1. PREDICTION OF CLINICAL SIDE EFFECTS FROM PRECLINICAL DATA: THE COX-2 STORY
Garret A. FitzGerald
Institute for Translational Medicine and Therapeutics, University of Pennsylvania School of Medicine, Philadelphia, PA, USA, 19104-6160
Early studies of the human pharmacology of nonsteroidal antinflammatory drugs (NSAIDs) selective for inhibition of COX-2, rofecoxib and celecoxib, revealed that they depressed biosynthesis of prostacyclin (PGI2), a compound with platelet inhibitory properties in vitro, while variably depressing the platelet agonist, thromboxane (Tx) A2, raising the possibility of a class specific hazard, particularly in patients predisposed to cardiovascular risk. Studies in mice deficient in the I prostanoid receptor and mice harboring a COX-2 mutation that mimicked drug action established the importance of a PGI2 as a restraint, not only on stimuli to platelet activation in vivo, but also on factors that elevate blood pressure, influence vascular remodeling and promote atherogenenesis. Selective deletion of COX-2 in cardiomyocytes resulted in congestive failure and a predisposition to arrhythmia. Before the withdrawal of rofecoxib, observational studies variably detected a cardiovascular risk at a high dose of rofecoxib, but largely missed any signal from celecoxib or valdecoxib. Seven placebo controlled trials have revealed that celecoxib, rofecoxib and valdecoxib confer a cardiovascular risk, variably comprised of myocardial infarction, congestive heart failure, stroke and sudden cardiac death. Evidence of a similar hazard has accumulated for lumiracoxib, etoricoxib and diclofenac. Comparative trials amongst the NSAIDs are consistent with this hazard relating to the degree of selectivity for inhibition of COX-2. Randomized trials of celecoxib relate this hazard to drug dosage and to antecedent cardiovascular risk. Comparisons of inter and intra-individual differences reflect high variability in response to rofecoxib and celecoxib and suggest that up to 30% may be attributable to genetic variance. Prediction of the mechanism based cardiovascular hazard of novel and traditional NSAIDs selective for COX-2 was based on human and rodent pharmacology confirmed by placebo controlled and comparative randomized trials1. Pharmacoepidemiological approaches variably detected a signal from a high dose of a long lived and highly selective drug – rofecoxib – where there was considerable experience of drug exposure. Traditional approaches to drug safety might be usefully informed by mechanistic information derived from basic and human pharmacology.
REFERENCE
- Grosser T., Fries S. and FitzGerald G. A. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006 Jan; 116(1):4–15.
2. PREDICTION OF SUBSTRATE PROPERTIES FOR ABC-DRUG TRANSPORTERS: THE CHALLENGE CONTINUES
Gerhard F. Ecker1, Michael Demel1, Rita Schwaha1, Dominik Kaiser1, Oliver Krämer2, Peter Ettmayer2, and Eric Haaksma2
1Department for Medicinal Chemistry, University of Vienna, Wien, Austria, 1090, 2Boehringer Ingelheim Austria GmbH, Wien, Austria, 1121
Membrane-bound multidrug transporters of the ABC-family actively extrude a large variety of therapeutically administered drugs from malignant cells and are responsible for multiple drug resistance in cancer patients. Additionally, they are involved in tissue protection, absorption and brain uptake. Furthermore, there is increasing evidence that cholestatic forms of drug-induced liver damage result from a drug- or metabolite-mediated inhibition of hepatobiliary transporter systems, such as ABCB1, ABCB4, ABCG2, ABCG5 and ABCG8. Therefore, interaction with ABC-transporters determines the clinical usefulness and ADMET properties of drugs.
Most of the clinically relevant ABC-transporters show a polyspecific ligand recognition pattern. ABCB1, ABCC1 and ABCG2, the three key transporters involved in multiple drug resistance in tumour therapy, efflux a broad panel of structurally and functionally diverse compounds including polypeptides, hormones, and lipids. Due to the lack of high resolution protein structure information the prediction of substrate properties of compounds of interest has to rely on ligand-based models. However, the promiscuity of these transporters requires more sophisticated algorithms than those routinely used in pharmacoinformatics. Especially non-linear methods such as artificial neural networks and similarity-based approaches showed good performance in in silico screening runs for identification of new P-glycoprotein modulators. Furthermore, combined pharmacophore modelling/ QSAR studies gave first insights into strategies for designing compounds with selectivity for either of the two polyspecific transporters ABCB1 and ABCG2.
Prediction of substrates is further challenged by the limited data available. One of the most interesting data sets is the one derived from the NCI60 cancer screen [1], which includes substrate properties of 1400 compounds on 48 human ABC-transporter. When combining autocorrelation vectors and counter propagation neural networks these data gave good results for classification of compounds into substrates and non-substrates at least for some of the transporters.
Financial support provided by the Austrian Science fund (#L344-N17) and the Austrian Research Promotion Agency (#B1–812074)
REFERENCE
- Szakacs, G., Annereau J.-P., et al. (2004). Predicting drug sensitivity and resistance: Profiling ABC transporter genes in cancer cells. Cancer Cell 6(2): 129–137.
Text Not Available
4. TIME-SERIES BEHAVIOUR OF QSAR ADMET MODELS: THE NEED FOR REGULAR UPDATING OF IN SILICO MODELS
Han Van de Waterbeemd
DECS - Global Compound Sciences, AstraZeneca, Macclesfield, United Kingdom, SK10 4TG
In recent years predictive modeling of ADMET data has become well established as a strategy to improve compound quality in guiding projects towards compounds with lead-like and drug-like properties.Tools used include mechanistical approaches such as physiologically-based pharmacokinetic (PBPK) modeling, molecular modeling of metabolising enzymes such as P450s and its ligands, expert systems for metabolite and toxicity prediction, and in particular using quantitative structure-activity relationships (QSAR) to model ADMET and physical chemistry data. QSAR models can be based on data from the literature, such as bioavailability, volume of distribution, oral absorption. For some properties it appears better to generate in-house data, such as solubility, distribution coefficients, plasma protein binding, metabolic stability, etc, to build predictive models. QSAR models age over time and get less predictive as new chemical space is explored in projects [1,2]. Several approaches to keep QSAR models up-to-date will be discussed and compared. Important strategies include the use of correction libraries, regular updating of global models, and use of various types of local models build with e.g. a lazy QSAR approach. Due to the scale of the task automated procedures will be favoured in the future (AutoQSAR) [3–5].
REFERENCE
- Rodgers, S.L., Davis, A.M., Van de Waterbeemd, H., Time-series QSAR analysis of human plasma protein binding data, QSAR Comb. Sci. 26 (2007) 511–521.
- Gavaghan, C.L., Hasselgren-Arnby, C., Blomberg, N., Strandlund, G., Boyer, S., Development, interpretation and temporal evaluation of a global QSAR of hERG electrophysiology screening data, J. Comp. Aid. Mol. Des. 21 (2007) 180–206.
- Rodgers, S.L., Davis, A.M., Tomkinson, N.P., Van de Waterbeemd, H., QSAR modelling using automatically updating correction libraries: Application to a human plasma protein binding model, J. Chem. Inf. Mod. 47 (2007) 2401–2407.
- Bruneau, P., McElroy, N.R., logD7.4 modeling using Bayesian regularised neural networks. Assessment and correction of the errors of prediction, J. Chem. Inf. Model. 46 (2006) 1379–1387.
- Cartmell, J., Enoch, S., Krstajic, D., Leahy, D.E., Automated QSPR through competitive work-flow, J. Comp. Aid. Mol. Des. 19 (2005) 821–833.
5. APPLICATION OF NUCLEAR IMAGING TO EVALUATE HEPATOBILIARY DRUG DISPOSITION
Kim L. R. Brouwer
Division of Pharmacotherapy & Experimental Therapeutics, University of North Carolina School of Pharmacy, Chapel Hill, NC, USA, 27599-7360
Imaging agents may be useful tools to assess hepatic uptake and biliary excretion capabilities in humans. Impaired hepatic uptake or biliary excretion due to drug interactions, disease states or genetic variations may alter hepatobiliary drug disposition and profoundly influence systemic or hepatic drug exposure. 99mTc-mebrofenin (MEB), a hepatobiliary imaging agent, is taken up rapidly by hepatocytes (OATP-mediated) and extensively excreted into bile (MRP-mediated). Using a customized oroenteric tube fitted at the distal end with a polyethylene balloon to occlude the intestine during bile collection, the biliary clearance (Clbiliary) of MEB in healthy humans, calculated as the ratio of mass in bile (corrected for gallbladder ejection fraction to account for variable gallbladder contraction) and the area under the blood concentration-time profile (AUC), was 16.1±3.2 ml/min/kg. The effect of ritonavir, an inhibitor of multiple transport proteins, on MEB hepatobiliary disposition was examined. Healthy volunteers received 2.5 mCi MEB i.v. with or without 200 mg ritonavir administered orally 2 hr earlier. Blood, urine and scintigraphic images of the liver were obtained over 180 min; duodenal secretions were aspirated continuously via the oro-enteric tube. Cholecystokinin-8 was infused i.v. (0.02 μg/kg; 30 min) at 120 min to stimulate gallbladder contraction. MEB was quantified by gamma counting, the biliary recovery of MEB was corrected for gallbladder ejection fraction, and hepatic exposure (mCi/min/g liver) was calculated from liver images. MEB AUC, Clbiliary and hepatic mean residence time (MRT) were determined by noncompartmental analysis. Although MEB systemic exposure was not significantly altered, ritonavir significantly increased the hepatic MRT of MEB (39±8.9 vs. 46.1±7.6 min). These results demonstrate that quantitative measurement of hepatic exposure by nuclear imaging is capable of determining potentially important changes in drug disposition that might not be apparent from systemic concentrations alone. In addition to assessing hepatic exposure and biliary excretion, nuclear imaging also may be useful to explore intestinal secretion. For example, we have shown that following a 2.5-mCi i.v. dose of 99mTc-sestamibi (MIBI), a compound that exhibits intermediate biliary clearance in healthy volunteers (5.5±1.2 ml/min/kg), a fraction of the administered dose appears in the intestine distal to the occlusive balloon. These results clearly demonstrate the ability to differentiate drug secretion into the intestines from drug excretion into bile in humans using this methodology. Taken together, these results highlight the applications of nuclear imaging in examining human hepatobiliary and intestinal drug disposition. The use of MEB and MIBI as probes may be particularly applicable to assess the potential for drug interactions or disease state alterations in drug disposition.
ACKNOWLEDGMENT
Supported by NIH GM41935 and GCRC RR00046.
6. MAGNETIC IMAGING TECHNOLOGIES: RELEVANCE FOR DRUG DEVELOPMENT
Werner Weitschies
Biopharmaceutics and Pharmaceutical Technology, University of Greifswald, Greifswald, Germany, 17498
Reliable bioavailability as a basic precondition of efficient oral drug treatment results from a complex interplay between the behaviour of the dosage form, that means in vivo disintegration and in vivo drug release, drug or dosage from transport within the gastrointestinal organs, luminal gastrointestinal conditions, function of passive and/or active transport mechanisms and metabolic processes along the ‘first-pass’ uptake route. Thereby, oral drug absorption is influenced by processes that take place on the macroscopic and microscopic scale. Variations in gastrointestinal transit and the regional conditions for specific drug absorption are together with first pass metabolism believed to be major sources of erratic drug absorption that results in unpredictable plasma profiles (1, 2). Using modern magnetic imaging techniques like Magnetic Marker Monitoring (3) and Magnetic Resonance Imaging that provide high spatial and temporal resolution insights into the in vivo fate and behaviour of dosage forms as well as the macroscopic gastrointestinal luminal conditions dosage forms meet during gastrointestinal transit are becoming increasingly possible (4, 5). In combination with the determination of plasma profiles imaging studies provide new understandings of food effects as for example dose dumping events (6). Furthermore, combined pharmacokinetic and imaging studies have the potential to identify absorption sites.
REFERENCE
- Kimura T, Higaki K. Gastrointestinal transit and drug absorption. Biol Pharm Bul. 2002;25:149–64.
- Martinez MN, Amidon GL. A mechanistic approach to understanding the factors affecting drug absorption: A review of fundamentals. J Clin Pharm. 2002;42:620–43.
- Weitschies W, Kosch O, Mönnikes H, Trahms L. Magnetic Marker Monitoring: An application of biomagnetic measurement instrumentation and principles for the determination of the gastrointestinal behavior of magnetically marked solid dosage forms. Adv Drug Deliv Rev. 2005;57:1210–1222.
- Schiller C, Fröhlich CP, Giessmann T, Siegmund W, Mönnikes H, Hosten N, Weitschies W. Intestinal fluid volumes and transit of dosage forms as assessed by magnetic resonance imaging. Aliment Pharmacol Ther. 2005;22:971–979
- Faas H, Schwizer W, Feinle C, Lengsfeld H, de Smidt C, Boesiger P, Fried M, Rades T. Monitoring the Intragastric Distribution of a Colloidal Drug Carrier Model by Magnetic Resonance Imaging. Pharm Res. 2001;18:460–466
- Weitschies W, Wedemeyer RS, Kosch O, Fach K, Nagel S, Söderlind E, Trahms L, Abrahamsson B, Mönnikes H. Impact of the intragastric location of extended release tablets on food interactions. J Control Rel. 2005;108:375–385
Abstract Not Available
8. TRANSPORT MEDIATED TOXICOLOGY: THE MITOCHONDRIAL COMPARTMENT
Jashvant (Jash) Unadkat
Department of Pharmaceutics, Univ. of Washington, Seattle, WA, USA, 98195
Hepatic toxicity is one of the most important reasons for drug failure in during development. Transporters are increasingly recognized as playing a significant role in the hepatotoxicity of drugs by allowing their entry into the hepatocytes and into intracellular organelles. To illustrate this phenomenon, I will present our studies on the role of nucleoside transporters in mediating hepatic toxicity of nucleoside drugs. Two families of nucleoside transporters have been identified namely, concentrative (CNTs) and equilibrative (ENTs) nucleoside transporters. Although these transporters function in the plasma membrane, we have recently identified that the human equilibrative nucleoside transporter, hENT1, is expressed on the mitochondrial membrane as well. This dual expression on the plasma and the mitochondrial membrane enhances the mitochondrial toxicity of nucleoside drugs such as filauridine (1). Fialuridine, a drug developed for hepatitis B, resulted in hepatic failure and death in 5 of 15 patients treated with this drug. However, this toxicity was not observed in preclinical studies in rodents even at 1000-fold higher doses than those administered to the hepatitis B patients. To decipher the mechanisms by which this dramatic interspecies difference in fialuridine toxicity occurs, we determined if rodents express ENT1 on the mitochondrial membrane. We found that they do not (2). We also discovered that this lack of expression of rodent ENT1 on the mitochondrial membrane is due to a lack of the mitochondrial targeting signal found in hENT1. This difference in ENT1 mitochondrial expression between rodents and humans may explain why rodents do not predict the mitochondrial toxicity of fialuridine observed in humans. This example illustrates the hitherto unappreciated role of interspecies differences in the expression of intracellular transporters and the role of such expression in clinically significant toxicity of drugs. The mitochondrial expression of hENT1 cannot completely explain the clinically significant mitochondrial toxicity caused by the anti-HIV nucleoside drugs such as zidovudine, stavudine or didanosine. This is because these drugs do not have a high affinity for hENT1. We have recently discovered a novel nucleoside transporter, present in the mitochondrial membrane, which is capable of transporting dideoxynucleosides. Thus, both hENT1 and this novel transporter may explain the mitochondrial toxicity of a variety of nucleoside drugs. As many drugs produce mitochondrial toxicity, the effect of drugs on human mitochondrial transporters and enzymes should be considered when evaluating or elucidating the mechanisms of toxicity of drugs. In addition, the mitochondrial compartment could be a drug target for treatment of certain cancers.
ACKNOWLEDGMENT
Supported by NIH GM5447.
REFERENCE
- Lai Y, Tse CM, and Unadkat JD. Mitochondrial expression of the human equilibrative nucleoside transporter (hENT1) results in enhanced mitochondrial toxicity of antiviral drugs. J Biol Chem 2004; 279:4490–4497.
- Lee EW, Lai Y, Zhang H, Unadkat JD. Identification of the mitochondrial targeting signal of the human equilibrative nucleoside transporter 1 (hENT1): Implications for interspecies differences in mitochondrial toxicity of fialuridine. J Biol Chem. 2006;281:16700–6.
9. THE PREGNANE X RECEPTOR HUMANIZED MOUSE
Xiaochao Ma, Jie Cheng, and Frank J. Gonzalez
Laboratory of Metabolism, National Cancer Institute, Bethesda, MD, USA, 20892
The pregnane X receptor (PXR), a member of the nuclear receptor superfamily is widely considered a xenosensor on the basis of its activation by a wide variety of structurally diverse xenobiotics that include clinically-used drugs. Most importantly, PXR is responsible for regulation of the cytochromes P450 3A(CYP3A) forms that metabolize many drugs. Typical rodent models do not predict drug-drug interactions mediated by human PXR because of species differences in response to PXR ligands. Introducing a BAC clone containing the complete human PXR gene into Pxr-null mice resulted in a PXR-humanized mouse that accurate expresses the functional human PXR in a tissue- specific manner. PXR is selectively expressed in the liver and intestine, the same tissue expression pattern as CYP3A. Treatment of PXR-humanized mice with the PXR ligands mimicked the human response, since both hepatic and intestinal CYP3As were strongly induced by rifampicin, a human-specific PXR ligand, but not by pregnenolone 16alpha- carbonitrile, a rodent-specific PXR ligand. In rifampicin-pretreated PXR-humanized mice, an 60% decrease was observed for both the maximal midazolam serum concentration (Cmax) and the area under the concentration-time curve, as a result of a 3- fold increase in midazolam 1alpha-hydroxylation. These results illustrate the potential utility of the PXR-humanized mice in the investigation of drug-drug interactions mediated by CYP3A and suggest that the PXR-humanized mouse model would be an appropriate in vivo tool for evaluation of the overall pharmacokinetic consequences of human PXR activation by drugs.
10. ANIMAL MODELS AND INTESTINAL DRUG DISPOSITION
Martin F. Fromm
Clinical Pharmacology and Clinical Toxicology, Institute of Experimental and Clinical Pharmacology and Toxicology, 91054 Erlangen, Germany
The gut wall is the first barrier for xenobiotics after oral drug administration. Drug metabolizing enzymes (e.g. CYP3A4) are expressed in the enterocytes and contribute to low bioavailability of drugs and to interindividual variability in drug disposition. In addition, several efflux transporters (e.g. P-glycoprotein, MRP2, BCRP) are expressed in the apical membrane of enterocytes. They limit access of orally administered drugs into the systemic circulation by pumping substrates back into the gut lumen. Moreover, induction and inhibition of intestinal drug transporters are newly recognized mechanisms underlying drug-drug interactions. Mice deficient in the expression of one or more intestinal efflux transporters have been generated and have been proven to be a valuable tool for the investigation of the importance of intestinal efflux transporters for drug absorption and drug-drug interactions. For example, it has been shown that bioavailability of P-glycoprotein substrates such as digoxin, HIV protease inhibitors or paclitaxel is considerably increased in P-glycoprotein deficient mice compared to P-glycoprotein expressing animals. After intravenous administration these mice can also be used to determine the extent of drug secretion from the systemic circulation into the gut lumen. Similar to the situation in humans (e.g. with digoxin) substantial drug secretion into the gut lumen can occur after intravenous drug administration in wild-type mice. However, in P-glycoprotein deficient animals this direct intestinal secretion is considerably reduced. Finally, comparison of the extent of drug-drug interactions between knock-out mice and P-glycoprotein expressing mice is a valuable tool to investigate the mechanisms underlying drug-drug interactions (e.g. digoxin- quinidine). Similarly, mice deficient in Bcrp or Mrp2 have been contributed significantly to our understanding of intestinal drug absorption.
11. VALUE OF ANIMAL MODELS DEFICIENT IN MULTIPLE DRUG TRANSPORTERS
Alfred H. Schinkel
Division of Experimental Therapy, The Netherlands Cancer Institute, Amsterdam, Netherlands
Our research focuses on proteins that affect drug resistance or susceptibility in tumors and the pharmacokinetics of anticancer and other drugs. Insight into these systems may improve pharmacotherapy of cancer and other diseases. Using knockout mice, we have established that the apical ABC multidrug transporters P-glycoprotein (P-gp/Abcb1a) and Breast Cancer Resistance Protein (Bcrp1/Abcg2) have important roles in limiting the oral availability of a range of substrate drugs, toxins and carcinogens, in mediating their hepatobiliary and intestinal excretion, and in restricting their penetration in a range of critical tissues, including brain and fetus. We found that BCRP actively secretes many xenobiotics into breast milk, including drugs and dietary carcinogens. This has direct implications for the use of drugs and pesticides during breast-feeding or in the treatment of dairy cattle, as contamination of milk with drugs, pesticides and other xenobiotics can present a major health risk for breast-fed infants and dairy consumers. Bcrp1 is also responsible for pumping vitamin B2 (riboflavin) into milk, but its absence is tolerated by suckling pups due to the continued milk secretion of the riboflavin-derived co-factor flavine adenine dinucleotide (FAD), which is not Bcrp1-dependent. Studies in Mrp2 (Abcc2) knockout mice indicate that Mrp2 has similar functions as P-gp and BCRP in oral availability and hepatobiliary excretion of substrate compounds. The extensive overlap between the transported substrates of P-gp, BCRP and MRP2, suggests possible redundant functions of these transporters. Indeed, using compound knockout mice, we found that the pharmacokinetics of the anticancer drug paclitaxel can be affected as much by P-gp as by Mrp2. Cytochrome P450 3A (CYP3A) enzymes metabolize >50% of drugs, and their substrates overlap extensively with those of P-gp, BCRP and MRP2. To investigate the functions of CYP3A, we generated Cyp3a knockout (Cyp3a-/-) mice, lacking all functional Cyp3as. Despite a large genomic deletion and many hypothesized Cyp3a functions, Cyp3a-/- mice were viable and fertile. However, the anticancer drug docetaxel displayed severely impaired detoxification, with 7–18-fold higher plasma docetaxel exposure after oral and intravenous administration. Our data suggest that CYP3A evolved primarily for xenobiotic detoxification. The importance of intestinal versus hepatic first-pass metabolism is subject to extensive discussion. We therefore generated Cyp3a-/- mice expressing human CYP3A4 in intestine or liver. Strikingly, intestinal CYP3A4 alone was sufficient to virtually abrogate docetaxel entry from the gut, whereas hepatic CYP3A4 was more important in systemic docetaxel clearance. The Cyp3a-/- mice thus present powerful models for CYP3A-mediated drug-, toxin- and carcinogen metabolism studies. It will be of great interest to study the overlap in function between CYP3A and the ABC multidrug transporters in compound knockout mice.
12. THE BK CHANNEL FROM PHYSIOLOGY TO DRUG ACTION
Peter Ruth
Pharmacology & Toxicology, Institute of Pharmacy, University of Tübingen, Tübingen, Germany
Mouse knockout models are important for biomedical research, because they help us to understand disease pathogenesis and allow for the development of therapeutic strategies to study the efficacy of novel treatments prior to the conduct of costly and time-consuming human clinical trials. The major benefit of the knockout technology is that it enables the analysis of the function of a protein produced from a specific gene in vivo. These mouse models also provide a global view of the target tissues of drug action following acute and repeated drug treatment, thus enabling the prediction of potential side-effects in the early phase of preclinical studies. Since genes rarely function in isolation, the knockout of one gene may result in a cascade of effects on expression of other genes. Such developmental compensation may confound interpretation of the phenotype that is observed. The more sophisticated gene knockout strategies now permits inactivation of genes in an inducible and tissue-specific restricted fashion. The evaluation of the large conductance, Ca2+- and voltage-activated (BK) K+ channel as a potential drug target by using BK channel knockout mice showed that the BK channel could serve as a new target for the treatment of several diseases, for example elevated blood pressure and overactive urinary bladder. However, unexpected complications as severe CNS dysfunctions resulting from modulation of BK channel activity are predicted from the phenotype of BK channel knockout mice and may limit the use of such compounds. Moreover, the phenotype of the constitutive knockout mice with respect to airway physiology was not compatible with the idea of its important contribution as suggested previously since developmental compensations superimposed the virtual function of the BK channel in this tissue. However, developmental compensations could be mostly eliminated by an inducible and tissue selective deletion of the BK channel. It is anticipated that further improvements of transgene architecture will lead to models that should provide a tremendous saving in time in the development of novel pharmacologically active molecules.
13. TOXICOGENOMICS: UTILITY IN HYPOTHESIS GENERATION AND TESTING
Ruth A. Roberts
Safety Assessment, AstraZeneca, Macclesfield, United Kingdom
Toxicogenomics can be used to examine and dissect toxicant-induced alterations in cellular biochemical networks. Generally, toxicogenomics comprises genomics supported by proteomic or metabonomic data. This facilitates a broader approach to hypothesis generation and testing within key areas such as drug development and mode of action analyses. Toxicogenomics used in hypothesis generation can often throw new light on old problems. For example, Nie et al (Mol Carcinogenesis 45, 914–933 2006) generated a large gene expression database from liver samples treated with >100 compounds and mined this to determine gene expression signatures for nongenotoxic carcinogens. They found five biochemical pathway networks linked to cancer all of which were linked by one gene, c-myc. These data enable the development of a hypothesis around c-myc that can be tested in subsequent experiments. Similarly, Marin-Kuan et al (Tox Sci 89, 120–134 2006) were interested in how Ochratoxin A (OTA) causes kidney tumours; toxicogenomics showed that genes associated with DNA synthesis/repair and damage were modified during tumour development. Subsequently, Arbillaga et al (Toxicol Applied Pharmacol 220 216–224 2007) hypothesised that OTA acts via a non-DNA reactive mechanism. Their data showed an increase in intracellular ROS level and oxidative DNA damage and a concomitant regulation of genes involved in mitochondrial electron transport chain and oxidative stress responses. However, genes implicated in DNA damage response were unaltered, supporting their hypothesis of a DNA non-reactive mechanism of OTA genotoxicity. We used toxicogenomics to understand the mode of action (MOA) of peroxisome proliferators (PPs), a class of rodent liver carcinogens that act via PPAR alpha (PP activated receptor alpha) (Hasmall et al Tox Sci 68 304–314 2002). Since PPAR alpha is a transcription factor, we hypothesised the MOA is via regulation of genes that remain to be identified. Toxicogenomic analyses of wild type versus PPAR alpha null mice showed down regulation of hepatic lactoferrin (LF) in response to PPs. Since LF has been reported to repress tumor necrosis factor alpha (TNF alpha), LF down-regulation by PPs may permit TNF alpha levels to rise, enhancing hepatocyte survival and proliferation. These data suggest that the regulation of iron binding proteins by PPAR alpha ligands plays a role in PP-mediated liver growth, but not in peroxisome proliferation. In summary, early work on genomics often suffered from lack of inter-experiment and inter-laboratory variability, which undermined trust in the technology and belief in its utility. Similarly, early experiments in the field were hampered by insufficient server space and lack of appropriate bioinformatics tools to permit appropriate analysis of such large data sets. Today, the majority of these issues are resolved although there is some residual scepticism; will toxicogenomics ever deliver on its early claims and promise? Several recent papers provide evidence that toxicogenomics has extensive utility for both hypothesis generation and testing and is a key tool in toxicology.
14. TOXICOGENOMICS
Philippe Beaune, N. Pallet, C. Narjoz, S. Ellero, D. Anglicheau and I. de Waziers
University of Paris Descartes, INSERM, U775, Molecular Basis of the Response ot Xenobiotics, 45 rue des Saints-Peres, Paris cedex 06, 75270, France
Toxicogenomics will be here considered as all the “without-a-priori” approaches that can be used to understand the mechanisms underlying the effects of xenobiotics on a living system and potentially to find biomarkers able to early detect deleterious effects of xenobiotics. It comprises all the “omics” approaches but genomic, proteomic and metabonomic are the most current. In this lecture the example of cyclosporine renal toxicity, over expression of CYP2C9 in hepatic cell line, HepG2, and pesticide effects on human adipocytes will be used to illustrate these approaches and their use in toxicology.
Cyclosporine is an efficient immuno-suppressor widely used in renal transplantation. However it is nephrotoxic in the long-term. The mechanism of this toxicity is not very well understood and no early biomarkers are available. Human tubular renal cells in primary culture were treated with cyclosporine and the effect on the global transcriptome was analysed with a microarray. The highest gene variations were then checked by real-time PCR. It was shown that an initial reticulum stress initiate an epithelio-mesenchymal transition quite similar to what is observed in vivo. This ER stress could be used as early biomarker of renal toxicity in renal transplantation and as a therapeutic target.
Using the same approach, over-expression of CYP 2C9 in HepG2 cell line was shown to lead also to an ER stress with the increase of numerous HSP proteins. It was also shown that at least two networks of genes were consistently perturbed by this over-expression. Finally the CP2C9 over-expression also modify the intracellular metabolic profile, mainly lipids and glutathione. As a whole, this over-expression is not very toxic to the cells which recover after an initial ER stress. This system was also used to test the toxicity of drugs after metabolism. Finally, the effect of two hydrophobic pesticides were studied on human adipocytes in culture (dioxine and). After treatment, the metabonomic profiles were studied and showed variations in CYP, lipid expression. In conclusion these approaches allow to develop and test hypothesis, to decipher mechanisms of toxicity and to find biomarkers that have then too be tested in prospective studies.
15. TOXICOGENOMICS IN PRECLINICAL DRUG SAFETY EVALUATION
Eric Boitier
Drug Safety Evaluation, Sanofi-Aventis Recherche & Développement, Vitry sur Seine, France
The process of risk assessment has undergone many refinements over the years. Recently, Toxicogenomics (TGx), the application of transcriptomics to toxicology, has allowed an unprecedented, hypothesis-free insight into molecular mechanisms underlying toxicological effects. In this talk we will present how we are currently using gene expression data 1) in support of safety risk assessment of novel drugs under development and the design of follow-up rescue strategies for the back-up compounds, and 2- as an adjunct to the conventional Genetic Toxicology battery.
1-As an attempt to decipher the molecular drivers responsible for the unexpected liver toxicity observed during the clinical trials with the active metabolite of a compound X, a 5-day TGx study was performed in rats with compound X. Gene expression profiling was performed on the liver from control and treated rats. Pathway mapping of the modulated genes showed a marked up-regulation of the phase I and II metabolizing enzymes, a putative establishment of oxidative stress coupled to a delayed acute phase response, which could represent the key inducers of the liver damage. Comparison of the liver gene expression profiles induced by compound X to Gene Logic gene expression compendium (Gene Logic, USA) revealed that this compound could potentially be an enzyme inducer and liver enlarger, have genotoxic and promoting properties and induce hepatitis and liver necrosis. When using the Iconix gene expression database (Iconix Biosciences, USA), compound X was predicted as a potent Ah receptor agonist. In order to tentatively rescue the therapeutic target of high interest for us, four back-up compounds were evaluated in a similar TGx study design. Hepatotoxicity predictions were carried out on the liver gene expression profiles and related likely liver pathologies were suggested. One of the four compounds presented the most favourable safety profile. In addition, this compound presented no AhR induction properties. All in all, the TGx studies allowed the identification of the putative mode of toxicity of compound X and the successful rescue of the therapeutic target.
2-Interpretation of positive chromosome aberration results in terms of relevant risk to humans is difficult due to the limited insight into the underlying molecular mechanisms. TGx is expected to advance our understanding of genotoxic mechanisms involving direct or indirect interaction with DNA. We exposed human lymphoblastoid TK6 cells to 14 anticancer drugs with either DNA reactive (alkylating or oxidative agents) or non-DNA reactive (topoisomerase inhibitors, antimetabolites and mitotic spindle inhibitors) genotoxic properties for 4-h and examined them immediately or after a 20-h recovery period. Gene expression profiling allowed us to classify the drugs according to their mechanisms of action. The molecular signature is composed of 28 marker genes mainly involved in signal transduction and cell cycle pathways. Our results suggest that these marker genes could be used as a predictive model to classify genotoxins according to their direct or indirect interaction with DNA.
16. APPLICATION OF TOXICOGENOMICS TO UNDERSTAND TOXICITY OF CHEMICAL MIXTURES
Rob Stierum1, Peter Hendriksen1, Marijana Radonjic1, Andreas Freidig1, Brian Lake2, and John Groten3
1Biosciences, TNO Quality of Life, Zeist, Netherlands, 3700 AJ, 2Centre for Toxicology, School of Biomedical and Molecular Sciences, University of Surrey, Guildford, Surrey, United Kingdom, GU2 7XH, 3Biosciences (current adress: NV Organon, Part of Schering Plough Corporation, Oss, The Netherlands), TNO Quality of Life, Zeist, Netherlands, 3700 AJ
To understand the effects of chemical mixtures, mixture toxicology relies on statistical inference of compatibility of the data with effect addition (dissimilar action) or dose addition (similar action). If the responses measured for the mixtures differ from the responses predicted upon dose response curves of the individual chemicals, interactions occur, with synergism and antagonism being more or less than predicted, respectively. This approach is limited to a few parameters only and often involves parameters such as body weight, which provides little insight into interactions at the mechanistic level. Here, we present approaches involving the application of toxicogenomics in mixture studies, together with pathway-based bioinformatics available at TNO. The first study involves a study on mixtures of food additives. Recently, the ILSI Europe Acceptable Daily Intake Task Force concluded that for butylated hydroxytoluene (BHT), curcumin (CC), propyl gallate (PG) and thiabendazole (TB), the possibility for mixture effects was not excluded for the liver. The effects of mixtures of these additives on hepatic gene expression and conventional endpoints were therefore investigated. Additives were administered for 28 days in the diet to Male Sprague-Dawley rats, as single compounds and in mixtures. Liver samples were analyzed on Affymetrix Rat 230A arrays. Results showed that treatment with TB caused the largest transcriptome changes compared to the other three compounds, when applied alone as well as in mixtures, and therefore may have the most prominent effect on hepatic biochemistry. In contrast, treatment with CC and PG alone caused only modest transcriptome changes, while a mixture of these additives showed the relative highest number of newly differentially expressed genes for all mixtures tested. Further, pathway analysis indicated the possible activation of pathways related to transcription in this CC and PG mixture. This possible mechanistic interaction was not noticed by statistical inference of conventional endpoints. The second study involves effects of methyl mercury, benzene and trichloroethylene, in rat liver and kidney. Chemicals were administered daily for 14 days at the Lowest-Observed-Adverse-Effect-Level or at lower concentrations, individually or in mixtures. Compounds had strong antagonistic effects on each others gene expression profiles. As an example, trichloroethylene was found to induce a gene set under control of PPAR alpha; interestingly this effect was suppressed upon mixture exposures. Mixture exposure also resulted in novel differentially expressed genes that were not or modestly differentially expressed by the individual chemicals. These included genes such as Id2, Nr2f6, Tnfrsf1a, Mdm2 and Nfkb1 in the liver, involved in cellular proliferation and apoptosis. These results show a benefit of toxicogenomics studies to better understand mechanistic implications in mixture exposure scenarios. Financial support provided by the United Kingdom Food Standards Agency (T01021 and T01040) and the American Chemistry Council (RSK-0103) is greatly appreciated.
Abstract Not Available
18. FORECASTING OF HUMAN DRUG RESPONSE FROM ANIMAL AND IN VITRO DATA: A REALITY CHECK
Urs A. Meyer
Dept of Pharmacol/Neurobiol, Univ of Basel Biozentrum, Basel, Switzerland, CH-4056
Considerable advances in our understanding of molecular mechanisms causing disease and drug response allow better prediction of clinical drug effects and of interindividual variation in drug response. The increasing commercial availability of animal models and probes and tools for in vitro studies in human cells and tissue allows the assessment of genetic, host and environmental factors that contribute to this variability.
Some of the questions to be asked preclinically are: How variable are the human drug targets and metabolic pathways? How much of the variation is genetic and constant throughout life and how much is host-specific (e.g. age, body mass, diseases, etc.) or environmental (e.g. diet, smoking, etc.) ? What are the biomarkers of variation, of efficacy and toxicity for this drug and this disease? How will this information affect the design of clinical trials and ultimately the use of this drug.
I will highlight recent examples of preclinical studies regarding the regulation of drug-metabolizing enzymes in regard to genotype-phenotype relationships and regulation of these enzymes by xenosensors / nuclear receptors and by cross-talk with endogenous pathways of lipid and energy homeostasis. These data will be used to forecast clinical responses to the xenobiotics studied.
REFERENCE
- Meyer, U.A. (2007) Endo-xenobiotic crosstalk and the regulation of cytochromes P450. Drug Metab Rev 39, 639–646,
- Podvinec, M., Meyer, U.A. (2006) Prediction of cis-regulatory elements for drug-activated transcription factors in the regulation of drug-metabolizing enzymes and drug transporters. Expert Opin Drug Metab Toxicol 2, 367–379.
- Oscarson, M., Zanger, U.M., Rifki, O.F., Klein, K., Eichelbaum, M., Meyer, U.A. (2006) Transcriptional profiling of genes induced in the livers of patients treated with carbamazepine. Clin Pharmacol Ther 80, 440–456.
- Blattler, S.M., Rencurel, F., Kaufmann, M.R., Meyer, U.A. In the regulation of cytochrome P450 genes, phenobarbital targets LKB1 for necessary activation of AMP-activated protein kinase. Proc Natl Acad Sci U S A. 104, 1045–1050.
Abstract Not Available
20. PHARMACOGENETICS IN MEN AND MICE - WHAT PGX DATA ARE REQUIRED FROM PRECLINICAL EXPERIMENTS?
Julia Kirchheiner
Department of Pharmacology of Natural Products & Clinical Pharmacology, University of Ulm, Ulm, Germany, 89081
The understanding of genetic mechanisms causing drug response largely depends on knowledge from animal models such as studies with knockout mice. However, genetic variability not always includes a complete knockout of function of the gene but rather means variability within gene expression, activity or regulation of gene expression. Pharmacogenetic factors are more and more being studied within the early phases of drug development, and certainly, knowledge from animal or preclinical studies is very precious for further studies on drug response predictors in humans. However, prediction of pharmacogenetic influences on drug response from preclinical studies is still very difficult.
In order to continue with clinical testing of the impact of genetic polymorphisms in humans, certain preclinical data are prerequisite. Preclinical pharmacology experiments will reveal the molecules involved in drug metabolism, drug transport, and important mechanisms for possible drug-drug interactions can be identified at this level already. On the target site of drug action, the primary and secondary drug targets can be identified preclinically, and followed up later in the clinical stage with regard to genetic variability in humans.
In conclusion, pharmacogenetic data from preclinical studies mostly include information on molecules somehow participating in drug effects, whereas the proper genetic polymorphisms within these molecules still have to be functionally studied during clinical drug development.
21. PREDICTION OF DRUG METABOLISM AND DRUG-DRUG INTERACTIONS IN PATIENTS
Uwe Fuhr
Department of Pharmacology, Clinical Pharmacology Unit, University of Cologne, Cologne, Germany, D-50931
A reasonable prediction of the metabolic stability of a NCE and its potential to be involved in drug-drug interactions is essential to avoid a major fraction of early attrition in clinical development. Available preclinical test systems include laboratory animals, primary human hepatocytes, human liver microsomes, and recombinant human enzymes. To predict drug clearance in humans, allometric scaling is used for animal in vivo data. Different specificity of human and non-human enzymes is probably a major reason for limited precision of such predictions. In vitro parameters obtained with human enzyme systems include in vitro half-life and intrinsic clearance. By in vitro – in vivo extrapolation, in vivo pharmacokinetic parameters such as bioavailability and in vivo half-life can be calculated, taking the abundance of individual drug metabolizing enzymes, liver size and often empirical correction factors into account. Such predictions may also address interindividual variation. The use of human hepatic enzymes is valuable primarily for drugs metabolized by hepatic cytochrome P450 enzymes but generally is more reliable than animal data. Identification of the type of metabolites (phase I, phase II) in animal studies is reasonable to select the appropriate in vitro model with human enzymes which may be used in high throughput settings. Forecasting drug interactions is possible only with systems containing human enzymes. Selective substrates and inhibitors along with the enzymes are available primarily for cytochrome P450 enzymes. Any system including expressed enzymes may be used to assess enzyme inhibition and enables a reliable semi-quantitative prediction of drug-drug interactions caused by competitive as well as mechanism-based inhibition. In contrast, cellular systems with intact regulation of enzyme expression are required to assess enzyme induction. Because in both hepatocytes and liver slices regulation are compromised, the predictive power for enzyme induction is limited. Despite many open questions, these approaches reduced the attrition rate attritutable to metabolic and pharmacokinetic properties of NCEs in the last to decades from half of the cases to 10 %. Although this is a major success, further improvement of quantitative prediction is still expected to generate additional value and save costs for drug development.
REFERENCE
- Ito, K., Houston, B.J. (2005) Prediction of human drug clearance from in vitro and preclinical data using physiologically based and empirical approaches. Pharmaceut Res 22, 103–112
- Baranczewski, P., Stañczak A., Sundberg, K., Svensson R., Wallin, Å., Jansson, J., Garberg P., Postlind, H. (2006) Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacol Reports 58, 453–472
- Shiran M.R, Proctor, N.J., Howgate, E.M., Rowland-Yeo, K., Tucker, G.T., Rostami-Hodjegan, A (2006) Prediction of metabolic drug clearance in humans: In vitro-in vivo extrapolation vs allometric scaling. Xenobiotica 36, 567–580
22. PREDICTING EXPOSURE IN THE OUTLIER
G.T. Tucker
Acedemic Unit of Clinical Pharmacology, University of Sheffield, Sheffield, United Kingdom, S10 2JF
The construction of physiologically-based pharmacokinetic (PBPK) models has typically incorporated average values of physiological and metabolic parameters. However, outcome in the average individual does not predict that across a population and, therefore, in those individual patients at the extremes of risk. Such risk is likely to reflect a combination of patient characteristics (demographic, physiological, genetic, environmental) and is unlikely to be anticipated adequately during the experimental clinical development programme. Incorporating PBPK modelling and simulation into a virtual population can provide ‘early warning’ in this respect, and allows ‘what if’ scenarios to be explored in the safety of the computer. For example, what if a highly metabolised drug were co-administered with several other drugs that, individually, are weak enzyme inhibitors? What if the patient who is genetically a poor metaboliser with respect to a relevant enzyme develops renal impairment? Is the kinetic behaviour of drug X going to be different in Caucasian and Asian people? Similar considerations apply when designing clinical trials in infants and children. Prior incorporation of our rapidly increasing knowledge of physiological development and the ontogeny of drug metabolising enzymes and transporters into a virtual population PBPK model offers a rigorous approach to the selection of safe and effective dosing regimens in children. The state-of-the science is such that the implementation of this ‘bottom up’ approach to modelling and simulation, can effectively predict variability in pharmacokinetic behaviour and drug exposure. The greater challenge is to predict variability in intrinsic drug effect and to link this to the simulation of exposure in different populations.
23. IS HEPATOTOXICITY PREDICTABLE?
Jack Uetrecht
Dept of Pharmacy, Univ of Toronto, Toronto, ON, Canada, M5S 3M2
The most common type of idiosyncratic drug reaction (IDR) leading to withdrawal of a drug is hepatotoxicity. If there were a way to predict the risk that a drug candidate would cause IDRs it would have a profound effect on the process of drug development. Animal testing has not been effective in predicting IDR potential although it is likely that many candidates that would have caused IDRs were not developed because they caused toxicity in animals. Most IDRs appear to be caused by reactive metabolites; therefore, screens for reactive metabolites probably improve drug safety, but there are some IDRs such as ximelegatran-induced hepatotoxicity that don't appear to involve a reactive metabolite. A major issue is whether most idiosyncratic liver toxicity is immune-mediated or represents metabolic idiosyncrasy. If metabolic idiosyncrasy is involved we would need to screen for polymorphisms in the metabolic pathways involved, yet no polymorphism in a metabolic pathway has yet been identified that explains the idiosyncratic nature of liver toxicity. One possibility are pathways affecting mitochondrial function and an animal model has been described in which a mouse heterozygous for mitochondrial SOD is susceptible to hepatotoxicity caused by troglitazone. Although quite interesting, to date others have not been able to reproduce this result. If hepatotoxicity is immune-mediated and the danger hypothesis is correct, there may be biomarkers of cell stress that predict a drug candidate's potential to cause IDRs, but the mechanisms of IDRs caused by different drugs are likely different so one pattern of biomarkers will not detect IDR potential for all drugs. It is likely that only through a better understanding of the basic mechanisms of IDRs that better predictive screens will be developed. At the present time we do not even know whether most idiosyncratic liver toxicity is immune-mediated, and if not, why there is a delay between starting a drug and the onset of liver damage.
Abstract Not Available
25. HOW TO CONDUCT SAFER CLINICAL DRUG INVESTIGATIONS
Alasdair M. Breckenridge
Medicines and Healthcare products Regulatory Agency, London SW 8 5NQ, United Kingdom
The aim of most clinical drug investigations is to examine the risk benefit balance of a medicine in a specific group of subjects. Early stage clinical investigations, such as those conducted for the first time in man, will rely heavily on the interpretation of the known pharmacology of the compound as well as on other preclinical information for the selection of size and frequency of dose, rate and route of administration, and choice of subjects. Further, the place where the investigation is to be carried out and the available facilities are of prime importance in ensuring safety. Scrutiny of all aspects of the protocol by a competent regulatory authority and an ethical committee is mandatory and the investigation must be conducted by an appropriately qualified and experienced investigator. The early development and validation of biomarkers to monitor drug safety is a valuable tool in pursuit of safety Safety monitoring in clinical investigations which are carried out in later stages of development pose an additional and separate set of problems., largely related to how and who reports adverse reactions in clinical trials . Phase 3 clinical trials will provide a more precise assessment of drug efficacy than of safety, because the size of a trial is usually predicated by efficacy rather than safety statistical considerations. Hence the investigation of safety of a new compound will continue after marketing authorisation has been granted. This assessment can be done either passively, by the submission of reports of adverse drug reactions, or in the case of higher risk compounds by active surveillance eg with the use of patient registers . In general, a risk based approach to safety monitoring should be adopted so that reporting requirements are matched with the level of knowledge about the drug. An important regulatory tool is the risk management plan, detailing not only what is known about the safety of a medicine at the time of licensing, but also what is not known, what needs to be known and how this information should be obtained.
Abstract Not Available
27. THE MOLECULAR PHARMACOGENETICS OF ANTIDEPRESSANTS
Ulrik Gether
Dept. of Neuroscience and Pharmacology, Universitry of Copenhagen, Faculty of Health Sciences, Copenhagen, Denmark, 2200 N
The biogenic amines, dopamine, norepinephrine and serotonin, play a key role as neurotransmitters in the brain, and alterations in their function are tightly coupled to psychiatric diseases such as schizophrenia, bipolar disorder, depression and anxiety. The biogenic amines exert their effects through binding to pre- and postsynaptic receptors, and their action is rapidly terminated via presynaptic transmembrane transporters mediating reuptake of the transmitters from the synaptic cleft. The transporters, which belong to the class of neurotransmitter:sodium symporters and include the dopamine transporter (DAT), the serotonin transporter (SERT) and the norepinephrine transporter (NET), have received particular attention as targets for the action of antidepressant medication and for psychostimulants such as cocaine and amphetamine. A major research goal of our the laboratory is to investigate the molecular mechanisms underlying drug action at the biogenic amine transporters and to explore how genetic variation in the transporters, as well as in the associated functional pathways, contributes to disease development, disease course and drug efficacy in patients. We seek to reach this goal within a multidisciplinary research center – Center for Pharmacogenomics – where molecular and cellular studies of drug action are performed together with patient sampling and genetic analyses. Recent efforts have provided novel insight into the structural basis for drug recognition by biogenic amine transporters and functional consequences of non-synonymous polymorphisms. In parallel, genome-wide association (GWA) studies and targeted SNP analyses are carried out on well-characterized samples of patients with major depression. Selected results from our studies will be presented.
28. PHARMACOGENOMIC PREDICTORS OF ANTIDEPRESSANTRESPONSE
Alessandro Serretti
Psychiatry, University of Bologna, Bologna, Italy
Up to 60% of depressed patients do not respond completely to antidepressants and up to 30% do not respond at all. Among the many reasons leading to non response, such as inadequate treatments and comorbid conditions, genetic liability plays an important role. Genetic factors contribute in fact for about 50% of the antidepressant response. This means that the knowledge of the patient genetic profile may predict antidepressant response thus leading to alternative treatments since the beginning.
However this will be the final goal and a genetic profile will be a routine test in everyday clinical practice only when all genes influencing response will be discovered and validated. Studies are still underway and we have preliminary but sound results. During the recent years the possible influence of a set of candidate genes as genetic predictors of antidepressant response efficacy were investigated and will be reviewed here. A growing number of gene variants were independently associated with short term SSRIs antidepressant efficacy. They include the functional polymorphism in the upstream regulatory region of the serotonin transporter gene (5-HTTLPR), the A218C gene variant on the tryptophan hydroxylase gene (TPH), the C(-1019)G variant in the 5HT1A receptor, some variants in the 5HT2A receptor, the G-protein beta3-subunit (Gbeta3) C825T gene variant, Catechol-O-methyltransferase (COMT) gene variant, Angiotensin I and II converting enzyme I/D (ACE I/D), the Norepinephrine Transporter (NET), Dystrobrevin-binding-protein 1 (DTNBP1), the glucocorticoid receptor-regulating cochaperone (FKBP5) and the Circadian Locomotor Output Cycles Kaput (CLOCK). The effects of 5-HTTLPR were further investigated with a new variant. The “long” alleles (16-A, 16-D and 16-F) and the “short” alleles (14-A, 14-B) have different consensus sequences for some transcriptional factors binding and resulted in different SSRI response. Further, a symptomatology dissection evidenced a specific effect on anxiety and core depressive symptoms of 5-HTTLPR. Similarly, CLOCK and PER3 variants evidenced specific effects on symptomatology (CLOCK on insomnia, PER3 on various personality aspects). Marginal associations were reported for ADRB1, BDNF, APOE, MAOA, 5HT3A, 5HT6, DAT, CRHR1, TPH2, PDE and IL-1beta. DRD2, DRD4, SERT-STin2, 5HT1B, MDR1P, NOS gene variants were not associated with outcome. Although in its preliminary phase, the results obtained in the pharmacogenetics of antidepressants are promising for an individualized therapy aimed to reduction of the resistant depression phenomenon.
29. PHARMACOGENOMICS OF ANTIDEPRESSANTS: AN INDUSTRY PERSPECTIVE
Irina A. Antonijevic1, Joseph Tamm1, Wiktor Mazin2, Roman Artymyshyn1, Jan B. Vistisen3, Birgitte Søgaard3, and Christophe Gerald1
1Translational Research, Lundbeck Research USA, Paramus, NJ, USA, 07652, 2Technical University of Denmark, Lyngby, Denmark, 2800, 3H. Lundbeck A/S, Valby, Denmark, 2500
It is recognized that the diagnostic criteria for major depressive disorders encompass pathophysiologically heterogeneous groups of patients. Attempts to include biologically-based diagnostic criteria are discussed for the new diagnostic manual (DSM-V). However, clinically valid phenotypes which allow clinicians to distinguish biological subgroups and subsequently to tailor treatment strategies are not available yet. In recent years, data are emerging suggesting that molecular markers, including genomic (transcription) and genetic (polymorphisms) markers, when combined with good phenotyping, can help identify biologically distinct subtypes of major depression. However, a systematic approach and a replication of findings are often not pursued, thereby limiting the use of most molecular markers published so far.
On one hand, the identification of molecular markers of depression can advance our understanding of distinct biological pathways leading to depressive disorders. On the other hand, such clinical markers can be utilized to develop animal models that reproduce certain biologically robust findings in depression and thereby help to profile novel treatment targets for specific subtypes of depression.
Our Translational Research efforts in depression and anxiety research focus on the understanding of the human disease, the translation into animal models and the discovery of markers predictive for the treatment response to existing and new antidepressants. With regard to molecular markers, we examine transcription and metabolite profiles in blood samples, as these are easy to obtain, particularly in humans. In addition, polymorphisms, mostly SNPs, in candidate genes, related to drug metabolism and drug targets, are explored.
Using sophisticated methods such as dynamic modeling, we have identified groups of patients who share a similar transcription (and metabolite) profile. In the next step, we searched for animal models that share a similar transcription pattern as human patient populations and examined drug effects.
Furthermore, transcription (and metabolite) profiles in patients form the basis for the detection of new candidate genes. This approach differs somewhat from many existing publications, as it attempts to first identify biologically more homogenous patient groups before investigating genetic polymorphisms that may be associated with a particular patient segment's disease vulnerability. Advantages and disadvantages of this approach will be presented.
30. CLINICAL RELEVANCE OF PHARMACOGENOMICS IN DEPRESSION
Ingolf Cascorbi
Institute of Pharmacology, University Hospital of Schleswig-Holstein, D-2405 Kiel, Germany
Depression is the fourth-leading cause of the global disease burden and the leading cause of disability. Almost every second patient fails to respond adequately to the initially administered antidepressant drug. Moreover, many patients treated with antidepressants suffer from class-specific adverse drug reactions (ADRs) including anticholinergic effects, vomiting, weight-gain, loss of libido and others. In Germany, an incidence of 1.4% has been estimated for severe ADRs during antidepressant treatment. Treatment failure and particularly ADRs are often dose-dependent. The pharmacokinetics of antidepressants are largely determined by the activity of drug-metabolizing cytochrome P450 (CYP) enzymes that are subject to extensive inter-individual variability leading to about 20-fold variability in the acquired plasma concentration. Most antidepressant drugs are substrates of the well established polymorphic CYP2D6 enzyme. Moreover, CYP2C19 is involved in the metabolism of many tricyclics. Carriers of two active CYP2D6 alleles with a normal EM phenotype have retrospectively been identified to be at lower risk of suffering from side effects than those with an IM phenotype while treated with amitriptyline or nortriptyline. An increased risk of side effects associated with venlafaxine treatment was also reported for CYP2D6 PMs. Moreover, a significant overrepresentation of CYP2D6 UMs was found among non-responders to antidepressant therapy. Tricyclics, however, have been overcome to some extent by SSRIs and by SNRIs such as venlafaxine, having a higher tolerability.
Pharmacodynamic variability due to genetic polymorphisms of catecholamine and 5-HT transporters or receptors may also contribute to interindividual differences in the dose-response relationship of antidepressants. E.g. for the 5-HT transporter SLC6A4, certain studies demonstrated a better response to SSRIs in carriers of the high active variant. In 5-HT receptors the results are less pronounced and not consistent. On the other side, particularly the 5-HT transporter SCL6A4 was reportedly associated with environment-triggered susceptibility of depression. Moreover SNPs in the cAMP response element CREB were shown to be associated with suicidal ideation.
The clinical use of pharmacogenetics is still limited by the lack of prospective studies, investigating the putative beneficial consequences of patient-tailored pharmacotherapy. Due to the rareness of e.g. CYP2D6 and CYP2C19 PM double-carriers (<1% in all ethnicities) but also of homozygote CYP2D6 ultra rapids, very large numbers of patients have to be enrolled to reach sufficient statistical power. Therefore, pharmacogenetics is currently applied only in rare cases with unexpected plasma levels and appearance of clinical symptoms. The increasing knowledge on genetic variability and the fastness, reliability and cost-effectiveness of the new genotyping technologies, however, will facilitate a better understanding of the influence of genetics to antidepressant drug response. The current data does not explain sufficiently the entire variability in drug response, but could give an orientation to avoid severe treatment failures in clinical praxis particularly for tricyclic antidepressants and venlafaxine.
31. GENETIC VARIATION OF BIOGENIC AMINE TRANSPORTERS AND THEIR RELEVANCE TO CNS DISORDERS
Wolfgang Sadee
Pharmacology, College of Medicine The Ohio State University, Columbus, OH, USA, 43210
Serotonin (SERT), dopamine (DAT), and norepinephrine (NET) transporters, and vesicular amine transporters such as VMAT2 play key roles in multiple CNS functions, as primary drug targets, and CNS disorders. Each of these genes consists of a complex structure with multiple introns and exons, and harbors numerous polymorphisms. Rare mutations have been identified using linkage analysis in family studies, while countless attempts have been made to characterize frequent variants with the use of large clinical association studies, for example, a promoter repeat polymorphism in SERT (SERT-LPR), and a 9/10 repeat in the 3′-untranslated region of DAT. However, discrepant results between studies raise questions about the relevant genetic variants and the true penetrance of these genes. As nonsynonymous SNPs are rare in these highly conserved genes, we have developed a new approach to finding regulatory polymorphisms that affect both transcription and mRNA processing, the latter apparently more frequent than had been suspected. The approach involves measurements of allelic mRNA expression, followed by SNP scanning of the gene locus to find polymorphisms associated with mRNA expression. Applied to SERT, we were unable to detect any effect of the SERT-LPR on mRNA expression in the pons of human autopsy brain tissues (1), raising questions about the relevance of this promoter variant. Using the same method, we have found that DAT is subject to significant cis-acting regulatory factors that still need to be identified to maximize the success of clinical studies. Recently, the dopamine D2 receptor has been shown to physically interact with DAT, modulating its functions presynaptically. Studying the D2 receptor, we have identified two intronic SNPs that alter the ratios between S and L splice isoforms, expressed pre- and postsynaptically, respectively (2). These results suggest that genetic variants in D2 can alter DAT activity, which needs to be explored further. Before we can fully exploit genetic biomarkers for the biogenic amine transporters, a clear understanding of the underlying molecular genetic mechanisms is needed.
REFERENCE
- Lim J-E. Papp A, Pinsonneault J, Sadée W, Saffen D. Allellic expression of serotonin transporter (SERT) mRNA in human pons: lack of correlation with the polymorphism SERTLPR. Molec. Psychiatry 11:649–62 (2006).
- Zhang Y, Bertolino A, Fazio L, Blasi G, Rampino A, Romano R, Lee M-L T, Xiao T, Papp A, Wang D, and Sadée W. Novel polymorphisms in human dopamine D2 receptor gene affect gene expression, splicing, and neuronal activity during working memory. Proc. Natl. Acad. Sci. USA. in press (2008).
Abstract Not Available
33. GENETIC POLYMORPHISMS OF OATP1B1 AND BCRP; IMPLICATIONS FOR INTERINDIVIDUAL DIFFERENCE OF PHARMACOKINETICS OF DRUGS
Yuichi Sugiyama and Hiroyuki Kusuhara
Department of Molecular Pharmacokinetics, The University of Tokyo, Tokyo, Japan, 113 - 0033
Xenobiotic transporters are closely involved in the disposition of drugs. The presentation will focus on the effect of genetic polymorphisms of OATP1B1 and BCRP on drug disposition. OATP1B1 mediates the hepatic uptake of a variety of anionic drugs. We reported for the first time that healthy volunteers with homozygous *15 allele (388A > G and 521T > C), causing the reduction in transport efficacy per protein molecule1), exhibit greater systemic exposure of pravastatin following oral administration2). Thereafter, our group and others have accumulated clinical data showing that OATP1B1*5 or *15 alleles are associated with interindividual variation of the systemic exposure of acid-type statins 3–5) except for fluvastatin 6). Pharmacokinetic consideration suggests that hepatic concentration of statins is not decreased so much as expected from the reduction in the hepatic uptake because of increased systemic exposure, resulting in weak attenuation of cholesterol lowering effect. Subjects with -11187GA or AA genotypes exhibited attenuated acute cholesterol lowering effect of pravastatin7). As for OATP1B1*15, there was no significant change in the acute effect by pravastatin7), while retrospective analysis suggests that the effect of pravastatin was significantly attenuated in patients with *15 allele by 8 weeks, but finally, the serum cholesterol level reached same values 1 year later8). The SNPs are also associated with interindividual variation of other drugs, such as fexofenadine9), repaglinide10), nateglinide11), atrasentan12), valsartan and temocapril13). BCRP actively extrudes drugs into the bile and lumen in the liver and intestine, respectively. Among the SNPs, a SNP, 421C > A, causing a reduced protein expression14), has been associated with interindividual variation of drug disposition. Subjects with CA or AA genotypes exhibit higher plasma concentrations of rosuvastatin than those with CC genotype15), but not for pitavastatin3). Unlike OATP1B1, the impact of the SNP of BCRP is different among statins. In addition, the SNP is also associated with greater systemic exposure of topotecan16) and sulfasalazine17) following oral administration. Clinical studies have supported the hypothesis that the genetic polymorphisms of OATP1B1 and BCRP are one of the factors causing interindividual variation in drug disposition.
REFERENCE
- Iwai, M., et al. Pharmacogenetics 14:749, 2004.
- Nishizato, Y., et al. Clin Pharmacol Ther 73:554, 2003.
- Ieiri, I., et al. Clin Pharmacol Ther 82:541, 2007.
- Pasanen, M. K., et al. Clin Pharmacol Ther 82:726, 2007.
- Pasanen, M. K., et al. Pharmacogenet Genomics 16:873, 2006.
- Niemi, M., et al. Clin Pharmacol Ther 80:356, 2006.
- Niemi, M., et al. Pharmacogenet Genomics 15:303, 2005.
- Takane, H., et al. J Hum Genet 51:822, 2006.
- Niemi, M., et al. Br J Clin Pharmacol 59:602, 2005.
- Niemi, M., et al. Clin Pharmacol Ther 77:468, 2005.
- Zhang, W., et al. Br J Clin Pharmacol 62:567, 2006.
- Katz, D. A., et al. Clin Pharmacol Ther 79:186, 2006.
- Maeda, K., et al. Clin Pharmacol Ther 79:427, 2006.
- Kondo, C., et al. Pharm Res 21:1895, 2004.
- Zhang, W., et al. Clin Chim Acta 373:99, 2006.
- Sparreboom, A., et al. Cancer Biol Ther 4:650, 2005.
- Yamasaki, Y., et al. Clin Pharmacol Ther 2008.
34. CARDIOVASCULAR DRUG TRANSPORT: RELEVANCE FOR THERAPY
Heyo K. Kroemer
Department of Pharmacology, Ernst-Moritz-Arndt University, Greifswald, Germany, 17487
Cardiovascular diseases (CVD) remain the leading cause of death in Western countries. Consequently, numerous drugs have been developed which target the heart, blood vessels or peripheral blood cells. Patients with high prevalence CVDs, however, respond to drug treatment in a highly variable manner. One yet unexplored factor is the modification of intracellular concentrations of drugs in cardiovascular cells by expression of transport proteins. Several ABC-type export transporters have been identified in human heart, among them BCRP (Meissner et al, 2006) and MRP5 (Dazert et al, 2003). Regulation is disease dependent; while expression of P-gp is decreased in heart failure, BCRP is significantly enhanced. Aside from export proteins, expression of uptake transporters may facilitate modification of drug concentrations in cardiovascular cells. In fact, OCTN2 (Grube et al, 2006a) and OATP2B1 (Grube et al, 2006 b) have been identified in human heart. Notably, OATP2B1 is a high affinity uptake transporter for atorvastatin, which is of interest in view of the controversial discussion regarding the role of statins in heart failure.
Aside from expression in cardiac cells transport proteins may modify drug concentrations in peripheral blood cells as highlighted in a recent review (Koeck et al, 2007). Most interestingly, Jedlitschky and colleagues (2004) described a high expression of MRP4 in human platelets. Expression was restricted to dense granules and several lines of evidence suggested, that MRP4 may play a role in concentrating ADP in dense granules thereby facilitating platelet aggregation. In summary, cardiovascular drug transport may contribute to variable drug action in CVD treatment.
REFERENCE
- Dazert P, Meissner K, Vogelgesang S, Heydrich B, Eckel L, Bohm M, Warzok R, Kerb R, Brinkmann U, Schaeffeler E, Schwab M, Cascorbi I, Jedlitschky G, and Kroemer HK (2003) Expression and localization of the multidrug resistance protein 5 (MRP5/ABCC5), a cellular export pump for cyclic nucleotides, in human heart. Am. J. Pathol. 163:1567–1577.
- Kock K, Grube M, Jedlitschky G, Oevermann L, Siegmund W, Ritter CA, and Kroemer HK (2007) Expression of adenosine triphosphate-binding cassette (ABC) drug transporters in peripheral blood cells: relevance for physiology and pharmacotherapy. Clin. Pharmacokinet. 46:449–470.
- Grube M, Kock K, Oswald S, Draber K, Meissner K, Eckel L, Bohm M, Felix SB, Vogelgesang S, Jedlitschky G, Siegmund W, Warzok R, and Kroemer HK (2006a) Organic anion transporting polypeptide 2B1 is a high-affinity transporter for atorvastatin and is expressed in the human heart. Clin. Pharmacol. Ther. 80:607–620.
- Grube M, Meyer zu Schwabedissen HE, Prager D, Haney J, Moritz KU, Meissner K, Rosskopf D, Eckel L, Bohm M, Jedlitschky G, and Kroemer HK (2006b) Uptake of cardiovascular drugs into the human heart: expression, regulation, and function of the carnitine transporter OCTN2 (SLC22A5). Circulation 113:1114–1122.
- Meissner K, Heydrich B, Jedlitschky G, Meyer zu Schwabedissen HE, Mosyagin I, Dazert P, Eckel L, Vogelgesang S, Warzok RW, Bohm M, Lehmann C, Wendt M, Cascorbi I, and Kroemer HK (2006) The ATP-binding cassette transporter ABCG2 (BCRP), a marker for side population stem cells, is expressed in human heart. J. Histochem. Cytochem. 54:215–221.
Abstract Not Available
36. DEVELOPMENTAL EXPRESSION OF HUMAN HEPATIC MONOOXYGENASE AND CONJUGATING DRUG METABOLIZING ENZYMES
Ronald N. Hines
Depts. of Pediatrics and Pharmacology/Toxicology, Children's Research Institute, Medical College of Wisconsin & Children's Hospital & Health System, Milwaukee, WI, USA, 53226
Significant changes in drug metabolizing enzymes (DME) expression occurs during ontogeny. Such changes have a profound effect on an the fetus' and child's susceptibility to adverse drug reactions and therapeutic efficacy. To gain a better understanding of DME ontogeny, enzyme content and activity were measured in 240 human liver samples representing ages from 8 wks gestation to 18 yrs. Where possible, both quantitative western blotting and activity assays with probe substrates were performed. Data has been obtained on the phase I enzymes, CYP3A4, 3A5, 3A7, 2C9, 2C19, 2E1, 2B6, 2D6, FMO1 and FMO3, and the phase II enzymes, SULT1A1, 1E1 and 2A1, and UGT2B7. Although oversimplified, the DME can be grouped into one of three categories. CYP3A7, FMO1 and SULT1E1 are expressed at their highest level during the first trimester. All three enzymes either remain at high concentrations, or decrease during gestation and are silenced or expressed at low levels within 1–2 years after birth. These data cause one to query whether or not these enzymes have an important endogenous function during development. CYP3A5, CYP2C19 and SULT1A1 are expressed at relatively constant levels throughout gestation. Postnatal increases in CYP2C19 are observed within the first year, but not for CYP3A5 or SULT1A1. CYP2C9, 2E1, 3A4, FMO3 and SULT2A1 are not expressed or are expressed at low levels during the 2nd and 3rd trimester. However, substantial increases in expression are observed within the first 1–2 years after birth. As will be demonstrated by other speakers in this symposium, these data have been useful for developing predictive physiological-based pharmocokinetic models.
A common characteristic of several DME, considerable interindividual variability is observed in the onset or increase in expression during the immediate postnatal (1 to 6 months), resulting in a window of hypervariability. This also is a time when the spectrum of enzymes being expressed undergoes the greatest changes. In addition, it is interesting that enzymes within a family that appear to share some regulatory mechanisms in the adult exhibit very different developmental expression patterns and presumably, mechanisms regulating ontogeny. Finally, some insight into these regulatory mechanisms has been obtained. Preliminary evidence is consistent with HNF1α being important for the expression of DME during early gestation. Members of both the C/EBP and PAR transcription factor families appear to be important for the onset of several DME during the third trimester and after birth, respectively.
Abstract Not Available
38. ONTOGENY OF DRUG METABOLIZING ENZYMES AND TRANSPORTERS IN HUMANS: THERAPEUTIC CONSEQUENCES
Evelyne M. Jacqz-Aigrain
Paediatric Pharmacology and Pharmacogenetics, Hopital Robert Debré, 88 Boulevard Sérurier, Paris, France
Recent research has demonstrated the central task of phases I and 2 metabolizing enzymes (cytochromes P450 and UDP glucuronyltransferases) and export pumps (phase 3) in xenobiotic defense. They play important roles in drug disposition and protection against xenobiotics, as their function is to reduce the entrance of harmful substances and to eliminate their detoxification products. The profile of appearance of metabolic activities differs with enzymes and may be influenced by environmental and pharmacogenetic factors. The therapeutic consequences of immaturity, delayed maturation and/or disequilibrium between metabolic reactions are important in terms of efficacy and/or toxicity during development. In contrast, data on the developmental pattern of efflux pumps are very limited. The two major transporters families that influence pharmacokinetics are the ATP-binding cassette (ABC) transporters and the solute carrier (SLC) transporters. ABC transporters, encoded by a large transporters gene family, are export pumps highly expressed in the apical membrane of intestinal cells, hepatocytes, cells of kidney proximal tubules and in the blood-brain barrier. Their basal expression vary widely among individuals. In addition to physiological and environmental factors, expression and function are modified by genetic polymorphisms. This may be illustrated with MDR1/ABCB1. MDR1 mediates the transport of numerous drugs and may modulate drug response. Components of daily nutrition can influence MDR1 activity (grapefruit juice, dietary salt). Pharmacogenetic polymorphisms have been identified in the ABCB1 genes. The factors that modulate the developmental pattern of Pgp expression in various tissues are unknown. Data in mouse have shown marked changes in Pgp expression during embryogenesis and post-natal maturation, with a five-fold increase in intestinal expression from birth through adulthood. Levels of hepatic and renal Pgp expression were similar at birth and in adult animals. The CYP3A Pgp complex is expressed in enterocytes, with potential individual differences in biovailability of xenobiotics. In adults, CYP-3A isoforms and Pgp levels were similar in samples from the stomach, jejunum and ileum. In children aged 1-month to 17 years, a significant expression of CYP3A and Pgp mRNA was evidenced in the duodenum. Age related maturation of the CYP3A-Pgp complex will be illustrated with example (cyclosporine, digoxin). Like ABC transporters, SLC transporters are important in drug disposition and response: more than 300 genes are grouped into 43 families, playing an important role in cellular physiological processes, including exporting or importing drugs. SLC19A1 (RFC1) is a folate transporter with high affinity for reduced folates, able to transport and mediate resistance to methotrexate. Genetic polymorphisms in the SLC19A1 gene have been associated with variable expression of the protein and alteration in therapeutic efficacy. The impact of RFC1, MDR1, MRP2 pharmacogenetic polymorphisms on the pharmacokinetics of methotrexate in paediatric patients with acute lymphoblastic leukaemia will be presented as an example. In conclusion, metabolizing enzymes and transport proteins constitute barriers to protect the body from toxic compounds. During ontogeny, the factors that modulate their developmental pattern are largely unknown, although animal studies identified hormonal and nutritional factors.
39. INCORPORATING INFORMATION ON CYP ONTOGENY INTO MODELS FOR PREDICTING DRUG DISPOSITION IN CHILDREN: THE IN SILICO CHILD. - HOW FAR AWAY ARE WE FROM REALITY?
Trevor N. Johnson
Simcyp Limited, Sheffield, United Kingdom, S2 4SU
Building in silico physiologically-based pharmacokinetic (PBPK) models for predicting drug disposition in children is dependent on the availability of a range of information in this population including demography, developmental physiology, pharmacogenetics, and ontogeny of drug metabolising enzymes and transporters. Information on CYP ontogeny is available from a number of in vitro and in vivo studies. The main problems with the in vitro data has been the limited availability of paediatric liver tissue which often has led to large gaps in the data for certain age groups, compromised viability of tissue and hence large variability in results. In addition the range of antibodies used to measure CYP abundance and variation in probe substrates used to measure activity caused obstacles in integrating the results from different reports. Data from in vivo studies must be interpreted correctly to unravel the effects due to enzyme ontogeny from the effects of changes in liver size, protein binding, and in the case of urinary ratios, the renal function [1]. A systematic review of the available in vitro information on CYP ontogeny has been undertaken with weighting for size of data points and variability of results in each study. The derived ‘best fit’ models of CYP ontogeny have been incorporated into a paediatric PBPK model [2] and these have been further verified using sensitivity analysis. This was achieved by changing the ontogeny model within the PKPB model and comparing predicted with observed clearance (CL) values for a range of drugs. A comparison of the performance of two ontogeny models of CYP3A4 on the prediction of midazolam (MDZ) CL is shown in .
Figure 1 Prediction of weight related MDZ systemic CL by incorporating either hyperbolic (A) or biphasic (B) CYP3A4 ontogeny patterns into the overall PBPK model. Solid squares plus error bars are in vivo data, grey symbols are PBPK models predictions, solid line is median prediction and dashed line 5th and 95th percentile.
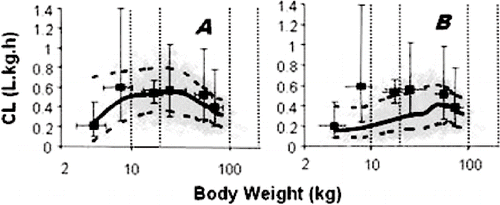
Others key areas pertinent to PBPK modelling where more research are required in the paediatric population include biliary excretion, transporter ontogeny and tissue composition. Moving towards the reliable fully mechanistic PBPK models provides prior information on key drug exposure parameters in neonates, infants and children such as CL, Cmax,and Tmax. In addition it enables estimation of two other key parameters, Vd and T1/2. Although collating information and model building needs time and effort, it may ultimately pay off by reducing the number of PK studies needed in the development of safe medicines for children.
REFERENCE
- Johnson TN, Rostami-Hodjegan A, Tucker GT, Prediction of drug clearance from in vitro data for eleven drugs in neonates infants and children using Simcyp Paediatric. Clin Pharmacokinet 2006; 45 (9) 931–956.
- Johnson TN, Tucker GT, Rostami-Hodjegan A, Ontogeny of CYP2D6 and CYP3A4 in the first year of life. Clin Pharmacol Ther (AOP Nov 28th).
Abstract Not Available
Abstract Not Available
42. THIOPURINE TREATMENT IN CANCER THERAPY: METABOLIC ASPECTS AND CLINICAL CONSEQUENCES
Matthias Schwab
Dr. Margarete Fischer-Bosch Institute of Clinical Pharmacology, Auerbachstrasse 112, Stuttgart, 70376, Germany
The thiopurines azathioprine (AZA), 6-mercaptopurine (6-MP) and 6-thioguanine (6-TG) are essential components of treatment protocols for childhood ALL and other diseases (e.g., inflammatory bowel disease). In the past 25 years, insights into thiopurine pharmacology have been gained and led to the development of strategies for improving efficacy and reducing toxicity. Polymorphisms in genes that encode drug metabolizing enzymes as well as drug transporters have profound effects on patient outcome including drug toxicity. One of the best examples is the thiopurine methyltransferase polymorphism (TPMT) and its clinical relevance for thiopurine therapy. TPMT deficient and heterozygous subjects are at high risk of developing severe hematotoxicity. However, it is unlikely that one single gene will affect exclusively disease and/or treatment outcome, and therefore, a more comprehensive approach will be to consider genetic polymorphisms in entire biological/pharmacological pathways. Other candidate genes in thiopurine metabolism and transport such as ITPA, NDPK and ABCC4 and ABCC5 may be of importance for better prediction of thiopurine response. Moreover, recently developed ‘-omics appoaches’ (e.g., genomics, transcriptomics, proteomics) to identify putative genes for predicting drug response will complement each other.
ACKNOWLEDGMENT
Supported by the Robert Bosch Foundation, Stuttgart, Germany
REFERENCE
- Teml A., Schaeffeler E., Herrlinger KR., Klotz U., Schwab M (2007). Thiopurine treatment in inflammatory bowel disease: clinical pharmacology and implication of pharmacogenetically guided dosing. Clin Pharmacokinet. 46:187–208.
- Schaeffeler E, Eichelbaum M, Reinisch W, Zanger UM, Schwab M (2006). Three novel thiopurine S-methyltransferase allelic variants (TPMT*20, *21, *22) – association with decreased enzyme function. Hum Mutat 27:976.
- Stanulla M, Schaeffeler E, Flohr T, Cario G, Schrauder A, Zimmermann M, Welte K, Ludwig WD, Bartram CR, Zanger UM, Eichelbaum M, Schrappe M, Schwab M (2005). Thiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemia. JAMA 293:1485–9.
43. IRINOGENETICS
Federico Innocenti
Medicine, University of Chicago, Chicago, IL, USA, 60637
This talk will discuss the current challanges in the application of pharmacogenetic testing in the clinic, using irinotecan as an example. Irinotecan is widely used in the treatment of metastatic colorectal cancer and extensive small-cell lung cancer. Its use is limited by severe toxicities, such as neutropenia and delayed-type diarrhea. Irinotecan is converted to its active metabolite SN-38. SN-38 is further metabolized to SN-38G by various hepatic and extrahepatic UGT1A isozymes, mainly UGT1A1. Reduced glucuronidation activity of the UGT1A1 enzyme has been linked with elevated levels of SN-38, leading to toxicities. UGT1A1*28 is an extra TA repeat in the UGT1A1 promoter region and is the variant most frequently associated with interpatient variability in irinotecan pharmacokinetics and toxicities. This information led to the revision of the irinotecan label by the FDA. Recently, UGT1A1*6 seems to contribute to the risk of toxicity of irinotecan in Asian patients. Irinogenetics (the pharmacogenetics of irinotecan) is one of the few promising examples of the application of pharmacogenetics to individualized drug therapy.
Abstract Not Available
Abstract Not Available
Abstract Not Available
47. TREATMENTS TO AID SMOKING CESSATION
Robert West
Department of Epidemiology and Public Health, University College London, London, United Kingdom
Approximately 95% of serious attempts to stop smoking fail, most of these within the first week. An important contributor to this is a powerful feeling of need or urge to smoke, often in situations where smoking would normally have occurred. The root causes of this are changes that take place in the central nervous system as a result from repeated ingestion of nicotine from cigarettes. Treatments to aid smoking cessation focus on reducing or eliminating this pharmacologically-instigated motivation to smoke. It has been known for decades that nicotine replacement therapies (NRT) in the form of patch, chewing gum, lozenge, nasal spray and inhaler can reduce the motivation to smoke during a quit attempt and improve the chances of success. More recently it has been found that starting nicotine patch use two weeks before stopping smoking can improve success rates and that combining different forms of NRT can also enhance success rates. These products are safe and, possibly because they do not deliver nicotine in such a rapid and flexible manner as cigarettes, few smokers become dependent on them. The anti-depressants, bupropion (Zyban) and nortriptyline have also been found to reduce motivation to smoke during a quit attempt and aid abstinence. The treatment that is currently yielding the highest success rates is varenicline (Chantix in the US and Champix elsewhere). This is a partial agonist binding strongly to the nicotinic acetylcholine receptor made up of alpha4 and beta2 subunits. No serious adverse effects of this medication have been observed to date and while nausea is a common side effect it is generally well tolerated. Quite a wide array of other medications are under development or being evaluated including CB1 antagonists (e.g. rimonabant) and nicotine vaccines. As yet none of these has reached the point where they can be submitted for marketing approval. At least as important as developing new medications is finding ways of encouraging more smokers to use existing treatments and to use them as directed to get the maximum benefit from them.
Abstract Not Available
Abstract Not Available
Abstract Not Available
Abstract Not Available
52. PHARMACOGENETICS OF PROTEIN DRUGS
Magnus Ingelman-Sundberg
Section of Pharmacogenetics, Karolinska Institutet/Physiology and Pharmacology, Stockholm, Sweden, SE-17177
The accumulating knowledge of human genomic variation is being utilized for the development of personalized medicine, with the aims to decrease the number of adverse drug reactions and to increase the efficacy of drug treatment. Considerable pharmacogenomic research has focused on understanding the molecular mechanisms behind ADRs, and finding biomarkers that identify people at risk. For immune-mediated toxicities, much focus has been placed on the MHC Class I genes. In light of the rapidly increasing field of development of protein drugs for treatment of e.g rheumatoid arthritis and cancer the issue of protein pharmacogenomics has recently been highlighted. This area includes pharmacogenomic biomarkers for predicting the response rate of antibody treatment of cancer and anti-inflammatory action by protein drugs for cytokine inactivation. This lecture will give an update of the field including several clinically useful examples.
53. FOG-2 ACTS IN CONCERT WITH GATA-4 IN THE TRANSCRIPTIONAL REGULATION OF THE CYP2C19 GENE
Jessica Mwinyi, Rasmus Steen Pedersen, and Magnus Ingelman-Sundberg
Department of Physiology and Pharmacology, Section of Pharmacogenetics, Karolinska Institutet, Stockholm, Sweden, SE-17177
CYP2C19 is an important enzyme involved in the metabolism of several important drugs such as omeprazole, proguanil and escitalopram. In the past many studies have investigated the influence of genetic polymorphisms on CYP2C19 enzyme activity. The physiological regulation of CYP2C19 however, is not well understood. Recently we preliminary reported that CYP2C19 gene expression is upregulated by the transcription factor GATA-4 via a double binding site within the first 200bp of the CYP2C19 5′-flanking region (Mwinyi et al. 2007). The transcription factor family GATA is known to act in concert with different co-regulators, which can inhibit or enhance the effect of GATA (Lowry et al. 2006). In this study we investigated, whether the multi-zinc finger protein Friend Of GATA 2 (FOG-2), an important co-factor of GATA-4 in several tissues (Lin et al. 2004), plays a significant role in CYP2C19 regulation. For this purpose luciferase gene reporter assays were conducted in two human liver - derived cell lines, Huh7 and HepG2. A 435bp long fragment of the CYP2C19 5′-flanking region was subcloned upstream of the luciferase gene into the pGL3 basic vector (2C19_-435). The CYP2C19 construct was transiently co-transfected with mouse pcDNA1.1-GATA-4 and with stepwise increasing amounts of pcDNA3-FOG-2 or with pcDNA3.1 empty vector as negative control. Moreover a mutated pcDNA3-FOG-2 construct expressing a FOG-2 protein lacking the essential protein domains for an interaction with GATA was included in the transfection experiments. Co-transfections were conducted in triplets. Cells were harvested after 48 hours and luciferase activity was measured. Co-transfection of 2C19_-435 and GATA-4 caused an up-regulation of luciferase activity in both cell lines (p < 0.05 in Huh7 cells). A strong loss in luciferase activity was observed, when 2C19_-435 was co-transfected with both GATA-4 and FOG-2 (p < 0.01, Huh7 cells). The suppressive effect correlated in both cell lines with the amount of pcDNA3-FOG-2 transfected. The suppressive effect disappeared totally when co-transfecting mutated pcDNA3-FOG-2 together with GATA-4 and 2C19_-435 (p < 0.03 in Huh7 and HepG2 cells compared to co-transfection with wild type FOG-2) indicating that an interaction of GATA-4 and FOG-2 is essential for an effective inhibition of FOG-2 on CYP2C19 expression. In this study we show that CYP2C19 expression is inhibited by FOG-2 via interaction with GATA-4. This study moreover identifies a protein with a suppressive effect on CYP2C19 expression.
REFERENCE
- Mwinyi, J., Hofmann, Y., Ingelman-Sundberg M.: Promoter mediated regulation of CYP2C19 by GATA-4. Basic Clin. Pharm Toxicol 2007: 101 (s1), 158–207.
- Lowry, JA, Mackay, JP.: GATA-1: one protein, many partners. Int. J. Biochem. Cell. Biol. 2006;38:6–11.
- Lin, AC, Roche, AE, Wilk, J, Svensson, EC.: The N termini of Friend of GATA (FOG) proteins define a novel transcriptional repression motif and a superfamily of transcriptional repressors. J. Biol Chem. 2004;279:55017–23.
54. MODULATION OF UGT ENZYMATIC PROPERTIES BY NATURALLY OCCURRING HUMAN UGT1 SPLICE VARIANTS
Judith Bellemare, Hugo Girard, Éric Lévesque, and Chantal Guillemette
Canada Research Chair in Pharmacogenomics, Laboratory of Pharmacogenomics, CHUQ Research Center, Faculty of Pharmacy, Laval University, Quebec, QC, Canada, G1V 4G2
UDP-glucuronosyltransferases (UGTs) are responsible for the glucuronidation pathway and play an essential role in the elimination and detoxification of a diversity of endogenous substances and xenobiotics. Our recent findings showed that the UGT1 locus encodes one additional exon (exon 5b) located in the common region shared by all UGT1. An alternative splicing process creates 9 splice variant proteins (referred to as isoform 2 or UGT1A-i2) containing the exon 5b instead of the exon 5 (or 5a). We initially showed that while i2 proteins lack transferase activity, they modify UGT1A-i1 mediated activity. Then, we fully characterized the influence of i2 proteins on i1 enzymatic function through detailed enzyme kinetics. This was achieved for two highly related UGTs using recombinant human UGT1A7 and UGT1A8 expressing the classical UGT-i1 isoform compared to microsomes from HEK293 cells expressing both i1 and the corresponding i2 splice variant (i1+i2). The relative expression levels of i1 and i2 proteins (i1/i2) in microsomes were 1.0/0.5 for UGT1A7-i1+i2 and 1.0/0.7 for UGT1A8-i1+i2. Using high affinity (irinotecan's active metabolite SN-38) and low affinity (4-methylumbelliferone (4-MU)) substrates, we show that the presence of i2 markedly inhibits UGT1A7-normalized activity by 5.0 to 6.5-fold (Vmax) in recombinant UGT1A7-i1+i2 compared to UGT1A7-i1 (p < 0.05), but has no influence on Km values. For UGT1A8 recombinant proteins, i2 also decreases drastically the apparent Vmax of the 4-MU glucuronidation reaction by 9.5-fold (p = 0.02). Conversely to UGT1A7, the presence of i2 substantially enhanced the affinity for 4-MU in UGT1A8-i1+i2 (Km = 176 uM) compared to UGT1A8-i1 (Km = 528 uM; p = 0.01). These significant changes on enzyme kinetics could be mediated by several mechanisms. Co-immunoprecipitation experiments using microsomes containing both i1 and i2 proteins support the formation of i1+i2 protein complexes. Further immunofluorescence experiments confirm that i1 and i2 co-localized within cells. Results indicate that the modulatory effect by i2 proteins is potentially mediated through functional interactions between i1 and i2 proteins leading to a decline of enzyme activity. However, for UGT1A8, the effect is more complex, decreasing glucuronidation rates while increasing affinity. Collectively, these findings delineate a role for UGT1A-i2 splice variants as a potential modulatory mechanism of glucuronidation.
ACKNOWLEDGMENT
Supported by NSERC.
55. DISTRIBUTION OF HUMAN FLAVIN-CONTAINING MONOOXYGENASE 3 POLYMORPHISMS IN DIFFERENT ETHNIC GROUPS
Mao Mao1, Marja-Liisa Dahl1, Arzu Günes1, Norio Yasui-Furukori2, and Maria G. Scordo1
1Department of Medical Sciences, Clinical Pharmacology, Uppsala University, Uppsala, Sweden, SE-75185, 2Department of Neuropsychiatry, Hirosaki University, Hirosaki, Japan, 035–8562
The human flavin-containing monooxygenase isoform 3 (FMO3) catalyzes the metabolism of many nucleophilic heteroatom-containing compounds, including drugs such as clozapine, ranitidine and sulindac, environment toxicants and endogenous compounds. The FMO3 gene is polymorphic, and a number of allelic variants causing reduction in the enzyme's catalytic efficiency have been described. Genetically determined interindividual variability in the enzyme activity may affect the therapeutic outcome of FMO3 substrates. Although the FMO3 allelic distribution has been evaluated to some extent in the three major ethnicities, frequency data in ethnic subgroups are still scant. In order to investigate the distribution of FMO3 polymorphisms in different ethnic groups, four populations of randomly selected healthy volunteers from Sweden (410 subjects), Italy (279 subjects), Turkey (300 subjects) and Japan (300 subjects) were genotyped for three common single nucleotide polymorphisms in the coding region of FMO3 gene (E158K, V257M and E308G) using TaqMan allelic discrimination assay. The frequency of the K158 allelic variant was high in the four populations (SWE 44.3%, ITA 33.5%, TUR 35.8%, JAP 22.7%). The G308 variant was the second most common (SWE 22.4%, ITA 10.8%, TUR 6.0%, JAP 21.0%) and the M257 variant was the rarest (SWE 7.1%, ITA 6.3%, TUR 6.3%, JAP 14.5%). The allelic frequencies were similar among the Italians and the Turks, but much lower compared to the Swedes (P < 0.001). Similarly individuals carrying variant haplotypes were more frequent among the Swedes (SWE 27.3%, ITA 16.5%, TUR 17.3%). The G308 variant was cis-linked to the K158 variant in all the four populations. The heterozygosity and homozygosity for the E158K/E308G variant were considerably higher among the Swedes (23.4%, 6.6%) and the Japanese (32.0%, 4.7%) than among the Italians (15.1%, 1.1%) and the Turks (8.0%, 0.0%). Hence, the distribution of common FMO3 polymorphisms varies not only inter-ethnically between Caucasians and Asians, but also intra-ethnically among Caucasian groups from North to South Europe. FMO3-mediated metabolic capacity may exhibit large variability between individuals and across populations.
56. FUNCTIONALLY OVERLAPPING ROLES OF BCRP1 AND MRP2 IN THE ELIMINATION OF METHOTREXATE AND ITS MAIN METABOLITE 7-HYDROXYMETHOTREXATE
Maria L.H. Vlaming1, Zeliha Pala2, Anita Van Esch1, Els Wagenaar1, Olaf Van Tellingen3, and Alfred H. Schinkel1
1Department of Experimental Therapy, Netherlands Cancer Institute, Amsterdam, Netherlands, 1066 CX, 2Department of Pharmacology, Istanbul University, Istanbul, Turkey, 3Department of Clinical Chemistry, Netherlands Cancer Institute, Amsterdam, Netherlands, 1066 CX
The multidrug transporters Bcrp1 (Abcg2) and Mrp2 (Abcc2), both members of the ATP-binding cassette (ABC) transporter superfamily, are present in the apical membranes of hepatocytes and epithelial cells of small intestine and kidney, where they play a role in the excretion of both endogenous and exogenous compounds into bile, feces and urine. Bcrp1 and Mrp2 show substantial overlap in substrate specificity, and can both influence the pharmacokinetics of many (anti-cancer) drugs.
To investigate the relative and possibly overlapping roles of Bcrp1 and Mrp2 in physiology and pharmacology, we generated Bcrp1;Mrp2-/- mice. The double knockout mice are viable and fertile and show no aberrations under standard housing conditions, except for mild hyperbilirubinemia, due to absence of Mrp2 in the liver. Bcrp1;Mrp2-/- mice also have a ∼30% increased liver weight.
The generated Bcrp1;Mrp2-/- mice were used to investigate the roles of Bcrp1 and Mrp2 in the elimination of the widely used anti-cancer and anti-rheumatic drug methotrexate (MTX) and its toxic metabolite 7-hydroxymethotrexate (7OH-MTX) after intravenous injection. Methotrexate was administered i.v. at a dose of 50 mg/kg to female wild-type, Bcrp1-/-, Mrp2-/- and Bcrp1;Mrp2-/- mice and plasma levels of MTX and 7OH-MTX were analyzed by HPLC. Compared to wild-type, the plasma areas under the curve (AUC) for MTX were 1.6-fold and 2.0-fold higher in Bcrp1-/- and Mrp2-/- mice respectively, whereas the plasma AUC was 3.2-fold increased in Bcrp1;Mrp2-/- mice, suggesting an additive effect of both transporters in MTX plasma elimination. When the 7OH-MTX levels in plasma of these mice were analyzed, we found no significant difference in Bcrp1-/- mice, but a 6.2-fold increased plasma AUC in Mrp2-/- mice, suggesting an important role for Mrp2 in elimination of 7OH-MTX. The plasma AUC for 7OH-MTX in Bcrp1;Mrp2-/- mice was even 11.8-fold increased, indicating that Bcrp1 can partly compensate for absence of Mrp2 in elimination of 7OH-MTX in mice.
These findings clarify the influence of Bcrp1 and Mrp2 on MTX and 7OH-MTX pharmacokinetics, and show the value of ABC transporter compound knockout mice for studying the pharmacokinetics of various endogenous and exogenous toxic compounds in vivo.
57. CHARACTERIZATION OF ENZYMES AND TRANSPORTERS INVOLVED IN THE DISPOSITION OF OSELTAMIVIR AND ITS ACTIVE FORM
Mototsugu Ito1, Hiroyuki Kusuhara1, Kenzo Yamatsugu1, Motomu Kanai1, Masakatsu Shibasaki1, Masakiyo Hosokawa2, Tsunenori Kondo3, and Yuichi Sugiyama1
1Department of Molecular Pharmacokinetics, The University of Tokyo, Tokyo, Japan, 113‐0033, 2Faculty of Pharmaceutical Sciences, Chiba Institute of Sciences, Choshi, Japan, 288-0025, 3Tokyo Women's Medical University, Tokyo, Japan, 162–8666
Oseltamivir (Tamiflu) is an ethyl ester prodrug of Ro 64–0802, a selective inhibitor of influenza virus neuraminidase. In Japan, abnormal behavior has been reported in influenza patients prescribed oseltamivir although the relationship between this abnormal behavior and oseltamivir medication remains an open question. Based on this background, there is growing interest in the transporters and enzymes involved in the disposition of oseltamivir and Ro 64–0802. Recently, we demonstrated that P-gp limits the penetration of oseltamivir through the blood-brain barrier (Ref 1). Oseltamivir is largely converted to Ro 64–0802 in the liver, and then predominantly excreted into the urine by tubular secretion as well as glomerular filtration. In this study, we characterized the role of human drug transporters in the pharmacokinetics of oseltamivir and Ro 64–0802. Oseltamivir and Ro 64–0802 were synthesized according to the methods reported previously (Ref 2). Cellular accumulation of oseltamivir and Ro 64–0802 was determined in HEK293 cells expressing hepatic and renal drug transporters. The amount of oseltamivir and Ro 64–0802 associated with cell and buffer specimens were quantified by LC/MS. Oseltamivir was significantly taken up by organic cation transporter (OCT) 1, organic anion transporters (OAT) 3, and organic anion-transporting polypeptide (OATP) 1B3, while Ro 64–0802 was significantly taken up by OAT1, OAT2, OAT3, OCT1, and OCT2. Neither OATP1B1 nor 1B3 accepted Ro 64–0802 as a substrate. Furthermore, we investigated the physiological roles of renal uptake transporters using human kidney slices, following the protocol reported previously (Ref 3). Uptake of Ro 64–0802 by human kidney slices was significantly inhibited by probenecid (1 mM) (50h}6 % of the control), but not by tetraethylammonium (1 mM), indicating that OAT1 and/or OAT3 plays a more important role in the renal uptake of Ro 64–0802. This finding is consistent with a clinical drug interaction study in which probenecid, but not cimetidine, inhibits the renal elimination of Ro 64–0802, resulting in a 2.5-fold increase in the systemic exposure (area under the curve) (Ref 4). On the other hand, the urinary excretion of oseltamivir and Ro 64–0802 is not different in P-gp (Mdr1a/1b), breast cancer resistance protein (Bcrp), multidrug resistance-associated protein (Mrp) 2, Mrp 4, and Na+/Phosphate cotransporter (Npt) 1-knockout mice compared with that of control mice. Other unknown transporter(s) in the apical membrane of renal epithelia might be thus involved in their urinary excretions.
REFERENCE
- Ose A. et al. P-glycoprotein restricts the penetration of oseltamivir across the blood-brain barrier. Drug Metab Dispos. 2007, in press.
- Yamatsugu et al. A concise synthesis of Tamiflu: third generation route via the Diels? Alder reaction and the Curtius rearrangement Tetrahedron Lett. 48:1403–1406, 2007
- Nozaki Y. et al. Characterization of the uptake of organic anion transporter (OAT) 1 and OAT3 substrates by human kidney slices. J Pharmacol Exp Ther 321:362–369, 2007
- Hill G. et al. The anti-influenza drug oseltamivir exhibits low potential to induce pharmacokinetic drug interactions via renal secretion-correlation of in vivo and in vitro studies. Drug Metab Dispos. 30:13–9, 2002.
58. ENDOPLASMIC RETICULUM STRESS INITIATES TUBULAR CELL DEDIFFERENTIATION AND DEATH DURING CYCLOSPORINE A TREATMENT
Nicolas Pallet1, Marion Rabant1, Eric Thervet1, Philippe Beaune1, Christophe Legendre2, and Dany Anglicheau1
1U775, INSERM, Paris, France, 75006, 2Service de transplantation rénale, Hopital Necker, Paris, France
Chronic nephrotoxicity related to cyclosporine (CsA) therapy leads to interstitial fibrosis and tubular atrophy and the tubular epithelium plays a central role of the in the pathogenesis of chronic nephropathies, especially through epithelial to mesechymal transition. cDNA microarrays screening on tubular cells led us to demonstrate that CsA induces endoplasmic reticulum stress (ER stress), since GRP78, protein disulphide isomerase, CHOP and HERP expression is increased at protein and mRNA levels during CsA exposure. ER stress was also observed in rat kidneys treated with 15/mg/kg CsA. CsA and various ER stress inducers (thapsigargin, A23187, DTT, glucose starvation) trigger marked tubular phenotypic changes (TPC) reminiscent of an incomplete epithelial to mesenchymal transition with cells loosing cell-cell contact, down regulation of E-cadherin expression, nucleo-cytoplasmic redistribution of ß-catenin, and increase of the expression of the fibroblast marker HSP47. ER stress and TPC are also observed when cyclophilin A gene expression is targeted by siRNA. Cyclophilin A is an isomerase implicated in nascent protein folding, that is inhibited by CsA. When CsA is co-incubated with Salubrinal, an ER stress inhibitor, tubular phenotypic changes do not appear since cell shape remains polygonal, E-cadherin is not down regulated, HSP47 expression level is indetectable and β-catenin do not translocate into the nucleus. Moreover, immunohistochemical tubular detection of GRP78, used as a surrogate marker of ER stress, on protocol biopsies performed on transplanted patients is significantly associated with tubular atrophy and interstitial fibrosis. ER stress in tubular cells that initiates tubular cell phenotypic changes suggestive of an incomplete epithelial to mesenchymal transition. Salubrinal, in inhibiting ER stress, blocks tubular epithelial phenotype alterations during CsA exposure. This study suggests that ER stress may initiates epithelial-to-mesenchymal transition during CsA treatment and its detection could serve as an early biomarker of interstitial fibrosis and tubular atrophy.
59. GENETIC POLYMORPHISMS OF CYP1A2 AND CYP2C19 INFLUENCE LEFLUNOMIDE TREATMENT IN RHEUMATOID ARTHRITIS PATIENTS
Petra Bohanec Grabar1, Iztok Grabnar2, Aleš Mrhar3, Blaž Rozman4, Dušan Logar4, Matija Tomšič4, Sonja Praprotnik4, and Vita Dolžan1
1Institute of Biochemistry, Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia, 1000, 2Faculty of Pharmacy,University of Ljubljana, Ljubljana, Slovenia, 1000, 3Faculty of Pharmacy, University of Ljubljana, Ljubljana, Slovenia, 1000, 4Department of Rheumatology, University Medical Centre Ljubljana, Ljubljana, 1000
Leflunomide is a disease-modifying antirheumatic drug that is used for the treatment of rheumatoid arthritis (RA). Upon oral absorption, it is converted to the active metabolite (A77 1726) that inhibits de novo synthesis of pyrimidine ribonucleotides. In vitro studies have demonstrated that cytochromes P450 (CYPs), mainly CYP1A2 and CYP2C19 may be involved in leflunomide activation (1). The aim of our study was to investigate the hypothesis that genetic polymorphisms of CYP1A2 and CYP2C19 influence the A77 1726 serum concentrations and the response of leflunomide treatment in RA patients. The main criterion for selection of RA patients was current or past leflunomide treatment. Among 112 RA patients, 62 were taking ongoing leflunomide treatment, while 50 patients discontinued the treatment within the first year due to inefficacy (N = 7), toxicity (N = 37) or both (N = 6). Trough steady-state A77 1726 serum concentrations were determined by validated HPLC with UV detection in a subset of 67 patients taking ongoing leflunomide treatment. A genotyping approach was used to determine CYP1A2 -163C>A, CYP2C19*2 and CYP2C19*17 genotypes in 112 RA patients. Mean values of A77 1726 serum concentrations were not significantly different between CYP1A2 -163CC and CYP1A2 -163CA+AA genotypes (43.9 ± 39.5mg/L and 33.7 ± 28.9 mg/L, respectively). However, carriers of the CYP2C19*2 allele had significantly lower mean values of A77 1726 serum concentrations as compared to patients with CYP2C19*1/*1 genotype (18.4 ± 12.8 mg/L vs. 43.7 ± 33.7 mg/L, P = 0.009). On the other hand, CYP2C19 genotypes did not inluence the treatment response (P = 0.258), while an association between CYP1A2 C-163A polymorphism and leflunomide toxicity was observed. Patients with the CYP1A2 -163CC genotype had higher risk for leflunomide-induced toxicity as compared to carriers of the CYP1A2 -163A allele (P = 0.001, OR = 11.933, 95% CI = 2.793–50.980). Our results suggest that genetic polymorphisms of CYP2C19 had an impact on the A77 1726 serum concentrations, while the CYP1A2 -163C>A polymorphism influenced the leflunomide toxicity in RA patients.
REFERENCE
- Kalgutkar AS, Nguyen HT, Vaz AD, Doan A, Dalvie DK, McLeod DG, et al. In vitro metabolism studies on the isoxazole ring scission in the anti-inflammatory agent lefluonomide to its active alpha-cyanoenol metabolite A771726: mechanistic similarities with the cytochrome P450-catalyzed dehydration of aldoximes. Drug Metab Dispos 2003;31:1240–50.
60. CLASSIFICATION OF CYTOCHROME P450 1A2 SUBSTRATES AND NON-SUBSTRATES BY MACHINE LEARNING TECHNIQUES AND BINARY QSAR
Poongavanam Vasanthanathan1, Olivier Taboureau1, Chris Oostenbrink2, Nico P. E Vermeulen2, Lars Olsen1, and Flemming Steen Jørgensen1
1Biostructural Research, Department of Medicinal Chemistry, Faculty of Pharmaceutical Sciences, University of Copenhagen, Copenhagen, Denmark, 2LACDR-Division of Molecular Toxicology, Dept of Pharmacochemistry, Vrije Universiteit, Amsterdam, Netherlands, NL-1083 HV
The cytochrome P450 (CYP) superfamily plays an important role in the biotransformation of a huge number of endogenous and exogenous compounds in humans. A large number of compounds can be screened for inhibition in short time by high throughput screening (HTS). There is an urgent need for computational methods to interpret and understand these large amounts of data. Here, we provide the first application of binary QSAR and machine learning techniques, like support vector machine (SVM), random forest, decision tree, and kappa nearest neighbors (kNN) methods for classification of a large, structurally diverse, data set of 7,469 drug-like compounds into substrates and non-substrates of the cytochrome P450 1A2 enzyme. Such a classification is useful, for example, in the early phase of the drug discovery process. Descriptors, including physical properties, Kier and Hall connectivity and kappa shape indices, pharmacophore feature descriptors, charge/surface descriptors, and Volsurf pharmacokinetic descriptors using four standard probes were used to cover various aspects of the chemical properties of the ligands. The percentage of correctly classified compounds in the training set (test set in brackets) were for random forest: 99(75) %; for kNN: 78(72) %; for SVM: 83(75) %; for binary QSAR 70(66) %; for a decision tree model 96(72) %. All the models have a balanced prediction of substrates and non-substrates. Finally, we tried to classify substrates and non-substrates by developing a model using the Lipinski “Rule of five” descriptors by a decision tree method (Matthews correlation coefficient is 0.43, 71% correctly predicted). The tree model suggests that substrate-like compounds have the following properties: the number of hydrogen bond acceptors below or equal to 6; number of hydrogen bond donors around 2; logarithmic partition coefficient (water/octanol) greater than or equal to 3.7; molecular weight between 198 to 516. Compounds that fall outside these ranges are considered to be non-substrates. This final model can be potentially useful for a very rapid computational filter to predict cytochrome P450 1A2 substrates in large HTS libraries.
61. THE ROLE OF LYSINE- AND ARGININ-RESIDUES FOR THE TRANSPORT ACTIVITY OF OATP1B3
Hartmut Glaeser, Kathrin Mandery, Martin F. Fromm, and Jörg König
Institute of Experimental and Clinical Pharmacology and Toxicology, Friedrich-Alexander-University Erlangen-Nuremberg, Erlangen, Germany, 91054
The uptake transporter OATP1B3 (organic anion transporting polypeptide 1B3) encoded by the SLCO1B3 gene plays an important role in the hepatic disposition and elimination of many drugs (e.g. pravastatin) and endogenous compounds (taurocholate). However, at present little is known about the structure, transport mechanisms and catalytic amino acids. Therefore, the importance of Lys- and Arg-residues for the protein expression, localization and transport activity was investigated in order to gain better insights into mechanisms underlying the OATP1B3-mediated transport. After a selection of several conserved Lys and Arg residues we mutated the Lys28 and Arg580 to Lys28 > Gly (A82 > G, A83 > G), Lys28 > Arg (A82 > C, A83 > G) and Arg580 > Gly (A1738 > G) and Arg580 > Lys (G1739 > A), respectively. Following transient transfection of these cDNAs into HEK293 cells the influence on protein expression, subcellular localization and transport activity was investigated using Western Blot, immunofluorescence, and cellular uptake assays performed with the substrate sulfobromopthalein (BSP). Western Blot analysis of all investigated mutants showed at most a change in protein amount of ∼25 %. Moreover, immunofluorescence analysis indicated that the changes of Lys28 to Arg and Gly as well the changes of Arg580 to Gly and Lys did not lead to altered membrane localization. However, in contrast to the Lys28 mutants, which have no effect on transport, Arg580 > Gly and Arg580 > Lys were characterized by a significant decrease of BSP uptake (Arg580 >Gly: 28 % of wild type (WT) activity, Arg580 > Lys: 12 % of WT activity) indicating that this conserved amino acid might play an pivotal role for the transport activity of OATP1B3. Taken together, these data indicate that the Lys28 residue in OATP1B3 plays a minor role for substrate recognition and transport of organic anions. In contrast, the Arg580 residue is important for the transport activity of OATP1B3.
62. COMPARISON OF HEPATIC AND INTESTINAL GLUCURONIDATION
Helen Cubitt, J. Brian Houston, and Aleksandra Galetin
School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester, United Kingdom, M13 9PT
Increasing awareness of the importance of phase II enzymes, specifically uridine diphosphate glucuronosyltransferases (UGTs), indicates a need to incorporate their contribution into clearance prediction. For some drugs, significant intestinal metabolism may explain the under-prediction of in vivo clearance observed when based solely on hepatic in vitro data. This study aimed to compare intestinal and hepatic glucuronidation for a number of substrates using alamethicin activated human intestinal (HIM) and hepatic (HLM) microsomes. Intrinsic glucuronidation clearance (CLint) was obtained for 10 compounds using a substrate depletion approach and corrected for experimentally determined microsomal fraction unbound (fu). The in vitro extent of glucuronidation (fmUGT) was determined using separate incubations with either NADPH (CYP) or UDPGA (UGT) cofactors in both HIM and HLM. Intestinal UGT CLint was 17-fold and 33–fold higher than hepatic UGT CLint for troglitazone and raloxifene, respectively. In contrast, hepatic UGT CLint was 6–fold higher than intestinal UGT CLint for gemfibrozil. For most of the compounds investigated, the in vitro fmUGT values were in good agreement between HLM and HIM; with the exception of troglitazone, where a 9-fold difference in the extent of glucuronidation was observed (74% and 8% in HIM and HLM, respectively). The hepatic UGT CLint values were scaled using a microsomal recovery of 40 mg protein/g liver, a liver weight of 21 g liver/kg and modelled with the well-stirred liver model. On average, a 2-fold underprediction of in vivo CLint was observed; with the exception of troglitazone, where a 44-fold underprediction was seen. Current study has shown that alamethicin activated microsomes represent a useful tool to assess intestinal and hepatic glucuronidation. The use of corresponding CYP and UGT cofactors allowed assessment of the fraction glucuronidated in vitro. In addition, current study has indicated a need for the incorporation of intestinal glucuronidation into clearance prediction, in particular for compounds with a contribution of intestine specific UGTs (UGT1A8, 1A10) to their clearance (e.g., troglitazone).
63. THE PROTECTIVE ROLE OF GLUTATHIONE S-TRANSFERASE T1 IN UNCOMPLICATED MALARIA
Isa Cavaco1, Andreas Martensson2, Achuyt Bhattarai2, Anders Björkman2, Vera Ribeiro1, and José Pedro Gil2
1Dqbf, CBME/IBB, University of Algarve, Faro, Portugal, 8005-139, 2Department of Medicine, Malaria Research Laboratory, Stockholm, Sweden, SE-171 76
The oxidative stress inside the erythrocyte plays an important role in malaria pathology and the antioxidant system is determinant to the progression of disease. Gluthatione S-transferase (GST) is a superfamily of enzymes involved in the antioxidant defence mechanism against reactive oxygen species. In this context, we analysed the known polymorphic deletion in GSTM1 and GSTT1 in the context of uncomplicated Plamodium falciparum malaria. GSTT1 was chosen because it is highly expressed in erythrocytes, so the presence of the GSTT1 isoform would protect the parasite against oxidative stress during the erythrocytic stage. As control, we also analysed GSTM1 in the same group of patients, since it is only expressed in lymphocytes and not expressed in the erythrocytes. By a multiplex amplification of GSTM1 and GSTT1 we analysed in a first study a group of patients with uncomplicated malaria (n = 1515) and a control group of patients without P. falciparum (n = 370), both from Zanzibar islands. In order to confirm the results obtained in the islands, a group of 816 patients from Nyamisati village, situated by the Rufiji River Delta, coastal Tanzania, was also analysed. This area is holoendemic for malaria and the patients were followed up for approximately 7 years (1993–1999), with full characterised malaria clinical history, levels of parasitemia and malaria therapeutics. The results show that in Zanzibar islands, the GSTT1 null genotype is present in a significantly higher frequency (45.4%) in patients without malaria when compared with malaria patients (21.5%) (OR = 0.328 (0.259–0.417)). For GSTM1 we didn't observe significant differences between the two groups (37% vs 36.5%). In the group of patients from Nyamisati village it was observed that the patients without a functional GSST1 are significantly less prone to malaria episodes. The results obtained both in Zanzibar and in Tanzania suggest that GSTT1 may be an important factor in malaria pathology, since the lacking of the GSTT1 gene revealed to be a protective factor in uncomplicated malaria. We may hypothesise that the lacking of GSTT1 can further lead to non viable erythrocyte, and/or might contribute to the elimination of parasited RBCs through the oxidative stress associated to the phagocytosis processes.
64. EXCRETION OF 5 (AND 6)-CARBOXY-2′,7′-DICHLOROFLUORESCEIN (CDF) BY MULTIDRUG RESISTANCE - ASSOCIATED PROTEINS (MRPS) IN RAT HEPATOCYTE CULTURES
Lucy C. Ellis1, G.M. Hawksworth1, and Richard J. Weaver2
1Dept of Med & Therapeutics, Univ of Aberdeen, Aberdeen, United Kingdom, AB25 2ZD, 2Centre for Biopharmacy Research, Servier Research & Development Ltd, Slough, United Kingdom, SL3 6PJ
The multidrug resistance associated proteins are ATP-dependent transporters responsible for the efflux of a wide range of substrates, including endogenous compounds e.g. bilirubin, drug metabolites e.g. paracetamol glucuronide and fluorescent dyes e.g. 5 (and 6)-carboxy-2′,7′-dichlorofluorescein (CDF) (Konig et al, 1999). We have developed a method for quantifying efflux by Mrps in hepatocytes cultured in a sandwich configuration of collagen (Type 1). This method enables us to distinguish efflux by Mrps on the sinusoidal membrane, on the canalicular membrane, and excretion by diffusion. For each CDF-DA concentration (0 - 750μM), 3 plates of cells (6cm) were used to determine efflux of CDF. Vanadate (10μM), an ATPase inhibitor, was added to the first plate, 30 minutes prior to efflux determination. For efflux determination, all plates were incubated with HBSS + calcium for 10 minutes and then CDF-DA for 10 minutes at 37°C. Plate 1 (vanadate treated) was then incubated with HBSS + calcium, plate 2 was incubated with HBSS – calcium and plate 3 was incubated with HBSS + calcium for 10 minutes. Efflux by Mrp2 is the difference between efflux in the absence of calcium (plate 2) and efflux in the presence of calcium (plate 3). Efflux by all Mrps except Mrp2 is the difference between efflux in the presence of calcium (plate 3) and efflux after a treatment with vanadate (plate 1). Diffusion of CDF is the efflux after treatment with vanadate, which inhibits all Mrps. Excretion of 10μM CDF is predominately via Mrp2 (0.09 ± 0.02 nmoles/mg/min) with other Mrps (0.02 ± 0.01 nmoles/mg/min) and simple diffusion (0.06 ± 0.01 nmoles/mg/min) playing lesser roles (n = 4). This method of quantifying efflux disrupts bile canaliculae leaving the hepatocytes intact. Bile canaliculae will re-form after a 24h incubation with fresh medium, so efflux determinations can be repeated on the same cells. Efflux of 10μM CDF decreases slightly after the first bile canalicular disruption (CDF efflux day 4 – 0.70 ± 0.01 nmoles/mg, day 5 – 0.54 ± 0.02 nmoles/mg, n = 4) but is then maintained (CDF efflux day 6 – 0.58 ± 0.03 nmoles/mg, day 7 – 0.50 ± 0.05 nmoles/mg, n = 4). The method described here is unique in its ability to distinguish between sinusoidal efflux, canalicular efflux and diffusion of CDF in intact hepatocytes.
65. XENOBIOTIC METABOLISM AND TRANSPORT IN HUMAN NORMAL BRAIN: REGIONAL AND CELLULAR CARTOGRAPHY FOR A PUTATIVE ROLE IN THE CEREBRAL FUNCTIONS
Marie-Anne Loriot1, Fabien Dutheil1, Sandrine Dauchy2, Ivan Bièche3, Lucille Mellottée1, Olivier Cloarec4, Véronique Sazdovitch5, Charles Duyckaerts5, Isabelle De Waziers1, Xavier Decleves2, and Philippe Beaune1
1Biochemistry, INSERM UMRS 775-Paris Descartes, Paris, France, 75006, 2Laboratory of pharmacokinetics, INSERM U705 CNRS UMR 7157, Paris, France, 3INSERM UMR 735, Saint-Cloud, France, 4Technology Servier, Orléans, France, 5Neuropathology, INSERM U546, Paris, France
The metabolism and transport of xenobiotics in human brain likely modulates the response to drugs and environmental toxics. In this way, cerebral cytochrome P450 enzymes (CYPs), the major phase 1 drug-metabolizing enzymes superfamily, and ABC transporters could play a crucial role. The expression of CYPs and ATP-binding cassette (ABC) transporters at the human brain has been extensively investigated but their relative expression remain unknown. The general objective of the study is the characterization of the distribution of these proteins in the whole normal brain as well as 16 cerebral localizations. First, the mRNA expression profile of 24 distinct cytochromes P450 of CYP1, CYP2 and CYP3 families and CYP46A1, 7 transporters (ABCB1, ABCC1, ABCC2, ABCC3, ABCC4, ABCC5 and ABCG2) was studied. Nuclear receptor (NR) mRNA, involved in the regulation of CYP and ABC proteins, (AhR, CAR, ER, FXR, LXR, PPAR, PXR and RXR) were also studied according to their role in the CYP and ABC protein expression regulation. Secondly, some major CYPs were studied in order to characterize the presence of proteins at cellular and subcellular levels in some brain regions. Analysis of the mRNA levels of each of these CYP isoforms, transporters and NRs was performed on total RNA from pooled specimens of human normal brain regions using real time RT-PCR. CYP, transporter and NR primers were previously validated for their sensitivity and their specificity. Western blotting and immunohistochemistry were used to determine cellular and sub-cellular locations of several CYP proteins.Among the 24 CYP mRNA studied, 6 account for more than 90% of isoforms tested in the brain (CYP46A1, 2J2, 2U1, 1B1, 2E1 and 2D6). CYP46A1 mRNA was widely expressed in all areas of the brain. Our results revealed the existence of specific CYP pattern expression depending on the brain regions. Among the 14 NR mRNA studied, 6 account for more than 95% of isoforms tested in the brain (LXRb, RXRb, PPARd, RXRa, PPARa and PPARg). The characterization of several CYP1A1, 2D6, 2J2, 2U1 and 46A1 proteins in some brain regions was then confirmed. Our results constitute an original description of CYPs and ABC transporters through the human brain. This cartography will broaden the understanding of the possible implication of these CYPs and transporters in the physiological functions and pathological processes in relation with their apparent selective presence in different areas.
66. PROMOTION OF HEPATOCARCINOGENESIS BY PERFLUOROALKYL ACIDS BY AN ESTROGENIC MECHANISM
Abby D. Benninghoff, Cari H. Buchner, Gayle A. Orner, Jerry D. Hendricks, and David E. Williams
Environmental and Molecular Toxicology, Oregon State University, Corvallis, OR, USA, 97330
Previous studies have shown that perfluorooctanoic acid (PFOA) promotes hepatocarcinogenesis in rainbow trout via an estrogenic mechanism. In rodents, PFOA and related perfluoroalkyl acids (PFAAs) exert hepatic toxicity via peroxisome proliferation, though humans are generally insensitive to this mechanism of toxicity. The rainbow trout is a useful animal model that represents the lack of sensitivity to peroxisome proliferators observed in humans. Recently, we demonstrated that PFOA and many other structurally related PFAAs are weak xenoestrogens in trout and bind to the liver estrogen receptor. In the present study, we determined whether multiple PFCs enhance liver carcinogenesis in trout via an estrogenic mechanism. Fry were initiated with the potent liver carcinogen aflatoxin B1 (AFB1, 10 ppb) and then fed either custom control diet or the following experimental diets for 30 weeks (daily ration 2 to 5% of body weight): 2000 ppm PFOA, 1000 ppm perfluorononanoic acid (PFNA), 200 ppm perfluorodecanoic acid (PFDA), 100 ppm perfluorosulfonate (PFOS), 2000 ppm 8:2 fluorotelomer alcohol (8:2 FtOH), 5 ppm 17β-estradiol (E2) or 2000 ppm clofibrate (CLOF), a classic peroxisome proliferator. Incidence, multiplicity and size of liver tumors in trout fed diets containing E2, PFOA, PFNA, PFDA and PFOS were significantly higher compared to AFB1-initiated animals fed control diet only, whereas CLOF was without effect and 8:2 FtOH exposure caused minor enhancement of tumorigenesis. To elucidate the mechanism by which these chemicals promote liver cancer in trout, a custom rainbow trout oligo DNA microarray was used to obtain hepatic gene expression profiles for each experimental diet after two weeks of exposure. E2, PFOA, PFNA and PFDA regulated many transcripts in common, including genes associated with vitellogenesis, regulation of transcription, signal transduction and immune response. Hierarchical clustering and Pearson correlation analyses showed that the expression profiles for E2 and PFOA, PFNA, PFDA and PFOS were highly similar (r > 0.7), although a number of genes were altered only in animals exposed to the perfluoroalkyl carboxylic acids. Notably, transcripts involved in the complement pathway and the coagulation cascade were repressed strongly in animals exposed to PFOA, PFNA and PFDA. 8:2 FtOH increased expression of only a few estrogen-responsive genes as well as genes involved in oxidative stress response. Overall, these data suggest that multiple PFAAs can promote liver cancer via an estrogenic mechanism. These findings highlight the need for further research to better assess the risk these environmental chemicals may pose to human health. Supported by NIH grants ES07060, ES03850 and ES00210.
67. A HOLISTIC APPROACH, USING UPLC-MS/MS AND CHEMICALLY INTELLIGENT SOFTWARE TO FULLY PROFILE METABOLIC BIOTRANSFORMATIONS OF NEW CHEMICAL ENTITIES
Amy E. Bartlett1, Jose Castro-Perez2, and John Shockcor2
1Waters Corporation (MS Technologies), Manchester, United Kingdom, 2Waters Corporation, Milford, MA, USA
In recent years the costs and time involved in developing new chemical entities in drug discovery has burgeoned and there is a trend to employ holistic approaches, involving automation and chemically intelligent software to improve efficiency and productivity. In the important arena of metabolite identification, when often faced with information rich LC/MS data, such approaches are particularly valuable. In vitro incubations of Nefazadone and Indinavir were performed in rat liver microsomes to generate phase I metabolites for identification. Samples were analysed using a Synapt HDMS system for exact mass acquisition with MSE scanning capabilities (giving full scan MS and CID fragmentation information in a parallel fashion in a single injection). Using this instrumentation, and an automated software algorithm for metabolite identification, a holistic approach is demonstrated to identify the in vitro rat metabolites of Nefazodone and Indinavir. This tool enables low and high energy exact mass data mining, aided by mass defect filtering tools to provide intelligent and automated software interpretation of metabolic profiles. The automated metabolite profiling process is completed through the use of a fragmentation interpretation software tool, based on systematic bond disconnections, for software driven assignment of metabolite structure from fragmentation patterns.
68. A RAPID METHOD FOR STEREOSPECIFIC ANALYSIS OF AMPHETAMINE-TYPE STIMULANTS USING SIMULTANEOUSLIQUID-LIQUID EXTRACTION AND CHIRAL DERIVATIZATION
Wan Raihana Wan Aasim1, Siew Hua Gan1, and Soo Choon Tan2
1Department of Pharmacology, Universiti Sains Malaysia, Kubang Kerian, Kelantan, Malaysia, 2Veterinary Forensic Laboratory, Universiti Sains Malaysia, Pulau Pinang, Malaysia
Amphetamine-type stimulants (ATS) are chiral amine drugs which are stereospecifically metabolised by cytochrome P450 enzymes. A rapid and sensitive method for the stereospecific analysis of amphetamine (AM), methamphetamine (MA), 3,4-methylenedioxyamphetamine (MDA), 3,4-methylenedioxymethamphetamine (MDMA) and 4-hydroxy-3-methoxymethamphetamine (HMMA) in human urine was developed. The liquid-liquid extraction and chiral derivatization of these commonly abused ATS were combined into a single step, resulting in a simplified and rapid sample preparation procedure.
Two mL urine samples were first alkalinised by a solution of saturated sodium chloride in sodium hydroxide (NaCl/NaOH). Simultaneous extraction and derivatization was performed by adding a solution containing hexane/ethyl acetate (1:1 v/v), derivatization reagent (R)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (R-MTP) in hexane (1:100 v/v) and triethylamine. Samples were mixed thoroughly, centrifuged and the organic layer subjected to a second derivatization with N-methyl-N-tri-methylsilyltrifluoroacetamide (MSTFA) to enable separation and analysis of HMMA which possesses a polar hydroxyl group that is not derivatized by R-MTP. Analysis was by gas chromatography – mass spectrometry using electron impact ionization in selected ion monitoring mode.
To identify experimental parameters influencing the sensitivity of the method, a 2-level, fractional factorial experimental design was used. Seven parameters were studied : (1) use of NaCl/NaOH solution, (2) volume of extraction solvent, (3) percentage of triethylamine used, (4) volume of derivatization reagent, (5) amount of time for sample mixing, (6) sample incubation temperature and (7) sample incubation time. Next, the stereoselectivity of the method was assessed by analysing samples (n = 15) containing racemic analytes and calculating the R/S enantiomer ratios. Finally, recovery and linearity were determined.
The results indicate that the use of a NaOH/NaCl solution decreased the sensitivity of the method whereas increasing the amount of R-MTP had a large positive effect on sensitivity. The other parameters did not have any significant effect on sensitivity. The highest sensitivity was achieved when the NaOH/NaCl solution was omitted and 100 uL of R-MTP was used. All samples analysed had R/S enantiomeric ratios close to one. This indicates that the chiral derivatization occurred completely and accurately. Calibration curves for all the analytes indicated good linearity with R2 > 0.990. Recoveries of the analytes were all above 70%.
The described method is a simple and rapid sample preparation method with good sensitivity, stereospecificity, linearity and recovery. This method shows potential for use in disposition and metabolism studies for chiral ATS drugs where accurate quantitative determination of enantiomers of ATS and their metabolites is required.
69. IDENTIFICATION OF PROCESSES INVOLVED IN BILASTINE BIOAVAILABILITY IN RATS
Ana Gonzalo and MarÍa Luisa Lucero
Department of R&D and Innovation, Faes Farma, S.A., Leioa, Spain, 48940
Introduction. After oral administration a drug can undergo a first pass process which may comprise either intestinal or pulmonary metabolism and/or P-glycoprotein (P-gp) efflux transport. Many drugs can be affected by both mechanisms resulting in enhanced bioavailability (F). Therefore, early identification of mechanisms involved in drug bioavailability is highly recommended. Objectives. To evaluate the contribution of the liver and/or lungs to metabolism and the possible influence of P-gp on bilastine pharmacokinetics. Method. Pharmacokinetics of bilastine was assessed in male Wistar rats after intra-arterial, intravenous and hepatic-portal administration of 10 mg/kg (metabolism study), and after an oral administration of 20 mg/kg (P-gp study). A total of 40 male rats were used. Weight of selected animals ranged from 250 to 400 g. The animals were distributed into three groups for metabolism assessment (n = 10 for each administration route) and blood samples were collected up to 24 hours. Animals corresponding to P-gp study were distributed into a control group (n = 5) and another group to be pretreated with valspodar (n = 5) in order to inhibit P-gp expression. In this case, blood samples were collected up to 6 hours post-dose. For quantitative bilastine determination, plasma samples were analyzed by a validated, sensitive HPLC/fluorescence method, by using protein precipitation for sample clean-up. Pharmacokinetic data analysis was carried out with RSTRIP v.5. and WinNonlin v.1.5 software tools. Liver (Eh) and lung (Ep) extraction ratios were calculated based on intra-arterial, intravenous and hepatic-portal areas under the curve (AUCs) normalized by dose relationships. Results. For bilastine, Eh was 0% and Ep was 0.65%, showing there is not a first pass-metabolism. In the valspodar pretreated group AUC and Cmax showed a significant increase (p<0.05) when compared to control animals, suggesting that bilastine may be a P-gp substrate. Conclusions. Results suggest the intrinsic capacity of the liver and lung to metabolize bilastine is practically nil. Thus, bilastine bioavailability is mainly dependent on P-gp activity. Moreover, no interaction between both studied processes is expected. Acknowledgments. This work was supported in part by the Ministry of Industry, Tourism and Commerce of Spain (PROFIT) and the Department of Industry, Commerce and Tourism of the Basque Government (INNOTEK).
70. INTESTINAL ABSORPTION OF STEMONININE TYPE STEMONA ALKALOIDS
Ge Lin1, Xin Zhou2, Li Zhang3, Zhong Zuo3, and Yang Ye4
1Department of Pharmacology, Chinese University of Hong Kong, Shatin NT, Hong Kong, 2Depatment of Pharmacy, Long Hua Hospital, Shanghai, China, 3School of Pharmacy, Faculty of Medicine, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong, N/A, 4Department of Natural Product Chemistry, Shanghai Institute of Materia Medica, Shanghai, China
Radix Stemonae derived from three species of Stemona genus (S. tuberose; S. sessilifolia; S. jepanica), is one of the most commonly used antitussive traditional Chinese medicines. Various stemona alkaloids have been identified and demonstrated as the main active ingredients of the herb1. However, except for our previous study on a few tuberostemonine type stemona alkaloids2, oral absorption of bioactive stemona alkaloids is largely unknown. The present study reports the intestinal absorption of five stemoninine type alkaloids, namely stemoninine, protostemonine, stemospironine, stemoninoamide and bisdehydrostemoninine, which are the main stemoninine type alkaloids isolated from the herb. Caco-2 monolayer cell model was used for the bidirectional transport study. The integrity of the cell monolayer was determined by the measurement of transepithelial electric resistance (TEER) before and after transport experiments, and only samples collected from the cells with TEER values higher than 600 W·cm2 were subjected to the analysis by HPLC-UV (for bisdehydrostemoninine) and HPLC-ELSD (for all other alkaloids).
The results showed that except for bisdehydrostemoninine, all four stemoninine type alkaloids were well absorbed with relatively high permeability [Papp(AB) 41.98 ± 1.95 — 71.30 ± 4.00×10−6cm/s] and no involvement of efflux transporters with an efflux ratio close to 1 [Papp(BA): 30.59 ± 1.42 — 69.63 ± 0.71×10−6cm/s]. Bisdehydrostemoninine, the only alkaloid containing pyrrole moiety (two double bounds in the original pyrrolidine of stemoninine type alkaloid) among five alkaloids tested, also permeated with no obvious efflux but to a relatively much less extent [Papp(AB): (6.35 ± 0.71)×10−6cm/s, Papp(BA): (6.30 ± 0.87)×10−6cm/s]. The current results demonstrated that all stemoninine type stemona alkaloids tested were absorbed passively. Moreover, the intestinal absorbability varied among alkaloids with different structures. Those with pyrrolidine moiety exhibited good intestinal absorbabilities, while the permeability was significantly reduced when the pyrrolidine moiety was replaced by pyrrole ring in the structure of stemoninine type stemona alkaloids.
REFERENCE
- H. Greger. Structural, relationships, distribution and biological activities of stemona alkaloids. Planta Med. 2006, 72: 99–113.
- P.H.H. Leung, L. Zhang, Z. Zuo and G. Lin. Intestinal absorption of stemona alkaloids in Caco-2 cell model. Planta Med. 2006, 72: 211–216.
71. EFFECTS OF SOLUBILITY, EFFLUX TRANSPORTERS, AND FOOD ON PLASMA EXPOSURE TO TANDUTINIB (MLN0518) AND ITS VARIABILITY IN RATS
Johnny J. Yang1, Mark N. Milton1, Cindy Q. Xia1, Jing-Tao Wu1, Christina Pligavko1, Sonja Komar2, Kym Cardoza2, Frank W. Lee1, and Shimoga Prakash1
1Drug Metabolism and Pharmacokinetics, Millennium Pharmaceuticals, Inc., Cambridge, MA, USA, 02139, 2Non-Clinincal Development Science, Millennium Pharmaceuticals, Inc., Cambridge, MA, USA, 02139
Tandutinib (MLN0518) is a novel small-molecule inhibitor of FLT3, platelet-derived growth factor receptor (PDGFR), and c-kit tyrosine kinase receptors. It is being developed for the treatment of acute myeloid leukemia (AML). Tandutinib showed oral bioavailability of approximately 20% and exposure variation range from 30% to 120% (Coefficient of Variation, CV) in rats. Investigational studies in rats were conducted to explore the cause of the low plasma exposure.
Tandutinib is (a) a weak base with a pH-dependent solubility profile, (b) metabolically stable in rat microsomes, and (c) a substrate of efflux transporters, P-glycoprotein (P-gp) and breast cancer resistance protein (Bcrp). Tandutinib showed high plasma clearance with low oral bioavailability in rats. Renal clearance of tandutinib was low. In rats with a 10 mg/kg oral dose, a 5 mg/mL dosing formulation on average increased plasma exposure (area under the plasma curve, AUC) by 2-fold and reduced the Coefficient of Variation (CV) value to 40.3% compared to that of 10 mg/mL formulation (CV of 66.7%). Oral co-administration of GF120918 (an inhibitor of both P-gp and Bcrp) with tandutinib resulted in an 8-fold increase in AUC and a 3-fold reduction of CV value. P-gp inhibitors TPGS and ketoconazole increased AUC 1.5–2-fold. In fed rats, exposure to tandutinib was markedly decreased with the Cmax and AUC being approximately 8-fold and 6-fold lower, respectively, compared to fasted animals. CV values for both Cmax and AUC were doubled as well when compared to fasted animals. In summary, solubility, efflux transporters, and food all appeared to contribute to the low plasma exposure and variation of tandutinib in rats. Therefore, selection of a formulation or salt form with improved solubility, addition of efflux transporter inhibitors in the formulation, and fasting condition would be helpful in improving tandutinib oral bioavailability and exposure variability.
72. PREDICTION OF THE IN VIVO BEHAVIOUR OF MODIFIED RELEASE FORMULATIONS OF METOPROLOL FROM IN VITRO DISSOLUTION PROFILES USING THE ADAM MODEL (SIMCYP V8)
Sebastian Polak1, Masoud Jamei1, David B. Turner1, Jiansong Yang1, Sibylle Neuhoff1, Geoffrey T. Tucker2, and Amin Rostami-Hodjegan2
1Scientific Development, Simcyp Ltd, Sheffield, United Kingdom, S2 4SU, 2Academic Unit of Clinical Pharmacology, University of Sheffield and Simcyp Limited, Sheffield, United Kingdom, S2 4SU
The Advanced Dissolution, Absorption and Metabolism (ADAM) model is able to predict the rate and extent of intestinal drug absorption and metabolism and their associated inter-individual variability [1]. The capabilities of the model have now been extended in Simcyp® V8 (http://www.simcyp.com) to handle modified release formulations, representing up to four in vitro dissolution profiles per compound (viz. gastric and intestinal profiles in fasted and fed states).
To investigate model performance three formulations of metoprolol were investigated. Physiochemical and in vitro parameters were collated from the literature and entered into the ADAM model. To mimic a clinical study [2], ten virtual trials with seven fasted and CYP2D6 extensive metabolisers (male:female - 4:3; age 33–47) were simulated. The virtual subjects were each given a single 100 mg oral dose of metoprolol in three different formulations (fast, moderate, slow release). The predicted PK parameters and profiles were compared with reported in vivo values ( and ).
Table 1 Predicted vs. observed PK parameters
Table 2 Predicted vs. observed PK parameter ratios for different formulations
The ADAM model successfully predicted the PK parameters for all three modified release formulations indicating its utility for planning and conducting bioavailability and bioequivalence studies. Since the model also provides measures of inter-individual variability it can be used to optimise the design of clinical studies with respect to statistical power.
REFERENCES
- Jamei M, et al. (2007) Novel Physiologically-Based Mechanistic Model for Predicting Oral Drug Absorption: The Advanced Dissolution, Absorption, and Metabolism (ADAM) Model. EUFEPS&COST Conference on Bioavailability and Bioequivalence: Focus on Physiological Factors and Variability. October 1–2, Athens, Greece.
- Sirisuth, N, Eddington N.D. (2002). The influence of first pass metabolism on the development and validation of an IVIVC for metoprolol extended release tablets. Eur. J. Pharm&Biopharm. 53(3): 301–309.
73. ENHANCEMENT EFFECT OF SODIUM CAPARATE ON THE INTESTINAL ABSORPTION AND BIOAVAILABILITY OF SALVIANOLIC ACID B
Limin Zhou, Moses S. S. Chow, and Zhong Zuo
School of Pharmacy, Faculty of Medicine, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong
Our previous studies consistently demonstrated that salvianolic acid B, one of the major hydrophilic active components in Saliva miltiorrhiza Bge, exhibit very limited oral permeability and bioavailabilities, which may be due to its hydrophilic properties and poor intestinal permeability. The current study aims to enhance intestinal absorption and bioavailability of salvianolic acid B by sodium caparate, one of the well known absorption enhancer for hydrophilic compounds. The potential cytotoxicities of sodium caparate towards Caco-2 cells were first evaluated by MTT assay. The intestinal absorptions of salvianolic acid B were investigated using in vitro Caco-2 monolayer model and rat single pass in-situ intestinal perfusion model with and without the treatment of sodium caparate. For the in vivo evaluations, salvianolic acid B with and without the addition of sodium caprate were administered orally to Sprague Dawley rats (n = 5 for each group) to obtain their pharmacokinetics profiles. The blood samples were collected at 10, 20, 30, 40, 60, 90, 120, 180, 240 and 360 min post dosing. The plasma concentrations of salvianolic acid B were determined by a developed and validated LC-MS-MS method. Win-Nonlin was used to calculate the pharmacokinetics parameters and student t-test was used to conduct the statistical analysis of the data. It was found that sodium caprate up to 0.25% (w/v) was safe to the Caco-2 cells. In the in vitro Caco-2 monolayer model, salvianolic acid B (100 μM) could only be detected in the basolateral side after the treatment with sodium caparate with a Papp value of (41.4±4.4) x 10−6 cm/s in the presence of 0.25% (w/v) of sodium caparate. Although no significant difference was found in the Papp value of salvianolic acid B at both 200 and 500 μM in the rat in-situ intestinal perfusion model treated with sodium caparate, the accumulated amount of salvianolic acid B in the mesenteric blood did demonstrate a trend of increase with the addition of sodium caprate (0.5% w/v). When 50 mg/kg salvianolic acid B was administered with 100 mg/kg sodium caprate, the area under the curve of its plasma versus time profile was significantly increased from 137.96 to 194.98 μg/ml·min with no significant changes on the Cmax and Tmax. In conclusion, sodium caprate was proved to be able to enhance intestinal permeability and in vivo bioavailability of salvianolic acid B.
74. APPLICATION OF DIFFERENT IN VITRO MODELS TO THE PREDICTION OF HEPATIC CLEARANCE IN DRUG DISCOVERY
Christopher R. Jones1, Philip Arundel1, Rachel Moran1, Alison Hunter2, and Romuald Laine1
1Discovery DMPK, AstraZeneca, Macclesfield, United Kingdom, 2Physical Chemistry, AstraZeneca, Macclesfield, United Kingdom
In drug discovery, rat hepatocyte intrinsic clearance (Clint) data is scaled to an unbound in vivo Clint (Clintub, in vivo) (Equation 1). This is used to predict the observed in vivo hepatic metabolic clearance, calculated through rearrangement of the well stirred model (Equation 2); where SF = scaling factor for hepatocytes, LBF = liver blood flow, CL = total in vivo clearance and fub = the unbound fraction in blood.(1)
(2)
The accuracy of the Clintub value and hence the quality of the in vitro to in vivo correlation (IVIVC), can be improved by accounting for non-specific binding within the hepatocyte incubation (fuinc) (Equation 3).(3)
(4)
(5)
Fuinc can be measured experimentally or predicted from intrinsic physical-chemical properties such as lipophilicity (logP) and ionisation state (Equation 4, Austin et al., 2005). These properties also drive the extent of binding within plasma. As such, we have assumed that a function of the unbound fraction in plasma (fup), in this case the square root, can be used to estimate the magnitude of fuinc (Equation 5), and have investigated whether this empirical approach can improve the predictability of the in vitro experiment and strength of the IVIVC. For this analysis, a set of compounds, covering a broad spectrum of physical-chemical properties (calculated logP = 1.0–5.8; 64 neutral and 86 basic compounds), were taken from a number of typical discovery projects. For each compound the blood to plasma concentration ratio was assumed to equal unity. The predicted values for Clintub,in vivo were generated from a measured Clint, an appropriate hepatocyte SF and either a correction for fuinc based on Equation 4 or 5; or by assuming fuinc = 1; or by excluding all binding parameters. These predictions were correlated with the observed Clintub,in vivo. With all binding parameters excluded the IVIVC (R2 = 0.20) and predictability (15% within 2 fold of the observed Clintub, in vivo) were weak. Improvements were observed when fuinc was assumed to equal one giving R2 = 0.54 (17.3% within 2 fold of the observed Clintub, in vivo), or an estimate of fuinc was included by applying Austin's approach to calculated logP values, then R2 = 0.46 (29% within 2 fold of the observed Clintub, in vivo). For this set of compounds the √fup approach gave the strongest IVIVC (R2 = 0.71) and most accurate prediction (63% within 2 fold of the observed Clintub, in vivo). In conclusion, the most robust model for prediction of in vivo metabolic clearance included all binding parameters. By utilising the √fup as an estimate of fuinc it is possible to improve the IVIVC. This approach avoids the necessity of a measured logP or fuinc value for scaling and may also be applied across species for scaling to man.
REFERENCE
- Austin, R.P., Bartin, P., Mohmed, S., and Riley, R.J., (2005). The binding of drug to hepatocytes and its relationship to physicochemical properties. Drug Metabolism and Disposition, 33:419–425.
75. OPTIMAL SCREENING ASSAY SETUP FOR ASSESSMENT OF METABOLIC CLEARANCE IN EARLY DRUG DISCOVERY
Erik Sjögren1, Joakim Nyberg2, Mats Magnusson2, Andrew Hooker2, and Hans Lennernäs1
1Department of Pharmacy, Uppsala University, Uppsala, Sweden, 75123, 2Division of Pharmacokinetics and Drug therapy, Uppsala University, Uppsala, Sweden
The assessment of metabolic stability, i.e. metabolic clearance, early in drug discovery is an important factor when determining if a new molecular entity will be suitable as a drug. This is commonly done with the “in-vitro t½ method” [1] where a mono exponential decay is fitted to a single depletion curve with an initial concentration regarded to be well under the reactions overall Km. The aim of this study was to find a more optimal method design for the metabolic stability screening assay using the reactions for mixed enzyme systems included in the SIMCYP [2] database as parameter, Vmax and Km, distribution. Optimization was performed by finding the sample times and concentrations giving the lowest uncertainty (SE) of the model parameter estimates. The incubation time was limited to a maximum of 40 min, also 0.1 μM as a lower limit of quantification and a proportional noise of 7.5 % were assumed. The optimal design was performed using a population nonlinear mixed effect modeling approach [3] and was optimized with the software PopED 2.0 [4] using ED-optimal design with a discrete parameter distribution. NONMEM [5] was used for estimating the model parameters with the concentrations measured at the computed optimal doses and times.
REFERENCE
- Obach, R.S., Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos, 1999. 27(11): p. 1350–9.
- Proctor, N.J., G.T. Tucker, and A. Rostami-Hodjegan, Predicting drug clearance from recombinantly expressed CYPs: intersystem extrapolation factors. Xenobiotica, 2004. 34(2): p. 151–78.
- Retout, S., S. Duffull, and F. Mentre, Development and implementation of the population Fisher information matrix for the evaluation of population pharmacokinetic designs. Comput Methods Programs Biomed, 2001. 65(2): p. 141–51.
- Foracchia, M., et al., POPED, a software for optimal experiment design in population kinetics. Comput Methods Programs Biomed, 2004. 74(1): p. 29–46.
- Beal, S.L. and L.B. Sheiner, NONMEM User's Guide.. Technical report. University ofCalifornia, San Francisco., 1998.
76. DO NON-HUMAN PRIMATE STUDIES ADD VALUE IN THE PREDICTION OF HUMAN CLEARANCE OF SMALL MOLECULAR THERAPEUTICS?
Kevin Beaumont1, Kathryn Chapman2, Peter Eddershaw3, Ian Gardner1, and Malcolm Rowland4
1Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development-Sandwich, Sandwich, United Kingdom, 2National Centre for the Replacement, Refinement and Reduction of Animals in Research (NC3Rs), London, United Kingdom, 3UCB-Group, Cambridge, United Kingdom, 4School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester, United Kingdom, M13 9PL
The selection of compounds with promising pharmacokinetic properties is a key consideration for the reduction of ADME-related attrition. One of the most important pharmacokinetic parameters to predict is human clearance, as this plays a part in determining both oral bioavailability and half-life.
Human clearance prediction methodologies fall into two broad categories: extrapolation of in vitro information and scaling of clearance from pre-clinical species (including non-human primates). The choice of prediction methodology relies heavily on the predominant clearance pathway involved for each compound.
Many published studies have examined the retrospective prediction of known clinical agents using a range of prediction methodologies. One of the most complete sets of compounds with published intravenous clearance data is that of Jolivette and Ward1. The present investigation has interrogated this 103 compound set to investigate the requirement for non-human primate studies (NHP) in the prediction of human clearance. As a general method, single species scaling (SSS) of rat, dog and NHP clearance produced similar success rates (within +/- 2 –fold) for human clearance prediction, although NHP tended to slightly higher success rates over the whole dataset. When P450 cleared compounds were considered, human liver microsomal (HLM) scaling tended to be more predictive than SSS, with NHP SSS tending to lower success than rat SSS. For renally cleared compounds, rat and NHP SSS tended to similar success rates. Coincidence of predictions from 2 different methods for single compounds tended to drive to greater success rates. From this analysis, using a consideration of clearance pathway, it is possible to propose a human clearance prediction paradigm that minimises the need for NHP studies for successful outcomes.
REFERENCE
- J. Pharm Sci (2005) 94(7) 1467–1483
77. COMPARISON OF STRATEGIES FOR ASSESSING METABOLIC STABILITY IN VITRO
Philip J. Butler, Caroline Weir, Michael Griffin, and Clive Dilworth
Cyprotex, Macclesfield, United Kingdom
Metabolic stability assays provide important information on the metabolic liabilities of new drug candidates. Compounds are typically rank ordered according to their intrinsic clearance (CLint). Highly cleared drugs may be unfavourable as they are likely to be rapidly cleared in vivo resulting in a short period of action and poor in vivo exposure. A number of in vitro systems are available to assess metabolic stability of a compound. Liver microsomes are perhaps the most widely used in vitro system for assessing CLint, primarily due to convenience and low cost. However, recent publications suggest that human liver microsomes under-predict metabolic clearance (Ito and Houston, 2005; Brown et al., 2007). Liver S9 fraction is becoming a more frequently used system for determining metabolic stability as it comprises of both the microsomal and cytosolic fractions of the cell, incorporating the majority of phase I and phase II enzymes and thus may provide a more complete metabolic profile. Alternatively, by supplementing with the co-factor UDPGA (uridine diphosphoglucuronic acid) and alamethicin, liver microsomes can also be used to assess phase II glucuronidation. The aim of this study was to demonstrate the use of microsomes, S9, and supplemented microsomes as enzymatic systems for predicting in vivo CLint. The in vitro CLint of test compounds was determined based on disappearance of compound as analysed by LC-MS/MS. The data presented in this study validates the use of a 384-well based metabolic stability assay to assess the contribution of both phase I and phase II metabolism in determining CLint. This allows for direct evaluation of microsomes, S9, and supplemented microsomes as suitable enzymatic systems for determining CLint. This study also examines the in vitro to in vivo correlation of hepatic metabolic clearance using microsomes, S9, and supplemented microsomes to determine the impact of phase II metabolism on in vivo CLint predictions.
REFERENCE
- Brown, H.S., Griffin, M., Houston, J.B. (2007). Evaluation of cryopreserved human hepatocytes as an alternative in vitro system to microsomes for the prediction of metabolic clearance. Drug Metab Dispos. 35(2):293–301.
- Ito, K. and Houston, J.B. (2005). Prediction of human drug clearance from in vitro and preclinical data using physiologically based and empirical approaches. Pharm Res. 22(1):103–112.
78. INCORPORATING INTER-INDIVIDUAL VARIABILITY INTO THE PREDICTION OF PHENYTOIN CLEARANCE FROM ALBUMIN-ADJUSTED IN VITRO DATA
Thomas M. Polasek1, Andrew Rowland1, David J. Elliot1, Kathleen M. Knights1, Peter I. Mackenzie1, Sebastian Polak2, Amin Rostami-Hodjegan3, and John O. Miners1
1Dept of Clinical Pharmacol, Flinders Univ, Adelaide, Australia, AU-5042, 2Simcyp Limited, Sheffield, United Kingdom, S24SU, 3University of Sheffield and Simcyp Limited, Sheffield, United Kingdom, S10 2JF
Mechanistic modelling and simulation can be employed to predict the pharmacokinetics (PK) of drugs on the basis of in vitro data (in vitro-in vivo extrapolation or IVIVE). In this study, the clearance of phenytoin (PHT) was predicted from kinetic studies with CYP2C9, and the impact of inter-individual variability assessed using the Simcyp® population-based ADME simulator (V7.1). The Michaelis-Menten constants (Km and Vmax) of PHT hydroxylation were determined in the absence and presence of fatty acid free human serum albumin (HSA-FAF) using human liver microsomes (HLM) and rCYP2C9 (Escherichia coli-expressed) as the enzyme sources. Virtual trials (n = 10) were run for a single 300 mg PHT oral dose, with population demographics matched to the participants of clinical studies (European Caucasians, aged 18–44 years, 1:0.3 males to females). Addition of HSA-FAF to incubations reduced the apparent Km of PHT hydroxylation by HLM and rCYP2C9 by approximately 75%, to 4.7 μM and 4.1 μM, respectively, with a minor effect on Vmax. Using rCYP2C9 data generated in the presence of HSA-FAF, mean simulated values of AUC0–96 hr (186 mg/L.hr), time-averaged CLint.liver.0–96 hr (10.8 L.hr−1), CLpo (1.9 L.hr−1), Cmax (5.0 mg.L−1), Tmax (5.7 hr) and t½β (17.5 hr), were consistent with those reported in vivo. Distribution plots of PK parameters were similar for albumin-adjusted IVIVE using either enzyme source, and identified ‘extreme’ individuals with values outside the reported ranges. PHT clearance was under-predicted from experiments performed in the absence of HSA-FAF. These results indicate that accurate IVIVE for single PHT dosing is achievable, and provide further support for incorporating inter-individual variability into the prediction of PK from in vitro data.
79. PREDICTION OF GLUCURONIDATION (UGT) CLEARANCE USING ALAMETHICIN ACTIVATED HUMAN LIVER MICROSOMES
Peter J. Kilford1, Rowan Stringer2, Bindi Sohal2, J. Brian Houston1, and Aleksandra Galetin1
1School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester, United Kingdom, M13 9PT, 2Novartis Horsham Research Centre, Horsham, United Kingdom, RH12 5AB
An increasing number of compounds are metabolised via a direct phase II pathway (e.g., glucuronidation) in parallel to elimination by phase I enzymes. Therefore, there is a need to evaluate the use of microsomes for the prediction of clearance for such compounds. In this study the clearance for nine compounds with differential contribution of CYP and UGT pathways was determined. In order to investigate the importance of metabolism via glucuronidation the intrinsic clearance (CLint) was determined in the presence of either CYP (NADP) or UGT (UDPGA) cofactors, both individually and combined. For incubations with UDPGA human liver microsomes were activated with alamethicin at a concentration of 50 μg/mg protein. In the presence of UDPGA the CLint,UGT ranged from 8.0 to 479 μL/min/mg protein for ketoprofen and raloxifene, respectively. In the presence of NADP the CLint,CYP ranged from 3.9 to 3056 μL/min/mg protein for naloxone and propofol, respectively. The CLint with combined cofactors present was approximately additive when compared to the sum of the individual CLint,CYP and CLint,UGT. In the presence of both cofactors the CLint ranged from 13.0 to 3355 μL/min/mg protein for naloxone and propofol, respectively. For each of the compounds investigated the fraction metabolised by UGT (fmUGT) and CYP (fmCYP) enzymes was calculated from the individual cofactor data. The in vitro clearance data indicated a varying contribution of glucuronidation to the clearance of the compounds studied with the fmUGT ranging from 0.09 to 0.83 for propofol and gemfibrozil, respectively. The CLint values obtained from incubations were corrected for nonspecific microsomal binding and scaled using a human microsomal scaling factor of 40 mg/g protein and an average liver weight of 21 g liver/kg. A good correlation between the predicted and observed glucuronidation CLint was obtained using data with only UDPGA present. In addition, predicted CLint obtained from data in the presence of both cofactors correlated well with the observed total CLint. The current study indicates the applicability of both individual and combined cofactor conditions in the assessment of clearance for compounds with a differential contribution of CYP and UGT enzymes to their elimination.
80. INVESTIGATION OF THE INTESTINAL GLUCURONIDATION ACTIVITIES OF 7,2′, 7,3′ AND 7,4′-DIHYDROXYFLAVONES
Yin Cheong Wong1, Li Zhang1, Ge Lin2, and Zhong Zuo1
1School of Pharmacy, Faculty of Medicine, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong, N/A, 2Department of Pharmacology, The Chinese University of Hong Kong, Shatin NT, Hong Kong
It was found that one of the major factors causing the low oral bioavailabilities of flavonoids was their extensive intestinal first-pass metabolisms including glucuronidation. A series of our previous structure-glucuronidation activity relationship studies from both selected mono-hydroxyflavones and di-hydroxyflavones demonstrated that the positions of hydroxyl group did affect the intestinal glucuronidation activity of individual hydroxyflavones. The present study aims to evaluate further the intestinal glucuronidation metabolism of di-hydroxyflavones, namely 7,2′, 7,3′ and 7,4′-dihydroxyflaovnes so as to compare the effect of -OH substitution position from different ring of flavone on their glucuronidation activity. Glucuronidation activities of three selected dihydroxyflavones, namely 7,2′, 7,3′ and 7,4′-dihydroxyflaovnes were investigated by incubating each dihydroxyflavone at various concentrations with rat intestinal microsome or human jejunum microsome under the optimized conditions. LC/MS/MS methods were developed to identify both the parent flavones and their metabolites and the amount of glucuronides formed was quantified by HPLC/UV. The metabolism kinetic profiles and parameters such as Vmax, Km and intrinsic clearance were obtained and compared with that obtained from our previous studies on 3,7-, 5,7-, 6,7-, 7,8- dihydroxyflaovnes and 7-hydroxyflavone. The results showed that two metabolites were identified for all studied di-hydroxyflavones studied. 7,4′-Dihydroxyflavone demonstrated similar glucuronidation activities as that of the 3,7-dihydroxyflavone and 7-hydroxyflavone with intrinsic clearances less than 100 μl/min/mg protein. 7,2′-Dihydroxyflavone demonstrated similar glucuronidation activity as that of 5,7-dihydroxyflavone, 6,7-dihydroxyflavone, and 7,8-dihydroxyflavone with intrinsic clearances between 150∼230 μl/min/mg protein. 7, 3′ Di-hydroxyflavone demonstrated the greatest intrinsic clearance with 1737.2 μl/min/mg protein, which is at least 10 times greater than the rest of the dihydroxyflavones studied. Similar as we found before, human jejunum microsome did demonstrate higher glucuronidation activities than rat intestinal microsome for 7,2′, 7,3′ and 7,4′-dihydroxyflaovnes as well. In conclusion, one additional hydroxyl group on B-ring could enhance the glucuronidation activity of flavones, with 7,3′-dihydroxyflavone as the most potent substrate for glucuronidation.
ACKNOWLEDGMENT
CUHK Direct (Grants 2041074 & 2041155).
82. INTERACTION OF ALCOHOL WITH ACETAMINOPHEN
Alla K. Voronina Jr.1 and Valentina N. Kovalenko2
1General Toxicology, SI “Institute of Pharmacology and Toxicology, Academy of Medical Sciences of Ukraine”, Kyiv, Ukraine, 03057, 2General Toxicology, IS, Kyiv, Ukraine, 03057
Background. acetaminophen is widely used as an analgesic and antipyretic agent, and it is assumed to be safe when taken in recommended doses. But in isolated cases in which alcohol has presumably played a synergistic role with acetaminophen in production liver toxicity after ingested drug for therapeutic reasons. It is known that P-4502E1 metabolizes acetaminophen, and that inducers such as ethanol can induce it. The purpose of this study was to clarity the effects of ethanol ingestion and chronic ethanol consumption on the hepatotoxicity of acetaminophen. Methods. the influence of alcohol on the hepatotoxicity of acetaminophen was studied in male albino rats. Rats were treated acetaminophen with ethanol and under alcoholism by intragastric administration. The liver tissue was collected, washed with cold saline, blotted dry and was homogenized. The homogenates were centrifuged, the pellets were resuspended and stored at –800C until use. Results. preliminary studies in mice support the concept that acetaminophen hepatotoxicity may be enhanced by ethanol ingestion. A significant reduction in the LD50 was seen in the alcohol-treated mice. In treated ethanol rats (3.2 mg/kg b. w.), the activities of aniline-N-hydroxylase (a marker of cytochrome Ð-450 2E1 activity) in liver microsomes significant increased after acetaminophen (dose - 400 mg/kg b. w.) administration compared with control animals. Among these glutathione and protein SH-groups contents as well as liver glutathione reductase and catalase activity were decreased. The rate of induced malonodialdehyde (MDA) formation, hydroperoxides and super oxide anion contents were increased in comparison to the control. Alanine aminotransferase (AlAT) activity elevated in blood serum of 1,7 fold the upper limit of normal. After chronic ethanol consumption feeding for 6 months, aniline-N-hydroxylase, glutathione reductase, AlAT activities and MDA formation was increased significantly, whereas content of glutathione were decreased compared with control. After parallel administration of acetaminophen (dose – 500 mg/kg) the activities of aniline-N-hydroxylase followed up 65 % and the serum ALAT activities increased of 5,6 fold. In acetaminophen-treated animals, ethanol-fed rats showed a significant decreased hepatic glutathione levels. Conclusion. our results suggest that ethanol may be an additional risk factor for developing acetaminophen hepatotoxicity. The observation that acetaminophen and alcohol intensify the induction of CYP2E1 in a synergistic manner may help to understand and to prevent an additional risk factor for developing acetaminophen hepatotoxicity.
83. EFFECTS OF ANDROGRAPHIS PANICULATA EXTRACT AND ANDROGRAPHOLIDE ON HEPATIC CYTOCHROME P450 EXPRESSION AFTER IN VIVO ADMINISTRATION TO RATS AND IN VITRO IN RAT AND HUMAN LIVER MICROSOMES AND HEPATOCYTE CULTURES
D. Pekthong
Laboratoire de Toxicologie Cellulaire, EA3921, UFR Medecine Pharmacie, Besancon, France, 25030
The expression of cytochrome P450 (CYP) is regulated by both endogenous factors and foreign compounds including drugs and natural compounds such as herbs. When herbs are co-administrated with a given drug in popular medicine it can lead to drug-herb interaction that can be clinically significant. The ability of Andrographis paniculata Wall.ex Nees extract (APE) and Andrographolide (AND), the most medicinally active phytochemical in the extract, to modulate hepatic CYP expression was examined in different models, i.e. in vivo in rats, in vitro in rat and human liver microsomes and in vitro in rat and human hepatocyte cultures. In vivo, APE and AND exhibited minimal capacity to inhibit CYP enzymes, except CYP2C11. A 30% decrease of CYP2C11-dependent activity was obtained at 0.5 and 2.5 g APE/kg rat /day, as compared to control rat group. The dose levels used for APE correspond respectively to 5 and 25 mg AND/kg/day. When administrated at these doses, the pure compound AND also decreased CYP2C11-dependent activity by 50%, as compared to the control group. In vitro, in microsomes from both rat and human livers, CYP2C-dependent activity was found to be inhibited by APE, with respective apparent Ki values of 8.21 and 7.51 μM and a mixed type mode of inhibition. Primary cultures of rat and human hepatocytes were then used to study the mechanism of action of APE and AND on CYP expression and activity. For this purpose, cultures were treated with 50μM APE or AND. In rat hepatocyte cultures, APE and AND treatment resulted in significant decrease in CYP1A2, CYP2C11 and CYP3A1 activities, but had no effect on CYP2E1. In human hepatocytes, APE significantly decreased CYP2C9 and CYP3A4 at the mRNA, apoprotein and activity levels. By contrast, AND exhibited minimal capacity to inhibit any CYP enzyme. This may be due to the presence of several other components present along with AND in the extract of Andrographis paniculata. In conclusion, it cannot be excluded from the present study that APE could cause herb-drug interactions in humans through CYP2C9 and CYP3A4 inhibition.
84. VALIDATION OF CYTOCHROME P450 TIME DEPENDENT INHIBITION ASSAYS FEATURING A TWO TIME POINT IC50 SHIFT APPROACH TO FACILITATE KINACT ASSAY DESIGN
Elke S. Perloff, Andrew K. Mason, Shangara S. Dehal, Andrew P. Blanchard, Ling Morgan, Thuy Ho, Andre Dandeneau, Ronell M. Crocker, Catherine M. Chandler, Nathalie Boily, Charles L. Crespi, and David M. Stresser
BD GentestSM Contract Research Services, BD Discovery Labware, Woburn, MA, USA, 01801
Objective. Quantitative endpoints for the in vitro assessment of time-dependent inhibition (TDI) of cytochome P450 often include KI and kinact determinations. A significant challenge in kinact assay design is selecting preincubation time points to adequately define the slope of the inactivation rate curve. A two preincubation time point IC50 shift assay was designed to facilitate the selection of kinact assay parameters and applied to the validation of TDI assays for six P450 isoforms. Methods. P450 inhibition assays included IC50, Ki, IC50 shift, KI, and kinact endpoints and were validated in pooled HLM using LC/MS/MS with stable-labeled isotope internal standards for the following enzyme/substrate/direct inhibitor/time-dependent inhibitor combinations: CYP1A2/ phenacetin/ alpha-naphthoflavone/ furafylline; CYP2B6/ bupropion/ ketoconazole/ ticlopidine; CYP2C9/ diclofenac/ sulfaphenazole/ tienilic acid; CYP2C19/ S-mephenytoin/ S-benzylnirvanol/ S-fluoxetine; CYP2D6/ dextromethorphan/ quinidine/ paroxetine; CYP3A4/ testosterone, midazolam/ ketoconazole/ azamulin, verapamil, diltiazem. For IC50 shift experiments, increasing concentrations of inhibitors were incubated with HLM with and without NADPH for 10 or 30 min prior to 10x dilution into a secondary incubation containing probe substrate at a concentration near the Km. For KI/kinact experiments, increasing concentrations of inhibitors were incubated with HLM with NADPH for multiple time points prior to 10x dilution into a secondary incubation containing probe substrate in excess of Km (∼5x). IC50 values were calculated by linear interpolation. IC50 “shift” was calculated as the ratio of the IC50 values in absence and presence of NADPH. KI and kinact values were determined by non-linear regression (SigmaPlot v8.0 with EK module v1.1). Results. IC50, Ki, IC50 shift, KI, and kinact values obtained in this validation compare well with literature data. The two-time point IC50 shift assay design proved to be a robust preliminary experiment to facilitate selection of preincubation times for the KI/kinact assay. Rapid acting inhibitors (e.g. azamulin) showed no change in IC50 shift between the 10 min and 30 min preincubation times suggesting a short maximum incubation time (<10 min) with close spacing of time points for the follow-up KI/kinact study. Slow acting inhibitors (e.g. verapamil) showed a substantial change in IC50 shift between the 10 min and 30 min preincubation times suggesting a longer maximum incubation time (30 min) with wider spacing of time points for the follow-up KI/kinact study. Conclusions. A suite of in vitro assays to characterize time-dependent inhibition of P450 isoforms by new NCEs has been successfully validated using a two preincubation time point IC50 shift experiment to facilitate the selection of appropriate KI/kinact assay parameters eliminating resource intensive trial-and-error steps from kinact assay design.
85. DIFFERENTIAL TIME- AND NADPH-DEPENDENT INHIBITION OF CYP2C19 BY ENANTIOMERS OF FLUOXETINE IN VITRO
David M. Stresser, Elke S. Perloff, Andrew Mason, Thuy Ho, Charles Crespi, Andre Dandeneau, Ling Morgan, and Shangara Dehal
BD GentestSM Contract Research Services, BD Discovery Labware, Woburn, MA, USA, 01801
Fluoxetine [±-N-methyl-3-phenyl-3-[(α, α, α -trifluoro-p-tolyl)oxy]propylamine)] is a widely used selective serotonin reuptake inhibitor useful in treating depression and other serotonin-dependent disease conditions. Racemic fluoxetine and its (R)- and (S)- enantiomers are potent reversible inhibitors of CYP2D6 and the racemate has been shown to be a mechanism-based inhibitor of CYP3A4. Racemic fluoxetine also demonstrates time- and concentration-dependent inhibition of CYP2C19 catalytic activity in vitro. In the course of enhancing our laboratory's time-dependent inhibition assay for CYP2C19, we tested model compounds that might serve as a reference (positive control) inhibitors to assess time-dependent inhibition of CYP2C19. In this study, we compared fluoxetine, its (R)- and (S)-enantiomers, ticlopidine and the reversible CYP2C19 inhibitor S-benzylnirvanol. In a reversible inhibition protocol (30 min preincubation with liver microsomes without NADPH), we found (R)-, (S)- and racemic fluoxetine to be a moderate inhibitors with IC50 values of 17, 67 and 27 μM. However, when the preincubation was supplemented with NADPH, IC50-values shifted to 4.1, 3.4 and 1.8 μM. Thus, (S)-fluoxetine demonstrated a 20-fold shift in the IC50 value. Follow up KI and kinact determinations were found to be consistent with these data, showing a good negative correlation between the shifted IC50 and the kinact/KI ratio [e.g. KI = 46 μM and kinact = 0.064 min-1 for (S)-fluoxetine); KI = 5.3 μM, 0.018 min-1 for (R)-fluoxetine]. By contrast, ticlopidine showed an approximate 2-fold shift in IC50-value and S-benzylnirvanol exhibited no shift, as expected. Although the (S)-enantiomer exhibits a much lower affinity for CYP2C19 inactivation relative to the (R)-enantiomer, it exhibits a more rapid rate of inactivation. From a practical viewpoint, (S)-fluoxetine appears to be a highly suitable reference inhibitor for studies examining time-dependent inhibition of liver microsomal CYP2C19. These data may also have implications for explaining inhibition of S-mephenytoin metabolism in healthy volunteers following administration of fluoxetine.
86. QUANTUM MECHANICAL CONSIDERATIONS ON THE MECHANISM OF THE P450 CONVERSION OF 11-DEOXYCORTICOSTERONE TO ALDOSTERONE
Luc Roumen1, Bram van Hoof1, Koen Pieterse1, Peter A. J. Hilbers1, Ralf Plate2, Erica M. G. Custers1, Marcel de Gooyer2, Jos F. M. Smits3, Ilona Beugels3, Judith Emmen3, Harry C. J. Ottenheijm3, Dirk Leysen3, and J. J. R. Hermans3
1BioModeling and bioInformatics, Eindhoven University of Technology, Eindhoven, Netherlands, 2Department Medicinal Chemistry, NV Organon, Oss, Netherlands, 5340 BH, 3Department of Pharmacology and Toxicology, University Maastricht, Maastricht, Netherlands
The final steps of the biosynthesis of the steroid aldosterone involve C11b- and C18-hydroxylations by cytochrome P450 11B1 and 11B2 (figure), and C18-aldehyde production by 11B2 [1]. Generally regioselective substrate and intermediate conversions are driven by enzyme-ligand sterical and energetic factors. To evaluate in how far energetic considerations play a role in determining the sequential conversions of DOC, we have analysed multiple steroid conformations using Quantum Mechanics calculations. Therefore, we have conducted: (1) conformational analyses for the prediction of the optimal steroid configurations and (2) Fukui index calculations to determine the inherent reactivity of the individual carbon atoms for prediction of carbon oxidation preference. Molecule geometries were optimised using the QM package Gaussian03. Mulliken population analysis was used to determine the electron density distribution. Accurate computation of the ionic state required for the Fukui index of a nucleophilic attack, was conducted using the B3LYP functional with the 6–31+g basis set. For the lowest energy conformations of all four steroids, the Fukui index indicated that C19 and in some cases C12 were more reactive than C11 and C18. We conclude that the 11B1 and 11B2 active site conformations must prohibit C12 and C19 interactions with the active site heme. For DOC, the Fukui index of C11 (0.088) is higher than that of C18 (0.043), indicating that B is more likely to be produced than 18OH-DOC. The resulting conformation of B portrays an unfavourable C18 Fukui index (-0.016), but the resulting 18OH-DOC conformation possesses a favourable C11 Fukui index (0.020). This can indicate two things: (1) either B is the most abundant product from DOC hydroxylation whereas 18OH-B and aldosterone are formed via the less abundant 18OH-DOC or (2) active site-ligand interactions result in a conformational change in the hydroxyacetyl group of B to allow further conversion to 18OH-B and aldosterone. In a next step the complexity of the steroids was reduced by neglecting the A and B rings. Conformational space analyses of the simplified steroids and their calculated Fukui indices suggest that the hydroxyacetyl group of 18OH-B requires an anti-periplanar conformation for conversion into aldosterone as well as an internal hydrogen bond with the C18 hydroxyl group.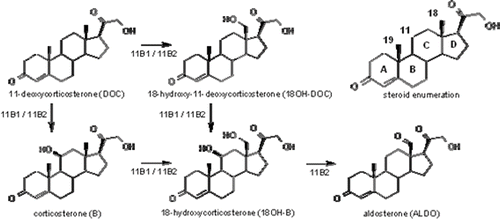
REFERENCE
- A Fisher, EC Friel, R Bernhardt, C Gomez-Sanchez, JM Connell, R Frazier, E Davies, Effects of 18-hydroxylated steroids on corticosteroid production by human aldosterone synthase and 11beta-hydroxylase, J Clin Endocrinol Metab (2001), 86 (9), p4326–p4329.
87. EFFECTS OF RHEUM RIBES ETHANOLIC EXTRACTS ON CYTOCHROMES P450 1B1 (CYP1B1) GENE EXPRESSION AND GLUTATHIONE-S-TRANSFERASE (GST) ACTIVITY IN HL-60 CELLS
Pembegul Uyar1, Fevzi Ozgokce2, Nusen Coruh3, and Mesude Iscan4
1Graduate Program of Biotechnology, Middle East Technical University, Ankara, Turkey, 2Department of Biological Sciences, Yuzuncu Yil University, Van, Turkey, 3Department of Chemistry, Middle East Technical University, Ankara, Turkey, 4Department of Biological Sciences, Middle East Technical University, Ankara, Turkey
This study was conducted to demonstrate whether Rheum ribes L. extracts affect the expression levels of CYP1B1 gene in HL-60 cells and to investigate the susceptibility of the relation between antiproliferation and CYP1B1 expression, and, also, GST activity. Dried and pulverized plant samples were extracted by ethanol at a ratio of 1:12 (w/v). Human Myeloid Leukemia (HL - 60) cell line was used as a model system for the studies. HL-60 cells were cultured in the presence of various concentrations of extracts for 72 hr. The percentage of cell viability was determined by metabolism of the tetrazolium salt XTT (2,3 - bis(2-methoxy-4-nitro-5-sulfophenyl) -5-[(phenylamino) -carbonyl] -2H-tetrazolium hydroxide). In the present study, R. ribes was found to inhibit the survival of human promyelocytic leukemia HL-60 cells in a concentration- and time-dependent manner. ED50 values of ethanolic shoot and root extracts were calculated as 120.01 ± 0.65 μg/ml and 104.15 ± 0.54 μg/ml, respectively. HL-60 cells were plated at a density of 1x105 cells/ml into T75 flasks. After overnight growth, cells were pretreated for 16 h with R. ribes extracts, dissolved in DMSO and diluted with RPMI 1640, to a final concentration 100 μg/ml of growth medium. The final concentrations of dimethyl sulfoxide in the culture medium were < 0.1%. At the end of treatment, RNA isolation and protein extraction were performed for all flasks. Isolated RNAs of both treated and non-treated cells were then reversely transcribed to cDNAs using Moloney Murine Leukemia Virus Reverse Transcriptase (M-MuLV-RT). cDNAs were amplified using CYP1B1 and β-actin primer sets. R. ribes shoot and root extracts affect HL-60 CYP1B1 gene expression, differentially with respect to non-treated control groups. GST activity of both treated and non-treated cells were measured in the presence of CDNB as subtrate in ELISA plate reader at 340 nm. There was a variation in the GST activity of R.ribes ethanolic shoot and root extracts treated HL-60 cells in comparison to non-treated control groups.
88. AGE-RELATED CHANGES IN CYTOCHROME P450S IMMUNOLOCALIZATION IN NORMAL AND REGENERATING RAT LIVERS
Piotr Czekaj, Aleksandra Bryzek, Danuta Plewka, and Ryszard Wiaderkiewicz
Dept of Histology, Med Univ of Silesia, Katowice, Poland, PL-40–752
Cytochromes P450 participate in the metabolism of exogenous compounds. The aim of the study was to show to which extent age, induction, chemical damage and regeneration may modify localization of P450 isoforms within the liver. Study was performed on male Sprague-Dawley rats presenting 11 age categories: fetuses (16-, 18-, 20-day old), newborns, adolescent animals (0.5-, 1-, 2-month old), mature ones (4-months old) and aging ones (20-, 24- and 28-month old). Within particular age groups 3 experimental subgroups were separated: one control subgroup (C), two inductor ones (BNF, Et) and - in adult rats - one subgroup of experimental hepatocarcinogenesis (CDE). Rats from BNF and Et subgroups were administered classical inducers of cytochrome P450 isoform: b-naphthoflavone (50 mg/kg of b.w.) and ethanol (6 g/kg of b.w., for three weeks in isocaloric diet), respectively. Rats from CDE subgroup were fed ad libitum for three weeks with choline-deficient ethionine-supplemented diet (CDE), which injures hepatocytes and stimulates liver regeneration process. Localization of P450 isofroms CYP 1A1, 1A2, and 2E1 within the liver acinus was analyzed immunohistochemically. It was shown that liver P450s were expressed during the last days of pregnancy and the first hours after the birth. During the late phase of fetal life CYP-positive cells were located in hepatoblasts lying around big, thin-walled blood vessels. Zone-like expression of P450 cytochromes was formed together with structural and functional separation of the liver acinus. Zone 3 of the liver acinus was the most important localization of studied isoforms appearance within the age range from 0.5 to 28 months. Unlike the other P450 isoforms subjected to chemical induction, CYP1A1 expression stimulated by BNF was revealed mainly within the 1 and 2 zone. In livers sampled from rats fed with CDE diet CYP2E1 was located within the 2 and 3 zone. Expression of 2E1 was heterogenous within the zones. Local effects of CYP 1A1 and 1A2 expression were difficult to predict in association with disappearance of zone-like localization of these isoforms, and intense histological changes caused by CDE diet. Supported by KBN, Grant no 3P05D03623
89. EXPRESSION OF HEPATIC CYTOCHROMES P450 CYP3A AFTER LONG-TERM TREATMENT WITH A GnRH RECEPTOR AGONIST – DALARELIN
Piotr Czekaj, Aleksandra Suszka-Šwitek, Aleksandra Bryzek, Danuta Plewka, and Anna Wiaderkiewicz
Dept of Histology, Med Univ of Silesia, Katowice, Poland, PL-40–752
Synthetic analogs of GnRH are used in long-term treatment of many diseases leading to desensitization of hypophysis, and - as a result - to repression of hypothalamic-pituitary-gonadal axis. However neuroendocrine mechanisms of the expression of hormone-dependent CYP3A representatives, a mechanism of synthetic GnRH analogs action, as well as, side-effects caused by their long-term administration are poorly recognized. The aim of the study was to indicate, whether a long-term administration of dalarelin - a GnRH receptor agonist and a new GnRH derivative - to adult female rats influences CYP3A protein expressions and localization within the liver. Study was performed on Sprague-Dawley rats weighing 200–220 g. Rats from experimental groups were administered dalarelin acetate (6 mg/kg of b.w.), intraperitoneally – for 1, 2 and 3 months. Simultaneously, rats from control groups were administered placebo – sodium-acetate buffer, pH 7.5. Localization of CYP3A cytochromes within the liver acinus was analyzed immunohistochemically. After the total RNA and microsomal proteins were isolated, CYP3A1, CYP3A2 and CYP3A9 mRNA (RT-PCR) and proteins (Western blotting) were identified and quantified by densitometry. Zone 3 of the liver acinus was the most important localization of studied isoforms appearance in both controls and experimental animals. In livers sampled from females administered 1, 2 and 3 months with dalarelin there was tendency for induction of CYP3A1 and reduction of related to female sex hormones CYP3A9 expression level; the expression of strongly affected by androgens CYP3A2 remained practically undetectable. It was concluded that GnRH agonist dalarelin changed the expression pattern but not localization of the CYP3A subfamily isoforms, influencing P450 expressions through the repression of the female sex hormone synthesis. It potentially can be connected with an activation of some xenobiotics; changes in the metabolism rate of drugs administered parallely with GnRH analogs and some endogenous substrates; and toxic effects in organs.
90. THE ROLE OF HEPATIC CYTOCHROME P-450 ENZYMES IN METABOLISM OF IMIDAZOACRIDINONE ANTITUMOR AGENT, C-1311 (SYMADEX): STUDIES WITH THE HEPATIC NADPH:CYTOCHROME P450 REDUCTASE NULL MICE
Agnieszka Potega1, Sebastian Ronseau2, Colin Henderson2, Jerzy Konopa1, and Zofia Mazerska1
1Department of Pharmaceutical Technology and Biochemistry, Chemical Faculty, Gdansk University of Technology, Gdansk, Poland, 80–952, 2Cancer Research UK Molecular Pharmacology Unit, Biomedical Research Centre, Dundee, United Kingdom, DD1 9SY
Antitumor agent, 5-diethylaminoethylamino-8-hydroxyimidazoacridinone, C-1311, (SymadexR) developed in our laboratory, is under Phase II clinical trials. We showed earlier that C-1311 metabolism in vitro with rat liver microsomes led to four main metabolites [1]. However, the incubation of the studied compound with human E. coli recombinant cytochrome P450 isoenzymes (CYPs): 1A2, 2C9, 2C19, 2D6 and 3A4 did not result in significant amount of metabolic product [2]. Therefore, we suspected that cytochromes P-450 were not crucial enzymes involved in rat liver metabolism of SymadexR. In order to elucidate the role of hepatic CYPs in metabolism of C-1311 we compared drug elimination with blood and urine observed in wild-type (WT) and hepatic NADPH: CYP oxidorectase null-type (HRNTM) mice. Furthermore, metabolic transformations of C-1311 and its less active analog, C-1330 with liver microsomal fractions of HRN and WT mice were also compared. Mice were dosed at 50 mg/kg by ip injection, blood and urine samples were taken at 10, 20, 40, 60 minutes and 2, 4 and 6 hours and were analyzed by HPLC. WT and HRNTM mice liver microsomes were incubated with C-1311 and C-1330 and the obtained reaction mixtures were analysed by HPLC with UV-vis or MS/MS detection. The results revealed slightly slower elimination of C-1311 in blood and urine of HRNTM than of WT mice. In the presence of WT mice liver microsomes C-1311 was transformed to at least one product m/z 365 identified as C-1311 derivative with additional oxygen atom in aromatic ring and probably the second one, m/z 323, resulted from deethylation of the side chain. After incubation with HRN mice liver microsomes HPLC peaks of metabolites were lower. Similar relations between metabolite amounts with HRN and WT microsomes were found in the case of C-1330 compound. In addition, C-1330 turned out to be a prodrug of C-1311 as the metabolite m/z 351 equal to M+1 of C-1311 was found. In conclusion, the differences in elimination rates of C-1311 observed between HRNTM and WT mice in blood and urine indicates that liver cytochromes P450 are involved in metabolism of Symadex to a small extent. If so, dealkylation of the side chain and some oxidative transformation of aromatic ring occur. However, extra-hepatic transformations of C-1311 may play a significant role.
REFERENCE
- A. Wisniewska, A. Chrapkowska, A. Kot-Wasik, J. Konopa, Z. Mazerska, Metabolic transformations of antitumor imidazoacridinone, C-1311, with microsomal fractions of rat human liver enzymes, Acta Bioch. Polonica, 54, 831–838, (2007).
- A. Chrapkowska, E. Piatek, A. Wisniewska, J. Konopa, Z. Mazerska, The studies of imidazoacridinone antitumor agent, C-1311, in the field of metabolic transformations with cyto-chrome P450 isoenzymes, Abstracts of the 41th meeting of the Polish Biochemical Society, Bialystok, September 2006, Acta Bioch. Polon. Vol 53, Suppl.1/2006, 83 (poster)
91. TRANSCRIPTOMIC EFFECTS OF CYP2C9 OVEREXPRESSION IN HEPG2 CELLS
Céline Narjoz1, Sandrine Imbeaud2, Alain Paris3, Lawrence Aggerbeck2, Philippe Beaune1, and Isabelle de Waziers1
1INSERM U775, Paris, France, 75006, 2Gif/Orsay DNA Microarray Platform (GODMAP) - Centre de Génétique Moléculaire, Centre National de la Recherche Scientifique, 91198 Gif-sur-Yvette, France, 91198, 3Xénobiotiques, INRA/ENVT, UMR 1089, Toulouse, France, 31931
Microsomal cytochromes P450 (CYP) are highly inducible enzymes whose the accumulation could result in proliferation of smooth endoplasmic reticulum membranes and/or endoplasmic overload and/or unfolded protein responses. The aim of this work was to study the global cellular consequences of CYP2C9 over-expression. CYP2C9 over-expression was obtained after infection of a human hepatoma cell line (HepG2) with a recombinant adenovirus encoding CYP2C9 (Ad-2C9) or ß-galactosidase (Ad-LacZ) used as control. CYP2C9 over-expression was checked at the three levels: mRNA level by quantitative RT-PCR, protein level by immunoblotting and enzymatic activity (luciferin H hydroxylase, CYP2C9-specific). Microarray analysis, using pangenomic Agilent gene chips, was performed to study whole genome expression changes associated with CYP2C9 over-expression. Multivariate analysis (PLS-DA) was realized on 11 245 genes expressed in HepG2 cells and indicated that HepG2 Ad-CYP2C9 and HepG2 Ad-LacZ cells expressed different gene expression profiles. Modified Student's test allowed to identify 155 genes significantly regulated by CYP2C9 over-expression. Ingenuity Pathway Analysis mapped 6 significant networks with scores up to 19. The two most relevant networks were associated: i) with cellular functions and maintenance (endoplasmic reticulum stress response, oxidative stress response, post-translational modifications) and ii) with cellular assembly and organization (stabilisation of keratin filament, formation of cellular inclusion, fusion of endosome and trafficking of vesicles). Some kinases appeared to be involved in these networks: AKT and P38-MAPK in the first network and JNK in the second one. For few genes, mainly coding heat shock proteins, regulation of their expression by CYP2C9 over-expression was validated by quantitative RT-PCR and by western blot. Moreover, activation by phosphorylation of AKT was observed in HepG2 Ad-2C9 cells. When HepG2 cells overexpressed CYP2C9, cells underwent a stress however not deleterious cellular effect was observed, cells should developed an adaptative response to stress. Aim of this study consists in identifying wide cellular effects of CYP2C9 overexpression. Metabolic consequences of CYP2C9 over-expression is now under investigation.
92. BIOTRANSFORMATION ENZYMES ACTIVITIES IN LANCET FLUKE (DICROCOELIUM DENDRITICUM)
Barbora Szotakova, Veronika Krizova, Jiri Lamka, and Lenka Skalova
Faculty of Pharmacy, Charles University, Hradec Kralove, Czech Republic, CZ-50005
Lancet fluke (Dicrocoelium dendriticum), being causal organism of dicrocoeliosis known in animals and man, continuously attract attention of scientific community. Only detailed knowledge of the properties of this parasite, including his defence mechanisms, can lead to successful control of parasitosis in mentioned hosts. The only means so far generally accepted against dicrocoeliosis is the use of suitable anthelmintics, especially benzimidazole ones. Biotransformation enzymes can, to a certain extent, protect the parasitic worms against the effects of xenobiotics including anthelmintics. The present study was designed to evaluate drug metabolizing enzymes activities in lancet fluke subcellular fractions. Activities of oxidation enzymes, carbonyl-reducing enzymes and conjugation enzymes were tested in subcellular fractions of D. dendriticum homogenate. Several enzyme activities corresponding to main isoforms of cytochrome P450 were measured in microsomes-like fraction but no activity was detected. On the other hand, activity of catalase (using europium (III)–tetracycline–hydrogen peroxide system) and peroxidase (assayed by measuring the H2O2 dependent oxidation of o-phenylendiamine) was proven in subcellular fractions. From conjugation enzyme activities, UDP-glucuronosyl transferase activity towards p-nitrophenol in microsomes-like fraction and glutathione-S-transferase (GST) activity towards 1-chloro-2,4-dinitrobenzene in cytosol-like fraction were assayed. The apparent kinetic parameters for GST reaction were determined: V´max = 1.64 ± 0.25μM.min-1, K´m = 2.57 ± 0.38 mM. Activities of carbonyl-reducing enzymes were tested using metyrapone, acenaphthenol, daunorubicin, and oracin. The results proved the ability of D. dendriticum enzymes to reduce the carbonyl group of xenobiotics. In lancet fluke, many model substrates were metabolized and the specific activities of some biotransformation enzymes were similar to the activities found in mammal host organisms. These results document the ability of D. dendriticum to effectively metabolize different xenobiotics and by this way be protected against the toxic effect of xenobiotics.
ACKNOWLEDGMENT
This project was supported by the Grant Agency of Czech Republic, Grant No. 524/07/0611.
93. COMPARISON OF FLUBENDAZOLE METABOLISM AND BIOTRANSFORMATION ENZYMES ACTIVITIES IN HEALTHY SHEEP AND SHEEP WITH HAEMONCHOSIS
Lenka Skalova1, Barbora Szotáková1, Veronika Krížová1, and Jirí Lamka2
1Dept of Biochemistry, Charles Univ, Fac of Pharmacy, Hradec Kralove, Czech Republic, CZ-50005, 2Dept of Pharmacology and Toxicology, Charles Univ, Fac of Pharmacy, Hradec Kralove, Czech Republic, CZ-50005
Most pharmacokinetic and biotransformation studies have been performed in healthy animals and the potential of infection to change the condition of the organism has not been considered. However, parasitic infections can modify the host's ability to metabolize drugs and other xenobiotics by altering the biotransformation enzymes and these changes may have various pharmacological, toxicological or physiological consequences. Haemonchosis as a parasitosis caused by Haemonchus contortus, belongs to the most pathogenic endoparasitoses of ruminants bred on farms, simultaneously to the most studied model parasitoses in over the world. The aim of present study was to evaluate the effect of haemonchosis on the activities of the main biotransformation enzymes and metabolism of anthelmintic drug flubendazole (FLU) in sheep (Ovis aries). For this purpose, young rams at the age of 3 months were experimentally infected by 5500 L3 larvae of H. contortus ISE strain. Subcellular fractions of liver and small intestinal mucosa homogenates were isolated from non-infected, parasitologically negative animals (control group A), from H. contortus infected-animals after 6 weeks-infection (group B) and from H. contortus infected animals after 10 weeks-infection (group C). In hepatic microsomes from infected animals, most of cytochromes P450 activities were significantly decreased comparing to control group. Also thiobenzamide-S-oxidase (TBSO) activity, corresponding mainly to the activity of flavine monooxygenases, was diminished to 50 % in group B and C. On the other hand, helminthosis did not change the glutathione-S-transferase activity in liver as well as in small intestine. The effect of haemonchosis on reductases of carbonyl group was not unambiguous: some activities (e.g. pyridincarboxyldehyde reductase, acenaphenol dehydrogenase) were significantly increased in group B, other activities (e.g. DL-glyceraldehyde reductase) were significantly decreased in groups B and C comparing to control group A. The changes in FLU in vitro biotransformation were so mild that undesirable alterations in FLU pharmacokinetic by haemonchosis are not expected. This project was supported by Grant Agency of Czech Republic, grant No 524/06/1345
94. INVESTIGATION OF DRUG METABOLISM IN MAN: THE PROCESS FOLLOWING THE IMPLEMENTATION OF THE CLINICAL TRIAL DIRECTIVE 2001/20/EC
Stuart Cameron, H. Charles, L. McGregor, M. Bottacini, and C. Young
Pharmaceutical Metabolism, Charles River Laboratories Preclinical Services Edinburgh, Edinburgh, United Kingdom, EH33 2NE
The “Metabolites in Safety Testing” guideline published in 2002 triggered an intense debate in the scientific community on the criteria to be used to determine if a metabolite observed in man should be more extensively studied. Both the scientific community and the regulators converge on the approach of attempting to identify differences in drug metabolism in animals compared to humans as early as possible in drug development. Particular efforts should be made in the identification of metabolites that are unique to humans. Early identification of unique or “major” metabolites should then drive the process for toxicological and clinical assessment.
The clinical metabolism (ADME) study offers a practical basis for the investigation of the disposition and metabolism of the candidate drug and there is a clear scope for anticipation of this type of study in the development plan.
The implementation of the European Clinical Trial Directive (2001/20/EC) in May 2004 brought significant changes to the way that clinical metabolism studies in man are conducted within the European Union. In the period since the introduction of the new legislation, we have conducted 18 clinical metabolism studies in our Phase 1 unit, in Edinburgh for clients worldwide. The objective of this poster is to provide an overview of our experience in regulatory and manufacturing procedures, which have changed since May 2004, and to illustrate the critical steps in the organization and conduct of these studies.
95. PREDICTION OF THE INCREASED EXPOSURE TO DRUGS IN LIVER CIRRHOSIS: A SYSTEMS BIOLOGY APPROACH INTEGRATING PRIOR INFORMATION ON DISEASE WITH IN VITRO DATA ON DRUG DISPOSITION
Trevor N. Johnson1, Koen Boussery2, Geoff Tucker3, and Amin Rostami-Hodjegan3
1Simcyp Limited, Sheffield, United Kingdom, S2 4SU, 2Laboratory of Medical Biochemistry and Clinical Analysis, Ghent University, Ghent, Belgium, 3University of Sheffield and Simcyp Limited, Sheffield, United Kingdom, S10 2JF
Liver cirrhosis is characterized by many biochemical and physiological changes, including a decrease in the number of functional hepatocytes, decreased concentrations of plasma proteins, and the development of porto-systemic vascular shunts. Depending on the impact of these changes on specific drugs, ‘first-pass’ metabolism and systemic clearance will be variably affected. The Simcyp simulator incorporates many genetic, physiological, demographic and clinical attributes of patient populations pertinent to in vitro - in vivo extrapolation of xenobiotic absorption and disposition into databases that can be used for automated prediction of pharmacokinetics. The aim of this study was to build a Simcyp population library enabling prediction of changes in drug exposure related to the severity of liver cirrhosis, by integrating relevant information on demography, blood flow, CYP enzymes, liver size, protein binding, renal function, tissue composition and volume and gastric emptying. The information was gathered from the literature and incorporated into three population libraries corresponding to severity of liver cirrhosis (Child-Pugh scores A (mild), B (moderate) and C (severe)). The drugs studied were midazolam (oral, iv), caffeine (oral), theophylline (oral, iv), metoprolol (oral, iv), nifedipine (oral), quinidine (oral), diclofenac (oral), sildenafil (oral) and omeprazole (oral, iv). In vitro Vmax and Km values and in vivo clearance values were obtained from the literature. The characteristics of members of the simulated populations were matched as closely as possible to those of the subjects of the actual clinical studies. Predicted fold changes in clearance (systemic or oral) (liver cirrhosis vs healthy controls) were compared with observed changes ().
Figure 1 Comparison of fold increases in exposure -: cirrhosis vs healthy controls (mean ± SD).
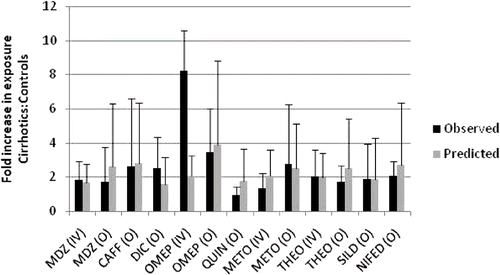
There was good agreement (lack of statistically significant difference, 2 tail paired t-test) between observed and predicted clearance ratios, with the exception of those for IV omeprazole (p < 0.05). Predicted CL ratios were within 0.8 to 1.25 fold of the observed CL ratios in 54% of cases (range 0.25 – 1.9 fold). Prior estimation of the likely extent of the influence of specific diseases on drug absorption and disposition should facilitate the selection of subsequent in vivo studies and their design with respect to safe dosage and patient numbers.
96. PHARMACOKINETICS OF FLUBENDAZOLE IN LAMBS AND ADULT RAMS AND EWES (OVIS ARIES)
Veronika Krizova, Jiri Lamka, Barbora Szotakova, Milan Nobilis, and Lenka Skalova
Faculty of Pharmacy, Charles University, Hradec Kralove, Czech Republic, CZ-50005
The aim of this study was to evaluate the age- and sex-differences in pharmacokinetic of anthelmintic drug flubendazole (FLU; methyl-[5-(4-fluorbenzoyl)-1H-benzimidazole-2-y1]-carbamate) in sheep. Three male and three female lambs (three-months-old) were used for the first pharmacokinetic study and five month later the same animals (as matured rams and ewes) were used for repetition of pharmacokinetic FLU study. During whole experiments, all animals were healthy, parasitologically negative, without any pharmacological treatment and bred under the same conditions. In both pharmacokinetic studies, single dose of FLU (30 mg per kg of body weight) in oral suspension were administered. Blood samples were taken before administration of FLU and than in 0.5, 1, 2, 4, 6, 8, 10, 12, 24, 36, 48, 60 and 72 hour intervals after FLU administration. Blood samples were centrifuged and plasma was frozen until the extraction and analysis. FLU and its metabolites were extracted into tert-butylmethyl ether and analysed by HPLC with albendazole as internal standard and mixture of methanol and sodium perchlorate (3:1) as mobile phase. The main detected metabolite was reduced flubendazole (FLU-R, compound with reduced keto group) and only traces of other metabolite (hydrolysed flubendazole) were found. Two enantiomers of FLU-R were detected on the chromatogram: (-)-enantiomer at retention time 14.2 min and (+)-enantiomer at 17.0 min. Considerable differences between lambs and mature sheep in plasmatic FLU-R concentrations were observed. In lambs, the Cmax value was achieved at 10 hours after FLU administration (300 pmol of (+)-FLU-R and 14 pmol of (-)-FLU-R per ml of male plasma and 210 pmol of (+)-FLU-R and 7 pmol of (-)-FLU-R per ml of female plasma) while in mature animals Cmax value was achieved at 36 hours after FLU administration (590 pmol of (+)-FLU-R and 22 pmol of (-)-FLU-R per ml of male plasma and 440 pmol of (+)-FLU-R and 24 pmol of (-)-FLU-R per ml of female plasma). In mature sheep high concentration of both isomers of FLU-R remained in plasma up to 72 h after FLU administration of experiment. These findings document that lambs in comparison to mature animals absorb and metabolise FLU much faster but in lower range.
ACKNOWLEDGMENT
This project was supported by Grant Agency of Czech Republic (Grant No 524/06/1345)
97. FREQUENCIES OF GSTM1 AND GSTT1 POLYMORPHISM IN DELHI POPULATION, INDIA
Satyender Singh1, Vivek Kumar2, S.S. Grover1, S. T Pasha1, S. K. Jain1, Arvind Rai1, Shiv Lal1, and B.D. Banerjee2
1National Institute of Communicable Diseases, Delhi, India, 110054, 2Department of Biochemistry, UCMS & GTB Hospital, Delhi, India, 110095
Epidemiological studies have estimated that approximately 80% of all cancers are related to environmental factors. Xenobiotic metabolism is the principal mechanism for maintaining homeostasis during the body exposure to enviromental carcinogen. Interindividual differences in sensitivity to chemical carcinogens may contribute to difference in susceptibility to human cancer subsequent to environmental exposures can be the result of several host factors, including differences in metabolism, DNA repair, altered expression of tumor suppressor genes and proto-oncogenes, and nutritional status. Glutathione-s-transferases (GSTs) are a polymorphic group of enzymes involved in phase-II detoxification of wide range of xenobiotics and chemotherapeutic agents. Polymorphism associated with genes coding for glutathione-s-transferases enzymes are known to influence metabolism of different carcinogens and have been associated with incidence of various type of cancers. Humans are polymorphic in their ability to detoxify intermediates, which in theory may explain differences in cancer risk as a result of exogenous exposure. In the present study, we determine the frequencies of GSTM1 and GSTT1 polymorphism in 206 unrelated healthy volunteers, who had been the resident of Delhi, India for more than five years. Out of the 206 individuals, 37.86% (78) were smokers. Consent and ethical clearance was obtained for the study. Information about the social habits and health problems were also recorded. Blood was collected and DNA extracted by Miller's salting out method with little modification. GSTM1 and GSTT1 polymorphisms were analyzed by PCR method, where as β-globin gene was used as an internal control. We found that 20.4% of the individuals had homozygotic GSTM1 null genotype, whereas, 28.88% had the GSTT1 null genotype. In 0.7% of individuals, concomitant lack of GSTM1 and GSTT1 genes were observed. In smokers, 63.2% had the GSTM1 null genotype and 68% had the GSTT1 null genotype. None of the homozygotic null genotype smoker found any known history of carcinoma. GSTM1 and GSTT1 deletion genotypes are genetic susceptibility biomarkers for cancer. The frequencies of GST polymorphisms in this study were found to be different from those observed in other Indian and South American, Asians populations. Our results highlight an impact of ethnicity and provide a basis for the future epidemiological and clinical further studies in Delhi and its surrounding areas population.
98. TISSUE DISTRIBUTION OF VINBLASTINE AND ITS METABOLITES USING MALDI IMAGING
Andrew McEwen1, Stuart Wood2, Hayley Bird1, Alan Lathall1, and Karen Dummer1
1Department of Metabolism and Pharmacokinetics, BioDynamics Research Limited, Northamptonshire, United Kingdom, 2BioDynamics, Northants, United Kingdom
Recent advances in vacuum matrix-assisted laser desorption ionisation (v-MALDI) mass spectrometry have enabled direct measurement of individual components (parent drug and metabolites) via direct analysis of tissue slices. In this study, MALDI techniques were been used to study the distribution of vinblastine and its major metabolite (desacetyl vinblastine) directly within tissues. After intravenous administration of vinblastine to rats, tissue samples were collected and placed on a conductive matrix and finally sprayed with matrix for direct vMALDI analysis. MALDI sampling occurred uniformly across the entire tissue sample regions selected and mass intensity images were created. By using different MS or MS/MS ions, it was possible to view images that were selective both for the parent drug and metabolites of interest. The objective of this study was to compare the distribution within key tissues of vinblasine and its major metabolite, desacetylvinblastine and to further evaluate the potential relevance of the physiological distribution of both drug and metabolite within key tissues. Vinblastine is an important anti-neoplastic agent which is often administered clinically as part of potentially curative multi-drug regimens. The data obtained showed a clear relationship between the within-tissue distribution of the drug and its metabolite with time and provides a basis to evaluate potential drug-drug interactions with several drugs simultaneously within tissues of interest.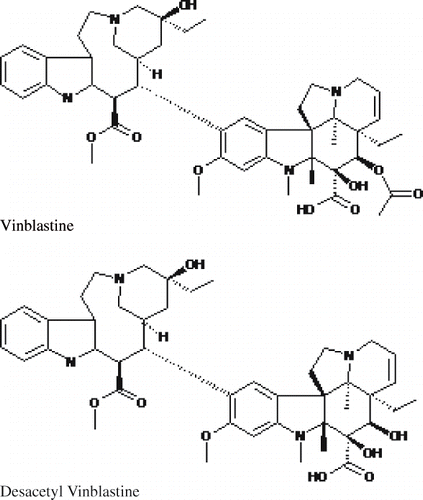
99. IN VITRO BINDING OF BN83495, A NOVEL STEROID SULPHATASE INHIBITOR, TO ERYTHROCYTES: BINDING KINETICS AND INTER-SPECIES COMPARISON
V. Ventura, J. Solà, C. Peraire, and R. Obach
Drug Metabolism, Pharmacokinetics and Immunology Department, Ipsen Pharma, S.A., Barcelona, Spain, 08980
BN83495 (STX-64, 667-COUMATE) is an irreversible steroid sulphatase inhibitor under phase I/II development for breast cancer by the Ipsen Group. BN83495 contains a sulphamate ester group (3-O-sulphamate) linked to an aryl ring (tricyclic coumarin-based). The 3-O-sulphamate group is indispensable for its activity and for its ability to bind to carbonic anhydrase II (CA II) resulting in sequestration and stabilization by red blood cells [1]. The results of the present study demonstrate that binding of BN83495 to erythrocytes is BN83495 blood concentration dependent. The affinity and capacity of the erythrocytes for BN83495 binding varies depending on the species, and might be altered by drug-drug interactions. In vitro binding of BN83495 to erythrocytes from dog, rat and humans was measured as the plasma to blood ratio (PBR) of radioactivity after incubation with 14C-BN83495 (blood concentrations from 100 to 50000 ng/ml). PBR remained constant up to BN83495 blood concentration of 1000 ng/ml in dog (PBR: 0.166), up to 5000 ng/ml in rat (PBR: 0.190), and up to 10000 ng/ml in humans (PBR: 0.051), showing saturation of the binding sites at higher concentrations. Apparent KD and Bmax parameters were estimated among species using a model for saturable binding equilibrium. KD values were 67, 104 and 56 nM, and Bmax values were 69.5, 123.0 and 208.3 μM in dog, rat and humans, respectively. KD in rat was higher than in the other species meaning a possible lower affinity of rat erythrocytes for BN83495. Bmax was clearly lower in dog meaning a lower binding capacity of dog erythrocytes for BN83495. Considering Bmax parameter, the species could be ranked following the same order as previously described for saturation of erythrocyte binding capacity (dog < rat < humans). No effect of Acetazolamide (AZM), a CA II inhibitor, was found up to 10 μM in rat blood incubations, but showing a displacement of 14C-BN83495 from the binding sites at AZM concentrations above 50 μM. In human blood, no effect of AZM was found up to 100 μM AZM (the highest concentration tested). These results suggest a species-dependent displacement of BN83495 from its erythrocyte binding sites and do not rule out the possibility of drug interactions with other CA II inhibitors. Very little inter-individual variability in BN83495 erythrocyte binding was observed in vitro in blood from human healthy volunteers.
REFERENCE
- Ireson C.R., Chander S.K., Purohit A., Parish D.C., Woo L.W.L., Potter B.V.L. and Reed M.J. (2004) Pharmacokinetics of the Nonsteroidal Steroid Sulphatase Inhibitor 667 COUMATE and its sequestration into Red Blood Cells in Rats. Br. J. Cancer 91(7):1399–404.
100. INTER-ANIMAL VARIABILITY IN INHALATION DOSING: IMPACT ON ADME STUDY DESIGN AND INTERPRETATION
Colin Webber1, Brian A. John1, Colin J. Hardy2, and Simon A. Moore3
1Drug Metabolism, Huntingdon Life Sciences Ltd., Huntingdon, England, PE19 2BG, 2Inhalation Toxicology, Huntingdon Life Sciences Ltd., Huntingdon, England, PE19 2BG, 3Aerosol Technology, Huntingdon Life Sciences Ltd., Huntingdon, England, PE19 2BG
Huntingdon Life Sciences is one of the leading contract facilities for respiratory safety assessment, a service for which there is currently increasing demand. This presentation focuses specifically on recently conducted inhaled dose ADME studies and seeks to address some common questions about such work. The review includes both rodent and non-rodent studies, in which aerosols of radiolabelled test compounds have been generated from dry powders, solutions or MDIs. Reproducing the aerosol characteristics of non-radiolabelled test compounds for the much smaller amounts of radiolabelled compound usually available is a key challenge when using dry powder aerosols. In our laboratories, sub-gram amounts of radiolabelled dry powders have been micronised by ball-milling (ambient or cryogenic) to routinely produce material with MMAD < 2 microns and inhalable fraction > 90%, broadly consistent with larger-scale non-radiolabelled batches. Inter-animal variability in inhalation dosing is also a major consideration in ADME study design and data interpretation. Achieved radioactive dose levels shed light on variability that is likely to be encountered during toxicology studies where achieved dose levels cannot be directly measured. Dose quantification for rodents in snout-only exposure chambers is achieved by radioactivity analysis of a small number of animals killed immediately after exposure, the mean total amount measured being taken to represent the dose received by each of the other animals on the chamber. This includes radioactivity deposited on the body surface, mainly around the snout, which typically represents about 25% of the total and will subsequently be ingested during preening. Our data confirm some inter-animal variation in both the relative proportions of deposited and inhaled radioactivity and the overall dose level achieved during a single exposure (CV typically >20%). Consequently, total apparent recoveries of radioactivity during rodent excretion balance studies, expressed as % estimated dose, are likely to show similar inter-animal variation. For non-rodents, dose quantification is achieved by rigorous analysis of the dose utensils used for each animal, subtracting residual radioactivity from the amount dispensed during exposure. This approach results in a more accurate individual dose assessment such that excretion recoveries usually show little variation. This poster includes specimen data from rats and dogs and reviews ways in which these studies may be designed and interpreted to ensure that conclusions are scientifically robust.
101. THE INFLUENCE OF SHORT-CHAIN ALPHA-TOCOPHEROL DERIVATIVE ON LIPID PEROXIDATION AND LIPID CONTENTS IN RAT LIVER MICROSOMES AND MITOCHONDRION UNDER DRUG AND CHEMICAL CAUSED HEPATHOTOXICOSIS
Ganna M. Shayakhmetova, Larysa B. Bondarenko, and Valentina M. Kovalenko
General Toxicology, SI “Institute of Pharmacology and Toxicology, Academy of Medical Sciences of Ukraine”, Kyiv, Ukraine
Background. Paracetamol intoxication causes hard liver damage and some times even lethal outcomes. Its hepatotoxic effect could be a result not only of covalent binding of its highly reactive metabolite N-acetyl-4 benzoquinon imine but also oxidative stress as a result of free radical processes activation. The aim of the present study was to compare the protective effects of alpha-tocopherol derivative with a truncated side chain – acetate-2,5,7,8 – tetramethyl-2-(4-methylpenten-3-yl)-6-oxychroman and alpha-tocopheryl acetate in rats with chemical and drug caused hepathotoxicosis. Methods. In experiments on Wistar male rats possibilities of paracetamol and combined ethanol-tetrachlormethane hepatotoxic effects correction with synthetic short-chain alpha-tocopherol derivative and pharmacopoeia preparation of alpha-tocopherol have been studied. Parameters of peroxydation processes, phospholipids and different fractions of cholesterol contents in liver microsomes and mitochondrion were investigated. Results. It was shown that the short-chain alpha-tocopherol derivative and pharmacopeia preparation of alpha-tocopherol had a membranotropic effect at paracetamol hepathotoxicosis model, normalizing free and total cholesterol levels in liver mitochondria fraction. In contrast to mitochondrion, in liver microsomes, where main processes of phospholipids and cholesterol biosynthesis take place, rats' paracetamol intoxication caused only clear tendency to changes in different fractions of cholesterol contents. At the same time paracetamol caused also tendency to decreasing in phospholipids contents in this fraction, presumably due to their oxidation and biosynthesis inhibition by products of lipid peroxidation processes. alpha-tocopherol and its derivative were able to stabilize hydoperoxides of membrane lipids limiting their degradation to peroxy radicals. Such processes could intensify these preparations effects on phospholipids metabolism. At combined ethanol-tetrachlormethane hepatotoxicosis synthetic short-chain alpha-tocopherol derivative normalized total cholesterol contents in microsomal fraction. Investigated compounds also decreased levels of dienes, trienes and TBA-products formation in rats' liver subcellular fractions with both models of hepathotoxicosis. Conclusions. The results of the present study indicate that derivatives of alpha-tocopherol may efficiently use as a protective agent against drugs and chemicals-induced hepatic damage. An antioxidative activity of short-chain alpha-tocopherol derivative and pharmacopeia preparation of vitamin E depended on the structure of chromane cycle but not on the length of side chain.
102. PRE-CLINICAL CANDIDATE SELECTION: RELATIONSHIP BETWEEN IN VITRO ADME AND IN VIVO PHARMACOKINETICS
Eva González1, Juana Peña2, Ágata Motos1, Anna Solans2, Roser Rosa1, Marcel·lí Carbó2, and Lluís Angel Gómez1
1Discovery Biology, Palau Pharma, Palau-solita i Plegamans (Barcelona), Spain, 08184, 2Pharmacokinetics and bioanalysis, Palau Pharma, Palau-solita i Plegamans (Barcelona), Spain, 08184
At the end of a Drug Discovery process it is essential to perform complementary in vitro and in vivo assays in order to choose the compound with the highest chance of success as a drug development candidate. In our study (within the process of pre-clinical candidate selection) the compound's metabolic stability was assessed in rat, mouse, dog and human liver microsomes at different concentrations. All the candidates, showed lower intrinsic clearance (Clint) in animal liver microsomes (7–62 μl/min/mg prot) than in human liver microsomes (<13 μl/min/mg prot). In all cases, Clint slightly decreased at higher compound concentrations (ratio Clint 1 μM/Clint 10 μM < 2.4) suggesting that non-linearity problems due to metabolism would not be expected. On the other hand, in vivo pharmacokinetic assays showed a good bioavailability (50–80%) in concordance with estimations from in vitro assays. However, in vivo clearances determined after intravenous administration (Cliv), were always higher than hepatic clearances predicted from in vitro microsomes (ratio Clhepatic/Cliv between 0.08 and 0.30), suggesting that metabolism was not the only route of elimination. In order to verify these results, an analysis of urine and faeces in rats were done in a selected compound. Urine elimination was about 2 % with a renal clearance of 0.58 ml/min. Qualitative analysis of faeces showed presence of parent compound in this matrix. The data suggests that differences between in vivo and in vitro clearances could be due to a biliary excretion. The relationship between in vitro and in vivo assays performed during the last steps of candidate selection give us enough confidence for the choice of a compound that would achieve an adequate pharmacokinetic profile in human.
103. OPTIMIZATION AND DOWNSCALING OF HEPATOCYTE CULTURE CONDITIONS FOR SCREENING OF COMPOUNDS
Lorena Dominguez1, Abadie Catherine1, Guenzi Jean-Philippe1, Bruno Heyd2, Georges Mantion2, Blanchard-Morand Nadege1, and Lysiane Richert3
1KALY CELL, Besançon, France, 25000, 2Centre de Transplantation Hépatique, Service de Chirurgie Viscérale et Digestive, Besançon,, France, 3UFR des Sciences Médicales et Pharmaceutiques, Laboratoire de Toxicologie Cellulaire, EA 3921, Besançon, France, 25200
The evaluation of drug-drug interactions, toxicity or metabolism of a given drug candidate as well as of many chemical compounds, compulsory due to the recent Reach law (2007), requires suitable in vitro models. Hepatocytes, in particular from human origin, in monolayer and in suspension, are one of these models, recognized for long to be predictive in the estimation of the metabolism and risk assessment in humans. Nevertheless, the limited access to primary human hepatocytes and the increasing demand for studies using both animal and human hepatocytes prompted us to downscale the cells used for metabolism and drug-drug interaction studies. We have assessed the possibility to downscale rat and human hepatocyte suspensions and monolayer cultures from 6 cm2 down to 2, 1 and 0.4 cm2, for the determination of the major rat and human cytochrome P450 (CYP)-dependent activities, i.e. CYP1A-, CYP3A-, CYP2B- and CYP2C- assays using HPLC separation followed by UV and fluorimetric assessment.
104. INTERACTION OF INHIBITORS OF CYCLIN DEPENDENT KINASES WITH HUMAN LIVER MICROSOMAL CYTOCHROMES P450
Michal Siller1, Pavel Anzenbacher1, Eva Anzenbacherova2, and Miroslav Strnad3
1Department of Pharmacology, Faculty of Medicine, Palacky University, Olomouc, Czech Republic, 2Department of Medical Chemistry and Biochemistry, Faculty of Medicine, Palacky University, Olomouc, Czech Republic, 3Laboratory of Growth Regulators, Faculty of Sciences, Palacky University, Olomouc, Czech Republic
Inhibitors of cyclin dependent kinases based on N6 substituted benzylaminopurine structure such as olomoucine and roscovitine have attracted much attention due to its ability to cause cell cycle arrest in the animal cell and act as potential antineoplastic drugs (1). As the information on the interaction of any new drug with the main enzymes of drug metabolism (cytochromes P450) is essential for understanding the possible mechanism of drug metabolism as well as for evaluation of possible drug-drug interactions, a systematic study on the inhibition of activities of nine cytochromes P450 (CYP) by roscovitine and olomoucine II has been performed. CYP1A2, 2A6, 2B6, 2D6, 2C9, 2C8, 2C19, CYP2E1 and 3A4 activities have been determined. Both compounds inhibited the CYP1A2, CYP2C9 and to a certain extent also the CYP3A4 activities. Roscovitine exhibits a rather specific inhibition of the CYP1A2 with Ki of 15 microM and of the CYP2C9 activity with Ki value of 40 microM. Olomoucine inhibits the CYP1A2 activity with Ki of 35 microM. The mixed mechanism of inhibition takes place in all cases studied. The results indicate that at least for roscovitine, a possibility of drug interactions cannot be ruled out when plasma levels reach range of tens of micromoles.
ACKNOWLEDGMENT
Support through the MSM6198959216 project is gratefully acknowledged.
REFERENCE
- Hajduch M, Kolar Z, Novotny R, Hanus J, Mihal V, Hlobilkova A, Noskova V, Strnad M. (1997) Induction of apoptosis and regression of spontaneous dog melanoma following in vivo application of synthetic cyclin-dependent kinase inhibitor olomoucine. Anticancer Drugs 8, 1007–1013.
105. INFLUENCE OF FORMOTEROL (AEROLAIZER) ON SEVERE EXACERBATION OF ASTHMA PATIENTS
Nadezhda G. Berdnikova
Chair of Clinical Pharmacology, Moscow Medical Academy named by Sechenov, Moscow, Russia, 109240
Hypothezis. We studied effectiveness and safety of formoterol (LABA – long acting beta2-agonist) in comparison with salbutamol (SABA – short acting beta2-agonist) for patients with severe exacerbation of asthma. Methods used. All patients were hospitalized due to severe exacerbation of asthma and took short course of prednizolone (30–40 mg/day) during 10–12 days. Patients were randomized in 2 groups. Group 1: n = 27, 45.3 ± 18.2 years old, mean FEV1 39.5 ± 6.4% pred., QTc 375 ± 6.4 ms, the mean number of SABA during 24 hours before hospitalization was 12.4 ± 6.2 puffs, duration of asthma was 8.3 ± 2.5 years; the patients were treated with formoterol 12 mcg x 2 daily and additionally 1–2 doses us per need. Group 2: n = 26, 55.4 ± 11.5 years old mean FEV1 41.2 ± 9.1% pred., QTc 388 ± 5.2 ms, the mean number of SABA during 24 hours before hospitalization was 14.6 ± 8.3 puffs, duration of asthma 6.8 ± 2.6 years: the patients were treated with salbutamol 2.5 mg with nebulaizer as per need. Nobody from asthma patients took LABA before hospitalization. Supporting data. There were no differences mean QTc in Group 1 (399 ± 2.7 ms) and Group 2 (398 ± 3.4 ms) at the end of hospitalization. Results. Formoterol is significantly more increased FEV1 at the end of hospitalization (78.3 ± 9.7% for Group1and 63.2 ± 7.6% for Group2: Mann Whitney test). The number of formoterol puffs in Group1 was 2.3 ± 0.4; the number of salbutamol inhalations in Group2 was 4.2 ± 0.8. Conclusions. Formoterol is safe and more effective than salbutamol in patients with severe exacerbation of asthma patients.
106. AUTOMATION OF HEPATOCYTE INCUBATIONS FOR THE PREDICTION OF IN VIVO HEPATIC CLEARANCE
Sandrine Simon1, Patrick Iaiza2, Frederic Delobel1, Tom Kissling2, Remo Hochstrasser2, Philippe Coassolo1, and Franz Schuler1
1DMPK Department, F. Hoffmann-La Roche Ltd., Basel, Switzerland, 4070, 2Discovery Technologies, F. Hoffmann-La Roche Ltd., Basel, Switzerland, 4070
Primary, fresh or cryopreserved hepatocytes are the most reliable in vitro tool for the prediction of in vivo hepatic clearance. Hepatocytes are increasing used in drug discovery in addition to or in place of hepatic microsomes as a primary in vitro assay to assess the metabolic stability of discovery molecules and candidate drugs. We evaluated the possibility to automate the incubation process of the assay. The accuracy of automatic pipetting devices with primary hepatocytes and the cell viability was determined in 96-well plates. Primary hepatocytes can be pipetted with good reproducibility (CV 4%) and excellent cell viability (>80%) using semi-automatic devices. Robot-compatible cell incubators were custom built and equipped with micro-titer-plate shakers. Cell viability and intrinsic clearance (Clint) of three probe substrates using incubation times up to three hours were determined in cells kept in suspension. Again, cell viability was excellent (>80%) and Clint of the probe substrates (Midazolam, Diclofenac, Bufuralol) were reproducible in rat hepatocytes. The intrinsic clearances of the probe substrates were two to eight fold higher than those determined in parallel incubations in cultured primary hepatocytes. The good viability and excellent reproducibility of Clint allowed the construction of a fully automated robotic incubation system for the determination of the metabolic stability of candidate drugs in primary hepatocytes.
107. IN VITRO RANDOM MUTAGENESIS SCREENING FOR ERBB2 MUTATIONS CONFERRING RESISTANCE TO THE SMALL MOLECULE KINASE INHIBITOR LAPATINIB (TYKERB®)
Sotiria Boukouvala1, Torsten Trowe2, Keith Calkins2, Richard E. Cutler Jr.2, Ryan Fong2, Roel Funke2, Steven B. Gendreau2, Yong D. Kim2, Nicole Miller2, John R. Woolfrey2, Valentina Vysotskaia2, Jing Ping Yang2, Mary E. Gerritsen2, David J. Matthews2, Peter Lamb2, and Timothy S. Heuer2
1Department of Molecular Biology and Genetics, Democritus University of Thrace, Alexandroupolis, Greece, GR-68100, 2Exelixis, Inc., South San Francisco, CA, USA, 94083-0511
Receptor tyrosine kinases (RTKs), like ErbB2, are key mediators of cell cycle regulation. Mutations in the kinase domain of ErbB2 have been detected in various tumors and the receptor is a therapeutic target for small molecule kinase inhibitors, such as the recently approved drug lapatinib (Tykerb®). However, a major drawback of cancer therapies with RTK inhibitors is the relapse of patients who develop secondary resistance to chronic treatment. Drug-resistance mutations have been reported in the clinic for several RTKs, but large-scale genotyping of tumors is impractical and cannot uncover the full spectrum of resistant variants. To identify prospective ErbB2 mutations conferring resistance to lapatinib, we used a sophisticated method1 involving random, target-specific saturation mutagenesis and in vitro screening of the mutants for drug resistance. The V659E oncogenic variant of ErbB2 cDNA was cloned into a retroviral vector and propagated inside repair-deficient bacteria. The randomly mutagenized plasmid DNA was used to generate retroviral particles that infected Ba/F3 cells, rendering them dependent of ErbB2 activity for survival and growth in the absence of IL-3. Lapatinib-resistant Ba/F3 clones were selected in soft-agar medium and DNA was isolated for sequencing of the entire intracellular domain of ErbB2. Seventeen point mutations, altering 16 amino acids, were identified repeatedly in 122 of the 125 lapatinib-resistant colonies recovered by multiple independent rounds of in vitro selection, indicating saturation of the screening. Mutations L755S and T733I have previously been reported in clinical samples, but not in the context of drug resistance. Mutations were most common in the ErbB2 kinase domain, including T798I which was found in 35 independent clones and is analogous to the gatekeeper mutation associated with clinical drug resistance of other RTKs. The resistance-conferring potential of the mutations was confirmed by site-directed mutagenesis of the original V659E ErbB2 cDNA construct and stable expression in Ba/F3 cells. Ten-point dose response cell viability assays and Western blot analyses determined highest lapatinib survival and ErbB2 phosphorylation IC50 values for the mutants isolated most frequently in the initial screens. The T798I mutation had the strongest resistance effect, with IC50 values consistent with ErbB2-independent lapatinib-induced toxicity. Computer-based structural modeling suggested that highly resistant mutations may act by direct steric interference of lapatinib binding to the inactive form of ErbB2 or by restricting conformational flexibility of the kinase. Our study describes the molecular mechanism of ErbB2 resistance to small molecule mediated kinase inhibition and provides a list of lapatinib-resistance mutations that may be therapeutically relevant.
REFERENCE
- Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 2003; 112:831–843.
108. USE OF QUANTITATIVE WHOLE BODY AUTORADIOGRAPHY TO DETERMINE THE DETAILED BRAIN DISTRIBUTION OF RADIOLABELLED TARGET MOLECULES
Stuart Cameron, A. Patterson, A. Cook, and C. Young
Pharmaceutical Metabolism, Charles River Laboratories Preclinical Services Edinburgh, Edinburgh, United Kingdom, EH33 2NE
Quantitative Whole Body Autoradiography (QWBA) is a powerful tool in determining the relative tissue distribution of radiolabelled drugs throughout the body of laboratory animals. It can also be used to determine drug distribution in localised sites and target tissues. A potential specialist use of this technique is to visualize and quantify the affinity of target molecules in selected regions of the brain. Identifying specific regions and their relative drug affinity can help elucidate their mode of action and/or understand their pharmacological effect. These effects can be influenced by the different stages of brain development. The rat is a standard laboratory animal used in toxicology and pharmacological assessment of new drugs. The brain morphology of the rat is very well understood and detailed brain atlases are available to map specific brain regions in histological investigations. Most existing brain atlases rely on standard sectioning techniques employed in histological examination. However these are not suitable for the examination and sectioning techniques employed in autoradiography. In this study, new born, immature and mature Sprague Dawley rats each received a single oral dose of carbon-14 labelled test item. The distribution of radioactivity in the whole body and in the major, distinct, brain regions was assessed by whole body autoradiography. Novel sectioning and spatial orientation techniques were developed to establish a brain map for autoradiography analysis. Up to 16 different regions of the brain could be individually identified and the levels of radioactivity in each region quantified. Applying these techniques allowed the differences in uptake of the test item to be established in 3 developmental stages of the brain (infant, juvenile and adult). The distribution of radioactivity to the detailed brain regions was also established and correlated against their known sensory functions. From this study a technique to reliably quantify the association of neurally active molecules to specific brain regions and sensory functions was developed. This methodology has potential for brain mapping other radiolabelled molecules in the brain and could help understand the complex relationship of each brain region with the pharmacological activity of new pharmaceutical candidates.
109. HIGH-THROUGHPUT CACO-2 PERMEABILITY ASSAY PLATFORM ALLOWS PREDICTION OF FRACTION ABSORBED AND MECHANISTIC TRANSPORTER STUDIES FOR ESTABLISHING IN VITRO IN VIVO CORRELATIONS
Suzanne Tilton, Xuena Lin, Xiao-Hui Chen, Bailin Zhang, and Jianling Wang
Metabolism and Pharmacokinetics/Global Drug Discovery, Novartis Institutes for Biomedial Research, Cambridge, MA, USA, 02139
It has been argued that selection of adequate ADME properties should be conducted in parallel with optimization of drug efficacy. To achieve this, highly efficient, accurate, as well as cost-effective assays are required. Permeability assays are fundamental in ADME profiling, as sufficient permeability through intestinal membranes can promote oral absorption, a highly desirable property for the majority of drug discovery programs. The Caco-2 cell-based permeability model is advantageous due to its human origin and expression of transporters prevalent in the human gastrointestinal tract. However, thus far, limited studies have described the adaptation of the Caco-2 assay from lower-throughput plate formats to 96-well filter plates with LC-MS/MS analysis. We describe a high throughput 96-well Caco-2 assay which has been correlated to experimental human fraction absorbed with nearly 100 validation standards and which has successfully measured thousands of new chemical entities (NCEs). Using validation standards reported to be actively transported, we establish an efflux “flag” for discovery compounds. Further, we present the reality of integrating permeability assessment with high throughput solubility and metabolic stability data for pharmacokinetic (PK) decision making and in vitro- in vivo correlations (IVIVC) in a discovery setting. In some instances, IVIVC may be improved with more in-depth mechanistic work, specifically efflux transporter inhibition (P-gp, MRP2, and BCRP) and dose-escalation studies. A case-study addresses how the use of the Caco-2 permeability assay and follow-up mechanistic assays made a significant impact in identifying potential development liabilities and integrating in vitro and PK results.
110. RECOMBINANT YEAST AND BACTERIA THAT EXPRESS HUMAN P450S: BIOREACTORS FOR DRUG–DISCOVERY, –DEVELOPMENT AND –BIOTECHNOLOGY
Thomas H. Friedberg1, Steven Hanlon2, Matthias Kittelmann3, Oreste Ghisalba4, and C. Roland Wolf5
1Biomedical Research Centre, University of Dundee, Dundee, United Kingdom, DD1 9SY, 2Safety and Technical Sciences, Hoffmann-LaRoche, Basel, Switzerland, CH 4070, 3Core Biotechnology Area, Novartis Pharma AG, Basel, CH-4002, 4Core Biotechnology Area, Novartis Pharma AG, Basel, Switzerland, CH-4002, 5Dept of Biomed Rsch Ctr, Univ of Dundee Ninewells Hosp & Med Sch, Dundee, Angus, United Kingdom, DD1 9SY
Background. Prediction P450-mediated metabolism in humans constitutes an essential part of drug development. A variety of recombinant expression systems have been employed, however only on a small scale, to predict P450 -mediated metabolism in man. Hypothesis: We argue that bacterial are ideally suited for semipreparative biooxidations, as they can be grown to high density and are technically not challenging. Methods. Human P450s were expressed in E. coli using published procedures. For biofermentations, E. coli were grown, if required in up to 100 l biofermenters, or employing BioWave bioreactors (Wave Biotech AG, Switzerland). Results. We demonstrated that the catalytic properties of several P450 isoforms when expressed in E. coli faithfully mimicked metabolic pathways of xenobiotics in human liver. The technical applicability of these systems for biofermentation is evidenced by the industrial applications presented here. These were conducted by two pharmaceutical companies, involving structurally diverse compounds, and utilized different bioreactor techniques. The strategies employed, resulted in highly efficient, semipreparative biooxidations of drugs and drug candidates. For example CYP3A4-mediated biooxidation of AAG561, a new drug compound in development by Novartis against anxiety and depression, revealed four metabolites in quantities of up to 13. 6 mg (1 litre culture). In another application, using optimized conditions, metabolism of amodiaquine was carried out in 5 ¥ 2L flasks each containing 80 nmol of CYP2C8. Metabolites were isolated by flash chromatography and 172 mg of desethyl-amodiaquine in >99% purity was recovered. Conclusions. The results presented demonstrate that E. coli expressing human P450s are ideally suited for semi-preparative production of drug metabolites for structural and drug safety studies, as will be increasingly required by regulators.
111. STEVIOSIDE AND SODIUM SALT OF MONOKETOCHOLIC ACID AS HYPOGLICAEMIC AGENTS
Aleksandar Raskovic1, Momir Mikov2, and Vida Jakovljevic3
1Dept of Pharmacology, Toxiclogy & Clinical Pharmacology, School of Medicine, University of Novi Sad, Republic of Serbia, Novi Sad, Serbia and Montenegro, 2School of Pharmacy, University of Otago, New Zealand, Dunedin, New Zealand, 3Dept of Pharmacology, Toxicology & Clinical Pharmacology, School of Medicine, University of Novi Sad, Republic of Serbia, Novi Sad, Serbia and Montenegro
To investigate the effect of a commercial preparation of stevioside and a synthetic compound, sodium salt of monoketocholic acid (MKC), applied per os and with the aid of osmotic pump, on glycemia in normoglycemic and diabetic Wistar rats. Hypotheses. Both substances have hypoglyceamic effect. Applied together, they have sinergistic effect. Methods. Diabetes was induced with alloxan, 100 mg/kg, i.p. Normoglycemic and diabetic rats were treated per os for five days either with saline solution (1ml/kg, control), stevioside (20 mg/kg), MKC (4 mg/kg) and combination of stevioside (20 mg/kg) and MKC (4 mg/kg). Apart from per os adminstration, stevioside and MKC were also applied with the aid of a subcutaneously implemented osmotic pump. During the treatment and after it, glycemia was measured and the rats that were treated per os were subjected to the oral glucose tolerance test (OGTT) at a dose of 1 g/kg. After that the animals were anesthesized with urethane (0.75 g/kg, i.p.) and sacrificed by cardiopunction, to determine C-peptide in the obtained serum. Results. In all three groups of normoglycemic rats the higest decrease of glucose level was observed on the fourth day of experiment. The combination stevioside + MKC showed a stronger hypoglycemic effect compared to individual treatments with stevioside and MKC (3.73:4.80:4.73 mmol/L). With the group of diabetic rats that received both substances through the osmotic pump the hypoglycemic action was also stronger compared to the individual treatments with stevioside and MKC (16.15:18.89:18.75 mmol/L). Treatment of healthy rats with both substances per os caused no statistically significant difference in glycemia, whereas with diabetic rats the combination of stevioside + MKC showed a statistically significant decrease of glycemia compared to control values. With both groups of rats, treatment with stevioside and MKC and their combination, prevented increase of glucose concentration in the OGTT. Conclusions. The application of stevioside by osmotic pump yielded a statistically significant increase in the concentration of C-peptide in serum of healthy rats. Compared to control, the concentration of C-peptide in diabetic rats was significantly higher after the treatment with either stevioside or its combination with MKC, irrespective of the mode of administration.
112. THE EFFECT OF GEMFIBROZIL ON REPAGLINIDE PHARMACOKINETICS PERSISTS FOR AT LEAST 12 H POSTDOSE
Aleksi Tornio, Mikko Niemi, Mikko Neuvonen, Jouko Laitila, Annikka Kalliokoski, Pertti J. Neuvonen, and Janne T. Backman
Department of Clinical Pharmacology, University of Helsinki, Helsinki, Finland
The antidiabetic repaglinide is metabolised by cytochrome P450 (CYP) 2C8 and 3A4. Gemfibrozil, which is a strong in vivo inhibitor of CYP2C8, has raised the area under concentration-time curve (AUC) of repaglinide 8-fold, when repaglinide was administered 1 hour after gemfibrozil (Niemi et al. 2003). The proposed mechanism of the gemfibrozil-repaglinide interaction is mechanism-based inhibition of CYP2C8 by a metabolite of gemfibrozil, gemfibrozil 1-O-β-glucuronide (Ogilvie et al., 2006). Thus, we studied the time-dependency of the gemfibrozil-repaglinide interaction. In a randomised 5-phase crossover study, 10 healthy volunteers ingested a single oral dose of 0.25 mg repaglinide either alone or 0, 3, 6 or 12 hours after the last dose of 600 mg gemfibrozil twice daily. Plasma concentrations of repaglinide, gemfibrozil and their metabolites, as well as blood glucose were measured for 9 hours postdose. When repaglinide was taken either simultaneously with, or 3, 6 or 12 hours after the last dose of the gemfibrozil treatment, gemfibrozil increased the AUC0-∞ of repaglinide 7.0-, 6.5-, 6.2-, and 5.0-fold, respectively (P < 0.001). The peak repaglinide concentration was increased about 2-fold (P < 0.001), and the half-life was prolonged from 1.2 hours to 2–3 hours (P < 0.001) during all gemfibrozil phases. The metabolite- to-repaglinide AUC ratios were decreased by gemfibrozil in all gemfibrozil phases, in particular that of the CYP2C8 specific metabolite M4. The pharmacodynamic effects of repaglinide were also increased by gemfibrozil treatment up to 12-hour dosing interval. In conclusion, gemfibrozil strongly interacts with repaglinide even when repaglinide is administered 12 hours after gemfibrozil, i.e. when the concentrations of both gemfibrozil and its 1-O-β-glucuronide have declined to about 5–10% of their peak. The main mechanism of this persisting interaction is likely the irreversible mechanism-based inhibition of CYP2C8 by gemfibrozil 1-O-β-glucuronide. A similar long-lasting interaction potential of gemfibrozil with other CYP2C8 substrate drugs can also be expected.
REFERENCE
- Niemi M, Backman JT, Neuvonen M and Neuvonen PJ (2003). Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia 46:347–51.
- Ogilvie BW, Zhang D, Li W, Rodrigues AD, Gipson AE, Holsapple J, Toren P and Parkinson A (2006). Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: implications for drug-drug interactions. Drug Metab Dispos 34:191–7.
113. THE EFFECT OF ST JOHN'S WORT ON THE PHARMACOKINETICS AND METABOLISM OF FINASTERIDE IN HEALTHY MEN
Anna Lundahl1, Mikael Hedeland2, Ulf Bondesson2, Lars Knutson3, and Hans Lennernäs1
1Department of Pharmacy, Uppsala University, Uppsala, Sweden, 751 23, 2Department of Chemistry, National Veterinary Institute (SVA), 3Department of Surgical Sciences, Uppsala University Hospital
The aim of the present study was to investigate the effect of 14 days St John's wort (SJW) (b.i.d 300 mg) pre-treatment on the pharmacokinetics and metabolism of finasteride, used in the treatment of benign prostata hyperplasia and male pattern baldness. Finasteride is mainly metabolized by CYP3A4 and two of the formed metabolites have been identified as ω-hydroxyfinasteride (M1) and finasteride-ω-oic acid (M3) (Carlin et al., 1992). The study consisted of two finasteride study days (T1 and T2) separated by a wash-out period and the SJW treatment. The study included 12 healthy male volunteers. In both treatments samples from blood, urine and bile were collected for further investigations of the metabolism and excretion of finasteride and its metabolites. Administration of finasteride (5mg) and collection of bile were done via a catheter with a multi-channel tubing system, Loc-I-Gut (Bergman et al., 2006). Finasteride, M1 and M3 were analysed by liquid chromatography-electrospray-tandem mass spectrometry (LC-ESI-MS/MS) in the Selected Reaction Monitoring (SRM) mode. The LOQ of all three analytes was 2 ng/mL in urine and 0.5 ng/mL in plasma. The LOQs of finasteride, M1 and M3 in bile were 2.5, 5.0 and 2.7 ng/mL respectively. The SJW treatment resulted in reduced finasteride plasma AUC0–24h in all treated individuals with mean ± SD of 113 ± 35 and 46 ± 5.6 (ng x h)/mL in T1 and T2 respectively (p < 0.001, n = 12). Also Cmax and t1/2 was significantly decreased after SJW treatment. The plasma AUC0–24h of M3 was unchanged after SJW treatment, but Cmax was significantly increased (p < 0.001, n = 12) and t1/2 was significantly decreased (p < 0.001, n = 12). This was probably a result of a more rapid formation and induced clearance as a consequence of induced CYP3A4 expression. M1 was not detected in the plasma, urine or bile samples. In bile, unchanged finasteride was excreted in very small amounts and less than 1% of the dose was found as M3. There was no significant difference in between the treatment groups. No unchanged finasteride was excreted into urine. Approximately 20 % of the dose was excreted as M3 both in T1 and T2. In conclusion, treatment with SJW induced finasteride metabolism and finasteride plasma AUC0–24h, Cmax, and t1/2 were reduced. The plasma pharmacokinetics of M3 was also affected by SJW treatment. Other metabolic pathways for finasteride and its metabolites can also have been affected by SJW and further investigations in humans will be conducted.
REFERENCE
- Bergman E, Forsell P, Tevell A, Persson EM, Hedeland M, Bondesson U, Knutson L and Lennernas H (2006) Biliary secretion of rosuvastatin and bile acids in humans during the absorption phase. Eur J Pharm Sci 29:205–214.
- Carlin JR, Hoglund P, Eriksson LO, Christofalo P, Gregoire SL, Taylor AM and Andersson KE (1992) Disposition and pharmacokinetics of [14C]finasteride after oral administration in humans. Drug Metab Dispos 20:148–155.
114. IN VITRO EVALUATION OF P-GLYCOPROTEIN INHIBITION USING LOPERAMIDE AS A PROBE SUBSTRATE
Gayle Corkill, David Turner, Boris Pufong, Helen Gill, and Clive Dilworth
Cyprotex, Macclesfield, United Kingdom, SK10 2DR
Drug-transporter interactions are well documented and are now a concern for the regulatory authorities with draft guidance from the FDA recommending that transporter-based drug interaction studies are conducted for any new molecular entity. P-glycoprotein (P-gp, MDR1) is the most studied of the ABC transporter family and is thus the main focus of drug-transporter interactions. A vast range of clinically relevant drugs interact with P-gp (either as substrates or as P-gp inhibitors/inducers) which can lead to altered pharmacokinetics and pharmacodynamics of co-administered drugs. Of the in vitro methods developed so far the bi-directional transporter assay is regarded by the FDA as the definitive assay for identifying P-gp substrates and inhibitors. We have developed 96-well bi-directional transporter assays using MDR1-MDCK cells to determine if an investigational compound is a substrate (not detailed here) or an inhibitor of P-gp. Two assays have been developed based on the draft FDA guidance, a single point inhibition assay and an IC50 inhibition assay. The single point inhibition assay compares the efflux ratio of the probe P-gp substrate, loperamide, in the absence and presence of a single concentration of an investigational compound. A decrease in efflux ratio in the presence of the investigational compound suggests the compound is likely to be a P-gp inhibitor. The IC50 assay, investigating percent inhibition over a concentration range, has been developed as a more definitive assay for compounds that are identified as P-gp inhibitors in the single point inhibition assay. Both assays described here use LC-MS/MS end-point and have been validated using known P-gp inhibitors.
115. INTERACTION OF ANTIDIABETIC DRUGS WITH HEPATIC OATP AND OCT UPTAKE TRANSPORTERS
Jörg König, Iouri Bachmakov, Hartmut Glaeser, and Martin F. Fromm
Institute of Experimental and Clinical Pharmacology and Toxicology, Friedrich-Alexander-University Erlangen-Nuremberg, Erlangen, Germany, 91054
Oral antidiabetic drugs have to be taken up into hepatocytes before their subsequent metabolism, their intrahepatic action or before they are transported back into the blood for extrahepatic effects. Many studies have demonstrated that this transporter-mediated uptake is an important determinant of drug disposition and that inhibition of uptake by concomitantly administered drugs is a newly recognized mechanism of drug-drug interactions. Important transporter families are the OATP family (organic anion transporting polypetides) mediating the uptake of several organic anions (e.g. bile salts and drugs like statins) and the OCT family (organic cation transporters) for the uptake of cationic drugs (e.g. metformin). The aim of our study was to test the hypothesis whether oral antidiabetic drugs can inhibit transport mediated by hepatic uptake transporters of the OATP and OCT family. Using stably transfected cells recombinantly expressing the hepatic uptake transporters OATP1B1, OATP1B3, OATP2B1 or OCT1 we analyzed, whether the antidiabetic drugs repaglinide, rosiglitazone or metformin influence the uptake of model substrates and drugs. For the analysis of OATPs we used BSP (Bromosulfophthalein) and pravastatin, for OCT1 we used MPP+ (1-Methyl-4-phenylpyridinium) and metformin. Metformin did not inhibit the uptake of the OATP- or OCT-substrates. In contrast, OATP-mediated BSP and pravastatin uptake as well as OCT1-mediated MPP+ and metformin uptake was inhibited by rosiglitazone (IC50 values 5.2 to 30.4 μM) and repaglinide (IC50 values 1.6 to 5.6 μM). These results demonstrate that inhibition of transporter-mediated uptake by oral antidiabetic drugs have to be considered as a potential mechanism underlying drug-drug interactions.
116. HOW GREATLY IS THE MAGNITUDE OF DRUG-DRUG INTERACTION INFLUENCED BY ADMINISTRATION TIME OF INHIBITOR ?: SIMULATION OF INTERACTION AFTER CO-ADMINISTRATION OF MELATONIN WITH FLUVOXAMINE BY DYNAMO-PK ANALYSIS METHOD
Katsumi Iga, Akiko Kiriyama, and Akino Honbo
Pharmaceutical Sciences, Doshisha Women's College of Liberal Arts, Kyotanabe, Japan, 610-0395
Fluvoxamine (I) is known to cause common-metabolism based drug-drug interaction with various drugs. Interaction with melatonin (S) is a typical example. The AUC of S was increased 23 fold (RAUC = 23) in the administration of a single dose of I (iD = 50mg), 3 hr prior to S-administration (sD = 5mg). By the simulation, we have demonstrated that RAUC was due largely to the hepatic extraction ratio (Eh) of S, as well as the inhibition constant (Ki) of I. In the present study, we analyzed how administration-time difference between I and S (DT) (ex. I-administration 3 hr before S-administration, DT = −3 hr) influenced the RAUC, by a similar simulation method, where two-compartment model with time-dependent parameters and tanks-in-series hepatic extraction model (number of tanks, 10) were assumed. In either single or multiple administration of I, (i) DT decreased RAUC (at DT = 0, maximal RAUC), (ii) administration of I later than S-administration dramatically decreased RAUC. The decrease was dependent on the first-order absorption rate constants of S (sKa) and I (iKa) and time interval in the multiple administration of I (tau). Larger sKa, smaller iKa and longer tau gave smaller RAUC. This information might be useful to determine the administration timing in the DDI study.
117. THE EFFECTS OF MODEL INHIBITORS, ANTI-INFLAMMATORY ANALGESICS AND FEMALE SEX STEROIDS ON CYP1A2 ACTIVITY IN VITRO AND PREDICTIONS OF IN VIVO INTERACTIONS
Marjo J. Karjalainen, Pertti J. Neuvonen, and Janne T. Backman
Department of Clinical Pharmacology, University of Helsinki and Helsinki University Central Hospital, Helsinki (HUS), Finland, 00029
Background. The cytochrome P450 enzyme CYP1A2 is crucial for the metabolism of many drugs, e.g. tizanidine. The purpose of the study was to investigate the effects of several non-steroidal anti-inflammatory drugs (NSAID) and female sex steroids on CYP1A2 activity in vitro, and to compare these effects with those of model inhibitors. Methods. To study the effects of the compounds on CYP1A2 activity, 10 μM phenacetin was incubated with human liver microsomes (0.1 mg/ml of protein) for 30 min. The time-dependency of the inhibitory effect was studied by preincubating the inhibitor with microsomes for 30 min, followed by a coincubation with phenacetin (final concentration 20 μM) for 20 min. The in vitro study was followed by prediction of the interaction of each inhibitor with tizanidine in vivo based on a simple competitive inhibition model. Results. In vitro, fluvoxamine, tolfenamic acid and mefenamic acid inhibited CYP1A2 activity with mean IC50 (Ki) values of 0.029 (0.011) μM, 1.5 (1.4) μM and 3.4 (3.5) μM, respectively. The IC50 (Ki) values of ethinyl estradiol, rofecoxib, celecoxib, desogestrel and zolmitriptan were somewhat higher, i.e. 24 (11) μM, 8.7 μM, 34 (25) μM, 55 μM and 122 (60) μM, respectively. The IC50 (Ki) values of ciprofloxacin, etoricoxib and etodolac were 220 (145) μM, 240 μM and 240 μM, respectively. At 100 μM, the other tested NSAIDs and steroids inhibited CYP1A2 less than 35%. Preincubation increased the inhibitory effects of rofecoxib, progesterone and desogestrel. Using the free portal plasma inhibitor concentration as [I]in vivo, the effect of fluvoxamine and the lack of effects of tolfenamic acid and celecoxib on tizanidine pharmacokinetics in humans were well predicted. However, the effects of ciprofloxacin, rofecoxib and oral contraceptives were greatly underestimated even when using their total portal plasma concentration. Among the drugs lacking in vivo interaction data, mefenamic acid was predicted to increase the AUC of tizanidine by 35%, when the free portal plasma concentration was used in the predictions. The other tested drugs were predicted not to have any significant in vivo effect on CYP1A2. Conclusions. Besides rofecoxib, and possibly mefenamic acid, the other NSAIDs are not expected to significantly inhibit CYP1A2 in humans. Moreover, when based on the free portal plasma concentration of the inhibitor, the in vitro results can predict well some of the interactions with tizanidine. However, the competitive inhibition model markedly underestimates the effects of ciprofloxacin, rofecoxib and oral contraceptives on the AUC of tizanidine in vivo. The type of enzyme inhibition, free inhibitor concentration and accumulation of the inhibitor into the hepatocytes should be considered in extrapolations of in vitro results to humans. Further studies are warranted to clarify the mechanisms of CYP1A2 inhibition caused by ciprofloxacin and oral contraceptives in humans.
118. THE INFLUENCE OF PRE-TREATMENT WITH PROBIOTICS ON THE IN VITRO ILEAL PERMEATION OF THE ANTIDIABETIC DRUG GLICLAZIDE, IN HEALTHY AND DIABETIC RATS
Hani Al-Salami1, Grant Butt2, Ian G. Tucker1, and Momir Mikov1
1School of Pharmacy, University of Otago, Dunedin, New Zealand, 9054, 2Department of Physiology, University of Otago, Dunedin, New Zealand, 9054
Background. Probiotics have been shown to influence gut motility and be beneficial in a wide range of conditions including infections, allergies, and metabolic disorders such as diabetes. The pathogenesis of T1D remains unclear but the generally accepted explanation is that it is a chronic inflammatory disease triggered in genetically susceptible individuals by a primary insult initiated in the gut. Gliclazide is a second generation sulphonylurea with extrapancreatic effects such as enhancing glucose uptake by muscles. Hypothesis: Pre-treatment with probiotics for three days changes the permeation of the antidiabetic drug gliclazide through the ileal mucosa of healthy and diabetic rats. Methods. Eight male Wistar rats (age 2–3 months, weight 350 ± 50g) were randomly allocated into a healthy and a diabetic group (4 rats per group and 8 chambers per rat i.e. n = 32), and administered probiotics (75 mg/Kg) twice a day for three days. Rats with blood glucose > 20 mmol/L, serum insulin < 0.04 μg/L, 2 to 3 days after alloxan injection (30 mg/Kg i.v.) were considered diabetic. Probiotic treatment started a day after rats became diabetics. Rats were sacrificed then a midline longitudinal incision was made and the distal ileum (10 cm) was immediately removed, flushed free of luminal contents then mounted in Ussing chambers. Gliclazide (200μg/ml) was administered for the measurement of the mucosal to serosal absorption Jss(MtoS) and serosal to mucosal secretion Jss(StoM). The study was approved by the Otago University Animal Ethics Committee. Results. Treatment of healthy rats with probiotics reduced Jss(MtoS) of gliclazide 6-fold (p < 0.01) and increased Jss(StoM) 2-fold (p < 0.01) resulting in net secretion while, in diabetic tissues, treatment with probiotics increased both Jss(MtoS) and Jss (StoM) fluxes of gliclazide to the comparable levels of healthy tissues and thus resulted in net absorption. Discussion. In healthy rats, the reduction in Jss(MtoS) after probiotic administration can be explained by the production of bacterial metabolites that upregulate the mucosal efflux drug transporters which controls gliclazide transport. In diabetic rats, the restored fluxes of gliclazide after probiotics treatment, suggests the normalization of the functionality of the drug transporters responsible for gliclazide transport. Conclusion. Probiotics may be beneficial as adjunct therapy in treating diabetes.
119. LACK OF SIGNIFICANT PHARMACOKINETIC INTERACTIONS BETWEEN THE ABCB1 SUBSTRATES SIROLIMUS AND EZETIMIBE IN HEALTHY SUBJECTS
Stefan Oswald, Luisa Borgwardt, Thomas Giessmann, Christiane Modess, and Werner Siegmund
Department of Clinical Pharmacology, University of Greifswald, Greifswald, Germany
Background. Hypercholesterolemia is a frequent finding in organ transplant recipients receiving immunosuppressive drugs such as sirolimus (SIR). To prevent increased cardiovascular morbidity and mortality in these patients, co-medication with lipid-lowering statins is recommended. However, treatment with statins is limited in many patients by insufficient cholesterol-lowering efficacy, drug interactions and serious adverse drug reactions (e.g. rhabdomyolysis). These patients may benefit from comedication with the cholesterol absorption inhibitor ezetimibe (EZE). Since SIR and EZE were shown to be substrates of the efflux transporter ABCB1 (P-glycoprotein), drug interactions between both compounds may occur. Therefore, we initiated a clinical study in healthy subjects to evaluate the clinical relevance of drug/drug interactions between SIR and EZE. Methods. Disposition of SIR (5 mg, po) and EZE (10 mg, po) alone and in combination was studied in a randomized, three-period, cross-over study in 24 healthy subjects (6 females, 18 males, age 21–34, body mass index 19.7–29.1) with at least 14 days wash-out between the study periods. Blood and serum, respectively, were sampled for 144 h, urine and feces for 5 and 10 days, respectively. EZE, EZE glucuronide and SIR were quantified using validated LC-MS/MS methods (limits of quantification: SIR 1 ng/ml in whole blood; EZE 0.05 ng/ml, 1 ng/ml and 10 ng/ml in serum, urine and feces, respectively). Results. EZE co-medication did not significantly affect Cmax (16.1 ± 5.29 ng/ml vs. 14.6 ± 4.23 ng/ml, p = 0.181), AUC (182 ± 98.2 ng×h/ml vs. 174 ± 111 ng×h/ml, p = 0.732) and half-life (142 ± 128 h vs. 172 ± 235, p = 0.884) of SIR. On the other hand, SIR tended to increase Cmax and AUC values of EZE (4.20 ± 2.98 ng/ml vs. 3.05 ± 1.90 ng/ml, p = 0.008; 73.9 ± 40.9 ng × h/ml vs. 68.3 ± 40.9 ng × h/ml, p = 0.073) which in turn resulted in a significantly increased urinary excretion of EZE glucuronide (0.79 ± 0.32 mg vs. 0.67 ± 0.24 mg, p = 0.004). Fecal excretion of EZE tended to be decreased (6.78 ± 2.43 mg vs. 7.46 ± 2.80 mg, p = 0.209). Conclusions. Cholesterol-lowering therapy with EZE is not expected to alter disposition of SIR significantly. The small influence of SIR on disposition of EZE as obviously caused be modulation of intestinal ABCB1 seems not to be of clinical relevance.
120. THE EFFECT AND CORRELATION ASPIRIN AND TICLOPIDINE ADMINISTERED ALONE AND IN COMBINATION ON BLEEDING TIME AND ACTIVITY OF CREATINE KINASE IN RATS
Zorica Stanojevic1, Radoslav Mitic1, Zoran Bukumiric1, Vojkan Nestorovic2, Snezana Stevic1, and Boban Bisevac2
1Institute of Pharmacology and Toxicology, Medical Faculty Pristina, Kosovska Mitrovica, Serbia and Montenegro, 2Institute of Physiology, Medical Faculty Pristina, Kosovska Mitrovica, Serbia and Montenegro
The combination of aspirin and ticlopidine reduce the incidence of haemorrhagic and vascular complications after intracoronary stenting. The purpose of this study was to evaluate the effects of aspirin and ticlopidine alone and in combination on bleeding time and serum activity of creatin kinase (CK) and correlation between this two variables in rats. The experiment was conducted on white laboratory rats, type Wistar. Thirty-two rats were divided in four groups and they received one of the following treatments for three days: group I - control, saline (1 ml/kg, i.p.); group II - aspirin (50 mg/kg/day i.p.); group III - ticlopidine (125 mg/kg/day i.p.) and group IV - aspirin+ticlopidine combination (50 mg/kg/day+125 mg/kg/day i.p.). Bleeding time and CK activity were determined at once after taking the sample of blood. Relationship between two measured variables was determined by calculating linear correlation coefficient (r). Bleeding time was significantly prolonged in aspirin, ticlopidine and aspirin+ticlopidine groups compared to control (p < 0,001). Also, bleeding time was significantly prolonged in group treated with aspirin+ticlopidine combination compered to aspirin (p < 0,001) and ticlopidine alone (p < 0,001). Serum CK activity was significantly decreased in aspirin, ticlopidine and aspirin+ticlopidine groups compared to control (p < 0.001). Between bleeding time and CK activity in control group of rats was noticed negative and middle correlation (r = −0,48); in group treated with aspirin negative and middle correlation (r = −,58); in group treated with ticlopidine positive and low correlation (r = 0,05); and in group treated with aspirin+ticlopidine combination negative and low correlation (r = −0,21). Based on obtained results it can be observed that prolonged bleeding time and decreased CK activity were more noticeable in group treated with aspirin+ticlopidine combination compared with groups treated with aspirin and ticlopidine alone. Also the negative correlation between bleeding time and CK activity in control, aspirin and aspirin+ticlopidine groups becomes positive only after the treatment with ticlopidine alone.
121. UTILITY OF CONTINUOUS INTRAPORTAL VEIN INFUSION FOR INDUCTION-MEDIATED DRUG INTERACTION STUDIES IN NONHUMAN PRIMATES
Amy Zhou, Pam McLawhon, Shirley Feng, Khanh Nguyen, Wendy Klopf, Michelle Lam, Melinda Runyan, Yeping Zhao, Dan Lu, Micaela B. Reddy, Werner Rubas, Mario Monshouwer, Jennifer L. Fretland, Philip D. Worboys, Danlin Wu, and Adrian J. Fretland
Department of Drug Metabolism and Pharmacokinetics, Roche - Palo Alto, Palo Alto, CA, USA, 94304
Induction of drug metabolism results in clinically significant drug-drug interactions. Considerable challenges in translating human in vitro data to risk for induction in vivo exist. Profound species differences in the receptors responsible for the induction of drug metabolism, particularly between rodents and humans, further complicate the translation of preclinical assessment to a clinical outcome. Other complications include species differences in gut flora, gut metabolism, and clearance routes. Due to the similarity in receptors and mechanisms of induction, use of intraportal vein (ipv) chronically cannulated nonhuman primates may provide a valuable preclinical model to assess the risk of induction in humans. Intraportal vein injection bypasses several confounding factors between humans and nonhuman primates, including differences in gut flora, gut wall metabolism, and absorption. To test the hypothesis whether the cynomolgus monkey ipv constant infusion model is appropriate to assess the risk of induction in human, a drug interaction study was conducted during a continuous infusion of model compounds. Cynomolgus monkeys were administered the model CYP3A substrate midazolam as a constant ipv infusion for 24 hours to establish steady-state plasma levels. After the initial infusion of midazolam, the classic CYP3A inducer rifampin was coadministered by ipv infusion for a period of 5 days. Sampling of plasma was performed at 24 hour intervals after the initiation of the rifampin infusion, and the total plasma concentrations of rifampin, midazolam, 1′-OH-midazolam, and 4-OH-midazolam were quantified by LC-MS/MS. After initiation of rifampin coadministration, there was a 1.7- and 3.6-fold reduction in midazolam plasma concentration after 24 and 48 hours, respectively. After 48 hours, there was no further reduction in the midazolam plasma levels. Interestingly, no corresponding increase in the amount of either 1′-OH-, or 4-OH-midazolam in plasma was observed. The total steady-state plasma levels of rifampin from the in vivo study, approximately 6 μM, were similar to the EC50 for induction of CYP3A8 mRNA in in vitro primary cynomolgus monkey hepatocyte incubations, 0.85 μM. These promising data suggest the cynomolgus monkey ipv infusion model is a valuable model for the in vivo assessment of induction in the drug development process.
122. COMPARISON OF CACO-2/TC7 CELL LINE (W/O INDUCTION) AND DIFFERENT HUMAN INTESTINAL SECTIONS
Arnaud M. Bruyere1, Francois Bouzom1, Xavier Decleves2, and Bernard Walther3
1Non Clinical Modelling, Technologie Servier, Orleans, France, 45000, 2Laboratory of pharmacokinetic, University Paris Descartes, INSERM U705, CNRS UMR 7157, Paris, France, 75006, 3Direction, Technologie Servier, Orleans, France, 45000
Purpose. To date, integration of both absorption and metabolism data with physiological models is not overcome. Indeed, caco-2 cell lines are commonly used to predict intestinal absorption of drugs. However, the functional expression of ATP-binding cassette (ABC) transporters and drug-metabolizing enzymes is not always correlated between caco-2 cells and human intestine. The TC7 Caco-2 clone is known to express the cytochrome P450 3A4 (CYP3A4) but the amount of CYP3A4 in TC7 cells is lower than in the intestine. The aim of our study was to select among different well-known inducers those allowing to reach similar CYP3A4, MDR1 and BCRP expression levels than that observed in human small intestine to integrate data in PBPK models. Materials and methods. TC7 cells were exposed for 48 hours to retinoic acid (a RXR agonist) and/or 1,25 OH-vitamin D3 (a VDR agonist) and CITCO (a CAR agonist) at three different times after sowing (day 7, 14 and 21). CYP3A4, MDR1 and BCRP gene expression in TC7 cells was evaluated by quantitative real time RT-PCR using the ΔΔCT method. Gene expression was also evaluated in intestinal healthy samples obtained from biopsies of six patients suffering intestinal diseases (2 duodenum, 3 ileum and 1 colon) in order to compare CYP3A4, MDR1 and BCRP expression to that observed in TC7 cells. Results. Gene expression analysis from intestinal biopsies revealed that CYP3A4 and BCRP were mainly expressed in duodenum and ileum whereas MDR1 was slightly detected in duodenum but mainly expressed in ileum and colon, its expression increasing from ileum to colon. Gene expression analysis of TC7 cells showed that 1,25 OH-vitamin D3 and CITCO did not modify any expression of the studied genes. In contrast, retinoic acid at day 7 of TC7 cell cultures increased by 5-fold the CYP3A4 mRNA levels, reaching those observed in the two different duodenum samples. This induction was stable until D14. Retinoic acid also induced by 4-fold the MDR1 gene expression in TC7 cells reaching levels close to those measured in the three different ileum samples but this induction was not stable from D14. Finally, retinoic acid did not induce BCRP gene expression in TC7 cells and mRNA levels were close to those observed in colon samples. Conclusion. These preliminary results evidenced that induction of CYP3A4 and MDR1 gene expression in caco-2 TC7 cells by retinoic acid may be an interesting alternative method making the caco-2 model more suitable for in vitro absorption and drug-drug interaction studies. CYP3A4 mRNA levels in induced TC7 cells are close to those in human duodenum and the induction of CYP3A4 went on day 7 to day 14, suggesting a possible impact on activity. Functional expression of these proteins is under investigation.
123. VALIDATION OF A SEMI-AUTOMATED METHOD FOR CYTOCHROME P450 INDUCTION ASSESSMENT IN CULTURED HUMAN HEPATOCYTES
Franca Pugnaghi, Craig Thomas, William Herron, Jonathan Jimenez-Novoa, and Lindsey Rollo
Dmpk, Aptuit, Edinburgh, United Kingdom, EH14 4AP
Hepatic cytochrome P450 enzymes are known to have a major role in the metabolism of new chemical entities (NCE). During their development it is important to estimate whether the compound will cause drug-drug interaction by being an inhibitor or an inducer of cytochrome P450. The inhibition studies are performed using liver microsomes or cDNA-expressed P450 enzymes. The industry standard to predict clinically significant induction of the cytochrome P450s is to determine the change in catalytic activity of the P450s in cultured primary human hepatocytes with and without exposure to potential inducers. To meet this need, 6 semi-automated assays for human marker substrate activities have been validated using an HPLC-MS/MS approach. The assays that were validated are: phenacetin O-deethylase (CYP1A2), coumarin 7-hydroxylase (CYP2A6), bupropion hydroxylase (CYP2B6), tolbutamide 4-hydroxylase (CYP2C9), (S)-mephenytoin 4-hydroxylase (CYP2C19) and testosterone 6β-hydroxylase (CYP3A4). The following inducers omeprazole (CYP1A2), dexamethasone (CYP2A6), phenobarbital (CYP2B6), rifampicin (CYP2C9, CYP2C19 and CYP3A4) were tested. Following 72 hr of exposure to the positive inducers, the hepatocytes were incubated in the presence of the specific cytochrome P450 probes for up to 4 hr. A change of the catalytic activity of the cytochrome P450 enzymes were measured as an increase of the production of acetaminophen, 7-hydroxycoumarion, hydroxybupropion, 4-hydroxytolbutamide, 4-hydroxymephenytoin and 6β-hydroxytestosterone. Variability of the induction was also tested using different donors of hepatocytes. Overall, the results demonstrate the utility of human hepatocytes for assessing the effects of NCEs on P450 enzymes.
124. EFFECT OF CRYOPRESERVATION ON INDUCTION OF DRUG METABOLIZING ENZYMES IN PRIMARY HUMAN HEPATOCYTE CULTURES
Adrian Little1, Stephen Ferguson2, Christie Baucam1, Jason Wright2, Cornelia Smith1, Shiloh Barfield1, Jasminder Sahi1, and Edward LeCluyse1
1CellzDirect, Inc., Durham, NC, USA, 27703, 2CellzDirect, Austin, TX, USA, 78754
Induction of the major cytochrome P450 (CYP450) enzymes can be clinically significant with regards to pharmacokinetics and efficacy of compounds that are auto-inducers of CYP450s. The FDA Draft Guidance for drug interactions (2006) accepts the use of fresh and/or cryopreserved human hepatocytes for in vitro studies as a predictor of drug-drug interactions. Cryopreserved hepatocytes that attach and form monolayers in culture are a valuable tool, as these are not dependant upon the vagaries of availability of fresh tissues. Earlier studies have demonstrated that drug metabolizing enzyme activity and expression are comparable in fresh vs. cryopreserved hepatocyte preparations from the same donor. However, there are limited reports on these same comparisons for CYP450 enzyme induction. For the current studies, hepatocytes isolated from transplant-reject livers were placed in primary sandwich culture and a portion of hepatocytes from the same donor preparation were cryopreserved. Induction studies with frozen cells were conducted after at least one month of cryopreservation. Cultured hepatocytes were treated with the prototypical inducers 3-methylcholanthrene (CYP1A inducer), phenobarbital (CYP2B inducer) and rifampicin (CYP3A inducer) for 3 days. Media was washed out and the probe substrates phenacetin, bupropion and testosterone (for CYP1A1/2, CYP2B6 and CYP3A4, respectively) added to the cultures and the hydroxylation products analyzed by LC-MS. In three separate hepatocyte preparations, CYP1A1/2 activity was induced 49.6 -, 42- and 43 – fold over vehicle control in fresh hepatocytes and 48.9 – 37 and 49 – fold in hepatocytes from the same liver after cryopreservation. Two studies comparing CYP3A4 activity revealed 4.9 – and 25.4 – fold increases over vehicle control after rifampicin treatment in the fresh cells and 6.4 – and 16.7- fold induction respectively, in the cryopreserved hepatocytes. Similar results were obtained with CYP2B6. These studies demonstrate that cryopreservation does not affect greatly the inducibility of human hepatocytes in culture and promotes the utility of cryopreserved hepatocytes in pre-clinical induction evaluations.
125. STRUCTURE-ACTIVITY RELATIONSHIPS FOR INDUCERS OF HUMAN CYP1A2 AND CYP3A4
Armin Kern and Michael Gerisch
BSP GDD-GED-DMPK Drug Metabolism, Bayer HealthCare AG, Wuppertal, Germany, D-42096
Many enzymes involved in drug metabolism are able to increase in amount and activity in response to xenobiotics. CYP induction and therefore enhanced metabolic clearance of the administrated drug may result in unwanted decrease or total loss of effectiveness of the drug itself and/or co-medication. To characterize CYP induction potential of a NCE the increase of CYP activity above baseline activity - indicating de novo synthesis of enzyme – was determined in a concentration-response curve. The critical concentration (NOEL) for induction was defined as the concentration at which the enzyme activity exceeded the baseline activity. Using a validated, clinically confirmed in vitro model of human hepatocytes in sandwich culture numerous structurally diverse inducers of CYP1A2 and CYP3A4 have been identified. A large set of induction data, generated during lead optimization support in different research projects was used to evaluate structure-activity relationships for induction of human CYP1A2 and CYP3A4.
126. COMPARISON OF DRUG DISPOSITION GENE EXPRESSION BETWEEN FA2N-4 IMMORTALIZED CELLS AND HUMAN HEPATOCYTES: EVALUATION OF FA2N-4 AS AN IN VITRO MODEL FOR CYTOCHROME P450 INDUCTION
Niresh Hariparsad1, Brain A. Carr1, Raymond Evers2, and Xiaoyan Chu2
1Drug Metabolism and Pharmacokinetics, Merck and Co., West Point, PA, USA, 19486, 2Drug Metabolism and Pharmacokinetics, Merck & Co., Rahway, NJ, USA, 07065
Primary or cryopreserved human hepatocytes are considered as the standard in vitro models to evaluate the propensity of drug candidates to cause induction of cytochrome P450 (CYP) and other drug metabolizing enzymes and transporters. The limited availability and interindividual variability observed with these models have led to the search for alternative systems. Fa2N-4 cells have been proposed as a tool to identify CYP3A4 inducers. To evaluate whether Fa2N-4 cells are a reliable surrogate for cryopreserved human hepatocytes we assessed the basal mRNA expression of 64 drug-disposition genes in Fa2N-4 cells. Significant differences were found in the mRNA levels of major drug metabolizing enzymes, nuclear receptors and transporters between Fa2N-4 cells and four batches of cryopreserved human hepatocytes. Importantly, the expression of constitutive androstane receptor (CAR) and several hepatic uptake transporters were significantly lower (>100-fold) in Fa2N-4 cells, while the expression of pregnane-X-receptor (PXR) was similar between Fa2N-4 cells and human hepatocytes. With nine selected compounds, which are known inducers of CYP3A4 either via activation of PXR, CAR, or both, we evaluated in vitro CYP3A4 induction using Fa2N-4 cells and cryopreserved human hepatocytes. No induction of CYP2B6 and -3A4 was observed in Fa2N-4 cells treated with selective CAR-agonists, which was in agreement with low expression of CAR in these cells. In addition, the CYP3A4 induction potency observed in Fa2N-4 cells was 2–10-fold lower than that observed in human cryopreserved hepatocytes. This includes compounds such as rifampicin and bosentan which are known substrates of OATP1B1. These findings highlight the limitations of Fa2N4-cells as a predictive induction model.
127. THE EFFECT OF CULTURE MEDIUM COMPOSITION ON CYTOCHROME P450 INDUCTION IN HUMAN HEPATOCYTES
S.T. Kingston, G. Crees, S. Peak, R. Graves, P. White, and J. Trafford
UK Human Tissue Bank, DeMontfort University, Leicester, United Kingdom, LE1 5XY
We recently participated in a collaborative study comparing cytochrome p450 induction using a standardised protocol (Richert et al. 2004) and recommended the use of HMM as a culture medium suitable for use in induction studies to a number of clients. However, over the past 12 months we have noticed considerable variation in the fold inductions routinely obtained with rifampicin in hepatocytes distributed to different clients. Consequently, we have compared a number of routinely used formulations for induction media on the same batches of hepatocytes. The results of these studies indicate that HMM medium maintains higher basal expression of Cytochrome p450 3A4 activity when compared to other Williams E based formulations. This in turn leads to a reduction in the fold induction levels observed with rifampicin. In contrast there appears to be no maintenance of 1A activity (as determined by EROD). On the basis of these results we would recommend Williams E medium be used during the course of induction experiments and that users of HMM medium should be aware that lower fold induction levels may be a response of the cells to the medium rather than an indicator of a poor response of the cells.
REFERENCE
- Richert L, Alexandre E, Lloyd T, Orr S, Viollon-Abadie C, Patel R, Kingston S, Berry D, Dennison A, Heyd B, Mantion G, Jaeck D. Liver Int. 2004 Aug;24(4):371–8.
128. HEPARG CELLS AS AN IN VITRO MODEL FOR CYTOCHROME P450 INDUCTION AND DRUG METABOLISM
Kajsa P. Kanebratt and Tommy B. Andersson
Development DMPK & Bioanalysis, AstraZeneca R&D Mölndal, Mölndal, Sweden, 43183
HepaRG is a highly differentiated cell line, which displays several hepatocyte-like functions including drug metabolising enzymes. The HepaRG cells differentiate into a hepatocyte-like morphology by treatment with dimethyl sulfoxide (DMSO). In the present study the stability over time in culture of the HepaRG cells was investigated together with drug metabolising and cytochrome P450 (P450) induction properties of the cell line. Differentiated HepaRG cells exhibits only minor changes in mRNA expression for drug metabolising enzymes, transporters, nuclear receptors, and liver specific factors over a period of 6 weeks in culture. A tendency towards lower expression of some genes could be a sign that the cell function starts to deteriorate at this time point. The metabolism of midazolam, naloxone, and clozapine in HepaRG cells was similar to human hepatocytes, indicating the function of CYP3A4, CYP1A2, and UGT enzymes. However, the metabolism of 7-ethoxycoumarin and dextromethorphan was low, confirming low levels of CYP2E1 and CYP2D6 in HepaRG cells. Exposure of HepaRG cells to prototypical inducers resulted in induction of CYP1A1, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP3A4 mRNA as well as phenacetin O-dealkylase, bupropion hydroxylase, diclofenac 4′-hydroxylase, and midazolam 1′-hydroxylase activities. The observed induction is consistent with the previously reported expression of the nuclear receptors PXR, CAR and AhR, which are necessary for a CYP induction response. To avoid problems with toxicity and solubility, the induction potency of test compounds was evaluated by calculating the concentrations leading to a 2-fold increase of baseline mRNA or enzyme activity levels (F2 values), instead of EC50 values from full dose-response curves. For CYP3A4 mRNA, the obtained F2 values were related to the in vivo exposure (AUC) of the inducer (AUC/F2). This score was then correlated with the decrease in AUC for a CYP3A probe drug, administered before and after treatment with the inducing agent. By using this method an excellent correlation (R2 = 0.863) was obtained, which implies that the degree of CYP3A induction in vivo can be predicted from CYP3A4 mRNA induction in HepaRG cells. The present study shows that the HepaRG cells is a valuable model to be used for drug metabolism studies as well as prediction of induction of drug metabolising P450 enzymes in vivo in humans.
129. EFFECTS OF CHEMICAL INHIBITORS OF CYP2E1 ON HUMAN CYTOCHROME P450 ENZYMES
Anne Matthews, Daniel S. Paul, and Michael Hall
Dept of Drug Metab, Huntingdon Life Sci Ltd., Cambridgeshire, United Kingdom, PE28 4HS
Cytochrome P450 (CYP) 2E1 catalyses the oxidation of several xenobiotics, including drugs such as chlorzoxazone and halogenated anaesthetics, and ethanol and other small organic molecules. Selective chemical inhibitors of CYP are used in vitro in the identification of individual enzymes involved in a compound's metabolism, as well as positive controls in CYP inhibition studies. The September 2006 FDA Draft Guidance on Drug Interaction Studies lists several chemical inhibitors that are “acceptable” for use with CYP2E1, but none that are “preferred”. This is partly due to a lack of inhibitory selectivity. The purpose of the present investigation was to evaluate the selectivity and relative potency of CYP2E1 inhibition of five compounds, namely diethyldithiocarbamate, 4-methylpyrazole, chlormethiazole, pyridine and trans-1,2-dichloroethylene. These compounds were incubated, at concentrations of 3, 30 and 300 μM, with human liver microsomes, co-factor and substrates selective for CYP1A2, 2A6, 2D6 (with the exception of trans-1,2-dichloroethylene), 2E1, and 4A11. Results from the incubations with all the above CYPs showed that diethyldithiocarbamate (300 μM) (an FDA “acceptable” inhibitor) was the least selective of the inhibitors investigated, as it inhibited CYP2A6 by 90% and CYP2D6 by 98% after a 30-minute pre-incubation period, compared with 70% inhibition of CYP2E1. Following evaluation of the data from all of the assays, it was found that the most potent and selective inhibitor of CYP2E1 was 4-methylpyrazole, with 72% inhibition after no pre-incubation period and 66% following a 30-minute pre-incubation and the least, trans-1,2-dichloroethylene.
130. THE PROTON PUMP INHIBITORS (PPIS) OMEPRAZOLE AND RABEPRAZOLE BUT NOT LANSOPRAZOLE AND PANTOPRAZOLE ARE IN VITRO TIME-DEPEND¬ENT INHIBITORS OF CYP2C19
Brandy Paris, Phyllis Yerino, Brian Ogilvie, and Andrew Parkinson
XenoTech, LLC, Lenexa, KS, USA, 66219
To assess their ability to cause metabolism-dependent inhibition of cytochrome P450 (CYP) enzymes, drug candidates are incubated for zero- and 30 min with NADPH-fortified human liver microsomes (HLM), followed by a short incubation (e.g., 5 min) with a CYP-selective marker substrate. In the present study, we evaluated the effect of protein concentration and substrate incubation time on the ability of a series of PPIs to function as inhibitors of CYP2C19, based on measurements of the 4′-hydroxylation of S-mephenytoin (40 μM, ≈ Km). This activity is commonly assessed by incubating 1.0 mg/mL HLM with S-mephenytoin for 30 min. Under these “high microsomal protein−long incubation time” conditions, we detected no metabolism-dependent inhibition of CYP2C19 by omeprazole, lansoprazole, pantoprazole or rabeprazole. In other words, incubating these PPIs with HLM for 30 min did not substantially enhance their apparent ability to inhibit CYP2C19. In contrast, when the concentration of HLM was decreased from 1.0 to 0.1 mg/mL and the incubation time with S-mephenytoin was shortened from 30 to 5 min, we identified omeprazole and rabeprazole, but not lansoprazole and pantoprazole, as time-dependent inhibitors of CYP2C19 (i.e., under “low microsomal protein−short incubation time” conditions, the inhibitory effect of omeprazole and rabeprazole on CYP2C19 increased significantly following a 30-min incubation with NADPH-fortified HLM). Further studies with omeprazole established that the increase in CYP2C19 inhibition was dependent on NADPH, suggesting the inhibition was metabolism-dependent and not simply time-dependent. Overnight dialysis did not reverse the metabolism-dependent inhibition of CYP2C19 by omeprazole, suggesting the enhanced inhibition was not due to formation of a metabolite that functioned as a more potent direct-acting inhibitor than the parent compound, but resulted from formation of an irreversible or quasi-irreversible complex between a metabolite and CYP2C19. From a study of the effects of omeprazole concentration on the time-course of CYP2C19 inhibition (based on an initial incubation of 5 to 60 μM omeprazole and 2.5 mg/mL HLM followed by a 25-fold dilution and subsequent incubation with 400 μM S-mephenytoin), we estimated KI and Kinact to be 9.1 μM and 0.046 min−1, respectively. A similar experiment conducted with 0.1 mg/mL HLM with no dilution provided similar estimates of these kinetic constants (KI = 1.6 μM; Kinact = 0.041 min−1). Like the racemic mixture, the individual enantiomers of omeprazole (R- and S-omeprazole [esomeprazole]) were both identified as metabolism-dependent inhibitors of CYP2C19, as was omeprazole sulfone. In contrast, omeprazole sulfide was not a metabolism-dependent inhibitor of CYP2C19. The results of this study demonstrate how in vitro incubation conditions impact the ability to detect metabolism-dependent inhibition of CYP enzymes. Furthermore, this study identified omeprazole and rabeprazole, but not lansoprazole and pantoprazole, as time-dependent inhibitors of CYP2C19.
Abstract withdrawn
132. COCKTAIL INCUBATION AND FAST LC/MSMS FOR CYP INHIBITION SCREENING
Marcella Martignoni, Lisa Vanoli, and Daniele Pezzetta
Pcd, Nerviano Medical Sciences, Nerviano (Milan), Italy, 20014
Midazolam and testosterone are CYP3A4 substrates commonly used for in vitro investigations into drug metabolism and drug-drug interactions. In order to assess their potential interaction at the active site of CYP3A4, they were incubated, either individually or in combination, with a recombinant CYP3A4 expression system. The formation of the main metabolites of midazolam (1OH- and 4OH-Midazolam) and testosterone (6bOH– 16bOH–, 2aOH and 2bOH-Testosterone) was followed and the inhibitory effect of nineteen specific enzyme inhibitors was investigated (erytromicin, digitoxin, ketoconazole, itraconazole, astemizol, dextromethorphan, ibuprofen, quinidine, clotrimazole, verapamil, budesonide, terfenadine, clozapine, diltiazem, progesterone, tamoxifen, paclitaxel, cyclosporine and nifedipine). The IC50 values were similar after both individual and combined incubations. In addition, when midazolam and testosterone were incubated together, the rank order of IC50 values based on the formation of their related metabolites was the same after individual incubations, demonstrating that the inhibitory potency was metabolite independent. Similarly when midazolam and testosterone were incubated individually, the formation of their respective metabolites was inhibited at the same extent, except for progesterone, which inhibited the formation of 1-hydroymidazolam and activated the formation of 4-hydroxymidazolam. These results provide experimental evidence to support the use of midazolam and testosterone in combination to screen new chemical entities for their potential inhibition of CYP3A4. Therefore, the in vitro CYP inhibition assay, so far conducted in our laboratory with two separate cocktail incubations, each including one probe substrate for CYP3A4, is now performed with a single incubation. Currently, midazolam and testosterone are incubated with a human recombinant cDNA expression system together with other five selective CYP probe substrates. Substrates and metabolites of melatonin (CYP1A2), bupropion (CYP2B6), amodiaquine (CYP2C8), diclofenac (CYP2C9), dextromethorphan (CYP2D6), midazolam and testosterone (CYP3A4) are analysed simultaneously in a single LC/MSMS run (2.45 min). In conclusion, this in vitro CYP inhibition assay, based on a single cocktail incubation and a fast LC/MSMS quantitation, can be used to screen large sets of discovery compounds with significant time savings and high effectiveness.
133. INHIBITORY EFFECT OF TANNIC ACID ON GLUTATHIONE S-TRANSFERASE ACTIVITY IN RABBIT LIVER AND KIDNEY
Serdar Karakurt1 and Orhan Adah2
1Biochemistry Graduate Program, Department of Biological Sciences, METU, Ankara, Turkey, 06531, 2Department of Biological Sciences, METU, Ankara, Turkey, 06531
Glutathione S-transferases (GSTs, EC 2.5.1.18) are multifunctional enzymes which have a crucial role in the biotransformation and detoxification of potential alkylating agents and xenobiotics during phase II reactions. It was demonstrated that bioactivation of toxic metabolites by GST may cause organ damages. Furthermore the resistance of cells and organisms to pesticides, herbicides and antibiotics was implicated in GST activities. GSTs have been shown to be over-expressed in tumor cells hence it increases the resistance for chemotherapeutic drugs. This study was designed to evaluate in vitro effects of tannic acid, hydrolysable polyphenol produced from the secondary metabolism of plants, for its ability to modulate rabbit liver and kidney cytosolic glutathione S-transferase activity.
Tannic acid was found to be the potent inhibitor of the glutathione S-transferase activity not only in liver but also in kidney with IC50 = 0.33 μM and IC50 = 0.24 μM, respectively when 1-chloro-2,4-dinitrobenzene was used as a substrate. In addition, the effect of tannic acid on GST activity was further studied to investigate the mode and the type of inhibition. For this purpose, various concentrations of tannic acid in the range of 0.05-0.35 μM were used in reaction mixtures containing different concentrations of 1-chloro-2,4-dinitrobenzene (0.2-2 mM). Lineweaver-Burk and Dixon plots were then generated from the resulting data and Km, Vmax and the inhibition constants (Ki) were determined. Tannic acid was shown to be a non-competitive inhibitor of hepatic GST with a Ki of 0.3 μM (Km remained unchanged while Vmax decreased) while it was uncompetitive inhibitor of kidney GST (both Km and Vmax were changed). These results indicate that tannic acid may modulate Phase II enzyme, glutathione S-transferase and influence the metabolic activation of xenobiotics mediated by this enzyme. Moreover, tannic acid due to its inhibitory effect on GST activity may have potency for use in cancer drug efficacy studies and as a chemoprotective agent against GST induced toxicities.
This study was designed to determine the effect of tannic acid on the total cytosolic GST activity of rabbit liver and kidney. However it was known that GST has isozymes, therefore further studies were needed to clarity the contribution of each isozymes to total GST and to analyze the modulation of GST isozymes by tannic acid.
134. IMPACT OF PERMEABILITY ON IN VITRO PREDICTIONS OF INTESTINAL AVAILABILITY IN THE QGUT MODEL
Michael Gertz1, Katherine S. Fenner2, Anthony Harrison2, John Davis2, J. Brian Houston1, and Aleksandra Galetin1
1School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester, United Kingdom, M13 9PT, 2Pfizer Global Research and Development, Sandwich, United Kingdom, CT139NJ
Intestinal first-pass metabolism contributes to the low oral bioavailability of various drugs and magnitude of drug-drug interactions. In addition to metabolism, permeability and efflux transporters are major contributing factors to the extent of intestinal availability (FG). In the current study FG values were predicted from in vitro permeability and clearance data for 20 CYP3A4 substrates using the QGut model. Predicted values were compared to in vivo FG estimates obtained from grapefruit juice (GFJ) interaction studies assuming complete inhibition of CYP3A4 in the intestine by GFJ constituents, no inhibition of hepatic enzymes and no change in the fraction absorbed. Single dose of GFJ administered with or up to 12h before drug intake was considered efficient to produce maximal inhibitory effects. The FG values were determined from the ratio of the area under the plasma concentration curve in control and GFJ group. For all the compounds investigated permeability data, Papp (A-B) were determined in both MDCK-MDR1 and Caco-2 cells at substrate concentrations of 0.1 or 2μM. A 100-fold range in Papp (A-B) values was observed, from 4 to 398nm/s for saquinavir and buspirone, respectively. Corresponding clearance data were determined in both human intestinal and liver microsomes and corrected for the respective population abundance of CYP3A4. A high inter-individual and inter-study variation was observed in the GFJ FG estimates (up to 4.5-fold for simvastatin). Weighted average mean FG estimates obtained from GFJ data were comparable to FG values obtained from i.v./oral studies, in particular for drugs eliminated predominantly via metabolism. Papp (A-B) values obtained under standardised conditions in MDCK-MDR1 and Caco-2 cells correlated well (R2 = 0.84), with permeability in MDCK-MDR1 being on average 2-fold higher than in Caco-2 cells. However, no significant difference in the subsequent FG predictions were observed indicating that Papp (A-B) values from both cell systems can be used inter-changeably in the QGut model. This agrees with the finding that the QGut model is less sensitive to permeability in comparison to clearance. The area of high intestinal extraction (FG < 0.25) was identified as problematic as it displayed the highest discrepancy between GFJ and i.v./oral estimates; an analogous trend was observed between FG predicted from in vitro data and in vivo values. Further refinement of the model is required to improve FG predictions.
135. CYTOCHROMES P450 IN HUMAN WHITE ADIPOSE TISSUE: EXPRESSION AND INDUCTION
Sandrine Ellero1, Chantal Benelli2, Corinne Barreau3, Luc Penicaud3, Eric Hugo4, Philippe Beaune1, and Isabelle de Waziers1
1U775, INSERM, Paris, France, 2U747, INSERM, Paris, France, 3UMR5241, CNRS-UPS, Toulouse, France, 4Department of cell and cancer biology, University of Cincinnati, Cincinnati, OH, USA, 45267-0521
White adipose tissue (WAT) represents a reservoir of lipophilic environmental pollutants, especially of those who are resistant to biological and chemical degradation. WAT is the key organ for energy homeostasis and is also an important endocrine tissue secreting obesity/diabetes-related hormones and cytokines. By modulating WAT differentiation, metabolism and function, the environmental pollutants could affect the physiological and pathological role of WAT, and influence the development of obesity/diabetes-associated diseases. Upon xenobiotic exposure, organisms can increase their metabolic activities to eliminate the chemicals more efficiently from the body, usually by activating the transcription of the P450 genes, mainly in the liver. The expression of cytochromes P450 was recently identified by Yoshinari et al. [1] in rat WAT. In the same study, the authors showed that the P450 regulation pathways were also active in the rat WAT, suggesting that the WAT could respond to xenobiotic exposition. In our present study, we identified for the first time, the expression of cytochromes P450 in human WAT and compared the cytochromes P450 regulation mechanisms in this tissue to those found in the human liver. We used three different human WAT models: human WAT explants, human primary cultures of adipocytes and an adipocyte cell line LS14. Human primary hepatocytes were used as a liver model. Cytochrome P450 mRNA expression of the families 1 to 4 was investigated by quantitative RT-PCR and protein expression by western-blotting. To compare the regulation mechanisms of the P450 in the two tissues, we treated simultaneously the WAT models and the hepatocytes with typical cytochromes P450 inducers, namely rifampicin, phenobarbital and 2,3,7,8-tetrachlorodibenzo-p-dioxine (TCDD), and with lipophilic environmental pollutants that can be found in the WAT, namely lindane, prochloraz and chlorpyrifos. Our results showed the presence of different isoforms of cytochromes P450 in human WAT. It also showed that TCDD and prochloraz were CYP1A1 and CYP1B1 inducers in the liver as well as in the WAT, but with reduced induction factors in WAT. The expected family 2 and 3 inductions by rifampicin and phenobarbital were found in the human hepatocytes but no induction was found in WAT. Thus, this study suggests that the AhR induction pathway, regulating the induction of the CYP1A and 1B, is functional in the human WAT, and that the two other pathways involving CAR and PXR, regulating the induction of the P450 of the families 2 and 3, are not.
REFERENCE
- Yoshinari et al., Expression and induction of cytochromes p450 in rat white adipose tissue, J. Pharmacol Exp Ther., 311(1):147–54, 2004.
136. PREGNANE X RECEPTOR INCREASES EXPRESSION OF S14, A PUTATIVE REGULATOR OF LIPOGENIC ENZYMES EXPRESSION, IN HUMAN HEPATOCYTES
Amélie Moreau1, Christelle Teruel2, Thierry Umbdenstock3, Yannick Parmentier4, Viola Tamasi5, Urs A. Meyer6, Patrick Maurel7, Marie-José Vilarem1, and Jean-Marc Pascussi1
1Hepatic Physiopathology, INSERM U632, Montpellier, France, FR-34293, 2Hepatic Physiopathology, INSERM U632, Montpellier, France, 34293, 3Technologie Servier, Orleans, France, 45007, 4Technologie Servier, Orleans, France, 5Dept of Pharmacol/Neurobiol, Univ of Basel Biocenter, Basel, Switzerland, 4056, 6Dept of Pharmacol/Neurobiol, Univ of Basel Biocenter, Basel, Switzerland, CH-4056, 7U632, INSERM U632, Montpellier, France, 34293
The Pregnane X Receptor (PXR, NR1I2) was isolated as xenosensor regulating both drug and xenobiotic responses. However recent studies have shown that activation of this receptor impacts lipid homeostasis and increases hepatic deposit of triglycerides. By using transcriptional profiling of genes deregulated in primary culture of human hepatocytes after PXR activation, we observed that S14 (Spot14 or THRSP) mRNA was up-regulated by rifampicin. Although the biochemical mechanism of its action is unknown, it is clear that S14 protein in general acts to transduce hormone- and nutrient-related signals to genes involved in lipogenesis such as fatty-acid synthase (FASN) and ATP citrate Lyase (ACLY). S14 was first identified as a thyroid-responsive gene and its promoter contains a Thyroid Hormone Receptor (TR) Binding Element (TRE). We confirmed by real-time PCR and western-blot analyses the up-regulation of S14 by several PXR agonists in human hepatocytes. Moreover, studies using knock-out mice or RNA interference showed that PXR is necessary for S14 activation by xenobiotics. Promoter analysis revealed that PXR:RXRα heterodimers bind to and transactivate the distal TRE of S14 promoter gene, suggesting that S14 is a direct transcriptional target of PXR. In addition, we observed a significant increased expression of FASN and ACLY forty-height hours after PXR activation, while expression of SREBP1c, PPARγ1–2 or ChREBP mRNA was unchanged. Accordingly, we find that rifampicin induced an increase of fatty acid content within human hepatocytes. These results suggest that PXR plays a role in hepatic lipogenesis in human, and corroborate the correlation between S14 expression and increased lipogenesis. In addition, these data suggest that the up-regulation of S14 by drug-activated PXR may promote aberrant hepatic lipogenesis and hepatic steatosis in human hepatocytes.
137. EXPRESSION AND REGULATION OF ORGANIC SOLUTE TRANSPORTER (OST α/β) IN RAT INTESTINE AND LIVER
Ansar A. Khan1, Edwin C.Y. Chow2, K. Sandy Pang2, and Geny M.M. Groothuis1
1Pharmacokinetics and Drug Delivery, University Centre for Pharmacy, University of Groningen, Groningen, Netherlands, 2Pharmaceutical Sciences, Leslie Dan Faculty of Pharmacy, University of Toronto, Toronto, Canada, ON M5S 3M2
The heterodimeric organic solute transporter (Ostα/β) has recently been identified as a basolateral bile acid transporter in ileum and bile duct epithelial cells. However, its regulation is poorly understood. The Ostα/β promoter is reported to contain a farnesoid X receptor responsive element (FXRE), a liver X receptor α responsive element (LXRαRE) and a hepatocyte nuclear factor 4α responsive element (HNF4αRE). We investigated whether Ostα/β is coordinately regulated with the apical bile acid transporter, ASBT, which mediates bile acid uptake in intestine and biliary epithelial cells. Previously, ASBT expression in rat ileum was reported to be under the positive control of Vitamin D receptor (VDR) and glucocorticoid receptor (GR) but not FXR. Methods. We used rat precision cut intestinal and liver slices to study the expression and regulation of Ostα/β by FXR, VDR and GR ligands in the rat jejunum, ileum, colon, and liver. Rat liver and intestinal precision cut slices were cultured in the presence of the FXR ligand, chenodeoxycholic acid (CDCA, also a weak VDR ligand), VDR ligands: 1,25(OH)2D3 and lithocholic acid (LCA, also a FXR ligand), and the GR ligand, dexamethasone (Dex) for 8 and 12 hrs. The mRNA expression of Ostα/β, HNF4α, VDR and FXR was evaluated by qRT-PCR. Results: Expression of Ostα/β mRNA was detectable at significant levels in rat liver and intestine, where it was higher in ileum compared to jejunum and colon. The segmental expression of Ostα/β of the intestine paralleled that of ASBT but not of FXR and VDR. However, these transporters are regulated differentially. In rat small intestine (jejunum, ileum and colon) and liver, CDCA, a stronger FXR than a VDR ligand, up-regulated Ostα/β expression in liver as well as in jejunum, ileum and colon, whereas1,25(OH)2D3, the VDR ligand, down-regulated Ostα/β in liver and all segments of the intestine. LCA, a VDR and FXR ligand, showed down-regulation of Ostα/β in the ileum but up-regulation in the jejunum, colon and liver, whereas Dex induced Ostα/β in liver, jejunum and colon but not in ileum. Both Dex and LCA up-regulated expression of the transcription factor, HNF4α in jejunum and colon but not in ileum; and the observations with Dex could be due to direct GR- and/or indirect HNF4α-effects inasmuch as HNF4α also contained a GRE promoter. Conclusions. In rat intestine Ostα/β is positively regulated by FXR and negatively regulated by VDR. The mixed effects of LCA, a ligand of both VDR and FXR, and Dex on Ostα/β expression may be due to different expression levels of VDR/FXR/HNF4α among intestinal segments.
138. ACTIVATION OF THE KEAP1-NRF2-ARE CELL DEFENSE SYSTEM BY THE REACTIVE METABOLITE OF ACETAMINOPHEN
Christopher E. Goldring1, Ian Copple1, Roz Jenkins1, Laura Randle1, Alvin Chia1, John Hayes2, Neil Kitteringham1, and Kevin Park3
1Department of Pharmacology & Therapeutics, The University of Liverpool, Liverpool, United Kingdom, L69 3GE, 2Ninewells Hospital and Medical School, Biomedical Research Centre, Dundee, United Kingdom, DD1 9SY, 3Dept of Pharmacology, University of Liverpool, Liverpool, United Kingdom, L69 3GE
Drug-induced liver injury (DILI) is a major type of adverse drug reaction (ADR), and accounts for over half of all cases of acute liver failure. The DILI associated with overdose of acetaminophen (APAP) is the single biggest cause of acute liver failure in the USA. APAP-induced hepatotoxicity is inextricably linked to the formation of a chemically reactive metabolite, N-acetyl-p-benzoquinoneimine (NAPQI), which causes chemical and oxidative stress, and inhibits the function of critical proteins within hepatocytes. Mammalian cells can defend themselves against chemical and oxidative stress via the inducible expression of phase II detoxification enzymes and antioxidant proteins. A major regulator of this adaptive response is the transcription factor Nrf2, which controls the inducible expression of many cytoprotective genes, through its action on the cis-acting antioxidant response element (ARE) regulatory motif. We have previously demonstrated that administration of APAP in vivo leads to activation of Nrf2 and induction of cell defence genes in mouse liver. In the current work, we show that NAPQI can directly activate the Nrf2 pathway in mouse liver cells, resulting in the induction of glutamate-cysteine ligase and a concomitant increase in cellular glutathione. Nrf2-targeted RNAi inhibited these effects, demonstrating the Nrf2-dependence of this adaptive response. Furthermore, RNAi depletion of Keap1, the cytosolic repressor of Nrf2, enhanced Nrf2-dependent cell defence. The modification by chemical inducers of critical cysteine residues within Keap1 has been proposed as the molecular basis of Nrf2 activation. Here, we show that NAPQI, and the model cysteine-reactive electrophiles 2,4-dinitrochlorobenzene (DNCB) and 15-deoxy-n-(12,14)-prostaglandin J2 (15d-PGJ2), triggered a dose-dependent nuclear accumulation of Nrf2, whereas the lysine-reactive molecule trimellitic anhydride (TMA) did not. To examine the molecular basis of Nrf2 activation by this panel of electrophiles, the residue-selectivity of Keap1 modification was determined using mass spectrometry. These data demonstrate modification of specific cysteines within the intervening region of Keap1 by the reactive metabolite of APAP, as well as DNCB and 15d-PGJ2, in a cellular context. This is consistent with the proposal that direct modification of Keap1 can provide a trigger for the activation of the Nrf2-driven cellular defence response during drug-induced liver injury. Using the model cell system described here, we are currently investigating the site-selectivity of Keap1 modification by other Nrf2 inducers, with the ultimate aim of understanding the precise chemical nature of the switch that controls the activation of this critical transcription factor.
139. EFFECTS OF DEXAMETHASONE ILLICIT TREATMENTS UPON CATTLE LIVER DRUG METABOLIZING ENZYMES GENE EXPRESSION AND REGULATION
Mery Giantin1, Rosa M. Lopparelli1, Pascal G.P. Martin2, Arnaud Polizzi2, Clara Montesissa1, Licia Ravarotto3, Thierry Pineau2, and Mauro Dacasto1
1Dipartimento di Sanità Pubblica, Patologia Comparata ed Igiene Veterinaria, Università degli Studi di Padova, Padova, Italy, I-35020, 2Laboratoire de Pharmacologie et Toxicologie, Institut National de la Recherche Agronomique - UR66, Toulouse, France, 35131, 3Istituto Zooprofilattico delle Venezie, Padova, Italy, 35020
In the European Community, the use of growth promoters (GPs) to increase animal performances in cattle is forbidden (Dir. CEE 96/22 and 96/23); nonetheless, the illicit use of GPs, like anabolic steroids, ß-agonists or corticosteroids like dexamethasone (DX), still represents a major concern in this food-producing species (4). Nowadays, increasing importance is attributed to “omic” methodologies useful to define biomarkers (BMs); in this respect, an increasing interest toward the setting-up of BMs to assist official analytical methods in illicit GPs screening was recently signalled in cattle (2–3). Illicit GPs may affect drug metabolizing enzymes (DMEs) post-transcriptionally (1); therefore, the pre-transcriptional effects of two illicit protocols containing DX, upon relevant DMEs genes and transcription factors, were investigated.
Small aliquots of liver tissue were obtained from male beef cattle, untreated or administered either with DX (given im or per os) or a cocktail consisting of DX (per os) and 17β-oestradiol (E2, im). Routes of administration and dosage regimens were similar to those illegally used in the field. Seventeen candidate genes were selected according to their relevance in drug metabolism, including both phase I and II DMEs (cytochrome P450s and glutathione- or glucuronyl-transferases, respectively) as well as transcription factors. For each gene, the corresponding bovine sequence was obtained and a gene-specific quantitative real-time PCR assay was optimised and validated.
As a whole, in the liver of treated cattle the expression of 8 target genes, out of the 17 tested, were significantly influenced by GPs: six when DX was administered alone and 7 in the case of DX plus E2. Moreover, an overall and significant down-regulation of CYP2B6 and CYP2E1 gene expression profile was recorded, independently from the GP, the route of administration and the dosage regimen adopted.
Present results indicate that some GPs (DX, E2) are likely to modulate liver DMEs at the pre-transcriptional level in cattle; besides, the gene expression profiling might represents a feasible approach for the screening of GPs used illicitly. This study represents a preliminary work for the development of a set of hepatic biomarkers to be used in the field monitoring, that should be improved increasing the number of candidates and validating them on a greater amounts of controls.
ACKNOWLEDGMENT
Supported by grants from Ministero della Salute (IZS.VE.009/03RC), Università degli Studi di Padova (CPDA054599) and Università Italo-Francese (Programma Galileo 2006).
REFERENCE
- Dacasto M., Montesissa C., Nebbia C.: Illegal drug treatments and drug metabolism: biomarkers or not? Vet. Res. Commun., 30 (Suppl. 1), 113–9, 2006.
- Gardini G., Del Boccio P., Colombatto S. et al.: Proteomic investigation in the detection of the illicit treatment of calves with growth-promoting agents. Proteomics, 6, 2813–22, 2006.
- Reiter M., Walf V.M., Christians A. et al.: Modifications of mRNA expression after treatment with anabolic agents and the usefulness for gene expression-biomarkers: Anal. Chim. Acta, 586, 73–81, 2007.
- Serratosa J., Blass A., Rigau B. et al.: Residues from veterinary medicinal products, growth promoters and performance enhancers in food-producing animals: a European perspective. Rev. Sci. Tech. Off. Int. Epiz., 25, 637–53, 2006.
140. TRANSCRIPTOME AND STEROL PROFILE OF RIFAMPICIN-TREATED HUMAN HEPATOCYTE: A TIME-COURSE STUDY
Tadeja Rezen1, Katalin Monostory2, Jean-Marc Pascussi3, Jure Acimovic4, Ingemar Bjorkhem5, and Damjana Rozman1
1Institute of Biochemistry, University of Ljubljana, Center for Functional Genomics and Bio-Chips, Ljubljana, Slovenia, SI-1000, 2Chemical Research Center, Hungarian Academy of Sciences, Budapest, Hungary, HU-1025, 3Hepatic Physiopathology, INSERM U632, Montpellier, France, FR-34293, 4Chemical Research Center, Hungarian Academy of Sciences, Budapest, Hungary, H-1025, 5Division of Clinical Chemistry, Karolinska University Hospital at Huddinge, Stockholm, Sweden, S-14186
Rifampicin is a human PXR activator and is used as an antibiotic to treat certain mycobacterial infections. However, PXR activation has other desired, but also undesired effects. For example, it affects cholesterol homeostasis and is used the treatment of cholestasis. The goal of our study was to determine changes in expression of genes involved in cholesterol homeostasis and other liver processes in primary human hepatocytes at 12, 24 and 48 hours after rifampicin application. Transcriptome analyses were performed using a custom cDNA microarray Steroltalk. The Steroltalk microarrays are original tools which were developed for use in systemic studies of the crosstalk between cholesterol homeostasis and xenobiotic metabolism. The array includes genes involved in both processes, such as enzymes of cholesterol synthesis and metabolism, phase I drug metabolism, nuclear receptors, transporters and regulators. We also measured the sterol profiles and drug-metabolizing activities of hepatocytes at 48 hours after rifampicin application. Transcriptome analyses showed that the biggest difference was between 12 hour time point and 24 – 48 hour time points. This indicates that the primary response to the drug happened before 12 hours, while later we observed the secondary effects. The important difference was that some genes of cholesterol biosynthesis were up-regulated at 12 hours, but not at 24 hours. The induction of drug metabolizing genes was still increasing at 48 hours and the number of genes with changed expression was also increased. Many genes involved in cholesterol homeostasis and regulation were differentially expressed as well as certain genes involved in glucose and fatty acid homeostasis. Sterol profiles and drug-metabolizing activities of CYP2s and CYP3As were in accordance with the transcriptome data. This study for the first time elucidates the time-course of changes in cholesterol homeostasis in the presence of a PXR activator, rifampicin, and how the Steroltalk microarrays crucially contributed to these discoveries.
ACKNOWLEDGMENTS
This abstract and work it concerns was generated in the context of the STE-ROLTALK project, funded by the European Community as contract No. LSHG-CT-2005–512096.
141. HEPATOCYTE-LIKE CELLS DERIVED FROM HUMAN EMBRYONIC STEM CELLS VIA DEFINITIVE ENDODERM DISPLAY DRUG METABOLISING ACTIVITY
Barbara Küppers-Munther1, Monica Ek2, Josefina Edsbagge1, Nico Heins1, Gabriella Brolén1, Marie Rehnström1, Elisabet Athley1, Malin Darnell3, Tommy B. Andersson3, Inger Johansson2, Magnus Ingelman-Sundberg2, and Petter Björquist1
1Cellartis AB, Göteborg, Sweden, 41346, 2Department of Physiology and Pharmacology, Section of Pharmacogenetics, Karolinska Institutet, Stockholm, Sweden, 3Global Development DMPK & Bionalaysis, Astra-Zeneca R&D, Mölndal, Sweden, 43183
Human embryonic stem cells (hESC) can differentiate into hepatocyte-like cells displaying a characteristic hepatic morphology and expressing various hepatic-related genes. Therefore, hESC offer a potentially unlimited supply of human hepatocytes which can be used for studies of drug metabolism and hepatotoxicity. To be useful in such studies, hepatocyte-like cells need to be functional and express significant levels of drug metabolising enzymes and transporters. Using a stepwise differentiation protocol, we direct hESC to differentiate into hepatocyte-like cells via the definitive endoderm (DE) and a progenitor stage. These DE-derived hepatocyte-like cells have a typical hepatic morphology, express a number of liver-related genes and show typical liver functions such as glycogen storage and indocyanine green transport. Moreover, they metabolise three probe drugs for CYP1A2, 3A4 and 2C9. In conclusion, our results represent an important step towards a future unlimited source of functional human hepatocytes useful in drug metabolism and toxicity testing.
142. THE ROLE OF MRP2 FOR THE HEPATOBILIARY EXCRETION OF AMANITA MUSHROOM TOXIN DEMETHYLPHALLOIN
O. Gavrilova, K. Meerkamp, J. Geyer, and E. Petzinger
Institute of Pharmacology and Toxicology, University of Giessen, Giessen, Germany, 35392
The role of mrp2 for the hepatobiliary excretion of the Amanita mushroom toxin demethylphalloin O. Gavrilova, K. Meerkamp, J. Geyer, E. Petzinger The Amanita mushroom toxin phalloidin is selectively taken up by hepatocytes where it cause cholestasis by preventing F-actin depolymerization. In 1977 we already showed that physiological concentrations of bile acids inhibit the response of rat hepatocytes to phalloidin intoxication. We suggested a bile acid carrier performing uptake of phalloidin. In the meantime, several OATP-carriers (organic anion transporting polypeptide) were cloned which show overlapping transport activities for a broad spectrum of physiological substrates (bile acids, eicosanoids, steroid conjugates), drugs (e.g. digoxin, CRC220, pravastatin, fexofenadine), and toxins (ochratoxin A). Very recently, rat Oatp1b2 and human OATP1B1 and OATP1B3, which are expressed at the basolateral membrane of hepatocytes were shown to transport phalloidin with Km of 5.7–39 μM. Furthermore, it has been demonstrated, that the main canalicular efflux carriers in hepatocytes, i.e. mrp2, mdr1, and bsep are lost in the canalicular membrane during phalloidin induced cholestasis. Based in this data, we were interested whether one of these canalicular ABC-carriers could excrete phalloidin into bile in subtoxic concentrations. Therefore, we analyzed hepatobiliary elimination of the phalloidin derivative [3H]-demethylphalloin ([3H]-DMP) into the cannulated bile duct of Wistar and mrp2-deficient TR(−) rats, as well as in gall bladder cannulated mdr1a,b (−/−) and FVB mice. After application of 0.03 mg [3H]-DMP/kg b.w. wild-type Wistar rats and FVB mice excreted 18% and 12% of the applied dose into bile during 120 min, respectively. In contrast, TR(−) rats showed a significantly reduced excretion of [3H]-DMP into bile (8%), whereas [3H]-DMP excretion was unchanged in the mdr1a,b (−/−) knockout mice. Furthermore, TR(−) rats excreted much higher levels of [3H]-DMP into the urine (8% of the applied dose) compared to Wistar rats (0.1%). In all animals bile flow was constant during time, indicating that the applied subtoxic dose did not significantly induce cholestasis in these animals. In conclusion: Mrp2 is the major efflux carrier for the hepatobiliary phalloidin excretion on rats. However, mrp2 and other efflux carriers are lost in the canalicular membrane of hepatocytes at toxic doses of phalloidin (0.3–0.5 mg/kg b.w.) leading to the hepatic accumulation of phalloidin and to intrahepatic cholestasis. Institute of Pharmacology and Toxicology, Justus-Liebig-University Giessen, Frankfurter Str. 107, D-35392 Giessen; [email protected]
143. MRP2 TRANSPORT FUNCTION IN RAT HEPATOCYTES AND THE EFFECT OF THE LIVER DIGESTION ENZYME
J. Sinclair1, C. Henderson1, I. J. Martin2, M. H. Grant1, and J. N. Tettey1
1Strathclyde University, Glasgow, United Kingdom, G4 0NR, 2Dept of Pharmacol, Organon as part of the Schering-Plough Corporation, Lanarkshire, United Kingdom, ML1 5SH
Isolated rat hepatocytes are a valuable whole cell in vitro tool for drug metabolism and disposition studies. Several modifications of the original two-stage collagenase perfusion technique for the preparation of hepatocytes (Seglen, 1972) have been reported. However, there are very limited published data on the influence of isolation procedures on xenobiotic efflux from hepatocytes. The objective of the present study was to examine the effect of the digestion enzyme used during preparation on the efflux properties of isolated rat hepatocytes. Hepatocytes were isolated from male Sprague Dawley rats (180–220 g) using collagenase type II (CII) as described previously (Grant et al, 2000). Modifications of this technique using collagenase A/trypsin inhibitor (CA/TI) and collagenase/dispase (C/D) were also investigated. Hepatocytes were seeded, using Williams' E medium (supplemented with 5% (v/v) bovine serum albumin), at 3 ¥ 106 cells/plate onto 60 mm plates pre-coated with rat-tail collagen and then incubated in 5% CO2/ 95% O2 at 37°C. At 3 h the medium was replaced, and this was repeated every 24 h. Transport function of Mrp2, a canalicular multispecific organic anion transport protein, was evaluated using the substrate carboxydichlorofluorescein (CDF). Biliary excretion measurements were carried out at 3, 24, 48 and 72 h as previously described (Zhang et al 2005). In brief, intracellular and total (cellular and canalicular) hepatocyte accumulation of CDF was measured spectrofluometrically as arbitrary units (ABU) and the biliary excretion index (BEI) determined as follows:
In 48 h cultured hepatocytes, intracellular CDF accumulation was significantly lower (p < 0.05) in cells prepared with C/D (24.4 ± 1.4 ABU/mg protein) compared with CII and CA/TI (39.0 ± 1.1 and 36.5 ± 4.1 ABU/mg protein respectively); at 72 h it was significantly lower (p < 0.05) in C/D (32.9 ± 1.0 ABU/mg protein) than in CII (52.4 ± 4.0 ABU/mg protein) isolated hepatocytes. However, the inclusion of dispase in the perfusion medium had no significant effect on CDF BEI measurements. At 48 h, for example, the BEI was 53.6 ± 4.6 and 47.9 ± 2.9% for CII and C/D isolated hepatocytes respectively. It was not possible to determine CDF BEI in CA/TI isolated hepatocytes since the intracellular and total CDF accumulation were statistically identical. The choice of digestion enzyme used in the preparation of isolated rat hepatocytes may therefore influence Mrp2 transport function and the formation of bile canaliculi in 72 h cultured rat hepatocytes.
REFERENCE
- Seglen P. Preparation of isolated rat liver cells. Biochemical and Biophysical Acta 264: 398–410, 1972.
- Grant MH, Anderson K, McKay G, Wills M, Henderson C, MacDonald C. Manipulation of the phenotype of immortalised rat hepatocytes by different culture configurations and by dimethyl sulphoxide. Human Experimental Toxicology 19: 309–317, 2000.
- Zhang P, Tian X, Chandra P, Brouwer K. Role of glycosylation in trafficking of Mrp2 in sandwich-cultured rat hepatocytes. Molecular Pharmacology 67: 1334–1341, 2005.
144. THE USE OF SANDWICH-CULTURED HEPATOCYTES (B-CLEAR®) TO ASSESS THE INHIBITION OF BILE ACID TRANSPORT AND CHOLESTATIC POTENTIAL OF DRUG CANDIDATES
Kenneth R. Brouwer1, John H. Ansede2, William R. Smith1, Robert L. St. Claire III1, and Cassandra H. Perry1
1Qualyst, Inc., Raleigh, NC, USA, 27607, 2Research and Development, Qualyst, Inc., Raleigh, NC, USA, 27607
Drug induced liver injury is the most common reason for withdrawal of FDA approved drugs from the market. Recent literature data suggests that hepatic transport proteins may be an important site of toxic interactions. Inhibition of the hepatic uptake and biliary excretion of bile acids (cholestasis) by drugs is one of the most common clinical liver toxicities. Sandwich-cultured hepatocytes (B-CLEAR®) maintain many of the structural and functional attributes of hepatocytes in vivo, and have been used as an in vitro model to predict the in vivo biliary excretion of drug candidates. The B-CLEAR® technology maintains the expression and function of key uptake and efflux transporters and is the optimal system to evaluate and predict the potential of a compound to cause transporter based liver toxicity. The effect of several test compounds on the accumulation, BEI (biliary excretion index), and Clbiliary (in vitro biliary clearance) of d8-taurocholate were evaluated in B-CLEAR®-RT (rat) and the results are summarized in .
Table 1 The effect of test compounds on the accumulation (hepatocytes), BEI and Clbiliary of d8-taurocholate as a percentage of control (no inhibitor)
The accumulation, BEI and Clbiliary of d8-taurocholate were 15.1 ± 2.47 pmol/mg protein, 88.3 ± 1.32% and 37.1 ± 9.54 mL/min/kg, respectively. Erythromycin-estolate and troglitazone, two compounds that are known to cause in vivo cholestasis, inhibited the uptake of d8-taurocholate into hepatocytes in comparison to controls (no inhibitor). Cyclosporine and glyburide, and to a much lesser extent; erythromycin-estolate, nefazodone and troglitazone showed a dose-dependent decrease in the BEI of d8-taurocholate. All compounds, especially at the higher concentration, showed a considerable decrease in Clbiliary of d8-taurocholate. Salicylate, buspirone and several macrolide antibiotics that have not been associated with in vivo cholestasis showed no effect on the accumulation, BEI or Clbiliary of d8-taurocholate.
145. OPTIMISATION OF THE CULTURE CONDITIONS OF RAT AND HUMAN HEPATOCYTE FOR CYTOCHROME P450 EXPRESSION AND INDUCIBILITY
Lysiane Richert
KaLy-Cell et Laboratoire de Toxicologie Cellulaire UFR SMP, Besançon, France, 25000
Primary cultures of hepatocytes are widely used for the study of the expression and inducibility of cytochrome P450 (CYP). Cell microsomes prepared from these cultures can be used for the determination of CYP activity, but important quantity of cells is then required. An important goal is to decrease cell number needed both for rat (ethic request) and human material (scarce material). For this purpose, an interesting tool is the use of direct incubation of specific substrates on hepatocyte monolayers. We compared CYP1A, 2C and 3A-dependent activities, both at the basal level and after treatment with CYP reference inducers (b-napthoflavone: NF, dexamethasone: Dex or rifampicin: Rif), using cell microsomes or cell monolayers for incubation. Moreover, two different formats were compared for monolayer: dish (3,5.106 cells/dish) versus 24-well support (0,3.106 cells/well). Basal activities for the three CYPs measured in cell microsomes (expressed per mg microsomal protein) were consistently 6- to 7-fold higher than activities measured in cell monolayers (expressed per mg cell protein). When using CYP reference inducers, the fold increase of the specific activities when assessed on cell microsomes and on cell monolayers – two different formats – were equivalent both in rat and human hepatocytes. Taken together, these data showed that 24-well is a very usefulness format for the determination of CYP activity both in rat and human hepatocytes. We also determined in primary cultures of human hepatocytes, an optimised treatment time in terms of CYP induction response. For this purpose, cultures were treated with 3 concentrations of NF or Rif, 24h or 48h after cell seeding. CYP-dependent activities and mRNA expression were evaluated after 72h of treatment (activities) or 24h, 48h and 72h of treatment (mRNA expression). Concentration-dependent induction of CYP1A2 after NF treatment and CYP2C9 and 3A4 after Rif treatment were obtained at mRNA and activity levels. We found an equivalent or slightly higher response to inducers both at the mRNA and activity levels, when the treatment started 48h after cell seeding, depending on the inducer used and depending on the human donor. In this condition, the duration of treatment for optimised response at the mRNA level is 24 or 48h depending on the inducer used and on the human donor.
146. AN INHIBITOR OF P38 MAP KINASE ACTIVATES ERK AND JNK MAP KINASES IN PRIMARY CULTURES OF HUMAN HEPATOCYTES
Radim Vrzal1, Pavla Henklova1, Barbora Papoukova2, Petr Bednar2, Petra Jancova1, Eva Anzenbacherova1, Jitka Ulrichova1, Patrick Maurel3, Petr Pavek4, and Zdenek Dvorak1
1Department of Medical Chemistry, Faculty of Medicine and Dentistry, Palacky University, Olomouc, Czech Republic, 77515, 2Department of Analytical Chemistry, Palacky University, Faculty of Science, Olomouc, Czech Republic, 77200, 3U632, INSERM U632, Montpellier, France, 34293, 4Department of Pharmacology and Toxicology, Pharmaceutical Faculty, Charles University, Hradec Kralove, Czech Republic, 50005
Mitogen-activated protein kinases (MAPKs) are the members of the signal transduction system involved in the control of gene expression and various events in eukaryotic cells. They were extensively studied in cancer-derived cell lines; however, the studies in non-transformed human cells are scarce.
In this work, we studied the effect of SB203580 (4-(4-Fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole), a pharmacological inhibitor of p38 MAPK, on activation and inhibition of p38 MAPK transduction partway in primary human hepatocytes (in vitro model of differentiated cells) in comparison with several tumor cell lines (proliferating non-differentiated in vitro model). The effect of selected compound was monitored by immunoblotting technique with primary antibodies detecting phosphorylated/activated kinases.
We show that SB203580 activates ERK and JNK kinases in primary cultures of human hepatocytes. The levels of ERK-P, JNK-P and c-Jun-P were increased by SB203580. In contrast, SB203580 activated ERK but not JNK in HepG2, HL-60, Saos-2 and HaCaT human cancer cell lines.
We tested, whether the effects of SB203580 are due to metabolism. Using liquid chromatography/mass spectrometry, we found one minor metabolite in human liver microsomes but not in HepG2 cells. These data imply that biotransformation could be responsible for the effects of SB203580 in human hepatocytes.
Finally, our study is the first report on the effects of MAPK activators (sorbitol, anisomycin, EGF) and MAPK inhibitors in primary human hepatocytes. We observed differential effects of these compounds in human hepatocytes and in cancer cells, implying the cell-type specificity of these processes and the essential differences between the role and function of MAPKs in normal and cancer cells.
ACKNOWLEDGMENT
This work was supported by the grant from the Ministry of Education, Youth and Sports of the Czech Republic MSM 6198959216
147. JNK PHARMACOLOGICAL INHIBITOR SP600125 IS A PARTIAL AGONIST OF HUMAN ARYL HYDROCARBON RECEPTOR AND INDUCES CYP1A1 AND CYP1A2 GENES IN PRIMARY HUMAN HEPATOCYTES
Zdenek Dvorak1, Radim Vrzal1, Pavla Henklova1, Petra Jancova1, Eva Anzenbacherova1, Patrick Maurel2, Petr Pavek3, Petr Bednar4, and Jitka Ulrichova1
1Department of Medichal Chemistry, Faculty of Medicine and Dentistry, Palacky University, Olomouc, Czech Republic, 77515, 2U632, INSERM U632, Montpellier, France, 34293, 3Department of Pharmacology and Toxicology, Pharmaceutical Faculty, Charles University, Hradec Kralove, Czech Republic, 50005, 4Department of Analytical Chemistry, Palacky University, Faculty of Science, Olomouc, Czech Republic, 77200
In this report we demonstrate that SP600125, a pharmacological inhibitor of c-Jun-N-Terminal kinase (JNK), is a partial agonist of human aryl hydrocarbon receptor (AhR).
SP600125 induced CYP1A1 and CYP1A2 mRNAs in primary human hepatocytes and CYP1A1 mRNA in human hepatoma cells HepG2. This effect was abolished by resveratrol, an antagonist of AhR. Consistently, SP600125 inhibited CYP1A1 and CYP1A2 genes induction by a prototype AhR ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in human hepatocytes. SP600125 displayed typical behavior of a partial agonist in HepG2 cells transiently transfected with a reporter plasmid containing two inverted repeats of the dioxin responsive element or with a plasmid containing 5′-flanking region of human CYP1A1 gene. SP600125 transactivated the reporter plasmids with EC50 of 0.005 mM and 1.89 mM, respectively. TCDD-dependent transactivation of the reporter plasmids was inhibited by SP600125 with IC50 values of 1.54 mM and 2.63 mM, respectively. We tested, whether the effects of SP600125 are due to metabolism. Using liquid chromatography/mass spectrometry, we observed formation of two minor monohydroxylated metabolites of SP600125 in human hepatocytes, human liver microsomes but not in HepG2 cells. These data imply that biotransformation is not responsible for the effects of SP600125 on AhR signaling.
In conclusion, we demonstrate that SP600125 is a partial agonist of human AhR, which induces CYP1A genes.
ACKNOWLEDGMENT
This work was supported by the grant from the Ministry of Education, Youth and Sports of the Czech Republic MSM 6198959216 and by the grant from the Czech Scientific Foundation GACR 303/07/0128.
REFERENCE
- Dvorak Z., Vrzal R., Henklova P., Jancova P., Anzenbacherova E., Maurel P., Svecova L., Pavek P., Ehrmann J., Havlik R., Bednar P., Lemr K., Ulrichova J. (2007) JNK inhibitor SP600125 is a partial agonist of human aryl hydrocarbon receptor and induces CYP1A1 and CYP1A2 genes in primary human hepatocytes. Biochem Pharmacol (in press).
148. A NEW IN VITRO METHOD FOR PREDICTION OF MYELOTOXICITY
Caroline Haglund, Anna Åleskog, Rolf Larsson, and Elin Lindhagen
Clinical Pharmacology, Department of Medical Sciences, Uppsala University, Uppsala, Sweden, 751 85
In anticancer drug development, much focus is put on the anti tumor properties of the drug candidates, but it is also important to gain early information about potential side effects. One of the most common dose limiting adverse effects in cancer treatment is myelotoxicity.The aim of this study was to develop an in vitro method for measuring drug effects on erythropoesis, trombocytopoesis and granulocytopoesis preclinically, and thereby finding potential myelotoxic properties of a drug candidate. The source of myeloid progenitors was CD34+ stem cells from umbilical cord blood. The cells were plated in 384-well microtiterplates prepared with drugs in liquid culture supplemented with specific cytokines for each lineage. After 7 days of proliferation and differentiation the cells were analyzed using the non-clonogenic fluorometric microculture cytotoxicity assay (FMCA). The FMCA is based on measurement of fluorescence generated from hydrolysis of fluorescein diacetate to fluorescein by cells with intact plasma membranes and can be used in a high-throughput setting. Flow cytometry and morphological examination to determine the degree of differentiation in each lineage was also performed. The cells proliferated at a high rate and were viable after 7 days culture in the 384-well microtiterplate. The automated FMCA was able to measure cell viability and thereby cytotoxicity of drugs and proved to be well suited for this application. Flow cytometry and morphological evaluations showed that the cells had begun to differentiate towards their specific lineage, although the cells were in very early stages. Method validation is ongoing using drugs with known myelotoxic profile. Preliminary data indicate that this method can detect myelotoxic effects of drugs. However, in many cases drug sensitivity seems to be similar in all three lineages, and possibly more mature cells could be used to be able to detect differences between the lineages. In conclusion, this method is a simple and reproducible alternative to CFU-GM in studying myelotoxicity preclinically.
149. EXPERIMENTAL VALIDATION OF SITE OF METABOLISM PREDICTION TO REDUCE METABOLIC CLEARANCE IN EARLY DISCOVERY
Alfred Zimmerlin and Markus Trunzer
Metabolism And Pharmacokinetics (MAP), Novartis Pharma, Basel, Switzerland
Introduction. The metabolic stability of a drug candidate is an important consideration in determining its potential for human use and to ascertain that sufficient exposure is reached in animal models. Relatively high throughput assays using human liver microsomes allow the estimation of oxidative clearance rate of drug candidates at early discovery stages. In silico Site Of Metabolism (SOM) prediction using MetaSite helps predicting metabolic pathways and identifying soft spots in metabolically unstable compounds. We here report on a new method combining SOM prediction and an automated experimental search and confirm procedure allowing fast and efficient establishment of Structure Metabolism Relationships (SMR) in early stages of drug discovery. Methods. Predictions were done with MetaSeer, an extension of MetaSite, which generates and exports the structures of the predicted metabolites, their probability ranking as well as their mono-isotopic molecular masses. Compounds were tuned on the LC-MS machine and the compound specific energy and fragmentation were used to detect the predicted metabolites. Human liver microsomal incubation extracts were analyzed by LC/MS and predicted metabolites were selectively detected. All procedures from the import of drug candidate structures into MetaSeer to the LC/MS output as a kinetic readout were automated and integrated in our regular high throughput screen for metabolic stability in animal liver microsomes i.e. no additional samples need to be generated to extract information on pathways. Results and Discussion. A test set of 20 marketed compounds was selected based on the existence of a documented main CYP dependent metabolic pathway in human liver microsomes (MDL Metabolite database). These main pathways were all among the top 3 MetaSeer predictions with as much as 60% predicted as most likely (Rank 1). For 80% of the compounds the main pathway was also detected and confirmed by the LC/MS procedure described here, using parent compound tuning parameters. These very encouraging initial results suggested that if a main pathway is present and is responsible for microsomal high clearance of a drug candidate it was likely to be predicted and confirmed experimentally using the new procedure. Indeed, for a test set of 96 randomly selected high clearance compounds belonging to 32 research programs, 162 metabolites of the 288 predicted metabolites could be positively confirmed in human microsomal incubations (i.e. 44% of the predictions are probably incorrect). For more than 84% (81/96) of the compounds at least one of the predicted metabolites could be confirmed experimentally, providing a good rationale for targeted structural modifications likely to reduce metabolic clearance Conclusions. Monitoring the three most likely metabolites predicted by MetaSeer in microsomal incubations allowed to confirm or invalidate the in silico predictions of metabolic pathways of highly cleared compounds in most cases. For 84% of the compounds at least one of the predicted metabolites could be confirmed experimentally. The combination of In Silico prediction and high throughput experimental confirmation allows to focus synthetic efforts (and resources) to stabilize compounds and reach adequate pharmacokinetic properties in both human and animal models.
150. COMPARISON OF HUMAN PHARMACOKINETIC VARIABILITY FOR DRUGS AMONG THE FOUR CLASSES IN THE BIOPHARMACEUTICS CLASSIFICATION SYSTEM (BCS)
Johnny J. Yang1, Cindy Q. Xia1, Mingxiu Hu2, Hua Yang1, and Frank W. Lee1
1Drug Metabolism and Pharmacokinetics, Millennium Pharmaceuticals, Inc., Cambridge, MA, USA, 02139, 2Department of Biostatistics, Millennium Pharmaceuticals, Inc., Cambridge, MA, USA, 02139
Low variability in plasma exposure is a desirable feature for a therapeutic agent from the aspects of efficacy and toxicity. The high plasma exposure variability is often the result of poor drug solubility, low permeability or high metabolism. It is our interest to know what is the most important factor that contributes high plasma exposure variability in clinical.
Seventy-four (74) drugs were selected from the four different classes of Biopharmaceutics Classification System (BCS). Healthy volunteers' plasma exposure data of these 74 drugs were collected from literature. There were no marked differences in Coefficient of Variation (CV, mean value of 36%) of area under plasma concentration curve (AUC) for drugs among the four different classes. Approximately 75–80% of the 74 drugs showed CV of AUC values less than 50%, while approximately 20–25% of the 74 drugs exhibited greater than 50% CV.
The low CV of some low soluble and low permeable drugs could be due to the improved clinical formulation that has enhanced drug dissolution rate or permeability. Quantitative prediction of PK variability based on drug solubility, permeability, and metabolism properties remains a major challenge. Other factors such as formulation and food effect should also be considered. To delineate and estimate the contribution of major factors to exposure variation of a drug, multiple regression methods and inhibition and induction methods were exemplified by further analysis of cyclosporine and carvedilol studies from literature.
151. SIMULATION OF SUBLINGUAL AND GASTROINTESTINAL ABSORPTION OF NIFEDIPINE
Viera Lukacova, M.B. Bolger, S. Ray Chaudhuri, and W.S. Woltosz
Simulations Plus, Inc., Lancaster, CA, USA, 93534
Purpose. To develop an integrated model for drug absorption from the oral cavity. The model simulates combined absorption of a drug from both the oral cavity and the gastrointestinal tract. Methods. GastroPlusTM 5.3 with the PBPKPlusTM Module (Simulations Plus, Inc., Lancaster, CA) was used to simulate adult human plasma concentration time (Cp-time) profiles of Nifedipine when administered as a 1 mg intravenous (IV) infusion, 10 mg oral capsule (PO), 10 mg oral solution held sublingually (SL) for 7 min and then swallowed, and 10 mg oral solution held sublingually for 20 min and expectorated. A physiologically-based pharmacokinetic model (PBPK) based on adult human physiology was used in all simulations. Tissue/plasma partition coefficients were calculated using in silico physicochemical properties (ADMET Predictor™, Simulations Plus, Lancaster, CA) combined with in vitro measured values where available. Cp-time data from an IV infusion dose were used to fit the systemic clearance model, which was then used in all PO and SL dose simulations. Cp-time profiles after expectorated SL and PO doses were used to calibrate the absorption and first pass extraction of drug from the oral cavity and the gastrointestinal tract, respectively. The combination of these absorption parameters were then used to simulate the SL dose swallowed after 7 min. Results. The Cp-time profiles for all the dosage forms were successfully modeled with a single absorption-pharmacokinetic model incorporating sublingual and gastrointestinal absorption with physiologically-based pharmacokinetics. Conclusion. The developed generic model can be used for simulation of a variety of dosage forms that deliver drug into the oral cavity. Integration of the new model with the well-established gastrointestinal absorption model (ACAT) provides a mechanistic interpretation of complex formulation issues.
152. SIMULATION OF GABAPENTIN ABSORPTION AND BIOAVAILABILITY IN PEDIATRIC PATIENTS
Viera Lukacova, M.B. Bolger, and W.S. Woltosz
Simulations Plus, Inc., Lancaster, CA, USA, 93534
Purpose. To fit an absorption-pharmacokinetic model for simulation of Gabapentin in adult and pediatric populations. The model will be able to describe the nonlinear dose dependence of absorption mediated by an amino acid transporter as well as age-dependent renal clearance of Gabapentin. Methods. GastroPlusTM 5.3 with the PBPKPlusTM Module (Simulations Plus, Inc., Lancaster, CA) was used to simulate adult human plasma concentration time (Cp-time) profiles after oral administration of Gabapentin in adults (doses ranging from 400 mg to 1600 mg) and children (400 mg dose). A physiologically-based pharmacokinetic model (PBPK) was used in all simulations. Tissue/plasma partition coefficients were calculated using in silico physicochemical properties (ADMET Predictor™, Simulations Plus, Lancaster, CA) and the Rodgers (2005, 2007) algorithm. The renal clearance of Gabapentin was estimated to be equal to glomerular filtration rate (GFR) times fraction unbound in plasma (Fup) in both populations. Experimental GFR values from literature were used for both populations. The nonlinear dose dependent absorption of Gabapentin was simulated by incorporating an intestinal influx transporter and in vitro affinity measurements of Gabapentin interaction with LAT1. Results. The fitted absorption-pharmacokinetic model was able to simulate the non-linear dose-dependent pharmacokinetics in an adult population. The adult model was successfully scaled to pediatric physiologies to estimate Gabapentin absorption and pharmacokinetics in children. For both populations the renal clearance estimated as GFR * Fup provided an accurate estimate of the clearance for the drug which is not extensively metabolized in humans and the urinary secretion is governed only by glomerular filtration. Conclusions. In vitro data and/or in vivo Cp-time profiles from adult populations can be successfully used to predict the Gabapentin Cp-time profiles in pediatric patients if the organ physiology for a given age is accompanied by scaling of the gastrointestinal tract parameters and glomerular filtration rate to the same age.
REFERENCE
- Rodgers T., Leahy D., Rowland M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci. 2005 Jun;94(6):1259–76. Erratum in: J Pharm Sci. 2007 Nov;96(11):3151–2.
153. USE OF POCKET TIPS TECHNOLOGY IN A PERKIN ELMER EVOLUTION P3 DISPENSER FOR CYP450_RI ASSAYS
Consuelo Tudela, José Pérez del Palacio, Antonio Jimenez, Laura Fernandez, Guillermo Tarazona, Manuel Sánchez, and Fernando Pélaez
Centro de Investigacion Basica de España (CIBE), Merck Sharp & Dohme, Madrid, Spain
Organic solvents are often required for the solubilization of lead compounds prior to test in any in vitro screens since most of them have a lipophilic structure. However, it is well known that organic solvents can severely affect the activity of some CYP isoforms involved in drug metabolism. In fact, Dimethyl Sulphoxide (DMSO), the solvent of choice for solubilization of chemical libraries for High Throughput Screening (HTS) applications, substantially inhibits some cytochrome subfamilies. Due to this low DMSO tolerance, it is absolutely required to transfer a very low volume of the test compound solutions dissolved in 100% DMSO to the assay plates. A usual practice to avoid having to invest in expensive nanoliter dispenser instruments is to prepare intermediate solutions of DMSO sample stocks with other organic solvents, which allows dispensing microliters instead. We had previously shown that a mixture DMSO:MeOH could be the best choice to balance compound solubility and the signal noise-to-ratio required for CYP activity assays. However, mixing DMSO with other solvents contributes to compound precipitation and increases variability and error, affecting data quality. Moreover, handling MeOH in automatic workstations involves some issues, being necessary to saturate the system with this solvent before dispensing the samples. Due to these issues, we have continued exploring the possibility of pipetting sub-microliter volumes of the samples in the CYP inhibition assays. Also, transferring small volumes directly from the compound library to assay plate is more cost effective, due to the savings in reagents and consumables and the reduction in time. Furthermore, DMSO solutions can be stored at room temperature for a longer time, making easier the compounds handling. We have evaluated the PocketTips® nanoliter dispensing technology (nAscent BioSciences Inc.) using the Evolution P3® (EP3) liquid handling station from Perkin Elmer for CYP activity assays. PocketTips® are disposable pipette tips containing a “pocket”, which is an open capillary molded on the inside surface that load and transfer precise nanoliter amounts of the sample. Using 250nl PocketTips, the samples can be transferred directly from DMSO dilutions to the assay plates, achieving 0.25% DMSO final concentration, a suitable concentration. Our results show that PocketTips® technology provides accurate, precise and reproducible sub-microliter pipetting. The results obtained with a set of CYP inhibitors are comparable to those achieved with the conventional system previously used.
154. HEPAC2: SERUM-FREE, STANDARDIZED, VALIDATED AND RE-USABLE PRIMARY HUMAN HEPATOCYTES FOR THE ANALYSIS OF XENOBIOTICS
Anett Ullrich1, Donna Beer Stolz2, Ewa C.S. Ellis3, Dirk Koczan4, Stephen C. Strom3, George K. Michalopoulos3, Jan G. Hengstler5, Dorothee M. Runge6, Berg Christine1, and Dieter Runge1
1PRIMACYT Cell Culture Technology GmbH, Schwerin, Germany, 2Cell Biology, Center for Biological Imaging, University of Pittsburgh, Pittsburgh, PA, 15213, 3Pathology, University of Pittsburgh, Pittsburgh, PA, 15213, 4Immunologie, Universität Rostock, Rostock, Germany, 5Leibniz Research Centre for Working Environment and Human Factors, Dortmund, Germany, 6Diagnostische Systeme und Technologien GmbH, Schwerin, Germany
Human hepatocytes are the in-vitro system of choice to study drug-induced processes in man. Here, we present HEPAC2: A standardized and validated culture system in which functional human hepatocytes can be maintained serum-free for several weeks. Anabolic and catabolic hepatocellular functions and cellular vitality are monitored daily. Albumin and urea are produced on a relatively constant level for up to 2–3 weeks, while the cells remain viable. An extensive analysis of RNA-expression profiles has been performed using Affymetrix microarrays. Based on this, a standard protocol was established that allows repeated exposure of hepatocytes to test substances for studying drug metabolism. In this protocol hepatocytes are exposed to test pieces for 24 hrs. Subsequently, the culture medium is replaced by medium without the test substance and the same exposure scenario is repeated in intervals of 4 days. As a first model substance we used acetaminophen (APAP) to assay the feasibility of this system. High doses of AAP (2815 mg/l) diminished urea production by 15–30% and albumin secretion by 70–80% and led to a complete loss of glycogen. These effects were reversible. After removal of AAP, secretion of urea and albumin returned to control levels, the glycogen stores were refilled. Within one cell culture this exposure scenario could be repeated 4–5 times without loss of reproducibility. A multi-center study has been initiated to evaluate HEPAC2 in different laboratories. A second protocol has been initiated to assay the cytochrome P450 (CYP) induction potential of xenobiotics. Here, the hepatocytes are cultured in the presence of prototypical inducers like rifampicin or beta-naphthoflavone or the substance(s) of interest for 48 hrs to induce CYP activities. After 48 hrs, CYP activities are detected by conventional assays, e.g. testosterone-6-beta-hydroxylase or ethoxyresorufin-O-deethylase. Then, hepatocytes are cultured in the absence of inducers or test substances, allowing CYP activities to return to basal levels, before a new induction is started. At least 2–3 induction cycles can be initiated that lead to CYP activities at identical levels as determined during the first induction. In conclusion, these data demonstrate the suitability of our long-term culture system to serve as a tool for repetitive screening of drug-mediated changes on hepatocellular functions. It may be used to assay long-term effects of drugs and thereby become an alternative to animal testing. In addition, HEPAC2 may be combined with artificial devices as a support system during liver failure.
155. INCREASED SENSITIVITY FOR DRUG-INDUCED HEPATOTOXICITY USING A NOVEL HUMAN IN VITRO CO-CULTURE MODEL
Louise Sivertsson, Ylva Edling, Angelica Butura, Magnus Ingelman-Sundberg, and Monica Ek
Department of Physiology and Pharmacology, Section of Pharmacogenetics, Karolinska Institutet, Stockholm, Sweden
Hepatotoxicity is the most common unexpected drug-induced adverse event and is a major cause for disapproval or withdrawal of drugs from the market. There are few pre-clinical markers that are predictive of liver toxicity in humans and a major problem with the existing human in vitro methods is their lack of sensitivity and predictability. It is therefore of high importance to develop sophisticated in vitro test systems that can detect relevant predictive biomarkers for hepatotoxicity. Drug-induced hepatotoxicity in many cases requires the involvement of monocytes or liver resident macrophages, the Kupffer cells. These cells mediate inflammatory processes through the release of several pro-inflammatory cytokines and chemokines. In contrast to in vivo models, the influence of inflammatory cells in liver toxicity has rarely been studied in vitro. We set up a human in vitro co-culture model in order to study the inflammatory involvement of monocytes in drug-induced hepatotoxicity. We studied drug-induced toxicity in single cultures and co-cultures of a hepatoma (Huh-7) and a monocytic (THP-1) cell line. As a model drug we used troglitazone, a known hepatotoxic substance, in comparison to a non-hepatotoxic substance, rosiglitazone. Both substances are PPARg agonists aimed for treatment of type II diabetes. We investigated the viability of both cell types and the gene expression of inflammatory as well as stress-related genes by using a MTT assay and real-time PCR technique, respectively. We also evaluated the effect of antioxidants on cell viability. Our results show that co-cultures of Huh-7 cells and THP-1 cells have an increased sensitivity for troglitazone induced cytotoxicity as compared to single cultures (40% vs. 80% viability at 24h treatment). Rosiglitazone did not significantly affect the viability. Moreover, troglitazone treatment increased gene expression of the pro-inflammatory cytokines TNFα, IL-1b, IL-6 and the stress-related genes DDIT3/CHOP, MT2A, CXCL2 and CXCL10 in THP-1 cells and DDIT3/CHOP, MT2A, CXCL2, CXCL10, HspA6 and catalase in Huh-7 cells. Pre-treatment with the antioxidant catalase caused protection of the THP-1 cells in co-cultures treated with troglitazone whereas no significant effect was apparent with Huh-7. Addition of catalase on rosiglitazone-treatment did not have any effect. In conclusion, a human co-culture system with a hepatoma and a monocytic cell line may provide a sensitive and important in vitro system for predicting drug-induced hepatotoxicity.
156. CHARACTERIZATION OF DRUG DISTRIBUTION IN TISSUES USING A RAPID EQUILIBRIUM DIALYSIS DEVICE
Mark G. Qian1, Susan Chen1, Debra Liao1, Tai-Nang Huang2, Jing-Tao Wu1, and Frank W. Lee3
1Dmpk, Millennium Pharmaceuticals, Inc., Cambridge, MA, USA, 02139, 2Dept. of DMPK, Linden Bioscience, Lexington, MA, USA, 02421, 3Dmpk, Millennium Pharmaceuticals, Inc., Cambridge, MA, 02139
In the discovery of new drugs for targeting solid tumors, the fast characterization of the distribution of a drug in selected tissues may be desirable for insightful information of the drugs' preferential binding to the target (tumor) and undesired distribution in susceptible organs (e.g., brains and heart muscles). Regular PK parameters, e.g., apparent volume of distribution, are indicative of only overall extravascular distribution of drugs with limited or lack of information on the nature and pattern of distribution in different tissues. Traditional distribution dialysis2 is a powerful tool but suffers from tedious and lengthy operation and effect of volume shift. This work details our efforts of using a new multiplex dialysis device for tissue binding assay with focus on shortening the incubation time, reducing the volume shift, and increasing the sample throughput and reproducibility on a more automation friendly sample format.
The apparatus used was a rapid equilibrium dialysis (RED) device. The dialysis membrane has a 8,000 daltons cutoff-mass. The testing compounds were prepared in DMSO and diluted to 5 mM with blank plasma. Tissues of interest, including liver, heart, brain, kidney, lung, and tumor were homogenized in buffer. Both tissue homogenates and compound-spiked plasma were further diluted by a factor of 10 and placed in the membrane chamber (300 mL, red color coded chamber) and buffer chamber (500 mL, white color coded chamber), respectively. All experiments were conducted in triplicates unless otherwise noted. The device assembly was vortexed for 4 hours in an incubator maintained at 37oC. At the end of dialysis, an aliquot of 50 mL samples was collected separately from both chambers. For radioactive compounds, the samples were directly ready on a Wallac Microbeta counter. The quenching factor of each compound in plasma and tissue homogenates was adjusted against the controls. For non-radioactive compounds, LC/MS/MS methods were used to measure the concentrations of the testing compounds. A protein precipitation method was used to clean up samples. Several model compounds were tested. The preliminary results indicated that the dilution of homogenate is necessary in order to reach equilibrium in a reasonable timeframe. With 5 to 10-fold dilution, the incubation time is shortened down to less than 4 hours. In addition, due to the short incubation time and the use of low cutoff mass membrane, the volume shift was reduced to a less significant level that in general doesn't require correction. The presentation will summarize the detailed results of using the device for achieving faster and reproducible tissue binding assay.
REFERENCE
- J. Clausen and MH Bickel, J of Pharm. Sci., 1993 (82), 345.
157. THE INVESTIGATION OF CYTOTOXIC EFFECTS OF BORON COMPOUNDS ON HEALTY HUMAN LYMPHOCYTE
Zerrin Canturk1, Yagmur Tunali1, and Zafer Gulbas2
1Pharmacy Department Pharmaceutical Microbiology, Anadolu University, Eskisehir, Turkey, 2Medical Deparment, Osmangazi University, Eskisehir, Turkey
Our aim in this study is to investigate the effects of boron compounds, whose effects have been found on leukemic cells recently, on healty human lymphocyte. We have done research into effects of boron compound to be used as a chemotherapeutic agent with MTT and Neutral Red cytotoxicity tests. Lymphocyte was incubated with the different concentrations of boric acid and sodium tetra borate. Boric acid and sodium tetra borate were tested in the concentration range of 100 μM to 1000 μM (100, 250, 500, 750, 1000 μM concentration).The cell proliferation was measured after 24 h, 48 h, using the MTT and NR assays. According to these results, the cytotoxic effect was found in boric acid at a concentration of 100 μM dose %7, 250 μM dose %19 and sodium tetra borate at a concentration of 100 μM dose %5, 250 μM dose %5 of lymphocyte. Sodium tetra borate was found to be less cytotoxic than boric acid, at a concentration of 100 and 250 μM.
According to the Neutral Red test, the cytotoxic effects haven't been seen in boric acid and sodium tetra borate at a concentration of 100 μM. Besides, even a slight increase in the cytotoxic effect hasn't been detected among the results of 48th hours. When similiar concentrations were applied to leukemic cell series, we showed in our previous study that cytotoxic effect reached to %50 in boric acid, and %40 in sodium tetra borate. According to the our results, while boric acid and sodium tetra borate have a cytotoxic effect on leukemic cells, they affects human healty lymphocyte minimally. Therefore, boron compounds could be used as a chemotherapeutic agent; however, there is a need of making studies with boron compounds on different sort of cell groups.
158. MEASUREMENT OF PLASMA PROTEIN BINDING: A COMPARISON OF CONVENTIONAL, 96 WELL PLATE AND RAPID EQUILIBRIUM DIALYSIS (RED) METHODOLOGIES
Nasreen Malik, Anthony P. Glazier, Anthony P. Chadwick, Georgina Adjin-Tetty, and David J. Lankester
Metabolism, Covance Laboratories Ltd., Harrogate, United Kingdom
Plasma protein binding studies play an important role in the development of candidate drugs as they provide information on the biologically available free fraction of a drug, the concentration of which usually provides a better correlation with therapeutic and/or toxic responses than total drug concentration. Equilibrium dialysis is one of the most widely used methods for determining the plasma protein bound and free fractions of a drug. The aims of this work were to compare binding data generated using conventional equilibrium dialysis equipment with a 96-well plate format method and the Pierce Rapid Equilibrium Dialysis (REDTM) device. [14C]Warfarin, [14C]phenytoin and [14C]caffeine were used as model test substances which typically display high, intermediate and low levels of plasma protein binding. Investigations were performed using rat, dog, mouse and human plasma using up to three test substance concentrations. The 96 well plate and REDTM methodologies where both found to be robust and produce reproducible data which was comparable to that generated using the conventional apparatus. In addition, the REDTM device was found to significantly reduce the time to equilibrium and offer much enhanced ease of operation. In summary, these methodologies were found to be attractive alternatives to conventional equilibrium dialysis methodologies and provided robust higher-throughput methods for use in plasma protein binding studies, thus offering potential screening capacity for multiple compounds with rapid production of data.
159. DIFFERENT ACTIVITY OF ESSENTIAL OIL OF CORIANDER (CORIANDRUM SATIVUM L. APIACEAE) ON OXIDATIVE PROCESSES UNDER IN VITRO AND IN VIVO CONDITIONS
Isidora Samojlik1, Biljana Bozin2, and Kornelija Ðakovic-Svajcer1
1Dept. of Pharmacology and Toxicology, Medical Faculty, Novi Sad, Yugoslavia, YU-21000, 2Dept. of Pharmacy, Medical Faculty, Novi Sad, Yugoslavia, YU-21000
Today, botanical products are becoming increasingly popular as alternative medicines used alone or in combination with therapeutic drugs, thus raising the potential of herb-drug interactions. Coriander seed (Coriandrum sativum L. Apiaceae) is used as very common spice in commercial foods, dishes and beverages, while its essential oil is usually used, alone or in combination, in the treatment of various gastrointestinal disturbances [1]. This work investigates the probable protective-antioxidant action of essential oil of coriander assessed by the corresponding in vitro and in vivo tests. In the in vitro experiments, essential oil was tested as potential scavenger of stable 2,2-diphenyl-1-picrylhydrazil (DPPH) radical and hydrogen peroxide (H2O2), as well as inhibitor of lipid peroxidation (LP) in Fe2+/ascorbate and Fe2+/H2O2 systems of induction [2]. In vivo experiments were related to antioxidant systems (activities of glutathione peroxidase (GSHPx), peroxidase (Px), catalase (CAT), xanthine oxidase (XOD), reduced glutathione (GSH) content and intensity of lipid peroxidation (LPx)) in liver homogenate of mice after their chronic treatment with essential oil (in appropriate form for per os application in the dose recalculated from human daily dose), or in combination with carbon tetrachloride (CCl4) [3]. Gas chromatography-mass spectrometry (GC-MS) analysis of essential oil revealed that dominant component was linalool (74,6%) followed by camphor (5,9%), geranyl acetate (4,6%) and p-cimene (4%). Essential oil neutralized H2O2 in the solution (IC50 = 4.05 μg/ml) and DPPH (IC50 = 53.5 μg/ml) radical in a dose-dependent manner. Inhibition of LP was detected in Fe2+/ascorbate system of induction ranging from 16.2 to 33.67%. In other tested system essential oil exibited strong prooxidant actvity (from –167.53 to –257.79%). After 5 days treatment with coriander essential oil, the activity of liver Px and LPx in mice was increased, which was in accordance to previous in vitro results. Other biochemical parameters were not changed. The application of CCl4 in mice treated with essential oil caused the reduction of GSH content only, while other effects of its toxicity missed. Although in vitro results point on potential prooxidative capability of essential oil of coriander, in vivo examination shows the possibility of its preventive action in such effect caused by other substances. This implicates the caution in the usage of phytopreparation, especially regarding its dose, bearing in mind the possible prooxidative effect of other drugs combined with it.
REFERENCE
- Blumenthal, M. (Ed): The Complete German Commission E Monographs – Therapeutic Guide to Herbal Medicines. American Botanical Council, Austin, Texas 1998.
- Bozin B, Mimica-Dukic N, Samojlik I, Jovin E. Antimicrobial and Antioxidant properties of Rosemary and Sage (Rosmarinus officinalis L. and Salvia officinalis L., Lamiaceae) Essential Oils. J. Agric. Food Chem. 2007;55:7879–7885.
- Popovic M, Kurinovic B, Trivic S, Mimica-Dukic N, Bursac M. Effect of Celery (Apium graveolens) Extracts on some Biochemical Parameters of Oxidative Stress in Mice Treated with Carbon Tetrachloride. Phytother. Res. 2006;20:531–537.
160. CYTOPROTECTION AGAINST OXIDATIVE STRESS INDUCED TOXICITY IN HEPATOCYTES BY HYDROGEN SULFIDE
Mohammad A. Eghbal1, Mahdieh Anoush2, Hossein Hamzeiy1, Peter P. Pennefather3, and Peter J. O'Brien3
1(1) School of pharmacy, (2) Drug Applied Research Center, Tabriz University of Medical Sciences, Tabriz, Iran, 2Pharmacology & Toxicology, Faculty of Pharmacy, Zanjan University of Medical Sciences, Zanjan, 3Pharmaceutical Sciences, Universiy of Toronto, Toronto, ON, Canada, M5S 2S2
Introduction. Hydrogen sulfide (H2S) is responsible for the smell of rotten eggs. It is an environmental and industrial pollutant that at high concentrations (more than 150 ppm) is highly toxic and often fatal. Physiological concentrations of H2S (10 - 130 μM) have recently been suggested to have an intracellular signaling role (Wang, R., The gasotransmitter role of hydrogen sulfide. Antioxid Redox Signal. 2003, 5, 493–501. ). Here, we have studied the protective effects of H2S against oxidative stress induced cytotoxicity by various agents towards freshly isolated rat hepatocytes. Methodology. Hepatocytes were isolated by liver perfusion with collagenase. Lipid peroxidation was determined by measuring thiobarbituric acid-reactive substances. Cytotoxicty in hepatocytes was assessed by trypan blue exclusion. Results and conclusion. H2S protected against cytotoxicity caused by either Cu2+, Cd2+, Hg2+ As3+ or the Fe3+- and/or Zn-8-hydroxyquinoline complexs. The cytoprotective effect of H2S was preceded by a decrease in hepatocyte lipid peroxidation induced by Cu2+. Preincubating hepatocytes with H2S also protected hepatocytes against hypoxia induced cell death. H2S prevented MNNG-induced cytotoxicity in a concentration dependent manner. Our data suggest that H2S could be protective against oxidative stress induced cell injury caused by various kinds of threats.
161. ENDOPLASMIC RETICULUM STRESS INITIATES TUBULAR CELL DEDIFFERENTIATION AND DEATH DURING CYCLOSPORINE TREATMENT
Nicolas Pallet1, Marion Rabant1, Eric Thervet1, Philippe Beaune1, Christophe Legendre2, and Dany Anglicheau1
1U775, INSERM, Paris, France, 75006, 2Service de transplantation rénale, Hopital Necker, Paris, France
Chronic nephrotoxicity related to Cyclosporine (CsA) therapy leads to interstitial fibrosis and tubular atrophy and the tubular epithelium plays a central role of the in the pathogenesis of chronic nephropathies, especially through epithelial to mesechymal transition. cDNA microarrays screening on tubular cells led us to demonstrate that CsA induces endoplasmic reticulum stress (ER stress), since GRP78, protein disulphide isomerase, CHOP and Herp are expressed at protein and mRNA levels during CsA exposure. ER stress was also observed in rat kidneys treated with 15/mg/kg CsA. CsA and various ER stress inducers (thapsigargin, A23187 and DTTox) trigger marked tubular phenotypic changes (TPC) reminiscent of an incomplete epithelial to mesenchymal transition with cells loosing cell-cell contact, down regulation of E-cadherin expression, nucleo-cytoplasmic redistribution of ß-catenin, and increase of the expression of the fibroblast marker HSP47. ER stress and TPC are also observed when cyclophilin A gene expression is targeted by siRNA. Cyclophilin A, an isomerase implicated in nascent protein folding, is inhibited by CsA. When CsA is co-incubated with Salubrinal, an ER stress inhibitor, tubular phenotypic changes do not appear since cell shape remains polygonal, E-cadherin is not down regulated, HSP47 expression level remains indetectable and β-catenin do not translocate into the nucleus. Moreover, Immunohistochemical detection of GRP78 used as a surrogate marker of tubular ER stress in protocol biopsies performed on 60 transplanted patients is significantly associated with tubular atrophy and interstitial fibrosis. In conclusion, CsA induces an ER stress in tubular cells that initiates tubular cell phenotypic changes suggestive of an incomplete epithelial to mesenchymal transition. Salubrinal, in inhibiting ER stress, blocks tubular epithelial phenotype alterations and reduces cell death during CsA exposure. This study suggests that ER stress may initiates epithelial-to-mesenchymal transition during CsA treatment and its detection could serve as an early biomarker of interstitial fibrosis and tubular atrophy.
162. A NOVEL PANEL OF DOUBLE ‘HUMANISED’ & KNOCKOUT PXR AND CAR MOUSE MODELS
Jillian Ross1, Nico Scheer2, and Roland Wolf3
1CXR Biosciences Limited, Dundee, United Kingdom, DD1 5JJ, 2Artemis Pharmaceuticals GmbH, Cologne, Germany, 51063, 3CXR Biosciences Ltd, Dundee, United Kingdom
The interactions of chemicals with constitutive androstane receptor (CAR) and pregnane X receptor (PXR) are complex, not only in the fact that they regulate common genes, but also because a single chemical agent can often interact with both receptors. In order to fully understand the relative importance of the receptors in the efficacy and safety of drugs, we have created a novel panel of mouse models in which the murine genes have been exchanged for their human counterparts in the same locus (huPXR, huCAR) or knocked out (PXRKO, CARKO). In an attempt to resolve issues with receptor cross-talk and redundancy, the following models have also been created; huPXR/huCAR, PXRKO/CARKO, huPXR/CARKO and PXRKO/huCAR. Using this panel, “species-specific”, CAR- and PXR-dependent differences have been demonstrated in the regulation of P450s, Phase II enzymes and transporters in response to treatment with non-genotoxic carcinogens. Programme funded by ITI Life Sciences.
163. EFFECT OF TARAXACUM OFFICINALE WATER EXTRACT ON INOS GENE EXPRESSION IN RAW264.7 MACROPHAGES: ROLE OF HEME OXYGENASE-1 VIA ACTIVATION OF AKT AND NRF2 SIGNALING
Sung Su Yea1, Chun Yeon Choi1, Gook-Hee Cho1, Won Hee Jang1, Dae-Hyun Seog1, Yeong-Hong Park1, Young-Sun Song2, and Kwang-Hyeon Liu3
1Dept. of Biochemistry and Biohealth Product Research Center, College of Medicine, Inje University, Busan, South Korea, 614–35, 2School of Food and Life Science and Biohealth Product Research Center, Inje University, Gimhae, South Korea, 621–749, 3Dept. of Pharmacology, Inje University, Busan, South Korea, 614–735
Taraxacum officinale has been known to have various biological activities, such as anti-carcinogenic, anti-oxidative, and anti-inflammatory effect. It also has been known that T. officinale produced an inhibition of nitric oxide (NO) production. In the present study, we investigated the effect of T. officinale water extract (TOWE) on NO production and its mode of action in activated macrophages. Lipopolysaccharide (LPS) induced NO production in RAW264.7 macrophages, which was markedly inhibited by TOWE in a dose-dependent manner. TOWE also decreased the expression of inducible nitric oxide synthase (iNOS) protein and mRNA in LPS-stimulated RAW264.7 cells. To investigate the inhibitory mechanism, we examined the effect of TOWE on the expression of heme oxygenase-1 (HO-1) which has been known to be a cytoprotective and anti-inflammatory enzyme as well as a negative regulator for iNOS expression. Enzyme activity of HO-1 was increased by the treatment of TOWE. TOWE induced the expression HO-1 mRNA and protein in a dose-dependent manner. TOWE also activated upstream signaling of HO-1 gene expression, such as nuclear translocation of Nrf2 and phosphorylation of AKT. Collectively, these results suggest that decreased NO production by TOWE in RAW264.7 macrophages is due to the inhibition of iNOS gene expression and is mediated, at least in part, through induction of AKT/Nrf2-mdeiated HO-1 expression.
164. OXIDATIVE STRESS IN THE MECHANISM OF ORGANOPHOSPHATES NEUROTOXICITY
Valerii Tonkopii
Institute of Limnology, Russian Academy of Sciences, 196105, St. Petersburg, Russia.
The toxic effect of some organophosphates (OPs) is not limited to inhibition of cholinesterase: following the cholinergic crisis changes in non-cholinergic neurotoxic parameters, such as specific damage to cell membranes, are observed. The present study was undertaken to elucidate the relations between lipid peroxidation, OPs toxicity and delayed, long lasting, non-cholinergic changes. We studied the influence of OPs intoxication on lipid peroxidation in rat cerebral hemispheres. The level of lipid peroxidation was measured as the amount of common phospholipids, peroxidate lipids and malondialdehyde (MDA) in reaction with thiobarbituric acid. Results were compared to those with pre-treatment with atropine and reversible cholinesterase inhibitor - galanthamine alone or together with different antioxidants ( a-tocopherol and oxymetacyl). OPs caused a rapid, dose-dependent increase of peroxidate lipids and MDA 15–30 days after intoxication. The level of lipid peroxidation correlated with the rate of conditioned reflex reaction. With paraoxon and sarin pre-treatment with atropine and galanthamine totally prevents the all symptoms of intoxication and changes in lipid peroxidation. Comparatively such type of prophylaxis in soman and malathion poisoned rats didn't normalize the biochemical and physiological parameters. The protective effect of antioxidants against soman and malathion - induced lipid peroxidation was shown. Therefore soman and malathion- associated lipid peroxidation is likely to arise mainly as a primary change which may, however, play a significant role in delayed neurotoxicity and conditioned reflex activity.
165. RECOMBINANT HUMAN ERYTHROPOIETIN PROTECTS HUMAN TUBULAR CELLS AGAINST CYCLOSPORINE TOXICITY
Nicolas Pallet1, Dany Anglicheau1, Philippe Beaune1, and Frank Martinez2
1U775, INSERM, Paris, France, 75006, 2Service de transplantation rénale, Hopital Necker, Paris, France, 75015
Introduction: human recombinant erythropoitein (rhEPO) is known to protect tubular cells from various injuries such ischemia-reperfusion or cisplatin treatment. Since calcineurin inhibitors use mediates renal allograft tubular injury and significantly alter graft function, we investigated whether rhEPO could protect human tubular cells from cyclosporine (CsA) toxicity. Methods. Primary cultured human tubular cells were exposed 72 hours to increasing CsA and/or various rhEPO concentrations and cell viability was measured with MTS test. Results. CsA induced a concentration-dependant decrease in cell viability with a CsA LC50 of 6 μM. rh EPO co-incubated with CsA increased tubular cells viability in a concentration dependant manner with a maximum effect of 50 UI/ml. This concentration increased cell viability around 30 %, at each CsA concentration level. Since EPO receptor was expressed in tubular cells in culture at mRNA level, we tested whether EPO cytoprotective effects could be mediated trough EPO receptor activation. rhEPO seems to activate Akt signaling because we found that rhEPO induced a transient phosphorylation of Akt at Thr 308 (+40% 10 to 20 minutes post stimulation). Akt is known to activate anti apoptotic pathways through EPO receptor activation and PI3 kinase signaling. Moreover, we observed that CsA transiently induced autophagy in tubular cells at 48 through LC3BII detection in western blot and autophagosomes visualization with electronic microscopy. Autophagy is a protective mechanism during various stresses but may lead to cell death when stress is sustained. Interestingly, EPO alone do not induce autophagy but when added to CsA, autophagy was more prolongated than with CsA alone. Indeed, at 72 hours post stimulation, LC3BII expression was observed under CsA+EPO treatment but not under CsA alone. Conclusion. our data support a cytoprotective effect of rHuEPO on CsA induced tubular toxicity. One can hypothetize that these effects may be mediated trough Akt signaling activation and autophagy regulation.
166. AUTOPHAGY PROTECTS TUBULAR CELL AGAINST CYCLOSPORINE TOXICITY
Nicolas Pallet, Dany Anglicheau, and Philippe Beaune
U775, INSERM, Paris, France, 75006
A major side effect of the powerful immunosuppressive drug cyclosporine (CsA) use is the development of a chronic nephropathy of which mechanism are not fully understood. Recent data suggest that tubular cells play a central role in the pathogenesis of chronic nephropathies. We have showed CsA was responsible for endoplasmic reticulum (ER) stress in tubular cells. Autophagy has recently been described to be induced by ER stress and to alleviate its deleterious effets. In this study, we demonstrate that CsA induced autophagy in primary cultured human tubular cells through LC3BII expression and autophagosomes visualization with electronic microscopy. Autophagy was dependant of ER stress since Salubrinal, an inhibitor eIF2α dephosphorylation that protects cells against ER stress, totally inhibited LC3BII expression. Furthermore, autophagy inhibition during CsA treatment with 3-methyl adenine, an autophagosome inhibitor, significantly increased tubular cell death. Finally, immunohistochemical analysis on rat kidneys demonstrated that LC3B staining was positive on injured tubular cells, suggesting that CsA induced autophagy in vivo. Taken together, these results demonstrate that CsA, through ER stress induction, activates autophagy as a protection against cell death.
167. HEPATIC ATP-DEPLETION PRECEDES THIOACETAMIDE-INDUCED LIVER DAMAGE
Sven Gottschalk1, Tom S. Chan1, Valérie-Ann Raymond1, Dieter Leibfritz2, Claudia Zwingmann1, and Marc Bilodeau1
1Département de médicine, Université de Montreal, Montreal, QC, Canada, H2X1P1, 2Department of Organic Chemistry, University of Bremen, Bremen, Germany
Introduction. Acute thioacetamide (TAA; CH3-CS-NH2) administration is widely used in animal models to investigate mechanisms of toxin-induced liver injury, regeneration and fibrosis. TAA is metabolized by micosomal CYP2E1 to TAA-S-oxide, which in turn is oxidized by CYP2E1 to TAA-S,S-dioxide. This highly reactive metabolite covalently modifies macromolecules in all cell compartments. It has been shown that TAA-bioactivation is subject to saturation kinetics and CYP2E1 has less specificity for TAA-S-oxide than for TAA. However, the exact cause leading to cell death and whether covalent binding to macromolecules is responsible for hepatic necrosis still remains unclear. Aim. Cellular energy metabolism in mouse liver after TAA-administration was followed to elucidate a mechanism that may pre-dispose liver cells to die from necrosis. Methods. BALB/c mice were injected with a non-lethal hepatotoxic dose of TAA (200 mg/kg, ip) and sacrificed 1–6, 12, 18 and 24hrs after TAA-injection. Standard assays were used for serum-ALT/AST and caspase-3 determinations. ATP/ADP/AMP and GSH/GSSG were analyzed by HPLC. NMR analysis: Mice were injected with [U-13C]glucose (500 mg/kg, ip) 45 min prior to sacrifice. Ex vivo 13C-NMR-spectra of water soluble metabolites from liver-extracts were recorded. Flux of 13C through pyruvate carboxylase (PC) and pyruvate dehydrogenase (PDH) was followed up by 13C-isotopomer analysis. Results. One hour after TAA-injection the hepatic energy charge ([ATP]+[ADP]/2)/([ATP]+[ADP]+[AMP]) dropped to 68±8% control (P<0.001) as indicated by decreased ATP (63 ± 13%control, P<0.005) and increased ADP and AMP (130 ± 4, P < 0.01 and 214 ± 28, P < 0.001; % controls, respectively). Neither the energy charge nor the ATP-levels recovered at any time-point up to 24hrs. In fact, ATP further declined to 27 ± 4% control (P < 0.001) at 24hrs. Analysis of 13C-NMR spectra suggests diminished flux trough PC (65 ± 17% control) and decreased glycolytic activity (fractional enrichments in [3-13C]lactate and [3-13C]alanine: 72 ± 3 and 35 ± 4% controls, respectively) at 1hr, while PDH-flux was at control-level. GSH-levels were unchanged up to 12hrs and decreased at 18 and 24 hrs (37 ± 9 and 28 ± 7% controls, P < 0.001, respectively). The ratio GSH/GSSG was not different from controls at any time-point. Histological evidence and serum-ALT/AST activities indicate that TAA-induced liver injury does not occur before 6 hrs after injection. No significant Caspase-3 activity was found up to 24 hrs after TAA-treatment, ruling out the involvement of apoptosis. Conclusions. Our results demonstrate a deterioration in hepatic energy status and energy metabolism early and prior to any identifiable signs of TAA-induced liver damage. We suggest that the decrease of ATP – due to an unknown mechanism – pre-disposes livers cells to undergo necrosis (through energy failure) instead of apoptosis (which requires well maintained ATP-stores). The absence of any evidence for a redox status change also supports this hypothesis.
168. EVALUATION OF THE NEW SOFIE SYSTEM FOR DETECTION AND ANALYSIS OF LOW LEVEL RADIO LABELLED COMPOUNDS ON-LINE WITH RADIO FLOW ANALYSERS
Keith Hall1 and Huw W. Loaring2
1LabLogic Systems Ltd., Sheffield, United Kingdom, 2LabLogic Systems Ltd., Sheffield, United Kingdom, S10 2QJ
Recent moves towards the use of increasingly lower levels of radioactivity in samples for metabolite profiling whilst still favouring the speed and convenience of on-line detection have led to the development of SoFie. On-line radio detection of compounds separated by both HPLC and the newer fast LC/Rapid resolution techniques remains an important tool for researchers. The challenge is to detect ever decreasing levels of activity whilst remaining in the highly controlled and regulated environment of the trusted software systems. Here we show that the new SoFie system, controlled by the Laura radio chromatography system offers up to 8 times the level of sensitivity of standard on-line detection. This eliminates the need for fraction collecting which is always time consuming and potentially destructive to the delicate sample as further processing is required for counting on plate readers. The SoFie system also offers enhanced resolution as well as improved limits of detection.
169. PHASE I AND PHASE II METABOLITE IDENTIFICATION ON A TRIPLE-QUADRUPOLE MASS SPECTROMETER USING 2 MILLISECOND DWELL TIMES
Laurance Lee
Thermo Fisher Scientific, San Jose, CA, USA, 95134
Methods to identify potential drug metabolites on a triple-quadrupole mass spectrometer include full scans in the first or third quadrupole, product scans, common neutral loss scans, and MRM/SRM (multiple reaction monitoring or selected reaction monitoring) experiments. These methods can be further improved by application of enhanced resolution and/or data dependent full product MS/MS scans. The relatively slower cycle time of full scans (full MS, product or neutral loss) as survey scans necessitate application of multiple injections for a comprehensive examination of metabolites. Alternately, surveying for metabolites using SRM scans can take advantage of the instrument's sensitivity and cycle time to identify and quantify expected metabolites. In silico metabolism prediction software and general knowledge of metabolism rules may provide hundreds of potential precursor to product transitions, depending on the structure of the parent drug. While prior applications of the SRM survey scan method have been reported using dwell times as fast as 5 milliseconds, shorter dwell times can further improve instrument productivity. We report demonstration of workflows that use the SRM survey scan with 2 millisecond dwell times per channel, followed by data-dependent full product MS/MS scans on a hepatocyte incubation of donezepil in plasma. Comparison of the application of 5 millisecond versus 2 millisecond dwell times will be reported in terms of sensitivity and limits of detection. With this faster dwell time, as many as 300 SRM transitions can be followed in one scan event, with sufficient scans under a peak for reliable, sensitive quantitation. Multiple experiments will be described that will take advantage of the faster instrument cycle time, including more comprehensive examination of potential metabolites, polarity switching experiments, enhanced resolution SRMs, SRMs utilizing noise-reducing FAIMS, and experiments using fast chromatography on a UPLC. Found metabolites will be reported using commercially available software.
170. THE DISPOSITION, METABOLISM AND EXCRETION OF DORIPENEM AFTER A SINGLE INTRAVENOUS DOSE OF 500 MG IN HEALTHY MALE SUBJECTS
M. Vermeir1, F. Cuyckens1, R. Hurkmans1, I. Goris1, N. Nelissen1, J. De Raedemaecker1, D. Desai-Krieger2, I. Cirillo2, L. M. Kao2, and G. Mannens1
1Johnson & Johnson Pharmaceutical Research and Development, a division of Janssen Pharmaceutica N.V., Beerse, Belgium, 2Johnson and Johnson Pharmaceutical Research and Development, L.L.C., Raritan, NJ, USA, 08869
Doripenem ((+)-(4R,5S,6S)-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-3-[[(3S,5S)-5-[(sulfamoyl-amino)methyl]-3-pyrrolidinyl]thio]-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid) is a broad-spectrum injectable carbapenem antibiotic with activity against Gram-positive and Gram-negative pathogens, including strains resistant to multiple antibiotic classes. The compound has potential therapeutic value for the treatment of hospital-based infections. In a clinical study, 8 healthy men received a single 500-mg dose of Doripenem, containing 50 μCi of 14C-Doripenem, administered as a 1-hour intravenous infusion. Urine, faeces and plasma were collected up to one week after dosing. Radioactivity levels were measured by liquid scintillation counting, and metabolite profiles were investigated by radio-HPLC and LC-MSMS. The 14C-Doripenem-related radioactivity was almost exclusively excreted in urine. At 7 days after dosing, urinary excretion accounted for 95.3% of the administered radioactivity, whereas only 0.72% of the dose was recovered in faeces. The largest fraction of radioactivity was excreted in urine within 4 hours after dosing (85.1% of the dose). Doripenem was not extensively metabolized in humans. The major metabolic pathway was the cleavage of the β-lactam ring, which formed the dicarboxylic acid metabolite, Doripenem-M-1. A total mean of 97.2% of the radioactivity recovered in urine was excreted as unchanged Doripenem (78.7 ± 5.7%) and Doripenem-M-1 (18.5 ± 2.6%). Three minor metabolites identified in urine were oxidized Doripenem-M-1 and the taurine and glycine conjugates of Doripenem-M-1, each representing less than 3% of the administered dose. Unchanged Doripenem and Doripenem-M-1 accounted for 80.7% and 12.7%, respectively, of the area under the plasma total radioactivity-versus-time curve. The results showed that Doripenem is predominantly eliminated as unchanged drug via the urine, with only a fraction metabolized to Doripenem-M-1 and other minor metabolites.
171. ELUCIDATION OF THE CHEMICAL STRUCTURE OF NEW PHASE I METABOLITES OF POTENTIAL ANTIFUNGAL DRUG, 3-(4-BROMOPHENYL)-5-ACETOXYMETHYL-2,5-DIHYDROFURAN-2-ONE, IN MOUSE URINE
Milan Nobilis1, Milan Pour2, Petr Senel2, Jan Pavlik2, Lenka Skalova3, and Jirí Kuneš2
1Department of Pharmaceutical Chemistry and Drug Control, Charles University, Faculty of Pharmacy, Hradec Kralove, Czech Republic, CZ-500 05, 2Department of Inorganic and Organic Chemistry, Charles University, Faculty of Pharmacy, CZ-500 05 Hradec Králové, Czech Republic, 3Department of Biochemical Sciences, Charles University, Faculty of Pharmacy, Hradec Kralove, Czech Republic, CZ-50005
3-(4-Bromophenyl)-5-acetyloxymethyl-2,5-dihydrofuran-2-one (LNO-18–22) belongs to a novel group of potential antifungal drugs, derived from a natural 3,5-disubstituted butenolide, (-)incrustoporine, as the lead structure. This lipophilic compound is characterized by high in vitro antifungal activity and low acute toxicity. A new HPLC bioanalytical method involving a direct injection or liquid-liquid extraction of incrustoporine analogues from mouse urine was developed and employed for the separation and identification of principal phase I and phase II metabolites using photodiode-array and mass spectrometric (ESI or APCI with IT) detection [1]. In this study, the chemical structure of newly found LNO-18–22 phase I metabolites was proposed based on their characteristic UV, MS spectra and their chromatographic behaviour. UV spectra of olefin-18, its arenoxide, HO-olefin-18a and HO-olefin-18b were found to be very similar (maximum at 300–307 nm corresponds to 3-(substituted aryl)-5-methylene-2,5-dihydrofuran-2-one type of chromophore), the molecular masses of these metabolites (267 Da) indicate the presence of an additional oxygen in the molecules of olefin-18-arenoxide, HO-olefin-18a and HO-olefin-18b in comparison with the molecular mass of olefin-18. To confirm the proposed chemical structures, more experiments are needed.
ACKNOWLEDGMENT
This work was supported by the Ministry of Education of the Czech Republic (projects No. MSM 0021620822 and Centre for New Antivirals and Antineoplastics 1M0508).
REFERENCE
- Nobilis M., Pour M., Pavlík J., Šenel P., Kuneš J., Vopršalová M., Kolárová L., Holcapek M.: “Metabolic profiling of a potential antifungal drug, 3-(4-bromophenyl)-5-acetoxymethyl-2,5-dihydrofuran-2-one, in mouse urine using high-performance liquid chromatography with photodiode-array and mass spectrometric detection.” Journal of Chromatography B 853 (2007) 10–19.
172. OPTIMISATION AND INTERPRETATION OF REACTIVE METABOLITE IN VITRO TRAPPING ASSAYS FOR USE IN EARLY DRUG DISCOVERY
Richard Weaver1, Sima Khan1, Cathy MacDonald1, Rob Riley1, Karine Litherland2,Iain Beattie2, and Christopher J. Smith3
1Discovery DMPK, AstraZeneca R & D Charnwood, Loughborough, United Kingdom, LE11 5RH, 2Medicinal Chemistry, AstraZeneca R & D Charnwood, Loughborough, United Kingdom, LE11 5RH, 3Drug Metabolism Pharmacokinetics, AstraZeneca, Cheshire, United Kingdom, SK10 4TG
Dealing with the risk associated with compounds that have the potential to form reactive metabolites has received much interest in the literature in recent years. The presence of reactive metabolites (RMs) does not simply equate to toxicity directly but the formation of these, often electrophilic, species is probably not desired owing to their inherent chemical reactivity with nucleophiles. Specifically, reaction with endogenous nucleophilic peptide residues in proteins or genetic material e.g. SH and NH2 protein sites and DNA bases, may result in immunological or genotoxic consequences. Additionally, there has been indirect evidence that compounds that form reactive metabolites tend to have higher incidence of idiosyncratic (type B) reactions (IDRs) versus those that do not form reactive metabolites. Cold-trapping techniques with hard and soft nucleophiles of reactive metabolites are often used in early screening strategies. The advantage of ‘front-loading’ these screens in early discovery enables many compounds to be screened, to develop structure-activity relationships and design out the liability. The authors have investigated the use of human liver microsomes with stable-labelled glutathione and cyanide with marketed drugs and in-house NCE compounds. The end-points were constant neutral loss scanning (CNL) and selected reaction monitoring (SRM) on a triple quadrupole mass spectrometer and full scan acquisition on a time of flight mass spectrometer (ToF). The success rate of the two instruments were compared. The RM hit rate was assessed with 100 randomly chosen compounds with both soft and hard nucleophiles. The hit rate was 25% (SRM) and 33% (ToF) of the compound set using 50 μM substrate with glutathione. The hit rate was 22% (SRM) and 18% (ToF) of the compound set using 50 μM substrate with cyanide. Additionally, the quantitative evaluation of glutathione adducts by inductively coupled plasma (ICP) mass spectrometry was determined for a subset of drugs.
173. DETERMINING THE LOCATION OF A DOUBLE BOND USING MASS SPECTROMETRY IN A SPECIES SPECIFIC METABOLITE FOUND IN DOG PLASMA
Cheng Hong Zhang, Matthew Baumgardner, Qin Yue, Bianca M. Liederer, Jason Halladay, V. Sashi Gopaul, S. Cyrus Khojasteh, and Cornelis Hop
Drug Metabolism & Pharmacokinetics, Genentech, Inc., South San Francisco, CA, USA, 94080
Objective. A major metabolite (M1) was identified in the plasma of dogs given a single IV dose administration of the parent compound I. This metabolite was only observed in the dog and appeared to be formed as the result of metabolic desaturation on the piperidine ring. The objective is to determine (1) the location of the double bond in this metabolite and (2) the drug metabolizing enzyme(s) responsible for it. Samples Analyzed. Plasma samples (9-hr time point) collected from Dog # 1, 2, & 3 following a single IV dose administration of I at 3 mg/kg were pooled (0.5 mL each) and protein precipitated with acetonitrile. The protein free filtrates were dried under nitrogen and reconstituted in water:acetonitrile (1:1). LC-MS Analysis. Analysis was performed on the Finnigan LTQ coupled to an Ultra high pressure liquid chromatograph (UPLC, Accela) and a TriVersa™ Nanomate® and ESI Chip™ system. Samples were first analyzed on the LTQ system by MS and MSn to confirm the presence of M1. The HPLC interval where M1 eluted was isolated from the 9-hr plasma sample by fraction collection and reanalyzed by infusion. An aliquot of the extract was then analyzed by the Finnigan LTQ-Orbitrap MS coupled to a UPLC (Accela) to determine accurate mass. Summary of Results. The combination of high resolution and ion trap mass spectrometry analyses revealed that M1 is most likely an unsaturated metabolite. The MS/MS fragmentation pattern associated with the fluorinated piperidine ring of the parent and metabolite showed a loss of HF for I but distinctively absent for M1, a determining factor to identify the position of the double bond at C2 and C3 in the piperidine ring. However, the possibility of the double bond being elsewhere in the ring cannot be excluded without an authentic standard. Of further interest, the metabolite which was only observed in the in vivo samples of the dog but not the monkey has not been detected in human liver microsomes fortified with NADPH. The enzyme responsible for this biotransformation pathway is still under investigation.
174. IN VITRO HYDROLYSIS OF ESLICARBAZEPINE ACETATE, A NEW ANTIEPILEPTIC DRUG
Ana Isabel Loureiro, Carlos Fernandes-Lopes, Lyndon Wright, and Patrício Soares-da-Silva
Department of Research & Development, BIAL, S Mamede do Coronado, Portugal, 4745-457
Eslicarbazepine acetate (ESL), is a novel antiepileptic drug which shares with carbamazepine and oxcarbazepine the dibenzazepine nucleus bearing the 5-carboxamide substituent, but is structurally different at the 10,11-position. This molecular variation results in differences in metabolism, namely by preventing the formation of toxic epoxide metabolites such as carbamazepine-10,11 epoxide. Metabolism of ESL forms two major compounds, 10-hydroxy carbamazepine (S(+) and R(-)-licarbazepine) and oxcarbazepine (OXC), but their relative amounts are strongly dependent on the species and its ability to shift this redox equilibrium to either one side. It was found that ESL was almost instantly hydrolysed to the corresponding S(+) and R(-)-licarbazepine compounds in mouse, rat, rabbit and in human liver microsomes. In this study the hydrolysis of ESL has been investigated in vitro using several mouse tissues, including small intestine and stomach mucosa, liver, kidney, brain homogenates and plasma. To characterize the hydrolysis of ESL human liver and intestinal microsomes were used. In mouse, results indicate that hepatic and intestinal hydrolase activities are the major contributors for ESL hydrolysis. Kidney, stomach and plasmatic hydrolases also contribute however to a lesser extent. Mouse brain hydrolases are not involved in ESL hydrolysis. In human, ESL hydrolysis occurs to approximately the same extent in liver and intestinal microsomes which may indicate a high contribution of first-pass hydrolysis in ESL bioavailability.
175. THE METABOLISM OF THE CDK INHIBITOR AZD5438 IN HUMAN SYSTEMS: POTENTIAL FORMATION OF REACTIVE INTERMEDIATES
Chris R. J. Pollard1, Timothy Schulz-Utermoehl1, Sunil Sarda1, Julia J. Young1, and Catherine J. Duckett2
1Global Development DMPK & Bioanalysis, AstraZeneca, Macclesfield, United Kingdom, SK10 4TG, 2School of Physical and Geographical Sciences, Keele University, Staffordshire, United Kingdom, ST5 5BG
AZD5438([4-(3-Isopropyl-2-methyl-3H-imidazol-4-yl)-pyrimidin-2-yl]-(4-methanesulfonyl- [U-14 C]phenyl)-amine) is a novel inhibitor of cyclin dependent kinase (CDK) that was being targeted for the treatment of cancer. Preliminary in vitro and in vivo metabolism studies of AZD5438 suggested that this compound could potentially generate reactive electrophilic metabolites that could covalently modify critical protein components leading to organ toxicity. To assess the potential formation of reactive metabolites of AZD5438, incubations with human hepatocytes were performed. Clinical plasma samples were also assessed. Analysis of the resultant metabolites by HPLC with mass spectrometric and radiometric detection was employed to detect and characterise any reactive species. The formation of a sulphate conjugated metabolite of AZD5438 that had the potential to undergo a loss of HSO4- to form an electrophilic carbo-cation intermediate was observed both in incubations with primary human hepatocytes and in human plasma samples. It is suggested that this metabolite was formed following sulphonation of a hydroxymeth1 metabolite. Incubation of AZD5438 with recombinant human cytochrome P450 (CYP450) enzymes demonstrated that several CYP450 enzymes were capable of producing the hydroxymeth1 metabolite. However, CYP2D6 produced this metabolite exclusively. Co-incubation of recombinant CYP2D6 with human cytosol or various recombinant human sulfotransferase enzymes (SULT1A1*2, 1A2*1, 1A3, 1E and 2A1) in the presence of AZD5438 or the synthetic hydroxymeth1 metabolite, failed to generate the sulphate conjugated metabolite of AZD5438. The covalent binding of AZD5438 to hepatic proteins was investigated using several test systems. AZD5438 underwent metabolism to a potential reactive intermediate. Both Phase I and II enzymes appear to be involved.
176. EVALUATION OF THE MULTIPLE DEPLETION CURVES METHOD, IMPLICATION FOR AN ACCURATE ASSESSMENT OF INTRINSIC CLEARANCE AND ENZYME KINETICS IN EARLY DRUG DISCOVERY
Erik Sjögren, Johan Gråsjö, and Hans Lennernäs
Department of Pharmacy, Uppsala University, Uppsala, Sweden, 75123
The use of multiple depletion curves for estimation of Vmax, Km and CLint [1] was thoroughly evaluated by means of experimental data and through a series of Monte Carlo simulations. The enzyme kinetics of 7 compounds was determined with the multiple depletion curves method (MDCM), the traditional initial formation rate of metabolite method (IFRMM) and the “in-vitro t½ method” [2] and the parameter estimates from the three methods were compared. The impact of a change in enzyme activity during the incubation period on the parameter estimates and the possibility to correct for this was also investigated. The MDCM showed good overall agreement to the IFRMM. Correction for a change in enzyme activity was possible and resulted in a better concordance in CLint estimates. The robustness of the method towards different rate of substrate turn over and choice of start concentrations was also demonstrated through Monte Carlo simulations. Further, the drawbacks associated with the “in-vitro t½ method” and its inherent assumptions were demonstrated both experimentally and by simulations. This study demonstrates that the MDCM is a robust and efficient method for estimating enzyme kinetic variables with high accuracy and precision. The method has potential to be applied for a wide range of applications, from pure enzyme kinetics to in vitro based PK predictions of compounds with multiple and/or unknown metabolic pathways.
REFERENCE
- Bousquet-Melou, A., et al., the loss of metabolic capacities of cultured hepatocytes: application to measurement of Michaelis-Menten kinetic parameters in in vitro systems. Xenobiotica, 2002. 32(10): p. 895–906.
- Obach, R.S., Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos, 1999. 27(11): p. 1350–9.
177. IMMUNOSUPPRESSIVE THERAPY ADJUSTED TO THE CYCLOSPORIN METABOLISM
Katalin Monostory1, Katalin Andor1, Pal Szabo1, Eniko Sarvary2, Zsuzsa Gerlei2, Janos Fazakas2, Zoltan Mathe2, Ibolya Gaal2, Jeno Jaray2, and Laszlo Kobori2
1Biochemical Pharmacology, Hungarian Academy of Sciences, Budapest, Hungary, H-1025, 2Transplantation and Surgical Department, Semmelweis University, Budapest, Hungary, H-1823
Success of organ transplantation and increasing graft and recipient survival is partly due to the improvement of immunosuppressive therapy. Cyclosporine A (CYA) widely used in liver or kidney transplantation selectively suppresses T-cell immunity. Therapeutic drug monitoring of CYA is essential because of the narrow therapeutic window and high interindividual variety in bioavailability, metabolism and elimination of CYA. Immunoassay techniques (FPIA, EMIT) offer simple and high throughput determination of CYA blood concentrations; however, the accuracy highly depends on the selectivity of the antibodies used in immunoassay procedures. Some of CYA-metabolites cross-react with the antibodies of immunoassays, which lead to false results of CYA concentration. An analytical system using two-dimension chromatography in combination with tandem mass spectrometry (HPLC-MS/MS) was developed for the measurements of blood concentrations of CYA and some of its hydroxylated metabolites, which may contribute to the toxicity of the parent compound. The association between CYA metabolites and liver function parameters (alkaline phosphatase, γ-glutamyltransferase, transaminases: GOT, GPT and serum bilirubin) was also investigated in liver transplants (n = 51). The concentration of 1,9-dihydroxy-CYA showed strong correlation with γ-glutamyltransferase and serum bilirubin levels. Additionally, the alterations in 1,9-dihydroxy-CYA concentrations seemed to cause parallel changes of these parameters. The immunosuppressive therapy for the patients with continuously increased levels of 1,9-dihydroxy-CYA was modified. Replacement of CYA to tacrolimus resulted in the elimination of 1,9-dihydroxy-CYA and normalization of γ-glutamyltransferase and serum bilirubin levels. Although our HPLC-MS/MS method is not considered to be applicable for high throughput screening of CYA blood levels, but may provide information on the alterations in CYA metabolism and may lead to the optimization of immunosuppressive therapy.
178. ABSORPTION AND DISPOSITION OF THE IMMUNOMODULATORY COMPOUND FTY720 IN HEALTHY VOLUNTEERS
Markus Zollinger1, Hans-Peter Gschwind1, Yi Jin1, and Stefan Hartmann2
1Drug Metabolism and Pharmacokinetics, Novartis Pharma AG, Basel, Switzerland, 2Clinical Development and Medical Affairs, Novartis Pharma AG, Basel, Switzerland
FTY720 (fingolimod) is currently in clinical development as a promising new treatment of multiple sclerosis.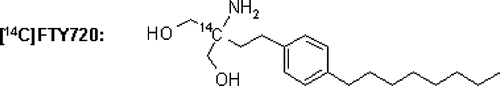
The compound is metabolically phosphorylated to FTY720-P, which acts as a potent agonist at four of the five known sphingosine-1-phosphate receptors, while the parent compound shows low or no activity. To investigate the absorption and disposition of FTY720 in humans, four healthy male subjects received a single oral dose of 5 mg [14C]FTY720 hydrochloride. FTY720 and FTY720-P were quantified in blood. Radioactivity and metabolite profiles were determined in blood, urine, and feces. Metabolite structures were assigned by mass spectrometry and/or by comparison with synthetic reference compounds. FTY720 was well tolerated without serious adverse events or discontinuation due to an adverse event. The absorption was slow (tmax = 8–36 h) but nearly complete (≥ 85% of dose). The apparent volume of distribution of FTY720 was large (mean Vz/f = 18.5 L/kg; high bioavailability f). The biotransformation of FTY720 occurred essentially by three pathways: (i) by reversible phosphorylation to FTY720-P, (ii) by oxidation at the terminal methyl group of the octyl chain to the carboxylic acid metabolite M1 (octanoic acid derivative), followed by losses of two-carbon units through β-oxidation to the carboxylic acid metabolites M2 (hexanoic acid derivative), M3 (butyric acid derivative), and M4 (acetic acid derivative), and (iii) by formation of nonpolar ceramide analogs. The phosphorylation of FTY720 appeared to occur in a strictly stereospecific way as only the pharmacologically active (S)-enantiomer of FTY720-P was observed in blood, as opposed to the inactive (R)-enantiomer. The subjects were systemically exposed mainly to FTY720, FTY720-P, and metabolite M3, with minor contributions by metabolite M2 and the ceramide analogs. FTY720 (mean t½λz = 137 h) and its metabolites were cleared slowly from the body. The elimination of FTY720 occurred predominantly by oxidative metabolism (first step catalyzed mainly by CYP4F2), while FTY720-P was eliminated predominantly by dephosphorylation back to FTY720. [14C]FTY720-related radioactivity was excreted very slowly, mainly via the kidney in the form of metabolite M3. FTY720 and FTY720-P were not detected in urine. Fecal excretion of radioactivity was minor (10% of dose up to 240 h, on average) and represented mainly FTY720 and FTY720-P. Excretion of radioactivity in urine and feces was still incomplete after 10 days (62% of dose on average) but continued beyond this time point, reaching 89% of dose by day 34.
179. IN VITRO SULFATION OF PICEATANNOL BY HUMAN LIVER CYTOSOL AND HUMAN RECOMBINANT SULFOTRANSFERASES
Michaela Miksits1, Katrin Wlcek1, Michael Sulyok2, Rainer Schuhmacher2, and Walter Jaeger1
1Department of Clinical Pharmacy & Diagnostics, University of Vienna, Vienna, Austria, AT-1090, 2Department IFA-Tulln, Christian Doppler Laboratory for Mycotoxin Research, University of Natural Resources and Applied Life Sciences, A-3430 Tulln, Austria
Piceatannol (3,3´,4,5´-tetrahydroxy-trans-stilbene) is a naturally occurring stilbene present in the skins of grape, rhubarb and sugar cane. Piceatannol is a promising chemopreventive agent with antileukemic activity and is being extensively studied in various cancers including colorectal and lung cancer as well as in cardiovascular disease. Hence, there is no information about metabolism of piceatannol in humans. As recent data from our lab demonstrated extensive in vitro and in vivo sulfation of the structural analog resveratrol we hypothesize that sulfation might also have a major impact in piceatannol biotransformation. Indeed, using human liver cytosol three metabolites (M1-M3) could be detected whose structures were identified by LC/MS/MS as piceatannol disulfate and monosulfates. The kinetics of M1 formation in human liver cytosol exhibited a pattern of substrate inhibition with a Ki of 20.41 ± 9.79 μM and a Vmax/Km of 7.67 ± 1.80 μl/mg protein/min. Contrary to M1, formation of M2 and M3 showed sigmoidal kinetics. The values of Km and Vmax were 26.87 ± 3.03 μM and 117.3 ± 4.08 pmol/mg protein/min for M2 and 35.73 ± 2.80 μM and 81.88 ± 2.46 pmol/mg protein/min for M3, respectively. Incubation in the presence of human recombinant sulfotransferases (SULTs) demonstrated that M1 was equally formed by SULT1A1*1 and SULT1B1 and only to a smaller extent by SULT1A1*2. M2 was preferentially catalyzed by SULT1E1, 1A3 and 1A1*2. The formation of M3, however, is mainly catalyzed by SULT1A2*1 and SULT1A3. The isoforms SULT1A1*1, 1A1*2, 1E1 and 1B1 exhibited only low catalytic activities for M3 formation. In conclusion, the results have elucidated the metabolism of piceatannol sulfation in human liver, which must be taken into consideration following dietary piceatannol uptake.
180. THE INFLUENCE OF PROBIOTIC TREATMENT ON SULPHASALAZINE (SSZ) METABOLISM BY GASTROINTESTINAL MUCUS AND CONTENTS IN RAT
Hee Ji Lee1, J. Paul Fawcett1, Richard Easingwood2, Liz Girvan2, Esther Kostan1,and Momir Mikov1
1School of Pharmacy, University of Otago, Dunedin, New Zealand, 9054, 2Department of Anatomy and Structural Biology, University of Otago, Otago Centre for Electron Microscopy, Dunedin, New Zealand, 9054
Aims. To investigate the metabolic activities of ileum and colon mucus layers and contents towards SSZ. To investigate the influence of probiotic treatment on the activities of ileum and colon mucus and contents towards SSZ. Hypotheses. Both gastrointestinal mucus and contents can metabolise SSZ. Probiotic treatment increases the azoreductase mediated cleavage of SSZ by both mucus and contents. Methods. Pretreat rats (n = 5 per group) with 1 ml/kg saline (control) or 1 ml/kg probiotic suspension (treatment) containing LAFTI® B94 CGS (2 × 109 cfu/ml Bifidobacterium animalis subsp. Lactis) and LAFTI® L10 CGS (3 × 109 cfu/ml Lactobacillus acidophilus) twice daily for 3 days. Separately incubate SSZ (100 μg/ml) with 10 % (w/w) suspension of rat ileum and colon contents and mucus for 3 hr sampling every 30 min. Determine the extent of SSZ metabolism by measuring the concentrations of SSZ, sulphapyridine and 5-aminosalicylic acid in samples by HPLC. Take transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images to confirm the presence of bacteria in the mucus layer of washed ileum and colon. Measure enzymatic activity of the luminal contents and mucus biofilm specifically the activities of azoreductase, nitroreductase, β-glucuronidase and β-glucosidase. Results. After 180 minutes 36%, 80% and 5.5% of SSZ was metabolised by the contents of ileum and colon and the colonic mucus, respectively while SSZ was metabolized by the colon mucus the ileal mucus was inactive. Probiotic treatment caused a 78% increase in SSZ metabolism by colon contents ( p < 0.01) and a 62% decrease in SSZ metabolism by ileum contents (p < 0.01). Activities of all enzymes were present in both contents and mucus of the ileum and colon but probiotic treatment affected the enzymatic activities differently. SEM revealed bacterial colonies embedded in the ileal and colon mucus. Conclusions. Colon mucus contributes to the metabolism of SSZ which is likely to be clinically significant in relation to its local antiinflammatory effects. The ileal mucus microbiota does not reduce SSZ in vitro and further studies are required to assess in situ metabolism of SSZ by gastrointestinal mucus. Probiotic treatment increases SSZ metabolism in the colon contents and mucus suggesting probiotics may be given to patients taking SSZ to increase its therapeutic efficacy.
181. XENOBIOTIC METABOLISM BY GUT FLORA CONDITIO SINE QUA NON FOR APPROPRIATE METABOLIC PROFILING
Momir Mikov and Svetlana Golocorbin-Kon
Dept. of Pharmacol & Toxicol, Serbia, Novi Sad, Yugoslavia, 21000
Aims. The purpose of these paper is to review, develop, validate and apply different approaches for an improved qualitative and quantitative monitoring and the role of gut microflora metabolic activity. Background. Xenobiotic metabolism by gut flora in the past has been usually ignored during drug development and other xenobiotic metabolism studies.Gut flora metabolic reserach within last two decades has been expanded concerning different aspects of gut flora metabolic activity, variability and composition and proposed more fully determination of the diversity and the role of gut flora and in the same time expanded probiotic and prebiotic approach. A considerable part of interindividual and inttraindividual metabolic differences can be ascribed to gut flora composition variability. Metabolic transformations of different xenobiotics within gut contents by gut flora could be as important as the one by gut wall and liver. It is not only that xenobiotic metabolites produced by gut flora can influence the function of the liver and body but also bacterial constituents and products of bacteria metabolism can pass from the gut lumen to the portal system and influence the liver function. Results. In in vivo and in vitro studies in mice we examined the effect of gut microflora effect on paracetamol, verapamil and sulfasalazine metabolism. Apart from supfasalazine metabolic profile change after antibiotic pretreatment, verapamil metabolic profile and availability was also affected, bioavailability was increased and metabolite production decreased. Paracetamol metabolic profile was affected by gut flora and there were variability between different gut segment contents in conventional mice. Antibiotic pretreatment changed paracetamol metabolic profile, with significant decrease of paracetamol thiomethyl metabolites. In germ-free animals, paracetamol thiomethil metabolites were not detectable. Discussion: Xenobiotics interfere in the gut contents ecology and it can be considered with the same methodological approach as in different ecosystem contamination investigations. Approaches to gut flora in vitro and in vivo metabolism of xenobiotics could be: 1. The direct action of gut flora in static incubations and flow models 2. The action of pure cultures, mixed cultures, continuous cultures and cell-free bacterial products 3. The difference in metabolism after oral and parenteral drug application 4. The difference between conventional and conventional animals treated with antibiotics 5. The difference between conventional and gnotobiotic animals . The in vitro system for studying the metabolic interaction of xenobiotic metabolism of liver and gut microflora previously developed, as well as a new metabonomics approach could be a precious tools for the revealing answers about the role and importance and interaction of intestinal microflora and liver for xenobiotic metabolism. Conclusion. In in vitro and in vivo studies on conventional, antibiotic pretreated and germ free animals it has been confirmed the importance of gut flora for metabolic profiling of xenobiotics. Gut flora metabolic profiling should be a compulsory part of initial xenobiotic metabolism profiling studies in order to provide scientists and regulatory bodies with the comprehensive information. Also, a harmonized evaluated methods for the monitoring of gut microflora role in xenobiotic metabolism and toxicity should be established.
182. IN VITRO METABOLISM OF RGH-188
Norbert Kirschner1, Larisza I. Gémesi1, Mónika Vastag2, Éva Ágai-Csongor3, György Domány3, Margit Kapás1, and Károly Tihanyi4
1Pharmacokinetics, Gedeon Richter PLC, Budapest, Hungary, 2In Vitro Metabolism, Gedeon Richter PLC, Budapest, Hungary, 3Medicinal Chemistry I., Gedeon Richter PLC, Budapest, Hungary, 4Pharmacology and Drug Safety, Gedeon Richter PLC, Budapest, Hungary
RGH-188 (trans-4-{2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl}-N,N-dimethylcarbamoyl-cyclohexylamine) is a new orally active dopamine D3/D2 receptor antagonist/partial agonist antipsychotic1–2 synthesized at Gedeon Richter Plc. and being developed in co-operation with Forest Laboratories, Inc. The compound is currently in clinical Phase II. studies for the indication of schizophrenia and bipolar mania. Objectives and Methods. The aims of the in vitro metabolism studies were to assess the metabolic stability of RGH-188 using liver microsomes of humans, monkeys, dogs, rats and mice, to explore enzyme induction and inhibition potencies of RGH-188 using human liver microsomes and/or hepatocyte cultures, to determine human CYP isozyme profile of RGH-188 by recombinant systems, to compare in vitro metabolite profile of the compound in different species and to identify the structure of detected metabolites. Biotransformation of RGH-188 was studied by mouse, rat, dog, cynomolgus monkey and human liver microsomal fractions using radiolabelled substance. Samples were analysed by HPLC-UV, radio-HPLC, ion trap LC/MS and LC/MS/MS methods. Results. RGH-188 showed high metabolic stability for each investigated species. It was characterized as a weak competitive inhibitor of human CYP2D6 and CYP3A4 isozymes. RGH-188 did not induce CYP3A4 and CYP1A2 in human hepatocytes. The drug was metabolized by CYP3A4 and to a lesser extent by CYP2D6. The metabolite profiles in liver microsomal fractions were qualitatively similar but quantitatively different for the various species. Eight metabolite compounds were observed in each species: desmethyl-RGH-188, di-desmethyl-RGH-188, 4-OH-phenyl-RGH-188, N-oxide of RGH-188, 4-OH-phenyl-desmethyl-RGH-188 and three cleavage products. Desmethyl-RGH-188 was the main in vitro metabolite, representing the highest amount of converted radioactivity in all investigated species.
REFERENCE
- B. Kiss, I. Laszlovszky, A. Horváth, É. Schmidt, Gy. Bugovics, Sz. Orosz, K. Hornok, I. Gyertyán, É. Ágai-Csongor, Gy. Domány, K. Tihanyi, Zs. Szombathelyi, RGH-188, an atypical antipsychotic with dopamine D3/D2 antagonist/partial agonist properties: in vitro characterisation. Int J Neuropsychopharmacol. 9 (Suppl. 1.), S221 (2006).
- I. Gyertyán, B. Kiss, K. Sághy, J. Laszy, Gy. Szabó, É. Ágai-Csongor, Gy. Domány, K. Tihanyi, Zs. Szombathelyi, RGH-188, an atypical antipsychotic with dopamine D3/D2 antagonist/partial agonist properties: behavioral characterisation. Int J Neuropsychopharmacol. 9 (Suppl. 1.), S222 (2006).
183. PHARMACOKINETICS AND METABOLISM OF A NEW NONOPIOID ANALGESIC PYRODAZOL
Oleg E. Iadlovskyi1, Alla K. Voronina Jr.2, Tatiana A. Bukhtiarova1, and Anatoliy M. Demchenko1
1Department of Anti-inflammatory and Analgesic Drugs, SE Institute of Pharmacology and Toxicology, Kyiv, Ukraine, 2General Toxicology, SI “Institute of Pharmacology and Toxicology, Academy of Medical Sciences of Ukraine”, Kyiv, Ukraine, 03057
The pharmacokinetics and metabolism of a new nonopioid analgesic pyrodazol-b derivant of 6,7-dihydro-5H-pyrrolo-(1,2a)-imidazole has been studied in rats. A high degree of pyrodazol absorption being administered orally as well as its ability to persist in a body and penetration through hystohaematic barriers including brain were shown. The mane pharmacokinetics parameters of pyrodazol administered orally comprised: Cmax = 0,140 g/ml; Tmax = 39,6 min; ÌRÒ = 80,7 min; ÀUÑ = 11,7 gmin/ml. By chromatomasspectlal method, in samples of serum of animals blood, are revealed pyrodazol and 7 its basic metabolites. They are characterized, time of deduction, (t, min), mass number of a molecular ion (m/z) and mass characteristics of transformations of affiliated ions (t, m/z). The possible ways of metabolic transformations of pyrodazol are reactions of dearylatoin, demethylation, digydroxilation, acetylation and conjugations with glutathione and glucuronide.
184. METABOLISM AND DISPOSITION OF THE ORAL ABSORPTION ENHANCER 8-(N-2-HYDROXY-5-CHLORO-BENZOYL)-AMINO-CAPRYLIC ACID (5-CNAC) IN HEALTHY POST-MENOPAUSAL SUBJECTS
Hans-Peter Gschwind1, Helene Sabia2, Felix Waldmeier1, Ulrike Glaenzel1, Bernard Wirz1, Marie-Noelle Bizot Carrier3, and Leslie Choi4
1Exploratory Development, Novartis Pharma AG, CH-4002 Basel, Switzerland, 2Exploratory Development, Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA, 07936-1080, 3Exploratory Development, Novartis Pharma, S.A.S., F-92500 Rueil-Malmaison, France, 4Exploratory Development, Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA, 07936-108
5-CNAC (8-(N-2-hydroxy-5-chloro-benzoyl)-amino-caprylic acid) is used an excipient and as a delivery agent to enhance the oral absorption of salmon calcitonin (sCT). 5-CNAC exerts no pharmacological effects. 5-CNAC was developed by Emisphere's Eligen® Technology and licensed in by Novartis. The present study was performed to investigate the absorption, pharmacokinetics, metabolism and excretion (ADME) of 14C-radiolabeled 5-CNAC in six healthy post-menopausal female subjects, following a single oral dose of 200 mg [14C]5-CNAC di-sodium monohydrate salt. The compound was administered in a gelatin capsule, without sCT. Subjects were dosed in a fasted state. There were no serious adverse events, and none of the subjects discontinued the study prematurely. Absorption was rapid (tmax = 0.5 h; Cmax = 2.3 ± 0.86 μg/mL) and virtually complete. Unchanged 5-CNAC contributed to 6% of the 14C-AUC0–24h in plasma; the rest of the 14C-AUC0–24h was due to metabolites. Elimination of 5-CNAC was rapid (t½ = 1.5 ± 1.1 h) and occurred mainly by metabolism. 5-CNAC appeared to undergo a substantial first pass metabolism, mainly by stepwise degradation of the octanoic acid side chain of 5-CNAC, analogous to the fatty-acid ß-oxidation pathway. In plasma at 0.5 hours post-dose, apart from 5-CNAC, six metabolites were observed. Three of them (M5, M3, M2) were main metabolites: the 2-O-glucuronide of the butanoic acid metabolite of 5-CNAC (M5) and of 5-CNAC itself (M3), and the butanoic acid metabolite of 5-CNAC (M2). Overall, about 30%, 21%, and 8% of 14C-AUC0–24h were accounted for by the M5, M3, and M2, respectively. 5-CNAC-related radioactivity disappeared from plasma with a terminal half-life of 14 ± 4 hours. 5-chlorosalicylic acid was observed as a minor metabolite which disappeared from plasma beyond 24 hours post-dose almost in parallel to total radioactivity. The biotransformation of 5-CNAC involves essentially two major metabolic pathways, namely: (i) stepwise degradation of the octanoic acid side chain of 5-CNAC, followed by 2-O-glucuronidation and (ii) direct conjugation of 5-CNAC at the 2-hydroxy group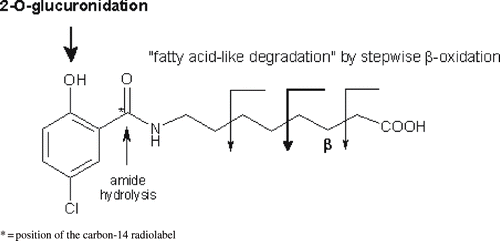 .
.
Excretion of the radioactivity was almost complete (∼90% of dose) after 1 day, and occurred predominantly via urine. Fecal excretion was minor (≤2%). In urine and feces, <0.08% and ∼0.3% of dose were excreted as unchanged 5-CNAC, respectively. By 7 days, the mass balance was close to complete (94.0 ± 3.5%).
Overall, 5-CNAC is predominantly cleared by metabolism, primarily through ß-oxidation pathway (68% of dose), and additionally through glucuronidation at the 2-O-phenolic position of 5-CNAC and its metabolites (79%).
185. METABOLISM AND DISPOSITION OF LORCASERIN, A NOVEL SELECTIVE SEROTONIN 5-HT2C RECEPTOR AGONIST, IN RATS, MICE, MONKEYS AND HUMANS
Weichao G. Chen1, Jay Xu2, Walter Gwathney2, Warren Prosser3, Jeff Edwards1, Michael Morgan1, and Paul Maffuid1
1Dmpk, Arena Pharmaceuticals, Inc, San Diego, CA, USA, 92121, 2Structural Chemistry, Arena Pharmaceuticals, Inc, San Diego, CA, USA, 92121, 3Clinical Operation, Arena Pharmaceuticals, Inc., San Diego, CA, USA, 92121
Lorcaserin, (1R)-8-Chloro-2,3,4,5-tetrahydro-1-methyl-1H-3-benzazepine, a novel selective serotonin 5-HT2C receptor agonist, is currently under development for the treatment of obesity. Metabolism and disposition of [14C]lorcaserin were investigated in Sprague-Dawley rats, CD-1 mice, Cynomolgus monkeys and humans. Rats, mice and monkeys received a 10 mg/kg oral dose, whereas humans received a single 10 mg oral dose. Plasma, urine and fecal samples were analyzed for mass balance and metabolite profiling. Mass balance of radioactivity was achieved in all species. In all four species, drug-related material was primarily excreted in the urine (69.9%, 87.7%, 83.3% and 92.3% of dose in rats, mice, monkeys and humans, respectively), and only small amounts of administered radioactivity were excreted in the feces (25.5%, 14.1%, 2.5% and 2.2% of dose in rats, mice, monkeys and humans, respectively). The high percentage of urinary elimination of total radioactivity suggested that the primary elimination route for lorcaserin and its metabolites was renal; fecal elimination was not a major route of elimination. In addition to unchanged lorcaserin, metabolite M1, lorcaserin sulfamate, was identified as the major circulating metabolite in the plasma of all four species. In rats and mice, lorcaserin sulfamate was found to be the major metabolite in the urine (37% and 58% of dose in rats and mice, respectively). In monkeys and humans, the N-carbamoyl glucuronide of lorcaserin was identified as the major metabolite in urine, and represented 24% and 36% of total administered dose in monkeys and humans, respectively. While lorcaserin sulfamate was the major metabolite in human plasma, it was only a minor metabolite in urine, representing approximately 3% of dose. Lorcaserin parent drug excreted in urine only represented a small percentage of doses in all four species. Additional metabolites excreted in urine were identified as either glucuronide or sulfate conjugates of oxidative metabolites. These results suggested that lorcaserin was extensively metabolized through multiple pathways including N-carbamoyl glucuronidation, N-sulfonation and oxidation followed by phase II conjugation. Since the metabolism and disposition of lorcaserin is not dependent on any single metabolic pathway lorcaserin is unlikely to be susceptible to drug-drug interactions caused by marketed drugs.
186. METABOLISM OF ANTITUMOR 9-AMINO-1-NITROACRIDINE DERIVATIVE, C-1748 (CAPRIDINE-BETA), IN HEPG2 TUMOR CELLS
Anita Wisniewska1, Joanna Koprowska1, Magdalena Niemira1, Ewa Augustin1, Agata Kot-Wasik2, Jerzy Konopa1, and Zofia Mazerska1
1Department of Pharmaceutical Technology and Biochemistry, Chemical Faculty, Gdansk University of Technology, Gdansk, Poland, 80–952, 2Department of Analytical Chemistry, Chemical Faculty, Gdansk University of Technology, Gdansk, Poland, 80–952
Antitumor agent 9-ethylamino-4-methyl-1-nitroacridine,C-1748, belongs to a new set of antitumor derivatives developed in our laboratory and was selected to preclinical studies. Metabolic pathway of a representative 9-amino-1-nitroacridine with rat microsomes was proposed earier [1]. Compound C-1748, which is less toxic than its analog without 4-methyl group, C-857, was shown to be less reactive with human and rat liver microsomes as well as with human E.coli recombinant CYPs [2]. Therefore, we hypothesized that slight metabolism of C1748 is a probably reason of its reduced toxicity in animals. All previous results were obtained in model enzymatic systems. In this work we present the studies on metabolic transformations of C-857 and C-1748 in human hepatoma line HepG2 cells. The investigations aimed to evaluate (i) whether any new metabolic products are formed in tumor cells in comparison with metabolites identified earlier with microsomal and hepatic recombinant enzymes, and (ii) whether the differences in HepG2 metabolism between C-857 and C-1748 are similar to those observed earlier for enzymatic transformations. HepG2 cells were incubated with specified concentration of C-857 and C-1748 for 3, 4, 6 and 12 hours. Metabolites were extracted with methanol and RP-HPLC analyses of extracts were performed with UV-vis and ESI-MS detection. The results demonstrated that several new metabolites were formed after HepG2 metabolism of the studied compounds in comparison with model enzymatic system. On the other hand, at least one common metabolite was found in the cell as well as with all enzymatic systems studied earlier. It is a derivative with additional pyrazole ring closed between nitrogen atoms at positions 1 and 9 of acridine ring. Metabolites of C-857 extracted from HepG2 cells were observed after 3 and 4 hours of incubations, whereas 6 and 12 hours of incubations gave significantly lower concentrations of all products. There was not the case of C-1748 where significant amounts of metabolites were formed until 12 hours. In conclusion, derivative C-1748 is less than C-857 reactive in HepG2 cell and metabolites of C-1748 were much more stable than those of C-857. The above results together with our earlier studies indicate better selectivity of this compound than other 9-amino-1-nitroacridines towards tumor cells.
REFERENCE
- K. Gorlewska, Z. Mazerska, Pawel Sowinski and Jerzy Konopa, Products of metabolic activation of the antitumor drug ledakrin (Nitracrine) in vitro, Chem. Res. Toxicol., 14, 1–10, (2001)
- A. Wisniewska, A. Chrapkowska, J. Konopa and Z. Mazerska, Selectivity in CYP mediated metabolism of antitumor 9-amino-1-nitroacridine derivative, C-1748 (Capridine-beta), an agent under Phase I clinical trials, Books of Abstracts, 15th International Conference on Cytochromes P450, Bled, Slovenia, June 2007, p. 190.
187. STUDIES ON HUMAN GASTROINTESTINAL BACTERIAL AZOREDUCTASE, THE ENZYME ACTIVATES AZO PRO-DRUGS IN THE TREATMENT OF INFLAMMATORY BOWEL DISEASE (IBD)
Chan-Ju Wang1, Chris Hagemeier1, Nawreen Rahman1, Lori Hu1, Michael Coughtrie2, George Macfarlane3, Ed Lowe4, Edith Sim1, and Isaac Westwood1
1Pharmacology, University of Oxford, Oxford, United Kingdom, OX1 3QT, 2Pathology, University of Dundee, Dundee, United Kingdom, DD1 9SY, 3Neuroscience & Microbiology, University of Dundee, Dundee, United Kingdom, DD1 9SY, 4Biochemistry, University of Oxford, Oxford, United Kingdom, OX1 3QU
The anti-inflammatory agent 5-ASA is given for IBD as a pro-drug formulation and 5-ASA is released in situ following activation by azoreductases in intestinal microflora. Given the scant information about azoreductase specificities for azo pro-drugs, the aim of this study is to characterise azoreductase structurally in relation to pro-drug activation. Azoreductases have been identified in several human intestinal bacteria including Clostridium perfringens, Salmonella typhimurium and P. aeruginosa1. The gene PA0785 has been cloned and overexpressed in E. coli. The purified recombinant enzyme exhibits azoreductase activity and activates pro-drugs Balsalazide and Sulfasalazine. Each recombinant protein molecule has an estimated molecular weight of 23,050 Da and one non-covalently bound co-factor flavin mononucleotide (FMN). This enzyme (paAzoR1) is a flavodoxin-like protein with an apparent molecular weight of 110 kDa by gel filtration chromatography, indicating that paAzoR1 is likely to associate to form tetramers. The three-dimensional structure of paAzoR1 with a substrate Methyl Red was solved to 2.18 Å resolution by X-ray crystallography. The protein is a dimer of dimers in crystal lattice, with two spatially separated active sites per dimer, and the active site of paAzoR1 was shown to be well-conserved hydrophobic pocket formed between two monomers. FMN serves as redox centre in electron transfer from NAD(P)H to the azo pro-drugs. The structure and spectral properties of paAzoR1 demonstrate the hydrophobic interaction between FMN, the active site in the protein and the substrate, and help to identify key residues in the enzyme for potential improvement of activity with existing and novel azo pro-drugs.2
188. IN VITRO - IN VIVO EXTRAPOLATION OF THE CLEARANCE OF DRUGS BY GLUCURONIDATION: SCALING FACTORS FOR RECOMBINANT UGT1A1 DATA
Sibylle Neuhoff1, Zoe E. Barter2, Karen Rowland-Yeo1, Geoffrey T. Tucker2, and Amin Rostami-Hodjegan2
1Scientific Development, Simcyp Ltd, Sheffield, United Kingdom, S2 4SU, 2Academic Unit of Clinical Pharmacology, University of Sheffield and Simcyp Limited, Sheffield, United Kingdom, S2 4SU
Increasing numbers of drugs have been shown to be metabolised directly by UDP glucuronosyltransferases (UGT), prompting the implementation of screens for UGT activity in drug development. These screens primarily utilise recombinantly expressed enzymes owing to a lack of specific inhibitors for use with liver microsomes or hepatocytes. Focus is on polymorphic UGT1A1, which exhibits a high degree of inter-individual variability with respect to hepatic and intestinal expression, and which metabolises important drugs such as irinotecan. Quantitative in vitro-in vivo extrapolation (IVIVE) using enzyme kinetic data and relevant scaling factors has been applied effectively to drugs metabolised by cytochromes P450. However, in the case of UGTs, lack of information on their abundance in human liver and the relative enzyme activity in recombinant systems precludes accurate quantitative prediction of clearance by glucuronidation in vivo. The aim of this study was to derive a scaling factor for UGT1A1 that accounts for differences in intrinsic activity per unit enzyme and abundance of the enzyme in human liver microsomes (HLM) and recombinant systems (rUGT). Values of Vmax (pmol/min/mg microsomal protein) and Km (μM, corrected for non specific microsomal binding) for three substrates metabolised by UGT1A1 to different extents (bilirubin, estradiol and etoposide) determined using both HLM (n = 10–15 livers) and rUGT1A1 SupersomesTM (BD Biosciences) were collated from the literature. Scalars for each substrate were calculated from the ratio of intrinsic clearance by HLM relative to rUGT1A1. The values were 1.05 for bilirubin (Patten et al., 2001), 1.03 for estradiol (Patten et al., 2001) and 0.79 for etoposide (Watanabe et al., 2003; Wen et al., 2007). The consistency of these values indicates that a mean (substrate-independent) value of 0.92 ± 0.29 (SD) may be used as a system specific hepatic scaling factor to facilitate extrapolation of in vitro data obtained using rUGT1A1 Supersomes to estimate the in vivo clearance of other UGT1A1 substrates.
Patten et al., (2001) Analysis of UGT enzyme levels in Human liver microsomes using specific anti-peptide antibodies, probe substrate activities and recombinant UGT enzymes. BD Biosciences (http://www.bd.com ).
Watanabe et al., (2003) Glucuronidation of etoposide in Human liver microsomes is specifically catalyzed by UDP-glucuronosyltransferases 1A1. Drug Metabolism and Disposition 31: 589–595.
Wen et al., (2007) UDP-Glucuronosyltransferase 1A1 is the principal enzyme responsible for etoposide glucuronidation in Human liver and intestinal microsomes: structural characterization of phenolic and alcoholic glucuronides of etoposide and estimation of enzymes kinetics. Drug Metabolism and Disposition 35: 371–380.
189. SIGNIFICANCE OF CYP2E1 AND NQO1 POLYMORPHISMS IN THE RISK OF DEVELOPMENT OF CHILDHOOD ACUTE LYMPHOBLASTIC LEUKEMIA
Gulen Ulusoy1, Tugba Boyunegmez Tumer1, Orhan Adali1, Gurses Sahin2, and Emel Arinç1
1Biochemistry Graduate Programme and Department of Biological Sciences, Middle East Tech Univ, Ankara, Turkey, N/A, 2Oncology Department, Dr. Sami Ulus Children's Hospital, Ankara, Turkey
Acute lymphoblastic leukemia (ALL) is a stem cell disorder characterized by overproduction of lymphoblasts in the bone marrow that eventually spill into circulation, and is the most frequent form of cancer affecting the children. The molecular etiology of childhood ALL is likely to involve interactions between environmental factors and genetic make up. Understanding these interactions between various predisposing genes for the risk of developing childhood leukemia is of considerable importance. Therefore, investigation of the polymorphisms of drug metabolizing enzymes is of crucial importance to determine the molecular etiology of the disease. Two such genes are CYP2E1 and NQO1. CYP2E1 is a susceptible gene, especially for its capacity to bioactivate many procarcinogens, while NQO1 has a protective role in metabolism of carcinogenic metabolites, involving those produced by CYP2E1. Several studies have been published on the effect of either CYP2E1 or NQO1 on childhood ALL, however the results were inconsistent. In this context we have carried out a multiloci study on the effect of three polymorphisms in CYP2E1, namely CYP2E1*5B located in 5′-flanking region, *6 in intron 6, and *7B in promoter region, and NQO1*2 polymorphism located in exon 6, in the risk of development of childhood ALL in 160 patients and 204 healthy controls by PCR-RFLP technique. None of the polymorphisms showed a significant association with the disease when investigated separately, however, presence of CYP2E1*5B and *6 alleles together significantly increased the risk of childhood ALL 2.9- fold (95% CI, 1.0–8.6; p < 0.05). Furthermore, co-presence of two CYP2E1 polymorphisms (either CYP2E1*5B and *6 or CYP2E1*6 and *7B) and NQO1*2 polymorphism together on the same individual increased the risk of childhood ALL further, 4.8-fold (95% CI, 1.0–24.0; p < 0.05) suggesting a combined effect. Co-presence of CYP2E1 and NQO1 polymorphisms as risk factors in the development of childhood ALL is meaningful in the sense that CYP2E1 activates carcinogens like benzene, producing quinone metabolites which accumulate in the bone marrow and NQO1–showing low activity in NQO1*2 allele- detoxifies these quinone metabolites. Studies on the polymorphism of multiple loci and genes, like this study, may be helpful in overcoming the inconsistencies in determining the risk factors for childhood ALL.
190. ABCB1 HAPLOTYPES DIFFERENTIALLY AFFECT THE PHARMACOKINETICS OF THE ACID AND LACTONE FORMS OF SIMVASTATIN AND ATORVASTATIN
Jenni E. Keskitalo, Kaisa J. Kurkinen, Pertti J. Neuvonen, and Mikko Niemi
Department of Clinical Pharmacology, University of Helsinki, Helsinki, Finland, FIN-00290
The cholesterol-lowering efficacy of simvastatin and atorvastatin has been associated with polymorphism of the ABCB1 gene encoding P-glycoprotein. Our aim was to investigate the frequencies of ABCB1 c.1236C>T, c.2677G>T/A, and c.3435C>T SNPs in the Finnish population, and to compare the effects of two common ABCB1 haplotypes, c.1236C–c.2677G–c.3435C (CGC) and c.1236T–c.2677T–c.3435T (TTT), on the pharmacokinetics of simvastatin and atorvastatin in a prospective genotype panel study. We genotyped 534 healthy white Finnish volunteers for the ABCB1 c.1236C>T, c.2677G>T/A, and c.3435C > T SNPs. The variant allele frequencies (with 95% confidence intervals) were: c.1236T 46.4% (43.5–49.4%), c.2677T 47.3% (44.3–50.3%), c.2677A 3.3% (2.4–4.5%), and c.3435T 59.9% (57.0–62.8%). The frequency of the CGC haplotype was 34.4% (31.6–37.3%) and that of the TTT haplotype was 42.7% (39.8–45.7%). Of these 534 volunteers, 12 subjects with the CGC/CGC genotype and 12 with the TTT/TTT genotype ingested a single 20 mg dose of simvastatin and 20 mg dose of atorvastatin with a washout period of at least one week. Plasma concentrations of simvastatin and simvastatin acid were measured for 12 hours, and those of atorvastatin and its metabolites for 48 hours. Subjects with the TTT/TTT genotype had a 60% (P = 0.039) larger mean area under the plasma simvastatin acid concentration-time curve from 0 to 12 hours (AUC0–12 h) than subjects with the CGC/CGC genotype. The AUC0–12 h of simvastatin lactone was similar in both groups. The AUC0-inf of atorvastatin acid, but not lactone, was larger (55%; P = 0.023) and its half-life longer (24%; P = 0.025) in subjects with the TTT/TTT genotype than in subjects with the reference CGC/CGC genotype. Furthermore, the AUC0-inf of 2-hydroxyatorvastatin acid, but not lactone, was larger (49%; P = 0.018) and the AUC0–48 h of 4-hydroxyatorvastatin lactone was larger (42%; P = 0.034) in subjects with the TTT/TTT genotype than in those with the CGC/CGC genotype. These results indicate that the ABCB1 haplotypes affect the pharmacokinetics of active simvastatin acid and atorvastatin acid, with limited effects on their lactones, potentially modulating the benefit-to-risk ratio of statin treatment.
191. THE INFLUENCE OF GENETIC AND NON-GENETIC FACTORS ON PHENPROCOUMON ASSOCIATED BLEEDINGS
Karen May1, Stefanie Dorow1, Jacek Szymanski2, Marion Hippius3, Iselore R. Reimann3, Jörg Hasford4, Dieter Rosskopf5, Bernd Drewelow6, Petra Thuermann2, and Werner Siegmund1
1Clinical Pharmacology, University of Greifswald, Greifswald, Germany, 2Clinical Pharmacology, University of Witten/Herdecke, Witten/Herdecke, Germany, 3Clinical Pharmacology, University of Jena, Jena, Germany, 4Biometry and Epidemiology, Ludwig-Maximilian University Munich, Munich, Germany, 5Pharmacology, University of Greifswald, Greifswald, Germany, 6Clinical Pharmacology, University of Rostock, Rostock, Germany
The anticoagulant treatment with phenprocoumon is characterised by a high variability in the dose regimen and associated with high risk for bleeding complications. The dose variability is at least in part attributed to polymorphisms of the metabolizing enzyme CYP2C9 and of the target enzyme vitamin K epoxide reductase complex 1 (VKORC1). However, there is only limited information whether the genetic variability and further non-genetic variables are also a risk factor for phenprocoumon associated bleeding complications. Methods. The CYP2C9*1,*2,*3 haplotypes, the VKORC1 polymorphisms 1173C>T, 4811G>A, 10179A>G and non-genetic factors (age, gender, comedication) were assessed in 90 patients with phenprocoumon associated gastrointestinal bleedings in comparison to 73 patients without bleedings after phenprocoumon (National Pharmacovigilance study) and an age and gender matched regional control group (n = 153, SHIP study). Results. The CYP2C9 haplotypes did not influence the phenprocoumon doses. The frequency of CYP2C9*2 and *3 among patients with phenproucomon associated gastrointestinal bleedings was significantly higher than in the regional control group. The frequency of the VKORC1 polymorphisms was identical in all groups. Carriers of at least one 1173T allele required significantly higher doses of phenprocoumon (17.9 ± 8.7 mg versus 15.3 ± 8.9; p = 0.023). Age, gender, comedication of CYP2C9 substrates and NSAIDs were not additional risk factors for bleedings. Conclusions. The risk for phenprocoumon associated bleedings seems to be related to the CPY2C9 but not to the VKORC1 genotype.
192. GLOBAL ANALYSIS OF GENETIC VARIATION IN SLCO1B1
Marja K. Pasanen, Pertti J. Neuvonen, and Mikko Niemi
Department of Clinical Pharmacology, University of Helsinki and Helsinki University Central Hospital, Helsinki, Finland
Organic anion transporting polypeptide 1B1 (OATP1B1), encoded by the SLCO1B1 gene, is an uptake transporter located on the basolateral (sinusoidal) membrane of human hepatocytes, and it is involved in the hepatic uptake of a broad array of endogenous and foreign compounds, including drugs. Several sequence variations (single nucleotide polymorphisms, SNPs) have been discovered in the SLCO1B1 gene encoding OATP1B1, some associated with altered transport activity in vitro and in vivo. Given the known interindividual and interethnic differences in drug disposition involving the OATP1B1 transporter and its functional role in the pharmacokinetics of many drugs, we wanted to characterize the SNP and haplotype variability of SLCO1B1 at the population level within and between different ethnic groups. Distribution of SLCO1B1 alleles was determined in 941 individuals from 52 populations comprising Africa, Middle East, Asia, Europe, Oceania, and the Americas. DNA samples were genotyped at 12 variant sites spanning the entire gene by TaqMan 5′nuclease allelic discrimination assays.
Large differences existed in allele frequencies between different populations. The low-activity variant c.521T>C occurred with a high frequency in the American, Middle Eastern, and European populations (24%, 20%, and 18%, respectively); in Oceania and Subsaharan Africa, the frequencies were small (0% and 1.9%, respectively). The African continent (Subsaharan Africa and North Africa) displayed the highest diversity as all investigated SNPs were found there. Moreover, the c.521C allele (r = 0.505, P < 0.001) and the *1B (c.388G–c.521T; r = -0.405, P = 0.006) and *15 (c.388G–c.521C; r = 0.510, P<0.001) haplotype frequencies correlated significantly with the latitude in the northern hemisphere, suggesting that a selective pressure may have acted on SLCO1B1 during human dispersal favoring low-activity variants in the north. Overall, SLCO1B1 genetic distances correlated significantly with geographic distances between populations, assuming likely routes of human migration out of Africa via five waypoints (r = 0.235, P = 0.001). SLCO1B1 diversity was greater within than between populations and genetic variation in SLCO1B1 was generally similar to that observed for other autosomal markers.
193. IMPACT OF CYP 2D6 POLYMORPHISMS ON RISPERIDONE PLASMA LEVELS AND RESPONSE TO ACUTE ANTIPSYCHOTIC TREATMENT
Matej Kastelic1, Igor Locatelli2, Jure Koprivšek3, Blanka Kores Plesniˇar3, Aleš Mrhar2, Iztok Grabnar2, and Vita Dolžan1
1Institute of Biochemistry, Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia, 1000, 2Faculty of Pharmacy, University of Ljubljana, Ljubljana, Slovenia, 1000, 3Department of Psychiatry, Teaching Hospital Maribor, Maribor, Slovenia, 2000
The majority of antipsychotic drugs, risperidone and haloperidol included, are metabolized by cytochrome P450 2D6. Genetic polymorphism of CYP2D6 results in a large interindividual variability in the metabolic capacity for CYP2D6 substrates, however the evidence that CYP2D6 polymorphism may predispose to poor response or extrapyramidal adverse effects upon antipsychotic treatment is less consistent. The aim of our study was to evaluate the influence of CYP2D6 genotype on risperidone plasma levels and clinical response to acute antipsychotic treatment. In total 65 Slovenian patients acutely treated with haloperidol or risperidone either for the first episode of schizophrenia spectrum disorders or for the psychotic episode after tapering their maintenance treatment were genotyped for 12 CYP2D6 deficiency alleles and CYP2D6 gene duplication. Steady state plasma concentrations of risperidone and its active metabolites; namely (+)- and (–)-9-hydroxyrisperidone, were determined. Psychopathological symptoms were assessed with the Brief Psychiatric Rating Scale (BPRS) and with Clinical Global Impression (CGI) on day 8 of treatment. Both measurements were repeated after 3 weeks. Tardive dyskinesia symptoms were assessed with the Abnormal Involuntary Movement Scale (AIMS), akathisia with the Barnes Akathisia Scale (BARS), and parkinsonism with the Simpson Angus Scale (SAS). When analysing association between the CYP2D6 genotype and clinical response, illness duration, drug type, and dose were considered as covariates. The frequency of wild type CYP2D6*1 allele was 0.615. The frequencies of alleles coding for moderately reduced activity were 0.038 (*10) and 0.115 (*41), respectively. The frequencies of nonfunctional alleles were 0.177 (*4), 0.015 (*3), 0.008 (*8), while *5, *6, *11, *12, *14 and *15 alleles were not found. CYP2D6 duplication alleles were found in two patients. Among 65 patients, four (6.2%) carried two nonfunctional CYP2D6 alleles (PMs), seven (10.8%) carried one of CYP2D6 alleles associated with reduced activity and one non-functional allele (IMs) and 54 (83.1%) had one or two functional alleles (EMs). Polymorphisms in the CYP2D6 gene correlated with the risperidone to 9-hydroxyrisperidone plasma concentration ratio. Population pharmacokinetic modelling revealed that nonfunctional CYP2D6 alleles were associated with significantly lower formation clearance of (+)-9-hydroxyrisperidone. However, no influence of CYP2D6 genotype on treatment response and extrapyramidal adverse affects was found. In conclusion, in Slovenian schizophrenia patients CYP2D6 genotype may not be the major factor that determines the clinical response to acute antipsychotic treatment.
194. MDR1 GENE POLYMORPHISMS AND RESPONSE TO ACUTE ANTIPSYCHOTIC TREATMENT
Matej Kastelic1, Jure Koprivšek2, Laura Mandelli3, Alessandro Serretti3, Blanka Kores Plesnicar2, and Vita Dolžan1
1Institute of Biochemistry, Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia, 1000, 2Department of Psychiatry, Teaching Hospital Maribor, Maribor, Slovenia, 2000, 3Institute of Psychiatry, University of Bologna, Bologna, Italy, 40123
Multidrug resistant protein (MDR1) gene codes for P-glycoprotein (P-gp) that transports drugs against a concentration gradient across the blood–brain barrier and reduces their bioavailability in the brain. Most second generation antipsychotics are P-gp substrates and differential entry of antipsychotics into the brain might explain interindividual differences in treatment response and adverse effects, even if plasma concentrations do not differ. In the present study we investigated the influence of MDR1 C3435T and G2677T/A polymorphisms on the response to acute antipsychotic treatment of schizophrenia. In 65 patients acutely treated with haloperidol or risperidone either for the first episode of schizophrenia spectrum disorders or for the psychotic episode after tapering their maintenance treatment MDR1 C3435T and G2677T/A were genotyped by real time PCR assay and sequence specific PCR, respectively. Psychopathological symptoms were assessed with the Brief Psychiatric Rating Scale (BPRS) and with Clinical Global Impression (CGI) twice: 8–12 days after the first dose of antipsychotic and after 4 weeks. Adverse effects were assessed with the Simpson-Angus Extrapyramidal Side Effects Scale (EPS), the Barnes Akathisia Scale (BARS) and the Abnormal Involuntary Movement Scale (AIMS). The distribution of 3435 CC, CT and TT genotypes (MDR1ex26) was 0.135, 0.423 and 0.442, respectively and the distibution of G2677T/A (MDR1ex21) genotypes was: GG 0.135, GT 0.519, GA 0.058, TT 0.269 and TA 0.019. Both polymorphisms were in Hardy-Weinberg Equilibrium (HWE) and they were in strong linkage disequilibrium (LD) (D' = 0.85. Chi-sq = 71.1 d.f. = 2 p < 0.0001). Drug type and high dosages of treatment were associated with higher BPRS and CGI baseline scores as well as with higher final BPRS scores, however efficacy of treatment defined as percent reduction of BPRS and CGI scores upon the two assessments was not dependent on these variables. Akathisia (BARS) was dependent on drug dosage, extrapyramidal symptoms (EPS) were more severe in patients treated with haloperidol and at a high dosage, and tardive dyskinesia (AIMS) was dependent on illness duration and was more severe in patients treated with haloperidol and high dosages. We did not find any association between MDR1 variants and efficacy of treatment neither with extrapyramidal adverse effects. In conclusion, neither MDR1 C3435T nor G2677T/A were associated with the treatment response in schizophrenic patients acutely treated with antipsychotics.
195. EFFECTS OF CYP3A4*18 ON REPAGLINIDE'S PHARMACOKINETICS
Ruzilawati Abu Bakar, Mohd Suhaimi Ab Wahab, and Gan Siew Hua
Department of Pharmacology, Universiti Sains Malaysia, Kelantan, Malaysia
The cytochrome P450 (CYP) 3A subfamily plays an important role in the metabolism of various endogenous and exogenous compounds. A number of allelic variations in CYP3A4 gene are known to affect catalytic activity including CYP3A4*4, CYP3A4*5 and CYP3A4*18. The aim of the present study is to investigate the effects of the CYP3A4*18 allele on the pharmacokinetic of repaglinide in healthy Malaysian subjects. One hundred and twenty-one subjects received oral administration of repaglinide (4 mg) and were genotyped by polymerase chain reaction-restriction fragment length polymorphism for CYP3A4*18. Serum repaglinide was extracted by liquid-liquid extraction and measured by high-performance liquid chromatography. The pharmacokinetics of repaglinide was studied during the first 4 h after the dose. The frequency of the CYP3A4*18 allele in the Malaysian population is 2.07%. Subjects who were heterozygous for CYP3A4*18 had significantly (p = 0.04) lower mean elimination rate constant of repaglinide than subjects of the normal group (Mann-Whitney test). In conclusion, the present study shows that genetic polymorphisms in CYP3A4 may play a role in the interindividual variability of repaglinide pharmacokinetics.
196. ABCC2 GENOTYPE FREQUENCIES AND HAPLOTYPES IN HEALTHY CAUCASIAN SUBJECTS
Sandra Lächelt1, Sierk Haenisch1, Werner Siegmund2, and Ingolf Cascorbi1
1Dept. of Pharmacology, University Hospital of Schleswig-Holstein, 24105 Kiel, Germany, 2Dept. of Clinical Pharmacology, University of Greifswald, 17487 Greifswald, Germany
Multidrug resistance-related protein 2 (ABCC2, MRP2, cMOAT), a member of ABC-membrane transporters, plays an important role in active excretion of various organic anion conjugates of endogenous and exogenous substances. It also contributes to the phenomenon of drug resistance. ABCC2 is found in liver, gut, kidney, placenta and blood-brain barrier. It exhibits several hereditary polymorphisms which at least partly could be conducive to the broad interindividual variation of its expression. In this study we investigated the distribution of 13 SNPs (single nucleotide polymorphisms) in the ABCC2 gene in a large group of 374 healthy Caucasian subjects. Genotyping was performed by PCR-RFLP and pyrosequencing techniques. Frequencies of haplotypes and diplotypes were calculated by using the Phase 2.0 program and plot of linkage disequilibrium (LD plot) by Haploview 4.0 program. The allele frequencies were 18.3% (–24T), 21.1% (c.1249A), 1.4% (c.1446C>G), 0.1% (c.3542G>T), 4.5% (c.3563T>A), 34.2% (c.3972T) and 4.4% (c.4544G>A); the polymorphisms –23G>A, c.1457C>T, c.3561G>A, c.2302C>T, c.2366C>T, and c.4348G>A were not found in our population. For the detected polymorphisms 12 haplotypes occurring in 24 diplotypes could be estimated. The wild-type haplotype CGCGTCG was the most common (39.3%), followed by CACGTCG (19.8%), TGCGTTG (17.5%), CGCGTTG (15.2%) and CGCGACA (4.4%). All other haplotypes occurred with frequencies lower than 2%. The statistical analysis identified the –24T allele to be in linkage disequilibrium with 3972T by 95.6% (X2 = 438.38; p < 0.001) and 3563A with 4544A by 97.8 % (X2 = 748; p < 0.001). The 1249A allele was weakly linked with other variant alleles. All SNPs were present in one haploblock. These data suggest that the investigation of five ABCC2 SNPs namely -24C>T, 1249G>A, 3563T>A, 3972C>T and 4544G>A covers >98% of ABCC2 haplotypes and should be considered in studies on the correlation of ABCC2 variants with phenotypical data.
197. POLYMORPHIC METABOLISM OF CYCLOPHOSPHAMIDE: INFLUENCE ON CLINICAL EFFICACY OF CHOP AND R-CHOP THERAPY OF FOLLICULAR LYMPHOMA
Sierk Haenisch1, Sebastian Böttcher2, Igor Mosyagin1, Christiane Pott2, Michael Kneba2, Eva Hoster3, Wolfgang Hiddemann3, Michael Unterhalt3, and Ingolf Cascorbi1
1Dept. of Pharmacology, University Hospital of Schleswig-Holstein, D-24105 Kiel, Germany, 2Second Dept. of Medicine, University Hospital of Schleswig-Holstein, D-24116 Kiel, Germany, 3Dept. of Internal Medicine III, University of Munich – Hospital Grosshadern, D-81377 Munich, Germany
Follicular lymphoma (FL) is the second most frequent lymphoma subtype in Western countries. Treatment regimens like CHOP (cyclophosphamide, doxorubicin, vincristine and prednisone) frequently induce remissions that, however, are inevitably followed by relapses. Very recently we and others demonstrated that the addition of the monoclonal antibody rituximab (R) increases remission rate and duration. Cyclophosphamide is metabolized by Cyp2C9 and 2C19 to the active metabolites 4-hydroxycyclophosphamide and phosphoramide mustard. The activity of both isoenzymes depends on well-described polymorphisms. The current study investigated the influence of Cyp2C9 and 2C19 polymorphisms on clinical efficacy of CHOP and R-CHOP therapy schemes. 224 FL patients (74 CHOP vs. 150 R-CHOP) were genotyped for the genetic variants Cyp2C9*2 and *3 and Cyp2C19*2 and *17. The overall response rate (ORR) was 95% (92% vs. 97%), the complete remission rate was 14% (11% vs. 16%) and the median time to treatment failure was 45 months in the CHOP group and not yet reached in the R-CHOP group, respectively. In patients treated by CHOP the ORR was in tendency lower for carriers of at least one Cyp2C19*2 allele than for individuals not exhibiting the *2 allele (83% vs. 96%, p = 0.07). However, the time to treatment failure was not significantly influenced by 2C19 genotype. 2C9 variants did not modulate the efficacy of CHOP scheme. In the R-CHOP group none of the tested polymorphisms did influence the clinical outcome. These data suggest that the previously described modulation of cyclophosphamide activation by CYP2C19*2 might have clinical relevance when FL patients are treated by CHOP, thus suggesting a dose-response relation for cyclophosphamide in FL. The effect is not measurable once patients are treated with R-CHOP. Regarding the efflux transport of doxorubicin, vincristine and prednisone further studies are in process to investigate the influence of genetic variants of ABC-transporters on efficacy of these constituents in CHOP and R-CHOP therapy.
198. DISTRIBUTION OF ALCOHOL DEHYDROGENASE2 (ADH2), ALDEHYDE DEHYDROGENASE2 (ALDH2), AND CYTOCHROME P450 (CYP2E1) GENOTYPES IN FIVE POPULATIONS
Sun Wook Woo, Tae Sun Kang, Hyun Joo Park, Sun Kyung Jin, Yoonsook Lee, and Jaesook Roh
Department of clinical pharmacology, National Institute of Toxicological Research, Korea Food & Drug Administration, Seoul, South Korea, 122–704
Alcohol is mainly metabolized by three enzymes, alcohol dehydrogenase 2 (ADH2), aldehyde dehydrogenase 2 (ALDH2), cytochrome P450 2E1 (CYP2E1). Alcohol is first oxidized to acetaldehyde by the alcohol dehydrogenase (ADH) enzyme, especially by ADH2 and cytochrome P4502E1, and then acetaldehyde is oxidized to acetate by the aldehyde dehydrogenase (ALDH), ALDH2 enzyme of which mostly influence on this oxidation. It has been reported that Variations in these genes have been found to influence alcohol consumption, alcohol-related tissue damage, and alcohol dependence. In this study, we analyzed the frequencies of genetic mutation in ADH2, ALDH2, and CYP2E1 and compared these frequencies among five ethnic groups which include Korean, Japanese, Chinese, European, and African. The five populations differ in allele frequencies, The ADH2 (*2), ALDH2 (*2), and CYP2E1 (c2) frequencies are 19.7%, 13.9% and 20.9% in Korean respectively. Each allele frequency is similar to that of Asian (Japanese: 26.1%, 22.7%, 20.5% and Chinese: 23.3%, 15.6%, 28.9% respectively); however, shows significant difference with African and European (African: 0%, 0%, 0% and European: 0%, 0%, 5.9% respectively). The allele at each locus (ADH2*2, ALDH2*2, CYP2E1 cl) encodes a subunit that either metabolizes ethanol to acetaldehyde more rapidly or slows the conversion of acetaldehyde to acetate. Taken as a whole, our data suggest that genetic differences in the enzymes that metabolize alcohol can substantially affect the risk for alcoholism.
199. THE ROLE OF SULT1A1 AND EPHX1 POLYMORPHISMS AS RISK MODIFIERS IN CHILDHOOD ACUTE LYMPHOBLASTIC LEUKEMIA IN TURKISH POPULATION: A CASE-CONTROL STUDY
Tugba Boyunegmez Tumer1, Gulen Ulusoy1, Orhan Adali1, Gurses Sahin2, and Emel Arinc1
1Department of Biological Sciences, Middle East Tech Univ, Ankara, Turkey, N/A, 2Oncology Department, Dr. Sami Ulus Children's Hospital, Ankara, Turkey
In recent years, epidemiological studies suggest that environmental chemicals bioactivated by certain DMEs may play an important role in the etiology of development of childhood acute lymphoblastic leukemia (ALL). Epoxide hydrolase (EPHX1) and Sulfotransferase 1A1 (SULT1A) are susceptible DMEs in this respect, since they play a dual role in the detoxification and activation of a broad spectrum of environmental and tobacco derived carcinogens like benzene and polyaromatic hydrocarbons. Both of these enzymes have several SNPs, one of them is a histidine to arginine substitution in exon 4 (His139Arg) of the EPHX1 gene increases in vitro enzyme activity by 25%. In case of SULT1A1 gene, arginine to histidine substitution (Arg213His-SULT1A1*2) significantly reduces enzyme activity and thermostability. These two polymorphisms have been shown to modify risk of various chemically-induced cancer types like lung, gastric and bladder cancers. However, as far as we know, there is no study on the associaton of SULT1A1*2 and EPHX1 exon 4 polymorphisms with childhood ALL. This is the first report evaluating the clinical significance of SULT1A1 and EPHX1 genetic polymorphisms for the risk of developing childhood leukemia. To test the hypothesis that variant alleles of both enzymes may modify the childhood ALL risk, we determined SULT1A1 Arg213His genotype and EPHX1 His139Arg genotype by using PCR/RFLP technique in 138 healthy adult controls and 119 ALL patients. We observed that, when compared with the wild type genotype, the variant SULT1A1 genotype (Arg/His and His/His) and variant EPHX1 genotype (His/Arg and Arg/Arg) were slightly more frequent in the control samples than ALL patients ie. 29% vs 33% for EPHX1 gene (OR = 0.90, 95%CI: 0.55–1.48) and 39% and 42% for SULT1A1 (OR = 0.86, 95% CI: 0.51–1.46). However, the observed differences were not statistically significant. In addition, when both SULT1A1*2-His213 variant and EPHX1 Arg139 variant were considered together OR decreased to 0.71. It was again not statistically significant (95% CI 0.35-1.65). To conclude, our study does not provide support for the suggested modifying role of SULT1A1 and EPHX1 variants for childhood ALL in Turkish population. However, we cannot fully rule out the possible protective benefit of these two polymorphisms because of their general high frequency trend in the control samples. Further studies are warranted to elucidate modifying role of EPHX1 and SULT1A1 polymorphisms in childhood ALL in different populations.
200. GENETIC POLYMORPHISM OF MANGANESE SUPEROXIDE DISMUTASE AND CATALASE IN ASBESTOSIS
Alenka Franko1, Niko Arneric1, Metoda Dodic Fikfak1, and Vita Dolžan2
1Clinical Institute of Occupational Medicine, University Medical Centre Ljubljana, Ljubljana, Slovenia, 1000, 2Inst of Biochem, Univ of Ljubljana Fac of Med, Ljubljana, Slovenia, SI-1000
Asbestosis is among the most frequent diseases caused by asbestos exposure. The causal relationship between asbestos exposure and asbestosis is well established, but little is known about genetic factors that may influence the risk for asbestosis. Asbestos induces the production of reactive oxygen species (ROS) and ROS are known to be involved in the pathogenesis of asbestosis. Manganese superoxide dismutase (MnSOD) and catalase (CAT) represent the primary defense system against ROS. The genes coding for MnSOD and CAT are polymorphic. The aim of this study was to investigate whether MnSOD Ala –9Val and CAT –262C>T genetic polymorphisms represent a risk factor for asbestosis in workers exposed to asbestos. In total 262 cases with asbestosis and 265 matched controls with no asbestos disease were selected from the cohort of 2080 workers occupationally exposed to asbestos that presented to the Board for Recognition of Occupational Diseases between 1998 and 2004. The cumulative asbestos exposure and the pack-years of smoking were calculated for each case and control. Genomic DNA was extracted from the capillary blood samples collected on the FTA Mini Cards (Whatmann Bioscience). MnSOD polymorphism leading to Ala –9Val substitution in the mitochondrial targeting sequence and CAT –262C>T promoter polymorphism were genotyped using a real time PCR assay. To estimate the risk of asbestosis, a logistic regression analysis was used. The average cumulative asbestos exposure was 37.67 fibres/cm3-years in the workers with asbestosis and 11.23 fibres/cm3-years in controls (t = 4.78, p = 0.00). No difference in smoking was observed between the cases and the controls. In both groups, approximately forty-six percent were ever smokers (c2 = 0.01, p = 0.91). MnSOD Ala/Ala genotype was observed in 29.0% of cases and 21.4% of controls, Ala/Val genotype in 45.0% of cases and 53.3% of controls, and Val/Val genotype in 26.0% of cases and 25.3% of controls. CAT TT genotype was found in 8.6% of cases and 6.5% of controls, CT genotype in 41.8% of cases and 40.1% of controls, while CC genotype was found in 49.6% of cases and 53.4% of controls. The OR of asbestosis was 1.50 (CI 1.01–2.24) for MnSOD Ala/Ala genotype versus Ala/Val and Val/Val genotypes, and 1.36 (CI 0.70–2.62) for CAT TT genotype compared to CT and CC genotypes. The risk of asbestosis for MnSOD Ala/Ala genotype did not change considerably after adjustment by gender, age, smoking and cumulative exposure. The gender, age and smoking did not influence the risk of asbestosis for CAT TT genotype either, but the involvement of cumulative exposure changed OR from 1.36 (CI 0.70–2.62) to 1.91 (CI 0.93–3.91), suggesting a confounding effect. In conclusion, the key finding of this study was that MnSOD Ala/Ala genotype significantly increases the risk of developing asbestosis. A non-significantly elevated risk of asbestosis was also observed for CAT TT genotype.
201. DETECTION OF CYP2A6 VARIANT ALLELES COMMONLY FOUND IN ASIAN POPULATIONS BY DUPLEX NESTED PCR
Wardah Yusof and Siew Hua Gan
Pharmacology, Universiti Sains Malaysia, Kelantan, Malaysia, 15150
Background. The CYP2A6 gene encodes the principal enzyme involved in the metabolism of coumarin, nicotine, tegafur, halothane, disulfiram and artesunate. Numerous interindividual variations in CYP2A6 activities have been observed in human, which could be explained partly by the occurrence of CYP2A6 genetic polymorphisms. Simple duplex nested PCR method for CYP2A6*1B, CYP2A6*2, CYP2A6*4, CYP2A6*7, CYP2A6*8 and CYP2A6*9 variant alleles will be useful for detecting CYP2A6 single nucleotide polymorphisms (SNPs) which are prevalent in Asian population. Methodology & Results. Genomic DNA was isolated from peripheral blood of patients treated with artesunate by using a DNA extraction kit. Fragments from TATA box in 5´ flanking region, Exons 1 to 4 and Exon 8 to 3´ UTR of the gene were amplified using a first PCR step (PCR1). Products from PCR1 were then used to run in duplex PCR (PCR2) for detection of the selected variant alleles. This method was verified in 24 healthy Malaysian volunteers where all five CYP2A6 alleles were successfully amplified and confirmed by sequencing. Conclusion. We have developed and optimized a nested duplex PCR method for genotyping of CYP2A6 variants commonly found in Asian populations. Keywords: CYP2A6, genetic polymorphism, nested PCR.
202. STUDYING OF ASSOCIATIONS BETWEEN POLYMORPHISMS OF GENES CYP2C9, CYP2D6, MDR1 AND ATYPICAL DRUG RESPONSE AT RUSSIAN PATIENTS WITH CARDIOVASCULAR DISEASES
Dmitry A. Sychev Sr.
Institute of clinical pharmacology, Roszdravnadzor, Moscow, Russia, 109651
Studying of associations between polymorphisms of genes CYP2C9, CYP2D6, MDR1 and atypical drug response at Russian patients with cardiovascular diseases Sychev DA, Ignatev IV, Kazakov RE, Rameskaya GV, Miheeva Yu, Kropacheva ES, Panchenko EP, Kukes VG Institute of clinical pharmacology of Roszdravnadzor, Moscow, Russian Federation. Objective. We studied the effects of CYP2C9, CYP2D6 and MDR1 genes polymorphism on pharmacokinetics and pharmacodynamics in the population of Russian patients with cardiovascular diseases. Materials and methods. The study was taken on 245 patients with following conditions: CAD, congestive heart failure (CHF), arterial hypertension, permanent atrial fibrillation. All the patients were investigated for carrying allele variants of CYP2C9, CYP2D6 and MDR1 genes using PCR RFLP method, DNA was obtained from peripheral blood leucocytes. We established relation between carrying different allele variants of CYP2C9, CYP2D6 and MDR1 genes and pharmacokinetics, pharmacodynamics and incidence of warfarin, metoprolol and digoxin side effects. Results. We have shown, that warfarin clearances at CYP2C9*2 and CYP2C9*3 allele variants carriers were lower then at carriers genotype CYP2C9*1/*1: 2.4 ± 0.3 ml/min vs 3.5 ± 0.25 ml/min, p = 0.04 and 1.9 ± 0.3 ml/min vs 3.5 ± 0.5 ml/min, g = 0.02 accordingly. The episodes of excessive hypocoagulation (INR more 3) were more often at CYP2C9*2 and CYP2C9*3 allele variants carriers in the sum then at carriers genotype CYP2C9*1/*1: 85% vs 40%, p = 0.0001. But bleedings were more often only at CYP2C9*3 allele variant carriers then at non carriers CYP2C9*3: 50% vs 13%, p = 0.04. Identification of allele variants CYP2C9*2 and CYP2C9*3 predicts bleedings with high specificity (89%), but low sensitivity (44 %) at Russian patients, when warfarin was used. We compared genotyping (determining the «slow» allele CYP2D6*4), phenotyping (determinig the metoprolole and it's metabolite O-demethylmetoprolole concentration) and metoprolole pharmacodynamics in patients with chronic heart failure. The maximal concentration of metoprolol was statistically significantly higher, and maximal concentration of metabolite was below at CYP2D6*4 allele variant carriers then at non carriers CYP2D6*4: 110 ± 25 ng/ml vs 42 ± 16 ng/ml, p = 0.02. Physicians selected a low doze to CYP2D6*4 allele homozygous carriers more often. We assumed that the “target” metoprolole dose should be estimated according to the CYP2D6*4 allele carriage: 46 ± 7 mg per day vs 104 ± 12 mg per day, p = 0.03. We compared genotyping (determining the gene carriage by polymorphic marker C3435T of the MDR1 gene), phenotyping (determining the of steady state dygoxine concentration) and development of the glycosyde intoxication symptoms in patients with chronic heart failure and constant atrial fibrillation. The steady state digoxin concentration was higher at carriers genotype TT then at carriers genotype CC: 1.77 ± 0.17 ng/ml vs 1.17 ± 0.21 ng/ml, p = 0.02. At carriers genotype TT we observed symptoms of digoxin intoxication more often: 57% vs 13%, p = 0.0001. Identification of genotype predicts symptoms of digoxin intoxication with high specificity (84%) at Russian patients. Conclusion. The pharmacogenetic studies of CYP2C9, CYP2D6 and MDR1 are a forward-looking method for patient-tailored pharmacotherapy in cardiology which may be usful in clinical practice in Russian Federation.
203. C3435T POLYMORPHISM IN MDR1 GENE IS NOT ASSOCIATED WITH THE DEVELOPMENT OF PYELONEPHRITIS IN RUSSIAN REPRODUCTIVE AGE FEMALE PATIENTS
Elena A. Sokova
Institute of clinical pharmacology, Roszdravnadzor, Moscow, Russia, 109651
Background. the chronic pyelonephritis, especially combined with gestosis, is known to make worse the outcome of pregnancy for the both mother and fetus. The pyelonephritis is the nephroepithelial disfunction that includes different transport mechanisms interruptions. P-glycoprotein, MDR1 gene product, has been shown to be an important factor in endothelial xenobiotic transport. A number of single nucleotide polymorphisms (SNPs) have been identified throughout the ABCB1 gene, they may influence P-gp expression level and function. The importance of haplotype as well as genotype analysis of SNPs is continuously increasing for identification of genetic variants underlying susceptibility to human diseases development. C3435T substitution is able to decrease MDR1 mRNA stability and so far to reduce P-glycoprotein level in cell membranes, so this attracts the most attention at the present time. Objectives. to determine, whether C3435T polymorphism in MDR1 gene is associated with the development of pyelonephritis in Russian reproductive age female patients. Methods and results. The comparative analysis of the allele/genotype frequencies of MDR1 C3435T polymorphism was undertaken in Russian reproductive age female patients with pyelonephritis (N1 = 43, average age 22,5) and healthy subjects (N2 = 65, average age 19,2). DNA was isolated from peripheral blood samples by standard phenol-proteinase K method. PCR-RFLP method was used for C3435T SNP detection. The differences in allele/genotype frequencies were not statistically significant in case and control groups (p = 0,11 for genotypes and p = 0,96 for alleles). Conclusions. C3435T polymorphism in MDR1 gene is not a risk factor for the pyelonephritis development in Russian reproductive age female patients.
204. STEVIOSIDE AND SYNTHETIC BILE ACID DERIVATIVE SODIUM SALT OF MONOKETOCHOLIC ACID AS HYPOGLICAEMIC AGENTS IN DIABETIC RAT MODEL
Aleksandar Raskovic, Vida Jakovlejvic, Svetlana Golocorbin-Kon, and Momir Mikov
Dept. of Pharmacol & Toxicol, Serbia, Novi Sad, Yugoslavia, 21000
Aims. To investigate the effect of a commercial preparation of stevioside and a synthetic compound, sodium 3α,7α-dihydroxy-12-keto-5β-cholanate (MKC) (monoketocholic acid), applied per os and with the aid of micro-osmotic pump, on glycemia in normoglycemic and diabetic Wistar rats. Hypotheses: Both substances have hypoglyceamic effect. Applied together, they have sinergistic effect. Methods. Diabetes was induced with aloxan, 100 mg/kg, i.p. Normoglycemic and diabetic rats were treated per os for five days either with saline solution (1ml/kg, control), stevioside (20 mg/kg), MKC (4 mg/kg) and combination of stevioside (20 mg/kg) and MKC (4 mg/kg). Apart from per os adminstration, stevioside and MKC were also applied continuously for two weeks with the aid of a subcutaneously implanted micro-osmotic pump. During the treatment and after it, glycemia was measured and the rats that were treated per os were subjected to the oral glucose tolerance test (OGTT) at a dose of 1 g/kg. After that the animals were anesthetized with urethane (0.75 g/kg, i.p.) and blood was sampled by cardiopunction to determine C-peptide in the obtained serum. Procedures were approved by Animal Ethics Committee of Medical Faculty Novi Sad. Results. In all three groups of normoglycemic rats the highest decrease of glucose level was observed on the fourth day of experiment. The combination stevioside + MKC showed a stronger hypoglycemic effect compared to individual treatments with stevioside and MKC (3.73:4.80:4.73 mmol/L). With the group of diabetic rats that received both substances through the osmotic pump the hypoglycemic action was also stronger compared to the individual treatments with stevioside or MKC (16.15:18.89:18.75 mmol/L). Treatment of healthy rats with both substances per os caused no statistically significant difference in glycemia, whereas with diabetic rats the combination of stevioside + MKC showed a statistically significant decrease of glycemia compared to control values. The application of stevioside by osmotic pump yielded a statistically significant increase in the concentration of C-peptide in serum of healthy rats. Compared to control, the concentration of C-peptide in diabetic rats was significantly higher after the treatment with either stevioside or its combination with MKC, irrespective of the mode of administration. Conclusions. Treatment with stevioside and MKC have hypoglycemic synergistic effect in diabetic rats and improve glucose tolerance in healthy as well as in diabetic animals.
205. PREDICTION OF THE PHARMACOKINETICS OF ANIONIC DRUGS USING KINETIC MODELS APPLIED TO ISOLATED RAT HEPATOCYTES
Phil Gardiner and Stuart W. Paine
Discovery DMPK, AstraZeneca R&D-Charnwood, Loughborough, United Kingdom, LE11 SRH
The pharmacokinetics of acid drugs in human are typically characterised by low clearance and low Vss (0.1-0.25L/Kg). Nevertheless, there are a significant proportion of acid drugs (∼20%) that have Vss values in excess of 0.25L/Kg. In the present study, the disposition of 7 marketed acid drugs and 3 AstraZeneca acid compounds, including established substrates of hepatic basolateral uptake transporters, with a range of Vss and clearances, have been profiled in preclinical species. In addition to calculating the pharmacokinetic parameters from plasma, liver and muscle tissue were collected and their contribution to total Vss measured in order to determine the physiological basis for the volumes of distribution. The compounds were also evaluated in suspended hepatocytes using cell and media concentration-time data fitted to a model incorporating active uptake, permeation, binding and metabolism. This ‘uptake sensitive’ model has been assessed for its ability to predict the in vivo pharmacokinetics for these compounds. The tissue levels measured revealed that the volumes of acid drugs could be explained by the levels observed in the liver and for those compounds that had a higher Vss in the preclinical species this was due to selective distribution to the liver. The physiologically based prediction of Vss correlated (all predictions within 2-fold) with the measured values. Hepatic clearance was generally well predicted by the ‘uptake sensitive’ model (within 2-fold), in contrast to predictions based on standard methods. Predictions of Vss using the model also correlated with the measured values and all predictions were within 2-fold with the exception of Tolmetin, which was within 3-fold and Telmisartan which was under-predicted, despite a predicted value of 0.5L/kg. This data suggests that uptake may be expected to have little overall impact on half-life but the ability to estimate the free-intracellular hepatic concentration of uptake substrates has major benefits in terms of predicting pharmacokinetics, potential CYP mediated DDI's and efficacy of hepatically targeted therapeutics.
206. THE IMPACT OF DRUG PERMEATION, UNBOUND FRACTION AND EFFLUX ON CENTRAL NERVOUS SYSTEM DRUG DISPOSITION
Raj K. Badhan, J. Brian Houston, and Jeffrey I. Penny
Centre for Applied Pharmacokinetic Research, School of Pharmacy, Manchester University, Manchester, United Kingdom, M13 9PT
Drug distribution throughout the central nervous system (CNS) is governed by drug permeation at the blood-brain and blood-cerebrospinal fluid barriers, drug free fraction and active transport. To further define this relationship, physiologically-based pharmacokinetic models of the rat and human CNS have been developed. The rat model was employed to validate model structure before construction of a human CNS model. The models incorporated brain interstitial fluid (ISF), chorodial epithelial, total cerebrospinal fluid (CSF) (rat model), cranial CSF and spinal (cervical, thoracic and lumbar) CSF (human model) components and physiological blood/plasma, ISF and CSF flows, permeability-surface area (PS) product and unbound drug fractions in blood/plasma, brain and CSF.
The rat CNS model predicted brain to plasma unbound drug concentration ratio (Kpuu brain) and unbound CSF to unbound plasma concentration ratio (Kpuu CSF) to within 3-fold of the observed Kpuu ratios, confirming the validity of the CNS model structure.
The human model identified high fu blood (≥0.5) and low fu brain (≤0.2) as fundamental for a Kpuu brain ≥ 1. At fu brain < 0.02, Kpuu brain reached 1000 and was insensitive to changes in PS between 1 and 100 mL/h/kg. Increasing P-glycoprotein efflux ratio (ER) from 0 to 80 reduced Kpuu brain by up to 2 orders of magnitude (fu brain 0.04–0.2). This reduction was less pronounced at a fu brain of 0.005 to 0.04, and yielded a Kpuu brain of between 1 and 10 over the range of PS studied.
Simulations using the human model suggested changes in unbound fraction in blood, CSF and brain had little impact on lumbar CSF Kpuu over a range of PS from 1 to 1000 mL/h/kg. P-glycoprotein active efflux at the chorodial epithelium increased Kpuu CSF by more than 10-fold (ER range: 1 to 40). Kpuu CSF was insensitive to change in PS (1 to 100 mL/h/kg) over the range of efflux ratios studied.
Candidate drugs that are substrates for efflux transporters and demonstrate low brain permeation may potentially demonstrate successful brain accumulation (Kpuu > 1), providing fu brain is low (0.005-0.04), and should not be routinely discarded during pre-clinical drug development.
207. PHARMACOKINETICS OF LEVOCETIRIZINE IN SUBJECTS WITH VARYING DEGREES OF RENAL FUNCTION
Margherita Strolin Benedetti1, Christian Otoul2, Rhys Whomsley3, and Jean-Baptiste Watelet4
1Non Clinical Development, UCB Pharma SA, Nanterre, France, 92000, 2Global Pharmacometrics, UCB Pharma S.A., Braine-l'alleud, Belgium, B-1420, 3Non Clinical Development, UCB Pharma S.A., Braine l'Alleud, Belgium, B-1420, 4Department of Otorhinolaryngology, Ghent University, Ghent, Belgium, B-9000
Levocetirizine (L), the eutomer of cetirizine, is a potent second generation H1-antihistamine eliminated mainly by the kidney. The pharmacokinetics and the plasma protein binding (PPB) of L after a single oral dose of 5 mg have been investigated in 18 subjects with normal renal function and a wide range of renal impairment based on measured creatinine clearance (CLcr). Plasma and urine were collected up to 72h and 96h, respectively, after administration and L determined by LC-MS/MS. PPB (at 1h) was measured by equilibrium dialysis. Pharmacokinetic parameters were calculated by non-compartmental analysis. Renal clearance of L by tubular secretion {CLr(ts)} was computed using CLr(ts) = CLr-fu*CLcr, where fu is the unbound fraction in plasma. The extent of exposure (AUC) was increased and the plasma elimination half-life (t1/2) prolonged in subjects with renal impairment. The cumulative urinary excretion (fe) decreased from 70.4% in healthy volunteers to 28.3% in severely impaired subjects. Decrease in total body clearance(CL/F) and renal clearance(CLr) highly correlated with decrease in creatinine clearance (CLcr) ( R2 = 0.85 and 0.83, respectively). CLr was greater than fu*CLcr, demonstrating tubular secretion. The decreases in CLr(ts) and in renal clearance by glomerular filtration {CLr(gf)} also highly correlated with the decrease in CLcr { R2 = 0.68 and 0.95, respectively}, but the slope of regression line of the CLr(ts) was significantly steeper (p = 0.013) than that of CLr(gf). The relationship between the non renal clearance and CLcr showed that some L clearance still remains when CLcr approaches zero (intercept significantly different from zero, p = 0.002). This was confirmed in subjects with end-stage renal disease (CLcr = 0) who showed a mean CL/F of 7 mL/min. Patients with mild to severe impaired renal function require reduced daily doses of L and/or longer dosing intervals compared to unimpaired patients. Although urinary elimination of L decreased progressively with extent of renal impairment, an appreciable non-renal (metabolic) clearance persists when CLcr approaches zero. The decay of Clr(ts) would appear to be more rapid than that of Clr(gf). PPB did not significantly change in the renal impaired subjects, which is of consequence e.g. for the estimation of free drug concentrations and receptor occupancy, related to the efficacy of the drug.
208. INFLUENCE OF A GRANISETRON ON PHARMACOKINETICS OF A MEDICINE IN PATIENTS WITH A DUODENAL ULCER
Svetlana Yu. Serebrova, Alexey K. Starodubtsev, Svetlana N. Kondratenko, Galina A. Belyakova and Vera D. Starodubtseva
Chair of Clinical Pharmacology, Moscow Medical Academy named by Sechenov, Moscow, Russia, 109240
Hypothesis. Many pati.ents with an exacerbation of peptic ulcer disease have a decrease in efficiency of the omeprazole and lansoprazole first doses and a decrease of their bioavailability. In these patients the effect of omeprazole and lansoprazole on intragastric pH comes at low their doses, but Cmax can decrease at Tmax. We assume that fluctuations of stomach pH with high amplitude due to violations of motor activity in these patients can lead to partial dissolution of proton pump inhibitors and their effect on gastroduodenal mucosa chemoreceptors. We also assume that this effect is realized with serotonin and its 5-HT3 receptors. Using granisetron we studied if blockade of 5-Í″3-dependent positive feedback of self-regulation of serotonin formation can influence pharmacokinetics of omeprazole in healthy volunteers and in patients with duodenal ulcers. Methods used. We researched pharmacokinetics of omeprazole (after single 20 mg oral dose) in 10 healthy volunteers and 11 patients with early remission of peptic ulcer disease twice: with preliminary reception of 1 mg granisetron and without it. Concentrations of omeprazole were measured by high-performance liquid chromatography with spectrophotometric detection at 302 nm). Supporting data. The serotonin plasma concentrations measured by immunenzymatic assay (in vitro immunoenzimatic test-system, Labor Diagnostika Nord GmbH & Co. KG, Nordhorn) in the most of our patients with early (< 2 month) remission of peptic ulcer disease were maximum degrees that could be shown by our measure device (≥152.27 ng/mL, references meanings are 1.80–7.50 ng/mL), but they were 9.26 ± 2.14 ng/mL in healthy subjects. Results. There were significant differences in volunteers and patients for Cmax (639.8 ± 155.7 and 299.0 ± 64.7 ng/mL respectively), AUC(0-t) (946.6 ± 122.0 and 578.5 ± 26.5 ng·hr/mL respectively), Clt (20.8 ± 2.3 and 33.0 ± 2.5 L/hr respectively) and Vz (41.4 ± 8.3 è 52.4 ± 2,3 L respectively). After previous taking of granisetron in volunteers group Cmax was 913.3 ± 200.4 ng/mL, AUC(0-t) was 1379.9 ± 249.9 ng·hr/mL (p < 0.05), Clt was 15.6 ± 2.9 L/hr, Vz was 23.0 ± 7.9 L (p < 0.05); in patients group Cmax was 171.3 ± 0.2 ng/mL (p < 0.05), AUC(0-t) was 475.6 ± 202.4 ng·hr/mL (p<0.05), Clt was 44.7 ± 23.7 L/hr, Vz was 87.9 ± 59.6 L (p < 0.05). Conclusions. Granisetron increases bioavailability of omeprazole in healthy subjects and decreases bioavailability in patients with early remission of peptic ulcer disease.
209. PK/PD AND DOSE SETTING OF MONOCLONAL ANTIBODIESFOR NDAS IN JAPAN
Teruyo Arato
Office of Biologics 1, Pharmaceuticals and Medical Devices Agency, Tokyo, Japan, 100-0013
The development of therapeutic antibodies is rapidly increasing and several monoclonal antibodies genetically engineered have been approved for the last several years in Japan. Therefore, new drug application dossiers and review reports were surveyed to investigate the current situation of PK/PD and dose setting of these monoclonal antibodies.The immunoassays were commonly used for analytical methods of blood concentration. Monkeys were used for nonclinical PK studies of all antibody products genetically engineered. Clinical PK studies were mostly conducted in patients. The elimination rate of some antibodies, such as tocilizumab, in patients was higher than that in healthy volunteers. The half-lives of monoclonal antibodies increased with the degree of humanization (mouse<chimeric<CDR-grafted), with increasing duration of dosing. On the other hand, immunogenicity decreased with the humanization, and the presence of anti-antibody antibodies may affect the PK.The approvals of these antibodies are 3–5 years behind the USA/EU and they have been approved based on a bridging strategy or by using foreign clinical data. The ethnic differences in PK parameters and efficacy/safety of these antibodies were not observed, and the standard “Dosage and Administration” of almost all these products were same between Japan and the USA/EU. As a rare case, for etanercept, even though the PK parameters in Japanese populations were similar to the foreign study populations, the different dose was set in Japan, because of the efficacy. It is difficult to detect the frequency of rare adverse events, because the number of patients enrolled in clinical trials is too small to detect it. Therefore, these antibodies were approved with mandatory post-marketing requirements of the collecting more information of the safety.
210. EFFECT OF CAFFEINE ON QUINIDINE TRANSPORT TO THE CENTRAL NERVOUS SYSTEM IN RATS
Velibor Vasovic, Vida Jakovljevic, Branko Banic, Ana Sabo, Sasa Vukmirovic, and Boris Milijasevic
Department of Pharmacology, Medical Faculty of Novi Sad, Novi Sad, Serbia, Novi Sad, Serbia and Montenegro
The aim of this research was to study the effect of caffeine on quinidine transition through the blood-brain barrier (BBB) to central nervous system (CNS). Study was performed on Wistar rats of both sexes, b.w. 270–350 g, divided in two groups, each of 30 animals. The control group received s.c. physiological solution 30 min before quinidine administration, while test group received (s.c.) caffeine 25 mg/kg b.w. Quinidine 25 mg/kg b.w. was administrated by catheterization of the right arteria axillaris. Blood samples were taken from jugulary vein 30, 60, 90, 120, and 240 s after quinidine administration. After blood sampling, CNS was rinsed with 5 ml of distilled water injected into the left heart ventricle. Rats were sacrificed in the same time intervals as for blood sampling. Brain tissue was divided into the brainstem (truncus encephalicus), cerebellum, left and right cerebral hemispheres. After weighting, the particular CNS parts were homogenized. Qunidine concentrations in brain and blood samples were determined by standard spectrophotofluorimetric method. In all time intervals caffeine decreased quinidine transport from blood to brainstem compared to control (except in the 60th s, statistically significant in the 90th s). Decrease in quinidine transport from blood to cerebellum was observed in all time points, except in 240th s (statistically significant in the 60th, 90th and 120th s). Observed decrease in quinidine transition to truncus encephalicus and cerebellum (together-first CNS compartment) in presence of caffeine may be explained by effect of caffeine (also a cation) on quinidine transport through the BBB. Further investigations should show whether there is competition for the carrier, having in mind possible active transport of qunidine. In test group decrease in quinidine transition from blood to the left cerebral hemisphere compared to control, was observed in all time points except in 60th s (statistically significant in the 90th s). Similar was observed for the right cerebral hemisphere. Decrease was not statistically significant. The hindered passage of quinidine from blood to second CNS compartment (cerebral hemispheres) under the influence of caffeine was less pronounced than in the case of the first compartment. This may be explained in terms of the local conditions of blood circulation, and regional properties of the BBB, yielding higher concentrations of quinidine in the first compartment of the control animals. The quinidine concentration was higher in brain than in blood. Increased level in brain was registered with a latency compared to blood levels what could be explained by the involvement of active transport of quinidine, as well as by the fact that at higher physiological pH values, quinidine is mainly present in its undissociated form. The regional differences observed in quinidine transition to the brain indicate that there are some local factors influencing active transport.
211. THE EFFECT AND CORRELATION OF ACTIVITY AND HYPERICIN CONCENTRATION OF DOMESTIC COMMERCIALLY AVAILABLE ST. JOHN'S WORT PRODUCT IN MICE
Zoran Bukumiric1, Vida Jakovljevic2, Radoslav Mitic1, Vladimir Janjic3, Snezana Stevic1, Zorica Stanojevic1, Sasa Vukmirovic2, and Jelena Cvejic4
1Institute of Pharmacology and Toxicology, Medical Faculty Pristina, Kosovska Mitrovica, Kosovska Mitrovica, Serbia and Montenegro, 2Department of Pharmacology, Medical Faculty of Novi Sad, Novi Sad, Serbia, Novi Sad, Serbia and Montenegro, 3Psychiatry Clinic, Clinical Center, Kragujevac, Serbia and Montenegro, 34000, 4Department of Pharmacy, Faculty of Med, Novi Sad, Vojvodina, Yugoslavia, YU-21000
St. John's Wort (Hypericum Perforatum) has been used in our traditional medicine for treatment of different diseases for a long time. In last couple of years use of different St. John's Wort based products is in constant increase. Usually it is a self-medication, patients buying such products in pharmacies or in health food stores, without consulting doctors about severity of disease and adequate dosage for their condition. The aim of research was to examine the efficacy of domestic commercially available St. John's Wort product (HySD) by forced swimming and tail suspension tests in mice. The aim was also to compare hypericin concentrations in different St. John's Wort products, as well as to determine correlation of hypericin concentration and effect in mentioned tests. Methods. Experiment was carried out on NMRI mice b.w. 25–30g. Animals were given water solution of St. John's Wort capsules of domestic origin HySD (standardised by manufacturer to contain 300mcg of hypericin per capsule) intraperitonealy (i.p.) 200mcg/kg of hypericin. Control substances were: imported commercially available St. John's Wort product HyQ (200mcg/kg of hypericin) i.p., sertraline 10 mg/kg i.p. and maprotiline 10mg/kg i.p. Substances were administrated 24h, 5h and 1h before the experiment. Modified forced swimming (Behavioral despair) and tail suspension tests were used in experiment. Each animal was control to itself. Analysis of animal behavior was performed by specially designed program, and results were statistically analyzed with paired t-test. Hypericin concentration was determined by HPLC-DAD method. Results. Maprotiline 10 mg/kg i.p. statistically significant decreased immobilization (p < 0.01) in forced swimming test. Sertraline 10mg/kg i.p. statistically significant decreased immobilization (p < 0.01) in tail suspension test. HyQ imported St. John's Wort tablets (200mcg/kg of hypericin) i.p. statistically significant decreased immobilization (p < 0.01) in tail suspension test. HySD domestic St. John's Wort product did not statistically significant decreased immobilization in forced swimming, or in tail suspension test. Amount of hypericin in HySD was 179mcg, although it was claimed to contain 300mcg of hypericin per capsule. Correlation was not established between hypericin concentration and decrease of immobilization in the forced swimming and tail suspension tests in examined groups of animals. Conclusions. According to results it can be concluded that domestic commercially available St. John's Wort product (HySD) did not have significant antidepressive effects in mentioned tests, probably because hypericin concentration differed from what was claimed to contain. For this reason it should be obligatory to consult a physician before using cantarion in treatment of depression.
212. ASSESSING SPECIES DIFFERENCES IN PREGNANE X RECEPTOR (PXR) ACTIVATION IN STABLE CELL LINES
Kyle Kramer, Elsa Ponce, and Judy Raucy
Puracyp, Inc, Carlsbad, CA, USA, 92010
When considering drug safety assessment, PXR is undeniably one of the most important nuclear receptors. Activation of this receptor is responsible for unwanted adverse drug reactions (ADRs) and induction of major drug metabolizing enzymes. To determine the potential for ADRs caused by PXR activation, in vivo animal studies or experiments conducted with animal derived materials have been approaches most often utilized. However, reliance upon data derived from these sources to predict human responses may be ineffective. For this reason, the present study examined species differences in the activation of PXR utilizing a novel approach. Three stable cell lines were constructed in hepatic-based cells that contained PXR from rat, monkey, or human. These cell lines were also stably integrated with CYP3A promoters linked to the luciferase reporter gene. Cell lines were treated with several prototypical PXR activators. Twenty-four hours later, activation was measured and compared to solvent control treated cells. Those cell lines harboring monkey PXR (MkB3) exhibited responses that more closely paralleled those generated in the DPX2 cells (human PXR), when compared to those from cells transformed with rodent PXR (rPXR). For example, rifampicin (10 uM) produced 18.5 + 0.84, and 13.3 + 0.1- fold activation over solvent treated cells in MkB3 and DPX2 cells, respectively, but did not cause enhanced luciferase activity in rPXR cells (1.36 + 0.02- fold), In contrast, dexamethazone (10 uM) produced 5.0 + 0.31, 1.7 + 0.1, and 1.3 + 0.03 fold increases in luciferase activity in rPXR, MkB3 and DPX2 cell lines, respectively. Of the agents tested, pregnenolone 16α-carbonitrile (PCN) was the only compound to produce activation in the rPXR cells with a negligible effect in the MkB3 or DPX2 Cells. Interestingly, the natural product, genistein (100 uM) was a more potent activator of PXR than rifampicin in both DPX2 (20.2 + 0.42-fold above control) and MkB3 cells (40.5 + 0.54-fold) but only caused a modest activation (2.8 + 0.04-fold) in the rPXR cell line. Comparisons between DPX2 and MkB3 cells revealed, only quantitative differences in response with the PXR activators tested. However, between rPXR and the other two cell lines, qualitative differences were observed. In conclusion, to predict ADRs stemming from PXR activation, cell lines harboring animal and human PXR may be used to assess species differences, decreasing the need for live animals.
213. SIMULTANEOUS QUANTIFICATION OF FOUR TRACERS: RACLOPRIDE, PIRENZEPINE, MDL-100907, AND SB-221284 FOR THE USE IN IN VIVO RECEPTOR OCCUPANCY STUDIES
Kara Schmelzer1, Darren Craig1, Douglas Bonhaus2, and Ali Tabatabaei1
1Dmpk, Acadia Pharmaceuticals, San Diego, CA, USA, 92121, 2Biosciences, Acadia Pharmaceuticals, San Diego, CA, USA, 92121
The atypical antipsychotics share the common attributes of working on serotonin and dopamine receptors. However, a comprehensive mechanism of action of these agents is unknown. In order to better elucidate the mechanism by which these drugs produce their therapeutic and the undesired effects, a robust method for measuring in vivo receptor binding is needed. The authors developed a liquid chromatography / triple quad mass spectral detectors (LC/MS/MS) method to measure the brain distribution of receptor occupancy using tracers targeting dopamine D2, serotonin 5-HT2A, 5-HT2C and muscarinic M1 receptors. The tracers are raclopride, MDL-100907, SB-221284 and pirenzepine, respectively. All four non-radiolabeled tracers were detectable in discrete rat brain areas after intravenous administration. These tracers demonstrated a differential brain distribution corresponding to the regional differences in their respective receptor densities. Oral pretreatment of various antipsychotic agents that occupy these receptors decreased this differential distribution in a dose-dependent manner. Our results demonstrate the utility of LC/MS/MS to quantify the in vivo receptor occupancy of antipsychotic and other agents acting in the CNS.
214. UPTAKE OF REPAGLINIDE IN HEK293 CELLS EXPRESSING OATP1B1 AND PLATED HUMAN HEPATOCYTES
Laura K. Hinton1, Jennifer S. Thomas2, Kathryn E. Kenworthy2, Graham I. Somers3, Gary Manchee3, Aleksandra Galetin1, and J. Brian Houston1
1School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester, United Kingdom, M13 9PT, 2GlaxoSmithKline, Ware, United Kingdom, 3GlaxoSmithKline, Stevenage, United Kingdom
A number of recent clinical studies have indicated the importance of the hepatic uptake transporter OATP1B1 on the disposition and efficacy of a wide range of therapeutic drugs. In order to assess the fraction of repaglinide transported via OATP1B1, uptake studies were performed in human embryonic kidney cells (HEK293) expressing OATP1B1 and plated human hepatocytes. Hepatic uptake in both systems was investigated using 3 min incubation time and over 0.2–100 μM range of repaglinide concentrations. The contribution of passive permeability was assessed by performing the experiments at 4°C over the range of repaglinide concentrations. Repaglinide clearance via passive diffusion (Pdiff) and hepatic uptake (CLuptake) were obtained in the HEK293 expression system and in plated human hepatocytes. In addition, relative importance of the active hepatic uptake in comparison to the passive process was estimated over the range of repaglinide concentrations investigated. In OATP1B1 HEK293 cells passive permeability of repaglinide was fairly rapid (160 nm/s) and linear over the range of concentrations at 4°C. In this cell line, repaglinide CLuptake was 7-fold greater than via the passive diffusion (120 and 19 μL/min/mg protein, respectively). In plated human hepatocytes, the rate of passive uptake of repaglinide at 4°C and Pdiff were comparable to the OATP1B1 expression system; CLuptake was 5-fold greater than the passive component. Assessment of the contribution of the passive permeability and active uptake to the overall repaglinide uptake indicated that, at clinically relevant concentrations (0.1–0.2 μM), active uptake is the predominant process in both cell types. Data from HEK293 cells expressing OATP1B1 indicated that the active process contributes 87% to total repaglinide uptake. The contribution of the active process to the total repaglinide uptake in hepatocytes was comparable (83% at 0.1 μM) suggesting that OATP1B1 is the predominant transporter involved in the hepatic uptake of this compound.
215. INTERINDIVIDUAL VARIABILITY OF ORGANIC CATION TRANSPORTER OCT1 (SLC22A1) AND OCT3 (SLC22A3) EXPRESSION IN HUMAN LIVER
Anne T. Nies1, Elke Schaeffeler1, Kathrin Klein1, Ulrich M. Zanger1, Reinhold Kerb1, Hermann Koepsell2, Dietrich Keppler3, and Matthias Schwab1
1Dr. Margarete Fischer-Bosch Institute of Clinical Pharmacology, Stuttgart, Germany, 70376, 2Institute of Anatomy and Cell Biology, University of Würzburg, Würzburg, Germany, 97070, 3Div Tumor Biochemistry, Deutsches Krebsforschungszentrum, Heidelberg, Germany, 69120
An important function of hepatocytes is the biliary elimination of endogenous and xenobiotic small molecules, many of which are organic cations and cationic drugs. The first step of hepatobiliary elimination is the hepatocellular uptake of substances from blood which is mediated by a number of transporters in the sinusoidal membrane. Organic cation transporter 1 (OCT1, SLC22A1; reviewed in ref. 1) has been identified in the sinusoidal membrane of rodent and human hepatocytes (2, 3). We here provide evidence that, in addition to OCT1, organic cation transporter 3 (OCT3, SLC22A3) is also expressed in the sinusoidal membrane of human hepatocytes. Because OCT1 and OCT3 may decisively determine the disposition and action of drugs such as metformin and oxaliplatin, we analyzed OCT1 and OCT3 expression in 150 non-tumor liver tissue samples of a white population by quantitative real-time PCR analysis (TaqMan). Standardized OCT1 and OCT3 mRNA expression showed a 41- and 11-fold variability, respectively. Expression of both transporters did not correlate with sex or age. The ratio of OCT1/OCT3 mRNA expression varied from 4 to 150. Genotype analyses are currently being performed to identify polymorphisms accounting for variability of OCT1 and OCT3 expression.
ACKNOWLEDGMENT
Supported by the Robert Bosch Foundation, Stuttgart, Germany.
REFERENCE
- Koepsell H, Lips K, Volk C (2007) Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res 24: 1227–1251.
- Meyer-Wentrup F, Karbach U, Gorboulev V, Arndt P, Koepsell H (1998) Membrane localization of the electrogenic cation transporter rOCT1 in rat liver. Biochem Biophys Res Commun 248:673–678.
- Nies AT, Herrmann E, Brom M, Keppler D (2007) Vectorial transport of the plant alkaloid berberine by double-transfected cells expressing the human organic cation transporter 1 (OCT1, SLC22A1) and the efflux pump MDR1 P-glycoprotein (ABCB1). Naunyn Schmiedeberg’s Arch Pharmacol, published online Dec 19, 2007. DOI: 10.1007/s00210-007-0219-x.
216. IN VITRO EVALUATION OF TRANSPORTER-MEDIATED DRUG-DRUG INTERACTIONS
Bo Feng and Rouchelle Mireles
Dept of PDM, Pfizer Inc, Groton, CT, USA, 06340
A number of uptake and efflux transporters have been identified in human liver and kidney. Transporter-mediated hepatic and renal uptake and efflux can be important determinants of hepatic and renal drug elimination, and transporter-mediated drug-drug interactions (DDIs). Two case studies are presented here to identify the transporters potentially involved in hepatic and renal drug disposition and elimination, and evaluate the propensity of compounds to cause DDIs at the level of transporters in vitro. Transporter-transfected HEK 293 and MDCK cells and membrane vesicles containing efflux transporters were used in the transporter assays in vitro. For case 1, compound X for oncology target was discontinued from development due to conjugated hyperbilirubinemia and/or increases in ALT and AST. In vitro hepatic uptake transporter assays have suggested that compound X was a potent OATP1B1 inhibitor with an IC50 of 1.7 uM. In addition, compound X was identified as a substrate of OATP1B1, thus hepatic accumulation of compound X could occur. For hepatic efflux transporter assays, compound X was characterized as a MRP2 inhibitor with IC50 of 52 uM, which may contribute to the observed hyperbilirubinemia. Compound X was also identified as a potent bile salt export pump (BSEP) inhibitor with IC50 of 8.5 uM, which may cause hepatic damage. For case 2, compound Y is for the treatment of type 2 diabetes, and metformin and compound Y are preferred to be dosed concurrently in MD and POC studies. Metformin has been known to interact with human OCT2, a major renal organic cation transporter. In vitro renal transporter studies have indicated that compound Y was not able to inhibit hOCT2-mediated uptake of metformin, which suggested that it is unlikely that compound Y will cause DDI with metformin. These studies have demonstrated the utility of these in vitro transporter assays to better predict the propensity of compounds to cause DDIs in hepatic and renal clearance, and to better understand the clinical observations of toxicity in liver.
217. FUNCTIONAL EVALUATION OF SLC TRANSPORTERS IN CRYO-PRESERVED HUMAN HEPATOCYTES
Eric Sands1, Vina Ketty1, Yong-Hae Han2, Joanne Bourgea1, Chris J. Patten1, Charles L. Crespi1, and Guangqing Xiao1
1BD Biosciences, Discovery Labware, Woburn, MA, USA, 01801, 2Metabolism and Pharmacokinetics, Bristol-Myers Squibb, Princeton, NJ, USA, 8540
The purpose of this study was to evaluate the function of Sodium Taurocholate Co-transporting Polypeptide (NTCP), Organic Anion Transporting Polypeptide (OATP), and Organic Cation Transporter (OCT) in cryopreserved human hepatocytes, and to examine the impact of various drugs on hepatic transporter-mediated uptake. Cryopreserved human hepatocytes were prepared using the Percoll™ method. Hepatocytes were suspended in Krebs-Henseleit buffer (KHB) and uptake assays were performed using an oil-filtration method. The function of NTCP, OATP, and OCT1 was characterized at both 37C and 4C by using relatively specific substrates and inhibitors. The transport activity in cryopreserved hepatocytes prepared from different donors was investigated. Time-, temperature-, and concentration-dependent uptake of Taurocholate, Estrone-3-Sulfate, and Tetraethylammonium (TEA) were observed in cryopreserved human hepatocytes. The transporter-mediated uptake was decreased in the presence of various inhibitors. Donor-to-donor distribution on the uptake of Taurocholate, Estrone-3-Sulfate, and TEA was found. There was no significant correlation between the uptake activities of each substrate. Our results indicated that significant function of NTCP, OATP, and OCT1 remained in cryopreserved human hepatocytes. Our data also demonstrated that the major transporter (s) for each substrate is (are) different, and the uptake activity of each substrate reflects the transport function of the corresponding transporter (s). Cryopreserved human hepatocytes can be used to study drug transport in the liver by using specific substrates and specific inhibitors. Cryopreserved human hepatocytes can also be useful for studying hepatic transporter involved drug-drug interactions.
218. GENOTYPING OF HUMAN MDR1 IN CACO-2 AND MDCK-MDR1 CELL LINES REVEALS IMPORTANT DIFFERENCES THAT MAY IMPACT ON THEIR USE AS IN VITRO TOOLS FOR STUDYING DRUG TRANSPORT
Rachel Jupp1, Graham Belfield1, Anna-Lena Ungell2, and Constanze Hilgendorf2
1Molecular Biology, AstraZeneca R&D Charnwood, Loughborough, United Kingdom, UK LE11 5RH, 2Discovery DMPK and Bioanalytical Chemistry, AstraZeneca R&D Mölndal, Mölndal, Sweden, 43183
The human colonic epithelial cell line Caco-2 is a widely used model of epithelial drug transfer and also MDR1 (P-gp, ABCB1) mediated drug transport. In recent years, recombinantly transfected cell lines e.g. MDCK-MDR1 have been preferentially used, as they offer the advantage of being able to distinguish a specific transporter activity (i.e. human MDR1) from any native background transporter activity the cell line possesses. Many sequence polymorphisms have been reported for the human MDR1 gene, with a number altering gene expression and/or transporter activity. We isolated the MDR1 coding sequences from both Caco-2 and MDCK-MDR1 cell lines to establish which MDR1 variant forms existed in the two cell lines. We discovered that the MDCK-MDR1 cell line contained the most common Caucasian polymorphic form. However the Caco-2 line (containing two inherited copies of MDR1) was heterozygous, containing two different polymorphisms at three positions within MDR1. These differences are summarised in .
Clinical and in vitro data has suggested that the G2677T – Ala893Ser and C3435T polymorphisms may affect MDR1 expression and/or function. Therefore the MDCK-MDR1 and Caco-2 cell lines have a different complement of MDR1 proteins which could in turn influence their behaviour in drug transport studies. Furthermore, both cell types contain mRNA copies which appear to be cryptically spliced, which could produce a variety of different product sizes and activities, albeit at much lower abundances than the full length MDR1 products they also produce. We believe this could be evidence of a mechanism of post transcriptional gene regulation in MDR1. In conclusion, we have identified MDR1 DNA sequence differences between two commonly used in vitro tools that could in some circumstances lead to differences in compound efflux and be a source of data variability.
219. THE ORPHAN CARRIER SLC10A4 IS EXPRESSED IN CHOLINERGIC NEURONS OF THE RAT CENTRAL AND PERIPHERAL NERVOUS SYSTEM
E. Petzinger, S. Burger, C. Fernandes, B. Döring, S. Rafalzik, R. Gerstberger, and J. Geyer
Institute of Pharmacology and Toxicology, University of Giessen, Giessen, Germany, 35392
The orphan carrier Slc10a4 is expressed in cholinergic neurons of the rat central and peripheral nervous system S. Burger1, C. Fernandes1, B. Döring1, S. Rafalzik2, R. Gerstberger2, E. Petzinger1, J. Geyer1 The solute carrier family SLC10 was formerly referred to as the “sodium bile acid cotransporter family”. Indeed, the founding family members are bile acid carriers which belong to the class of sodium-coupled cotransporters at the plasma membrane of hepatocytes and ileal cells. They maintain the enterohepatic circulation of bile acids between the liver via Ntcp (Slc10a1) and the ileum via Asbt (Slc10a2) and participate in the homoeostasis of cholesterol. In 2004 we cloned a new member of this carrier family which is referred to as Slc10a4 (GenBank accession no. AY825923). Phylogenetic analysis revealed that Slc10a4 and Ntcp emerged from a common ancestor gene, but both proteins clearly differ in function. In contrast to Ntcp, Slc10a4 showed no transport activity for bile acids and sulfoconjugated steroid hormones when expressed in Xenopus laevis oocytes and HEK293 cells. Gene expression analysis by real-time quantitative PCR revealed that Slc10a4 expression is highest in the brain. High SLC10A4 expression was also detected in human digestive organs pointing to an additional role of this protein outside the brain. By employing a polyclonal rabbit antibody directed against a C-terminal epitope of the rat P4 protein we localized the Slc10a4 protein in cholinergic neurons in many regions of the rat central nervous system. The protein co-localized together with typical marker proteins for acetylcholine transport like ChT1 and VAchT. Additionally, we detected Slc10a4 expression in neurons and fibers of the myenteric plexus of the enteric nervous system. In conclusion: Although the functional properties of Slc10a4 are so far unknown, we assume that Slc10a4 is an orphan carrier at cholinergic neurons or may show regulatory function for cholinergic transmission by neuronal and non-neuronal cells. 1 Institute of Pharmacology and Toxicology, Frankfurter Str. 107, and 2 Institute of Physiology, Frankfurter Str. 100, Faculty of Veterinary Medicine, Justus-Liebig University of Giessen, D-35392 Giessen.
220. TRANSCRIPTIONAL REGULATION OF RAT HEPATIC OATPS
Gabrielle M. Hawksworth1, David E. Cowie1, and Richard J. Weaver2
1Dept of Medicine and Therapeutics and School of Medical Sciences, University of Aberdeen, Aberdeen, United Kingdom, AB25 2ZD, 2Centre for Biopharmacy Research, Servier Research & Development Ltd, Slough, United Kingdom, SL3 6PJ
Hepatobiliary transporters are involved in the systemic elimination of therapeutic agents, and therefore play a key role in their bioavailability, pharmacokinetics and toxicity. Nuclear hormone receptors such as the pregnane X receptor (PXR) and constitutive androstane receptor (CAR), which act as transcriptional regulators of the cytochrome P450 superfamily of enzymes, have been implicated in the transcriptional regulation of the hepatic transporters, Oatps and Mrps. Quantitative RT-PCR (Taqman) was used to determine changes in RNA levels of Oatp1a1, Oatp1a4 and Oatp1b2 in vivo and in vitro in male Sprague - Dawley rats after treatment with PXR and CAR activators. PXR ligands resulted in a dose-dependent increase of Oatp1a4 mRNA in vivo, with maximal induction occurring at 50 mg/kg dexamethasone (12703 ± 1985 copies/ng RNA compared with control, 1540 ± 193 copies/ng RNA) and 80mg/kg pregnenolone-16a-carbonitrile (8667 ± 993 copies/ng RNA compared with control values of 1540 ± 193 copies/ng RNA). Neither Oatp1a1 nor Oatp1b2 transcript levels were significantly altered by PXR ligands. Treatment with the CAR activator, phenobarbital, or ligand, TCPOBOP (1,4-Bis-[2-(3,5-dichloropyridyloxy)] benzene), did not significantly alter any of the Oatp levels. This PXR-dependent effect on Oatp1a4 was also seen in vitro using isolated hepatocytes in a Matrigel™ sandwich culture configuration. 48h treatment with 10mM dexamethasone (7959 ± 826 copies/ng RNA compared with a mean control value of 1795 ± 307 copies/ng RNA) or 5mM RU486 (13235 ± 4183 copies/ng RNA compared with control, 1795 ± 307 copies/ng RNA) elevated Oatp1a4, but not Oatp1a1 or Oatp1b2 levels. GAPDH mRNA levels were unaltered by treatment or vehicle in vivo or in vitro. The data demonstrate that Oatp1a4 is the only PXR-inducible Oatp isoform in rodent liver and that CAR does not play a major role in the transcriptional regulation of rat hepatic Oatps, suggesting that receptor cross talk may be less relevant for rat hepatic transporters than for P450s.
221. CHARACTERIZATION OF TRANSPORTER EXPRESSION IN PRIMARY CULTURES OF HUMAN HEPATOCYTES
Pilar Prentiss, Maciej Czerwinski, David B. Buckley, Kevin C. Lyon, Greg Loewen, and Andrew Parkinson
XenoTech LLC, Lenexa, KS, USA, 66219
Primary human hepatocytes are a widely accepted in vitro system for analysis of hepatic drug metabolism and transport. Although the contribution of the uptake and the efflux transporters to the hepatic clearance of xenobiotics is recognized, the details of their expression in this system are not well documented. The mRNA expression of the organic anion transporting peptides (OATP1B1, OATP1B3, OATP2B1), sodium-dependent taurocholate co-transporting polypeptide (NTCP), multidrug resistance protein (MDR1), multidrug resistance associated protein (MRP2) and bile salt export pump (BSEP) was analyzed in cultures of freshly plated primary hepatocytes over six days. Additionally, the response of the cells to prototypical agonists of the constitutive androstane receptor (CAR), pregnane X receptor (PXR) and farsenoid X receptor (FXR), phenobarbital, rifampin, and chenodeoxycholic acid, respectively, was analyzed following a 72-h treatment period. The mRNA expression was determined by reverse transcription-polymerase chain reaction (RT-PCR). Transporter expression reached the highest levels at 3 to 4 days in culture, followed by a period of slight decline. Notably, BSEP and NTCP mRNA expression increased significantly by 72 hours post-plating, indicating restoration of hepatic function. Phenobarbital (750 μM) and rifampin (10 μM) induced the mRNA expression of OATP1B1 (2.4–3.5 fold), OATP2B1 (1.8–2.4 fold), MDR1 (3.7–4.2 fold), and MRP2 (4.0–4.2 fold). Hepatocytes did not respond to chenodeoxycholic acid (10 μM) treatment with increased levels of BSEP mRNA. However, this response was potentially masked by the increase in BSEP mRNA expression observed over time, resulting from synthesis and disposition of endogenous bile acids. Based on these results, we conclude that primary human hepatocytes are a suitable system for the analysis of inductive potential of new chemical entities toward hepatic uptake and efflux transporters.
222. STRUCTURAL REQUIREMENTS FOR DRUG INHIBITION OF THE HUMAN ORGANIC CATION TRANSPORT PROTEIN OCT1 (SLC22A1)
Gustav Ahlin1, Johan Karlsson2, Jenny Pedersen1, Lena B. M. Gustavsson3, Rolf Larsson4, Par Matsson1, Ulf Norinder5, Christel A.S. Bergstrom1, and Per Artursson1
1Department of Pharmacy, Uppsala University, Uppsala, Sweden, SE-75124, 2AstraZeneca R&D, Molndal, Sweden, SE-431 83, 3Dept of Discovery/DMPK, AstraZeneca R&D-Lund, Lund, Sweden, SE-221 87, 4Clinical Pharmacology, Department of Medical Sciences, Uppsala University, Uppsala, Sweden, 751 85, 5AstraZeneca R&D, Sodertalje, Sweden
The organic cation transport protein OCT1 is a hepatic sinusoidal drug transport protein that transports several registered cationic drugs, including the antidiabetic drug metformin and the anticancer agents oxaliplatin and imatinib. In this study we explored the chemical space of registered oral drugs with the intention of examining the inhibition pattern of OCT1 and to generate predictive computational models of OCT1 inhibition. In total, 191 structurally diverse compounds were studied/investigated/examined/considered/observed in HEKs293 cells stably that had been transfected with wild type human OCT1. Those compounds that inhibited the transport of the model substrate 4-(4-(dimethylamino)styryl)-N-methylpyridinium (ASP+) uptake by 50% or more were defined as OCT1 inhibitors. The assay identified 50 novel inhibitors and confirmed 12 previously reported inhibitors/ones. Enrichment of OCT1 inhibitors was seen in several therapeutic drug classes, including antidepressants, antihistamines, steroids and a-receptor blocking agonists. High lipophilicity and having a positive charge were found to be the two key physicochemical properties for OCT1 inhibition at a physiological pH, whereas [the existence of] a dipole moment and hydrogen bonding energy were negatively correlated to OCT1 inhibition. The experimental data was used to generate computational models for classification of OCT1 inhibitors; the final OPLS-DA model correctly predicted 82% of the inhibitors and 88% of the non-inhibitors of the test set. The simplicity, accuracy and speed of the in silico model obtained will hopefully make it applicable as a virtual screening filter in the early drug discovery process.
223. ARE THE COMMONLY USED DRUG TRANSPORT INHIBITORS, ELACRIDAR, MK-571, AND KO143, AS SELECTIVE AS OFTEN ASSUMED?
Ilaria Badagnani, Mario Monshouwer, and Adrian J. Fretland
Drug Metabolism and Pharmacokinetics, Roche Palo Alto, Palo Alto, CA, USA, 94304
Both uptake and efflux transporters play an important role in the transport of new chemical entities (NCEs) across the hepatocyte and in their hepatobiliary disposition. Availability of inhibitors for MDR1, multi-drug resistance associated proteins (MRPs), and breast cancer resistance protein (BCRP) have provided the opportunity to examine the contribution of these efflux transporters to the disposition of NCEs during drug discovery and development. However, there is little information on whether elacridar, MK-571, and Ko143, selectively inhibit MDR1, MRPs, and BCRP, respectively, as well as their selectivity for major uptake transporters. Therefore, the inhibitory profiles of these compounds was determined for the major liver uptake (human and rat organic anion transporting polypeptides or OATPs/Oatps) and efflux transporters (human MRP2 and human BCRP) involved in drug disposition. Inhibition of human OATPs (OATP1B1, 1B3, and 2B1) and rat Oatps (Oatp1a1 and Oatp1b2) was performed using stably transfected chinese hamster ovary cells (CHO) or human embryonic kidney cells (HEK), and the radiolabeled model substrates, estradiol-17β-glucuronide (E17BG) and estrone-3-sulfate (ES). Inhibition of human MRP2 and BCRP utilized membrane vesicles isolated from insect Sf9 cells expressing the corresponding transporter protein, and the model substrates E17BG and ES, respectively. The commonly used MDR1 inhibitor, elacridar, inhibited OATP1B1, OATP2B1, and MRP2, with a Ki of 1.7 μM, > 1 μM, and > 15 μM, respectively. Elacridar, also, potently inhibited BCRP (Ki = 12.3 ηM). The reported MRP inhibitor, MK-571, was a potent inhibitor (Ki < 2 μM) of all human OATPs, rat Oatps, as well as BCRP. Its potency was far greater for these transporters than for MRP2 (Ki = 12.2 μM). Ko143 inhibited all OATPs/Oatps tested (Ki = 3–5 μM), except for OATP1B3. Additionally, Ko143 did not inhibit MRP2, but potently inhibited BCRP (Ki = 25.9 ηM). These data suggest that elacridar, Ko143, and especially MK-571, are not selective inhibitors of the efflux transporters MDR1, BCRP, and MRPs, respectively. All three inhibitors interacted with human and rat OATPs/Oatps, which are uptake transporters for a wide range of xenobiotics in the liver. Therefore, caution should be exercised when using elacridar, MK-571, and Ko143, to assess the contribution of efflux transporters to the disposition of NCEs both in vitro and in vivo.
224. IN VITRO AND IN VIVO EVALUATION OF HEPATOTOXIC POTENTIAL OF TROGLITAZONE, ROSIGLITAZONE AND PIOGLITAZONE BY INHIBITION OF BILE SALT EFFLUX TRANSPORTERS
Jane Barber1, B. Kevin Park2, and John R. Foster1
1Safety Assessment, AstraZeneca, Cheshire, United Kingdom, SK10 4TG, 2Department of Pharmacology & Therapeutics, The University of Liverpool, Liverpool, United Kingdom, L69 3GE
Troglitazone (Trog) is known to cause cholestasis in humans and has previously been shown to be a potent inhibitor of the major bile salt efflux pump (Bsep). Bsep inhibition by Trog has been described in vitro in vesicles and sandwich culture systems, and inferred in isolated perfused rat livers. ‘Non-cholestatic’ analogues of Trog, Rosiglitazone (Rosi) and Ciglitazone, are reported to have equal potencies against Bsep inhibition in vitro, suggesting a class effect of thiazolidinediones on bile acid (BA) transporter inhibition. Despite these findings a full understanding of how Bsep inhibition in vitro translates into hepatotoxicity in vivo is not known. We aimed to investigate this link. We observed similar potencies against Bsep following 25min incubations in our primary rat hepatocyte sandwich culture system with Trog, Rosi and Pioglitazone (Pio). However following 24hr incubations the potency of Rosi and Pio decreases whilst Trog remains the same. Interestingly, following 24hr treatment Trog was cytotoxic at the highest dose tested (100uM) whereas Rosi and Pio were not. This data suggests a compensatory mechanism for BA transport occurs following Rosi and Pio treatment. We therefore tested the ability of the drugs to inhibit the multidrug resistance protein 2 (Mrp2), a protein able to compensate for impaired BA efflux via Bsep. Whereas Trog inhibited Mrp2 transport potently, Rosi and Pio were much less potent. Although Trog, Rosi and Pio are equi-potent against Bsep following acute doses, the inhibition by Trog of Mrp2, helps elucidate mechanisms of Trog induced hepatotoxicity that does not appear to occur in individuals following treatment with Rosi and Pio. A link between transporter inhibition by Trog seen in vitro to transporter interactions in vivo was investigated. A single 400mg/kg dose showed significant elevations of BAs in plasma 24hr post dose. To assess accompanying interactions with BA transporters we used IHC to stain liver sections for Bsep and Mrp2. We observed a clear induction and/or re-localisation of both Bsep and Mrp2 in all dosed rats. In addition a striking change in the pattern of Mrp2 staining occurred; canalicular membranes appear wider and more branched. Vesicle-like structures appear alongside membranes suggesting transport of Mrp2 to the membrane or internalisation into the cytoplasm. In conclusion the data suggests that hepatotoxic effects following Trog treatment may be due to effects on Bsep and Mrp2 resulting in the inability of hepatocytes to compensate for the inhibitory effects of the drug on bile salt efflux via Bsep.
225. PREDICTION AND IDENTIFICATION OF DRUG INTERACTIONS WITH THE HUMAN ATP-BINDING CASSETTE TRANSPORTER MULTIDRUG-RESISTANCE ASSOCIATED PROTEIN 2 (MRP2; ABCC2)
Jenny Pedersen1, Par Matsson1, Christel Bergstrom1, Janet Hoogstraate2, and Per Artursson1
1Department of Pharmacy, Uppsala University, Uppsala, Sweden, SE-75124, 2Discovery DMPK&BA, AstraZeneca R&D Södertälje, Södertälje, Sweden
Multidrug-resistance associated protein 2 (MRP2/ ABCC2) is an ATP-binding cassette (ABC) transporter highly expressed the human liver, and its physiological role is to take part in the hepatic detoxification process through biliary efflux. MRP2 has been shown to transport intrinsically anionic drugs such as methotrexate and pravastatin, as well as anionic conjugates of drugs and endogenous compounds such as bile salts. Inhibition of MRP2 in the hepatocyte can result in disruption of the lipid homeostasis and toxic accumulation of compounds in the liver, which is a major cause of withdrawal of drugs from the market.
In this study1, the chemical space of registered oral drugs was explored for MRP2 inhibitors using a dataset of 191 structurally diverse drugs and drug-like compounds. The inhibition of MRP2-mediated transport of estradiol-17β-D-glucuronide was studied in inverted Sf9 membrane vesicles. Twenty-seven previously unknown MRP2 inhibitors were identified, and the results indicate an overlapping but narrower inhibitor space for MRP2 compared with the two other major ABC efflux transporters P-gp (ABCB1) and BCRP (ABCG2). In addition, 13 compounds were shown to stimulate the transport of estradiol-17β-D-glucuronide. The experimental results were used to develop a computational model able to discriminate inhibitors from non-inhibitors according to their molecular structure, resulting in a predictive power of 86% for the training set and 72% for the test set. The inhibitors were in general larger and more lipophilic and presented a higher aromaticity than the non-inhibitors. The developed computational model is applicable in an early stage of the drug discovery process, and is proposed as a tool for prediction of MRP2 mediated hepatic drug interactions and toxicity.
REFERENCE
- Pedersen, J.; Matsson, P.; Bergstöm, C. A. S.; Norinder, U.; Hoogstraate, J.; Artursson, P., Prediction and Identification of Drug Interactions with the Human ATP-Binding Cassette Transporter Multidrug-Resistance Associated Protein 2 (MRP2; ABCC2). Submitted 2008.
226. IDENTIFICATION AND PREDICTION OF INHIBITORS OF THE THREE MAJOR HUMAN ATP-BINDING CASSETTE TRANSPORTERS P GP, MRP2 AND BCRP
Par Matsson1, Jenny Pedersen1, Ulf Norinder2, Christel Bergstrom1, and Per Artursson1
1Department of Pharmacy, Uppsala University, Uppsala, Sweden, SE-75124, 2AstraZeneca R&D, Sodertalje, Sweden
Specific inhibitors of ABC-transporters can be used to delineate transport processes in complex experimental systems, whereas multi-specific inhibitors are useful in primary screening for ABC transport in drug discovery settings. In this paper1, we studied the inhibition patterns of the three major human ABC transporters P-gp (ABCB1), BCRP (ABCG2) and MRP2 (ABCC2), using a dataset of 126 structurally diverse drugs. Specific and general inhibitors of the three ABC-transporters were identified (n = 24 and 22, respectively), using inhibitor concentrations in the vicinity of the Km-values for each transporter's probe substrate. Computational models were developed that describe the structural features of specific inhibitors for each transporter, inhibitors affecting several transporters, and compounds that did not inhibit any of the transporters. The multi-specific inhibitors affecting all three ABC transporters were found to be more lipophilic and aromatic than specific inhibitors and non-inhibitors. A computational model predicting ABC transporter inhibition was developed from easily interpreted molecular descriptors. The model correctly classified 82% of the ABC transporter inhibitors and 85% of the non-inhibitors in the external test set. This model can be used to predict the liability for ABC transporter inhibition at an early stage of the drug discovery process.
REFERENCE
- Matsson, P.; Pedersen, J.; Norinder, U.; Bergstrom, C. A. S.; Artursson, P., Identification and Prediction of Inhibitors of the Three Major Human ATP-Binding Cassette Transporters P gp, MRP2 and BCRP. In Department of Pharmacy: in manuscript 2008.
227. P-GLYCOPROTEIN LIMITS ORAL AVAILABILITY, BRAIN PENETRATION AND TOXICITY OF AN ANIONIC DRUG, THE ANTIBIOTIC SALINOMYCIN
Jurjen S. Lagas1, Rolf W. Sparidans2, Robert A.B. Van Waterschoot1, Els Wagenaar1, Jos H. Beijnen3, and Alfred H. Schinkel1
1Division of Experimental Therapy, The Netherlands Cancer Institute, Amsterdam, Netherlands, 2Department of Pharmaceutical Sciences, University of Utrecht, Utrecht, Netherlands, 3Department of Pharmacy and Pharmacology, Slotervaart Hospital, Amsterdam, Netherlands
Salinomycin is a polyether organic anion that is extensively used as a coccidiostatic antibiotic in poultry and commonly fed to ruminant animals to improve feed efficiency. However, salinomycin also causes severe toxicity when accidentally fed to animals in high doses. In addition, humans are highly sensitive to salinomycin and severe toxicity has been reported. Multidrug efflux transporters like P-glycoprotein, BCRP and MRP2 are highly expressed in the intestine and can restrict oral uptake and tissue penetration of xenobiotics. The purpose of this study was to investigate whether the anionic drug salinomycin is a substrate for one or more of these efflux pumps. Surprisingly, salinomycin was actively transported by human MDR1 P-gp expressed in polarized MDCK-II monolayers, but not by the known organic anion transporting human MRP2 and murine Bcrp1. Using P-gp knockout mice we found a marked impact of P-gp deficiency on salinomycin pharmacokinetics. Oral administration of 1 mg/kg salinomycin resulted in a 3.1-fold higher AUC(0-∞) in P-gp deficient mice compared to WT mice (250.8 ± 27.3 versus 80.2 ± 12.3 hr.μg/L, P < 0.01). After intravenous administration of 1 mg/kg the AUC(0-∞) was 2.0-fold increased in P-gp-deficient animals (2813 ± 318.1 versus 1374 ± 253.2 hr.μg/L, P < 0.05) and plasma clearance was 2.2-fold lower in knockout mice (0.37 ± 0.10 versus 0.82 ± 0.29 l/hr.kg, P < 0.05). Furthermore, absence of P-gp resulted in a significantly increased brain penetration. P-gp-deficient mice also displayed clearly increased susceptibility to salinomycin toxicity. Thus far, P-gp was thought to mainly affect hydrophobic, positively charged or neutral drugs in vivo. Our data show that P-gp can also be a major determinant of the pharmacokinetic behavior and toxicity of an organic anionic drug. Variation in P-gp activity might thus directly affect the effective exposure to salinomycin and possibly to other anionic drugs and toxin substrates. Individuals with reduced or absent P-gp activity could therefore be more susceptible to salinomycin toxicity.
228. MODULATION OF BILIRUBIN-GLUCURONIDE TRANSPORT VIA MRP2/MRP3 USING SANDWICH CULTURE OF PRIMARY HUMAN AND RAT HEPATOCYTES
Katalin Jemnitz, Zsuzsa Veres, Regina Tugyi, and Laszlo Vereczkey
Biochemical Pharmacology, Chemical Research Center of the Hungarian Academy of Sciences, Budapest, Hungary
Hepatobiliary transport systems are essential for normal bile formation and hepatic elimination of various endo- and xenobiotics including bile salts and bilirubin. Drug induced transport defects might explain impaired hepatic trafficking of bilirubin and other bile constituents resulting in cholestasis and jaundice. Bilirubin is taken up from blood into hepatocytes by sinosuidal membrane transporters and then excreted into bile through the bile canalicular membrane mainly as bilirubin glucuronides (BG) by ABCC2 (MRP2), a member of the ATP-binding cassette transporter family. ABCC3 (MRP3), which is located in the basolateral membrane, transports bilirubin glucuronides into blood under conditions of impaired biliary bilirubin excretion. In this study we modeled this vectorial BG transport in vitro using primary human and rat hepatocytes cultured in sandwich configuration. Our aim was to demonstrate the effect of some cholestatic drugs and transporter modulators on the in vitro canalicular and sinusoidal disposition of BG, as an indicator of drug induced cholestatic processes using human and rat primary hepatocytes cultured in sandwich configuration. The in vivo effect of probenecid administration on BG biliary elimination was compared with the in vitro data. Canalicular and sinusoidal disposition of BG were assayed in the presence of probenecid (10–2500 μM), indomethacin (1–100 μM), rifampicin (10–100 μM), and benzbromarone (10–100 μM). All drugs decreased the canalicular efflux of BG more than the sinusoidal transport, therefore the biliary efflux index of BG decreased both in human and rat hepatocyte culture. However, rifampicin and probenecid highly increased the sinusoidal efflux providing a cytoprotective compensatory route for BG elimination, which occurs in vivo as well resulting in conjugated hyperbilirubinaemia. Modulators increased the mono/diglucuronide ratio of BG both transported by canalicular or sinusoidal transporters, which is considered as an early cholestatic marker. A single dose of 50 mg/kg probenecid ip decreased the BG biliary elimination in vivo as well, whereas a single 25 mg/kg dose had no significant effect. Our results show that all of the studied compounds shifted the elimination of BG towards the sinusoidal domain of hepatocytes by decreasing the activity of MRP2/Mrp2 in sandwich cultured hepatocytes. This effect is particularly true for rifampicin and probenecid, which markedly enhanced the sinusoidal elimination BG at the same time. This effect can be seen only in in vitro models allowing the study of canalicular and sinusoidal transport processes separately. In conventional hepatocyte cultures, in which the vectorial transport of BG cannot be studied, the enhanced sinusoidal transport masks the inhibition of MRP2, resulting in false negative results in drug interaction studies.
229. REGULATION OF OATP2B1 (SLCO2B1) TRAFFICKING BY PROTEIN KINASE C
Kathleen Köck, Markus Grube, Gabriele Jedlitschky, and Heyo K. Kroemer
Department of Pharmacology, Ernst-Moritz-Arndt University, Greifswald, Germany, 17487
The organic anion transporting polypeptide 2B1 (OATP2B1, SLCO2B1) as a multispecific organic anion transporter of the solute carrier family (SLC) mediates the uptake of endogenous compounds like estrone-3-sulfate (E1S), dehydroepiandrosteronesulfate (DHEAS), pregnenolonesulfat (PS) and drugs like atorvastatin, pravastatin or glibenclamide. In contrast to other important members of this transporter-family like OATP1B1 or 1B3 OATP2B1 is expressed in a variety of tissues like liver, intestine, heart and placenta. It has recently been implicated for several transport proteins of the SLC family, that posttranslational modification by activation of the protein kinase C (PKC) might influence trafficking. We therefore investigated whether activation of protein kinase C affects function and localization of OATP2B1.
In silico analyses using the internet based phosphorylation site prediction programs NetPhos1, GPS and DisPhos revealed the existence of several potential PKC phosphorylation sites within the OATP2B1 protein. In OATP2B1-overexpressing MDCKII cells activation of the PKC by phorbol esters resulted in a significant time and concentration dependent inhibition of OATP2B1-mediated E1S and DHEAS uptake. This inhibition was associated with a decrease of the maximal transport velocity from 288 pmol/min/mg protein (DMSO) to 165 pmol/min/mg protein (100 nM PMA), while the substrate-affinity remained statistically unchanged (9.5 ± 2.6 vs. 13.2 ± 4.4 μM). Immunofluorescence analyses and live cell imaging with GFP-tagged OATP2B1 demonstrates a rapid redistribution from the plasma membrane into intracellular, vesicle-like compartments after PKC activation. By immunofluorescence and western blot analyses we could show that the internalized transporter is targeted to the lysosomal pathway and partially degraded.
In conclusion, our data show that OATP2B1 function is regulated rapidly by phorbol ester treatment through alterations of trafficking processes, which may serve as a mechanism to regulate uptake and transepithelial transport of endogenous or exogenous OATP2B1 substrates.
230. DETOXICATION ROLE OF PLACENTA; IMPORTANCE OF EFFLUX TRANSPORTERS
Lenka Cygalova, Martina Ceckova, and Frantisek Staud
Department of Pharmacology and Toxicology, Faculty of Pharmacy, Hradec Kralove, Czech Republic
Aim. Drug efflux transporters P-glycoprotein (P-gp) and Breast cancer resistance protein (BCRP) have been shown to be highly expressed in many physiological tissues. In the placenta, they contribute to the protection of developing fetus against potentially harmful substances from mother. The intention of our study was to describe the role of P-gp and Bcrp in transplacental pharmacokinetics of Rhodamine 123 (Rho123; substrate of P-gp) and BODIPY FL Prazosin (BP; substrate of both P-gp and BCRP) employing dually perfused rat placenta. Methods. Rat term placenta was dually perfused either in an open-circuit system or with fetal perfusate recirculation. To study maternal-to-fetal and fetal-to-maternal clearances, substrates were added to the maternal or fetal reservoirs, respectively and their concentrations were measured in the fetal effluent. To study fetal-to-maternal ratio at steady state, both maternal and fetal sides were infused with equal concentrations of substrate and the fetal perfusate was recirculated for 60 min. Results. For both BP and Rho123, maternal-to-fetal clearances were significantly lower than those in the opposite direction (fetal-to-maternal clearances were 4 and 11-fold higher, respectively). Addition of P-gp and/or BCRP inhibitors decreased this asymmetry to 2.4 and 4.5, respectively. In addition, we demonstrate the potential of these transporters to pump their substrates against a concentration gradient from fetus to mother. Ratios of feto/maternal concentrations 60 min after the beginning of fetal perfusate recirculation were 0.47 for BP and 0.32 for Rho123. Inhibition or saturation of the transporters significantly increased these ratios. Conclusions. Our results show that P-gp and Bcrp are able to hinder the transport of potential toxins from maternal to fetal circulation. Moreover, their ability to actively remove substances already present in fetal circulation has been revealed.
ACKNOWLEDGMENT
This work was supported by 119007 C 2007 FaF of the Grant Agency of the Charles University in Prague, Czech Republic and by the grant No. 1A/8696-4 of the Ministry of Health, Czech Republic.
231. EXPRESSION OF DRUG-UPTAKE TRANSPORTERS IN NORMAL AND DILATIVE HEARTS
Markus Grube1, Sabine Ciecholewski2, Gabriele Jedlitschky1, Alexander Staudt3, Uwe Völker2, Stephan B. Felix3, and Heyo K. Kroemer1
1Department of Pharmacology, Ernst-Moritz-Arndt University, Greifswald, Germany, 17487, 2Department of Functional Genomics, Ernst-Moritz-Arndt-University, Greifswald, Germany, 17489, 3Klinik für Innere Medizin B, Ernst-Moritz-Arndt-University, Greifswald, Germany, 17487
Background. Uptake transporters like the organic anion transporting polypeptides (OATP; SLCO family) or the organic cation/anion transporters (OCT(N)/OAT; SLC22A family) have been shown to be involved in pharmacokinetic processes of many drugs. However, these studies mainly addressed the expression and function of these transporters in organs like intestine, liver or kidney, which are of high relevance for systemic drug bioavailability. In contrast much less is known about the transporter expression in organs like the heart, where they might affect local drug bioavailability. We therefore studied the cardiac mRNA expression of the uptake transporters OCT1, OCT2, OAT2, OCTN1, OCTN2, OATP2B1, OATP3A1, OATP4A1 and OATP5A1. Methods/Results. Cardiac transporter expression was assessed using the TaqMan low density array real-time PCR platform in heart samples obtained from patients suffering from inflammatory dilative cardiomyopathy (n = 16) and control hearts (n = 7). Besides the transport proteins, 6 reference genes (18SrRNA, GAPDH, TBP, HMBS, B2M and GUSB) were measured and the transporter expression was normalized to the mean expression of these reference genes. mRNA of all mentioned transporter genes - except OCT2, which shows no cardiac expression - could be detected in significant amounts in almost all samples; however, only OCTN2 (57 ± 25%) and OATP2B1 (62 ± 42%) were significantly down-regulated during dilative cardiomyopathy. In addition, the transporter expression was correlated with the left ventricular ejection fraction (EF) of the respective patients. Here, a significant negative correlation could be found for OATP5A1 (r = - 0.48; p = 0.021), while EF and OCTN2 expression were positively correlated (r = 0.46; p = 0.026). Conclusion. Taken together, drug-uptake transport proteins of the SLC22A and SLCO family are expressed in human heart. Moreover, some of these proteins like OCTN2 and OATP2B1 were down-regulated during inflammatory dilative cardiomyopathy. These findings may be of interest for cardiac uptake of certain drugs like verapamil, spironolacton (OCTN2) and atorvastatin (OATP2B1) as well as endogenous transporter substrates like the physiologically important carnitine (OCTN2).
232. EXPRESSION OF ABC-TRANSPORTERS IN MALIGNANT BREAST TISSUE
Martina Ceckova1, Antonin Libra2, Alena Viskova2, Sylvia Aust3, Martin Klimpfinger4, Franz Sellner5, Theresia Thalhammer3, and Frantisek Staud1
1Department of Pharmacology and Toxicology, Charles University in Prague, Faculty of Pharmacy, Hradec Kralove, Czech Republic, 2Generi-Biotech, Hradec Kralove, Czech Republic, 3Centre for Physiology and Pathophysiology, Medical University of Vienna, Vienna, Austria, 4Institute of Pathology and Bacteriology, Kaiser-Franz-Josef-Spital, Vienna, Austria, 5Clinic of Surgery, Kaiser-Franz-Josef-Spital, Vienna, Austria
Drug efflux transporters of the ATP-binding cassette (ABC) superfamily are membrane proteins able to pump its substrates out of the cells. In the cancer cells, ABC transporters are able to cause multidrug resistance, by decreasing the intracellular concentration of cytotoxic drugs and their metabolites. In the present work we studied possible alterations in the expression of ABC transporters between malignant and non-malignant breast tissue. Methods. Thirty-six specimens from ductal and lobular breast carcinomas, lymph node metastasis, mastopathy and normal breast tissue were derived from 27 patients. Expressions of ABCB1, ABCC1, ABCC2, ABCC3, ABCC4, ABCC5, ABCC11 and ABCG2 mRNAs were quantified by real-time RT-PCR. Results. All the transporters were found to be expressed in malignant as well as non-malignant samples. The major finding was a significant decrease (p < 0.01) in the expression of ABCG2 in malignant compared to mastopathy tissues. However, we observed no dependence in the expression of ABCG2 and all the other analyzed ABC transporters neither on the grade nor on the stage of the tumor. Furthermore no difference in expression of the transporters was observed in the estrogen receptor (ER), progesterone receptor (PR) or Her-2 Neu positive versus negative tumor samples. Conclusion: We conclude that in contrast to the other ABC transporters, the expression of ABCG2 is down-regulated in the malignant breast tissue and that this decreased expression does not correlate with grading, staging, ER, PR nor Her2-Neu status of the tumor.
ACKNOWLEDGMENT
This work was supported grant from Ministry of Health, Czech Republic No. 1A/8696-4 and by the Medizinisch-Wissenschaftlicher Fonds des Bürgermeisters der Stadt Wien P2312.
233. MODULATION OF MRP4 AND MRP5 EXPRESSION AFFECTS SENSITIVITY OF PANCREATIC CANCER CELLS TO NUCLEOSIDE ANALOG ANTICANCER AGENTS
Praveen K. Nambaru1, Kathleen Köck1, Christian Rimmbach1, Frank Ulrich Weiss2, Dieter Rosskopf1, Markus M. Lerch2, Heyo K. Kroemer1, and Christoph A. Ritter1
1Department of Pharmacology, University of Greifswald, Greifswald, Germany, 2Department of Internal Medicine A, University of Greifswald, Greifswald, Germany
Hypothesis. Pancreatic cancer is considered to be one of the malignancies most resistant to therapy. Several factors are associated with pancreatic cancer resistance and expression of ABC transport proteins is one of the major mechanisms of drug resistance. Expression of the MRP family members MRP1, MRP3, MRP4 and MRP5 in human pancreas and pancreatic carcinoma has been reported. However, the contribution of MRPs to chemoresistance of pancreatic cancer is not fully understood. Methods. The following human pancreatic cancer cell lines were used in this study: Colo-357, T3M4, Aspc-1, Dan-G, Panc-1, Patu-89O2, Patu-8988T and Patu-8988S and IC50 values of 5-fluorouracil (5-FU), 6-mercaptopurine (6-MP), 6-thioguanine and gemcitabine were assessed for all cell lines. 5-FU and 6-MP resistant sub-clones were selected from Patu-8988T cells by cultivation in the presence of increasing concentrations of 6-MP and 5-FU over a 6-month period. MRP4 and MRP5 expression and localisation was measured using quantitative RT-PCR and immunofluorescence microscopy. Cellular sensitivity to cytotoxic drugs was measured by crystal violet assay or cytofluorometric analysis of cell death. To study transport activity, radiolabelled [14C]-6-mercaptopurine and [14C]-5-fluorouracil were used and radioactivity was measured using scintillation beta-counter. Results. Our correlation analysis between MRP expression and cellular sensitivity to cytotoxic drugs revealed a significant relation between MRP5 expression and cellular sensitivity to 5-FU (r = 0.841, p < 0.01). Long-term treatment with 6-MP and 5-FU resulted in marked drug resistance with an increase in expression of MRP4 and MRP5 suggesting a role of MRP4 and MRP5 in acquired resistance. To further study the role of MRP5 and its transport profile, we overexpressed MRP5 in Colo-357 cells. MRP5 overexpression showed enhanced efflux and reduced accumulation of 5-FU which resulted in cellular resistance to 5-FU. Knock-down of MRP4 and MRP5 greatly affected the cellular sensitivity to 6-MP and 5-FU, respectively. Furthermore, transport assays with the MRP4 and MRP5 knock-down cells confirmed that the increase in sensitivity was indeed caused by enhanced accumulation and reduced efflux. Conclusions. Our results suggest that MRP5 is expressed and functionally active and may contribute to multidrug resistance in pancreatic cancer. Therefore, expression of MRP5 should be considered while selecting the chemotherapeutic regime in pancreatic cancer and modulation of MRP5 transport might help in overcoming resistance to 5-FU.
234. THE EFFECT OF DIFFERENT DRUGS ON HEPATOBILIARY TRANSPORTER MRP2 STUDIED BY FLUORIMETRIC METHODS IN SANDWICH HEPATOCYTE CULTURE
Regina Tugyi1, Katalin Jemnitz1, Zsuzsa Veres1, Endre Kiss2, and Laszlo Vereczkey1
1Biochemical Pharmacology, Chemical Research Center of the Hungarian Academy of Sciences, Budapest, Hungary, 2Department of Immunology, Eötvös Loránd University, Institute of Biology, Budapest, Hungary
Drug induced intrahepatic cholestasis often derives from modulation of the expression and/or of the activity of hepatobiliary transporters. Cholestatic effect of drugs is studied mostly by the transport of bilirubin-glucuronides, the main hepatic endogenous substrate. However, if a fluorescent exogenous substrate is used, then the effect can easily be visualized. In our experiments we have used 5,(6)-carboxy-2′,7′-dichlorofluorescein, to study the interaction of selected drugs with Mrp2, the main canalicular transporter of xenobiotics. The modulation of substrate elimination with rifampicin, probenecid, indomethacin, MK571 and benzbromarone was assayed. In sandwich cell culture, hepatocytes are plated on dishes precoated with collagen and 24 h after plating the cells are overlaid with another layer of collagen. In the next 48 h (rat) or 4–5 days (human), canalicular network is formed between hepatocytes. The incubation with drugs lasted for 10 min. For the fluorimetric measurements, following a substrate uptake period, cells were treated with standard or Ca2+ and Mg2+-free HBSS containing the modulators or the vehicle for 20 min. In standard HBSS the amount of substrate accumulated in the cells and transported into the canalicular network can be measured together, whereas incubation in Ca2+ and Mg2+-free HBSS, which resulted in the opening of the canalicular network, the amount of substrate accumulated in the cells can be determined. To obtain the amount of substrate transported into the canalicular space, the amount of the substrate measured in the Ca2+ and Mg2+-free HBSS was subtracted from the same in the standard HBSS. Our results show that all drugs studied decreased the canalicular transport in a concentration dependent manner that could be visualized by confocal laser microscopy, but for quantitative analysis fluorimetric measurements were made in the presence and absence of Ca2+ and Mg2+. We have compared the concentration effect of the different drugs on the transport in rat and in human hepatocytes and have found that, for example in the case of indomethacin, the rat hepatocytes were ten times more sensitive than human hepatocytes (IC50 is about 1μM in the rat hepatocytes and 10 μM in the human hepatocytes).
235. EXPRESSION OF ABC TRANSPORTERS AND CYTOCHROMES P450 IN ISOLATED HUMAN BRAIN MICROVESSELS
Sandrine Dauchy1, Isabelle De Waziers2, Richard J. Weaver3, Francine Chassoux4, Catherine Daumas-Duport4, Pierre-Olivier Couraud5, Lucille Mellottee2, Philippe Beaune2, Jean-Michel Scherrmann1, and Xavier Decleves1
1Laboratory of pharmacokinetics, INSERM U705 CNRS UMR 7157, Paris, France, 2INSERM U775-S, Paris, France, 3Centre for Biopharmacy Research, Servier Research & Development Ltd., Slough, United Kingdom, SL3 6PJ, 4Hopital Ste Anne, Paris, France, 5INSERM U567/CNRS UMR 8104, Paris, France
The brain distribution of many endogenous and exogenous compounds depends on their ability to cross the blood-brain barrier (BBB). The BBB is a neurovascular unit whose cellular architecture prevents the brain penetration of numerous lipophilic drugs either by effluxing them from brain to blood by transporters or by metabolizing them by drug-metabolizing enzymes expressed in brain endothelial cells. The expression of drug efflux transporters, especially ATP-binding cassette (ABC) transporters at the human BBB has been extensively investigated but their relative expression at the BBB as well as that of cytochromes P450 (CYP), the major phase 1 drug-metabolizing enzymes superfamily, remain unknown. The aim of this study was to establish exhaustive expression profile of ABC transporters and CYP at the adult human BBB. We investigated gene expression of 9 ABC transporters and 23 CYP by quantitative real-time RT-PCR analysis of cerebral cortex samples and freshly isolated cortical microvessels from 11 patients with epilepsia or glioma. First, we validated the purity of our microvessel preparation by measuring marker gene expression of the cell types composing the BBB (endothelial cells, pericytes, astrocytes and neurons). Our results showed that ABCG2 (BCRP) and ABCB1 (MDR1/P-gp) were the main ABC transporters expressed in isolated microvessels and were expressed at a much higher level (15–25 fold) in microvessels than in total cerebral cortex. Furthermore, we found that CYP1B1 isoform represents more than 80% of all CYP isoforms detected in microvessels. A 15-fold increase in mRNA level of CYP1B1 was observed in microvessels as compared to total cortex, showing that CYP1B1 was mainly expressed at the BBB. The expression of BCRP, MDR1 and CYP1B1 proteins in microvessels was then confirmed by immunoblotting. In conclusion, this study establishes for the first time a full characterization of ABC transporters and CYP expression patterns at the adult human BBB and therefore provides useful information for understanding and predicting brain penetration of their substrates.
236. SIMULATION OF THE EFFECT OF P-GLYCOPROTEIN ON DRUG ABSORPTION IN THE HUMAN GASTROINTESTINAL TRACT
Sibylle Neuhoff1, Jiansong Yang1, Masoud Jamei1, Geoffrey T. Tucker2, and Amin Rostami-Hodjegan2
1Scientific Development, Simcyp Ltd., Sheffield, United Kingdom, S2 4SU, 2Academic Unit of Clinical Pharmacology, University of Sheffield and Simcyp Limited, Sheffield, United Kingdom, S2 4SU
Intestinal P-glycoprotein (P-gp) is considered to play a significant role in the oral bioavailability of some drugs by limiting their uptake from the gut. The aim of this study was to investigate the dose-dependency of drug efflux by P-gp and the relative roles of P-gp-mediated efflux and passive diffusion in drug absorption. A new algorithm considering nonlinear (saturable) drug efflux by intestinal P-gp was developed and integrated into the Advanced Dissolution, Absorption and Metabolism (ADAM) model (Simcyp®, Simcyp Limited, Sheffield, UK), which has been used to predict the rate and extent of intestinal drug absorption and metabolism and their associated inter-individual variability based on physiochemical and in vitro data. In addition to the existing physiological parameters in the ADAM model, including gastric emptying time, intestinal transit time, regional pH values and fluid dynamics, the new module considers regional expression of P-gp in the human gastrointestinal tract (Mouly and Paine, 2003; Troutman and Thakker, 2003). Three commonly used P-gp substrates, digoxin, quinidine and talinolol were used to assess the model. Physiochemical and in vitro parameters of the compounds were collated from the literature and entered into Simcyp Simulator V7.20 to predict the fraction absorbed (fa) in virtual populations (10 trials with 10 Caucasians in each trial). The simulations were performed with the drug in solution as well as in solid form (instant release formulation). The predicted fa values for digoxin (1 mg, p.o.), quinidine (500 mg, p.o.) and talinolol (50 mg, p.o.) were 0.54 ± 0.15 [Mean ± SD], 0.92 ± 0.07 and 0.45 ± 0.11, respectively. These values were in agreement with reported in vivo values for corresponding doses: 0.63 ± 0.11 for digoxin (Greiner et al., 1999), 0.76 ± 0.17 for quinidine (Darbar et al., 1997) and 0.55 ± 0.15 for talinolol (Trausch et al., 1995). When digoxin, quinidine and talinolol were given at low doses (e.g. 0.1 mg), P-gp was not saturated and the predicted fa values were 0.54 ± 0.15, 0.74 ± 0.10, and 0.45 ± 0.11, respectively. Thus, it was shown that an increased dose can lead to increased absorption due to the saturation of P-gp. However, P-gp saturation is unlikely to occur with compounds of poor solubility (e.g., intrinsic solubility of 0.001 mg/mL), especially those with low affinity for P-gp (high Km values). In addition, P-gp-mediated drug efflux can play a significant role for compounds with low passive permeability like talinolol, but not for compounds with high passive permeability such as quinidine.
REFERENCE
- Darbar et al., 1997 Clinical Pharmacology and Therapeutics 61: 292–300; Greiner et al., 1999 Journal of Clinical Investigation 104: 147–153;
- Mouly and Paine, 2003 Pharmaceutical Research 20: 1595–1599;
- Trausch et al., 1995 Pharmaceutics and Drug Disposition 16: 403–414;
- Troutman and Thakker, 2003 Pharmaceutical Research 20: 1210–1224
237. DRUG INTERACTIONS WITH THE HUMAN ORGANIC CATION TRANSPORTER HOCT2: IMPACT OF PHYSICOCHEMICAL PROPERTIES AND MOLECULAR STRUCTURE
Thomas F. Solbach, Jörg König, Martin F. Fromm, and Oliver Zolk
Institute of Experimental and Clinical Pharmacology and Toxicology, Erlangen, Germany
Organic cation transporters (OCTs) provide an essential pathway for the uptake of cationic compounds in the liver and the kidney, the first step in their elimination from the organism. Although many drugs have been identified which interact with human OCT2, structural elements required for an interaction with OCT2 are not well defined. To address this issue, HEK293 cells stably expressing hOCT2 were generated. [3H]MPP+ uptake in these cells was inhibited to varying extents by a diverse set of 48 structurally unrelated drugs. A subset of 25 of these molecules was used to determine IC50 values for inhibition of [3H]MPP+ uptake and to correlate these with physicochemical descriptors such as molecular weight, logP, pKa, volume, solvent accessible surface area, and topological polar surface area (TPSA). The most potent inhibitors were imipramine, fenfluramine, doxepine, amitryptiline, chlorpromazine, ipratropium, clonidine, diphenhydramine, propafenone, and sibutramine. The IC50 values for the uptake of 10 μM [3H]MPP+ were 6.0, 9.7, 13.3, 14.2, 14.4, 14.6, 16.4, 20.5, 25.1, 29.1 μM, respectively. Moreover, we found a significant correlation between observed IC50 and TPSA values (r = 0.66, p = 0.0004). Structural alignment of these compounds was used to construct a two point pharmacophore which consists of an ion pair interaction feature and a hydrophobic aromatic feature at a distance of 4.95 Å. We then tested phenylmethylamine and three derivatives which differed only in the distance between the aromatic ring (Ar) and the amine nitrogen atom (N). Most potent inhibition of OCT2-mediated MPP+-uptake was achieved with 2-phenylethylamine and 3-phenylpropylamine with Ar-N distances of 5.17 and 6.36 Å, respectively. In contrast, those compounds with a shorter (3.80 Å) or a longer Ar-N distance (7.76 Å) were significantly less potent OCT2 inhibitors. In conclusion, the following descriptors may be used to predict whether a compound is a potent inhibitor of OCT2: (1) amines that have a positive charge at physiological pH, (2) TPSA value <50, (3) a hydrophobic feature which contains an aromatic group, (4) and an Ar-N distance of about 5 Å.
238. COMPARISON OF SANDWICH-CULTURED PRIMARY HEPATOCYTES FROM RAT AND MOUSE: EXPRESSION AND FUNCTION OF MRP2 AND PGP
Viola Draheim1, Andreas Reichel1, Werner Weitschies2, and Ulla Mönning1
1Research Pharmacokinetics, Bayer Schering Pharma AG, Berlin, Germany, 2Institute of Pharmacy, University of Greifswald, Greifswald, Germany
Hepatobiliary elimination via ATP-dependent efflux transport proteins is a very important excretion pathway for drugs and their metabolites. Therefore, much effort is being spent on setting up in vitro assay systems allowing the investigation of the hepatobiliary transport behaviour of compounds. As they are easy to obtain, rodent hepatocytes are of particular value to establish biliary excretion assays on a routine basis. In the present study we have compared sandwich-cultured hepatocytes of rats and mice to assess their applicability for biliary excretion assays. After a morphological characterisation of the sandwich cultures, we investigated the protein expression of the canalicular efflux transporters Mrp2 (Abcc2) and Pgp (Abcb1) by Western blotting and performed functional studies using fluorescence microscopy. Differences between rat and mouse hepatocytes were already observed after isolation: Mouse hepatocytes were generally larger in diameter than those of rats, 24 μm versus 20 μm, respectively. After a few days in culture, differences in the morphology of the canalicular network became obvious. Whereas rat hepatocytes developed a complete and uniform network, the bile canaliculi in the sandwich culture of mouse cells were less uniform and more dilated. In contrast to rat hepatocytes, mouse hepatocytes formed their bile canaliculi more rapidly in sandwich culture. Western blot analysis demonstrated that the re-establishment of bile structures was associated with alterations in protein expression of Mrp2 and Pgp. While the expression of Mrp2 and Pgp in rat hepatocytes increased over time in culture, the content of Mrp2 and Pgp decreased in mouse hepatocytes. This expression behaviour was in principle reflected by the function of the transport proteins. Interestingly, Pgp did not regain its excretory function in the mouse hepatocytes. In summary, there are species-differences between sandwich-cultured hepatocytes from rat and mouse, concerning the morphology of the cell cultures as well as the expression and function of both efflux transporters. Thus, in vitro-in vivo correlations across rodents should be avoided and a clear species (and gender) correspondence preferred. It remains open to what extent the observed differences relate to the presence (mouse) and absence (rat) of the gall bladder in these species.
239. THE INVOLVEMENT OF NAPHTHALENE IN THE INHIBITION OF P-GLYCOPROTEIN FUNCTION BY THE EXTRACT OF TOBACCO SMOKE IN ORAL EPIDERMAL CELLS
Wen-Chi Pan1, Chien-Chih Chen2, and Yune-Fang Ueng1
1Division of Basic Chinese Medicine, National Research Institute of Chinese Medicine, Taipei, Taiwan, 2Division of Medicinal Chemistry, National Research Institute of Chinese Medicine, Taipei, Taiwan
The involvement of naphthalene in the inhibition of P-glycoprotein function by the extract of tobacco smoke in oral epidermal cells. Pan, W.-C., Chen, C.-C., and Ueng, Y.-F.* National Research Institute of Chinese Medicine Effects of unsmoked tobacco (UT) and mainstream tobacco smoke (TS) extracts on P-glycoprotein (Pgp) function were studied in an oral epidermal carcinoma cell line, OECM-1. The cyclohexane extract was prepared to obtain the benzo(a)pyrene enriched fraction. After extraction by cyclohexane, the residue was further extracted by isopropanol to obtain the isopropanol extract. The Pgp-mediated rhodamine (Rh) 123 efflux was decreased when cells were treated with the isopropanol extract of TS (ITS) for 18 and 24 hours. Rh 123 accumulation was increased by an 18-hr pre-treatment of 100 ug/ml ITS. However, cyclohexane and isopropanol extracts of UT and the cyclohexane extract of TS (CTS) had no effects. The mixture of ITS and CTS at a ratio equivalent to their relative extraction yield also decreased Rh 123 efflux. ITS had no effects on the protein level of Pgp. HPLC and GC-MS analyses showed that benzo(a)pyrene and naphthalene were present in the CTS and ITS, respectively. Naphthalene decreased Rh 123 efflux, whereas benzo(a)pyrene and nicotine had no effects. The presence of naphthalene enhanced the cytotoxicity of CTS. These results suggested that a decrease of transporter function by smoke may accelerate the accumulation of its toxic constituents and enhance their toxicities in oral cells.
240. EFFECT OF MONOGLYCERIDES ON MULTIDRUG RESISTANCE-ASSOCIATED PROTEIN 2 (MRP2) WITHIN CACO-2 CELLS
Xi (Jessica) Jia and Kishor M. Wasan
Dept. of Pharmaceutical Scis, Univ of British Columbia, Vancover, BC, Canada, V6T 1Z3
Purpose. Oral administration of drugs had been the preferred route for many drugs; however, its limited bioavailability often creates serious concerns in oral drug development. In particularly for hydrophobic oral drugs, enhancing bioavailability had been one of the major tasks pharmaceutical companies have to consider in formulating their oral dosage forms. The effective delivery of orally administered drugs is usually assessed by their absorption in the gastrointestinal tract and pre-systemic clearance through hepatic first pass metabolism. The use of lipid excipients in a number of oral drug delivery systems have shown to enhance the bioavailability of many hydrophobic drugs. One proposed mechanism for this observation is the ability of these excipients to facilitate drug uptake by altering the activities of intestinal efflux transporters. Even though lipid excipients, such as Peceol®, have been shown to downregulate the activity and expression of one major intestinal efflux transporter, P-glycoprotein, the specific lipid components of these excipients that may illicit these effects have not been identified. Thus, the objective of this project was to investigate the effects of several lipid components (i.e. monoglycerides) on the efflux activity and protein expression of another intestinal efflux transporter, multidrug resistance-associated protein 2 (MRP2), within human colon adenocarcinoma cells (Caco-2). Methods. Non-cytotoxic concentrations of Peceol® and 1-monostearin, 1-monoolein and 1-monopalmitin were determined following 24 hours of incubation with Caco-2 cells by assessing cell membrane integrity and mitochondrial respiration. Accumulation studies were then conducted to assess the activity of MRP2 using a non-specific MRP2 fluorescent substrate, rhodamine 123 (Rh123). In addition, bidirectional efflux studies were completed using a specific MRP2 substrate, estradiol 17 β-D-Glucuronide, in a Transwell® semi-permeable cell culture support system. Western blot analysis was used to determine MRP2 protein expression. Results. Caco-2 cells were viable when treated with concentrations equal to or less than 0.25% v/v Peceol®, 1000μM 1-monopalmitin, 1000μM 1-monostearin, and 500μM 1-monoolein. Cells incubated with Peceol® at 0.25% v/v, 1000μM 1-monoplamitin and 500μM 1-monoolein resulted in a significant increase in Rh123 accumulation of 70%, 19%, and 18%, respectively, compared to non-treated controls. The efflux ratio of estradiol following 1-monoplamitin, 1-monostearin, 1-monoolein, Peceol® and MK571 (a specific MRP2 inhibitor) treatment resulted in a significant decrease of 39%, 46%, 24%, 25% and 36%, respectively, compared to untreated controls. Lastly, MRP2 protein expression was significantly decreased by 30%, 30% and 10%, respectively, compared to the control following incubation with 0.25% v/v Peceol®, 1000μM 1-monopalmitin and 500μM 1-monoolein. Conclusion. Taking together these findings suggest that 1-monopalmitin, 1-monoolein and Peceol® may reduce the activity of the MRP2 efflux transporters by downregulating MRP2 protein expression.