, fewer than 20% alignment gaps, missing data, and ambiguous bases were allowed at any position. There was a total of 15,170 positions in the final dataset. Evolutionary analyses were conducted in MEGA7 (Kumar et al., Citation2016).
Discussion
This article describes the resurgence of NDV sub-genotype VII.2 in Israel and the special circumstances that led to the discovery. The resurgence was discovered due to a failure to classify the viral pathogenicity by rRT-PCR. The cause of the diagnostic failure was a third mismatch in the nucleotide sequence between the viral template and the sequence of the reverse primer. the mismatch is located at the 3′ end of the reverse primer. The number of mismatches with primers and probes can affect the sensitivity of the rRT-PCR assay. Mismatches can reduce annealing, and this leads to reduced efficiency and sensitivity of the assay. A high number of mismatches (≥ 3) might lead to a failure to detect the virus. The position of the mismatch at the 3′ end may also contribute to the failure and lead to false-negative results (Fuller et al., Citation2009; Cattoli et al., Citation2010; Ferreira & Suarez, Citation2019).
Modifications were made to the F gene velogenic diagnostic assay to adjust it to the circulating field isolates. For the 2019–2020 VII.2 samples, the updated rRT-PCR test enabled the virulence classification that failed in the former test. For VII.1 samples, slightly lower Ct values were obtained with the updated rRT-PCR test. Although it may also fit other genotypes, the updated test was intended to detect the local circulating genotypes and was not tested on other worldwide genotypes.
Genotype VII is a large and genetically diverse group of virulent NDV (Dimitrov et al., Citation2016). Sub-genotype VII.2 originated in South-East Asia in 2010, spread to the Middle East and Eastern Europe and continued Westward spread into North-West Europe (BeNeLux region) in 2018 (Fuller et al., Citation2015; Miller et al., Citation2015; Steensels et al., Citation2020; Wiseman et al., Citation2018). The 2020 VII.2 virus from Israel has high nucleotide similarity to viruses from Jordan (Ababneh et al., Citation2018) and Pakistan, but the specific mutation in the reverse primer binding site was not discovered in virus sequences from other countries.
The vast genomic diversity of NDV poses a diagnostic challenge (Miller et al., Citation2010). The use of two different rRT-PCR assays, one for general NDV identification (M gene assay) and one for virulence differentiation (F gene assay), is an advantage that can prevent cases of unidentified infection. It is also recommended to use additional diagnostic methods such as virology and serology. It is important to be alert to abnormal rRT-PCR results (including a suspicious difference in Ct values between the two assays), as well as to inconsistencies between clinical findings and laboratory test results. Since NDV will continue to challenge us in the future, it is recommended to monitor and genetically characterize circulating strains. This will contribute to the ongoing efforts of diagnostic assay development and optimization.
Supplemental Material
Download MS Word (31.3 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Ababneh, M., Ferreira, H.L., Khalifeh, M., Suarez, D.L. & Afonso, C.L. (2018). First genome sequence of Newcastle Disease virus of genotype VIIi from Jordan. Microbiology Resource Announcements, 7, e01136-18.
- Afonso, C.L., Amarasinghe, G.K., Bányai, K., Bào, Y., Basler, C.F., Bavari, S., Bejerman, N., Blasdell, K.R., Briand, F.-X., Briese, T., Bukreyev, A., Calisher, C.H., Chandran, K., Chéng, J., Clawson, A.N., Collins, P.L., Dietzgen, R.G., Dolnik, O., Domier, L.L., Dürrwald, R., Dye, J.M., Easton, A.J., Ebihara, H., Farkas, S.L., Freitas-Astúa, J., Formenty, P., Fouchier, R.A.M., Fù, Y., Ghedin, E., Goodin, M.M., Hewson, R., Horie, M., Hyndman, T.H., Jiāng, D., Kitajima, E.W., Kobinger, G.P., Kondo, H., Kurath, G., Lamb, R.A., Lenardon, S., Leroy, E.M., Li, C.-X., Lin, X.-D., Liú, L., Longdon, B., Marton, S., Maisner, A., Mühlberger, E., Netesov, S.V., Nowotny, N., Patterson, J.L., Payne, S.L., Paweska, J.T., Randall, R.E., Rima, B.K., Rota, P., Rubbenstroth, D., Schwemmle, M., Shi, M., Smither, S.J., Stenglein, M.D., Stone, D.M., Takada, A., Terregino, C., Tesh, R.B., Tian, J.-H., Tomonaga, K., Tordo, N., Towner, J.S., Vasilakis, N., Verbeek, M., Volchkov, V.E., Wahl-Jensen, V., Walsh, J.A., Walker, P.J., Wang, D., Wang, L.-F., Wetzel, T., Whitfield, A.E., Xiè, J., Yuen, K.-Y., Zhang, Y.-Z. & Kuhn, J.H. (2016). Taxonomy of the order Mononegavirales: update 2016. Archives of Virology, 161, 2351–2360.
- Amarasinghe, G.K., Bào, Y., Basler, C.F., Bavari, S., Beer, M., Bejerman, N., Blasdell, K.R., Bochnowski, A., Briese, T., Bukreyev, A., Calisher, C.H., Chandran, K., Collins, P.L., Dietzgen, R.G., Dolnik, O., Dürrwald, R., Dye, J.M., Easton, A.J., Ebihara, H., Fang, Q., Formenty, P., Fouchier, R.A.M., Ghedin, E., Harding, R.M., Hewson, R., Higgins, C.M., Hong, J., Horie, M., James, A.P., Jiāng, D., Kobinger, G.P., Kondo, H., Kurath, G., Lamb, R.A., Lee, B., Leroy, E.M., Li, M., Maisner, A., Mühlberger, E., Netesov, S.V., Nowotny, N., Patterson, J.L., Payne, S.L., Paweska, J.T., Pearson, M.N., Randall, R.E., Revill, P.A., Rima, B.K., Rota, P., Rubbenstroth, D., Schwemmle, M., Smither, S.J., Song, Q., Stone, D.M., Takada, A., Terregino, C., Tesh, R.B., Tomonaga, K., Tordo, N., Towner, J.S., Vasilakis, N., Volchkov, V.E., Wahl-Jensen, V., Walker, P.J., Wang, B., Wang, D., Wang, F., Wang, L.-F., Werren, J.H., Whitfield, A.E., Yan, Z., Ye, G. & Kuhn, J.H. (2017). Taxonomy of the order Mononegavirales: update 2017. Archives of Virology, 162, 2493–2504.
- Berman, E.M. & Samberg, Y. (1991). A national medical database for poultry in Israel. Abstracts, S24.3. Presented at the XXIV World Veterinary Congress, Rio de Janeiro, Brazil.
- Cattoli, G., Fusaro, A., Monne, I., Molia, S., Le Menach, A., Maregeya, B., Nchare, A., Bangana, I., Maina, A.G., N’Goran Koffi, J.-N., Thiam, H., Bezeid, O.E.M.A., Salviato, A., Nisi, R., Terregino, C. & Capua, I. (2010). Emergence of a new genetic lineage of Newcastle disease virus in West and Central Africa – implications for diagnosis and control. Veterinary Microbiology, 142, 168–176.
- Diel, D.G., da Silva, L.H.A., Liu, H., Wang, Z., Miller, P.J. & Afonso, C.L. (2012). Genetic diversity of avian paramyxovirus type 1: proposal for a unified nomenclature and classification system of Newcastle disease virus genotypes. Infection, Genetics and Evolution, 12, 1770–1779.
- Dimitrov, K.M., Abolnik, C., Afonso, C.L., Albina, E., Bahl, J., Berg, M., Briand, F.-X., Brown, I.H., Choi, K.-S., Chvala, I., Diel, D.G., Durr, P.A., Ferreira, H.L., Fusaro, A., Gil, P., Goujgoulova, G.V., Grund, C., Hicks, J.T., Joannis, T.M., Torchetti, M.K., Kolosov, S., Lambrecht, B., Lewis, N.S., Liu, H., Liu, H., McCullough, S., Miller, P.J., Monne, I., Muller, C.P., Munir, M., Reischak, D., Sabra, M., Samal, S.K., Servan de Almeida, R., Shittu, I., Snoeck, C.J., Suarez, D.L., Van Borm, S., Wang, Z. & Wong, F.Y.K. (2019). Updated unified phylogenetic classification system and revised nomenclature for Newcastle disease virus. Infection, Genetics and Evolution, 74, 103917.
- Dimitrov, K.M., Ramey, A.M., Qiu, X., Bahl, J. & Afonso, C.L. (2016). Temporal, geographic, and host distribution of avian paramyxovirus 1 (Newcastle disease virus). Infection, Genetics and Evolution, 39, 22–34.
- Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution, 39, 783–791.
- Ferreira, H.L. & Suarez, D.L. (2019). Single-nucleotide polymorphism analysis to select conserved regions for an improved real-time reverse transcription–PCR test specific for Newcastle disease virus. Avian Diseases, 63, 625–633.
- Fuller, C., Löndt, B., Dimitrov, K.M., Lewis, N., van Boheemen, S., Fouchier, R., Coven, F., Goujgoulova, G., Haddas, R. & Brown, I. (2015). An epizootiological report of the re-emergence and spread of a lineage of virulent Newcastle Disease virus into Eastern Europe. Transboundary and Emerging Diseases, 64, 1001–1007.
- Fuller, C.M., Collins, M.S. & Alexander, D.J. (2009). Development of a real-time reverse-transcription PCR for the detection and simultaneous pathotyping of Newcastle disease virus isolates using a novel probe. Archives of Virology, 154, 929–937.
- ICTV. (2019). Genus: Orthoavulavirus – Paramyxoviridae – Negative-sense RNA viruses. International Committee on Taxonomy of Viruses (ICTV).
- Israel Veterinary Services and Animal Health. (2019). Israeli Veterinary services 2016-2018 Annual report (p. 193). Beit Dagan: Israel Veterinary Services and Animal Health.
- Israel Veterinary Services and Animal Health. (2021). Israeli Veterinary services 2020 Annual report (p. 127). Israel Beit Dagan: Veterinary Services and Animal Health.
- Kumar, S., Stecher, G. & Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33, 1870–1874.
- Miller, P., Haddas, R., Simanov, L., Lublin, A., Rehmani, S., Wajid, A., Bibi, T., Khan, T., Yaqub, T., Setiyaningsih, S. & Afonso, C. (2015). Identification of new sub-genotypes of virulent Newcastle disease virus with potential panzootic features. Infection, Genetics and Evolution, 29, 216–229.
- Miller, P.J., Decanini, E.L. & Afonso, C.L. (2010). Newcastle disease: evolution of genotypes and the related diagnostic challenges. Infection, Genetics and Evolution, 10, 26–35.
- OIE. (2018). Infection with Avian Influenza viruses. OIE Terrestrial Manual 2018.
- Saitou, N. & Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4, 406–425.
- Steensels, M., Borm, S.V., Mertens, I., Houdart, P., Rauw, F., Roupie, V., Snoeck, C.J., Bourg, M., Losch, S., Beerens, N., Berg, T.v.d. & Lambrecht, B. (2020). Molecular and virological characterization of the first poultry outbreaks of Genotype VII.2 velogenic avian orthoavulavirus type 1 (NDV) in North-West Europe, BeNeLux, 2018. Transboundary and Emerging Diseases, 68, 2147–2160.
- Tamura, K., Nei, M. & Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proceedings of the National Academy of Sciences of the United States of America, 101, 11030–11035.
- Wise, M.G., Suarez, D.L., Seal, B.S., Pedersen, J.C., Senne, D.A., King, D.J., Kapczynski, D.R. & Spackman, E. (2004). Development of a real-time reverse-transcription PCR for detection of Newcastle disease virus RNA in clinical samples. Journal of Clinical Microbiology, 42, 329–338.
- Wiseman, A., Ben Izhak, M., Tal, S., Wachtel, C., Berman, E., Yechezkel, E., Golan, E., Haddas, R., Lublin, A., Raviv, Z., Pirak, M., Klement, E. & Louzoun, Y. (2018). Epidemiological and genetic analysis of avian avulavirus-1 in Israel reveals parallel circulating strains and a new sub-genotype within genotype VI. Infection, Genetics and Evolution, 66, 159–170.