1. Stem Cells and Drug Discovery: The Beginning of a New Era?
Lee Rubin
Dept of Stem Cell and Regenerative Biology and Harvard Stem Cell Institute, Harvard University, Cambridge, MA
To most pharmacologists, stem cells have traditionally been thought of as being highly interesting from a scientific perspective, but virtually intractable for use clinically, at least at the current time. Recent advances, however, seem likely to change the way in which stem cells are used and increase their application to conventional drug discovery and compound validation efforts. First of all, there has been a startling advance made by Yamanaka and colleagues that facilitate the creation of embryonic stem cell lines from normal and diseased humans almost at will. This method is based on “reprogramming” in which the state of cell differentiation can be altered by altering the expression of an impressively small number of transcription factors. Second, as has now been shown by several groups, these reprogrammed cells, when derived from patients with specific diseases, can be differentiated into the very cells affected by the disease. This allows creation of more valid culture systems that should represent each disease more faithfully, thereby allowing for a clearer understanding of underlying pathology. In addition, these new cellular systems permit small molecule screens that are carried out in a biologically more relevant background. Finally, it should soon be possible to direct the newly created stem cells to differentiate into cardiac myocytes and hepatocytes, thereby providing new, potentially more predictive systems in which to assess the safety profile of potential drugs.
2. Integration of Drug Transporters as Important DMPK Determinants in Drug Development
Christoph Funk, Agnès Poirier, and Thierry Lavé
Department of Non-Clinical Drug Safety, Hoffmann-La Roche Ltd, Basel, Switzerland
Drug absorption, distribution, and elimination of new chemical entities are often mediated by multiple drug metabolism and transport processes. By improving metabolic stability, transport might become the rate-limiting step. A metabolically stable compound, which was eliminated in rats mainly as parent drug into bile, urine, and the gastrointestinal tract, was found to be a good substrate of MDR1 (ABCB1). The potential contribution of this ABC transporter in the elimination of this compound was important to predict in vivo pharmacokinetic properties and the drug-drug interaction potential in man. On the contrary, for many compounds, multiple, interrelated metabolic, and transport processes are involved in drug distribution and elimination. For a number of compounds, including many statins, OATPs mediate their uptake into liver tissue as the target organ. For such a compound, OATP1B1 was found to be the main transporter involved in hepatic uptake, followed by oxidative metabolism (CYP2C8). In vitro drug interaction studies suggested interactions at the level of hepatic uptake (OATP1B1 inhibition by statins and gemfibrozil) as well as by inhibition of CYP2C8. Accordingly, a reduced elimination, and thus enhanced drug exposure, was found in a drug-drug interaction study performed with gemfibrozil. As for drug metabolism, the quantitative extrapolation of in vitro transport data to in vivo represents a crucial step in understanding the overall relevance of individual processes. For selected model compounds (napsagatran and fexofenadine) in vitro transport parameters (Km, Vmax, and Pdif) were determined from hepatocyte uptake kinetic experiments by using a two-compartment model. This allowed the generating of reliable kinetic data, which could subsequently be used for physiologically based pharmacokinetic (PBPK) modeling of the in vivo pharmacokinetics of these compounds. In conclusion, the impact of major transporters on the pharmacokinetics and safety profile of potential clinical development candidates is currently studied early on in the process of drug research and development. By this, unfavorable compound properties can be recognized and eliminated during lead optimization and the improved mechanistic understanding helps to guide clinical compound development.
3. Intestinal Membrane Transport of Drugs and Nutrients
Hannelore Daniel
Nutrition and Food Sciences, Technische Universiaet Muenchen, Freising-Weihenstephan, Germany
Drug transport processes in the intestine that determine overall oral availability are mediated by import pathways, in which drugs utilize mostly transporters for nutrients, and by export pathways, in which xenobiotics mostly utilize ATP-dependent export pumps. By the capability of drugs to compete with nutrients and other xenobiotcs for shared transport processes, a variety of nutrient/drug and non-nutrient/drug interactions can be anticipated and have partly been described. Since nutrients are usually monomers and small molecules (except for cholesterol), compound size matters and not too many drugs are, therefore, known to share nutrient transport proteins. I shall be focusing on one of the most interesting nutrient transporters that has the unique capacity for transporting oligomers and this is the intestinal peptide transporter, PEPT1. It is an electrogenic H+-coupled symporter that transports di- and tripeptides and related peptidomimetic drugs, resulting in a high oral availability of its substrates. With a variety of techniques from HTS applications on chip-based platforms to cell cultures and in vivo bioavailability studies employing animal models, the drug transport features of PEPT1 will be demonstrated. In addition, a variety of nutrient-drug interations that may be important for drug availability shall be addressed. Natural xenobiotics, such as dietary polyphenols, also compete with drug transport processes. Flavonoids are secondary plant metabolites included in our diet, but are also provided in a growing number of supplements. They are suggested to interact with intestinal transport systems, including phosphoglycoprotein (P-gp), which mediates the efflux of a variety of xenobiotics back into the gut lumen. In human intestinal Caco-2 cells, we tested the effects of 14 different flavonoids on P-gp expression in vitro. Protein expression levels were quantified by Western blotting, flow cytometry, and real-time PCR. Except apigenin, all flavonoids were at increased P-gp protein expression when cells were exposed to the compounds over 4 weeks. Flavone was one of the most effective P-gp inducers in Caco-2 cells and its effects were, therefore, also assessed for changes in P-gp in vivo in the gastrointestinal tract of C57BL/6 mice. P-gp expression was significantly increased by flavone (400 mg/kg body weight × day over 4 weeks) in the small intestine, but not in the colon, which displayed intrinsically the highest expression level. The increase in P-gp expression caused by dietary flavonoids in intestinal epithelial cells in vitro and in vivo may serve as an adaptation and defense mechanism, limiting the entry of lipophilic xenobiotics into the organism.
References
- Lohner K, Schnäbele K, Daniel H, Oesterle D, Rechkemmer G, Göttlicher M, et al. (2007). Flavonoids alter P-gp expression in intestinal epithelial cells in vitro and in vivo. Mol Nutr Food Res 51:293–300.
- Rubio-Aliaga I, Daniel H. (2002). Mammalian peptide transporters as targets for drug delivery. [review] Trends Pharmacol Sci 23:434–440.
- Rubio-Aliaga I, Daniel H. (2008). Peptide transporters and their roles in physiological processes and drug disposition. [review] Xenobiotica 38(7–8):1022–1042.
4. Hepatic Drug Transport: From the Sinusoid to the Canaliculus and Back
Bruno Stieger
Department of Medicine, University Hospital, Zürich, Switzerland
The liver is a key organ for the distribution and elimination of many endogenous substrates and xenobiotics (e.g., drugs, toxins). Investigation of drug elimination has focused traditionally on metabolic processes, which are divided into phase I and II reactions. In recent years, research on drug elimination has pinned down the importance of transport systems, both for (hepato) cellular uptake of xenobiotics and export of xenobiotic metabolites out of (liver) cells. Therefore, drug elimination is now grouped into phase 0 (cellular uptake), phases I and II (metabolism), and phases IIIa and IIIb (cellular export for excretion). Drug uptake into hepatocytes involves, among others, members of the SLCO transporter family: organic anion–transporting polypeptides or OATPs. OATPs are sodium-independent transport systems, which transport bile salts, steroid metabolites, and a wide variety of drugs. The exact transport mechanism of the OATPs has not yet been worked out in full detail, but it is becoming clear that many of them work as anion exchangers, which are stimulated by a low extracellular pH. In addition, the main hepatocellular bile-salt uptake system, SLC10A1 (or NTCP), has also been shown to transport some drugs. After metabolism, drugs can be transported back via the basolateral membrane into the sinusoidal blood plasma for renal excretion or across the canalicular membrane for biliary elimination. As, in particular, across the cancaliular membrane, this exit step occurs against steep concentration gradients, and the transporters involved in efflux are members of the ATP-binding cassette (ABC) transporter superfamily. MRP2 and ABCG2 are two prominent canalicular exporters for phase II endproducts. Since drug-uptake systems also mediate bile-salt transport, it is not surprising that the main canalicular bile-salt export system, BSEP, is subject to interaction with drugs and drug metabolites. In susceptible patients, inhibition of BSEP will lead to intracellular accumulation of cytotoxic bile salts, resulting in drug-induced liver injury. It is, therefore, of interest to identify predisposing risk factors for acquired cholestasis due to drug-BSEP interaction. First, clinical studies have identified the p.V444A variant of BSEP to contribute to the risk of acquired cholestasis. In situations where BSEP function is impaired, bile salts will be exported back into the sinusoidal blood by MRP3 and MRP4, which act as salvage systems. These two transporters are also involved in the basolateral efflux of drugs.
5. An Immortalized Human Brain Endothelial Cell Line as a Model of Human Blood-Brain Barrier
Pierre-Olivier Couraud
Université Paris Descartes, INSERM U567 / CNRS UMR 8104, Paris, France
Brain microvascular endothelium, which constitutes the blood-brain barrier (BBB), differs from that of peripheral organs by low paracellular permeability due to highly impermeable intercellular tight junctions as well as by active influx transport of nutriments and efflux of xenobiotics. Modeling the BBB is a key issue for understanding the mechanisms of maintenance of BBB integrity and for facilitating drug screening in industrial R&D programs. A number of in vitro BBB models have been proposed for the last ten years, but establishment of a human model has proven to be a difficult goal: we recently produced and characterized a human brain endothelial cell line hCMEC/D3 which retains in culture a stable endothelial phenotype highly reminiscent of the human BBB. These cells express a variety of tight junction proteins and membrane transporters: we recently investigated the expression of the ABC-transporters and cytochromes P450 expressed by these cells and their regulation by the aryl hydrocarbon receptor (AhR). In addition, we could study the migration of activated lymphocytes across the BBB and elucidate the mechanisms of migration of neural precursor cells into the brain parenchyma in inflammatory situation. In conclusion, on the basis of the extensive characterization of the hCMEC/D3 cell line provided so far by us and others, we propose this cell line as a unique in vitro model of human BBB.
6. Biomarker Opportunities and Challenges within Drug R&D
Ina Schuppe Koistinen
Safety Assessment/Molecular Toxicology, AstraZeneca R&D, Södertälje, Sweden
Our improved understanding of the molecular bases of drug action and organ toxicity suggests that monitoring specific molecular responses may provide improved prediction of human outcomes and, in doing so, provide “bridging biomarkers” that may eliminate much of the current uncertainty in extrapolating from laboratory models to human outcome during drug R&D. The opportunities for efficacy and safety biomarkers in the drug-development process are huge. In the safety area, these include enabling the prediction or early detection of toxicity in the preclinical or clinical setting, patient stratification by identifying patients least likely to show an adverse event, and finally, to assist problem solving in order to better understand the mechanisms of toxicity and decrease the likelihood of late-stage failures. Regarding efficacy, biomarkers are needed for compound selection in the preclinical phase and in clinical development for the stratification of patient populations or the quantification of drug benefit to allow decision making on candidate drugs. It is acknowledged that there are significant limitations with the current safety biomarkers, in that they are either not available, not validated, or do not provide adequate information to improve the robustness of decision making. Safety biomarkers can be used throughout the development process and, depending on where they are to be used, they will require different levels of qualification and validation. Safety biomarker development involves a number of stakeholders, and when it comes to the qualification and validation of biomarkers, this needs to be performed mainly as part of consortia under regulatory guidance. Two main consortia have their focus on the qualification and validation of safety biomarkers: the Predictative Safety Testing Consortium (PSTC) in the United States and two projects within the Innovative Medicines Initiative (IMI), which is a new EU initiative. The talk will define biomarker needs at the different phases of drug R&D and describe AstraZenecas approach to discovery and qualification of biomarkers consisting of four steps: 1) hypothesis generation: Are patients well characterized, is the technology robust, and the statistical analysis rigorous?; 2) replication of findings in an independent population: Is this a true result?; 3) evaluation of biological and clinical plausibility: Does the result agree with the way the disease and drug are believed to work?; and 4) prospective validation in clinical trials: Is there clinical utility? In summary, the use of efficacy and safety biomarkers offers considerable utility to the drug R&D process. They have the potential to significantly change the preclinical and clinical development programs needed, to enable the earlier termination of drug-development projects, and provide evidence to bridge understanding between preclinical and clinical studies. Additionally, biomarkers have the potential to offer real clinical utility to monitor subjects/patients in both clinical trials and real-life clinical practice.
7. Biomarkers of Kidney Injury
Frank Dieterle
Translational Sciences, Novartis, Basel, Switzerland
Drug-induced kidney injury (DIKI) is not an uncommon adverse event in drug development and affects various classes of drugs, for example, in the field of oncology, immune suppression, or antibiotics. In many circumstances, DIKI, and especially its acute form, could be prevented or at least minimized by screening and monitoring with appropriate tools and earlier clinical intervention. At this time, the main problem is the late identification of acute kidney injury linked to the current standards in human kidney safety assessment, that is, serum creatinine (sCrea) and blood urea nitrogen (BUN). These are functional markers and very late indicators of renal injury. During recent years, a number of new urinary and serum biomarkers have emerged to monitor the integrity of the kidney and indicate injury earlier, are more sensitive, and some even allow to localize the injury. Until recently, none of these markers had been qualified preclinically or clinically, preventing their use for regulatory decision making in drug development, such as stopping the treatment, changing doses, or selecting patients for treatment. In 2008, seven urinary biomarkers—Kim-1, Clusterin, Albumin, TFF3, Total Protein, Cystatin C, and b2-Microglobulin—were accepted by the FDA and EMEA for monitoring acute drug-induced tubular or glomerular injuries in rat GLP studies and on a case-by-case basis under specific conditions for monitoring renal safety in early clinical trials. The feedback of the health authorities for this first-ever formal qualification of safety biomarkers will be discussed, as well as the lessons learned for the qualification of safety biomarkers. Using real-life data, it will be shown how these new tools can be used to enhance safety assessment in regular preclinical studies and how to bring a drug candidate into human clinical trials by using the biomarkers to guard kidney safety. Finally, the most recent efforts around renal safety biomarkers will be presented, which involve also consortia such as the Critical Path Institute’s Predictive Safety Testing Consortium (PSTC) and the European Innovative Medicine Initiative (IMI).
8. Novel Approaches to Generate Biomarkers of Idiosyncratic Hepatotoxicity
Paul B. Watkins
Hamner-UNC Center for Drug Safety Sciences, University of North Carolina, Chapel Hill, North Carolina, USA
Improved understanding the mechanisms that underlie idiosyncratic hepatotoxicity will lead to the identification of novel biomarkers and should solidify acceptance of biomarkers that are empirically discovered. A promising line of research is the study of patients who have actually experienced severe drug-induced liver injury (DILI). Several efforts are examining genomic DNA from registries of DILI patients, and there has been one stunning success using a whole-genome association (WGA) approach. However, it does not appear that WGA approaches will be successful in identifying susceptibility factors for most types of DILI, in part due to the relatively low sample sizes available for each drug or drug class. What is needed is an unbiased way to generate hypotheses that are testable in relatively small numbers of DILI patients. One approach is to use panels of inbred mice to model genetic variation in patient populations. This approach has been used in a study of paracetamol hepatoxicity. About one third of healthy adults treated with therapeutic doses of paracetamol develop mild hepatoxicity, as measured by elevation in serum alanine aminotransferase, aspartate aminotransferase, and glutathione-S-transferase. To generate specific hypotheses for a genetic basis for this susceptibility, 36 strains of inbred mice were administered 300 mg/kg of acetaminophen. WGA analysis and targeted sequencing determined that polymorphisms in five genes correlated with extent of liver injury in the mice. Variation in an orthologous gene, CD44, was found to correlate with susceptibility to paracetamol hepatotoxicity in two separate healthy volunteer cohorts. The ongoing “Collaborative Cross” project will generate up to 1,000 strains of inbred mice, greatly magnifying phenotypic variation across the strains. Another promising line of investigation is the application of metabolomics to prospective clinical trials. In healthy adults treated with recurrent doses of paracetamol, unbiased analysis of the urine metabolome distinguished individuals who would develop the mild liver injury days before it became manifest. Analysis of the specific predictive metabolites revealed that the urinary breakdown products of the paracetamol reactive metabolite (NAPQI) were higher in those that were going to develop liver injury, However, these paracetamol metabolites alone were not predictive without considering changes in the endogenous metabolome. Combining genetic and metabolomic biomarkers may improve the identification of patients susceptible to DILI. The inbred mouse panel and metabolomics/transcripomic approaches may have their greatest value in identifying previously unsuspected pathways involved in idiosyncratic hepatotoxicity. The aggregate activity of these pathways could reflect changes in the activities of any one of the multiple genes, and this may explain why WGA observations have not extrapolated well across different racial/ethnic populations. It will, therefore, be essential to build a consensus model of relevant pathways. To this end, a collaboration was recently announced between Entelos, the Food and Drug Administration, and the Hamner/UNC Center for Drug Safety Sciences to use the Entelos Physiolab® platform to create virtual models of liver physiology relevant to DILI. This platform, which will be publicly available, will be fully annotated and should quickly become a useful tool to generate specific hypothesis concerning DILI biomarkers.
9. Abstract Not Available
10. Biomarkers for Hepatotoxicity
B. Kevin Park
MRC Centre for Drug Safety Science, University Of Liverpool, Liverpool, UK
Drug-induced liver injury (DILI) is a significant cause of patient morbidity and mortality. It is also a major cause of attrition in the development of new medicines. Studies with model hepatotoxins have demonstrated that liver injury may result from either drug accumulation or metabolic activation in hepatocytes. However, the prediction of DILI remains difficult, particularly in those cases characterized by marked interspecies or interindividual variation. There is, therefore, a need to discover, develop, and validate new biomarkers in order to inform better the medicinal chemist and the clinician. Traditional biomarkers of DILI discovered and developed after the 1970s include leakage markers of cell death, such as transaminases (hepatocyte damage), alkaline phosphatase and ƒ×-glutamyl-transpeptidase (cholestasis), as well as markers of liver function (albumin, bilirubin, and prothrombin time). Recent attention has been focused on molecular biomarkers, which are more informative with respect to chemical stress, adaptation, and mechanisms of cellular damage. The ideal biomarker is one that is mechanism based, organ (cell) selective, and one that can be used in both laboratory models and in the clinic. For drug development, biomarkers are required that reflect and inform tissue damage. A number of biomarkers have been proposed that can be used to assess hepatic drug disposition and metabolism (6β-hydroxy-cortisol, drug-protein adducts), mitochondrial function (malate dehydrogenase, cytochrome c, arginase 1), adaptive cell defense (Nrf2, ophthalmic acid, taurine), apoptosis (caspase activity, Annexin V, keratin 18), and necrosis (hypoacetylated high-mobility group box protein 1), alongside immunological and markers of the inflammatory responses (hyperacetylated high-mobility group box protein, TNFα, and interleukins 4, 6, and 10). The integrated use of such biomarkers will be discussed in the context of understanding fundamental hepatology and translational applications (Antoine et al., 2008; Cummings et al., 2008; Halegoua-De Marzio and Navarro, 2008; Ozer et al., 2008).
References
- Antoine DJ, Williams DP, Park BK. (2008). Understanding the role of reactive metabolites in drug-induced hepatotoxicity: state of the science. Exp Opin Drug Metab Toxicol 4:1415–1427.
- Cummings J, Ward TH, Greystoke A, Ranson M, Dive C. (2008). Biomarker method validation in anticancer drug development. Br J Pharmacol 153:646–656.
- Halegoua-De Marzio D and Navarro VJ. (2008). Drug-induced hepatotoxicity in humans. Curr Opin Drug Discov Dev 11:53–59.
- Ozer J, Ratner M, Shaw M, Bailey W, Schomaker S. (2008). The current state of serum biomarkers of hepatotoxicity. Toxicology 245:194–205.
11. Humanized Mouse Models for PK and Safety Profiling of Compounds
Nico Scheer,1 Jillian Ross,2 Anja Rode,1 and C. Roland Wolf3
1TaconicArtemis, Cologne, Germany, 2CXR Biosciences Limited, Dundee, UK, 3Cancer Research UK Molecular Pharmacology Unit, Dundee, UK
The pathways that have evolved to protect mammals from toxic environmental chemicals have diverged remarkably between species, both in their multiplicity and substrate specificity. In addition, these enzymes are controlled by a number of transcription factors that form an adaptive response to environmental challenge, and the activation of these receptors is also subject to marked species variability. As a consequence, there are profound differences between animals and man in response to chemical agents, including therapeutic drugs. In order to create models that more closely reflect the human situation and that are more predictive of potential liabilities of drugs in development, we have established a large panel of mouse models humanized for particular pathways of drug metabolism and disposition. These pathways include the nuclear receptors, CAR, PXR, PPARalpha, and Ahr, the cytochrome P450-dependent mono-oxygenases, and the drug transporters, Mdr1 and Mdr2. In this presentation, the power and utility of these models as it applies to understanding pathways of chemical toxicity and metabolism and their generic application to early- as well as late-stage drug discovery will be described.
12. Use of Gene Knockout and Reporter Mice as Humanized Models of Drug Metabolism
Colin J. Henderson, Kenneth J. Ritchie, and C. Roland
Wolf Biomedical Research Institute, Cancer Research UK Molecular Pharmacology Unit, Dundee, UK
Genetically altered animals have proved invaluable in many research areas, including drug metabolism and toxicology. The ability to express foreign genes and delete or mutate genes in vivo—in a specific, spatiotemporal fashion, or globally in all cells—has vastly added to our knowledge of drug-metabolizing enzymes (DMEs)—how they are expressed and regulated, how they function, and how they interact with each other. In drug metabolism, as in other areas, the major limitation in using genetically altered animals is that there are often significant interspecies differences in DME function and regulation, resulting in the differential metabolism of compounds between species, thus making extrapolation of data to humans difficult and, in some cases, impossible. Historically, the mouse has been the favored model for transgenic studies and, in recent years, has been the focus for the generation of “humanized” animals for use in drug-metabolism studies. Glutathione transferase P1 (GSTP1) expression has been shown to be elevated in animal and human tumors and, in cell lines, made resistant to a variety of drugs, including anticancer agents. The role(s) of GSTP1 in tumorigenesis and drug resistance are unclear, and we have previously generated a GstP null mouse to investigate this further, showing significantly increased chemically induced skin and lung carcinogenesis in mice lacking GstP and a possible role for the enzyme in apoptosis through the regulation of Jun N-terminal kinase (JNK). Recently, we have crossed the GstP null mouse with the APCmin line to generate a model in which adenomas occur in significant numbers in the rectal region of the colon, reflecting more accurately the development of the human disease. The mouse not only has two GstP genes (GstP1 and GstP2)—most other species only have a single gene—they are also expressed in a different spatiotemporal manner to that of the human gene. Accordingly, we have made a transgenic mouse reporter line in which the human GSTP1 promoter and the entire GSTP1 gene have been fused to lacZ and inserted into the transcriptionally active ROSA26 locus to optimize expression of the fusion protein in vivo. Initial work with this reporter mouse suggests not only that authentic expression of human GSTP1 is recapitulated, but we have also shown induction of expression in the liver by the antioxidant, ethoxyquin, providing the first evidence that the human GSTP1 gene contains a functional antioxidant response element. Further, by crossing the [hGSTP1::lacZ]ROSA26 line with mice lacking murine GstP1/P2, we will effectively generate a mouse humanized for GSTP, allowing us to determine the function(s) of human GSTP without the potentially confounding expression of the corresponding murine genes. Thalidomide has recently been employed in an anticancer role, and the metabolism of this drug differs between species—in rodents hydroxylation by the P450 systems predominates, while in humans the main route is via hydrolysis. We will present data from the hepatic reductase null (HRN) mice, in which ablation of P450 function in the liver results in the essential humanization of thalidomide metabolism.
13. CYP3A4 Humanized Mice: Insights and Applications
Alfred H. Schinkel
Division of Molecular Biology, The Netherlands Cancer Institute, Amsterdam, The Netherlands
Cytochrome P450 3A (CYP3A) enzymes metabolize >50% of prescribed drugs and represent one of the most important detoxifying systems. As CYP3A activity shows high inter- and intrapatient variability, it can have a profound influence on variable drug efficacy and toxicity. To investigate the physiological and pharmacological roles of CYP3A, we generated Cyp3a knockout mice, and demonstrated a pronounced effect of Cyp3a deficiency on the oral bioavailability, intravenous (i.v.) clearance, and toxicity of the anticancer drug and CYP3A substrate, docetaxel. Using transgenic mice that specifically overexpressed human CYP3A4 in either the liver or intestine of Cyp3a knockout mice, we could further show a dominant role of intestinal CYP3A4 in restricting oral bioavailability of docetaxel, whereas hepatic CYP3A4 dominates the clearance of i.v. docetaxel. We expected that midazolam metabolism would be severely reduced in the absence of Cyp3a. Surprisingly, Cyp3a knockout mice still displayed marked hepatic midazolam metabolism in vitro and in vivo. We found that Cyp2c enzymes were upregulated in the liver of Cyp3a knockout mice and responsible for this metabolism. Compensatory changes in detoxifying systems may, therefore, occur in Cyp3a knockout mice. We then investigated the mechanism of upregulation of Cyp2cs and other detoxifying systems in Cyp3a knockout mice. Induction studies demonstrated an important role for the nuclear receptors, PXR and CAR, in Cyp2c upregulation. Diet-switch experiments indicated that food-derived xenobiotics are primarily responsible for the induction of Cyp2cs and other primary detoxifying systems in Cyp3a knockout mice. Apparently, CYP3A normally metabolizes food-derived activators of PXR and/or CAR. Analysis of tissue-specific CYP3A4 transgenic Cyp3a knockout mice revealed that not only hepatic, but also intestinal, expression of CYP3A4 could reduce the hepatic expression of detoxifying systems to wild-type levels. This implies that intestinal CYP3A4 can limit the hepatic exposure to food-derived activators of nuclear receptors, thereby regulating the expression of a range of detoxifying systems in the liver. These findings underscore the importance of intestinal CYP3A activity and could have implications for the prediction of drug exposure.
14. Potential Utility of Humanized CYP450 Mouse Models for Generating Human Drug Metabolites
Mark W. Powley1 and Xiao Yu2
Departments of 1Safety Assessment, and 2Drug Metabolism and Pharmacokinetics, Merck Research Laboratories, West Point, Pennsylvania, USA
Well-known species differences in drug metabolism present challenges to nonclinical safety evaluations of candidate drugs. These differences can result in circulating metabolites being observed uniquely or disproportionately in humans as well as nonclinical toxicities with unknown human relevance. The current paradigm for dealing with unique or disproportionate human metabolites involves the direct administration of synthesized metabolite to nonclinical species. This approach is resource intensive and yields questionable data. A more desirable approach is to identify alternative models capable of generating adequate plasma concentrations of metabolite following the administration of parent compound. One possible solution is to use genetically engineered mouse models (GEMMs) that express human CYP450 enzymes. These models may have the capacity to generate major human metabolites as well as eliminate or reduce the formation of mouse-specific metabolites. The goal is to apply data obtained in these models for regulatory decision making and, ultimately, improve the scientific relevance of nonclinical safety assessments involving human drug metabolites. Our current project focuses on using in vitro techniques to identify promising human CYP3A4-expressing GEMMs. Experiments involve incubating liver microsomes with substrates known to have unique or disproportionate CYP3A4 metabolites. Metabolite profiles are generated by LC/MS utilizing a high-resolution mass spectrometer (Q-TOF) with Metabolynx™ data-processing software. When combined with protein expression data, a comparison of metabolite profiles can be used to identify models that produce in vitro data more closely associated with humans vs. wild-type mice. GEMMs with a high probability of success will undergo a robust in vivo evaluation.
15. Knockout and Humanized Mouse Models for Sulfotransferases: Usage in Biotransformation and DNA Adducts Studies with Various Carcinogens
Hansruedi Glatt, Gisela Dobbernack, Korinna Wend, Nicole Schade, Carolin Müller, Bernhard Monien, Simone Florian, and Walter Meinl
German Institute of Human Nutrition (DIfE) Potsdam-Rehbruecke, Nuthetal, Germany
Previously, we expressed various human and rodent sulfotransferases (SULTs) in mammalian and bacterial target cells of standard in vitro genotoxicity tests. We detected numerous compounds that are activated by SULTs to mutagen. Thereby, we noticed large differences in the substrate specificity between orthologous SULTs from different species. Further, tissue distribution of various SULTs strongly differs between species. For example, the most abundant human SULT (SULT1A1) is highly expressed in numerous tissues, where the corresponding rat and mouse enzymes are concentrated to the liver. This led us to construct humanized mouse models by the chromosomal integration of human SULT genes (SULT1A1, allelic variants *1 and *2; SULT1A2; and SULT1B1, including long flanking regions) and disruption of endogenous murine genes (Sult1a1, Sult1d1). Human SULT proteins were expressed in the transgenic mice with a tissue distribution similar to that observed in humans. Immunohistochemistry was used to study the cellular and subcellular distribution of human SULTs in transgenic animals by using wild-type animals as a negative control. Transgenic animals were also used to study the DNA adduct formation of SULT-dependent mutagens. The heterocyclic amine, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), formed higher adduct levels in all nine investigated tissues of mice expressing the human SULT1A1-SULT1A2 cluster, compared to wild-type mice. Since the increase varied between tissues (from 1.1- to 17-fold), the tissue distribution of the adducts was substantially altered in the transgenic mice. Likewise, 1-hydroxymethylpyrene formed up to 30-fold higher levels of DNA adducts in many tissues of mice expressing human SULT1A1-SULT1A2, as compared to wild-type mice. In several tissues, the formation of adducts was only detected in transgenic, but not in wild-type, mice. In conclusion, cDNA-expressed SULT1A1 and/or SULT1A2 were shown to activate more than 100 promutagens in recombinant cells. A substantial role of these human enzymes was also corroborated in transgenic animal models with selected promutagens/procarcinogens. Currently, we are studying whether allelic variants of SULTs differ in the activation of promutagens in transgenic animals.
16. Adverse Drug Reactions: Who Cares?
Tom MacDonald
Clinical Pharmacology and Therapeutics, University of Dundee, Dundee, UK
Public alarm at publicity concerning adverse drug reactions has driven the drug-safety agenda to the point where safety is now the dominant issue for new therapeutic agents. In striving to find new patentable treatments, the pharmaceutical industry has increasingly made safety and tolerability the primary advantage of several new agents. Prominent examples include the COX2 inhibitors, SSRIs, antipsychotics, and selective aldosterone receptor antagonists. Channeling of “safe drugs to risky patients” has made the interpretation of observational safety studies more difficult and led to debate about the relative and absolute safety of new versus old medicines. The regulatory agencies have also developed an asymmetrical view of the evidence required to show benefit versus that required to demonstrate safety, and products have been suspended based on evidence from case reports of toxicity that represent poor quality data. The demonstration of safety is best done by carrying out large-scale, randomized studies. These are expensive and time-consuming. Most healthcare systems could facilitate the evaluation of the benefits and risks of new medicines prescribed in the setting of normal care. A culture change is required to work with innovators to promote the use of healthcare systems to provide affordable methods to define the risks and benefits of new treatments to patients in the real-world setting of heathcare provision. Patients, doctors, healthcare providers, and the pharmaceutical industry all care profoundly about the safety and efficacy of medicines. These stakeholders should work together to provide these data.
17. Epigenetic Regulation of Drug Metabolism Genes
Alvaro Puga1 and Michael Schnekenburger2
1Department of Environmental Health, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA and 2Laboratoire de Biologie Moleculaire et Cellulaire du Cancer, Hôpital Kirchberg, Luxembourg, Luxembourg
Metabolic bioactivation of the environmental procarcinogen, benzo[a]pyrene, is catalyzed, among others, by the substrate-inducible cytochrome P450 mono-oxygenase, CYP1A1. CYP1A1 gene induction requires transactivation by the transcriptional complex formed by the liganded Ah receptor (AHR) and its partner, ARNT. The 5’- flanking region of the CYP1A1 gene contains a transcriptional control region located in the proximal promoter region upstream of the transcriptional start site. AHR/ARNT binding sites are clustered in an enhancer domain located several hundred base pairs upstream of the proximal promoter to which the complex must bind to initiate the promoter chromatin remodeling needed for gene induction (Neuhold et al., 1989; Yanagida et al., 1990). Remodeling destabilizes a nucleosome poised over the promoter, allowing assembly of the general transcription machinery (Morgan and Whitlock, 1992). These observations provide clear evidence that chromatin structure plays an essential role in CYP1A1 transcription. In the ground uninduced state, the histone deacetylase, HDAC1, is bound to the Cyp1a1 promoter and is released in concert with the recruitment of p300 upon B[a]P stimulation (Wei et al., 2004). To follow-up to those studies, we have used ChIP assays and real-time PCR to map the architecture of histone modifications associated with the transition from ground to activated states in Cyp1a1 transcription. We found that Cyp1a1 induction is associated with the modification of specific chromatin marks, including hyperacetylation of histones H3K14 and H4K16, trimethylation of histone H3K4, and phosphorylation of H3S10 (Schnekenburger et al., 2007). HDAC1 and DNMT1 form complexes on the Cyp1a1 promoter of uninduced cells, but HDAC1 inhibition alone is not sufficient to induce Cyp1a1 expression, although it allows for the hyperacetylation of H3K14 and H4K16 to levels similar to those found in B[a]P-induced cells. These results show that by blocking the modification of histone marks, HDAC1 plays a central role in Cyp1a1 expression, and that its removal is a necessary, but not sufficient, condition for Cyp1a1 induction, underscoring the requirement for a concerted series of chromatin-remodeling events to complete the initial steps of gene transactivation by the Ah receptor. (Supported by NIH Grants R01 ES06273 and R01 ES010807)
References
- Morgan JE and Whitlock, JP, Jr. (1992). Transcription-dependent and transcription-independent nucleosome disruption induced by dioxin. Proc Natl Acad Sci U S A 89:11622–11626.
- Neuhold L, Shirayoshi Y, Ozato K, Jones J, Nebert D. (1989). Regulation of mouse CYP1A1 gene expression by dioxin: requirement of two cis-acting elements during induction. Mol Cell Biol 9:2378–2386.
- Schnekenburger M, Peng L, Puga A. (2007). HDAC1 bound to the Cyp1a1 promoter blocks histone acetylation required for transactivation by the Ah receptor. Biochim Biophys Acta 1769:569–578.
- Wei Y, Tepperman K, Huang M, Sartor MA, Puga A. (2004). Chromium inhibits transcription from polycyclic aromatic hydrocarbon-inducible promoters by blocking the release of histone deacetylase and preventing the binding of p300 to chromatin. J Biol Chem 279:4110–4119.
- Yanagida A, Sogawa K, Yasumoto K, Fujii-Kuriyama Y. (1990). A novel cis-acting DNA element required for a high level of inducible expression of the rat P-450c gene. Mol Cell Biol 10:1470–1475.
18. Abstract Not Available
19. Abstract Not Available
20. MicroRNAome Dysregulation during Chemical Carcinogenesis
Igor P. Pogribny and Frederick A.
Beland Biochemical Toxicology, National Center for Toxicological Research, Jefferson, Arkansas, USA
Environmental exposure to natural and man-made chemicals is one of the major causes of human cancer. It is widely believed that genotoxic alterations induced by some of these carcinogens are critical for tumorigenesis; however, it is also clear that these genotoxic alterations by themselves are not sufficient for tumor formation, which results from much broader alterations in cellular homeostasis, mainly from the inability of cells to maintain and control accurately the expression of genetic information. Currently, in addition to the genetic and epigenetic mechanisms in the transmission of genetic information, extensive studies have indicated the existence and importance of another mechanism of regulation of gene function mediated by microRNAs (miRNAs). miRNAs function as critical effectors of several canonical oncogenic and tumor-suppressor pathways, including controlling the balance between cell proliferation and apoptosis, which is frequently disrupted during carcinogenesis. We have conducted experiments to examine the role and contribution of miRNAs alterations in the regulation of apoptosis/cell-proliferation pathways during rodent liver carcinogenesis induced by two genotoxic carcinogens, tamoxifen and 2-acetylaminofluorene. Long-term exposure of rats to tamoxifen and 2-acetylaminofluorene resulted in the substantial alterations of the miRNA expression profiles. In the livers of tamoxifen-exposed rats, miR-34, miR-17-5p, and miR-21 were significantly upregulated, while miR-203, miR-195, miR-192, and miR-194 were downregulated. Likewise, increased expression of miR-34, miR-17-5p, and miR-21 was observed in the livers of rats exposed to 2-acetylaminofluorene. Interestingly, each of these miRNAs is involved in the regulation of apoptosis or cell proliferation, mainly through the p53-network, either by targeting genes involved in the p53-pathway, for example, miR-203, the downregulation of which is associated with the upregulation of deltaNP63 protein, or by being p53-responsive miRNAs, for example, miR-34, miR-195, miR-192, and miR-194. The disruption of apoptosis and cell proliferation was confirmed by the expression profiling of genes associated with the p53-signaling pathway, which showed a predominant upregulation of antiapoptotic growth-related genes and downregulation of proapoptotic genes. These data indicate a significant role of miRNAs alterations in disrupting the balance between cell proliferation and apoptosis during hepatocarcinogenesis.
21. Abstract Not Available
22. Abstract Not Available
23. Abstract Not Available
24. Translational PK/PD Modeling of Therapeutic Antibodies
Lene Alifrangis
Biomodelling, Novo Nordisk, Copenhagen, Denmark
The translation of safety and efficacy from preclinic to clinic of monoclonal antibodies (mAbs) is often challenging due to the highly species-specific nature of the targets and the specificity of the antibodies. This is of particular importance when planning the starting dose and dose escalations in first human dose (FHD) trials. Here, several case studies from the Novo Nordisk development program illustrate the utility of simulations of predicted human PK/PD for this purpose. Contrary to conventional allometric scaling, the prediction of human PK was not based on animal PK data. Rather, it was assumed that the general PK properties of fully human antibodies most likely will resemble that of endogenous immunoglobulins (IgGs). Typical IgG PK parameters to support a two-compartment model were collected from the literature. In addition to the general PK, the mAbs may undergo metabolism and binding specifically related to the target: target-mediated drug disposition (TMDD). The potential for TMDD was evaluated from in vitro data for target expression and affinity in humans and relevant animal species, combined with animal in vivo PK/PD. Not all membrane-bound targets are associated with significant TMDD in vivo, so a realistic estimation of TMDD is important; overprediction would lead to a too high starting dose and a potential safety issue, whereas underprediction would lead to too many dose levels in the FHD trial. Due to the lack of good preclinical in vivo efficacy models and data, the degree of target occupancy was used as a surrogate marker of efficacy. In 2 cases, in vivo PK/PD models for occupancy in transgenic animals were used to derive the PD part for the human PD simulation model. However, access to high-quality in vitro data on human cells or tissue was essential to validate the translation of the animal in vivo and/or in vitro data to humans. In conclusion, for mAbs binding to highly species-specific targets, predicted human PK/PD simulations were found to be powerful tools for decision making and to ensure compliance with regulatory requirements when planning the FHD studies. It was achieved by combining carefully planned PK/PD experiments and selected literature information in the model development.
25. Improved In Vitro Methods to Predict the Toxicity of Therapeutic Monoclonal Antibodies
Stephen Poole, Lucy Findlay, David Eastwood, Robin Thorpe and Richard Stebbings
Biotherapeutics Group, National Institute for Biological Standards and Control, Herts, UK
In 2006 a near-fatal “cytokine storm” occurred in 6 healthy volunteers during the phase I clinical trial of TGN1412, a therapeutic superagonistic CD28-specific monoclonal antibody, signaling a failure of preclinical safety testing. Subsequently, it was established that TGN1412 could stimulate a “cytokine storm” in vitro from human white blood cells, but only if presented to the cells by immobilization onto plastic or if the white blood cells were cultured over a monolayer of human endothelial cells. Data from the novel in vitro procedures suggest that the dose of TGN1412 given to the volunteers was close to the maximum immunostimulatory dose. In contrast to human white blood cells, TGN1412 was found not to be a superagonist in vivo or in vitro for white blood cells from the nonhuman primates used in the preclinical testing. The novel procedures are now being applied to emerging immunotherapeutics and to other therapeutics that have the potential to act upon the immune system.
26. Interspecies Differences in Drug Metabolism: Implications for Drug Development
Griff Humphreys
Pharmaceutical Candidate Optimization, Bristol-Myers Squibb, Princeton, New Jersey, USA
The safety and efficacy of a drug after administration to humans is determined by the mixture of the drug and its metabolites. As animal models are extensively used to predict the above behaviors of a new drug when given to humans, it is an obvious logical extension that drug development would be a more certain enterprise if the mixture of drug and metabolites was the same in humans as that found in all species used for toxicity and efficacy testing. Unfortunately, this is not the normal case, and while specific metabolism to form truly unique metabolites in a given species is rare, species-selective metabolism is quite normal and can lead to pronounced interspecies differences in metabolite exposure. Early knowledge of metabolite exposure differences can be key to effective decision making. On the efficacy side, the presence of an abundant active metabolite in an efficacy model, but not in humans, can lead to overprediction of pharmacodynamic effects and underprediction of effective dose when administered to humans. Conversely, the presence of an active metabolite in humans that is underrepresented the efficacy species leads to the opposite situation. Both scenarios can lead to significant delays or, in the extreme, could lead to discontinuation of a development program. The same scenarios as described with active metabolites can also occur with toxic metabolites. An animal-selective metabolite can lead to animal-specific toxicology, and a human-selective metabolite can lead to uncertainty regarding the extrapolation of animal toxicology findings, as outlined in the U.S. FDA guidance on Safety Testing of Drug Metabolites. This presentation will describe the background considerations of species-selective metabolism along with examples of how drug-development programs have been impacted when the property is observed.
27. Abstract Not Available
28. Species Differences in Drug Glucuronidation
Michael H. Court
Department of Pharmacology and Experimental Therapeutics, Tufts University School of Medicine, Boston, Massachusetts, USA
Species differences in drug metabolism are a major challenge for the pharmaceutical development industry and also for the practice of veterinary medicine. Glucuronidation, catalyzed by the UDP-glucuronosyltransferase (UGT) enzymes, is a major drug-metabolism pathway, which displays significant interspecies variability. The two most well-known and extreme examples of species differences involve the quaternary N-linked glucuronidation of tertiary amines and O-linked glucuronidation of simple phenols. Humans and higher primate species (chimpanzee) glucuronidate tertiary amine drugs, such as lamotrigine and trifluoperazine, extensively, while most commonly used laboratory animal species, including the rat, mouse, dog, and monkey glucuronidate these drugs poorly or not at all. Exceptions include the rabbit and guinea pig, which glucuronidate these compounds at a comparable rate to humans. These differences are likely a consequence of species differences in the activity of UGT1A4, the main enzyme that catalyzes tertiary amine glucuronidation in humans. Genome sequencing has demonstrated that UGT1A4 is a nonfunctional pseudogene in both the rat and mouse explaining low liver activity. UGT1A4 genes in other species studied, so far, appear to be intact, and so, low activity may be a consequence of poor UGT1A4 gene expression or protein-sequence differences affecting enzyme catalysis. Glucuronidation of simple phenols also shows substantial interspecies variability, with the most extreme example being the domestic cat. Cats are known to glucuronidate phenolic drugs, such as acetaminophen and acetylsalicylic acid, extremely slowly, resulting in accumulation and associated toxic effects. Over 10 years ago, we showed that the gene encoding UGT1A6, the major phenol, UGT, was a nonfunctional pseudogene in domestic cats. Since then, we have traced the evolution of UGT1A6 pseudogenization within the mammalian order Carnivora. We determined that fixation of the UGT1A6 pseudogene occurred approximately 11 million years ago, such that all extant Felidae (cat-like species) have dysfunctional UGT1A6. Of 44 additional taxa sampled, representing all modern Carnivora families, both the brown hyena (Parahyaena brunnea) and Northern elephant seal (Mirounga angustirostris) showed novel inactivating UGT1A6 mutations. The results are consistent with multiple independent UGT1A6 pseudogenization events within unrelated Carnivora species and implicate the carnivore diet, which is deficient in plant-derived phenols, as one factor influencing the evolution of UGT1A6.
29. Abstract Not Available
30. Evolution of Pharmacologic Specificity and Species Differences in the Pregnane X Receptor
Sean Ekins,1 Erica J. Reschly,2 Lee R. Hagey,3 and Matthew D. Krasowski2
1Collaborations In Chemistry, Jenkintown, Penssylvania, USA,2Department of Pathology, University of Pittsburgh, Pittsburgh, Pennsylvania, USA, 3Department of Medicine, University of California at San Diego, San Diego, California, USA
The pregnane X receptor (PXR) shows the highest degree of cross-species sequence diversity of any of the vertebrate nuclear hormone receptors. We have determined the pharmacophores for activation of human, mouse, rat, rabbit, chicken, and zebrafish PXRs, using a common set of 16 ligands. In addition, we have also compared in detail the selectivity of human and zebrafish PXRs for steroidal compounds and xenobiotics. The ligand activation properties of the Western clawed frog (Xenopus tropicalis) PXR and the putative ortholog of PXR and vitamin D receptor (VDR) cloned in this study from the chordate invertebrate sea squirt (Ciona intestinalis) were also investigated. Using a common set of small-molecule ligands, human, mouse, and rat PXRs share structurally similar pharmacophores consisting of hydrophobic features and widely spaced excluded volumes indicative of large binding pockets. Zebrafish PXR was found to have the most sterically constrained pharmacophore of the PXRs analyzed, suggesting a smaller ligand-binding pocket than the other PXRs. Chicken PXR possesses a symmetrical pharmacophore with four hydrophobes, a hydrogen-bond acceptor, as well as excluded volumes. Comparison of human and zebrafish PXRs for a wide range of possible activators revealed that zebrafish PXR is activated by a subset of human PXR agonists. The Ciona VDR/PXR showed low sequence identity to vertebrate VDRs and PXRs in the ligand-binding domain and was preferentially activated by planar xenobiotics, including 6-formylindolo-[3,2-b]carbazole. Last, the Western clawed frog (Xenopus tropicalis) PXR was insensitive to vitamins and steroidal compounds and was activated only by benzoates. In contrast to other nuclear hormone receptors, PXRs show significant differences in ligand specificity across species. By pharmacophore analysis, certain PXRs share similar molecular features, such as human, mouse, and rat PXRs, suggesting an overlap of function and, perhaps, common evolutionary forces. The Western clawed frog PXR, like that described for African clawed frog PXRs, has diverged considerably in ligand selectivity from fish, bird, and mammalian PXRs. The implications of this will be discussed.
31. Abstract Not Available
32. Pharmacoepigenetic Control of Drug Metabolism
Magnus Ingelman-Sundberg
Section of Pharmacogenetics, Department of Physiology and Pharmacology, Karolinksa Institutet, Stockholm, Sweden
The interindivudual differences in drug metabolism are extensive. At present, we do understand a major part of the true genetic reasons to such variability as copy number variations, in/dels, and SNPs. Still, the bases for a large extent of interindividual differences in enzyme expression or activity, as revealed to be inherited from in vivo studies, is extensive and remains to be elucidated. Such differences can be explained by epigenetic factors such as DNA methylation, post-translational modification of histones, and expression of ncRNAs such as microRNAs and RNAas. Among the P450s, such regulation is to be expected to be of importance for the variation in CYP1A2 and CYP3A4 expression, where no functional genetic polymorphism has been found. Today indeed, epigenetic regulation of the expression of CYP1A1, CYP1A2, CYP1B1, CYP2E1, CYP2W1, CYP3A4, CYP3A5, and CYP3A7 has been described and, in addition, putative regulatory CpG islands in the CYP2A6, CYP2C19, CYP2D6, CYP2J2, CYP2R1, and CYP2S1 genes have been identified. A true microRNA regulation of CYP1B1 expression has been described, which relates to its tumor expression and, in addition, the expression in PXR, with some influence on the CYP3A4 expression, has been clearly identified. In the lecture, an update on the knowledge about epigenetic regulation of phase I and II will be given and future direction in this novel research field outlined.
33. Role of OATP Transporters in the Disposition of Drugs
Mikko Niemi
Department of Clinical Pharmacology, Helsinki University and Helsinki University Central Hospital, Helsinki, Finland
Organic anion-transporting polypeptides (OATPs) form a superfamily of membrane influx transporters expressed in various tissues important for drug disposition (Niemi, 2007). Of the 11 human OATP transporters, OATP1B1, OATP1B3, and OATP2B1 are expressed on the sinusoidal membrane of hepatocytes and can facilitate the uptake of their substrate drugs into the liver. OATP1A2 is expressed on the luminal membrane of small intestinal enterocytes, in the cholangiocytes, and at the blood-brain barrier, potentially mediating drug transport at these sites. Several clinically used drugs have been identified as substrates of OATP transporters (e.g., many statins are substrates of OATP1B1). Certain drugs (e.g., cyclosporine) or dietary constituents may inhibit OATP transporters, causing pharmacokinetic drug interactions. Moreover, genetic variability in genes encoding OATP transporters can result in marked interindividual differences in pharmacokinetics. For example, a single-nucleotide polymorphism (c.521T>C, p.Val174Ala) in the SLCO1B1 gene encoding OATP1B1 decreases the ability of OATP1B1 to transport active simvastatin acid from portal circulation into the liver, resulting in markedly increased plasma concentrations of simvastatin acid and an enhanced risk of simvastatin-induced myopathy (Pasanen et al., 2006; SEARCH, 2008). The SLCO1B1 c.521C allele also raises the plasma concentrations of many other, but not all (fluvastatin), statins and that of the antidiabetic drug, repaglinide, the antihistamine, fexofenadine, and the endothelin A receptor antagonist, atrasentan (Pasanen et al., 2007; Niemi et al., 2006; Niemi et al., 2005a, 2005b; Katz et al., 2006). On the other hand, the SLCO1B1*1B allele (c.388G-c.521T) is associated with increased hepatic uptake and reduced plasma concentrations of certain OATP1B1 substrates, such as pravastatin and repaglinide (Maeda et al., 2006; Kalliokoski et al., 2008). More studies are needed to establish the roles of OATP1B3, OATP2B1 and OATP1A2 in drug pharmacokinetics in vivo in humans.
References
- Kalliokoski A, et al. (2008). Effects of the SLC01B1*1B haplotype on the pharmacokinetics and pharmacodynamics of repaglinide and nateglinide. Pharmacogenet Genom 18:937–942.
- Katz DA, et al. (2006). Organic anion-transporting polypeptide 1B1 activity classified by SLC01B1 genotype influences atrasentan pharmacokinetics. Clin Pharmacol Ther 79:186–196.
- Maeda K, et al. (2006). Effects of organic anion-transporting polypeptide 1B1 haplotype on pharmacokinetics of pravastatin, valsartan, and temocapril. Clin Pharmacol Ther 79:427–439.
- Niemi M. (2007). Role of OATP transporters in the disposition of drugs. Pharmacogenomics 8:787–802.
- Niemi M, et al. (2005a). Polymorphic organic anion-transporting polypeptide 1B1 is a major determinant of repaglinide. Clin Pharmacol Ther 77:468–478.
- Niemi M, et al. (2005b). Fexofenadine pharmacokinetics are associated with a polymorphism of the SLC01B1 gene (encoding OATP1B1). Br J Clin Pharmacol 59:602–604.
- Niemi M, et al. (2006). SLC01B1 polymorphism and sex affect the pharmacokinetics of pravastatin but not fluvastatin. Clin Pharmacol Ther 80:356–366.
- Pasanen MK, et al. (2006). SCL01B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet Genom 16:873–879.
- Pasanen MK, et al. (2007). Different effects of SLC01B1 polymorphism on the pharmacokinetics of atorstatin of rosuvastatin acid. Clin Pharmacol Ther 82:726–733.
- SEARCH Collaborative Group. (2008). SLC01B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 359:789–799.
34. Pharmacogenomic Biomarkers for Drug-Induced Hepatotoxicity
Ann Katherine Daly
Institute of Cellular Medicine, Newcastle University, Newcastle-upon-Tyne, UK
Hepatotoxicity is a common reason for discontinuation of drug development. A number of currently licensed drugs are also associated with idiosyncratic liver injury. This drug-induced liver injury (DILI) is relatively rare, but potentially serious, sometimes leading to death or requiring a liver transplant. Several studies on the genetics of DILI are in progress in Europe and the United States. Patient recruitment is generally low due to the rarity of the problem, but some clear associations are beginning to emerge. In particular, susceptibility appears to vary between different drugs. The UK-based DILIGEN study is mainly concerned with DILI due to the antimicrobials, co-amoxiclav and flucloxacillin. Candidate gene case-control studies and, in the case of flucloxacillin, genome-wide association studies, have been completed for 51 cases of flucloxacillin-induced injury and 52 cases relating to co-amoxiclav. Controls exposed to these drugs who have not suffered toxicity have also been recruited together with population controls. Samples have been genotyped for a range of candidate genes considered good candidates, mainly from previously reported associations with DILI. For flucloxacillin, both the candidate gene and genome-wide studies showed a very strong association between the presence of the HLA class I allele B*5701 and susceptibility to disease (odds ratio, 80.6; 95% CI: 22.8–284.9), with 84% of cases positive for B*5701, compared with 6% of controls. This finding has been replicated by further genotyping in an additional cohort. In the case of co-amoxiclav, HLA genotype also appears to contribute to susceptibility, but here, the association is weaker and is with HLA class II DRB1*15 (odds ratio, 2.5; 95% CI: 1.4–4.8), as was suggested in two earlier, smaller studies. Both antimicrobials mainly give rise to DILI, showing a cholestatic phenotype, but certain other drugs, in particular nonsteroidal antiinflammatory drugs, including diclofenac and ant-tuberculosis agents such as isoniazid, are more likely to induce a hepatocellular form of injury. We find no evidence for a strong HLA association for DILI induced by either diclofenac or isoniazid. For diclofenac-induced disease, UGT2B7 and ABCC2 genotypes are risk factors with a weaker contribution from IL-10. In the case of isoniazid, there have been a number of small published studies reporting somewhat contradictory findings, but in general suggesting that NAT2, SOD2, and various GST genotypes are predictors of risk, though effect sizes are small. In view of the range of phenotypes seen for DILI and the diverse nature of the drugs involved, current findings pointing to drug-specific markers of susceptibility are not surprising, but more general pharmacogenomic biomarkers may also emerge from ongoing larger studies, including that sponsored by the International SAEC and the U.S.-based DILIN study.
35. Abstract Not Available
36. The Ontogeny of Detoxification Enzymes in Pediatric Liver
Abby C. Collier
Department of Pharmacology, University of Hawaii School of Medicine, Honolulu, Hawaii, USA
Although the developmental profiles for most phase II metabolizing enzymes are not defined, it was hypothesized that the major phase II enzyme superfamilies develop differentially, and that within superfamilies, isoenzymes also develop at individually determined rates that may be environmentally and/or genetically determined. Using biochemical assays, ELISA, HPLC, and western blot, the development of protein expression and activity was assessed for UDP-glucuronosyl transferases (UGTs), sulfotransferases (SULTs) and glutathione-S-transferases (GSTs), as well as the associated deconjugation and antioxidant enzymes: beta-glucuronidase, aryl sulfatases A and B (ASA and B), aryl sulfatase C (ASC), glutathione reductase (GR), and glutathione peroxidase (GPx). Our sample population consisted of 27 normal pediatric livers (13 days to 20 years). Total UGT activity is low at birth and rises to adult levels at or around 20 months of age, while beta-glucuronidase activity is high at birth but decreases to a plateau by 4 months. The activity of UGT1A1, 1A4, and 1A6 each increase with age but show differential expression and activity profiles. Similarly, microsomal and cytosolic GST enzymes both have low activity at birth but show differential ontogeny, with microsomal GST activity peaking at 10 years, while cytosolic GST protein activity peaks by 5 months. The activity of GR declined significantly with age (P = 0.0077, r = −0.54, Spearman), and while not significantly linear, GPX activities were consistently lower in 6–10 year olds, compared to neonates or adolescents (P < 0.05). Changes in the activity levels and balance of total SULT, ASA and B, and ASC enzymes did not occur, but some evidence for both higher activity and greater protein expression of SULT2A1 in infants 3 months of age and under was observed. While involvement of multiple metabolic pathways (redundancy) for detoxifying chemicals is common in liver detoxification, some xenobiotics and endobiotics are restricted to one enzyme family or isoenzyme. For these compounds, young children and neonates are likely at greater risk of adverse reactions and toxicity.
37. Environmental Chemical Stress and Human Placental Xenobiotic- and Steroid-metabolizing Enzymes
Markku Pasanen
Department of Pharmacology and Toxicology, University of Kuopio, Kuopio, Finland
Only a few forms of CYP proteins, namely CYP1A1 and CYP2E1, have been demonstrated to be expressed at the protein level in the human placenta at term. CYP1A1 is the only placental xenobiotic-metabolizing enzyme for which both the expression and the inducibility due to maternal cigarette smoking have been demonstrated. At least at the mRNA level, the panel of distinct CYPs detectable during the first trimester seems to be wider than those present at term; at full term, only the mRNAs of CYPlA1, 2E1, 2F1, 3A3/4, 3A5, 4B1, and 19A1 have been detected. UDP-glucuronosyl transferase activity is expressed in the placenta throughout the entire gestation and is believed to play a major role in placental metabolic processing of foreign compounds. In the placenta, both the CYP and conjugative enzyme profiles may change during pregnancy (i.e., there are differences in the CYP enzyme profiles between first trimester placentas and their at term counterparts). Moreover, there seems to be a negative correlation between the protein content of the UGT1A family and gestational age. There is also evidence that the GST enzyme does not respond via enhanced expression to classical inducers of phase I metabolism, such as smoking or polycyclic aromatic hydrocarbons. Maternal cigarette smoking acts as a placental endocrine disruptor, and maternal diseases and drug therapies also can affect the expression of placental xenobiotic- and steroid-metabolizing enzymes in the placenta. Several studies on human placenta have demonstrated that increased expression in the oxidative metabolism of phase I (CYP1A1) enzymes does not automatically result in increased expression of conjugating enzymes of phase II. Potentially, this “imbalance” may result in an increase in the levels of active metabolites of phase I and elevated oxidative chemical stress in the placenta, which could cause detrimental effects on placental function and on the well-being of the foetus. (Grant: The Academy of Finland #122859)
38. Abstract Not Available
39. Drug Safety in the Elderly
Ulrich Klotz
Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Stuttgart, Germany
Because of multimorbidity (confounding disease factors), polypharmacy (causing more frequently drug interactions), age-related changes in pharmacokinetics (PK), and pharmacodynamics (PD), elderly patients (above 65 years, according to the WHO definition) are regarded as a population at risk in drug treatment. In Europe, 20% of elderly outpatients suffer from adverse drug reactions (ADRs) and approximately 5–10% of hospital admissions are related to ADRs, which account for considerable morbidity, mortality, and extra costs and which have steadily increased over the last two decades. NSAIDs, anticoagulants, cytostatics, immunosuppressants, diuretics, calcium-channel blockers, captopril, and digoxin are among the drugs most commonly associated with ADR-related hospitalizations. The most frequent ADRs were gastrointestinal bleeding, metabolic and hemorrhagic complications, digoxin toxicity, and hypokalemia. More than 80% of ADRs causing admission to hospitals or occurring in hospitals are type A reactions; as such, they can be predicted and prevented. Of the various factors that are consistently associated with ADRs in the elderly, polypharmacy and inappropriate drug use are considered as the most important. In addition, changes in PK and PD should be taken into account, which both should be counteracted by the corresponding dosage adjustments incorporating also pharmacogenetic peculiarities (Shi et al.). To increase drug safety in the elderly, more such patients need to be included in clinical trials and careful monitoring of all drug effects (including genetic biomarkers) is mandatory. Thereby, the general dosage recommendation “start low—go slow” should be followed.
Reference
- Shi S, Mörike K, Klotz U. (2008). The clinical implications of ageing for rational drug therapy. Eur J Clin Pharmacol 64:183–199.
40. Abstract Not Available
41. γ-Glutamylcyclotransferase and the Regulation of Glutathione Homeostasis
Philip Board
John Curtin School of Med Research, Australian National University, Canberra, Australia
Glutathione is a key compound in the maintenance of cellular redox balance and in the biotransformation of many xenobiotics. It has been conventionally thought that glutathione synthesis is primarily regulated by a feedback loop where GSH is a competitive inhibitor of γ-glutamylcysteine ligase, the first of two enzyme-catalyzed steps in the synthetic pathway. This view does not consider the potential role of γ-glutamylcyclotransferase (GGCT) that can compete with glutathione synthetase (GSH-S) for the available γ-glutamylcysteine and convert it to 5-oxoproline and cysteine. Little is known about GGCT, and to further investigate its role in glutathione homeostasis, we cloned a human cDNA and expressed the recombinant protein in Escerichia coli. The expressed protein was crystallized and its structure solved by X-ray crystallography. GGCT has a novel fold, and although there are large sequence differences, it has structural similarities to an enzyme that catalyses the cleavage of a γ-glutamyl bond in the synthesis of an aminoglycoside antibiotic by Bacillus circulans. We have also cloned another member of this structural family from humans, and current studies are aimed at determining its substrate preferences. Kinetic analysis of recombinant human GGCT and recombinant human GSH-S with γ-glutamylcysteine as a substrate suggests that newly synthesized γ-glutamylcysteine may be kinetically partitioned and preferentially converted to GSH. In GSH-S deficiency, the level of γ-glutamylcysteine rises and overcomes this kinetic barrier, causing the excess production of 5-oxoproline. Inhibitors of GGCT could be used therapeutically to prevent 5-oxoprolinuria in GSH-S-deficient patients. Variations in the level of expression of GGCT could also play a role in modulating glutathione synthesis. In our hands, stable overexpression of GGCT in HepG2 cells was not achieved because it resulted in cell death. However, we have noted high levels of expression in some tumors and in some cancer cell lines that must be resistant to this effect.
42. Glutathione S-transferase P, S-glutathionylation in Cancer
Kenneth David Tew
Department of Cell and Molecular Pharmacology & Experimental Medecine, Medical University of South Carolina, Charleston, South Carolina, USA
Correlative studies have shown that genetic differences of GST isozyme expression may contribute to cancer susceptibility and treatment. A body of data links aberrant expression of GST isozymes with the development and expression of resistance to cancer drugs. In particular, GST Pi (GSTP) is overexpressed in a number of different tumors, compared to normal tissues (including ovarian, non-small-cell lung, breast, colon, liver, pancreas, and lymphoma). Moreover, a significant range of anticancer drugs can cause an increased expression of GSTP in drug-resistant selected cell lines (and drug-treated patients). From a functional viewpoint, not all drugs used to select for resistance are substrates for GSTP with poor catalytic constants for GSTP conjugation reactions. So, why is GSTP so frequently overexpressed in these situations? There is recent evidence that GST isozymes, and GSTP in particular, have multiple functions in cells, many unrelated to thioether bond catalysis with chemical moieties. These include: 1) ligand biding and transport of heme, bilirubin, and nitric oxide; 2) protein: protein interactions with possible chaperone-like functions; 3) regulation of mitogen-activated protein kinases, particularly c-jun NH2 terminal kinase (JNK); and 4) mediation of the forward reaction of the post-translational process of S-glutathionylation. Of these, aberrant kinase-signaling pathways and altered protein S-glutathionylation patterns are both characteristic of the cancer phenotype. GSTP null animals have essentially normal development and lifespans. Mouse embryo fibroblast (MEF) cells isolated from wild-type or GSTP null animals differ in a number of characteristics related to signaling and growth. In particular, null animals have elevated c-jun NH2-terminal kinase (JNK) activity, compared to wild type, and this correlates with altered regulation of genes downstream of JNK. As a whole, the genetic absence of GSTP influences the capacity of stress kinases to regulate gene expression and this can have an impact on cell-proliferation pathways. Some low pKa cysteine residues in proteins are subject to the direct addition of GS—creating an S-glutathionylated residue, with an increase in both MW (of 605) and negative charge (glu residue). GSTP can facilitate the forward, and glutaredoxin, sulfiredoxin, or thioredoxin the reverse reaction. This cyclical characteristic is important in facilitating a sulfur-based regulatory pathway that can expedite response to stress conditions. Importantly, a number of phosphatases can be regulated by S-glutathionylation and this provides a conduit with phosphorus-based signaling pathways. Because the structure, function, and cell distribution of proteins can be affected by S-glutathionylation, the importance of GSTP in mediating this reaction could have significant consequences and may be a contributory factor in the high expression levels of GSTP in many tumors. These multiple functionalities contribute to recent rational efforts to target GSTP with small-molecule therapeutics. At least three drugs are in late-stage clinical testing. Two of these (NOV-002 and Telintra) are myeloproliferative agents. As the field progresses, the concept of designing new drugs that interfere with protein:protein interactions between GSTs and regulatory kinases provides a plausible approach in the search for novel cancer therapeutics.
43. Abstract Not Available
44. Selenium, Glutathione Peroxidases and Cancer
Regina Brigelius-Flohe
Biochemistry of Micronutrients, German Institute of Human Nutrition Potsdam-Rehbruecke, Nuthetal, Germany
A low intake of selenium has been shown to correlate with a higher incidence of cancer, and therefore, chemopreventive functions have generally been attributed to selenium. However, it is not known whether individual selenoprotein(s) contribute to cancer prevention or particular selenium compounds act independently from selenoprotein biosynthesis. We focus on the gastrointestinal glutathione peroxidase (GPx2), which is upregulated in an inflammatory status and in a variety of cancer cells and tissues with, so far, unknown consequences. A protective role of GPx2 can be deduced from two observations: 1) it can be induced by Nrf2 activators (Banning et al., 2005), and 2) it counteracts COX-2 expression in HT-29 cells (Banning et al., 2008a). On the other hand, the GPx2 promoter is activated by beta-catenin (Kipp et al., 2007), which makes it a target of a dysregulated Wnt pathway, but also reflects its putative physiological role in maintaining the proliferation and the self-renewal of mucosal epithelial cells. Thus, GPx2 might have a dual role in inflammation and carcinogenesis. To get more information, cells in which the GPx2 was stably knocked down by siRNA were tested for cancer-relevant processes (Banning et al., 2008b). siGPx2 cells had an increased capability to migrate in a wound-healing test, as well as to invade in a transwell assay. However, siGPx2 cells grew less anchorage independently in soft agar, and most interestingly, tumor development from siGPx2 cells was distinctly lower than from control cells when injected into nude mice. Migration and invasion of siGPx2 cells were inhibited by celecoxib, a cyclo-oxygenase-2 (COX-2)-specific inhibitor. The data show that GPx2 inhibits malignant behavior of tumor cells, such as migration and invasion, but is required for the growth of transformed intestinal cells and, thus, may facilitate tumor cell growth. The selected example already reveals that selenoproteins, depending on their physiological function, may display both the prevention of initiation and the promotion of tumor growth. In later stages, metastasis might be inhibited. Accordingly, the potential benefit of selenium will depend on the stage of tumorigenesis. At present, the effect of selenium and GPx2 is investigated in a model of inflammation-driven tumorigenesis. Preliminary data will be presented.
References
- Banning A, Deubel S, Kluth D, Zhou Z, Brigelius-Flohé R. (2005). The GI-GPx gene is a target for Nrf2. Mol Cell Biol 25:4914–4923.
- Banning A, Florian S, Deubel S, Thalmann S, Müller-Schmehl K, Jacobasch G, et al. (2008a). GPx2 counteracts PGE(2) production by dampening COX-2 and mPGES-1 expression in human colon cancer cells. Antioxid Redox Signal 10:1491–1500.
- Kipp A, Banning A, Brigelius-Flohé R. (2007). Activation of the glutathione peroxidase 2 (GPx2) promoter by beta-catenin. Biol Chem 388:1027–1033.
- Banning A, Kipp A, Schmitmeier S, Lowinger M, Florian S, Krehl S, Thalmann S, et al. (2008b). Glutathione peroxidase 2 inhibits cyclo-oxygenase-2-mediated migration and invasion of HT-29 adenocarcinoma cells but supports their growth as tumors in nude mice. Cancer Res 68:9746–9753.
45. Abstract Not Available
46. The Development of UGT1A1 and UGT1A6 in Pediatric Liver
Shogo J. Miyagi and Abby C.
Collier Department of Pharmacology, University of Hawaii School of Medicine, Honolulu, Hawaii, USA
The UDP-glucuronosyltransferases (UGTs) are arguably the most critical metabolic clearance pathway for drugs, dietary compounds, environmental chemicals, and hormones, although their postnatal development is relatively undefined. Within the UGT superfamily, UGT1A1 and UGT1A6 have some of the widest tissue distributions and range of chemical reactions. It was hypothesized that UGT ontogeny in the pediatric liver is dynamic, with each isoform developing individually through genetic and/or environmental mechanisms. Using the biochemical assays, ELISA, HPLC, and western blotting, enzyme development in children aged 13 days to 20 years was assessed (n = 27). Pooled samples of 50 adult livers were used for comparison (age: 22–71 years, equal ratio of males and females). Neither UGT1A1 nor UGT1A6 showed age differences in levels of protein when probed by western blot using specific antibodies. In contrast, when serotonin was used as a specific probe for UGT1A6, activity was age dependent, reaching apparent maximum adult activity of 2.4 ± 0.32 nmol/min/mg protein at 3.9 years of age with an 8-fold interindividual variation (range: 0.64–4.98 nmol/min/mg protein). These data demonstrate that the developmental regulation of UGT1A1 and UGT1A6 in the pediatric liver is unlikely to be purely transcriptional, since levels of protein do not change with age. However, for UGT1A6, since activity differs significantly with age and activity does not correlate to levels of protein, post-transcriptional and/or -translational effects on UGT1A6 proteins are likely developmental mechanisms. This may also be true for UGT1A1, particularly if activity results when using bilirubin as a probe substrate show the same lack of correlation to protein expression as UGT1A1 protein levels do with age. To our knowledge, this is the first description of the development of human hepatic UGT1A1 and UGT1A6 during childhood. In addition to providing insight into developmental changes in early life, knowledge of the rate and extent at which detoxification enzymes mature can assist with the development and use of drugs, in developing chemical safety guidelines, and for defining the mechanisms of steroid and neurochemical signaling.
47. Functional Coexpression of UGT1 Alternative Splice Isoforms and Evidence of Protein-Protein Interaction
Judith Bellemare, Mélanie Rouleau, and Chantal Guillemette
Canada Research Chair in Pharmacogenomics, CHUQ Research Center and Faculty of Pharmacy, Laval University, Quebec, Quebec, Canada
UDP-glucuronosyltransferases are major actors in phase II metabolism. Anchored in the reticulum endoplasmic membrane, biochemical evidence supports that they might oligomerize between each other and/or with other metabolic enzymes. This capacity of UGTs to physically interact may influence their enzymatic activity. We demonstrated that the UGT1 locus encodes for novel isoforms, isoform 2 or UGT1A-i2, by alternative usage of the final exon, 5. While i2 proteins lack transferase activity, they modify UGT1A-i1-mediated activity. The mechanism of such an inhibition is still unclear. Herein, we explored the influence of coexpressing various UGT1A i1 and i2 on enzyme kinetic parameters and their potential to interact together. Previous observations in human tissues demonstrated coexpression of multiple i1 and i2 forms, raising the possibility that classical i1 may interact with multiple i2 proteins. We tested various combinations of i1 and i2 based on expression data gained in human tissues. Data revealed that isoform 1 of UGT1A1 is able to interact with its homolog, UGT1A1-i2, as well as with various isoforms 2, namely UGT1A7, UGT1A8, and UGT1A9. Interactions between various other combinations of i1 and i2 were also observed. Enzymatic assays were conducted by using microsomes from cells overexpressing i1 and i2 from these UGTs. In all cases, the presence of an i2 protein significantly reduces the velocity from 20 to up to 82%, but without affecting apparent Km. Data would be consistent with the existence of a mixture of homodimeric (i1-i1 or i2-i2) and heterodimeric (i1-i2) enzymes, in which i2-i2 and i1-i2 subunits are inactive. These experiments are essential to gain information on whether the expression of various UGT1A isoforms i2 has a potential incidence on glucuronidation activity, and whether these new inhibitory isoforms may be involved in the glucuronidation variability in tissues. In conclusion, the potential for UGT1A-i1 and i2 to oligomerize drastically increases the possible mechanisms by which these proteins can be modulated and their potential as regulatory factors for elimination/detoxification of drugs. (This work is supported by NSERC.)
48. Interplay between CYP3A and P-Glycoprotein: The Case of Lopinavir
Robert A.B. van Waterschoot,1 Rob ter Heine,2 Els Wagenaar,1 Cornelia M.M. van der Kruijssen,1 Antonius E. van Herwaarden,1 Alwin D.R. Huitema,2 Jos H. Beijnen,2 and Alfred H. Schinkel3
1Division of Molecular Biology, The Netherlands Cancer Institute, Amsterdam, The Netherlands, 2Department of Pharmacy and Pharmacology, Slotervaart Hospital, Amsterdam, The Netherlands, 3Division of Molecular Biology, The Netherlands Cancer Institute, Amsterdam, The Netherlands
Both cytochrome P450 3A (CYP3A) and P-glycoprotein (P-gp; MDR1) are known to have a strong effect on the oral bioavailability of various drugs. It is noteworthy that CYP3A and P-gp have an extensive overlap in their substrates. It has, therefore, been hypothesized that it is the combined intestinal activity of CYP3A and P-gp that makes for efficient first-pass metabolism of many orally administered drugs. Theoretically, the function of P-gp would prevent the saturation of CYP3A and give the enzyme repeated access to its substrates. Overall, this would lead to a highly efficient intestinal metabolism. Lopinavir is currently one of the most widely used HIV protease inhibitors and is a known substrate for CYP3A and P-gp. Our aim was to investigate the individual and combined effects of CYP3A and P-gp on the pharmacokinetics of lopinavir. Moreover, since lopinavir in the clinic is always coadministered with ritonavir, we also evaluated whether ritonavir increases lopinavir oral bioavailability by inhibition of CYP3A and/or P-gp. We found a significant increase in lopinavir systemic exposure in P-gp−/- mice (~9-fold, compared to wild-type) after oral administration. Strikingly, an increase of more than 2,000-fold in lopinavir systemic exposure was observed in Cyp3a−/- mice. Notably, however, no significant differences were seen in systemic exposure between Cyp3a−/- and Cyp3a/P-gp−/- mice. Our study thus revealed that ritonavir boosts lopinavir exposure primarily through CYP3A inhibition and not through inhibition of P-gp. Further, a more detailed examination of the role of CYP3A revealed that both hepatic and intestinal CYP3A activity has a major impact on the oral bioavailability of lopinavir. Combined, these results clearly demonstrate that CYP3A, and not P-gp, is the major determinant of lopinavir pharmacokinetics. Further, as the impact of P-gp was only detectable in the presence of CYP3A, it appears that in the case of lopinavir, it is not P-gp that prevents the saturation of CYP3A, but rather, that CYP3A prevents the saturation of P-gp. These unexpected findings shed new light on the interplay between CYP3A and P-gp.
49. Hyperammonemia Induced by Valproic Acid Therapy: Urea Cycle Impairment by Intramitochondrial Valproyl-CoA
Catia C.P. Aires,1 Arno van Cruchten,2 Jos Ruiter,2 Lodewijk IJlst,2 Isabel Tavares de Almeida,1 Marinus Duran,2 Ronald J.A. Wanders,2 and Margarida F.B. Silva1
1Metabolism and Genetics Group, iMED.UL, Centro de Patogenese Molecular, Faculdade de Farmácia, Universidade de Lisboa, Lisbon, Portugal, 2Laboratory Genetic Metabolic Diseases, Department of Clinical Chemistry and Pediatrics, AMC, University of Amsterdam, The Netherlands
Valproic acid (2-n-propylpentanoic acid; VPA) is a commonly prescribed anticonvulsant drug that may induce a severe hepatotoxicity, often clinically characterized by hyperammonemia. Previous results obtained by our group suggested an interference of VPA with the urea cycle not to be primarily at the carbamoylphosphate synthase (CPS I) or ornithine transcarbamylase (OTC) level, the first enzymes of this pathway.
Aims: To elucidate the pathogenic mechanisms by which VPA and its metabolite, valproyl-CoA, interfere with the urea cycle, leading to hyperammonemia. We studied the effect of valproyl-CoA on the biosynthesis of N-acetylglutamate (NAG), which controls the flux through the urea cycle.
Methods: The amount of NAG was quantified in livers of control and VPA-treated rats (100 mg/Kg for 2 weeks; n = 12). The NAG synthase (NAGS) activity was measured after the incubation of sonicated mouse liver mitochondria (MLM) with VPA (0–10 mM) or valproyl-CoA (0–1 mM), plus 10 mM of glutamate, 1 mM of L-arginine and acetyl-CoA (0–2.5 mM), for 5 minutes at 30°C. The synthesized NAG was identified and quantified by LC-MS/MS, using N-acetyl-[13C5]-glutamate as the internal standard.
Results: The level of NAG in liver tissues of VPA-treated rats was found to be decreased, as compared with controls (7.1 ± 6.3 and 57.4 ± 51.6 nmol/g wet weight, respectively). Further, valproyl-CoA was found to be a stronger inhibitor of NAGS in MLM than the free acid, VPA. To characterize the inhibition mechanism on NAGS activity, a competition assay was done with valproyl-CoA and the substrate acetyl-CoA, and the kinetic parameters were calculated. Valproyl-CoA was shown to increase the Km and to decrease the Vmax of NAGS, in agreement with a mixed type of inhibition.
Discussion: The results presented in this work strongly demonstrate that VPA-induced hyperammonemia results from the mixed inhibition of valproyl-CoA on the NAGS activity. The reduced bioavailability of NAG accounts to a reduced flux through the urea cycle and to the subsequent accumulation of ammonia. This effect may be clinically significant, resulting in severe hyperammonemic encephalopathy. (Supported by FCT, SFRH/BD/22420/2005)
50. Toward the Complete Elucidation of the Mitochondrial ß-Oxidation Pathway of Valproic Acid and its Interference with the Oxidation of Branched-Chain Amino Acids
Paula B.M. Luis,1 Jos Ruiter,2 Lodewijk IJlst,2 Rob Ofman,2 Luisa Diogo,3 Paula Garcia,3 Marinus Duran,2 Jerry Vockley,4 Isabel Tavares de Almeida,5 Ronald J.A. Wanders,6 and Margarida F.B. Silva5
1Metabolism and Genetics Group, iMED.UL, Faculdade de Farmácia, Universidade de Lisboa, Lisbon, Portugal, 2Laboratory Genetic Metabolic Diseases, Department of Clinical Chemistry and Pediatrics, AMC, University of Amsterdam, The Netherlands, 3Hospital Pediátrico de Coimbra, Coimbra, Portugal, 4Department of Pediatrics, School of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA, 5Metabolism and Genetics Group, iMED.UL, Centro de Patogenese Molecular, Faculdade de Farmácia, Universidade de Lisboa, Lisbon, Portugal, 6Laboratory Genetic Metabolic Diseases, Department of Clinical Chemistry and Pediatrics, AMC, University of Amsterdam, The Netherlands
Valproic acid (VPA; 2-n-propylpentanoic acid) is a simple branched medium-chain fatty acid that is used worldwide for its anticonvulsant properties. VPA is a substrate for the fatty acid ß-oxidation (FAO) pathway; however, the enzymology of this pathway has not been completely elucidated yet. Both VPA and branched-chain amino acids (BCAAs) undergo ß-oxidation, generating structurally similar metabolites, which suggest that VPA might use key enzymes of the BCAA catabolic pathway. Previous results showed a high excretion of 3-hydroxyisovaleric and 2-methyl-3-hydroxybutyric acids in patients under VPA therapy, suggesting an interference of valproate on isovaleryl-CoA dehydrogenase (IVD) and 2-methyl-3-hydroxybutyryl-CoA dehydrogenase (MHBD) activities. Aims: To investigate the potential involvement of specific enzymes of the BCAA oxidative metabolism on the ß-oxidation of VPA. Studies were also performed to elucidate if human 2-enoyl-CoA hydratase (EH) is the only enzyme catalyzing the hydration step from ß-oxidation of VPA.
Methods: In vitro studies were performed by using human heterologously expressed IVD, isobutyryl-CoA dehydrogenase (IBD), short branched-chain acyl-CoA dehydrogenase (SBCAD), and MHBD, using valproyl-CoA and 3-hydroxyvalproyl-CoA as potential substrates, respectively. The different enzymatic activities were measured by using optimized HPLC procedures. EH immunoprecipitation in human liver homogenate was performed and supernatant activities were measured by HPLC, using 2-enoyl-CoA and Δ2-valproyl-CoA as substrates.
Results: Valproyl-CoA was converted to Δ2-valproyl-CoA by IVD, although SBCAD dehydrogenated valproyl-CoA at a much higher rate (Km = 356 μM and Vmax = 0.29 nmol/min/mg). IBD was not active with valproyl-CoA as the substrate. MHBD was found to dehydrogenate 3-hydroxyvalproyl-CoA into 3-ketovalproyl-CoA (Km = 35 μM and Vmax = 3.9 μmol/min/mg). In samples with immunoprecipitated EH, there was no detectable hydratase activity when using both 2-enoyl-CoA and Δ2-valproyl-CoA as substrates.
Discussion: This work demonstrates, for the first time, that besides SBCAD, MHBD is also involved in VPA ß-oxidation, next to its role in isoleucine catabolism. Further, EH is shown to be the only enzyme responsible for the hydration of Δ2-valproyl-CoA. The participation of certain mitochondrial enzymes in the biotransformation of VPA may contribute to the mitochondrial dysfunction and potential hepatotoxicity often reported in VPA-treated patients.
51. Human Cytochrome P450 1A2: Comprehensive Study of Eight Polymorphic Variants
Bernardo Brito Palma,1 Marta Silva e Sousa,2 Jeroen Lastdrager,3 José Rueff,2 Nico P. E. Vermeulen,3 and Michel Kranendonk2
1Department of Genetics, Universidade Nova de Lisboa, Faculty of Medical Sciences, Lisbon, Portugal, 2Department of Genetics, Universidade Nova de Lisboa, Faculty of Medical Sciences, Lisbon, Portugal, 3Division of Molecular Toxicology, Department of Pharmacochemistry, LACDR/Vrije Universiteit Amsterdam, Amsterdam, The Netherlands
Interindividual variability in xenobiotic and drug metabolism is extensive. Cytochrome P450s (CYPs) are responsible for the metabolism of a wide variety of clinically, physiologically, and toxicologically important compounds, displaying a central role in pharmacology and toxicology. CYP1A2 is involved in the metabolism of many drugs and is an important catalyst for the bioactivation of several classes of precarcinogens. To date, 36 CYP1A2 haplotypes are annotated at the Human Cytochrome P450 Allele Nomenclature Committee website (http://www.cypalleles.ki.se/cyp1a2.htm). The aim of this current study was to make a comprehensive evaluation of the effect of CYP1A2 polymorphism by studying eight polymorphic forms of human CYP1A2, namely: T83M, S212C, S298R, G299S, I314V, I386F, C406Y, and R456H. These variants were constructed through site-directed mutagenesis and coexpressed with human CYP reductase (CYPOR). The expression levels of CYPOR were, in all cases, equal. Western analysis did demonstrate similar levels of protein synthesis of the apoprotein for all CYP variants. However, the levels of holoprotein detected by CO-spectra were low and nondetectable for variants I386F and R456H, respectively. Subsequently, CYP1A2 activity was investigated by using three different fluorescent probe-substrates (7-ethoxy- and 7-methoxyresorufin and 3-cyano-7-ethoxycoumarine), two therapeutic drugs (clozapine and phenacetine), and three precarcinogens known to be bioactivated by CYP1A2 (4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone [NNK], 2-aminoanthracene [2AA] and 2-amino-3-methylimidazole(4,5-f)quinoline [IQ]). The heterogeneous activity data set of the holoenzyme expressible variants was then compared with the wild-type form, applying multivariant analysis. Both MDS and PCA multivariant analysis indicated a very significant dissimilarity of G299S and, to a lesser extent, variant T83M, relative to the wild-type CYP1A2 allele. Surprisingly, 7-ethoxyresorufin and NNK were the most discriminative compounds among the CYP1A2 variants of a heterogenous set of eight substrates. The detected disturbance of heme-binding by variants R456H and I386F as well as the altered enzyme activity of variants T83M and G299S could be rationalized through an interpretation of protein conformations, based on CYP1A2 crystal structures.
52. Relationship between Variants of Apical Sodium-dependent Bile Acid Transporter Gene (SLC10A2) and the Development of Colorectal Cancer
Cátia Carvalho,1 Vera Ribeiro,2 and José Estevens3
1Center of Molecular and Structural Biomedicine., Faro, Portugal, 2CBME-University of Algarve, Faro, Portugal, 3Hospital do Barlavento Algarvio, Portimão, Portugal
Colorectal cancer (CRC) is the most frequent type of neoplasia in developed countries and the second leading cause of death among cancers, killing more than 655,000 per year [1]. Some genes have been identified as associated to disease risk, particularly concerning familial CRC. However, little is known about genetic factors that could modulate the risk for its development. Epidemiological and experimental studies suggest that bile acids, in particular secondary bile acids, may play an important role in colorectal carcinogenesis [2]. The apical sodium-dependent bile salt transporter (ASBT, SLC10A2) is the major transporter involved in active bile acid uptake by the intestinal mucosa [3]. Structural variability in this transporter has been recently unveiled, with circa 13 known variants with variable effects in function and/or expression [4, 5], which can alter bile acid flow, leading to increased concentrations of cytotoxic secondary bile acids in the large intestine. In this study, we investigate a possible relationship between three variants (V98I, P142P, and A171S) in ASBT and the risk for the development of colorectal cancer (case-control study) in Portuguese Caucasian subjects. Novel PCR-RFLP genotyping methods were developed for the analysis of these variants, this being the first analysis of ASBT variants in the Portuguese population. We did not identify homozygous individuals for the mutant genotype for any of the variants. Individuals with the mutant allele for P142P variant were also not found in this study. The heterozygous genotype for each one of V98I and A171S variants individually seems to constitute a protective factor on the development of CRC [P = 0.699, OR = 0.589 (0.126–2.749) and P = 0.094, OR = 0.541 (0.266–1.102), respectively]. This hypothesis is strengthened by the observed association of the possible genotypes for both V98I and A171S variants and the development of colorectal cancer risk, showing that the incidence of the heterozygous genotype seems to confer a protective effect on the development of this neoplasia [VA/IA: P = 0.307, OR = 0.229 (0.023–2.300); VA/VS: P = 0.207, OR = 0.566 (0.246–1.305)].
53. Comparison of Intestinal and Hepatic Sulphation and Glucuronidation for a Range of Drugs
Helen Cubitt, Brian Houston, and Aleksandra Galetin
School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester, UK
Increasing awareness of the importance of conjugation enzymes, specifically sulphotransferases (SULTs) and uridine diphosphate glucuronosyltransferases (UGTs), indicates a need to incorporate their contribution into clearance prediction. The aim of the current study was to assess the extent of intestinal and hepatic sulphation in comparison to glucuronidation for a range of selected drugs. Intrinsic sulphation clearance (CLint,SULT) was obtained for six drugs in human intestinal and hepatic cytosol, using a substrate-depletion approach. In addition to sulphation, glucuronidation (CLint,UGT) and CYP (CLint,CYP) clearances were determined for all the compounds investigated in alamethicin-activated human intestinal and hepatic microsomes. The obtained CLint values were corrected for experimentally determined fraction unbound in either cytosol or microsomes. In order to allow valid comparison between the organs, the parameters were expressed per gram of tissue, using reported microsomal and cytosolic recovery estimates. Scaled CLint,SULT ranged from none to 6.4 mL/min/g intestine for diclofenac and quercetin, respectively, and from 1.5 to 17 mL/min/g liver, for raloxifene and quercetin, respectively. Raloxifene and salbutamol showed the lowest and highest extent of sulphation, ranging from 8 to 90% and from 1 to 80% in the liver and intestinal cytosol, respectively. Troglitazone was the only drug where intestinal and hepatic scaled CLint,SULT were comparable. For the other compounds, intestinal CLint,SULT represented 0–47% of the hepatic value (diclofenac and raloxifene, respectively). In contrast to sulphation, intestinal glucuronidation clearance (when scaled per gram of organ) was 28- and 4-fold greater, in comparison to the liver for raloxifene and troglitazone, respectively. The current study has shown the utility of human cytosol and microsomes in the assessment of the extent of intestinal and hepatic sulphation and glucuronidation in vitro. In addition, significant sulphation and glucuronidation in the intestine indicated a need for the incorporation of intestinal conjugation metabolic pathways into drug clearance prediction.
54. Effect of Valproic Acid on Extracellular Mitogen-activated Protein Kinase Pathways and Major Transcriptional Factors in Hepatoma Cell Lines and Primary Human Hepatocytes
Michal Bitman,1 Radim Vrzal,2 Katerina Pospechova,1 Lucie Svecova,3 Lenka Cygalova,3 Zdenek Dvorak,2 and Petr Pavek1
1Department of Pharmacology and Toxicology, Faculty of Pharmacy, Charles University, Hradec Kralove, Czech Republic, 2Department of Cell Biology and Genetics, Faculty of Science, Palacky University in Olomouc, Olomouc, Czech Republic, 3Department of Pharmacology and Toxicology, Faculty of Pharmacy, Charles University, Hradec Kralove, Czech Republic
Valproic acid is a widely used drug for the treatment of epilepsy and bipolar disorder. Valproic acid has been proven to affect numerous gene-expression regulatory mechanisms, including histone deacetylases (HDACs) and mitogen-activated protein kinase pathways (MAPKs). We showed earlier (Cerveny et al., 2007) that valproic acid has the potential to upregulate the expression of cytochrome P-450 CYP3A4 and P-glycoprotein genes, stimulates the rifampicin-mediated induction of CYP3A4 in primary human hepatocytes and hepatoma cell lines and transactivates CYP3A4 gene reporter construct in HepG2 cells via constitutive androstane receptor (CAR) and pregnane X receptor (PXR). In this study, we focused on the effect of valproic acid on the activation of major MAPKs in hepatoma cell lines and in primary human hepatocytes. The effect of valproic acid on ERK1/2, SAPK/JNK, and p38 MAPKs in hepatoma cell lines was evaluated by employing Western blot analysis using phospho-specific antibodies and PathDetect pathway cis-reporting assays (Stratagene, SA Biosciences, Frederick, Maryland, USA). Using the SuperArray reverse-transcriptase polymerase chain reaction (RT-PCR) method, we analyzed the effect of valproic acid on the expression of 86 nuclear receptors and transcriptional factors in primary human hepatocytes. As positive and negative controls, pharmacological inhibitors and activators of the MAPKs and siRNA methodology were employed. The activation of the CYP3A4 promoter was analyzed by using luciferase reporter assays in HepG2 cells and the mRNA expression level was monitored by RT-PCR in primary hepatocytes. We found that valproic acid activates the ERK1/2 (p44/42) MAPK pathway by employing Western blot analysis in HepG2 cells and primary human hepatocytes. Consistently, valproic acid activated Elk1 phosphorylation, a transcriptional factor phosphorylated by ERK1/2 kinases, in the PathDetect reporter assay. We also observed the activation of the p38 pathway in primary human hepatocytes. We found that the silencing of ERK1/2 suppressed the valproic acid–mediated activation of the CYP3A4 promoter in the HepG2 cell line. Our preliminary data suggest a potential role of ERK1/2 in the valproic acid–induced PXR-mediated transactivation of CYP3A4. (Supported by GACR 303/07/0128 and GAUK 170/50/85007)
Reference
- Cerveny L, et al. (2007). Valproic acid induces CYP3A4 and MDR1 gene expression by activation of constitutive androstane receptor and pregnane X receptor pathways. Drug Metab Dispos 35:1032–1041.
55. DNA Demethylation and Histone Deacetylation Inhibition in the Derepression of CYP46A1 Expression
Inês Milagre, Maria João Nunes, Maria João Gama, and Elsa Rodrigues iMed
UL-Molecular and Cell Biology of Eukaryotic Systems, Faculty of Pharmacy, University of Lisbon, Lisbon, Portugal
CYP46A1 is a neuron-specific cytochrome P450 responsible for the conversion of cholesterol into 24S-hydroxycholesterol. The flux of this oxysterol across the blood-brain barrier into the circulation is the major pathway for cholesterol elimination from the brain. Since the CYP46A1 promoter is localized in a CpG island, and its reporter constructs presented high luciferase activity even in cell lines where CYP46A1 mRNA is not detected, we have investigated whether transcription of the CYP46A1 gene is subject to modulation through coordinated regulation at both the histone and DNA levels. Transient transfection studies, using reporter CYP46A1 promoter constructs, were performed in HeLa and SH-SY5Y cells treated with different inhibitors of histone deacetylases (HDACs) and resulted in increased CYP46A1 promoter activity. In parallel, we have evaluated, by real-time PCR, whether the endogenous expression of the CYP46A1 gene could be upregulated by inhibitors of HDACs. Trichostatin A (TSA) markedly elevated endogenous CYP46A1 mRNA levels in a dose-dependent manner. The silencing of SP transcription factors through small-interfering RNAs led to a significant decrease of CYP46A1 expression induction by TSA, supporting our previous results that point to a key role of the SP family of transcription factors in the regulation of CYP46A1. To mimic the effect of endogenous DNA methylation, methylated and mock-treated reporter constructs were used for reporter gene analysis. Methylation of a reporter construct containing the SP responsive region led to an almost complete reduction of the promoter activity. Further, treatment with the DNA demethylating reagent, 5’-azacytidine, elevated endogenous CYP46A1 mRNA levels in a time- and dose-dependent manner, and a synergistic activation of the CYP46A1 gene by TSA was observed when cells were pretreated with the DNA demethylating agent. Our results show, for the first time, that coordinated changes in DNA methylation and histone modifications are involved in the regulation of the brain-specific expression of CYP46A1. [Supported by FCT and FEDER–projects POCTI/SAU-MMO/55919/2004, PTDC/SAU-GMG/64176/2006, and PhD grants SFRH/BD/ 27660/2006 (to IM), SFRH/BD/41848/2007(to MJN)].
56. CYP2C19 Transcriptional Regulation by Estrogen Receptor Alpha
Jessica Mwinyi, Rasmus Steen Pedersen, Isa Cavaco, Anna Persson, Sabrina Burkhardt, Souren Mkrtchian, and Magnus Ingelman-Sundberg
Section of Pharmacogenetics, Department of Physiology and Pharmacology, Karolinska Institute, Stockholm, Sweden
CYP2C19 is important for the biotransformation of many drugs, such as omeprazole, proguanil, and escitalopram. CYP2C19 activity is influenced by both genetic polymorphism and by several endogenous and exogenous compounds. An example is the use of oral contraceptives that drastically inhibits CYP2C19 activity in vivo. The underlying molecular mechanisms of this effect are still unknown. In this study, we hypothetizised that 17β-oestradiol (EE) and 17α-aethinyloestradiol (ETE; an active compound of oral contraceptives) regulate CYP2C19 expression via oestrogen receptor alpha (ERα) and examined the effect of the antioestrogens, 4-hydroxytamoxifen (4-OHT) and raloxifene (R), two selective estrogen-receptor modulators (SERMs). Four putative ERE half sites were identified by in silico analysis within the first 1.8 kb of the 59-upstream region of the CYP2C19 gene. A specific protein-DNA complex was observed by using nuclear proteins from Huh-7 cells at the ERE half site (–152/–147), which was successfully supershifted with an antibody against ERα. Chromatin immunoprecipitation analysis, using Huh-7 cells, showed a specific complex. After cotransfecting Huh7 cells with pcDNA3.1-ERα and a luciferase construct carrying a 1.6-kb-long CYP2C19 promoter fragment including the ERE half site (–152/–147), we observed strong downregulation upon treatment with EE or ETE at half-maximum concentrations of 10−7 and 10−8 M, respectively. Mutations introduced into the ERE half site (–152/–147) significantly inhibited these ligand-dependent effects. In contrast to this, 4-OHT and R were unable to affect the CYP2C19 promoter activity. A Flp-In™ CYP2C19 stable cell line was created and it was found that the competitive inhibition of CYP2C19 activity by ETE and EE occurred at much higher concentrations, namely with IC50 values of 10−7 and 10−6 M, respectively. These results indicate that both EE and ETE inhibit CYP2C19 activity, mainly through the inhibition of CYP2C19 gene expression via the classical ERα-dependent regulatory pathway, thus providing a new insight into the molecular mechanism behind the clinically established inhibitory effect of oral contraceptives on CYP2C19 activity.
57. A Biochemical, Toxicological, And Proteomic Analysis Investigating the Effect of Nrf2 Gene Deletion On Paracetamol-induced Hepatoxicity In Vivo
Laura E. Randle,1 Chris E.P. Goldring,1 Roz E. Jenkins,1 Daniela Denk,2 Daniel J. Antoine,1 Rowena L. Sison,1 Dominic P. Williams,1 John D. Hayes,3 Neil R. Kitteringham,1 and B. Kevin Park1
1CDSS, Department of Pharmacology and Therapeutics, The University of Liverpool, Liverpool, UK, 2Department of Veterinary Pathology, The University of Liverpool, Liverpool, UK, 3Ninewells Hospital and Medical School, Biomedical Research Centre, Dundee, UK
Drug-induced liver injury (DILI) is a major complication during drug development. The Keap1-Nrf2-ARE-signaling pathway plays an important role in the defense against chemical stress. Early studies using paracetamol (APAP) demonstrated that Nrf2 null mice are more susceptible to toxicity. They postulated that the increased sensitivity was associated with a decreased expression of drug-metabolizing and antioxidant genes. We have shown that APAP can induce Nrf2 translocation and defense of gene expression in vivo in a dose-dependent manner. The aim of this study was to investigate the consequence of Nrf2 deletion on the initiation and progression of APAP-induced toxicity, to determine the time dependency of Nrf2 activation. Currently, there are no global hepatic proteomic studies exploring Nrf2 null mice within the literature. Many studies have concentrated on investigating Nrf2-dependent changes at the RNA level; however, these may not necessarily be translated into changes at the protein level and function. We hope to establish an Nrf2 signature set to inform drug discovery and development. Nrf2 WT and null mice were administered APAP (300 mg/kg) or saline control (n = 6) at 0, 1, 3, and 5 hours. Serum ALT, CK18, and HMGB1 levels were determined, alongside oxidative stress biomarkers in serum and hepatic tissue (MRM analysis). GSH levels were also measured. Hepatic protein expression was analyzed by using iTRAQ labeling with LC-MS/MS analysis and RNA expression via QRTPCR. Serum ALT and histolopathology confirmed that null mice were more sensitive to APAP toxicity than WT mice. Basal GSH levels were significantly (P < 0.01) reduced in Nrf2 null mice liver, compared to WT. APAP induced a number of proteins in WT mice, which were not observed in null liver, suggesting that they are likely to be under Nrf2 control. We are comparing these changes to the nontoxic analog of APAP, metacetamol (AMAP). Identification of APAP and AMAP target proteins may provide additional information regarding the mechanism of APAP-induced toxicity and enable us to identify proteins involved with the initiation and/or the progression of toxicity.
58. In Vitro Uptake of the Magnetic Resonance Imaging Contrast Agent Gadoxetate Is Mediated by the Human Hepatic Uptake Carrier Organic Anion-Transporting Polypeptide 1B3
Mirko Leonhardt,1 Stefan Oswald,2 Ulrike Adam,1 Werner Siegmund,2 and Werner Weitschies1
Departments of 1Biopharmaceutics and Pharmaceutical Technology and 2Clinical Pharmacology, University of Greifswald, Greifswald, Germany
The use of magnetic resonance imaging (MRI) has emerged as a useful technique to detect and characterize hepatic lesions, hepatic cancer, or metastasis. The use of liver-specific contrast agents is often necessary to provide a secure diagnosis. Gadoxetate (Gd-EOB-DTPA, Primovist®) is a novel liver-specific contrast agent developed for tissue-contrast enhancement in hepatobiliary MRI. Although millions of applications have been performed to date, the distinct transport mechanism of gadoxetate into healthy human hepatocytes remains unknown. Therefore, it was the aim of this study to identify the hepatic uptake carriers of gadoxetate. HEK-293 cells transfected with human OATP1B1 (formerly known as OATP-C) and OATP1B3 (formerly known as OATP-8) and vector control cells were used for the in vitro studies. Radiolabeled 3H-bromosulfophthalein, a well-known substrate of both transporter proteins and gadoxetate, were used in a competition assay. In another experiment, the direct cellular accumulation of gadoxetate in HEK-293 cells was determined by using a validated LC-MS/MS method. Data from both experiments suggest that gadoxetate is a substrate of human OATP1B3. The uptake into OATP1B3 transfected HEK-293 was found to be up to 7-fold increased, compared to vector control cells. On the other side, the intracellular concentrations of gadoxetate were decreased in the presence of rifampicin, a potent inhibitor of all human OATPs, in a concentrated-dependent manner. A concentration of 1 μM of rifampicin modulated the uptake of gadoxetate into OATP1B3 expressing cells by up to 35%. No interaction of gadoxetate with OATP1B1 was detectable. In conclusion, the organic anion-transporting polypeptide, OATP1B3, and not OATP1B1, seems to be responsible for the uptake into human hepatocytes.
59. An LC-MS/MS Method for Determination of 3,6-Disinapoylsucrose, One Active Component of Polygala Tenuifolia, in Rat Plasma and Its Application for a Pharmacokinetic Study
Ying Chen, Rui-Le Pan, Xin-Min Liu, Geng-Nan Wang, Fu Ying, and Qi Chang
Research Center for Pharmacology and Toxicology, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
The roots of Polygala tenuifolia Willd (common name: Yuan-Zhi) has long been used as a sedative folk medicine in traditional Chinese medicine and is a main ingredient in the formula “Kaixinsan” with traditional reputed benefits for the treatment of insomnia, palpitations with anxiety, restlessness, and disorientation. 3,6-disinapoylsucrose, one of the major active components isolated from Yuan-Zhi, showed significant inhibition effects on potassium cyanide– and scopolamine-induced hypoxia and scopolamine-induced memory impairment. In order to explore the pharmacokinetic properties of the active component DSS, a specific and sensitive LC-MS-MS method was developed. An aliquot of 100 μL of plasma sample was mixed with forsythin, used as the internal standard, and then deproteinated by mixing with 300 μL of acetonitrile. After centrifugation, the supernatant was evaporated to dryness by nitrogen gas. The residue was reconstituted with 100 μL of mobile phase and 20 μL of the solution was analyzed by LC-MS/MS in positive electrospray ionization condition. Chromatographic separation was achieved on a C18 reversed-phase HPLC column (2.1 × 100 mm, 3 μm) eluted with a mobile phase consisting of methanol and 0.01% formic acid in 1 mM of ammonium acetate buffer (30:70, v/v) at a flow rate of 0.2 mL/min. A multiple-reaction monitoring (MRM) model was employed to measure the ion transitions: m/z 777.4 → 409.3 for DSS and 557.3 → 309.3 for forsythin, respectively. The method was linear over the studied concentration range of 0.5–1,000 ng/mL. The precision and accuracy were less than 10.23 and 11.35% for within-day and 9.08 and 6.21% for between-day determination, respectively. Extraction recovery was more than 86.61%. The lower limit of quantification (LLOQ) for DSS in rat plasma was 0.5 ng/mL. The present method was successfully applied to pharmacokinetic study for DSS after intragastric and intravenous administration in rats and for DSS after intragastric and intravenous administration in rats.
60. Development of a Sensitive LC-APPI-MRM Method to Measure 3-HYDROXY-B[a]P, a Biomarker of B[a]P Exposure
Andrew Baxter, Nik Newland, and Emmanuel Minet
Risk Characterization, British American Tobacco, Southampton, UK
Benzo[a]pyrene (B[a]P) and pyrene are found in complex PAH (polyaromatic hydrocarbons) mixtures, which result from the combustion of organic matter, such as fossil fuel and tobacco. B[a]P is generally found in low quantities but has been widely studied because of its positive activity in tumor models. Conversely, pyrene is not considered to be toxic. Yet, for the measurement of PAH exposure in humans, total hydroxypyrene is the most commonly used biomarker, despite the fact that B[a]P is more toxicologically relevant. Since PAHs do not ionize by using classical LC-MS sources, the most commonly used method to detect PAH metabolites are fluorescence based or involve derivatization. We describe here the development of a specific and sensitive LC-MS detection method for 3-hydroxy-B[a]P, using LC-APPI (atmospheric pressure photoionization)-MS/MS. Three independent serial dilutions of 3-hydroxy-B[a]P in synthetic urine were spiked with 1-hydroxy-pyrene, the internal standard. A 95% toluene, 5% anisole solution was used as a dopant. 3-hydroxy-B[a]P was resolved on a phenomenex Gemini C-18 column (Phenomenex, Torrance, California, USA). MRM acquisition was performed in the positive mode at 268 m/z for Q1. 3-hydroxy-B[a]P and gave two major fragments in Q3 at 239.1 and 237.1 m/z. The 239.1 fragment gave the best sensitivity. Detection of 3-hydroxy-B[a]P by LC-APPI-MRM is sensitive with an LOD of 250 pg/mL and LOQ of 600 pg/mL. LOQ was established with independent triplicates and an accuracy and precision of 100 ± 20% with a signal-to-noise ratio of at least 3/1. The sensitivity is within the range of occupational exposure to B[a]P; however, many studies conducted with smokers’ urine and using a range of techniques (e.g., GC-MS, fluorescence, Q-TOF, ± derivatization) were inconclusive. This is attributed to the very low level of B[a]P metabolites; for instance, 3-hydroxy-B[a]P is found in the low pg range in urine from smokers (<10 pg/mL). Therefore, additional refinements are needed to increase our sensitivity in urine from smokers, including the test of alternative dopants, solid-phase extraction, or screen of other B[a]P metabolites, such as 9-hydroxy-B[a]P and B[a]P-tetraols.
61. New Urinary DNA Adducts Derived from Styrene
Petr Mikeš,1 Marek Korinek,1 Igor Linhart,2 Jan Krouželka,2 Ludmila Vodicková,3 Jan Mráz,3 and Ludmila Dabrowská3
1Re&D VUFB, Prague, Czech Republic,2Department of Organic Chemistry, Institute of Chemical Technology, Prague, Prague, Czech Republic,3National Institute of Public Health, Prague, Czech Republic
Two adenine adducts, 3-(2-hydroxy-1-phenylethyl)adenine (N3αA) and 3-(2-hydroxy-2-phenylethyl)adenine (N3βA), were identified and quantified in the urine of mice exposed to styrene at 1,200 mg/m3 for 10 consecutive days (6 hours/day) by LC/MS. Excretion of these adducts amounted to approximately 0.8 × 10−5 % of the dose, with no apparent accumulation, and it ceased shortly after finishing the exposure. Human urine samples from occupationally exposed individuals at styrene exposure levels up to 90 mg/m3 were also analyzed; however, the presence of neither N3αA nor N3βA could be confirmed in any of these samples at the detection limit of 5 and 2 pg/mL for N3αA and N3βA, respectively. Assuming the same conversion of inhaled styrene to N3αA and N3βA in humans and mice, the expected urinary concentration of these adducts would amount to approximately 18 pg/mL for N3αA and 42 pg/mL for N3βA. Therefore, it can be inferred that the conversion of styrene to these adducts is at least by one order of magnitude lower in humans than in mice, and as a consequence, the damage to DNA caused by styrene 7,8-oxide, a reactive metabolite of styrene, should be much lower in humans than in mice. (This study was supported by grants 2B08051 and MSM 604 613 73 01 from the Ministry of Education, Youth and Sports of the Czech Republic.)
62. Absorption, Distribution, Metabolism and Excretion Of Zabofloxacin After a Single Oral Administration to Non-Fasted Male Rats
Shaun Johnson,1 Colin Webber,1 Clair Smart,1 Misba Tasawar,1 Brian John,1 Jim Rock,2 and J.M. Ryu3
1Huntingdon Life Sciences Ltd., Alconbury, Cambridgeshire, UK, 2Pacific Beach BioSciences, San Diego, California, USA, 3Dong-Wha Pharm Ind. Co. Ltd., Anyang, Korea
The pharmacokinetics, tissue distribution, metabolism, and excretion of zabofloxacin (1-cyclopropyl-6-fluoro-7-[8-(methoxyimino)-2,6-diazaspiro[3,4]oct-6-yl]-4-oxo-1,4-dihydro[1,8]naphthyridine-3-carboxylic acid hydrochloride, also known as DW-224a and PB-101, a new broad-spectrum fluoroquinolone-type antibiotic with enhanced in vitro activity against Streptococcus pneumoniae, including strains resistant to other antibiotics, was investigated in nonfasted male rats after oral administration of [14C]zabofloxacin hydrochloride (100 mg salt/kg) as a solution in purified water. In intact nonfasted SD rats, the total recovery of radiolabeled dose after 7 days was 93.2% and most of the dose (90.2%) was eliminated in feces, with about 2.9% of the dose excreted in urine. In bile duct–cannulated fasted SD rats, the highest proportion of the dose was also recovered in the feces (72.9%), with 7.0 and 5.8% of the dose excreted in urine and bile, respectively. The total circulating drug-related material (represented by 14C-radioactivity) increased to a Cmax of 0.91-μg equivalents of free base/mL after 1 hour. Tissue concentrations of drug-related material were generally maximal between 0.5 and 1 hour postdose, with the highest concentrations seen in the small intestine, liver, and stomach wall. Tissue concentrations declined, and at 24 hours, all tissues were below the level of quantitation, except for the skin, wall of the large intestine, and rectum. In the nonfasted pigmented (Lister-Hooded) rats, tissue concentrations were highest in the eyes, pigmented skin, and liver; after 24 hours, radioactivity in the eyes persisted up to, and including, 112 days postdose, indicating the binding of drug-related material to ocular melanin. Oral absorption of [14C]zabofloxacin hydrochloride was poor in nonfasted (<15%), compared to fasted, rats (30%).
63. Simulation of Cilostazol Absorption and Pharmacokinetics
Viera Lukacova, Michael B. Bolger, John R. Crison, and Walter S
Woltosz Simulations Plus, Inc., Lancaster, California, USA
Cilostazol absorption and pharmacokinetics were simulated by using GastroPlus™ 6.0 (Simulations Plus, Inc., Lancaster, California, USA). The program’s Advanced Compartmental and Transit (ACAT) model described the absorption of the drug, while pharmacokinetics was simulated with a physiologicall -based pharmacokinetics (PBPK) model. Human organ weights, volumes, and blood-perfusion rates were generated by the program’s internal Population Estimates for Age-Related (PEAR) Physiology™. Tissue/plasma partition coefficients were calculated by using a modified Rodgers (2007) algorithm from in vitro and in silico physicochemical properties (ADMET Predictor™; Simulations Plus). The metabolic clearance of cilostazol in the gut and liver was estimated from in vitro enzyme kinetic constants for CYP3A4, 3A5, 2C8, and 2C19 (Hiratsuka, 2007) combined with built-in in vitro values for the distribution of 3A4 in the gut (Paine, 1997) and the average expressions of all four enzymes in the liver (Inoue, 2006). The resultant model accurately reproduced in vivo plasma concentration-time profiles in humans for solid oral doses ranging from 25 to 300 mg. Simulations with doses from 10 to 1,000 mg showed a nonlinear dose dependency of bioavailability, with the maximum at ~50 mg/dose. For lower doses, the predicted fraction absorbed (Fa) was nearly 100% with predicted bioavailability affected mainly by saturable first-pass extraction (FPE). The predicted bioavailability increased by ~5% from 10 to 50 mg. For higher doses, limited solubility caused a decrease in Fa with increasing dose. This decrease in Fa was more significant than the further decrease in FPE, resulting in ~50% decrease in the predicted bioavailability from 50 to 1,000 mg. This study demonstrates that in the absence of intravenous data, mechanistic simulations of oral doses can help to estimate fraction-absorbed and first-pass extraction in the gut and liver, and to provide information about processes affecting bioavailability, which can be used in estimates of drug-drug interactions and/or formulation design.
64. Prediction of In Vivo Clearance from In Vitro Data: Protein Binding as a Source of Underprediction
Inge A.M. de Graaf, Marina J.H. de Jager, Johannes H. Proost, and Geny M.M.
Groothuis Pharmacokinetics, Toxicology and Targeting, Groningen Research Institute of Pharmacy, University of Groningen, Groningen, The Netherlands
In vitro models predicting the metabolic clearance of new chemical entities have contributed greatly to the selection of lead compounds with favorable pharmacokinetic properties in humans. However, in vitro–in vivo predictions are still not always accurate. An important determinant of the outcome of predictions and, therefore, a potential source of error, is the in vivo free fraction (fu) of a compound in plasma required to extrapolate the intrinsic clearance (obtained in vitro under protein-free conditions) to clearance in vivo. To investigate the relation between metabolic clearance and fu, we determined the metabolic rate of testosterone and methyltestosterone (at substrate concentrations << Km) in precision-cut liver slices in medium with increasing concentrations of plasma (0–100%) and albumin (0–4%) and measured fu by ultracentrifugation in medium supplemented with the same concentrations of plasma or albumin. In theory, at concentrations well below Km, the metabolic rate is proportional to the free concentration of a drug at the enzyme site. At low plasma/albumin concentrations, the metabolic rate was, indeed, found to be proportional to the extracellular free concentration. However, at higher plasma and albumin concentrations, the metabolic rate was higher than predicted from the free concentration. From the difference between the metabolic rate in slices incubated under protein-free conditions and in 100% plasma or 4% w/v albumin, an apparent free fraction of 0.19 and 0.24, respectively, for testosterone and 0.13 and 0.23, respectively, for methyltestosterone was calculated, whereas the measured free fractions for these compounds were 0.02 in 100% plasma and 0.09 in 4% w/v albumin. In conclusion, for both compounds used in this study, the intracellular free drug concentration at the enzyme site does not seem to correlate with the extracellular free fraction at physiological protein concentration. As a consequence, using the fu measured in plasma to calculate in vivo clearance from in vitro data, measured in protein-free medium, is an important source of underprediction of in vivo clearance.
65. How Good Is Prediction of In Vivo Metabolic Clearance from Established In Vitro Systems?
Joanne A. Foster, David Hallifax, and Brian J.
Houston School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester, UK
Statement of Hypothesis: Our aim was to conduct a systematic review to provide a large set of reference compounds with diverse physicochemical and pharmacokinetic properties for probing novel in vitro systems focusing on CLint in both HLMs and cryopreserved hepatocytes.
Methods: Data were sourced from research laboratories using cryopreserved hepatocytes (1, 2, 3, and 4) and HLMs, respectively (4 and 5).Other criteria included clearance values corrected for drug binding (6). For the majority of compounds, plasma clearance data were compiled from intravenous studies taken from Obach et al. (2008).
Supporting Data: The use of human in vitro systems as a tool in the prediction of drug metabolism is well established. However, with ever increasing need for less labor-intensive protocols and improved precision outcome, there is a driving force to strengthen well-established in vitro systems.
Results: We collated CLint values for 101 and 70 compounds in the hepatocyte and HLM systems. We found a general underprediction, with an average 29-fold in hepatocytes and a 12-fold in HLMs. Subsequent analysis recovered 50 compounds that were common in both in vitro systems. The data set was separated into two groups: high and low clearance. We found a 6-fold underprediction of in vivo CLint for HLMs and a 3-fold underprediction for hepatocytes and a 17-fold underprediction of in vivo CLint for HLMs and a 60-fold underprediction for hepatocytes for low and high CL, respectively.
Conclusion: This is, to our knowledge, the largest set of clearance values for compounds in both hepatocytes and HLMs. From this data set, we found there to be consistent underpredictions in both hepatocyte and HLM systems. However, although this data set is large, it may be compromised by intervariability between donors and laboratories. Future work will address this, involving substrate depletion analysis on a number of compounds, using the same donor in both in vitro systems. Judicious selection of compounds will provide appropriate calibrators to reduce imprecision and account for bias.
66. Involvement of Drug Uptake in Hepatic Clearance
Emilie Jigorel and J. Brian Houston
School of Pharmacy and Pharmaceutical Sciences, Center for Applied Pharmacokinetic Research (CAPkR), Manchester, UK
Evidence for the involvement of drug transporters in the hepatic clearance of compounds is increasing, and significant underpredictions of clearance may arise due to hepatic uptake when conventional drug-depletion protocols are employed. Therefore, in vitro models that include active transport are required to predict accurately and comprehensively hepatic clearance. Soars et al. explored a simple adaption of the conventional drug-depletion method in hepatocytes to predict hepatic clearance mediated by metabolism and/or active uptake. The loss of parent compound was measured in the media only, rather than in the overall hepatocyte incubation (media and cells). In this context, the aim of the present study was to compare intrinsic clearances (CLint) obtained with the media loss assay and the conventional assay by using a set of model drugs in freshly isolated rat hepatocytes in suspension. Comparison of the conventional assay and the media loss assay has led to the identification of two characteristic profiles. One profile was characterized by a higher CLint value obtained with the media loss assay than the conventional assay. For example, atorvastatin exhibited a 2-fold higher rate when using the media loss assay (28 ± 7.32 and 59.03 ± 14.72 μL/min/106 cells when using the conventional and the media loss assay, respectively). A biphasic depletion time profile was evident, which can be explained by the rapid uptake and slower metabolism of atorvastatin in hepatocytes. In the second profile, similar CLint values were obtained with the two methods and this may be explained by either a lack of active transport or transporter rate limitation in hepatic clearance. For example, erythromycin CLint was 7.38 ± 3.63 and 8.75 ± 4.93 μL/min/106 cells with using the conventional and the media loss assay, respectively. To conclude, differences in CLint predictions, depending on the method used, were observed, with some compounds confirming the role that uptake transporters can play in hepatic clearance. In this case, the media loss approach would provide the most accurate CLint value, which reflects hepatic uptake and/or metabolism.
67. Prediction of Intestinal and Hepatic First-Pass Effects in Humans using Data of Caco-2 Cell Permeabilty and Oral Clearance
Katsumi Iga, Akiko Kiriyama, and Akino Honbo
Pharmaceutical Sciences, Doshisha Women’s College of Liberal Arts, Kyotanabe, Japan
Cyp3A4 substrates eliminating from the liver after intravenous (i.v.) administration have the potential to be subject to intestinal and hepatic first-pass elimination after oral administration. Separate estimation of the intestinal bioavailability (Fg) and hepatic bioavailability (Fh) in the oral administration requires data of i.v. administration as well as of oral administration. In the present study, a new method, which employs data of Caco-2 cell permeability in place of total body clearance (i.v. data), was derived. In the theory, the following points were assumed: 1) Fg is determined by the ratio of intestinal intrinsic clearance (CLint,g) to the sum of intestinal permeability clearance (CLperm) and CLint,g; 2) CLperm is proportional to the permeability of Caco-2 cells (Papp,caco-2); and 3) CLint,g is proportional to the hepatic intrinsic clearance (CLint,h). Typical CYP3A4 substrates, nifedipine, midazolam, cyclosporin, tacrolimus, and saquinavir, which are known to exhibit the intestinal first-pass effects, were chosen to test the method. The values of Fg for nifedipine, midazolam, cyclosporin, tacrolimus, and saquinavir calculated by the present method, assuming Fa = 1, were 0.59, 0.51, 0.45, and 0.19, respectively. The values of Fh and F (absolute bioavailability = Fa × Fg × Fh) as well as Fg were in good agreement with those calculated by the conventional method. These findings suggest that the assumptions made in the present method were valid, and that the method would help us to understand intestinal extraction in relation to intestinal permeability and metabolism, and to analyze drug-drug interactions involving intestinal transport and metabolism.
68. Characterization of N-Acetyl Transferase Activity in Reconstructed Human Skin Models
Joan Eilstein, Guillaume Lereaux, Edwige Daronnat, Sanaa Dichich, Jean Roch Meunier, Jacques Leclaire, and Daniel Duche
Life Sciences Department, L’Oréal Research, Clichy, France
Skin is mainly considered as a physical barrier to its environment. However, it contains numerous metabolizing enzymes that give to it a potential role in terms of metabolism and detoxification. Cosmetics take care of skin aspect, integrity, and aging. The 7th European amendment to the cosmetic directive forbids the use of animal testing to evaluate the efficacy and safety of new cosmetics. This policy has forced the cosmetic industry to develop reconstructed human skin models as a tool for alternative methods to animal experimentation. For these reasons, the models have to be characterized and compared with a normal human skin (NHS) in terms of metabolic capabilities. In the present study, we characterized the N-acetyl transferase (NAT) activity particularly involved in aromatic amines detoxification at the skin level. Previous studies showed that NHS and reconstructed human tissues, Episkin™, Realskin, and SkinEthic (RHE), expressed NAT1 and NAT5 isoforms. Only NAT1 seems involved in xenobiotic detoxification processes. Thus, NAT1 activity of these models and NHS were quantified by using p-aminobenzoïc acid (PABA) as a substrate. Apparent Vmax, Km, and clearance were measured for each tissue. Results showed that even if the Vmax and Km between tissues were different, the clearance was equivalent. Besides, the NAT activity of NHS was detected with a high variability between samples. The NAT1 polymorphism could explain this result. This variability was not observed with skin models, which are reconstructed from a keratinocyte pool of several donors. In conclusion, these findings confirm that NAT activity is present and effective in skin and models.
69. Characterization of the UDP Glycosyltransferase 3A Family
Peter Ian Mackenzie, Robyn Meech, and John Miners
Department of Clinical Pharmacology, Flinders University School of Medecine, Adelaide, Australia
The UDP glycosyltransferases (UGTs), which are found mostly in the liver, kidney, and gastrointestinal tract, are important modulators of drug efficacy, and are our main defense against the myriad of chemicals that we are exposed to in our environment. They are also essential for the removal of lipophilic endogenous compounds. In humans, there are four UGT families: UGT1, UGT2, UGT3, and UGT8. We and others have shown that the 19 UGTs of the UGT1 and UGT2 families are drug-metabolizing enzymes that use UDP glucuronic acid to glucuronidate a myriad of lipophilic compounds. In contrast, the single member of the UGT8 family does not appear to be a drug-metabolizing enzyme, as the only activity attributed to this UGT is the transfer of galactose from UDP galactose to ceramide, an essential step in the synthesis of glycolipids. The UGT3 family contains two members, UGT3A1 and UGT3A2, which are 78% identical in sequence. We have recently shown that UGT3A1 is present in the liver and kidney and is an N-acetylglucosaminyltransferase. It uses UDP N-acetylglucosamine, instead of UDP glucuronic acid and other UDP sugars, to glycosidate ursodeoxycholic acid, 17α-estradiol, 17β-estradiol, and, to a lesser extent, the prototypic substrates of the UGT1 and UGT2 forms, 4-nitrophenol and 1-naphthol (Mackenzie et al., 2008). We have now characterized the substrate specificy of UGT3A1 further and shown that several substrates of the UGT1 and UGT2 families are also substrates of UGT3A1. These include etiocholanolone, borneol, and 7-hydroxycoumarin and the bioflavones, genistein, diadzein, and biochanin A. The relative importance of glucuronidation verses N-acetylglucosaminidation in the metabolism of these compounds will be discussed.
Mackenzie PI, Rogers A, Treloar J, Jorgensen BR, Miners JO, Meech R. (2008). Identification of UDP glycosyltransferase 3A1 as a UDP N-acetylglucosaminyl-transferase. J Biol Chem 283:36205–36210.
70. GSTpi Mediates MPTP-induced JNK Activation in the Nigrostriatal Pathway
Maria J. Gama,1 Margarida Castro-Caldas,2 Andreia Neves Carvalho,1 Elsa Rodrigues,1 Colin J. Henderson,3 and C. Roland Wolf4
1iMED.UL, Faculty of Pharmacy, University of Lisbon, Lisbon, Portugal, 2Dcv, Faculty of Sciences and Technology, New University of Lisbon, Monte de Caparica, Portugal, 3Biomedical Research Institute, Cancer Research UK Molecular Pharmacology Unit, Dundee, UK, 4Cancer Research UK Molecular Pharmacology Unit, Dundee, UK
The multifactorial etiology of idiopathic Parkinson’s disease (PD) is known to involve aging, genetic predisposition, and environmental aggressions. MPTP (1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine) is a neurotoxin that induces the selective loss of SNpc dopaminergic neurons, characteristic of PD, by a cascade of deleterious events, which involve the activation of the c-Jun N-terminal kinase (JNK). GSTP1 has been shown to block JNK by direct protein-protein interaction, preventing the subsequent trigger of the apoptotic cascade by these stress-activated kinases. In this study, we investigate the putative neuroprotective role of Gstpi in both C57BL/6 GstP1/P2(-/-) and wild-type mouse midbrain and striatum, upon single-dose MPTP administration. Tyrosine hydroxylase levels were assessed by immunohistochemistry as a measure of dopaminergic cell death. Gstpi expression was evaluated by both immunohistochemistry and Western blot assays. Identification of Gstpi-positive cells, in cryostat coronal mice brain sections, was assessed by double immunohistochemical staining techniques. Relative concentrations of pJNK/JNK and apoptotic proteins were estimated by Western blot, and JNK activities were measured by a commercial nonradioactive kit. Direct interaction of Gstpi and JNK was determined by coimmunoprecipitation assays. Our results show that Gstpi is actively and mostly expressed in glial cells in both C57BL/6 mouse midbrain and striatum. The observed neurotoxic response to a single dose of MPTP involves a decrease in the concentration of the antiapoptotic protein, Bcl-2, and an associated transient increase of JNK activity in the midbrain and in the striatum. The transient increase of the Gstpi relative concentration observed in the midbrain and in the striatum is suggestive of a defensive mechanism to be activated upon MPTP aggression. Moreover, our results show that MPTP-induced dopaminergic neuronal death is an early event, when comparing Gstpi null versus wild-type mice. Our findings are consistent with the hypothesis that Gstpi may be a major regulator of JNK activity and serve as an important protective factor of dopaminergic neurons in vivo. (Supported by FCT/MCTES–Portugal and FEDER: project POCI/SAU-FCF/58171/2004)
71. In Vitro and In Vivo Glucuronidation of Midazolam in Humans
Ruth Hyland,1 Toby Osborne,2 Anthony Payne,3 Sarah Kempshall,1 Y. Raj Logan,1 Khaled Ezzeddine,4 and Barry Jones1
1Pharmacokinetics, Dynamics and Metabolism, Pfizer Global R&D, Sandwich Kent, UK,2Department of Biochemistry, University of Sussex, Brighton, UK, 3Pharmaceutical Sciences, Pfizer Global R&D, Sandwich Kent, UK, 4Department of Chemistry, Loughborough University, Loughborough, UK
Midazolam is a benzodiazepine used as a probe for CYP3A4 in clinical and in vitro studies. A glucuronide metabolite of midazolam has previously been identified in vitro in human liver microsome incubations. The primary aim of this study was to understand the in vivo relevance of the glucuronidation pathway in man. To achieve this, an authentic standard of N-glucuronide was generated from microsomal incubations and isolated by using solid-phase extraction. NMR data confirmed the conjugation of the midazolam N-glucuronide standard to be on the -nitrogen of the imidazole ring. Enzyme kinetic behavior of the pathway was investigated in human liver microsomes and recombinant UGT enzymes. In vitro incubations confirmed UGT1A4 as the specific UGT enzyme of interest. The pathway exhibited atypical kinetics, and a substrate-inhibitory cooperative binding model was applied to determine Km (46 and 64 μM), Vmax (445 and 427 pmol/min/mg), and Ki.(58 and 79 μM) in HLM and rUGT1A4, respectively. Additionally, preliminary experiments were performed in human liver microsomes and rUGT enzymes, using1’-OH midazolam and 4-OH-midazolam as substrates, to investigate both O- and N-glucuronidation. In vivo, the direct glucuronide metabolite of midazolam was quantified in human urine samples from individuals who had received oral and intravenous midazolam. Midazolam glucuronide in the urine accounted for 1–2% of the administered dose, demonstrating that direct N-glucuronidation of midazolam does occur in vivo. Through pharmacokinetic modeling using Simcyp™, an increased role for UGT1A4-mediated metabolism is predicted under CYP3A-inhibited conditions. In summary, a more complete picture of midazolam metabolism and the enzymes involved has been elucidated.
72. The Expression of Recombinant Human CYP2C9 in Saccharomyces Cerevisiae and the Research of Genetic Polymorphism in Drug-Drug Interaction
Ping-Ping Ma,1 Hui-Juan Wang,1 Juan-Li Zhu,2 and Chao Chen2
1College of Life Science, Northwest University, Xi’an, China, 2National Engineering Research Center for Miniaturized Detection System, Shanxi Lifegen Co. Ltd., Xi’an, China
Objective: The aim of this work was to develop a broadly applicable assay system for studying the mechanism of human CYP2C9 polymorphic enzymes in drug-drug interaction.
Methods: Recombinant clones of human CYP2C9*1 and its mutant, CYP2C9*2, were provided by our lab; then, we expressed them in yeast cells, and the proteins were identified by Western blotting. The system was validated by comparing the Km value (2C9/diclofenac) with the FDA-recommended range. Then, using Vivid® fluorescent probe (BOMCC, Invitrogen, Carlsbad, California, USA) as a substrate to establish a fluorescence-based detecting system to evaluate CYP2C9-drug interaction, five chemical compounds were screened with both 2C9*1 and 2C9*2 enzymes.
Results: The exprsessed proteins were confirmed by Western blotting.Using diclofenac as a substrate, the Km of CYP2C9*1 was 5.34 ± 0.96 μM, which was in the FDA-recommended range, which means the system was credible. When using BOMCC as a fluorescent substrate, the Km and Vmax of CYP2C9*1 and CYP2C9*2 were 20 and 35 μM and 0.26 and 0.12 pmol/min/pmol P450, respectively. As to the tested five drugs, the inhibitory potency toward CYP2C9*1 was sulfaphenazole>ketoconazole>sertraline>tolbutamide>pioglitazone, but the potency toward CYP2C9*2 was sulfaphenazole>tolbutamide>ketoconazole>sertraline>pioglitazone. The data suggested that a genetic polymorphism was one of the factors compounding the drug-drug interaction problems.
Conclusion: The yeast expression system of CYP2C9, and its viarants, was constructed successfully. Besides, the detecting method of drug-drug interaction for CYP2C9 and its genetic polymorphism in vitro was initially established.
73. Hydroxylation of Tanshinone IIa in Human Liver Microsomes Is Specifically Catalyzed by Cytochrome P450 2A6
Hui-Xin Liu, Ying Hu, and Ling Yang
Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China
Salvia miltiorrhiza (Danshen) has been widely used in China for the treatment of coronary heart disease, cerebrovascular disease, bone loss, hepatitis, hepatocirrhosis, and chronic renal failure. Tanshinone IIa (6,7,8,9-tetrahydro-1,6,6-trimethylphenanthro[1,2-b]furan-10,11-dione), one of the major active constituents of S. miltiorrhiza, has been observed to possess various kinds of pharmacological activities, including antioxidant, prevention of angina pectoris and myocardial infarction, anticancer, antibacterial, and antiplatelet aggregation activities. As compared to the extensive research of the pharmacological activities of tanshinone IIa, few studies have dealed with its metabolism and pharmacokinetics. Hydroxylation is an important pathway in the metabolism of tanshinone IIa. However, the metabolites and primary cytochrome P450 (CYP) isozymes responsible for tanshinone IIa hydroxylation remain to be determined in humans. Here, we characterized tanshinone IIa hydroxylation by human liver microsomes (HLMs) and nine recombinant human CYP (rCYP) isozymes to identify what kinds of metabolites are present and which human CYP isozymes are involved. One hydroxyl metabolite was detected in reactions catalyzed by HLMs and rCYP2A6 and was identified as tanshinone IIb by comparing the tandem mass spectra and the chromatographic retention time with that of the standard compound. A kinetic study showed that tanshinone IIa hydroxylation by HLMs and rCYP2A6 followed Michaelis-Menten kinetics. The Km values of HLMs and rCYP2A6 for tanshinone IIa hydroxylation were 5.93 ± 0.44 and 5.22 ± 0.31 ìM, respectively. A CYP2A6 selective inhibitor (8-methoxypsoralen) inhibited tanshinone IIa metabolism almost completely, with no metabolite detectable, but other CYP-selective inhibitors exerted no inhibitory effects. Rates of tanshinone IIa hydroxylation correlated well (r >v0.98, P < 0.01) with rates of hydroxylation of the CYP2A6 probe substrate, coumarin, from 15 individual HLMs. In combination, we demonstrated that tanshinone IIa is metabolized by HLMs to a single metabolite (tanshinone IIb), and that tanshinone IIa hydroxylation is specifically catalyzed by CYP2A6 in HLMs, which makes tanshinone IIa potentially useful as an in vitro probe for CYP2A6 catalytic activities.
74. Memantine Inhibits CYP 2D2 in Rat Liver—Comparison with Inhibitory Influence of Fluoxetine
Ondrej Zendulka, Lucia Zahradnikova, Jan Jurica, and Alexandra Sulcova
Department of Pharmacology, Masaryk University, Brno, Czech Republic
Memantine (1-amino-3,5-dimethyladamantane) is indicated in Alzheimer’s disease with other applications proposed, too. Many psychotropics are substrates for CYP450 2D6 isoenzyme and modification of CYP’s activity can be crucial for their efficacy. Being not a substrate for CYP450, memantine was not supposed to interfere with its activity (Sonkusare et al., 2005). However, according to the literature available, at least one study focused toward the influence of memantine on CYP450 described its inhibitory effect on some of CYPs (Micuda et al., 2004). Thus, combining memantine with some other psychotropics can probably result in interactions on the CYP2D6 level. This study determined the influence of memantine on CYP2D2 activity in the rat and compared it with the effect of the known CYP2D2 inhibitor, fluoxetine. The model of isolated perfused rat liver and O-demethylation of dextrometorphan to asses the CYP2D2 activity was used. The 2D2 isoenzyme was selected as an analog of the human CYP2D6. Prior to the liver isolation and perfusion, drugs were administered intraperitoneally for 10 days as follows: memantine (5 mg/kg/day) and fluoxetine (5 and 20 mg/kg/day). Concentrations of the CYP2D2-specific metabolite, dextrorphan, in perfusion medium were significantly lower in memantine-treated animals than in controls. As was hypothesized, the combined treatment with memantine+fluoxetine showed a significantly higher inhibitory effect on CYP2D2 activity than memantine alone. The levels of dextrorphan were decreased by the combination of memantine+fluoxetine up to 68% in the 30th minute of perfusion, while memantine caused depression only up to 50%, when compared to control animals. The further, more-detailed discussion on the comparison between the inhibitory effects of memantine and fluoxetine on the activity of CYP2D2 will be provided. (The work was supported by the Czech Ministry of Education Project: MSM0021622404.)
References
- Micuda S, et al. (2004). Inhibitory effects of memantine on human cytochrome P450 activities: prediction of in vivo drug interactions. Eur J Clin Pharmacol 60:583–589.
- Sonkusare SK, et al. (2005). Dementia of Alzheimer’s disease and other neurodegenerative disorders-memantine, a new hope. Pharmacol Res 51:1–17.
75 High CYP3A Expression in the Human Hepatoma Cell Line Huh7 during Differentiation
Louise Sivertsson, Monica Ek, Magnus Ingelman-Sundberg, and Etienne P.A. Neve
Department of Physiology and Pharmacology, Section of Pharmacogenetics, Karolinska Institutet, Stockholm, Sweden
Drug-induced hepatotoxicity is one of the most important single causes for disapproval, limitations on use, or even withdrawal of drugs. Today, human liver-derived cell lines are frequently used in drug research. However, the use of these cell lines is limited, as they contain very low levels of drug-metabolizing enzymes, as compared to primary hepatocytes. Previously, we have shown that P450 expression is increased in human B16A2 cells during differentiation, and we hypothesized that the human hepatoma cell line, Huh7, when grown confluent, would differentiate into a more hepatocyte-like phenotype. Indeed, we show that growing Huh7 cells confluent up to 5 weeks generates a more metabolically competent cell line. In confluent Huh7 cells, the mRNA levels of several CYP450 enzymes, in particular CYP3A4 and CYP3A5, PXR and the liver-specific protein albumin were increased, as measured by quantitative real-time PCR. Western blot analysis revealed increased amounts of CYP3A4, NADPH CYP450 reductase and albumin with increasing time of confluence. In addition, using a luminescent activity method, we observed a time-dependent increase in catalytic activity for CYP3A4. After 4 weeks of confluence, the CYP3A4 activity level was 7-fold higher, as compared to control cells, an activity that could effectively be inhibited by adding 5 μM of ketokonazol. Treatment of Huh7 cells with 1% dimethylsulfoxide (DMSO) for 4 weeks did not further enhance the CYP3A4, NADPH CYP450 reductase, and albumin protein levels and CYP3A4 activity, compared to cells grown confluent for the same time without DMSO. Incubation of cells grown confluent for 4 weeks in the presence of the hepatotoxin, aflatoxin B1, a well-known substrate for CYP3A4, resulted in significantly higher cytotoxicity, as compared to control cells. Our results indicate that Huh7 cells grown confluent drastically increase the expression of catalytically active CYP3A4 as well as other drug-metabolizing enzymes. We propose that this hepatocyte cell model could be suitable as a model to study drug metabolism and toxicity.
76. Determination from a Large Cohort of Heart Failure Patients of mRNA Levels of Cardiac CYP450s Involved in the Metabolism of Drugs
Véronique Michaud, Martin Frappier, Francois Bélanger, and Jacques Turgeon
Faculty of Pharmacy, Université de Montréal, Montreal, Quebec, Canada
Background/Aims: Extrahepatic tissues, including the heart, express CYP450 isozymes. Previous studies, using a limited number of human heart samples, have indicated that CYP450 genes could be detected in cardiac tissues. The aims of our study were 1) to determine the relative levels of CYP450 mRNAs (CYP1A1, CYP2B6, CYP2C8, CYP2C9, CYP2E1, CYP2J2, and CYP4A11) in explanted hearts from patients with end-stage heart failure, 2) to compare CYP450 mRNA levels in these hearts to that measured in hearts obtained from healthy donors, and 3) to compare the relative levels of CYP450 mRNAs between left and right ventricular tissues.
Methods: Frozen tissues were obtained from human heart ventricles (79 hearts from patients with heart failure and 6 hearts from healthy organ donors ). Total mRNA was extracted and quantitative RT-PCR was performed, using primers and probe sets from Applied Biosystems for CYP1A1, 2B6, 2C8, 2C9, 2E1, 2J2, 4A11, and GAPDH (Applied Biosystems, Carlsbad, California, USA). Relative mRNA expression of CYP450s was determined by using the ΔCT and the ΔΔCT methods.
Results: We have confirmed that CYP2J2 mRNA is clearly the most abundant of all CYP450 isozymes found (62 to ~3 million times higher levels) in the left and right ventricles of human hearts; we have obtained data suggesting that CYP2J2, CYP1A1, CYP2E1, and CYP2C8 mRNA levels are diminished (3 to 10 times) in samples from human failing hearts, compared to normal hearts; we have demonstrated that CYP450 mRNAs are present at similar levels in both the left and right ventricles, with the exception of CYP2E1 (P = 0.04).
Conclusion: In conclusion, we performed a thorough investigation of CYP450 mRNA expression, using the largest cohort of human hearts ever analyzed. Our results indicate that various CYP450 isozymes may contribute to the local metabolism of drugs and control their intracellular levels in cardiac myocytes. Our results also suggest that the production of CYP450-mediated compounds is modulated in the heart of patients with heart diseases.
77. A Novel Analytical Method for the Resolution of Hydroxylated Metabolites of Testosterone
Daniela Gentile,1 Campbell Barkby,2 Andrew Simpson,1 and Stephen Madden1
1In Vitro Sciences, Charles River, Edinburgh, UK, 2Bioanalytical Chemistry, Charles River, Edinburgh, UK
6ß-hydroxylation of testosterone is a widely used marker of CYP3A4 activity in in vitro drug interaction studies. Bioanalysis is complicated by the fact that testosterone can undergo hydroxylation at multiple sites, and thus traditional bioanalytical methods have employed complicated solvent gradients and long run times to facilitate the resolution of these metabolites. Since the advent of UPLC, a number of methods have been published with significantly reduced run times, while maintaining resolution. We have developed a method, using conventional HPLC, that provides the resolution of 6ß-hydroxytestosterone and other hydroxylated metabolites in under 10 minutes. The method utilizes a narrow bore column (HALO C18 2.1 × 75 mm, 2.7 μm) and a simple gradient of formic acid and methanol. d7-6ß-hydroxytestosterone is used as an internal standard. The assay is linear in the range of 16–2,500 pmol 6ß-hydroxytestosterone. The method was succesfully used to characterize CYP3A4-mediated testosterone 6ß-hydroxylase activity in pooled human liver microsomes with a Vmax and Km of 1,970 pmol/min/mg and 85 μM, repectively. Common solvents were shown to have differential effects on enzyme activity. DMSO caused a 50% decrease in activity at 1% (v/v), methanol and ethanol decreased activity by only 10% at the same concentration, while acetonitrile caused a marginal activation. Significant inhibition of activity was observed with quercetin (60% at 10 μM), methoxypsoralen (68% at 6 μM), and ketoconazole (>90% at 0.5 μM). The IC50 for ketoconazole was 0.04 μM. This method is now being routinely utilized in in vitro inhibition (DDI) studies and its application to induction studies (human hepatocytes) is being investigated.
Figure 1. 4-iodo-2-fluoro-aniline and reactive intermediate (3-fluoro-4-iminocyclohexa-2,5-dienylidene)iodonium.
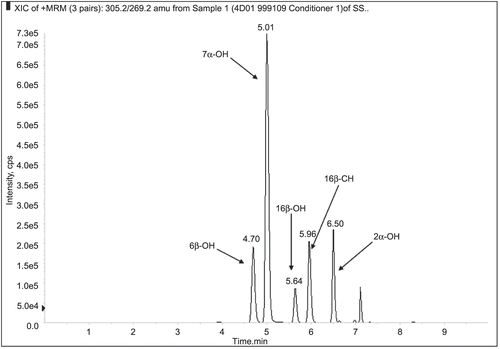
78. Codex™ MicroCyp Plate for Effective and Scalable Drug Metabolite Production
Erik J. de Vries, and Martin P. Mayhew
Metabolites Services, Codexis, Inc, Redwood City, California, USA
Conventional methods for P450 drug metabolite identification and production make use of a variety of commercially available biocatalysts, including microbial libraries, human and animal microsomes, and recombinant P450s. Many challenges to using P450s for milligram-scale drug metabolite production (e.g., poor stability, troublesome reconstitution, and high cost) have been overcome with Codexis’ Human Cytochrome Biocatalysts. Recent guidance published by the FDA strongly recommends all significant metabolites be characterized for safety earlier in the drug discovery process. Because larger amounts of drug metabolites are needed for toxicology and bioactivity tests, even more efficient P450 biocatalysts are needed. The Codex™ MicroCyp Plate contains 90 engineered P450 variants of the CYP102A1 enzyme from Bacillus megaterium, evolved for activity toward drug compounds. The enhanced activity and stability of these bacterial P450 variants along with lower biocatalyst costs allows for cost-effective metabolite production at the 100 mg to gram scale. This higher productivity make s the MicroCyps a viable biocatalyst system for lead compound diversification. Production of metabolites from several human P450 drug substrates from screening to scale-up are compared to reactions by using Codexis’ Human Cytochrome Biocatalysts. We will demonstrate the gram-scale conversion of diclofenac as a benchmark example. In addition, case studies of metabolite production from novel drug candidates, using the MicroCyps, are presented.
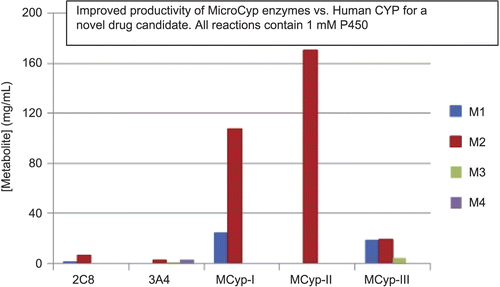
79. Comparison of P450-Glo™ Substrates with Testosterone in Determining Induction of CYP3A4 in Fresh and Cryopreserved Human Hepatocytes
Timothy A. Moeller,1 Mary Sobol,2 Dongping Ma,2 Ji-Young Lee,1 and Scott Heyward1
1Celsis In Vitro Technologies, Baltimore, Maryland, USA, 2Department of R&D, Promega Corp., Madison, Wisconsin, USA
Induction of P450 enzymes is an important determinant for a potential drug-drug interaction of new chemical entities (NCEs). They are critical data for informed drug development and the decision of lead compound selection. Subsequently, induction data are being generated in early drug discovery as a screening tool versus for solely IND submission. This necessitates a need for simple diagnostic tools to determine the induction potential of cytochrome P450s (CYPs) in a relevant biological system. The gold standard for determining the induction of CYPs is the use of hepatocytes, fresh or cryopreserved, with a positive control inducer and by measuring activity with specific substrates. For induction associated with PXR nuclear receptor, CYP3A4 activity is the preferred model with the use of testosterone or midazolam as a substrate and LC/MS quantitation of the metabolite formation. An alternative to LC/MS methods is to use fluorescent or luminescent substrates on a plate reader, reducing time and cost. P450-Glo™ substrates utilize the luciferin-luciferase reaction for the production of luminescence, and through new generations, have provided specific, more active substrates for CYPs. We have tested two P450-Glo™ CYP3A4 substrates, luciferin-6’-pentafluorobenzyl ether (Luciferin-PFBE), and the newest generation, luciferin-isopropyl acetal (Luciferin-IPA), along with the nonspecific luciferin-methyl 2-(6-methoxybenzo[d]thiazol-2-yl)-4,5-dihydrothiazole-4-carboxylate (Luciferin-MultiCYP) to measure the induction of CYP3A4 with rifampin in freshly isolated and plateable cryopreserved human hepatocytes, and compared them to testosterone metabolism. The results demonstrate that Luciferin-MultiCYP was not sufficiently sensitive to discern CYP3A4 induction due to the fact that the majority of the activity is from CYP1A2. Luciferin-PFBE did measure induction but was below the activity and induction measured by Luciferin-IPA. Luciferin-IPA provided the greatest amount of activity and was most aligned to the induction measured by testosterone. With improved specificity and sensitivity for CYP3A4, P450-Glo™ Luciferin-IPA offers a reduction in traditional analysis time, expense, and expertise, while maintaining relevant data for the measurement of induction in human hepatocytes.
80. CYP21 Catalyzed Production of 18-nor-17β-hydroxymethyl,17α-methylandrosta-1,4,13-trien-3-one: A New Player in the Fight against Doping
Andy Zöllner,1 Maria Kristina Parr,2 Calin-Aurel Dragan,1 Frank Peters,3 Hans Maurer,3 Wilhelm Schänzer,2 and Matthias Bureik1
1PomBioTech GmbH, Saarbrücken, Germany, 2Institute of Biochemistry, German Sport University, Köln, Germany, 3Department of Experimental and Clinical Toxicology, Saarland University, Homburg, Germany
Anabolic-androgenic steroids are some of the most frequently detected drugs in amateur and professional sports. Doping control laboratories are constantly developing new assays and methods to detect these steroids as well as their metabolites. Recently, a previously unknown metabolite of metandienone has been discovered (Schänzer et al., 2006). This metabolite, 18-nor-17β-hydroxymethyl,17α-methylandrosta-1,4,13-trien-3-one, could be detected in the urine of test persons via GC-MS up to 19 days after the administration of 5 mg of metandienone. This leads to a significantly increased detection period, compared to methods previously applied, for the determination of metandienone abuse. However, so far, it has not been possible to obtain a purified sample of this metabolite, for example, by chemical synthesis, and to confirm its structure via NMR. We have succeeded in producing this metabolite in a whole-cell biotransformation assay by using the fission yeast, Schizosaccharomyces pombe, as the host. Our strategy involved the use of recombinant fission yeast strains that express different hepatic and steroidogenic cytochrome P450 enzymes, such as CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A4, CYP4Z1, CYP11A1, CYP11B1, CYP11B2, CYP17, and CYP21. The CYP profiling study revealed that 18-nor-17,17-dimethylandrosta-1,4,13-trien-3-one, chemically derived from metandienone, is converted to the desired urinary metabolite by CYP21. Human CYP21 is a microsomal steroidogenic cytochrome P450 enzyme that converts progesterone to 11-deoxycorticosterone, as well as 17α-hydroxyprogesterone to 11-deoxycortisol, in the body. The current goal is to produce this metabolite in sufficiently high quantities for use as reference material for the unequivocal identification of metandienone abuse even after almost 3 weeks. The production of this metabolite, using our whole-cell biotransformation system, will be a valuable contribution to the fight against doping in sport.
Schänzer W, et al. (2006). Mass spectrometric identification and characterization of a new long-term metabolite of metandienone in human urine. Rapid Commun Mass Spectrom 20:2252–2258.
81. Cytochrome P450 Induction in HepaRG Cells Cultured in a Dynamic 3D Bioreactor
Malin Darnell,1 Thomas Schreiter,2 Therese Söderdahl,2 Ingrid Rossberg,2 Katrin Zeilinger,2 Jörg C. Gerlach,3 and Tommy B. Andersson1
1AstraZeneca R&D Mölndal, Clinical Pharmacology and DMPK, Mölndal, Sweden, 2Division of Experimental Surgery, BCRT, Charité Campus Virchow-Klinikum, Universitätsmedizin Berlin, Berlin, Germany, 3McGowan Institute for Regenerative Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA
Maintaining specific liver functions in cellular systems in vitro (e.g., drug-metabolizing enzymes) is still a major challenge. The currently available cell lines contain very low or no levels of the enzyme systems of interest. Isolated primary human hepatocytes have been used with reasonable results, but the stability of enzymes in culture is a problem, and the limited access to human tissue restricts their utilization. The recently developed cell line, HepaRG, exhibits promising features expressing drug-metabolizing enzymes and transporter proteins that resemble those found in primary human hepatocytes (Aninat, 2006; LeVee, 2006, Kanebratt and Andersson, 2008). In this study, cytochrome P450–dependent drug metabolism in HepaRG cells cultured in a three-dimensional (3D) culture system was investigated. To mimic the in vivo situation of cells in the human liver, the 3D model system consists of a multicompartment capillary membrane bioreactor for high-density cell culture. The HepaRG cells were differentiated into a hepatocyte-like morphology, either in a 2D culture before bioreactor inoculation or in the bioreactor, by adding 2% dimethyl sulfoxide (DMSO) to the culture medium. Cells were allowed to adapt to the 3D environment and to reach a stable functional level before starting experiments. CYP1A1/1A2, CYP2B6, CYP3A4, and CYP2C9 activities were determined by adding a CYP cocktail, including phenacetin, bupropion, midazolam, and diclofenac, to the circulating medium. Substrate and metabolite concentrations were analyzed at 10 different time points during 24 hours by using LC/MS. The ability to induce and inhibit CYP activities in HepaRG cells cultured in the bioreactor was also evaluated by adding rifampicin or ketokonazole, respectively. HepaRG cells differentiated in the bioreactor showed higher CYP activities, compared to cells differentiated in 2D culture prior to bioreactor inoculation. Phenacetin O-dealkylase (CYP1A1/1A2), bupropion hydroxylase (CYP2B6), and midazolam 1-hydroxylase (CYP3A4) activities were all induced by rifampicin, whereas CYP3A4 inhibition with ketokonazole decreased midazolam 1-hydroxylase activity. The present study shows that 3D cultures of HepaRG cells can be used to study both the induction and inhibition of drug-metabolizing enzymes.
82. Enzymatic Detoxification of Organophosphates to Daphnia magna
Valerii Tonkopii
Department of Pharmacology and Toxicology, Institute of Limnology, Russian Academy Of Science, St. Petersburg, Russia
It is well known that for many types of vertebrates, several enzymes participate in the metabolism of organophosphates (OPs). They include OP hydrolyzing enzymes, such as the CYP-type of mono-oxygenases (MOs), arylesterases (ArEs), and carboxylesterases (CaEs). The participation of these enzymes in the metabolism of OP to Daphnia magna is unknown. This study was conducted to assess the activity of D. magna hydrolyzing enzymes and the inducibility of these esterases by different modulators. The effect of some inducers (phenobarbital, a-naphthoflavone, 3-methylcholantrene, aldrin, dieldrin, and arochlor) and inhibitors (piperonyl butoxide and SKF-525a) on the OP hydrolyzing enzymes activity and toxicity of OP was studied. Two groups of OP were used: those not requiring metabolic activation before expressing their toxicity (dichlorvos, paraoxon, mevinphos, and chlorfenvinphos) and those requiring metabolic activation (parathion, malathion, diazinon, and fenthion). The activity of enzymes was determined after the pretreatment of hydrobionts with modulators in nontoxic concentration during 72 hours. Exposure of daphnids with inducers has been shown to markedly increase the activity of all investigated OP hydrolyzing enzymes. Enzyme inducers increased the acute toxicity of metabolically activated OP and decreased the toxicity of OP not requiring metabolic activation. The decrease in ArE and MOs activities was observed following exposure of SKF-525a and piperonyl butoxide. These inhibitors, in contrast to the inducers, reduced the toxicity of metabolically activated OP. The results suggest that D. magna contains different CYP-related enzymes. These enzyme activities may serve not only as a biomarker of OP exposure, but also as a predictor of toxicant effects.
83. Expression and Functional Analysis of Human Cytochrome 2C19 Polymorphic Alleles
Huijuan Wang,1 Yiwen Gao,2 Ping-Ping Ma,2 Juan-Li Zhu,2 and Chao Chen2
1College of Life Science, Northwest University, XI’an, China, 2Shaanxi Lifegen Co., Ltd., National Engineering Research for Miniaturized Detection Systems, Xi’an, China
Aim: The aim of this work was to construct a Saccharomyces cerevisiae–expressing system and develop an enzymatic detection method for human CYP2C19 polymorphic genes.
Methods: The prototype cDNA of CYP 2C19 was obtained by RT-PCR from human liver tissue and cloned into a pYES2/CT vector for galactose-inducible expression in yeast. The cloned cDNA was subsequently used as a template to introduce polymorphisms by site-directed mutagenesis and were cloned into the same vector. Transformed yeasts produced large quantities of microsome-bound 2C19 enzymes, as determined by Western analysis. The isolated microsomes were used to measure the kinetic constants of 2C19 enzymes in real-time assays, using a fluorogenic substrate CEC (3–cynao-7-ethoxycoumarin). The inhibition of CYP 2C19 prototype enzyme by a known inhibitor drug (tranylcypromine) was tested by the serial titration of drugs in the fluorogenic assays.
Results: Five cDNA of CYP2C19 corresponding to 2C19*1A (prototype) and variant alleles of 2C19*1B (991A>G, I331V), 2C19*6 (395G>A, R132Q), 2C19*9 (431G>A, R144H), and 2C19*13 (1228C>T, R410C) have been constructed for expression in yeast. The results showed that all enzymes possess robust activity, with the exception of 2C19*6; the 2C19*6 enzyme failed to show activity even at the highest enzyme concentration tested (3 mg/mL). The Km value of four catalytically active enzymes showed little difference, which were all close to 20 μmol/L; the Vmax value of the 2C19*1B and 2C19*13 enzymes were very similar to 2C19*1A, but the Vmax value of the 2C19*9 enzyme was 10-fold lower than that of 2C19*1A. Then, the inhibition testing of the 2C19*1A enzyme testified that tranylcypromine had a specific inhibition on CYP2C19.
Conclusion: The prototypical and four variant forms of human cytochrome P450 2C19 have been expressed in a functional form in yeast. The recombinant enzymes show robust activity in real-time fluorogenic assays, and the enzyme activity was specifically inhibited by a known inhibitor of the 2C19 enzyme. This work demonstrates the feasibility of large-scale analysis of CYP2C19 polymorphic enzymes in drug mtebabolism and drug-drug interaction study.
84. Human P450 Reductase Y181D Variant, Molecular Effects of FMN Deficiency
Michel Kranendonk,1 Chris C. Marohnic,2 Karen McCammon,2 Bettie Sue S. Masters,2 and José Rueff1
1Genetics, Universidade Nova de Lisboa, Faculty of Medical Sciences, Lisbon, Portugal 2Biochemistry, University of Texas Health Science Center at San Antonio, San Antonio, Texas, USA
A mutation causing Y181D substitution in P450 reductase (CYPOR) contributes to adrenal hyperplasia (Arlt et al., 2004). The Y181D variant, bacterially expressed, retained no measurable NADPH-cytochrome c reductase (NCR) activity (Arlt et al., 2004) and was unable to support CYP17- or CYP21-catalyzed steroidogenic reactions (Huang et al., 2005). The rat homolog (Y178D) was previously shown to be FMN deficient and amenable to functional rescue by the addition of excess FMN to the NCR assay (Shen et al., 1989). Homozygous Y181D mutations would likely cause a severe Antley-Bixler syndrome phenotype. Bacterially expressed and purified human Y181D was confirmed by HPLC analysis to lack FMN. Incubation with excess FMN failed to reconstitute the protein. Y181D NADPH-ferricyanide reductase (NFR) activity was reduced ~30%, compared to WT. NCR activity of Y181D was too slow to be measured. Titration of Y181D with FMN increased activity in the NCR assay from being undetectable without FMN to a maximum of 117 min−1 (~20% of WT), exhibiting a 2-μM FMN-activation constant (one-site model). Coexpression of CYPOR (WT or Y181D) with CYP1A2, at the physiological stoichiometry of 1 CYPOR per ~10 CYPs in the MK1A2_POR strain of Escherichia coli, demonstrated the compromised capacity of Y181D to support CYP1A2-catalyzed metabolism of the procarcinogens 2AA, IQ, and NNK in a whole-cell bioactivation assay. Membranes of the MK1A2_POR cells confirmed the FMN activation of Y181D NCR activity with a 1.6-μM activation constant. CYP1A2-catalyzed ethoxyresorufin-O-dealkylation (EROD) activity of the MK1A2_PORY181D membranes, undetectable in the absence of added FMN, increased to 37% of MK1A2_PORWT membranes with a 1.2-μM FMN-activation constant. In summary, we have demonstrated FMN deficiency in the purified Y181D variant of human CYPOR, shown functional rescue with excess FMN, and described the effects of the mutation on several CYPOR electron-transfer activities, including support of drug-metabolizing CYP1A2.
References
- Arlt W, et al. (2004). Congenital adrenal hyperplasic caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study. Lancet 363:2128–2135.
- Huang N, et al. (2005). Diversity and function of mutations in p450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis. Am J Hum Genet 76:729–749.
- Shen AL, et al. (1989). Structural analysis of the FNM binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. J Biol Chem 264:7584–7589.
85. Inhibitory Effects Of Three Antineoplastic Drugs On CYP2B6 Allelic Variants
Yiwen Gao,1 Xin Men,1 Jing Guo,1 Juan-Li Zhu,2 and Chao Chen1
1School of Life Science, Northwest University, Xi’an, China, 2National Engineering Research for Miniaturized Detection Systems, Xi’an, China
Allelic variants of CYP enzymes have been confirmed that could cause large variability in drug-metabolizing capacity, which might result in a different inhibitory effect of a drug among patients. So, evaluating the different inhibitory potential of drugs to CYP allelic variants could help to avoid the DDIs.
Aim: The aim of this work was to evaluate the different inhibitory potential of three antineoplastic drugs (ThioTEPA, raloxifene, and tamoxifen) to five CYP2B6 variants (CYP2B6*1, CYP2B6*4, CYP2B6*6, CYP2B6*9, and CYP2B6*13).
Methods: 1. Coexpression of five CYP2B6 variants with human cytochrome P450 reductase in Saccharomyces cerevisiae and preparation of yeast microsome. 2. Validation of the five CYP2B6 variants’ activity by fluorogenic probe was as follows: The activity of CYP2B6 variants was determined by using specific fluorescent substrate (Vivid® BOMCC, Invitrogen, Carlsbad, California, USA), and the Km and Vmax values were measured by fluorescent assay. 3. Determination of the three antineoplastic drugs inhibition was as follows: The inhibition assay was developed by incubating a mixture of enzymes, fluorescent substrate at the concentration equal to its apparent Km, the test drug, and other components necessary to the assay. The test drugs were 2-fold serial diluted from 128 to 0.0625 μM.
Results: 1. ThioTEPA can inhibit CYP2B6 to metabolize Vivid BOMCC with an IC50 of 5.64 μM to CYP2B6*1. The highest IC50 of ThioTEPA to the five variants is 16.6 μM to CYP2B6*6.2. Raloxifene can inhibit CYP2B6 to metabolize Vivid BOMCC with an IC50 of 12.71 μM to CYP2B6*1. The lowest IC50 of raloxifene to the five variants is 5.5 μM to CYP2B6*6.3. The IC50 of tamoxifen to CYP2B6*4,*9,*13 are greater than 128 μM, and the IC50 of tamoxifen to CYP2B6*1 is 83.31 μM.
Conclusion: 1. The recombinant CYP2B6 polymorphic enzymes could be used as tools for the metabolism-based drug-drug interaction screening. 2. The IC50 values of the three antineoplastic drugs to CYP2B6*1 are similar to the previosly reports. 3. There are obviously inhibitory differnces of ThioTEPA, raloxifene to CYP2B6*1 and CYP2B6*6, and the inhibitory potential of tamoxifen is similar to CYP2B6*1,*4,*9,*13. 4. In this work, these findings may show useful information in the prediction of potential metabolism-based drug-drug interactions with consideration of CYP2B6 polymorphic enzymes.
86. TCDD Potentiate CCl4 Hepatotoxicity Effects by Increasing CYP2E1 Hepatic Levels. Role of Aryl Hydrocarbon Receptor
Guillermo Elizondo, Alejandro Mejia-Garcia, Esmeralda Sanchez-Ocampo, Octavio Reyes-Hernandez, and Mineko Shibayama
Cell Biology, CINVESTAV, Mexico D. F., Mexico
CYP2E1 is highly conserved in mammals and is constitutively expressed in various tissues, with the liver having the highest level. It is the principal P-450 responsible for the metabolism of many low-molecular-weight compounds, including alcohols and industrial solvents. In vitro data have indicated that many of these chemicals, some of them toxins and carcinogens, are metabolized by CYP2E1. Previous microarray data suggested that CYP2E1 induction could be under aryl hydrocarbon receptor (AhR) control. In the present study, we evaluated the effect of AhR activation on CYP2E1 expression and the consequences on the hepatotoxicity mediated by CCl4 exposure. To do so, AhR-wild-type and AhR-null mice were treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) or TCDD+CCl4, and CYP2E1 mRNA and protein levels, transaminase activities, and liver histology studies were done. RT-PCR and Western-blot analysis revealed that TCDD treatment increased CYP2E1 mRNA and protein levels in an AhR-dependent manner. TCDD treatment also potentiates the alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities, and hepatic lesions induced by CCl4 exposure. All together, these data suggest that CYP2E1 induction may be under AhR control, and therefore, TCDD treatment increases CCL4 hepatotoxic effects. (This work was supported by CONACYT 48786.)
87. Variation in the Human Metabolism of Chlorpyrifos
Ernest Hodgson, Andrew D. Wallace, and Edward L
Croom Department of Toxicology, North Carolina State University, Raleigh, North Carolina, USA
The bioactivation and detoxification of chlorpyrifos, a widely used organophosphorothioate insecticide, was investigated by using human liver microsomes (HLMs) from 17 individual donors. Bioactivation leads to the formation of the potent acetylcholinesterase-inhibiting metabolite chlorpyrifos-oxon (CPO) and release of reactive sulfur able to inhibit CYP activity. CPO formation varied 57-fold between donors at a substrate concentration of 100 μM of chlorpyrifos and 19-fold at a substrate concentration of 20 μM. CPO formation in phenotyped individual HLM was significantly correlated with CYP2B6-, CYP2C19- and CYP3A4-related activity. CPO formation was best correlated with CYP2B6-related activity at low (20-μM) chlorpyrifos concentrations, which are more relevant to human exposure levels, while CYP3A4-related activity was best correlated with oxon formation at high concentrations (100-μM) of chlorpyrifos. Detoxication of chlorpyrifos to the 3,5,6-trichloro-2-pyridinol (TCP) metabolite was greater than CPO formation for most samples. TCP production varied 19-fold at the 100-μM substrate concentration and 9-fold at 20 μM. TCP production was correlated best with CYP3A4 activity at both 20 and 100 μM of chlorpyrifos. The greater importance of CYP2B6 for the formation of the toxic oxon at low substrate concentrations is, presumably, related to the Km of CYP2B6 (0.81 μM; Foxenberg et al., 2007), as compared to that for CYP3A4 (27 μM; Foxenberg et al., 2007). Calculations of percent total normalized rates identified CYP3A4, CYP2C19, and CYP2B6 as the major isoforms forming TCP and CPO. Use of the inhibitors, ketoconazole and ticlopidine, confirmed the importance of CYP2B6, CYP2C19, and CYP3A4 in the metabolism of chlorpyrifos to CPO and TCP.
Reference
- Foxenberg et al. (2007).
88. In Vitro Characterization of Hepatic Eugenol Bioactivation vs. Detoxification Pathways, a Comparative Study in Mouse and Human
Emmanuel Minet, Clive Meredith, and Eian Massey
Risk Characterization, British American Tobacco, Southampton, UK
The allylbenzene, eugenol, is found in cosmetics, essential oils, and herbal products and has been added to “certain” cigarettes as a component of clove. The US NTP program reported “no evidence” of eugenol-induced carcinogenicity in rats, but “equivocal” evidence for tumor induction in female mice. In in vitro tests for genotoxicity, eugenol has been demonstrated to be positive, on occasions. In contrast, a variety of studies in animal models and cell culture indicated that the structurally related compound, methyleugenol, is consistently carcinogenic. To further understand the underlying mechanisms of allylbenzene activation and detoxification, we have compared the oxidative and conjugative metabolism of eugenol and methyleugenol in human and mouse liver. Pooled human and mouse liver microsomes and S9 fractions were incubated with C14-eugenol and C14-methyleugenol (20 uM) in the presence of phase I and II enzyme cofactors. Soluble metabolites were quantified by radio-HPLC and identified by coelution with standards. Protein-bound metabolites were quantified following extraction of the insoluble fraction. P450-dependent formation of 1-hydroxy genotoxic precursors in mouse microsomes (40 pmol/min/mg prot) was twice the level detected in human (22 pmol/min/mg prot) and was also much higher in mouse S9 fractions, in comparison to human. Upon the addition of glucuronide to S9 fractions, eugenol was almost entirely conjugated and only traces of 1-hydroxy-eugenol were observed in both species. Glucuronidase treatment of the samples further confirmed that 1-hydroxy-eugenol is a trace metabolite in the human and mouse. In contrast, methyleugenol is not glucuronidated and formation of the 1-hydroxy remained unchanged in both species following the addition of UDPGA. Our data indicate that there is an increased potential for the formation of the genotoxic precursor, 1-hydroxy-eugenol, in the mouse, when compared to humans. Eugenol activation, however, appears to be prevented by direct glucuronidation in both species.
89. Interspecies Differences in Pharmacodynamic and Disposition of BIA 5-453, a Novel Dopamine-β-Hydroxylase Inhibitor
Bruno Igreja, Ana I. Loureiro, Carlos Fernandes-Lopes, Rita Machado, Leonel Torrão, Lyndon Wright, and Patrício Soares-da-Silva
Department of Research and Development, BIAL, S Mamede do Coronado, Portugal
BIA 5-453 ((R)-5-(2-aminoethyl)-1-(6,8-difluorochroman-3-yl)-1,3-dihydroimidazole-2-thione hydrochloride) is a long-acting dopamine-β-hydroxylase (DβH) inhibitor that decreases noradrenaline (NA) levels in sympathetically innervated tissues. It was designed to act as a reversible inhibitor of peripheral DβH with limited access to the brain (J Med Chem 49, 1191–1197, 2006,). In order to evaluate interspecies differences in BIA 5-453 disposition and pharmacodynamic efficacy, mouse, rat, and hamster were administered with BIA 5-453 (100 mg/kg, p.o.). Plasma and heart samples were collected 9 hours after administration. Circulating levels of BIA 5-453 and its N-acetylated metabolite (BIA 5-961) were quantified in plasma, using LC-MS/MS. To evaluate DβH inhibition, dopamine (DA) and NA in the heart were analyzed by LC-ED. BIA 5-453 decreased NA levels in the heart of all three species, with mouse presenting the largest decrease and hamster the smallest decrease in NA levels. BIA 5-453 also increased DA levels in the heart of all three species. DA increases in the heart was similar in rat and mouse, and higher than seen in hamster. Also, the levels of BIA 5-453 in plasma were similar in rat and mouse and the circulating levels of BIA 5-453 in hamster were significantly lower. On the other hand, the N-acetylated metabolite was much higher in rat plasma than in the other two species analyzed. In conclusion, a good correlation between the disposition of BIA 5-453 and DβH inhibition was established in the species analyzed and a significant interspecies difference in BIA 5-453 disposition and metabolism was observed.
Reference
- Alexandre Beliaer, David Learmonth, and Patricia Soaves-de-Silva, Journal of Medical Chemistry, Volume 49, Issue 3.
90. Pig Cytochromes P450 and UDP Glucuronosyltransferases are Models of their Human Counterparts
Pavel Anzenbacher,1 Eva Anzenbacherova,2 Jaroslav Matal,1 and Zuzana Matuskova1
Departments of 1Pharmacology and 2Medical Chemistry and Biochemistry, Faculty of Medicine and Dentistry, Palacky University, Olomouc, Czech Republic
Pig liver microsomal cytochromes P450 seem to be, at least in several cases, good models of their human orthologs. Namely, the pig CYP3A29 mimic rather closely the properties of the human CYP3A4 form, the pig CYP2A19 this of the human CYP2A6, and the pig and human CYP2E1 forms are also rather close in their function. This is probably not true for the CYP2C and CYP2D enzymes. Model substrates for the newly isolated pig CYP forms, 2A19 and 1A, revealed also a similarity in the metabolism by CYP forms of the man and the pig. Skatole is a relatively important substrate of pig CYP enzymes, as it contributes to the boar taint. Skatole metabolites are similar in human as well in pig microsomes and reconstituted systems with the isolated pig CYP1A, CYP2A19, and CYP2E1 enzyme preparations. UDP glucuronosyl transferases are the most important enzymes of the second phase of xenobiotic metabolism; experiments with two marker substrates of the form UGT1A6 have shown that these compounds are metabolized similarly in the pig and human microsomal fraction. (Work on this project was supported by the COST861 (1P05OC050) and MSM6198959216 (MSMT Czech Republic) projects.)
91. Retrospective Analysis of CYP Activity Levels in Microsomes Isolated from Human Donor Livers
Charles L. Crespi,1 Sweta Parikh,2 Zoe E. Barter,3 Teri Bordonaro,2 Edward Francis,2 and Christopher J. Patten1
1R&D, BD Biosciences, Woburn, Massachusetts, USA, 2BD Biosciences, Woburn, Massachusetts, USA, 3Academic Unit of Clinical Pharmacology, University of Sheffield and Simcyp Limited, Sheffield, UK
Human donor livers, originating from U.S.-based organ procurement organizations, have been the basis for the preparation of key reagents used for the characterization of drugs and drug candidates. Over time, the practices for liver transplantation have evolved, which has indirectly affected the nature of organs that are unused for transplantation and are made available for research use. We have prepared microsomes from, and characterized the enzyme activity levels of, CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A in over 300 donor livers, procured from 1985 to the present, and conducted detailed statistical analyses of activity distributions and trends. We have found the following: 1. CYP2B6 and CYP2C19 show the highest variability among donors (CV 150–175%), CYP2C9 (CV 47%) shows the lowest variability with CYP1A2, CYP2D6, and CYP3A4 (CV 82–85%), being intermediate. (Note: the CV values above are based on an assumption of normality.). 2. The age of donors did not change significantly with year of donation. The median age for females was 52, while for males it was 50. The levels of CYP activity did not vary with donor age for all CYPs except CYP2C19, where a statistically significant decrease in activity with age was observed. 3. The gender distribution was 60% male and 40% female. The gender difference was observed with CYP3A4. On average, females had 34% higher activity. While the average activity for CYP2C19 was 48% higher in females, this difference was not statistically significant. 4. No significant change in any enzyme activity was seen as a function of year of donation. Therefore, liver samples obtained over a large period of time are suitable for creation and predicting the properties of pools. Monte Carlo analyses for a 50-donor pool predicted an average CV for these six enzyme activities of 13%. The actual CV observed for four pools prepared in a manufacturing context was 12%. The implications of these observations on the design and performance of reagents derived from human liver will be discussed.
92. Blood-Brain Barrier Penetration: Evaluation of In Vitro Parameters to Predict the Unbound Concentration in Brain
Hinnerk Boriss,1, Katherine Tsaioun,2 and Robert A. Annand2
1Sovicell GmbH, Leipzig, Germany, 2Apredica, Watertwon, Massachusetts, USA
Early assessment of compound availability in the central nervous system (CNS) is essential for CNS drugs and useful for optimizing the toxicity profile of non-CNS drugs. The unbound concentration in the brain interstitial fluid (ISF) determines drug availability in brain. It is, therefore, critical to identify the prediction power of accessible in vitro and in silico parameters for predicting the free concentration in brain. In this article, we assess the contribution of molecular weight, polar surface area, brain membrane affinity, PAMPA-BBB, and plasma protein binding to predict the in vivo availability of free drug in brain, as determined by the distribution coefficient logBB and the brain-free fraction. The overall model fit has a P-value of less than 10–12, while the P-values for the individual parameters range from 10–3 to 10–8 and were used to rank the significance of each parameter. Strongest predictors were polar surface area and brain membrane affinity, followed by PAMPA-BBB and plasma protein binding. These findings highlight that both rate and extent measurements are required to predict CNS uptake.
93. Glucuronidation of S(+)-Licarbazepine and S(-)-Licarbazepine in Mouse Plasma
Ana I. Loureiro, Carlos Fernandes-Lopes, Lyndon C. Wright, and Patricio Soares-da-Silva
Department of Research & Development, BIAL, S Mamede do Coronado, Portugal
Eslicarbazepine acetate (ESL) research is a novel antiepileptic drug that has just completed phase III clinical trials as adjunctive therapy in partial epilepsy refractory to standard antiepileptic drugs. Metabolism of ESL consists primarily of hydrolysis to S-(+)-licarbazepine, which subsequently can generate minor amounts of oxcarbazepine (OXC) and R-(-)-licarbazepine, but their relative amounts are strongly species dependent. S-(+)-licarbazepine, R-(-)-licarbazepine, and OXC are subject to glucuronidation, followed by renal excretion. To date, the glucuronidation of S-(+)-licarbazepine and R-(-)-licarbazepine has not been evaluated in detail. The purpose of this study was to investigate the R(-) and S-(+)-licarbazepine glucuronidation in mouse plasma. NMRI mouse plasma was collected 2 hours after intravenous administration of 10 mg/kg of R(-) and S-(+)-licarbazepine. Glucuronides were evaluated by HPLC-MS/MS and quantification was performed following β-glucuronidase hydrolysis. The glucuronide of S-(+)-licarbazepine and R-(-)-licarbazepine in plasma was identified and quantified in mouse plasma. The plasma concentration of S-(+)-licarbazepine and R-(-)-licarbazepine were approximately the same. After deconjugation with β-glucuronidase, the plasma levels of R-(-)-licarbazepine increased by 3-fold, whereas the plasma levels of S-(+)-licarbazepine increased by 1.5-fold only. These findings suggest that glucuronidation of licarbazepine is enantioselective and the metabolism and disposition of S-(+)-licarbazepine in mouse considerably differs from that of R-(-)-licarbazepine.
94. A Novel Approach To The Detection and Quantification of Metabolites and Matrix Effects in Bioanalytical Assays
Robert Plumb1 and Ian Wilson2
1Pharmaceutical Business, Waters Corp., Milford, Massachusetts, USA, 2Dmpk, AstraZeneca, Macclesfield, UK
In this paper, we present a new rapid-approach simultaneous detection and quantification of drugs, metabolites, and matrix in a bioanalytical assay. The approach uses sub-2-μm particle LC coupled to a tandem quadrupole mass spectrometer with a novel collision cell design allowing the simultaneous collection of MRM and full-scan MS data. The high data capture rate of the MS system combined with the high resolution of sub-2-m particle LC allowed for high sensitivity and high specificity data collection. The ability to collect MRM and full-scan MS data was utilized to simplify methods development and evaluate the effect of plasma source on the quality of the data. This was achieved by monitoring the full-scan plasma profile and the phospholipid polar head groups. The assay sensitivity was not affected by the collection of the full-scan MS data and the high data capture rate allowed greater than 20 points to be collected across the 2-second-wide MRM and full-scan MS peaks. During the course of a bioanalytical assay, it is necessary to understand the effects of the biological matrix and drug metabolites on assay performance. Recent regulatory guidelines have required that drug metabolites with an exposure of greater than 10% of the parent must be quantified and characterized. In this study, two separate pharmaceuticals were analyzed in rat and dog plasma: a common steroid, fluticasone propionate, and a new novel drug entity. The results showed excellent sensitivity (5 pg/mL) from plasma and reproducibility. The dual-scan capability of the MS instrumentation allowed for the detection of the metabolites of the new drug entity ensuring that then coelution occurred and the metabolites were correctly quantified using both full-scan MS and parent ion scanning for common fragment ions of the drug molecule.
95. A Novel CYP3A Gene Cluster Humanized Mouse Model
Yury Kapelyukh,1 Jillian Ross,1 Nico Scheer,2 and C. Roland Wolf1
1CXR Bisciences Ltd, Dundee, UK, 2TaconicArtemis, Cologne, Germany
The cytochrome P450 isoenzyme, CYP3A4, is one of the major enzymes responsible for the metabolism of xenobiotics in humans. In order to improve the prediction of human metabolism from animal studies, we have developed a novel humanized mouse model for CYP3A, in which the murine genes from the Cyp3a subfamily were replaced with two of their human counterparts by introducing CYP3A4 and CYP3A7 into the Cyp3a locus (termed hCYP3A4/3A7_Cyp3a KO). An equivalent knockout for Cyp3a isoforms was also created (termed Cyp3a KO). Using RNA and protein analysis, we demonstrated that CYP3A4 was expressed constitutively in the liver and small intestine of hCYP3A4/CYP3A7_Cyp3a KO mice and was highly induced by treatment with 5-pregnen-3β-ol-20-one-16α-carbonitrile (PCN), a potent inducer of CYP3A4 expression. Interestingly, CYP3A7 RNA was also found in the liver of PCN-treated animals, while constitutive CYP3A7 RNA expression was below the limit of detection. Induction with PCN resulted in a significant increase in the rate of oxidation of CYP3A4-specific substrates by liver and intestinal microsomes from hCYP3A4/CYP3A7_Cyp3a KO mice. (This work was supported by ITI Life Sciences. ITI Life Sciences, Dundee Technology Park, Dundee, UK.)
96. Mass Spectrometric Identification of Differentially Regulated Proteins in an In Vitro Human Airway Epithelium System in Response to Treatment with a Novel Natural Product Mixture
Robert Streeper, Armando Diaz, D. Campos, S. Baek, R. Mussman, T. Long, and E.
Izbicka Cancer Prevention and Cure Ltd., San Antonio, Texas, USA
Objective: To identify novel protein biomarkers of the biological responses of human lung epithelium to treatment with natural product mixture, AirAide that has proven useful in human trials for the treatment of inflammatory diseases of the lung, such as asthma and COPD, in a three-dimensional human lung epithelial tissue model using high-resolution mass spectrometry.
Methods: We evaluated the effects of the irritant, urethane, used alone or concurrent with AirAide or fluticasone proprionate as a positive control in the human EpiAirway™ model. Drug concentrations corresponded to standard ranges used in human clinical treatment. Following topical exposure to the agents or vehicle for 5 hours, differentially expressed proteins were identified by using liquid chromatography electrospray ionization mass spectrometry (LC-ESIMS MS). Tissue lysate and media specimens were digested with trypsin and analyzed by LC-ESIMS. Data were analyzed by using Mascot search software.
Results: We have identified a number of heretofore unrecognized differentially expressed proteins in the samples. The identified proteins include proteins identified in prior genetic-sequencing efforts, several patented proteins having functions that have not been described in the literature, and a number of proteins involved in multiple cellular processes.
Conclusions: Our results support an extended MS analysis of biomarker profiles as a supplementary tool in the identification of target molecules and systems for the subsequent application of quantitative validation techniques for the identification and molecular characterization of tissue responses to pharmacologically active agents for treatment of inflammatory airway diseases using the EpiAirway™ tissue model.
97. Multiplexed Immunoassay Quantification of Inflammatory Biomarker Responses in an In Vitro Human Airway Epithelium System for Evaluation of a Novel Natural Product
Armando Diaz, D. Campos, S. Baek, R. Mussman, Robert Streeper, T. Long, and E. Izbicka
Cancer Prevention and Cure Ltd., San Antonio, Texas, USA
Objective: To develop a methodology for screening of AirAide, a natural product mixture that has proven useful in human clinical trials for the treatment of inflammatory diseases of the lung, such as asthma and COPD, in a three-dimensional human lung epithelial tissue model.
Methods: We evaluated the effects of the irritant, urethane, used alone or concurrent with AirAide or fluticasone proprionate in the human EpiAirway™ model. Agent concentrations corresponded to standard ranges used in human clinical treatment. Following topical exposure to the agents or vehicle for 18 hours, we measured levels of 32 cytokines and chemokines in culture media and tissue lysates in multiplexed Luminex immunoassays. ATP production was quantified in the lysates. Tissue morphology was evaluated in paraffin-embedded specimens stained with hematoxyllin-eosin (H&E). Intergroup differences were assessed by the Student t-test.
Results: Immunoassays assays indentified “signature patterns” for the agents based on highly significant (t < 001) differences. The patterns were heterogeneous; AirAid and fluticasone proprionate differentially modulated diverse patterns and types of inflammatory biomarkers. H&E staining has further shown a protective effect of counterirritants on tissue morphology.
Conclusions: The study provided a proof of concept for using the EpiAirway™ model for comprehensive screening of pharmaceutical and nutraceutical agents in conjunction with multiplexed immunoassay quantification of inflammatory biomarkers.
98. R-Alpha Lipoic Acid Enhances the Susceptibility of Human Colon Adenocarcinoma Cells to 5-Fluorouracil
Sherif Ibrahim1 and Patrick J. Sinko2
1Department of Pharmaceutics, Ernest Mario School of Pharmacy, Rutgers University, Piscataway, New Jersey, USA, 2Department of Pharmaceutics/Room 213, State University of New Jersey–Rutgers, Piscataway, New Jersey, USA
Purpose: Earlier observations have shown that mitochondrial abnormalities, especially defective oxidative phosphorylation, may play a role in the hampered ability of cancer cells to undergo apoptosis. Therefore, our aim was to investigate the effect of R-alpha lipoic acid (R-ALA), a mitochondrial coenzyme with a known ability to stimulate mitochondrial functions, on the cell death and apoptosis response of HT-29 human colon adenocarincoma cells to 5-FU treatment.
Methods: HT-29 cells were treated with either 5-FU (2 μM) or a combination of 5-FU (2 μM) and R-ALA (1 mM). Cytotoxicity analysis at 24 and 48 hours was performed with the AlamarBlue® (Invitrogen, Carlebad, California, USA) fluorescence cytotoxicity assay, and apoptosis levels at 8, 16, 20, and 24 hours were determined by measuring caspase-3 activation (DEVD-AFC cleavage). In addition, phase-contrast microscopy at 24 and 48 hours was used to visually confirm the results of the cytotoxicity and apoptosis assays.
Results: Cytotoxicity and apoptosis indices were always significantly greater for the combination treatment than for 5-FU alone. The differences were particularly dramatic at the later time points. At 48 hours, relative to controls, 5-FU-treated cells showed 73% viability, whereas 5-FU- and R-ALA-treated cells showed 8% viability. At 24 hours, relative to controls, 5-FU-treated cells showed 2-fold caspase-3 activation, whereas 5-FU- and R-ALA-treated cells showed 5.6-fold caspase-3 activation. Phase-contrast microscopy visually confirmed the cytotoxicity and apoptosis results. Conclusions: Treatment with R-ALA enhances the susceptibility of HT-29 human colon adenocarincoma cells to cell death and apoptosis in response to 5-FU treatment. This observation provides evidence that the stimulation of mitochondrial function may sensitize cancer cells to apoptosis in response to chemotherapeutic agents. Further studies will highlight the effects of mitochondrial stimulation on the cell death and apoptosis response of different cancer cell types to chemotherapeutic agents.
99. A Progress Curve Approach for the Assessment of Time-dependent Inhibition of CYP3A4
Howard J. Burt,1 Carolina Lager,1 Ruth Hyland,2 J. Brian Houston,1 and Aleksandra Galetin1
1School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester, UK 2Pfizer Global Research & Development, Sandwich, Kent, UK
Characterization of time-dependent inhibition requires the determination of kinact and KI, the maximum inactivation rate constant and the inhibitor concentration leading to 50% of kinact, respectively. A progress curve method involves the monitoring of enzyme activity over the time course of a simultaneous incubation with inhibitor and probe substrate. This “all-in” approach may potentially overcome some of the limitations of the commonly used two-step dilution method in the assessment of time-dependent inhibition. In the current study, in vitro inactivation parameters were obtained by using the progress curve method for three time-dependent inhibitors of CYP3A4, namely mibefradil, erythromycin, and diltiazem. Progress curves were generated by incubating recombinant CYP3A4 in the presence of an inhibitor (7 concentrations + control) and the probe substrate, quinidine, at 50 μM. At multiple time points over a 40-minute period, aliquots were removed and analyzed for both 3-hydroxyquinidine and inhibitor concentration, using LC-MS/MS. Estimates of kinact and KI were obtained by nonlinear regression analysis and were corrected for microsomal binding. Time-dependent inhibition was observed for mibefradil and erythromycin, resulting in kinact estimates of 0.139 and 0.0656 min−1 and KI(unbound) of 0.029 and 0.427 μM, respectively. The KI(unbound) for mibefradil and erythromycin were approximately 3- and 20-fold lower, respectively, than estimates obtained with the two-step dilution method. When inhibitor depletion was accounted for by using a time-averaged concentration, the mibefradil KI(unbound) estimate was reduced approximately 30-fold, whereas no significant change was observed in erythromycin KI(unbound). In contrast to these inhibitors, no time-dependent inhibition was observed in the case of diltiazem. This finding may be linked to reduced production of its N-desmethyl metabolite, which is known to inhibit CYP3A4 to a greater extent than the parent drug. In conclusion, this study demonstrates the potential of the progress curve method to assess CYP3A4 time-dependent inhibition. Further work on a wide range of inhibitors and an assessment of the predictive utility of the parameters obtained is required.
100. Development of a Population Pharmacokinetic Model to Characterize the Interaction Between Grapefruit Juice and Bilastine in Humans
Monica Rodriguez,1 María Luisa Lucero,2 Valvanera Vozmediano,1 and Nerea Leal1
1Modelling and Simulation, DynaKin, S.L., Derio (Vizcaya), Spain, 2Department of R&D and Innovation, FAES Farma, S.A., Leioa, Spain
The H1-receptor antagonist, bilastine, has been shown to be a P-glycoprotein (P-gp) substrate. Some substrates of P-gp also appear to be substrates of OATP. Fruit juices such as grapefruit may alter the bioavailability and exposure of drugs, mainly due to inhibition of OATP1A2 activity, but also by modulating P-gp activity with a net result that is difficult to predict. A classical noncompartmental approach has been already used to evaluate the interaction of bilastine with grapefruit juice (GFJ), but no information regarding absorption processes could be elucidated with this modeling approach. Population pharmacokinetic (POPPK) modeling is a helpful tool to characterize drug interactions contributing to the enrichment of previously developed models.
Aim: The aim of this work was to assess the effect of the consumption of GFJ on the bioavailability of bilastine in humans by using a population approach.
Methods: The present analysis has utilized information from healthy subjects enrolled in a phase 1 study conducted to evaluate the effect of GFJ on the single-dose (20-mg) PK of bilastine, administered with 240 mL of water or coadministered with GFJ. POPPK analysis was performed by using NONMEM. The model was constructed in order to allow all absorption-related parameters to vary except the bioavailability in the control group, which is fixed to one; this allows estimation of a relative bioavailability value and an absorption constant (Ka) value related to grapefruit coadministration.
Results: A two-compartment structural model parameterized in terms of intercompartmental clearance and with a first-order Ka was used to fit the concentrations of bilastine. PK parameters related to distribution and elimination remained unchanged with the coadministration of grapefruit. However, a relative bioavailability of 0.56 was obtained and the Ka was reduced from 1.57 to 1.12 h−1 with grapefruit consumption.
Conclusion: GFJ coadministration reduces the bioavailability and Ka of bilastine without altering the distribution and elimination parameters, which is in accordance with a nonmetabolized drug such as bilastine. Population-based assessment of drug interactions provides clinically relevant results that can be directly applied to target populations.
101. Effect of Quercetin on the Pharmacokinetics of Fexofenadine in Healthy Subjects
Kyoung-Ah Kim and Ji-Young Park
Department of Clinical Pharmacology and Toxicology, Anam Hospital, Korea University College of Medicine, Seoul, South Korea
Objective: The aim of the present study was to assess whether quercetin exhibited any inhibitory effect on P-glycoprotein (P-gp)-mediated drug disposition in humans by using fexofenadine as a P-gp substrate.
Methods: Twelve healthy subjects were enrolled for the study and treated daily for 7 days with 500 mg of quercetin or placebo 3 times a day. On day 7, a single dose of 60 mg of fexofenadine was administered orally. Plasma and urinary fexofenadine concentrations were measured and pharmacokinetic differences between placebo and quercetin phases analysis were assessed.
Results: The mean plasma concentrations of fexofenadine were significantly increased after quercetin treatment, compared to that of the placebo phase. The area under the time versus concentration curve (AUC) of plasma fexofenadine was increased by 55% by quercetin (2,005.3 vs. 3,098.6 ng*h/mL; P < 0.001), and similarly, the maximum plasma concentration (Cmax) during the quercetin phase was elevated by 68%, compared to that of the placebo phase (295.3 vs. 480.3 ng/mL; P = 0.006). Although the oral clearance of fexofenadine was decreased significantly by 37% after quercetin treatment (61.4 vs. 38.7 L/h; P < 0.001), no differences in the renal clearance and half-life were observed between placebo and quercetin phases.
Conclusion: The results of the present study showed that short-term use of quercetin elevated the plasma concentrations of fexofenadine, probably by the inhibition of P-gp-mediated efflux in humans. Therefore, caution should be necessary during the coadministration of quercetin and P-gp substrates, including fexofenadine.
102. Improved Predictions of Clinical Drug-Drug Interactions Using the Concept of PBPK (Simcyp) as Compared to [I]/Ki Approach
Jan Snoeys,1 V. Sinha,2 G. Mannens,1 and Mario Monshouwer1
1Drug Metabolism and Pharmacokinetics and 2Clinical Pharmacology Johnson & Johnson Pharmaceutical Research and Development, a division of Janssen Pharmaceutica N.V., Beerse, Belgium
Simcyp® ADME Simulator v.7 (Simcyp Ltd., Sheffield, UK), a software package that applies PBPK concepts, was used to simulate 93 clinical metabolic drug-drug interaction studies on the basis of in vitro input data collected from the literature. Results showed that the prediction of significant clinical drug-drug interactions (DDIs), defined as an AUC increase of the victim drug of at least 2-fold, was clearly better with the Simcyp Simulator, compared with the [I]/Ki approach with total plasma Cmax as input parameter for [I], since the DDI prediction sensitivity improved from 58 to 72%, whereas the specificity remained the same (96 vs. 94%). An additional drawback of the [I]/Ki approach is that for the evaluation of test compounds as perpetrator in a preclinical development phase, the human total plasma Cmax as input for I is not known. With PBPK tools, however, it is possible to simulate the human PK profile and hence DDI liability of drugs as a potential perpetrator, based on in vitro data in a preclinical stage. Overall, Simcyp simulations of CYP2D6 DDIs were most successful with a sensitivity of 90% (9/10) and a specificity of 86% (6/7). CYP3A4 DDIs were predicted with a sensitivity of 80% (16/20) and a specificity of 94% (17/18). CYP2C DDIs were predicted with a sensitivity of 75% (9/12) and specificity of 95% (18/19), and CYP1A2 DDIs were predicted with a sensitivity of 0% (0/3) and a specificity of 100% (18/18). An in vitro binding-factor sensitivity analysis showed that although binding factors (fup, fumic, and B/P) are crucial in the prediction of human PK profiles and hence AUC, they are less crucial in the prediction of fold increase in AUC of victim drugs due to metabolic inhibition. Based on literature data from a large compound set, it is concluded that Simcyp Simulator offers several advantages over [I]/Ki-based mathematical equations for clinical DDI predictions.
103. Methadone Inhibits CYP2D6 and UGT2B7 In Vivo: a Study using Codeine as the Substrate
Andrew Alexander Somogyi,1 Eloise Gelston,1 Janet K. Coller,1 Jason M. White,1 and Helmut Schmidt2
1Department of Clinical and Experimental Pharmacology, University of Adelaide, Adelaide, Australia, 2Institute for Clinical Pharmacology, Universitatsklinikum Frankfurt am Main, Frankfurt, Germany
Although not a substrate, methadone can inhibit CYP2D6- and UGT2B7-mediated metabolism in vitro. We aimed to determine whether this would occur in vivo by using codeine, whose O-demethylation to morphine is via CYP2D6 and glucuronidation to C6G is via UGT2B7; morphine metabolism to M3G and M6G is also via UGT2B7. Twelve subjects on methadone (Meth: dose 35–200 mg/day) and 9 on buprenorphine (Bup: dose 4–34 mg/day) maintenance treatment (all CYP2D6 genotypic extensive metabolizers) were given a single 60-mg dose of codeine, followed by the collection of a blood sample at 3 hours and all urine for 6 hours; samples were assayed by LC-MS. The urinary metabolic ratio for O-demethylation to morphine was significantly (P = 0.03) higher in Meth (2.8 ± 3.0) than Bup (0.6 ± 0.4) subjects and similarly for glucuronidation (Meth: 0.31 ± 0.23; Bup: 0.049 ± 0.027; P = 0.002). In 3-hour plasma samples, whereas morphine concentrations remained unchanged (Meth: 1.2 ± 0.4; Bup: 1.2 ± 0.6 ng/mL; P = 0.7), C6G, M3G, and M6G concentrations were, on average, 1.8-, 2.4- and 3.4-fold lower (P < 0.008), respectively, in Meth, compared to Bup, subjects. In contrast, plasma codeine and norcodeine concentrations were 3- and 6.3-fold higher (P < 0.0004), respectively, in Meth, compared to Bup, subjects. The mechanism for this somewhat unique interaction involving two important and distinct drug-metabolizing enzymes remains unclear, given that methadone is not a substrate for either enzyme. Although unlikely to be clinically important in methadone maintenance subjects due to tolerance and the weak analgesic effect of codeine, methadone is being increasingly used in pain treatment, and therefore, the potential for drug-drug interactions is far greater with an enhanced likelihood for adverse effects.
104. Theoretical Assessment of Metabolic Drug Interactions in Pediatric Population: the Impact of Age-Related Fractional Metabolism (fm) and its Disparity between Adults and Neonates
Trevor N. Johnson,1 Farzaneh Salem,2 Zoe E. Barter,3 and A. Rostami-Hodjegan4
1Simcyp Limited, Sheffield, UK, 2Academic Unit of Clinical Pharmacology, University of Sheffield, Sheffield, UK, 3Academic Unit of Clinical Pharmacology, University of Sheffield and Simcyp Limited, Sheffield, UK, 4University of Sheffield and Simcyp Ltd, Sheffield, UK
The most common approach to estimate the level of any metabolic drug-drug interaction (mDDI) is defined in the equation below:
where fm is the fraction of substrate clearance mediated by the inhibited metabolic pathway, [I] is the concentration of inhibitor at the enzyme site, and Ki is the inhibition constant for inhibitor obtained from in vitro studies. This equation applies only to orally administered drugs undergoing hepatic metabolism and it ignores the possibility of inhibition of gut “first-pass” metabolism and time-variant inhibitor concentration. In the presence of a strong inhibitor, the level of interaction is defined by (1/(1-fm)). Information on the ontogeny of various CYP enzymes in the liver points to variable rates of maturation (e.g., CYP1A2 and 3A4 time to reach half adult expression is 1.1 and 0.38 years, respectively). This information together with knowledge of renal function maturation may be used to estimate the fractional metabolism of drugs in neonates. The aim of this study was to investigate age-varying “fm” and the consequent influence on mDDIs in neonates, compared to adults. The results indicated some disparities between adults and neonates in potential for mDDI. For example, strong inhibition of CYP3A4 leads to a greater than 6-fold increase in exposure to alprazolam in adults, but less than a 4-fold increase in exposure at 8 weeks of age due a higher fractional renal elimination. Conversely, a hypothetical drug with an equal elimination via CYP3A4 and CYP1A2, which is not subject to substantial gut metabolism, was predicted to have a larger mDDI under inhibition of CYP3A4 in newborns (3-fold), compared to the expected level of interaction in adults (1.5-fold) due to reduced relative levels of CYP1A2. Considering the paucity of the data on mDDI in pediatrics, and the ethical difficulties in carrying out DDI studies in this population, modeling, and simulation, combined with in vitro–in vivo extrapolation of information on metabolic routes, could prove an essential tool in understanding the potential for mDDI in young children.
105. Modulatory Effects of Resveratrol on Liver, Kidney, and Lung Total GST, GST Mu, and NQO1 and CYP2B4, CYP2E1, and CYP3A Associated Enzyme Activities
Sevki Arslan,1 Mine Nuyan,2 and Emel Arinç3
1Department of Biology, Pamukkale University, Faculty of Arts and Sciences, Denizli, Turkey 2Biochemistry Graduate Programme and Department of Biological Sciences, Middle East Technical University, Ankara, Turkey, 3Biochemistry Graduate Programme and Department of Biological Sciences, Middle East Technical University, Ankara, Turkey
Cancer protective effects of flavonoids have been attributed to a wide variety of mechanisms, such as free radical scavenging and modifying phase I and II enzymes that activate or detoxify carcinogens. Among these flavonoids, recently known important flavonoid with well-known cancer chemoprevention effects is resveratrol. It was shown to modulate the risk of cardiovascular diseases and inhibit chemical carcinogenesis in rodents. One of the possible mechanisms by which resveratrol may exert their anticarcinogenic effects is through an interaction by certain CYPs and conjugation enzymes. In this respect, the focus of this study was to determine the effects of resveratrol on phase I and II enzymes. Male New Zealand rabbits were treated intragastrically with resveratrol at a dose of 25 mg/kg b.w. for 3 days. Kidney total GST and GST Mu activites increased nearly 1.50 fold in the resveratrol group. Also, total GST activity of the resveratrol group increased (1.6-fold) in lung. These results indicate that resveratrol reversed toxic and carcinogenic action of chemicals by increasing activities of total GST in kidney and lung and GST Mu in kidney. However, liver total GST and GST Mu activities were not changed by resveratrol. A 1.52-fold increase was observed in CYP2B4-associated benzphetamine N-demethylase activity in kidney. However, this activity was not changed in lung and liver. Similarly, CYP2E1-associated NDMA N-demethylase, p-nitrophenol hydroxylase activities, and CYP3A6-associated erythromycin N-demethylase did not change significantly in liver, kidney, and lung. Moreover, NQO1 activity of the resveratrol group did not change in these tissues. These results showed that CYP2B4, total GST, and GST Mu activities were significantly increased in kidney as a result of resveratrol. CYP2B4 is generally associated with detoxifying metabolism of xenobiotics. Therefore, detoxification of some toxic chemicals may be increased in humans that consume resveratrol. Moreover, not only toxicities of reactive oxygen species, but also toxic and neoplastic challenges of carcinogens may be prevented in humans consuming a resveratrol-rich diet due to induction of total GST and GST Mu in kidney.
106. Comparison of RapidFire® Ultra High throughtput LC/MS/MS with Traditional LCMS/MS for Cytochrome P450 Inhibition Testing
Elke S. Perloff,1 Shangara S. Dehal,1 Andrew K. Mason,1 Andrew P. Blanchard,1 William A. LaMarr,2 Can C. Ozbal,2 Vaughn P. Miller,2 Charles L. Crespi,1 and David M. Stresser1
1BD GentestSM Contract Research Services, Woburn, Massachusetts, USA, 2BioTrove, Inc., Woburn, Massachusetts, USA
Objective: Assessment of cytochrome P450 inhibition by NCEs has moved into earlier phases of drug discovery/development, and recent guidance from the FDA recommends routine in vitro IC50 determinations for at least six P450 isoforms. The resulting increase in the number of samples from P450 inhibition screens has led to a demand for higher throughput analysis options. The objective of this study was to apply RapidFire® LC/MS/MS technology (BioTrove, Woburn, Massachusetts, USA) to in vitro P450 inhibition testing and compare the results to traditional LC/MS/MS methods validated in-house.
Methods: Seven-point IC50 values for a range of inhibitors were determined in individual incubations in human liver microsomes, using the following FDA recommended probe substrates with previously validated methods (Perloff ES et al., 2009; Xenobiotica, in press): CYP1A2/ phenacetin (tacrine for RapidFire); CYP2B6/ bupropion; CYP2C8/ amodiaquine; CYP2C9/ diclofenac; CYP2C19/ S-mephenytoin; CYP2D6/ dextromethorphan; CYP3A4/ testosterone, and midazolam. Samples were analyzed individually by RapidFire technology as well as by traditional LC/MS/MS methods developed and validated in-house. Stable-labeled isotope internal standards were used for all probe substrate metabolites, except for 1-hydroxytacrine, where bucetin was employed. Percent remaining activity was calculated for each inhibitor concentration, and IC50 values were calculated by linear interpolation.
Results: IC50 values obtained by using RapidFire ultra high throughput LC/MS/MS analysis were consistent with the data obtained by using traditional LC/MS/MS methods validated in-house. Greater than 90% of corresponding IC50 values were within 2.0-fold of each other. A run time of approximately 7 seconds per injection with RapidFire technology compared to 2–4 minutes for traditional LC/MS/MS methods provided a significant decrease in analysis time.
Conclusions: P450 inhibition IC50 results obtained by using RapidFire were comparable to those obtained by using validated traditional LC/MS methods. The increased analysis speed represents a >20-fold improvement in cycle time, thereby permitting rapid data delivery to project teams and clients. Alternatively, ultra-rapid analysis allows the acquisition of more data points per unit time, providing for the option of conducting robust, multipoint assays typical of drug development.
107. Effect of Model Inhibitors on Albendazole Sulphoxidation in Lancet Fluke (Dicrocoelium dendriticum)
Barbora Szotakova, Hana Bartikova, Ivan Vokral, Vladimir Kubicek, Jiri Lamka, and Lenka Skalova
Faculty of Pharmacy, Charles University, Hradec Kralove, Czech Republic
Dicrocoeliosis, a helminthosis caused by lancet fluke (Dicrocoelium dendriticum), is, at present, considered a worldwide significant, but little investigated, parasitosis of farm, domestic, and wild animals, and a relatively rare parasitosis of man. Lancet fluke particularly parasitizes in biliary duct, gallbladder, and at the outlet of pancreas of its host, small ruminants. The only means, so far, generally accepted against fluke is pharmacotherapy and pharmacoprophylaxis practiced in endangered or attacked breeds with the use of suitable anthelmintics (preference is given to benzimidazole anthelmintics). The aim of this project was to study the metabolism of benzimidazole anthelmintic albendazole ([5-(propylthio)-1H-benzimidazol-2-yl] methylcarbamate; ABZ) in lancet fluke subcellular fractions and to characterize the responsible enzymes. The results showed that ABZ is metabolized via sulphoxidation in lancet fluke in vitro. The highest velocity of ABZ sulphoxide (ABZSO) formation was found in the mitochondria-like fraction, lower in the microsomes-like fraction, and none ABZSO arising in the cytosole-like fraction. Several model inhibitors of biotransformation oxidation enzymes were used to characterize the enzymes responsible for sulphoxidation of ABZ. Salicylhydroxamic acid, mercaptosuccinic acid, octylamine, methimazole (MET) and alpha-naphthyl thiourea (ANTU) were used in incubations of ABZ with fluke’s subcellular fractions. MET and ANTU, typical inhibitors of flavine mono-oxygenases (FMOs) in mammals, significantly inhibited the ABZSO formation in lancet fluke. The concentration-dependent inhibition was observed both in mitochondria- and microsomes-like fractions. Values of IC50 were calculated. Based on the inhibition study, FMOs seemed to be responsible for ABZSO formation, although no activity of thiobenzamide-S-oxidase (typical reaction catalyzed by FMO in mammals) was detected in lancet fluke. This contradiction indicate that lancet fluke’s FMO does not use thiobenzamide as a substrate or MET and ANTU inhibit other oxidase than FMO. The results demonstrate considerable differences in biotransformation enzymes properties in mammals and helminths. (This project was supported by the Czech Science Foundation, grant no. 524/07/0611.)
108. Evaluation of High throughput Screening Methods for Time-dependent Inhibition of Human Cytochrome P450s Utilizing RapidFire® Ultra LC/MS/MS Technology
Vaughn P. Miller,1 Can C. Ozbal,1 Elke S. Perloff,2 Andrew K. Mason,2 Shangara S. Dehal,2 Andrew P. Blanchard,2 David M. Stresser,2 Charles L. Crespi,2 and William A. LaMarr1
1BioTrove, Inc., Woburn, Massachusetts, USA, 2BD GentestSM Contract Research Services, Woburn, Massachusetts, USA
Objective: Recently, there has been a greater recognition of the importance of screening for time-dependent inhibition (TDI) of human cytochrome P450s earlier in drug discovery. This has led to the demand for higher throughput analysis options for this assay. The objective of this study was to evaluate single vs. multiconcentration methods for in vitro TDI screening and compare the results from RapidFire® (BioTrove, Inc., Woburn, Massachusetts, USA) LC/MS/MS technology to traditional LC/MS/MS analysis.
Methods: IC50 curve-shift assays were performed for a range of inhibitors in individual incubations of human liver microsomes, using FDA-recommended drug-probe substrates and previously validated methods (Perloff et al., 2009: Xenobiotica, in press). Samples were analyzed individually by RapidFire technology as well as by traditional LC/MS/MS methods.
Results: The RapidFire technology provided a >20-fold decrease in analysis time, compared to traditional LC/MS/MS methods. The utility of using a single-inhibitor concentration (e.g., 10 or 50 μM) with a 30-minute preincubation time was analyzed within the IC50 curve data. Significant changes in inhibition potential as a result of the preincubation were detectable regardless of experimental design.
Conclusions: Results from the RapidFire LC/MS/MS technology for the analysis of in vitro cytochrome P450 time-dependent inhibition screening were comparable to traditional LC/MS/MS methods. The data in this study suggest that a single-inhibitor concentration provides relevant outcomes for initial screening. As expected, full IC50 shift determinations improve robustness, but increase sample load. However, the >20-fold decrease in assay analysis times with using the RapidFire system may offset concerns with the turnaround time required for full inhibition curves for this important ADMET screen.
109. Recovery of CYP2C8 Activity after Gemfibrozil Administration: Estimation of CYP2C8 Half-Life using Repaglinide as an In Vivo Probe
Johanna Honkalammi, Janne T. Backman, Mikko Niemi, Aleksi Tornio, and Pertti J. Neuvonen
Department of Clinical Pharmacology, Helsinki University and Helsinki University Central Hospital, Helsinki, Finland
Cytochrome P450 (CYP) 2C8 participates in the metabolism of both endogenous and exogenous substances. Gemfibrozil 1-O-β-glucuronide is a mechanism-based inhibitor of CYP2C8 (Ogilvie et al., 2006), and the administration of gemfibrozil strongly inhibits the metabolism of the CYP2C8 substrate drug, repaglinide. In a previous study, the area under the plasma concentration-time curve (AUC) of repaglinide was increased up to 8.1-fold by a 3-day pretreatment with gemfibrozil. In another, recent study, the strong inhibitory effect of gemfibrozil on repaglinide metabolism persisted for the whole 12-hour postdose period studied (Tornio et al., 2008), consistent with mechanism-based inhibition of CYP2C8 by gemfibrozil 1-O-β-glucuronide. Our aim was to study the recovery of CYP2C8 activity after gemfibrozil administration, using repaglinide as a probe drug, in order to estimate the turnover half-life of CYP2C8. In a randomized five-phase crossover study, 10 healthy volunteers ingested 0.25 mg of repaglinide alone or after a 3-day pretreatment with 600 mg of gemfibrozil twice-daily. Plasma repaglinide, gemfibrozil, their metabolites, and blood glucose were measured. The AUC0-∝ of repaglinide was 7.7-, 2.9-, 1.4-, and 0.9-fold, compared to the control phase, when the last dose of gemfibrozil was taken 1, 24, 48, or 96 hours before repaglinide, respectively. Thus, significant (P < 0.001) inhibition was seen up to 48 hours after gemfibrozil administration. This finding was supported by the repaglinide metabolite data. Using the recovery of the oral clearance of repaglinide, the in vivo turnover half-life of CYP2C8 was estimated to average 20 hours. Based on these data, a safe margin for starting administration of substrates of CYP2C8 to patients previously treated with gemfibrozil would be about 3–4 days after the last gemfibrozil dose.
Ogilvie BW, et al. (2006). Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: implications for drug-drug interactions. Drug Metab Dispos 34:191–197.
Tornio A, et al. (2008). The effect of gemfibrozil on repaglinide pharmacokinetics persists for at least 12 hours after the dose: evidence for mechanism-based inhibition of CYP2C8 in vivo. Clin Pharmacol Ther 84:403–411.
110. Trans-resveratrol, a New Esterase Inhibitor of Eslicarbazepine Acetate Hydrolysis
Ana I. Loureiro, Carlos Fernandes-Lopes, Lyndon C. Wright, and Patricio Soares-da-Silva
Department of Research & Development, BIAL, S Mamede do Coronado, Portugal
Eslicarbazepine acetate (ESL) is a novel antiepileptic drug that shares with carbamazepine and oxcarbazepine the dibenzazepine nucleus bearing the 5-carboxamide substituent, but is structurally different at the 10,11-position. This molecular variation results in differences in metabolism, namely by preventing the formation of toxic epoxide metabolites, such as carbamazepine-10,11 epoxide. Metabolism of ESL originates (S(+)- and R(-)-licarbazepine) and oxcarbazepine (OXC), but their relative amounts are strongly dependent on the species. It was found that ESL was extensively hydrolyzed by esterases to the corresponding S(+) and R(-)-licarbazepine compounds in mouse, rat, rabbit, and human liver microsomes. Esterases are ubiquitous proteins responsible for the detoxification of xenobiotics. However, these enzymes also activate prodrugs. The hydrolysis of ESL was investigated in vitro by using human microsomes and some esterases inhibitors. Kinetic analysis of ESL hydrolysis was performed in human liver and intestine microsomes pools. The apparent Km, determined by fitting to the Hill equation, was 201 ± 9 and 166 ± 8 μM for human and intestinal microsomes, respectively. Dichlorvos (10 μM) and p-nitrophenyl acetate (1 mM) were inhibited by 100 and 80% ESL hydrolysis, respectively. The IC50 for trans-resveratrol inhibition was 0.76 and 3.9 μM, using ESL concentrations of 25 and 250 μM, respectively. However, trans-resveratrol failed to prevent ESL hydrolysis when given orally. These results suggest that ESL hydrolysis may occur in liver and intestine, and trans-resveratrol could be used under in vitro conditions as an esterase inhibitor. The extensive metabolism that trans-resveratrol undergoes after oral administration may explain its failure in prevent ESL hydrolysis when both compounds are given.
111. Characterization of Oxidative Deaminase Activity in the Reconstructed Human Epidermis Episkin™
Joan Eilstein, Eric Arbey, Florence Canivet, Jean-Roch Meunier, Jacques Leclaire, and Daniel Duché
Life Sciences Department, L’Oréal Research, 92585 Clichy, France
The skin is not just a passive physical barrier, but is also a biological structure involved in a wide range of metabolic activities. The reconstructed human epidermis Episkin™ was a tool developed for studying, in vitro, skin toxicity of chemicals and their metabolism as alternative methods to animal experimentations. This implies that the model is metabolically active. Adenosine deaminase (ADA) and adenylate deaminase (AMPDA) are enzymes involved in the irreversible oxidative deamination of adenosine and adenosine derivatives to inosine and inosine derivatives, respectively. These enzymes, which are present in normal human epidermis, have a critical role in the adenosine homeostasis and may be in the proliferation and maturation of certain types of mammalian cells. In this work, we reported the characterization of oxidative deaminase activity in Episkin™ by using adenosine derivatives as enzyme substrates (2’,3’-O-isopropylidene adenosine ). Substrate disappearance and the corresponding inosine derivatives apparition were detected by UV-HPLC and demonstrate that ADA and /or AMPDA are present and efficient in Episkin™.
112. Prediction of the Intestinal Availability and Associated Inter-individual Variability for Drugs with High Intestinal First-pass Metabolism
Michael Gertz,1 Anthony Harrison,2 John Davis,3 J. Brian Houston,1 and Aleksandra Galetin4
1School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester, UK, 2Pfizer Global Research and Development, Sandwich, UK, 3Pfizer Global Research and Development, Sandwich, UK, 4School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester, UK
Intestinal availability (FG) represents an important parameter for the prediction of drug-drug interactions and oral clearance. Estimation of FG from in vivo or in vitro data is associated with considerable interindividual variability and parameter uncertainty. In the current study, 9 CYP3A4 drugs with FG ≤ 0.5 were selected to address the interindividual variability and assess the impact of drug absorption and enzyme abundance on the prediction of FG. Intrinsic clearances were determined in 9 individual jejunal microsomes (BD Gentest, Franklin Lakes, New Jersey, USA) and corrected for microsomal binding and intestinal CYP3A4 abundance. The 6ß-testosterone hydroxylation activity in the individuals ranged from 269 to 2,520 pmol/min/mg (1346 ± 747). For all the drugs with rapid absorption (Tmax < 2 hours), intestinal CLuint values were corrected for CYP3A4 abundance restricted to duodenum and jejunum (48.1 nmol), otherwise total CYP3A4 abundance (70.5 nmol) was used. Additionally, enterocytic drug concentrations were estimated to assess potential saturation of intestinal CYP3A4 metabolism in vivo. Determined intestinal CLuint ranged over 3 orders of magnitude and showed the following rank order: atorvastatin<buspirone<midazolam<felodipine = tacrolimus<lovastatin<simvastatin = nisoldipine<saquinavir. The CLuint values were associated with a CV of ~60% and were highly correlated to the CYP3A4 activity of the individual donors (r2 = 0.85) and also hepatic microsomal CLuint previously determined (r2 = 0.81). FG predictions were within 2-fold of the observed values in the case of midazolam, lovastatin, nisoldipine, and tacrolimus, whereas atorvastatin and buspirone FG were overpredicted (350%). In contrast, predicted FG values were only 6–36% of the in vivo estimates for saquinavir and felodipine, respectively. For these drugs, the enterocytic concentration exceeded their Km (>10 times in the case of saquinavir), implying that saturation of intestinal CYP3A4 contributes to the underprediction observed from in vitro data. Predicted interindividual variability of FG from experimental data (7–60%) reflected the variability observed in vivo (16–54%). This study highlights the importance of drug absorption and enzyme abundance on the accurate prediction of FG.
113. Transcription Factor Nrf2 Regulates Murine Cytochrome P450 2A5 Constitutively and in Heavy Metal Induction by Lead and Methyl-Mercury
Virpi Lämsä,1 Anna-Liisa Levonen,2 Seppo Ylä-Herttuala,2 and Jukka Hakkola1
1Department of Pharmacology and Toxicology, University of Oulu, Oulu, Finland, and 2Department of Biotechnology and Molecular Medicine, A.I. Virtanen Institute for Molecular Sciences, Biocenter Kuopio, Kuopio University, Kuopio, Finland
Background: Nuclear factor erythroid-derived 2-like 2 (Nrf2) regulates a battery of redox-homeostasis stabilizing genes through antioxidant response element (ARE) activation. Several Nrf2-responsive phase II detoxification target genes, such as glutathione S transferases (GSTs), have been described, but not much is known of potential phase I target genes. Recently, we discovered that murine Cytochrome P450 2a5 (Cyp2a5) induction by cadmium is Nrf2-mediated in the liver through an ARE residing at about -2,4 kb upstream of the transcriptional start site.
Hypothesis: In relation to the Nrf2/ARE pathway, we have further considered the high inducibility potential of Cyp2a5 in the liver by chemicals often hepatotoxic and downregulatory to total CYP activity. Here, we present data regarding CYP2A5 induction by heavy metals PbCl2 and methyl-Hg. Further, we have evidence for Nrf2-mediated constitutive activation of Cyp2a5. Previous studies have associated Nrf2 only in constitutive regulation of certain GSTs.
Methods and Results: Studies were conducted in cultured primary hepatocytes derived from wild-type and Nrf2(-/-) C57BL/6 mice. Real-time quantitative PCR-based analysis revealed CYP2A5 mRNA induction only in wild-type cells with about 10- to 30-fold induction after 24 hours of exposure to PbCl2 or Me-Hg. Nuclear protein extracts from induced cells showed nuclear accumulation of Nrf2 within 1 hour of treatment, and Nrf2 binding to the 5’ -2,4 kb upstream ARE was detected by chromatin immunoprecipitation. Unexpectedly, Nrf2(-/-) primary hepatocytes expressed less than 1% of CYP2A5 mRNA, compared to wild-type cells. Consistently, CYP2A5 mRNA and protein levels were 10-fold lower in livers of Nrf2(-/-) mice. CYP2A5 was upregulated by adenoviral overexpression of human Nrf2. Vice versa, CYP2A5 downregulation was detected by adenoviral overexpression of hKeap1, a cytoplasmic Nrf2 sequestering protein linked to proteosomal degradation.
Conclusions: Results imply a broad role for Nrf2 in induction of CYP2A5 by hepatotoxic chemicals such as heavy metals. Demonstration of Nrf2-mediated constitutive activation suggests putative endogenous role for CYP2A5, which, thus far, has mainly been considered as a xenobiotic-metabolizing isoform.
114. Epigenetic Regulation of the Liver-specific Expression of Mouse Oatp1b2, Ntcp, Bsep, Abcg5, and Abcg8
Satoki Imai,1 Ryota Kikuchi,1 Hiroyuki Kusuhara,1 Shintaro Yagi,2 Kunio Shiota,2 and Yuichi Sugiyama3
1Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan, 2Graduate School of Agriculture and Life Sciences, The University of Tokyo, Tokyo, Japan, 3Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan
Background: Liver-specific expression of transporters is highly relevant with the efficient turnover of endogenous substrates, such as bile acids and sterols, as well as drug disposition in the body. However, the regulatory mechanism of the tissue-specific expression of these transporters has not been fully elucidated. We have recently demonstrated that the kidney-specific expression of OAT3 and URAT1 is negatively regulated by DNA methylation in their promoter regions. DNA methylation is one of the most well-characterized mechanisms underlying the epigenetic regulation of gene expression. The objective of the present study was to investigate the involvement of epigenetic systems in the expression of liver-specific transporters, mouse Oatp1b2/Slco1b2, Ntcp/Slc10a1, Bsep/Abcb11, and Abcg5/g8 by 1) analyzing the DNA methylation status around the transcriptional start site (TSS) in the liver, kidney, and cerebrum and 2) exploring the status of histone H3 acetylation in the upstream regions of TSS.
Methods: Bisulfite genomic sequencing and the chromatin immunoprecipitation (ChIP) assay were performed to examine the DNA methylation and histone H3 acetylation profiles, respectively.
Results: Bisulfite sequencing demonstrated that the CpG dinucleotides around the TSS of Slco1b2, Slc10a1, Abcb11, and Abcg5/g8 were significantly hypomethylated in the liver, where these transporters are predominantly expressed. On the other hand, most of the CpGs were hypermethylated in the kidney and cerebrum, where the expression of these transporters is negligible. The ChIP assay further revealed that histone H3 in the Slco1b2, Slc10a1, Abcb11, and Abcg5/g8 promoters was hyperacetylated in the liver, whereas acetylation in the kidney and cerebrum was minimal. The epigenetic signatures determined by DNA methylation and histone modification profiles are in good agreement with the liver-specific expression of Oatp1b2, Ntcp, Bsep, and Abcg5/g8.
Conclusions: These results suggest that epigenetic regulation is involved in the tissue-specific expression of Oatp1b2, Ntcp, Bsep, and Abcg5/g8.
115. Response of Nuclear Import Factor Encoding Genes to Oxidative Stress
Manraj Cheema, Supaporn Yimthaing, Nick Plant, and Kathryn Plant
Center for Toxicology, Faculty of Health and Medical Sciences, University of Surrey, Guildford, UK
One defining feature of the eukaryotic cell is the presence of a nuclear envelope surrounding the genetic material. Access into the nucleus is restricted to the nuclear pore complexes (NPCs), which perforate the nuclear envelope; in many cases, the regulated transport of molecules through these complexes can partly control their activity. Nuclear-cytoplasmic shuttling lies at the heart of the response to oxidants: Its key regulator, Nrf2, is predominantly cytoplasmic in naïve cells, but upon oxidative insult, translocates to the nucleus and activates genes encoding cytoprotective enzymes to eliminate oxidants. Other components of the transcriptional response to oxidative stress also translocate to the nucleus in a temporal manner during the stress response: Fyn kinase phosphorylates nuclear Nrf2 and triggers its translocation back to the cytoplasm for degradation, while Bach1 competes with Nrf2 to downregulate certain promoters. Proteins destined for the nucleus are characterized by the presence of a nuclear localization signal (NLS) and their transfer through the NPC is mediated by nuclear import factors. Karyopherin (importin) β1 mediates the transfer of many nuclear proteins and is able to directly recognize some NLSs, although others require an additional factor, karyopherin α (KPNA), to act as a molecular bridge between cargo and karyopherin β1. In higher eukaryotes, there are multiple versions of karyopherin α, which show specificity in both cargo recognition and tissue distribution. Using a reporter gene assay system, we have studied the promoters of two members of the karyopherin α family. We have shown that both KPNA2 and KPNA3 are sensitive to hydroquinone, showing an EC50 of 10–15 μM in the human hepatoma cell-line, Huh7: Under these conditions, cell death was minimal and a control plasmid showed no repression. Interestingly, others have shown that during oxidative insult, nuclear import factors become mislocalized into high-molecular-weight nuclear complexes. We postulate that nuclear transport is disrupted by prolonged oxidative insult, and that this will have a feedback effect on the localization of Nrf2 and its coregulators.
116. Cellular Stress Responses of Nongenotoxic Carcinogens in Mouse Embryonic Stem Cells
Ilse Huijskens,1 Haziz Jadaar,1 Martijs Jonker,2 Timo Breit,2 Leon Mullenders,3 Harry Vrieling,3 John Meerman,1 and Bob van de Water1
1Department of Toxicology, Leiden University, Leiden, The Netherlands, 2MicroArray Department, University of Amsterdam, Amsterdam, The Netherlands, 3Department of Toxicogenetics, LUMC, Leiden, The Netherlands
Nongenotoxic carcinogens cause tumorigenesis without generating direct DNA damage. Yet, the biological responses affected by nongenotoxic carcinogens are very diverse and the corresponding signaling pathways are not well known. In addition, there is the lack of a predictive model to identify a potential nongenotoxic carcinogen hazard of novel chemical entities. Our long-term goal is to assemble toxicogenomics-based cell reporter systems to detect potential nongenotoxic carcinogenic features of chemicals by using mouse embryonic stem (mES) cells as a model. Here, we evaluated the cellular stress responses of four established nongenotoxic carcinogens in mES cells, including cyclosporine A (CsA), diethylstilbestrol (DES), 17β-estradiol (E2), and Wyeth-14643 (WY). mES cells were exposed at three different concentrations for each compound, followed by Affymetrix microarray analysis (Affymetrix - High Wycombe, United Kingdom). CsA, DES, and WY were responsive in mES. Significantly expressed probe sets were identified that recognize two or three of the four nongenotoxic carcinogens. However, no genes were identified that predict all four nongenotoxic responses, since mES cells presumably were relatively nonresponsive to E2. We also compared the microarray data of the above compounds with gene expression profiles associated with the genotoxic stress responses mediated by cisplatin. Approximately 50% of the cisplatin-induced DNA damage stress response biomarkers were not significantly altered after the nongenotoxic exposures. The remaining biomarkers significantly change after nongenotoxic carcinogen exposures, but expression levels are considerably lower compared to cisplatin. Thus, the DNA-damaging biomarkers are qualified to discriminate the nongenotoxic carcinogens CsA, DES, E2, and WY from the genotoxicant, cisplatin.
117. Discrimination Between DNA-Damage and Pro-Oxidant Stress Responses in Mouse Embryonic Stem Cells Based on Gene Expression Profiling
Bob van de Water,1 M. Raamsman,2 M. Atallah,3 and H. Vrieling3
1Division of Toxicology, Leiden University, Leiden, The Netherlands, 2Division of Toxicology, Leiden Amsterdam Center for Drug Research, Leiden, The Netherlands, 3Department of Toxicogenetics, Leiden University Medical Center, Leiden, The Netherlands
Genotoxic effects of chemicals are the primary cause of genomic instability followed by the onset of carcinogenesis. Tissue stem cells are the primary site where chemical-induced mutation may initiate tumor formation. Our goal is to establish classifiers for the DNA-damage response (DDR) in mouse embryonic stem (mES) cells, which are highly susceptible for genotoxic stress. Using time- and concentration-dependency-based Affymetrix gene expression studies with cisplatin (Affymetrix, High Wycombe, United Kingdom), we carefully characterized the timing of the DDR in mES cells. Next, we determined the gene expression for a panel of other DNA-damaging agents, including etoposide, doxorubicin, mitomycin C, and methyl methanesulfonate. Since some of these compounds may also indirectly induce oxidative stress, we compared the cellular stress response of the DNA-damaging agents with four pro-oxidants, including H2O2, t-butyl-hydroperoxide, diethylmaleate, and menadione. Our Affymetrix data indicate drastic gene expression changes after genotoxicant, as well as pro-oxidant, exposure. The DDR in mES cells clearly involves a p53 response. Our detailed bioinformatics analysis resulted in the identification of gene sets that are solely induced upon DDR. In addition, gene sets were identified that are upregulated upon pro-oxidant exposure, but not after DNA damage. We have cloned the promoter regions of around 40 promising candidate reporter genes and have tested these in both luciferase and DsRed-reporter constructs to discriminate between DNA-damage and pro-oxidant stress responses. This will lead to the development of novel toxicogenomics-based in vitro reporter assays enabling selective in vitro high-throughput screening of DDR versus pro-oxidant responses in mES cells.
118. Tobacco Smoke Biomarkers’ Discovery, a Metabonomics Investigation of Urine Samples from Smokers of 1, 4, and 10 MG ISO-Tar Yield Cigarettes
Daniel Waterman,1 Frank Bonner,1 Francis Cheung,2 Michael McEwan,2 and Emmanual Minet
1Metabometrix Ltd, London, UK, 2Risk Characterization, British American Tobacco, Southampton, UK
The conventional pipeline for biomarker identification in smokers targets specific endo- or xenobiotics, with an initial method-development phase, followed by clinical validation. This approach, however, is limited by our understanding of the basic underlying physiological processes involved in biomarkers’ formation. Therefore, the focus of biomarker discovery is shifting toward nonhypothesis-driven methods, such as metabonomics. We investigated the metabolic effects of cigarette smoking by performing 1H-NMR spectroscopy analysis of urine from human volunteers. In the first phase of our study, we wanted to determine whether it is possible to discriminate smokers from nonsmokers based on their total metabolic profile. Urine samples were collected from three groups, each smoking a different type of cigarette (1-, 4-, and 10-mg ISO-tar yields). The “smoker” groups’ metabolic profiles were compared to those from samples collected from 43 nonsmokers. Treatment groups contained 42, 43, and 44 subjects, respectively, and samples were analyzed by using multivariate statistics to determine differences between smoking groups and the controls. PLS-DA (partial least square-determinant analysis) showed that the samples could be separated according to their group classification in a pair-wise manner. The 10-mg tar group separated most significantly from the control group, followed by the 4-mg smokers, then the 1-mg group. The loadings plots indicated that entities with peaks in the regions of 2.7 and 8.6 ppm contributed most significantly to this separation. STOCSY (statistical total correlation spectroscopy) analysis provided evidence that the peaks at 2.7 and 8.5 ppm were linked; these matched publicly available nicotine NMR reference profiles. These peaks were excluded from the data set and further O-PLS-DA (orthogonal-projection on latent structure-DA) was performed to show that without these putative nicotine-derived compounds, the groups could still be clearly differentiated. Models excluding nicotine-related peaks suggested the differentiation between smokers and controls was due to a singlet peak at 1.43 ppm and other NMR peaks in the aromatic region. Further work is required to identify these putative biomarkers.
119. Characterization of the Drug Metabolism Profiles of Isolated and Cultured Rat Hepatocytes using Transcriptomic (Superarray) and Proteomic (iTRAQ) Technologies and Involvement of PI3 Kinase/Akt Signaling in CYP Protein Degradation
Cliff Rowe, Christopher E. Goldring, Neil R. Kitteringham, Rosalind Jenkins, and B. Kevin Park
C.D.S.S. Department of Pharmacology and Therapeutics, The University of Liverpool, Liverpool, UK
Loss of metabolic competence in cultured hepatocytes is a major limitation to their use in metabolism and toxicological studies. Expression levels of major genes of drug metabolism are greatly reduced in primary culture at the level of translated protein and gene transcript. We have undertaken analysis of the drug metabolism profile of hepatocytes at the transcriptional and proteomic levels by utilizing real-time PCR and proteomic technologies in order to characterize these changes in hepatocytes cultured for 48 hours under de-differentiating conditions. Within the transcription profile of the 84 drug metabolism genes analyzed by SuperArray, 39 were downregulated, 9 were upregulated, and 34 were unchanged after 48 hours of culture (SA Biosciences, Fredrick, Maryland, USA). The highly abundant genes, such as CYP2Cs, exhibited the greatest downregulation (>1,600-fold for CYP2C7). Proteomic analysis of whole-cell extracts revealed that the largest fall in abundance was seen for CYP2C7, which retained just 20% of the quantity of freshly isolated hepatocytes. Fourteen cytochrome P450 proteins were identified, of which 9 were downregulated in 48-hoursold cultures (to <0.75 of levels detected in freshly isolated cells). To begin to identify mechanisms through which these changes occur, several kinase inhibitors were utilized. CYP1A declines rapidly in culture, and we used Western blots for this as a marker of the loss of metabolic competence. Inhibitors of p38 and ERK1/2 had no effect on CYP1A1/2 protein, although levels were better maintained in the presence of an inhibitor of PI3 kinase (PI-103) in a dose-dependent manner. An inhibitor of Akt had similar effects. Real-time PCR analysis indicated that the effect was not transcriptionally based, as both control and exposed cells contained only around 5% of the CYP1A2 mRNA of freshly isolated cells. These data implicate the PI3 kinase/Akt pathway in the degradation of CYP1A proteins, but not the silencing of transcription. By further defining the mechanisms by which cultured hepatocytes lose their metabolic competence, it may be possible to intervene to maintain the metabolic competence for the purposes of metabolism and toxicology studies.
120. Chelidonium majus caused Toxicity in Primary Human, Monkey, Canine, and Rat Hepatocytes
Hendrik Rudolf,1 Anett Ullrich,1 Kaja Ludwig,2 and Dieter Runge1
1PRIMACYT Cell Culture Technology GmbH, Schwerin, Germany, 2Klinik für Chirurgie, Klinikum Südstadt Rostock, Rostock, Germany
Herbal extracts of Chelidonium majus have been widely used in European countries and in Chinese herbal medicines for different applications, based on their antiviral, anti-inflammatory, and antimicrobial properties and their use against various liver disorders including cancer. Recently, several cases of liver disease have been related to C. majus intake. Therefore, we have used primary hepatocyte cultures of human, rat, canine, and monkey to elucidate the potential hepatotoxicity of C. majus. Water-soluble extracts were prepared and incubated with primary hepatocytes. Cellular viability and functions were analyzed by means of lactate dehydrogenase (LDH) and urea release and via the MTT assay. The amounts of different alkaloids present in C. majus extracts were determined by HPLC analysis. The liquid extract showed a significant concentration-dependent toxicity in human hepatocytes and at concentrations of 7.5 mg/mL for animal hepatocytes. In addition, C. majus led to a decrease in urea release. The decline in urea production varied between the different species with regard to cellular sensitivity and to its maximal extent. The data suggest that human hepatocytes seemed to be more sensitive to C. majus extracts than canine, rat, and monkey hepatocytes, with monkey and canine hepatocytes being the most insensitive cells. In addition, these results demonstrate the necessity to use human hepatocytes in order to evaluate possible toxicological effects of complex plant extracts in humans.
121. Differentiated Primary Human Hepatocytes used for Toxicity Testing of Xenobiotics as Alternative to Animal Studies
Anett Ullrich,1 Ewa C.S. Ellis,2 Donna B. Stolz,3 George K. Michalopoulos,4 Stephen C. Strom,2 and Dieter Runge1
1PRIMACYT Cell Culture Technology GmbH, Schwerin, Germany, Departments of 2Pathology, 3Cell Biology, and 4Pathology, University of Pittsburgh, Pittsburgh, Pennsylvania, USA
Hepatocytes are used as biosensors for pharmacological-toxicological assays to monitor the influence of xenobiotics on hepatocellular functions and metabolism. We established a long-term culture system in which hepatocytes are plated and cultured under serum-free conditions for up to 2–3 weeks. During this time, the hepatocytes retain their specific functions determined by the quantification of urea and albumin release and stay vital, as measured by the release of LDH and controlled by light microscopy. Due to the maintenance of functionality, the same hepatocytes can be used more than once for assaying the effects of xenobiotics on hepatocellular functions and viability. We used acetaminophen as a model substance to demonstrate the feasibility of our culture system. High doses of acetaminophen led within 24 hours to a concentration-dependent and reversible decrease in urea and albumin synthesis and to ultrastructural changes, which were also reversible. The drug-mediated effects did not even change between the first cycle of application on day 4 and the fourth cycle started on day 20. This model was also used successfully to screen the toxic potential of the anticancer drug cis-4-hydroxy-proline. The safety of the drug was documented and in line with the clinical data from a human study, while the data generated in animals were not consistent with the clinical data and let to false conclusions. The robustness of the long-term culture system with human hepatocytes is also shown by repetitive inductions of CYP450 activities. Prototypical inducers increase the enzyme activities to identical levels, regardless of whether induction is started on days 3, 7, or 11 of culture. Thus, with modifications in the protocol, the potential of xenobiotics on the induction of CYP450 activities can be assayed. The model can be used especially in the early stages of drug development. It allows repetitive analyses of metabolic and toxic effects of drug compounds, and the compound can be optimized before starting more time- and cost-intensive animal studies.
122. Directed Differentiation of Human Embryonic Stem Cells into Hepatocytes for In Vitro Applications
Josefina Edsbagge,1 Barbara Kuppers-Munther,1 Jenny Lindqvist,1 Carina Ström,1 Gabriella Brolén,1 Nico Heins,1 Marie Rehnström,1 Lisa Athley,1 Gustav Eriksson,1 Janne Jensen,1 Monica Ek,2 Louise Sivertsson,2 Malin Darnell,3 Tommy B. Andersson,3 Inger Johansson,2 Magnus Ingelman-Sundberg,2 and Petter Björquist1
1Cellartis AB, Göteborg, Sweden, 2Karolinska Institutet, Stockholm, Sweden, 3AstraZeneca R&D Mölndal, Clinical Pharmacology and DMPK, Mölndal, Sweden
Human hepatocytes are an essential tool for analyzing the nature of xenobiotics and predicting drug metabolism for the chemical and pharma industries. However, the demand for primary human hepatocytes is much greater than the availability. In addition, the quality is not good enough, and logistics are problematic, costly, and cause limitations to studies. The derivation of human embryonic stem cell lines, and their ability to become any cell type of the body, inspires hope in finding an endless source of human hepatocytes. Besides unlimited supply, hESC-derived hepatocyte-like cells offer the user to source cells differentiated from multiple hESC-lines, reflecting a variety of different genotypes. Here, we present a stepwise directed differentiation protocol via the definitive endoderm to hepatoblast-like cells and finally to hepatocyte-like cells exhibiting characteristics similar to human hepatocytes. The hESC-derived hepatocyte-like cells have a typical hepatic morphology, express a number of liver-related genes, and show liver functions, such as glycogen storage and indocyanine green transport. Moreover, they metabolize three probe drugs for CYP1A, 3A, and 2C. In conclusion, our results represent an important step toward a future unlimited source of functional human hepatocytes useful in drug metabolism and toxicity testing.
123. Dynamic 3D Bioreactor Culture Model for Long-Term Maintenance of Human Hepatocyte Metabolic Function
Thomas Schreiter,1 Malin Darnell,2 Therese Söderdahl,1 Nils Bohmer,1 Daniel Knobeloch,3 Andreas K.N. Nüssler,4 Jörg C. Gerlach,5 Katrin Zeilinger,1 and Tommy B. Andersson2
1Division of Experimental Surgery, BCRT, Charité Universitätsmedizin Berlin, Berlin, Germany 2AstraZeneca R&D Mölndal, Clinical Pharmacology and DMPK, Mölndal, Sweden, 3Department of Surgery, Charité Universitätsmedizin Berlin, Berlin, Germany, 4Department of Traumatology, TU Munich, MRI, Munich, Germany, 5McGowan Institute for Regenerative Medicine, University of Pittsburgh, Pennsylvania, USA
The liver represents the central target for drug toxicity, which creates the need for in vitro systems allowing the detection of toxic effects of drugs and their metabolites in the preclinical phase of drug development. We focus on bioreactor developments addressing the cellular needs of three-dimensional (3D) tissue conditions. The four-compartment bioreactor consists of interwoven bundles of capillaries for supply with nutrients and oxygen, providing a 3D scaffold for the cells residing in the extracapillary space (cell compartment). In this study, we investigated the pattern and long-term stability of cytochrome P 450 (CYP) activities in primary human hepatocytes (pHHCs) and two liver cell lines (C3A and Huh7) cultured in a miniaturized bioreactor prototype. The kinetics of CYP-dependent metabolism was investigated by analyzing specific metabolites transformed by CYP isoenzymes. After inoculation, cells were allowed to adapt to the 3D environment before starting experiments. Parameters for cell viability, such as glucose consumption and production of lactate and ammonia, were measured daily. CYP1A1/1A2, CYP2C9, and CYP3A4 activities were determined by incubation with suitable substrates (phenacetin, diclofenac, and midazolam). Concentrations of parent substances and their metabolites were analyzed at 10 time points within 24 hours by using liquid chromatography/mass spectrometry (LC/MS). CYP activities of C3A cells were stable in bioreactors for up to 100 days. pHHC cultured in bioreactors for 3–5 days produced markedly higher metabolite concentrations than C3A cells: about 20 times for CYP1A1/1A2 and CYP3A4, and about 1,000 times for CYP2C9. Moreover, two pHHC bioreactor runs showed outstanding CYP activities after 23 or 12 days culture duration. In conclusion, the 3D bioreactor technology allows long-term maintenance of liver cells for repeated studies in the same experimental setup. CYP450 activity assays revealed high reproducibility for the applied cell lines and comparable results for pHHC from different donors. Superior to 2D cultures of pHHC, the functionality of CYP enzymes can be maintained for more than 3 weeks in bioreactors. Thus, the system could provide a useful instrument for in vitro drug metabolism studies.
124. Effect of Cryopreservation on the Activity of Human Hepatic Uptake Transporters
Lassina Badolo1 and Marie-Michèle Trancart2
1Discovery ADME, Lundbeck, Denmark, 2Xenoblis, Rennes, France
Isolated hepatocytes have been used for several decades in in vitro drug metabolism studies and have proven to conserve high metabolizing enzyme activities. The two-step liver perfusion techniques widely used to isolate hepatocytes have been reported as stressful for cell membranes. Cryopreservation of hepatocytes is an additional stressful procedure, as the cell is exposed to a high concentration of DMSO (10%) and an extremely low temperature. Although cryopreserved hepatocytes well preserve cytochrome P450 enzyme activities, only a few studies have documented the presence of functional drug transporters in cryopreserved hepatocytes. The present study had the goal to assess the ability of cryopreserved hepatocytes to maintain functional uptake transport proteins. The presence of functional transport proteins in cryopreserved hepatocytes would be highly advantageous due to the limited access of freshly isolated human hepatocytes. It also allows the standardizing of these studies by using cells from the same donors in several experiments and will reduce considerably the number of animals used in these studies. Freshly isolated hepatocytes were incubated with either [H3]-estradiol-17-β-glucuronide (substrate for OATP 1B1), 3H taurocholate (substrate for NTCP), or [H3]-Mpp+ (substrate for OCT1) at eight different concentrations. The uptake of radioactive substrates into hepatocytes was assessed by measuring the total radioactivity in the cell pellet. Background uptakes at 4°C were subtracted from the uptake at 37°C. Kinetic parameters (Km and Vmax) were determined to evaluate changes in both affinity and maximal activity. In these experiments, cryopreservation of human hepatocytes did not change the affinity of OATP1B1 or OCT1 for their substrates (estradiol-17-β-glucuronide and Mpp+, respectively); a decrease in the Vmax was observed for estradiol-17-β-glucuronide, while no statistically difference was observed for Mpp+.
125. Glutathione Levels in Fresh and Cryopreserved Human Hepatocytes as Detected by the Bioluminescent GSH-Glo™ Assay Kit
Ji-Young Lee, Timothy A. Moeller, and Scott Heyward Celsis
In Vitro Technologies, Baltimore, Maryland, USA
Glutathione (GSH) is an L-γ-glutamyl-L-cysteinyl-glycine tripeptide thiol that acts in a vast number of biological reactions. Among other crucial roles, it has essential functions in protecting the liver by inactivating reactive electrophilic and toxic xenobiotics and metabolic derivatives. As the major intracellular antioxidant, it scavenges various reactive oxygen species that are known to be involved in specific disease states. Consequently, GSH depletion can lead to impairment of physiological defenses, resulting in cellular injury and death. Hepatocytes are responsible for most xenobiotic metabolism and glutathione synthesis. Freshly isolated primary hepatocytes contain sufficient GSH levels to assess the toxicity of drug candidates. However, although cryopreservation of these cells has been reported to decrease GSH concentrations the extent of reduction has never been fully explored. Using the Promega GSH-Glo™ kit (Promega, Madison, Wisconsin, USA), a very rapid, simple, and reproducible luminescence-based assay, GSH levels were compared among various formats of fresh and cryopreserved human hepatocytes. In most instances, nominal differences were detected in GSH levels between fresh and cryopreserved hepatocytes. As expected, concentrations were subject to interindividual variability. For cryopreserved cell lots, incubation at 37°C changed GSH levels in a time- and lot-dependent manner. In hepatocyte suspensions, GSH quantities were, on average, one third lower at 4 hours than at initial thaw. In contrast, plated cells exhibited a time-dependent rise of at least 5-fold over 3 days. This increase appears to be mediated by de novo synthesis of GSH, as determined by the reduction of GSH levels in the presence of L-buthionine-sulfoximine (BSO), which selectively inhibits γ-glutamylcysteine synthetase, the first, rate-limiting enzyme in this process. Indeed, for some cryopreserved lots, GSH levels equaled that observed in fresh cells at similar timepoints. These results indicate that, in certain conditions, cryopreservation does not significantly alter initial GSH concentrations or the ability for de novo GSH synthesis.
126. Human HepaRG Hepatocytes, an Alternative In Vitro Model for Assessment of Drug Metabolism Induction
Ruoya Li,1,Sebastien Antherieu,2 Miia Turpeinen,3 Sandrine Camus,1 Rozenn Josse,2 Jouko Uusitalo,3 Guguen-Guillouzo Christiane,4 Andre Guillouzo,4 Ari Tolonen,3 Nathalie Rougier,1 and Christophe Chesne1
1R&D, Biopredic international, Rennes, France, 2INSERM U620/U522, Rennes, France, 3Novamass Ltd., Oula, Finland, 4INSERM U620, Rennes, France
The human HepaRG cells represent the first human hepatoma cell line able to differentiate in vitro into hepatocyte-like cells and display hepatocyte functions. Several lines of evidence have demonstrated that HepaRG cells exhibit 1) a hepatocyte-like morphology; 2) a greater metabolic competence for phase I and II enzyme activities; 3) a concomitant expression of hepatic influx and efflux transporters; and 4) a good inducibility on drug-metabolizing enzymes. The main objective of the present study is to determine whether HepaRG cells remain responsive to inducers of metabolizing enzymes, nuclear factors, and transporters involved in chemical metabolism when maintained during three subcultures at confluence for several weeks. After seeding of HepaRG cells at high density (subcultures 14, 17, and 20), the differentiated HepaRG hepatocytes that resulted from three end-time point cultures were treated by the following procedure: 1) depletion of DMSO for reducing the basal activity; 2) induction of HepaRG with three CYP model inducers (rifampicin, omeprazole, and phenobarbital); and 3) incubation of treated cells with a cocktail of eight probe substrates. Then, the effects of inducers on the mRNA expression profile of target genes (phase I and II metabolizing enzymes, transporters, and nuclear receptors) were quantitatively measured by RT-qPCR analysis, while the CYPs metabolic activities were assessed by LC/MSMS. Further, the functional expression/activities of hepatic efflux transporters were evaluated by a specific fluorescent substrates assay. Overall, our data indicate that HepaRG cells retain a stable maintenance of metabolic activity and related gene expression with subcultures (P14 to P20) as well as good inducibility of.hepatic biomarkers in response to inducers. Therefore, these results suggest that HepaRG hepatocytes should represent a valuable model for cocktail substrate-based high-throughput screening in drug metabolism induction and for the evaluation of drug transporter induction and activity in vitro.
127. Maintenance of a Variety of CYP, UGT, and Drug Transporter Activities in Rat and Dog Hepatocytes Preserved in Abcellute Matrix
Ken Grime
Discovery DMPK, AstraZeneca, Loughborough, UK
For effective and reproducible ADMET studies, there is a fundamental requirement for high-quality hepatocytes that maintain good morphology and hepatocyte specific functionality from the time of isolation to assay. Founded in late 2001, Abcellute is a company that markets a novel cell preservation technology (Abcallute, Cardiff, United Kingdom). The Abcellute matrix permits the storage of fresh primary hepatocytes with the potential to deliver cells “as freshly isolated” up to several days after isolation. In drug metabolism and pharmacokinetic studies, successful in vitro–in vivo extrapolation across two preclinical species is used to increase confidence that novel therapeutic drugs will have appropriate disposition in man. There may, therefore, be a commitment to the regular preparation of freshly isolated rat hepatocytes and slightly less frequent access to isolated dog hepatocytes. In this regard, additional flexibility offered by “Abcellute hepatocytes” is welcome. To that end, work was undertaken to investigate the maintenance of several key phase I drug-metabolizing enzymes in Abcellute rat and dog hepatocytes. Ketoprofen was used to investigate the maintenance of UGT activity and estrone sulphate, and tiotropium was used to probe drug-transporter activities. Excellent maintenance of hepatocyte function was demonstrated to 7 days. Additionally, the rat hepatocyte intrinsic clearance of a large number of AstraZeneca new chemical entities (NCEs) was shown to be very similar to when the compounds were assayed with rat hepatocytes freshly isolated at AstraZeneca. These results will be discussed in detail.
128. TNFα Enhances Diclofenac-induced Apoptosis of Hepatocytes. An Explanation For Idiosyncratic Drug-induced Liver Injury?
Lisa Fredriksson, Bram Herpers, and Bob van de
Water Toxicology, LACDR, Leiden, The Netherlands
Adverse drug reactions related to hepatotoxicity have been responsible for the withdrawal of many marketed drugs. Today, no adequate strategies are available to predict this kind of safety, especially not since these drug-induced liver injuries (DILIs) often are idiosyncratic and, therefore, rare. We hypothesize that idiosyncratic DILI occurs due to cross-talk between reactive drug metabolites and the immune response signaling. To study this hypothesis, human hepatoma HepG2 cells were exposed to diclofenac, which can cause DILI in humans, in the presence or absence of the proinflammatory cytokine TNFα. Using cell-cycle analysis, we established that diclofenac only induced a mild concentration-dependent apoptosis (subG1G0) of HepG2 cells, but no necrosis. While TNFα itself was not cytotoxic, it strongly enhanced the diclofenac-induced apoptosis. This apoptosis was associated with the onset of caspase activation, as determined by the formation of cleaved caspase-3 and enhanced DEVDase activity. Moreover, apoptosis caused by diclofenac/TNFα was inhibited by the pan-caspase inhibitor, z-VAD-fmk. Cell death was independent of the TNFR-mediated activation of caspase-8, since a specific caspase-8 inhibitor did not block diclofenac/TNFα-induced apoptosis. In the HepG2 cells, TNFα also rapidly activates the IKK/NF-κB pathway, resulting in a transient translocation of GFP-tagged p65 unit of NF-κB transcription factor to the nucleus. Since NF-κB target genes typically have proinflammatory and prosurvival functions, we wondered whether the enhanced apoptosis under diclofenac/TNFα conditions was due to modulation of the antiapoptotic TNFR-dependent signaling. While TNFα alone readily activated the IKK-dependent phosphorylation of IκBα, this phosphorylation was affected by diclofenac pretreatment. We propose that diclofenac treatment will induce a cellular stress response that suppresses the TNFα-related antiapoptotic stress response. Currently, we are further investigating the downstream signaling components that underlie these effects. We anticipate that by unraveling the mechanism behind the increased hepatotoxicity observed after both reactive drug metabolite formation and an inflammatory stress response, we will be able to identify mechanism-based biomarkers that can predict idiosyncratic DILI in a preclinical drug development setting.
129. Development of High-throughput Human OCT2 Expressing Uptake Assay System
Zoltan Nagy, Viktoria Juhasz, Attila Nemeth, Peter Krajcsi, and Hristos Glavinas
R&D, SOLVO Biotechnology, Budaors, Hungary
Organic cation transporters are important for the elimination of many drugs and toxins from the body. The goals of this study were to establish a cell culture model that stably expressed OCT2 and that could be used to study the characteristics and drug interaction of this transporter, and to study drug-drug interaction in this assay system. In the present study, substrate-transporter interactions were investigated in Chinese hamster ovary cells stably transfected with the human orthologs of the principal organic cation transporter in the kidney, OCT2. We set up a high-througput cell-based uptake assay system with the selected CHO-KI monoclone stably expressing OCT2. Cell culturing and assay protocol was developed on a 96-well plate. Cell number and time course of culturing were optimized. Time dependence of the OCT2-mediated tetraethylammonium (TEA) uptake was established. The transport activity was pH dependent. The uptake of the model organic cation, TEA, was saturable (K+, ~70 μM; V+, ~400 pmol/mg of protein/min) and was inhibited by known OCT inhibitors (e.g., cimetidine, quinidine, quinine, and verapamil). Values for IC50 of verapamil and quinidine for TEA uptake were ~6 and ~30 μM, separately. Metformin was suggested as a superior substrate for renal OCT2, rather than hepatic OCT1, and renal OCT2 plays a dominant role for metformin pharmacokinetics. In order to validate our uptake assay system for a certain pharmacokineticaly relevant drug, metformin was also examined using our CHO-KI cells stably expressing OCT2. Collectively, all the data indicated that the developed high-throughput uptake assay system can serve as a useful and convenient tool in screening candidate drugs for interaction with OCT2 and for studying drug-drug interaction.
130. Quantitative Automated Cellular Imaging of Oxidative and Inflammatory Stress Responses in Xenobiotic-induced Hepatotoxicity
Bram Herpers, Lisa Fredriksson, and Bob Van de
Water Division of Toxicology, LACDR, Leiden, Netherlands
The liver is the major site for drug metabolism and, as such, the most heavily exposed organ to drug metabolites. Adverse drug reactions that compromise liver function are hypothesized to be caused by a negative interaction between these drug metabolites and existing inflammatory stress, which compromises cell function and thereby induces cell killing. To test this hypothesis, we set up a high-throughput imaging-based assay for the BD Pathway 855 system that allows the visualization of the two major stress responses occurring during drug metabolite formation and inflammation in the human HepG2 and the mouse Hepa1c1c7 hepatocyte cell lines. By fluorescently tagging the oxidative stress responsive transcription factor, Nrf2, the accumulation of reactive metabolite-induced stress can be monitored over time. The response to inflammatory stress is monitored through fluorescently tagged p65, a subunit of NF-kB. The onset of apoptosis is monitored by Annexin-V-Cy5 addition. The intracellular accumulation of Nrf2, the nuclear translocation of p65, and the apoptosis are induced by exposing the cells to different compound concentrations combined with various cytokines and TLR ligands. Using this system, we can quantify the rate of reactive metabolite formation through monitoring Nrf2 accumulation induced by the metabolism of drugs linked to adverse drug reactions, such as diclofenac, clozapine, and troglitazone, and link this degree of Nrf2 induction to shifts in cytokine-induced p65 translocation and to enhanced rates of apoptosis induced by drug-cytokine combinations. The combinations that indicate cross-talk between the two stress pathways will be used as starting points for gene expression analysis and siRNA-mediated knockdown of kinases and phosphatases that might mediate this cross-reaction to identify relevant signaling pathways that can be used as biomarkers for the prediction of hepatoxicity-related adverse drug reactions.
131. Physiologically Based In Silico Prediction of Intestinal Drug Absorption Used as a Tool to Justify the Waiver of In Vivo Bioequivalence Studies
Ivan Kovacevic,1 Jelena Parojcic,2 Marija Tubic-Grozdanis,3 and Peter Langguth3
1National Pharmacovigilance Center, Medicines and Medical Devices Agency of Serbia, Belgrade, Serbia and Montenegro, 2Faculty of Pharmacy, Belgrade, Serbia and Montenegro, 3School of Pharmacy, Johannes Gutenberg-University Mainz, Mainz, Germany
The ability to accurately predict intestinal drug absorption based solely on in vitro data provides an opportunity to simulate in vivo drug performance. There are several physiologically based mathematical models that were used, with a relatively high degree of accuracy, for human drug absorption predictions. These models are based on the interplay between the drug characteristics and the human physiology to simulate the processes involved in the drug intestinal absorption, thus providing the information about the underlying mechanisms and absorption limitations (1). The Biopharmaceutics Classification System (BCS) has been introduced as a scientific framework for classifying drug substances according to their aqueous solubility and intestinal permeability. It is based on the mechanistic assumptions that the rate and the extent of oral drug absorption are governed by drug solubility, intestinal permeability, and dissolution rate from the dosage form administered. One of the goals of BCS is to identify classes of drugs for which bioequivalence may be established based solely on the in vitro dissolution data, that is, which would be eligible for biowaiver (2, 3). At present, the biowaiver concept is adopted and recommended for the immediate release drug products containing high-solubility and high-permeability compounds (BCS class I drugs) (4, 5). Biowaiver extensions have also been discussed for BCS class III drugs, as well as for the class II drugs, under the presumption that they dissolve completely during the gastrointestinal passage (6, 7). The aim of the study was to use gastrointestinal simulation technology, based on the advanced compartmental absorption and transit (ACAT) model—GastroPlus® (Simulations Plus, Inc., Lancaster, California, USA), as a tool to investigate the possibility of the extension of biowaiver criteria, using several model drug substances (carbamazepine, losartan, omeprazole, and valproic acid) belonging to the different BCS classes (I, II, and III). Further, the relative performance of the ACAT model and its limitations for in vivo drug disposition prediction are discussed.
132. Utilization of In Silico Modeling to Determine Control Coefficients for Development of Multiple Drug Resistance Phenotype in Breast Cancer Cells Exposed to the Anticancer Agent Paclitaxel
Nick Plant
Center for Toxicology, University of Surrey, Guildford, UK
More than 1 million women are diagnosed with breast cancer every year, which represents approximately one quarter of all new cancers in women: Such a rate of diagnosis represents a lifetime risk of developing breast cancer of 1 in 8 for women born in the United States and 1 in 9 for women in the United Kingdom, Simulations Plus, Inc, Lancaster, California, USA. Fortunately, there are a number of established therapies for metastatic breast cancer (MBC), ranging from endocrine-based therapies for hormone-receptor-positive tumors through anthracyclines and taxanes to the recent development of novel biologics, such as trastuzumab (Herceptin). Despite this range of therapeutics, the response rate to first-line chemotherapies, such as anthracyclines and taxanes, is suboptimal, being reported as between 30 and 70%, falling to 20–30% for subsequent treatments with a median duration of response of 6 months. One area that often limits chronic therapeutic treatment, including anticancer chemotherapy, is the development of a multiple drug resistance (MDR) phenotype, and it has been estimated that MDR is involved in over 90% of treatment failures for MBC. A large amount of research has been undertaken to understand the development of the MDR phenotype during treatment of MBC, and several mechanisms have been identified those factors that will contribute: increased metabolism, increased export, altered interaction with target proteins, and altered biological response to that interaction. Based upon this large body of evidence, we have created both deterministic and stochastic models for the development of cellular resistance to paclitaxel (Taxol) during the treatment of MBC, using CellDesigner and COPASI. We are able to replicate the cyclical growth/catastrophe phases of microtubule flux and the disruption of this caused by paclitaxel binding. Using literature expression levels for each species within the model, we have, for the first time, been able to combine these disparate experimental approaches and determine the relative importance of each mechanism in the overall development of MDR. Such data are an important first step in the development of novel therapeutic strategies to mitigate MDR.
133. An OATP1B1 Inhibition Assay using Nonradiolabeled Pitavastatin: Important Results and Exploitation of the Assay as a Surrogate for Human Hepatocyte Active Uptake Intrinsic Clearance
Ken Grime,1 Matt G. Soars,2 and Robert J. Riley1
1Discovery DMPK, AstraZeneca, Loughborough, UK, 2Discovery DMPK, AstraZeneca, Leicestershire, UK
The organic anion-transporting polypeptides (OATPs), organic anion transporters (OATs), and organic cation transporter (OCTs) are responsible for the sodium-independent hepatic uptake of a plethora of organic substrates, with perhaps the most important, in terms of drug disposition, being the OATP family. Isolated hepatocytes have become the system of choice for obtaining quantitative information regarding hepatic drug uptake, and successful in vitro–in vivo extrapolations of primary pharmacokinetic parameters have been made (Soars et al., 2007; Paine et al., 2008). This piece of work primarily describes the utility of an inhibition assay utilizing nonradiolabeled pitavastatin and human OATP1B1 expressed in HEK293 cells. Screening of a wide range of chemical space across acidic, basic, and neutral charge types has revealed some surprising results, which will be discussed. Additionally, we investigated the possibility that the OATP1B1 assay could be used as a surrogate for human hepatocyte uptake intrinsic clearance and discuss the consequences of the findings. In the same context, in vitro rat data will also be described and discussed.
134. Comparison of the Binding of Drugs to Human Intestinal Fatty Acid Binding Protein and Albumin: Application to In Vitro–In Vivo Extrapolation
John O. Miners, Andrew Rowland, Peter I. Mackenzie, and Kathleen M. Knights
Clinical Pharmacology, Flinders University, Adelaide, Australia
Like albumin, human intestinal fatty acid binding protein (IFABP) avidly binds fatty acids and may thus serve as an alternative to albumin as a sequestrant of inhibitory long-chain unsaturated fatty acids in incubations of human liver microsomes (HLMs). This work aimed to: 1) characterize the binding of acidic, neutral, and basic drugs to IFABP and, for comparison, to bovine serum albumin (BSA), and 2) compare the effects of IFABP and BSA on the kinetics of zidovudine glucuronidation by HLM. His-tagged IFABP was expressed in Escherichia coli and purified by affinity chromatography. Each molecule of purified IFABP bound a single molecule of the fluorescent probe, 1-anilino-8-naphthalene sulfonate, or arachidonic acid, with Kd values similar to those reported for the rat protein. Basic drugs bound negligibly to IFABP. In contrast, all acidic drugs were investigated and the neutral compound β-estradiol bound to IFABP. The presence of a carboxylate group was not a requirement for binding. The binding of acidic and neutral to IFABP (0.5% w/v) was independent of concentration, with fractions unbound and ranging from 0.45 to 0.89. For all drugs investigated, however, fractions unbound in the presence of BSA were lower, compared to the same concentration (0.5% w/v) of IFABP, due to the higher binding capacity of albumin. For example, mean fractions unbound for torsemide in the presence of IFABP and BSA were 0.80 and 0.07, respectively. The kinetics of zidovudine glucuronidation by HLM were characterized in the absence and presence of BSA and IFABP (0.5–2.5% w/v). Each protein reduced the Km for zidovudine glucuronidation in a concentration-dependent manner, although a higher content of IFABP in incubations (2.5% vs. 1–1.5% for BSA) was required for a 10-fold reduction in this parameter. The results indicate that IFABP is likely to have advantages over BSA in microsomal kinetic studies with drugs that bind extensively to albumin.
135. Development of an In Vitro Assay for the Assessment of the Reactivity of Acylglucuronide and AcylCoA-thioester Metabolites
Ester Tor Carreras, Calaf Elena, Córdoba Mònica, De Luca Manel, Albertí Joan, and Salvà
Miquel Pharmacokinetics and Metabolism, Almirall, Sant Feliu de Llobregat, Spain
Drugs bearing a carboxylic acid moiety may be metabolized to reactive electrophilic intermediates capable of binding covalently to proteins. These modifications are thought to contribute to drug toxicity, either through alteration of the functionality of the modified protein or through antigen formation causing subsequent immune reactions. This report will focus on two important groups of electrophilic intermediates: xenobiotic acyl glucuronides (AG) and xenobiotic acyl-coenzyme A thioesters (acyl-CoA thioesters). A number of UDP-glucuronosyltransferases (UGTs) catalyze the formation of AG. These enzymes have the highest activity in the liver, thus constituting the major site of AG formation. In this study, in vitro reactions, using glutathione as a nucleophile, have been used to mimic the covalent binding of AG to proteins. On the other hand, it is known that AG intermediates are unstable at physiological pH and form positional isomers by intramolecular acyl migration (transfer of the acyl group from 1ß to the C-2, C-3, or C-4 position of the glucuronic acid ring). These isomers could also react covalently with proteins. Determination of the in vitro stability of the 1ß-isomer has been employed herein for assessing the reactivity of potential drug candidates. The alternative pathway by which xenobiotic carboxylic acids can be bioactivated is the formation of acyl-CoA thioesters. This reaction is catalyzed by a number of microsomal acyl-CoA synthetases. An in vitro screening model to determine the reactivity of AG and acyl-CoA thioesters to proteins has been developed. With this setup, a single incubation with human liver microsomes allows to generate acyl glucuronides, identify the ß-1-O-acyl-glucuronide, determine its chemical stability in buffer, and study the reactivity of the generated acyl glucuronides toward gluthathione. The novel assay was validated by using a series of reference compounds. Time dependency of acyl-CoA-thioester formation has also been studied by using ibuprofen as a positive control for the acyl CoA thioester reaction in human and rat liver microsomes.
136. DNA Damage after Exhaustive Exercise, using Comet Assay and Protective Role of Vitamin E
Diren Beyoglu,1 Gulden Z. Omurtag,1 Nuri Topsakal,2 Inês Franco do Couto Monteiro,1 H. Birol Cotuk,3 and Semra Sardas1
1Toxicology Department, Faculty of Pharmacy, 2School of Physical Education and Sports, and, 3Research Center for Sport Health Sciences, Marmara University, Istanbul, Turkey
Exercise appears to increase reactive oxygen species (ROS), which can result in damage in all cellular macromolecules, such as DNA, lipids, and proteins. The rise in oxygen consumption may increase the ROS production; leading to oxidative stress. Oxidative DNA damage may play an important role in biological processes, such as mutagenesis, carcinogenesis, and aging in humans. The comet assay, which detects DNA strand breaks and alkali-labile sites, has been used in the study. Genotoxicity tests reflect reversible damage resulting from a toxic interaction, either at the molecular target or at an analogous side target, which is considered to be pathogenically linked to cancer. The purpose of this study was to investigate if exercize induces DNA damage and to assess the protective effect provided by antioxidant supplementation. The study participants were competitive rowers (n = 10) and sport academy students (n = 10). Exhaustive exercize tests were performed by all participants. Vitamin E supplementation was given to all subjects for 2 months. At the end of vitamin E consumption, the comet assay was again carried out in all subjects. Mean tail %DNA for each cell was calculated as 100-head %DNA. Blood samples from all subjects were taken before the exercize and 24 hours after the exercize. The same application was carried out for all subjects after 2 months of vitamin E supplementation. The preliminary results of this study show that DNA damage increases after exhaustive exercise and vitamin E seems to have a slight decrease in the mean tail %DNA.
137. Inducibility and Activity of CYP1A1/CYP1B1, the Major Polyaromatic Hydrocarbon Activating Enzymes, is Maintained in Primary Human Bronchial Epithelial Cells after Differentiation In Vitro
Nik Newland, Katherine Hewitt, and Emmanuel Minet
Risk Characterization, British American Tobacco, Southampton, UK
Inhaled xenobiotics and organic pollutants such as PAHs (polyaromatic hydrocarbons) are, in part, activated in the lung. Differentiated primary human bronchial epithelial cells (HBECs) and the NCI-H292 lung carcinoma cell line have been used in vitro to investigate responses to inhaled xenobiotics associated with lung diseases such as COPD and lung cancer. Lung bioactivation of xenobiotics is, in part, driven by CYP1A1 and CYP1B1. Loss of P450s expression/inducibility in in vitro primary cell cultures has been documented in many cell types but has not been investigated in differentiated/polarized HBECs. Since P450s play a role in smoke xenobiotics-related lung injuries, HBECs differentiated in vitro should be characterized for phase I activity. HBECs from 3 donors and NCI-H292 control cells were cultured for 28 and 5 days, respectively, at an air-liquid interface. HBECs polarization and differentiation was evaluated by electron-microscopy and transepithelial resistance. CYP1A1/1B1-dependent oxidative metabolism was measured with a LMAXII luminometer following the oxidation of a Luc-CEE probe. When needed, CYP1A1 and CYP1B1 were induced with TCDD (10 nM) for 72 hours. Following TCDD induction and incubation with luc-CEE, CYP1A1/CYP1B1 activity is increased by 23-, 66-, and 22-fold in HBECs from 3 individual donors. Incubation with the CYP1A1/CYP1B1 specific inhibitor, alpha-naphthoflavone, prevented the oxidation of the reporter probe. Total RNA from NCI-H292 and HBECs cells was analyzed by QRT-PCR for CYP1A1 and CYP1B1 gene expression to further confirm the TCDD-dependent induction of CYP1A1/1B1. We conclude that differentiated HBECs polarize, form a tight monolayer, and maintain CYP1A1/1B1 inducibility after 28 days in culture. Further work is required to assess the expression and activity of other phase I and II enzymes.
138. Prediction of Xenobiotic Passage through the Blood-Brain Barrier using a Primary Rat Syngenic Coculture In Vitro Model
Nicolas Perrière
VigiCell, Villejuif, France
Context: One of the major issues for the development of pharmaceutical compounds is the capacity to cross or not the blood-brain barrier (BBB). Development of a reliable in vitro model is a key discriminating step before in vivo testing.
Aims: The aims of this work were to develop and characterize an in vitro model enabling 1) to determine the overall passage of a xenobiotic into the brain for screening purposes—notably, the challenge was to optimize the model while maintaining high performance and 2) to study the xenobiotic specific entry route (paracellular or transcellular, endocytosis, transporter-mediated efflux or influx). For mechanistic purposes, we studied transporter functionality as well as their transcripts by RT-PCR.
Methods: We used a previously described original primary model of rat BBB (Perrière et al., 2005, 2007). Culture protocols were optimized to preserve brain endothelial cell specificity in order to mimic the in vivo BBB environment.
Results: BBB endothelial cells present specific attributes, which are required to maintain brain homeostasis, such as tight junctions and functional efflux transporters: P-gp, Mrps, and Bcrp. 1) Low paracellular permeability for small hydrophilic compounds and 2) daunorubicin permeability studies (from apical to basolateral side and conversely) confirmed a polarized state of the endothelial monolayer with a ratio >20 (unpublished data). In addition, we tested four pharmaceutical compounds with different P-gp affinity in the presence or absence of PSC833, a specific P-gp inhibitor. The mRNA efflux transporter levels showed similar transcription patterns compared to in vivo assessments (unpublished data).
Conclusion: For screening purposes, we adapted this model in terms of 1)_ quality and reproducibility and 2) in terms of quantity and reactivity.
139. Short-Term Culture of Cryopreserved Lung Slices, a Model to Profile Smoke Xenobiotics Metabolism
Nik Newland, Andrew Baxter, and Emmanuel Minet
Risk Characterization, British American Tobacco, Southampton, UK
Activation of tobacco smoke xenobiotics, such as polyaromatic hydrocarbons (PAHs) and aza-arenes, is recognized as one mechanism of potential relevance in the etiology of smoke-related lung diseases. However, from the estimated thousands of chemicals present in cigarette smoke, only a limited number have been evaluated as potential lung carcinogens. Study of smoke xenobiotics bioactivation and in vitro assessment of potentially harm-reduced tobacco products requires an array of metabolically active models representative of smoke xenobiotics target tissues. Although in vitro preparations such as microsomes and S9 fractions are used as rate-optimized systems to evaluate xenobiotics metabolism, they do not represent the full physiology of the tissue of interest. Therefore, we wanted to evaluate whether cryopreserved lung slices, which conserve the overall tissue morphology, can be used as a tool to model smoke xenobiotics metabolism. We used methylcholantrene-induced and noninduced rat cryopreserved lung slices obtained from Biopredic (Biopredic, Rennes, France). To evaluate CYP1A1- and CYP1B1-dependent oxidative metabolism, the slices were incubated with a Luc-CEE luminogenic probe or with pyrene (10 uM) for periods of 3 and 20 hours, respectively, with or without alpha-naphthoflavone. Oxidation of the luminogenic probe was recorded with a LMAXII luminometer and showed a 14-fold increased activity between the induced and noninduced preparations. For pyrene, the culture media was collected and analyzed by LC-APPI (atmospheric pressure photoionization)-MS/MS. 1-OH-pyrene (1–0.8 uM) was detected in the media from induced slices following treatment with glucuronidase. This is indicative of both phase I and II metabolic activity. A limited amount of 1-OH-pyrene was detected in the noninduced slices (0.1 uM). Viability of the slices was evaluated by a LDH leakage assay, and no significant reduction in viability was detected following incubation with the probe substrates and inhibitor. We conclude that CYP1A1/1B1-dependent oxidative metabolism and UGT-mediated conjugation are active pathways in short-term cultured cryopreserved rat lung slices. Although further characterization of other phase I and II enzymes is required, this model constitutes a potential tool for the metabolic profiling of smoke xenobiotics.
140. The Organophosphorous Compound Diethyldithiophosphate Present Immunotoxic Activity in Human Cells
Libia Vega,1 Melquisedec Esquivel-Sentíes,1 and Guillermo Elizondo2
Departments of 1Toxicology and 2Cell Biology, CINVESTAV, México D. F., Mexico
Diethyldithiophosphate (DEDTP) is a dialkylphosphate (DAP) produced by the biotransformation of organophosphorous (OP) pesticides. Although the neurotoxic effects of OP pesticides have been studied in human populations, there is no information regarding the immunotoxic effects of these compounds or the possible effects produced by OP metabolites. To date, only one study describes the immunotoxic effects of DAPs on human lymphocytes (Lima and Vega, 2005). This report indicates that DAPs uncouple the activation and proliferation signals on human peripheral blood mononuclear cells (PBMCs), thus altering the secretion of cytokines, such as interleukin-2 (IL-2), and altering activation cell markers, such as CD25 (IL-2 receptor α) and CD69 molecules (early cell-activation marker). The aim of this study was to determine the effect of DEDTP on the activation pathway of PBMCs by evaluating the phosphorylation status of three protein kinases involved in IL-2 secretion (STAT5, SOCS3, and NFAT) and in the induction of cell proliferation (ERK, JNK, and P38). We used the incorporation of tritiated thymidine (3HT) and the induction of the high-affinity IL-2 receptor (CD25, CD122, and CD132 molecules) as proliferation and cellular activation parameters, respectively. PBMCs were incubated in supplemented RPMI medium at 37ºC. Cells were treated with DEDTP dissolved in DMSO with or without further αCD3/CD28 antibodies stimulation. DEDTP treatment alone induced expression of CD122 and CD132 molecules, but reduced CD25 expression. PBMCs also showed reduced 3HT incorporation without compromising cell viability. DEDTP treatment induced phosphorylation of all protein kinases evaluated. When DEDTP treated cells were activated with αCD3/CD28 antibodies, we observed an increase in cell proliferation not associated with the secretion of IL-2. These results indicate that DAP have an immunotoxic effect due to modification of the basal as well as the induced phosphorylation of protein kinases as the probable mechanism of action. (This work was supported by CONACyT 172660.)
141. Organophosphorous Pesticides Metabolites Show Genotoxic Effects Depending on the Metabolic Status of the Cells
Libia Vega,1 Francisco Leyva,2 Mahara Valverde,2 Guillermo Elizondo,3 and Emilio Rojas2
1Toxicology, CINVESTAV, México D. F., Mexico, 2Biomedical Research Institute, UNAM, México D. F., Mexico, 3Cell Biology, CINVESTAV, México D. F., Mexico
Human exposure to organophosphorous (OP) pesticides increases the frequency of sister chromatid exchanges, chromosomal aberrations, and micronucleus formations in peripheral blood mononucleated cells (PBMCs). OPs are biotransformed by cytochromes P450 (CYP) 2D6, CYP3A4, and A-esterases in hepatic cells into dialkylphosphate metabolites (DAPs), which have a longer half-life and elimination period than their parental compounds. There are reports on susceptibility to the toxic effects of OP pesticides, but scarce information exists regarding the toxicity of their metabolites (Lima and Vega, 2005). To determine if diethylthiophosphate (DETP) and diethyldithiophosphate (DEDTP), two of the major metabolites of OP pesticides, could present genotoxic capacity and further elucidate their possible genotoxic mechanisms, we treated WRL68, HepG2, HeLa, and PBMC with different concentrations of DETP and DEDTP dissolved in DMSO (1–100 μM) and determined their genotoxicity by using the single-cell gel electrophoresis assay (SCGEA, comet assay) measured as the olive tail moment. We also explore the possible participation of the oxidative stress generation and CYP450 enzymes in the genotoxicity induced by OP metabolites. Our results showed that both OP metabolites (DETP and DEDTP) produced DNA damage only in the hepatic cell lines without damaging DNA of HeLa cells or PBMC. The genotoxic effect of DETP and DEDTP on hepatic cells could be due to a secondary nondiffusible metabolite generated by CYP450 enzymes activity, since previous incubation of cells with sulconazole, a general inhibitor of CYP450 enzymes, inhibited the induction of DNA damage in hepatic cells. The incubation of PBMC or HeLa cells with the conditioned media of HepG2 and WRL68 cells exposed for 48 hours to DETP or DEDTP did not induce DNA damage, indicating that the biotransformation process per se directly induced DNA damage. The generation of possible secondary metabolites of OP pesticides should be taken in account when assessing risk to human populations exposed to OP pesticides, since not only the parental compound, but also the metabolites could be implicated in the health effects observed.
142. Assessment of the Irritation Potential of Dermal Products using Episkin®
Cameron Bain, Jon Welch, Daniela Gentile, and Stephen Madden
In Vitro Sciences, Charles River, Edinburgh, UK
ECVAM have approved the Episkin® model for in vitro assessment of skin irritation. We have evaluated this model by using a selection of compounds from the ECVAM validation and dermal products available over the counter (OTC) from a local pharmacy. Episkin inserts were placed in 12-well plates containing culture medium and transfered to an incubator at 37° in, 5% CO2 for 24 hours and allowed to equlibrate. Then, 10-mg solid or 10-μL liquid test materials were then applied to the surface of the Episkin (n = 3, per product). Solids were applied with a few drops of phosphate-buffered saline (PBS) to dampen the skin surface. After 15 minutes, the inserts were rinsed with PBS. The Episkin insert was then incubated in fresh culture medium for 42 hours at 37°C and 5% CO2. The inserts were then transfered to media containing MTT (0.03%, w/v) for 3 hours. Following incubation with MTT, Episkin discs were removed with a biopsy punch and placed in microcentrifuge tubes with extraction solvent and vortex mixed to extract the formazan. The absorbance of duplicate aliquots of formazan extract was measured on a plate reader at 550 nm. All materials selected from the ECVAM validation experiments produced the cited irritant (5% SDS, tris-isobutylphosphate, 1-decanol, and propyldisulfide) or nonirritant (PBS, naphthalene acetic acid, methyl stearate, isopropanol, and dipropylene glycol) classification. PanOxyl in two formulations, Acnegel (ethanol based) and Aquagel (water based), was classified as a nonirritant, as were the topical anesthetics, Vagisil® and Lanacane®, and the two antifungal treatments, Canesten AF® and Lamisil®. Ibuprofen was noniritating when applied neat and in formulation. Bazuka gel™ (salicylic acid, 26%) was classified as an irritant, corresponding to in vivo data for salicylic acid. Benzoyl peroxide, DEET, and 6% hydrogen peroxide were classified as nonirritants, despite being described as mild irritants. In conclusion, the Episkin irritation test has been validated for use in our laboratories. A number of test chemicals and OTC preparations were clearly classified as irritants or nonirritants from using the model.
143. Defluorination of Halogenated Aniline via Bioactivation by Glutathione: Evidence for the ipso Addition of Glutathione
Chenghong Zhang, Jane Kenny, Cornelis Hop, and Cyrus Khojasteh Drug
Metabolism and Pharmacokinetics, Genentec, Inc., South San Francisco, California, USA
Bioactivation of several halogenated anilines were investigated in human liver microsomes in the presence of NADPH and glutathione (GSH). Here, we present some SAR around the bioactivation of 4-iodo-2-fluoro-aniline and propose a mechanism of adduct formation. We observed that when 4-iodo-2-fluoro-aniline was incubated in human liver microsomes fortified with NADPH and GSH, it led to the formation of five GSH conjugates. Four of these metabolites had the addition of oxygen and GSH to parent with or without defluorination, possibly due to epoxide or benzene oxidation followed by quinine imine formation. The last conjugate was a GSH adduct of the defluorinated parent. There are two possible mechanisms for the formation of this metabolite, both formed via the same reactive intermediate: (3-fluoro-4-iminocyclohexa-2,5-dienylidene)iodonium (). The first proposed mechanism is that GSH attacks at the 2-C. The second involves the ipso addition of GSH at the amino-carbon (1-C,) followed by an attack on the 2-C by the thiol group in GSH. The last step of both mechanisms is loss of HF and 2 electron reduction to form 4-iodo-2-glutathione-aniline. A number of halogenated aniline analogs were investigated to define the mostly likely of the two proposed mechanisms. It was observed that defluorination takes place only when fluorine is on 2-C or 6-C. When 4-iodo-3-fluoro-aniline was incubated under the same conditions, no defluorinated GSH conjugate was detected. Interestingly, the rate of dehalogenated GSH conjugate formation increases when iodine on 4-C is substituted with other halogens and has a rank order of Br>Cl>I. No GSH conjugate was observed with 2,4-difluoro-aniline. We also determined that other halogens in position 2 could also lead to dehalagenation and GSH adduct formation. These data suggest that the mechanism involved is the ipso addition of GSH at the amino-carbon (1-C), followed by an attack on the 2-C by the thiol group in GSH.
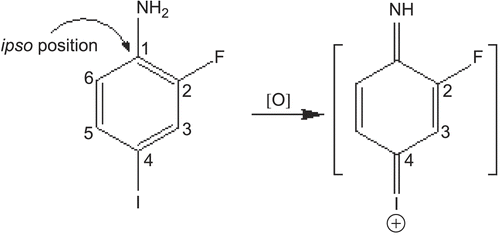
144. Determinants of Increased Sensitivity to Lovastatin-induced Apoptosis in Cisplatin-resistant Human Laryngeal Carcinoma Cells
Tamara čimbora-Zovko,1 Gerhard Fritz,2 and Maja Osmak1
1Ruđer Bošković Institute, Zagreb, Croatia, 2University of Mainz, Mainz, Germany
Cisplatin is one of the most effective and commonly used agents for the treatment of solid tumors. Nevertheless, acquired resistance to cisplatin represents a major obstacle to successful chemotherapy. We have observed that our cisplatin-resistant CA3ST and CK2 sublines display substantial alterations in cell morphology, adhesion, and cytoskeleton organization comparing to their parental human laryngeal carcinoma HEp-2 cells. In addition, these sublines were more sensitive to lovastatin-induced apoptosis. Since HMG CoA-reductase inhibitors have already entered clinical trials for several types of cancer, we explored the potential mechanisms of the increased sensitivity of the cisplatin-resistant cells to lovastatin. We used semiquantitative RT-PCR and Western blot to screen already known mechanisms that contribute to differential sensitivity of tumor cells to lovastatin, namely, the expression of HMG CoA-reductase, P-glycoprotein, Bcl-2, and survivin, but found no correlation with the degree of sensitivity. In addition, statins with different lipophilicity and metabolic pathways gave the sensitivity pattern similar to the one of lovastatin, suggesting that the difference in sensitivity is not due to higher intracellular concentration of the active compound. Nonetheless, lovastatin induced a substantial increase in RhoB in all cell lines tested, while the expression of Rac1 and Cdc42 was decreased, especially in cisplatin-resistant sublines. Lovastatin toxicity was suppressed by the addition of geranylgeranyl PPi and, to a lesser extent, farnesyl PPi. In accordance, the alterations in the expression of Rho GTPases were also diminished. Since RhoB expression is downregulated in the cisplatin-resistant sublines, we presumed that this difference in basal RhoB level provides the difference in sensitivity. However, the silencing of RhoB in HEp-2 cells with specific siRNA did not alter their sensitivity to lovastatin. Therefore, we conclude that increased susceptibility of cisplatin-resistant cells to lovastatin involves several geranylgeranylated proteins, among others Rac1 and Cdc42, which are presumably altered during the development of cisplatin resistance. Given that statins are safe and well tolerated even at higher doses, cancer treatment involving lovastatin could give superior results in patients that acquired resistance to cisplatin.
145. Resistance to Acetaminophen-induced Hepatotoxicity in Female CD-1 Mice
Yasuhiro Masubuchi, Junpei Nakayama, and Eriko Makita
Faculty of Pharmaceutical Sciences, Chiba Institute of Science, Choshi, Japan
Gender is a factor influencing hepatic drug metabolism and susceptibility of individuals to drug-induced hepatotoxicity in experimental animals and in humans. Recent studies presented a male-predominant acetaminophen (APAP) hepatotoxicity in C57BL/6 mice but not in CD-1 mice. In our preliminary experiments, a relatively low dose of APAP developed liver injury prominently in male CD-1 mice, as compared to females. Here, we investigated the mechanism underlying the resistance of female CD-1 mice to APAP-induced hepatotoxicity. Overnight-fasted CD-1 mice were given APAP (300 mg/kg) intraperitoneally. Hepatotoxicity was assessed by serum leakage of alanine aminotransferase (ALT). Glutathione (GSH) concentrations were determined by the dithionitrobenzoic acid-glutathione disulfide reductase recycling assay. Liver heme oxygenase-1 (HO-1) mRNA expression was assayed by real-time RT-PCR. Only a minor increase in serum ALT was observed in female CD-1 mice after the administration of APAP, which causes severe hepatotoxicity in male CD-1 mice. Early depletion of hepatic GSH, which has been observed prior to the initiation of hepatotoxicity, was found both in male and female mice, suggesting no sex difference in the metabolism of APAP into its reactive metabolite, N-acetyl-p-benzoquinone imine, as previously reported. On the other hand, the recovery of hepatic GSH was more rapid in female mice than in males, probably because of relatively active GSH biosynthesis in female mice. In fact, inhibition of the GSH biosynthesis by buthionine sulfoximine induced a potentiation of the APAP hepatotoxicity, particularly in female mice, resulting in abrogation of the sex difference in hepatotoxicity. In addition, early induction of liver HO-1 after the APAP administration was more prominent in female mice than males. In conclusion, the resistance of female mice to APAP-induced hepatotoxicity is strain independent. The hepatoprotective responses enhanced in female CD-1 mice may contribute to their resistance to APAP-induced hepatotoxicity.
146. Sperm Protein Adducts as Potential Biomarkers in Male Reproductive Toxicity of N,N-Dimethylformamide: In Vitro Study
Jaroslav Mráz,1 Hana Nohová,1 šárka Dušková,1 Jana Cimlová,2 and Petr Simek2
1Center of Public Health Laboratories, National Institute of Public Health, Prague, Czech Republic, 2Laboratory of Analytical Biochemistry, Biology Centre, Academy of Sciences of the Czech Republic, Ceské Budejovice, Czech Republic
The metabolic pathway related to hepatotoxicity of the industrial solvent N,N-dimethylformamide (DMF) involves methylisocyanate (MIC) as a reactive toxic intermediate. The adduct of MIC with glutathione, S-(N-methylcarbamoyl)glutathione (SMG), and its metabolic product, N-acetyl-S-(N-methylcarbamoyl)cysteine (AMCC), are unstable species, acting as a vehicle to transport the N-methylcarbamoyl moiety in the organism. DMF is a suspect male reproductive toxicant because of the recent finding of impaired sperm motility in DMF-exposed workers (Chang et al., 2004). We hypothesize that this adverse effect is associated with adduct formation in the sperm proteins, and that selected adduct species might be potential biomarkers of the cumulative dose of the toxicant in target tissues. Sperm protein protamines were used for the study. Here, we examined the pattern of N-methylcarbamoyl adducts following incubation with MIC, SMG, and AMCC (10 mM) in pure fish protamines and in human semen samples. The adducts at protein N-termini were converted by Edman degradation to specific hydantoins and detected by GC/MS, whereas the complete adduct pattern was obtained by HPLC/MS analysis of total protein hydrolysates. Adduct identification was facilitated by comparison with synthetic standards. From the expected adducts at N-termini of the fish and human protamines (i.e., Ala, Pro, and Met), those with Ala and Pro, but not with Met, were found. Several other N-terminal adducts detected in human semen were from proteins other then protamines. Lys was the major nonterminal amino acid reactive with MIC, SMG, and AMCC. The adduct levels in MIC-treated proteins were approximately 1 order of magnitude higher than those in proteins incubated with SMG or AMCC. The presented data show that the above adducts may be prospective biomarkers in a future in vivo male reproductive DMF toxicity study.
Reference
- Chang H-Y, et al. (2004). Sperm function in workers exposed to N,N-dimethylformamide in the synthetic leather industry. Fertil Steril 81:1589–1594.
147. Time-Dependent Effect of Chronic Exposure to Depleted Uranium In Rat On Xenobiotic Metabolizing Enzymes
Yann Gueguen,1 Line Grandcolas,1 Cédric Baudelin,1 Stéphane Grison,1 Patrick Gourmelon,2 Isabelle Dublineau,1 and Maâmar Souidi1
1Drph/Srbe/Lrtox, IRSN, Fontenay-aux-Roses, France, 2Drph, IRSN, Fontenay-aux-Roses, France
The use of depleted uranium (DU) in both civilian (e.g., nuclear industry, mill tailings, and phosphate fertilizers) and military applications results in the increase populations exposed to this compound. In order to evaluate the effect of internal chronic exposure to DU on xenobiotic metabolism, rats were exposed via drinking water (40 mg/L) during 3, 6, 9, or 18 months. This dose corresponds to twice the highest uranium concentration found naturally on Earth, in Finland well-water. Note that the exposure to DU had no influence on food consumption, body weight, or general health status of the rats. In the present work, liver and kidney gene expressions were analyzed by real-time RT-PCR. Hepatic cytochrome P450 activities were measured by testosterone metabolism in the microsomal fraction. Surprisingly, CYP3A1 and CYP3A2 kidney gene expressions were dramatically decreased by 73 and 97% of rats exposed during 6 months, whereas no significant changes were observed in the liver. On the contrary, CYP3A1 were increased by 3 or 9 times, respectively, in the liver or in the kidney after a 9-month chronic exposure. Shorter (3 months) or longer (18 months) chronic contaminations by DU did not reveal any variation of these gene expressions. This time-dependent effect may be due to the fact that the accumulation of DU in the rat organs is not gradually increasing with time, so it did not reach a steady state. Otherwise, the liver cytochrome P450 activities measured did not differ in all the groups studied, compared to sham-exposed rats. These results strengthen the previous data, which showed that CYP3A could be a target of uranium exposure. Moreover, the kidney is a known target tissue of uranium chemical toxicity, so the effects observed probably foreshadow a slight kidney defect, which could lead to a xenobiotic metabolic disorder.
148. Tiopronin Protects against Isoniazid-induced Hepatotoxicity in Rats through CYP2E1 Inhibition and GSTs Induction
Jiang Yue,1 Renxiu Peng,1 and Guicheng Dong2
1Department of Pharmacology, Medical College of Wuhan University, Wuhan, China, 2Baotou Teacher College of Inner Mongolia University of Science and Technology, Baotou, China
Isoniazid has been the first-line antitubercular drug since 1952. Isoniazid can cause mild to moderate elevation of transaminases in approximately 10–20% of patients and severe hepatotoxicity in approximately 0.5–2%. In rats, CYP2E1 is involved in isoniazid-induced hepatotoxicity via free radical generation, and GSTs can function as an intercellular free radical scavenger during the development of isoniazid hepatotoxicity. Here, we investigate, for the first time, the protective effects of N-(2-mercaptopropionyl)-glycine (tiopronin) on the hepatotoxicity of isoniazid. Rats were daily injected with isoniazid (100 mg/kg, i.p.) for 21 days and exposed to either tiopronin (60 mg/kg, i.p.) or saline from day 11 until day 21. DNA oxidative damage induced by isoniazid was analyzed in precision-cut liver slices by HPLC-MS/MS. Tiopronin coadministration completely abolished the increases in serum ALT and AST induced by isoniazid, which is consistent with histopathological examinations that the incidence of the vacuolation and patchy necrosis was significantly decreased in the tiopronin-isoniazid group. Compared with isoniazid exposure alone, tiopronin inhibited isoniazid-induced activities of chlorzoxazone hydroxylase and aniline hydroxylase (16–19%, P < 0.05), which are the typical probes of hepatic CYP2E1. In vitro, tiopronin concentration dependently inhibited CYP2E1-dependent aniline hydroxylation with an apparent IC50 value of 110 μM (Ki = 63.6 μM), but did not inhibit CYP3A-catalyzed erythromycin demethylation. Tiopronin cotreatment attenuated the suppression of cytosolic total GST, mu GST, and alpha GST (1.25- to 1.50-fold; P < 0.05) by isoniazid. In precision-cut liver slices, the generation of DNA adducts by isoniazid were significantly decreased by 56% (P < 0.05) after tiopronin coincubation. These results suggest that tiopronin can afford protective effects against isoniazid-hepatotoxicity in rats. Tiopronin may reduce free radical production from isoniazid by the inhibition of hepatic CYP2E1 and increase the scavenging of free radicals through cytosolic GSTs. (This work was supported by the National Natural Science Foundation of China, no. 30600773.)
149. An Improved Predicted MRM Method for In Vivo Metabolite Detection Method using a Hybrid LIT/Triple Quadrupole Mass Spectrometer
Elliott Jones,1 Hau-Fen Liu,2 and Ji Ma3
1Pms, Applied Biosystems, Foster City, California, USA, 2Application Laboratory, Applied Biosystems, Inc., Foster City, California, USA, 3Department of PKDM, Amgen, South San Francisco, California USA
LCMS-based metabolite analysis is a critical experiment in drug development. One of the most critical challenges is sensitivity and specificity in the detection of low-level metabolites in complex in vitro or in vivo sample matrix. Sensitivity and selectivity are the limitations of some of those technologies in cases in which low-level metabolite detection is warranted or matrix signals have been proven to be overwhelming. With the advent of the hybrid linear ion trap instrument, a series of unique experiments for metabolite analysis have become possible. The ability to use selective and sensitive pMRM (predictive MRM survey scans), followed by high-sensitivity ion trap MS/MS scans, provided a unique approach for metabolite analysis.
Experiment: Verapamil was dosed orally in rat at 1 mg/kg. Rat bile and urine samples were collected after 4 hours of dosage. Samples with a simple sample preparation were injected on a QTRAP® 5500 system (Applied Biosystems, Foster City, California, USA). The pMRM method included the unknown metabolite assessment, for which a wide array of theoretically predicted MRMs was generated in a blind fashion. The list was established by the combination of a series of all potential biotransformations and some basic fragmentation rules to cover all potential modifications for metabolite.
Results: In this work, verapamil samples from 1-mg/kg orally dosed rat bile and urine matrices were analyzed on a QTRAP 5500 system. A complete tabulation of the results, using a single-injection pMRM method in rat urine and bile samples, is shown in . A total of 24 and 19 potential metabolites with high-quality EPI (MS2) in bile and urine samples were detected, respectively, in one run via the pMRM methodology. The pMRM method found the most major metabolites in a single injection, based on the high level of sensitivity in this mode. No major metabolites were missed with the combination of pMRM and structurally related predicted dealkylation. Further, the range of metabolites found is inline with the metabolites detected at high levels in an in vitro incubation.
Table 1. The metabolites detected by pMRM and M IM from orally dosed bile sample.
150. High-Throughput Screening GSH Adducts using Hybrid Linear Ion Trap Systems Coupling with Fast Chromatography at Clinically Relevant Dose Concentration
Elliott Jones, Jr.,1 Claire Bramwell,2 Hesham Ghobarah,3 and Hau-Fen Liu4
1LCMS, Applied Biosystems, Foster City, California, USA, 2Applied Biosystems, Inc., Foster City, California, USA, 3LCMS, AB/MDS, Applied Biosystems, Inc., Foster City, California, USA, 4Application Laboratory, Applied Biosystems, Inc., Foster City, California, USA
The detection of reactive metabolites, such as GSH adducts, has become a common practice in early drug toxicity screening. In order to detect all potential GSH adducts, two or more injections were often used. Higher substrate concentrations (10–50 uM), which may be higher than clinical relevant concentrations and complex sample preparations, were often needed in order to achieve the sensitivity desired. With the new development of much faster scan speeds in both quadrupole and linear ion trap (LIT) trap mode, and fast switching between positive and negative ions, a novel new approach with one injection as a generic high-throughput screening method can be achieved with sufficient sensitivity to ensure an accurate result at clinical relevant concentrations. The acquisition method has significant advantages for the application of 1.8 ìm high-pressure chromatography with short (5- to 10-minute) run times. A second highly effective method of GSH detection possible on the hybrid linear ion trap system is pMRM (predictive MRM). This method uses the next-generation software MRM builder coupled with Excel™ to produce a complete set of potential MRM transitions for any GSH biotransformation that, including a positive MRM list, based on the neutral loss of 129 or 307, and a negative list based on the precursor of 272. Trazodone is a psychoactive compound with sedative, anxiolytic, and antidepressant properties. The treatment of the patient is started with low initial doses of 25 mg daily and the dose may be increased slowly to a 300-mg daily dose. An overdose of trazodone may cause severe side effects. The GSH formation of trazodone at different clinical relevant concentrations (2 and 20 μM) in human liver microsomes was evaluated by using the novel workflows outlined earlier. The results show the novel methods can detect trace-level metabolites not easily seen by standard LCMS workflows. Further, the complete GSH detection and structural confirmation at clinically relevant concentrations ensure better in vivo prediction.
151. Identification of Flubendazole Phase II Metabolites in Rat and Ovine Urine and Faeces
Milan Nobilis,1 Viktor Cvilink,2 Barbora Szotakova,2 Jiri Lamka,3 and Lenka Skalova2
1Bioanalytical Laboratories, Institute of Experimental Biopharmaceutics, Hradec Kralove, Czech Republic, 2Department of Biochemical Sciences, Faculty of Pharmacy, Charles University, Hradec Kralove, Czech Republic, 3Department of Pharmacology and Toxicology, Faculty of Pharmacy, Charles University, Hradec Kralove, Czech Republic
In our previous papers (Nobilis et al., 2007, 2008), the in vitro and in vivo phase I metabolites of anthelmintic flubendazole were studied in pig, pheasant, and sheep. In all mentioned species, (+)-reduced flubendazole was identified as the principal metabolite in various biomatrices, whereas hydrolyzed flubendazole and (-)-reduced flubendazole were found in only low concentrations. The reduction of the flubendazole carbonyl group and hydrolysis of its ethyl carbamoyl moiety form the prepositions for the conjugation reactions leading to polar phase II metabolites. To complete the information about the fate of flubendazole in various species, the conjugates of phase I flubendazole metabolites were searched in the urine of sheep and rats and feces of rats.Thirty milligrams of flubendazole per kilogram of body weight were administered to animals and the excretes were collected during 24 hours. Comparative HPLC-PDA-FLUOR analyses of the samples of urine and homogenized feces and of the same biomatrices after their enzymatic treatment using beta-glucuronidase afforded the qualitative and quantitative information about the phase II flubendazole metabolites. (This project was supported by the Ministry of Education of the Czech Republic, project no. MSM 0021620822.)
References
- Nobilis M, Jira T, Lísa M, Holcapek M, Szotáková B, Lamka J, et al. (2007). Achiral and chiral high-performance liquid chromatographic determination of flubendazole and its metabolites in biomatrices using UV photodiode-array and mass spectrometric detection. J Chromatogr A 1149:112–120.
- Nobilis M, Vybíralová Z, Krízová V, Kubícek V, Soukupová M, Lamka J, et al. (2008). Sensitive chiral high-performance liquid chromatographic determination of anthelmintic flubendazole and its phase I metabolites in blood plasma using UV photodiode-array and fluorescence detection. J Chromatogr B 876:89–96.
152. In Vitro Study of Metabolic Rate and Routes of Atazanavir by a Fast LC/MS Methodology with Improved Sensitivity
Jose Castro-Perez,1 Kate Yu,2 Henry Shion,3 John P. Shockcor,4 and Emma Marsden-Edwards5
1Waters Corp., Milford, Massachusetts, USA, 2Department of Pharmaceut Market Development, Waters Corporation, Milford, Massachusetts, USA, 3Waters Corp., Beverly, Massachusetts, USA, 4Department of Metabolic Profiling, Waters Corp., Milford, Massachusetts, USA, 5Department of Metabolic Profiling, Waters Corp., Manchester, UK
A typical metabolic study includes the estimation of both rate and routes of the parent drug, as the rate reflects the drug’s half-life and the routes of metabolism may indicate the possible toxic effect. We present here a simple and generic strategy for Met ID studies that utilizes UPLC and a new, highly sensitive, benchtop oaTOF mass spectrometer. An LC/TOF MSE data acquisition strategy incorporated with a software application manager with chemical intelligence allows metabolites to be positively identified with a single injection of the sample. Consequently, with one injection per time point, the rate of metabolism of the drug is obtained as well. An anti-HIV drug, atazanavir, was used as the parent drug for the study. The drug was incubated at 0.1 uM in human liver microsome (HLM) at 0, 15, 30, 60, 90, and 120 minutes. Performing in vitro Met ID studies at sub-uM concentrations is desirable, since the results reflect more closely with those generated at the “real dose” level. Because the enzymes are not saturated during the incubation, a clearer picture of the rate and route of clearance is obtained. A Waters ACQUITY UPLC/Xevo QTof system was used with an LC run time at 13 minutes (Warter, Milford, Massachusetts, USA). Data was acquired with the UPLC-MSE strategy, in which the mass spectrometer acquires data at fast speeds (up to 20 spectra/s) in two discrete scan functions: low-collision energy scan and high-collision energy scan. The mass spectrometer switches rapidly between the two scan functions (25-ms interscan delay). As a result, information for both intact molecule and fragments ion can be obtained from a single UPLC injection. The 60-minute sample was used for the identification of metabolites. Major metabolites identified were the hydroxylated metabolites (+16). A few metabolites that were resulted from dealkylation cleavages were also observed. The half-life of this drug was also estimated.
153. Strategies to Obtain Exposures of Metabolites in Preclinical Species through Plasma Pooling and Quantitative NMR without Using Radiolabeled Compounds and Chemically Synthesized Metabolite Standards
Abdul Mutlib, Karthick Vishwanathan, Katlene Babalola, Jack Wang, Robert Espina, Linning Yu, Adedayo Adedoyin, Rasmy Talaat, and JoAnn Scatina
Drug Safety and Metabolism, Wyeth Research, Collegeville, Pennsylvania, USA
The Safety Testing of Drug Metabolites guidance recently issued by the U.S. FDA Center for Drug Evaluation and Research has highlighted the importance of identifying significant circulating metabolites in human plasma as early as possible in drug development. Further, it has become important to demonstrate that these metabolites are presently at an equal or greater exposure level (AUC) in any one of the preclinical species used in safety testing. However, quite frequently, synthetic standards of metabolites are not available, and hence, obtaining their AUC values can be a challenge. In this presentation, we demonstrate how the combination of nuclear magnetic resonance spectroscopy, liquid chromatography/ultraviolet/mass spectrometry and plasma pooling methods were used to obtain reliable AUC values of metabolites present in the plasma of preclinical species. Plasma pooling methods were compared to the traditional approaches of obtaining exposure levels of circulating metabolites. The exposure values obtained via sample pooling were comparable to those obtained by the traditional method of analyzing samples individually. In the absence of synthetic chemical standards, estimations of AUC values of metabolites, using either sample pooling or traditional approaches, were achieved with UV detectors. In cases where the UV properties of metabolites were significantly different from their parent compounds, NMR was used as a quantitative tool to obtain exposure values. NMR was found to be useful in quantitating biologically generated metabolites, which were subsequently used as “reference standards” for further quantitative studies. It is proposed that a limited set of method verifications be conducted with these standards prior to their use in bioanalytical assays. A tiered approach to bioanalytical validation could be implemented by using these biologically produced metabolites. The tiered approach would allow quantitative information to be achieved in early development, using bioanalytical methods with limited validation, with validation criteria increasing as a compound progress in development. This strategic approach will lead to considerable savings for the companies as the use of radiolabeled compounds or chemically synthesized standards of metabolites will be circumvented.
154. Using MSM for Intelligent Metabolite Screening and Structural Elucidation with an LTQ Orbitrap
Yingying Huang,1 Ji Ma,2 Jae Schwartz,1 Robert Cho,2 Yan Chen,1 and Tim Carlson2
1Thermo Fisher Scientific, San Jose, California, USA, 2Department of PKDM, Amgen, South San Francisco, California, USA
The characterization of drug metabolites is an integral part of drug discovery and development. Described here is the use of MSM, utilizing multiple collision cells, dissociation methods, scan modes, mass analyzers, and detectors to perform intelligent metabolite identification experiments. A modified LTQ Orbitrap XL (Thermo Scientific, Weltham, Massachusetts, USA) with an HCD (higher energy collisional dissociation) collision cell was used. A linear ion trap (LIT) isolation mass window up to 600 amu for HCD scans was enabled, so that a large number of precursors could be excited simultaneously and their fragmentation information be captured in the HCD scan. The experiment was designed such that a high-resolution full scan was acquired followed by a high-resolution HCD MS/MS. By comparing the data from HCD with the full-scan MS, it is possible to mimic conventional neutral loss scanning, precursor ion scanning, and multiple reaction monitoring experiments. This was achieved by data mining, using accurate mass and high resolution. In parallel, the LIT acquired data-dependent MSn spectra. Using m/z 165.0910, a diagnostic fragment ion of verapamil, an accurate mass precursor ion analysis of 10-μM rat hepatocytes led to the identification of over 16 putative metabolites. Definitive structural elucidation of these putative metabolites was achieved by analysis of the data-dependent LIT CID MSn data collected in parallel. Similar analysis of 1-μM rat hepatocytes also led to the identification of all phase I and II metabolites. For in vivo sample analysis, an inclusion list of the accurate masses of the predicted metabolites was used to trigger data-dependant MSn even when the intensity of the precursor ion was 2–3 orders of magnitude less than the background ions. Metabolites of haloperidol were observed in urine/plasma/bile samples, using accurate mass precursor ion analysis. In the case when the CID MSn scan was not triggered automatically on some unpredicted metabolites due to matrix interference, a second run, using Orbitrap full scan plus LTQ (or Orbitrap) MSn with an inclusion list derived from the previous MSM experiment, was performed.
155. An In Vitro Metabolism Assay for the Simultaneous Assessment of Glucuronidation and Sulfation in S9 Fraction
Peter Eichhorn,1 Dolores Marín,1 Jordi Gràcia,2 Miquel Salvà,1 and Joan Albertí1
Departments of 1Pharmacokinetics and Drug Metabolism and 2Medicinal Chemistry, Laboratorios Almirall S.A., Sant Feliu de Llobregat, Spain
Among the phase II metabolic reactions, glucuronidation and sulfation may be the most prominent detoxification pathways for the conjugation of phenols and aliphatic alcohols, as well as primary and secondary amines. Transfer of the glucuronic acid and the sulfate residue, respectively, to the substrate considerably enhances its polarity and thus facilitates excretion of the formed metabolites. Standard in vitro protocols used for evaluating the relevance of glucuronidation commonly rely on incubations in hepatic microsomal fractions after pretreatment for activation of membrane-bound UDP-glucuronosyltransferases, while the sulfation, mediated by cytosolic sulfotransferases, usually involves the usage of the S9 fraction or the cytosolic fraction. In the present work, an in vitro screening model was developed to allow for the simultaneous assessment of glucuronidation and sulfation. To this end, incubations were performed with the liver S9 fraction, supplementing the medium with both cofactors, UDPGA and PAPS. Incubated samples were subsequently analyzed by UPLC-ESI-MS/MS in either the positive or the negative ion mode to unequivocally assign the metabolite identity. The proposed protocol was employed to rank a total of 20 compounds belonging to the same chemical series. Experimental conditions, namely incubation time and protein concentration, were properly adjusted so as to achieve low to moderate metabolic conversions. The test set, comprising both phenolic and aliphatic alcohols, was assayed in liver S9 fractions of human and rat origin with the objective of establishing structure-activity relationships.
156. Drug Metabolite Profiling in Parallel to Drug Quantification in Plasma of Treated Patients Using Liquid Chromatography/Mass Spectrometry
Bertrand Rochat
Faculte Biologie et Medecine, CHUV, Lausanne, Switzerland
Besides affecting the drug disposition of the parent drug, xenobiotic metabolizing enzymes may produce bioactive and toxic metabolites of clinical interest. Taking into account the capability of liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS), simultaneous determination of the parent drug and self-identified metabolites in patients’ plasma samples is currently feasible. The quantification of the parent drug and the profiling of its metabolites in plasma can be relatively easily established: First, the setup of generic LC-MS/MS methods, and second, LC-MS/MS metabolite identification, using either in vitro microsomal incubations or patient plasma samples, have to be performed. Partial method validation can be achieved and can reveal that precise determinations of metabolite levels in patient plasma are feasible. Correlation between the parent drug and the complete metabolite determination can reveal interindividual variables. Drug safety or efficacy can be prospectively or retrospectively associated with determined metabolite levels. Thus, in addition to the therapeutic drug monitoring (TDM) of a few compounds, LC-MS/MS technology can determine, in the same analytical run, a complete drug metabolic profile (tens of metabolites) enabling various correlation studies of clinical interest.
157. Microdosing and Normal Dose Metabolic Profile: A Comparison
Carmai Seto,1 Axel Besa,2 Takeo Sakuma,1 Jinsong Ni,3 Van Dinh,3 Fred Ouyang,3 Devin Welty,3 Gabriella Gabriella Szekely-Klepser,3 and Andrew Acheampong3
1MDS Analytical Technologies, Concord, Ontario, Canada, 2Applied Biosystems, Darmstadt, Germany, 3Allergan, Irvine, California, USA
Introduction: Microdosing provides a new approach for early assessment of human pharmacokinetics. Microdosing studies are designed to evaluate pharmacokinetics or the imaging of specific targets without inducing pharmacologic effects. Microdosing studies may not predict the pharmacokinetic behavior of the drug at clinical doses, since it is not known whether or not dose linearity is maintained between a microdose and a clinical dose. Nonlinearity of pharmacokinetics could be induced when absorption is dose dependent or when metabolism or elimination becomes saturated. The work presented investigates if there is a dose dependency of metabolism between a microdose and normal dose in rat.
Methods: For PK studies, atorvastatin, methimazole, ofloxacin, omeprazole, and tamoxifen were administered to jugular vein–cannulated male Sprague-Dawley rats via oral gavage. The compounds were administered at two different doses at 1.67 μg/kg (microdose) and 5.0 mg/kg (normal dose). For LC/MS/MS analysis,,multiple reaction monitoring information-dependent acquisition (MRM-IDA) was used to detect and characterized metabolites in plasma and urine. The analyses were conducted by using a QTRAP® 5500 LC/MS/MS system with automated method creation, using LightSight™ software ((QTRAP) Applied Biosystems, Foster City, California, USA/Light Sight Software, Applied Biosystems).
Preliminary Data: The type and the number of metabolites detected and confirmed in the plasma and urine from the microdose and the normal dose were compared. This was to determine if there is dose dependency of the metabolic processes of the compounds. Metabolites were detected and characaterized for atorvastatin, methimazole, ofloxacin, omeprazole, and tamoxifen, which were first identified in the plasma and urine collected from the rats given the normal dose of each compound. Then, a predicted MRM-IDA method was created for each compound with the information obtained from the normal dose and a biotransformation set that contained an extensive list of possible metabolites. These MRM-IDA methods were used to characterize and confirm the circulating metabolites in the plasma and urine collected from the microdose. The MRM scan mode allowed for the detection of the metabolites, and the full-scan MS/MS sensitivity of the linear ion trap allowed for the characterization and confirmation of these metabolites.
158. Vinylphenylmercapturic Acids: New Urinary Metabolites Derived from Styrene 3, 4-Oxide, a Reactive Metabolic Intermediate of Styrene
Jaroslav Mráz,1 Igor Linhart,2 Jan Scharff,2 Jan Krouželka,2 šárka Dušková,1 Hana Nohová,3 and L´udmila Vodicková1
1Center of Public Health Laboratories, National Institute of Public Health, Prague, Czech Republic, 2Department of Organic Chemistry, Institute of Chemical Technology, Prague, Czech Republic, 3Center of Public Health Laboratories, National Institute of Public Health, Prague, Czech Republic
The urine from mice exposed to styrene vapours (1,200 mg/m3, 6 hours) was analyzed for metabolites derived from styrene 3,4-oxide (3,4-STO), a reactive and highly mutagenic metabolic intermediate. Authentic samples of 2-, 3-, and 4-vinylphenols, N-acetyl-S-(3-vinylphenyl)cysteine and N-acetyl-S-(4-vinylphenyl)cysteine (3- and 4-vinylphenylmercapturic acids, VPMA), and 4-vinylcatechol were prepared to facilitate the identification of these potential metabolites in urine. 3- and 4-VPMA were identified by GC/MS/MS in the urine of exposed animals as new minor metabolites of styrene. The analytical procedure included extraction from acidified urine, followed by methylation. The ratio between 3- and 4-VPMA was approximately 1:4, while the ratio between total VPMA and the previously known hydroxyethylphenylmercapturic acids was approximately 1:300. The finding of VPMA indicates the capability of 3,4-STO to react with cellular thiol groups despite its rapid isomerization to 4-vinylphenol in an aqueous environment, as observed in model reactions with 3,4-STO. Interestingly, after the reaction of 3,4-STO with aqueous 0.1 M N-acetylcysteine, not 4-VPMA itself, but a small amount of N-acetyl-S-(6-hydroxy-4-vinylcyclohexa-2,4-dien-1-yl)cysteine, a probable precursor of 4-VPMA, was detected by HPLC-ESI-MS. Although the authentic sample of this compound was not available, MS/MS spectrum of its MH+ ion agreed well with the proposed structure. 4-vinylphenol, but not its positional isomers, were found in the extracts of urine from exposed mice, which was treated with glucuronidase/arylsulphatase to release conjugated phenolic metabolites. 4-vinylcatechol was found in both exposed and unexposed mice. Besides the above phenolic compounds, phenylethyleneglycol and 1-phenylethanol were clearly identified as major urinary metabolites resulting from vinyl oxidation, while several other phenolic compounds were only partially characterized. (This study was supported by grant nos. 2B08051 and MSM 604 613 73 01 from the Ministry of Education, Youth and Sports of the Czech Republic.)
159. Identification of the Human Esterases Responsible for the Enzymatic Hydrolysis of Aclidinium Bromide
Joan Alberti, Audrey Martinet, Francisco Jiménez, Sònia Sentellas, and Miquel Salvà
Department of Pharmacokinetics and Drug Metabolism, Laboratorios Almirall, S.A., Barcelona, Spain
Aclidinium bromide (3R-(2-hydroxy-2,2-dithiophen-2-yl-acetoxy)-1-(3-phenoxy-propyl)1-azonia bicyclo [2.2.2] octane bromide), a novel, long-acting muscarinic antagonist, is currently in clinical development for the treatment of COPD. The main metabolic pathway for aclidinium is the ester hydrolysis to two pharmacologically inactive metabolites, an alcohol and an acid derivative. The aim of this study was to identify the human esterases involved in the hydrolytic cleavage of aclidinium. Aclidinium hydrolysis was studied in sodium phosphate buffer (pH range, 2.0–9.0), human plasma, human pulmonary and liver subcellular fractions, and human recombinant cytochrome P450 (CYP450) isoforms. A panel of selective human esterase inhibitors, including p-chloromercuribenzoate (paraoxonase/arylesterase), diisopropylfluorophosphate and eserine (cholinesterases), bis(p-nitrophenyl) phosphate and sodium fluoride (carboxylesterases), iso-OMPA (butyrylcholinesterase), and BW248c51 (acetylcholinesterase), was used to study the enzymatic hydrolysis of aclidinium in diluted human plasma. In addition, purified endogenous or recombinant human esterases were also evaluated to identify the enzyme(s) responsible for aclidinium hydrolysis. Aclidinium was stable in aqueous acid solutions (pH <4), but was hydrolyzed at neutral and basic pH levels. The hydrolysis half-life of aclidinium in phosphate buffer at pH 7.4 at 37 °C was 1.2 hours. In human plasma, hydrolytic cleavage of the ester moiety of aclidinium was mainly enzymatic and very rapid, with a hydrolysis half-life of 2.4 minutes. The enzymatic hydrolysis of aclidinium was comparatively less extensive in human pulmonary and liver subcellular fractions, indicating that the enzymes responsible for aclidinium hydrolysis are mainly located in human plasma. Cleavage of the ester bond was not mediated by human CYP450. In human plasma, inhibition of aclidinium hydrolysis was mainly caused by specific chemical inhibitors of cholinesterases. Additional kinetic hydrolysis studies in diluted human plasma and with purified human butyrylcholinesterase showed a clear biphasic profile, reflecting the enzymatic and nonenzymatic (i.e., chemical) processes involved in the metabolism of aclidinium. Together, these results demonstrate that aclidinium is rapidly metabolized by esterase hydrolysis, and that butyrylcholinesterase is the major human esterase involved in the hydrolytic cleavage of aclidinium.
160. Mechanistic Investigation of the Stereoselective Carbonyl Reduction of Triadimefon in Hepatic Microsomes from Human and Seven other Vertebrate Species
John F. Kenneke,1 Christopher S. Mazur,1 and Thomas J. Sack2
1Office of Research and Development, National Exposure Research Laboratory, Ecosystem Research Division, U.S. EPA, Athens, Georgia, USA, 2Senior Service America, U.S. EPA, Athens, Georgia, USA
Understanding the toxic mode of action of the 1,2,4-triazole fungicides (i.e., conazoles) is important for improving and harmonizing conazole risk assessment. Relative to other conazoles, triadimefon [(RS)-1-(4-chlorophenoxy)-3,3-dimethyl-1-(1H-1,2,4-triazol-1-yl)butan-2-one] is unique with respect to tumorigenesis in rodents, and it has been proposed that triadimefon does not share a common mechanism of toxicity with other conazoles. We postulate that one reason for this difference is that while many conazoles are metabolized via an oxidative P450-mediated pathway, triadimefon is not. In studies conducted with rat hepatic microsomes, triadimenol [(1RS,2RS;1RS,2SR)-1-(4-chlorophenoxy)-3,3-dimethyl-1-(1H-1,2,4-triazol-1-yl)butan-2-ol)] was identified as the major metabolite (~80%) of triadimefon metabolism. Carbonyl reduction in triadimefon occurred stereoselectively with preferential formation of the significantly less toxic triadimenol B diastereomer (1R,2R and 1S,2S). Using chemical inhibitors of P450 and carbonyl reducing enzymes, both triadimefon depletion and triadimenol formation were found to be mediated by 11â-hydroxysteroid dehydrogenase type 1 (11â-HSD1). Studies examining lumenal NADPH production and inhibitor studies for glucose-6-phosphate translocation across the endoplasmic reticulum (ER) membrane implicated hexose-6-phosphate dehydrogenase (H6PDH) in the metabolism of triadimefon as well. These results ultimately associate triadimefon metabolism not only with steroidogenesis (i.e., 11â-HSD1), but also carbohydrate metabolism (i.e., H6PDH). In order to examine the role of 11â-HSD1 and H6PDH in the metabolism of triadimefon for other vertebrate species, a battery of kinetic- and mechanistic-based in vitro assays was conducted with rat, mouse, guinea pig, dog, minipig, rabbit, monkey, and human hepatic microsomes. Significant differences in the metabolism of triadimefon were observed across species, but not genders. Human hepatic microsomes, for example, produced significantly more of the toxic triadimenol A than either rat or mouse hepatic microsomes, suggesting that human health risk assessment of triadimefon, based upon rat and mouse toxicity studies, may be inaccurate. Further comparison and contrast between species will be presented along with the implications of these results.
161. Reduction of Flubendazole, Mebendazole, and other Carbonyl Bearing Xenobiotics in Lancet Fluke (Dicrocoelium dendriticum)
Hana Bártíková,1 Vladimír Kubícek,2 Viktor Cvilink,1 Milan Nobilis,3 Barbora Szotáková,1 Jirí Lamka,4 and Lenka Skálová1
Departments of1Biochemical Sciences, 2Biophysics and Physical Chemistry, 3Pharmaceutical Chemistry, and 4Pharmacology and Toxicology, Faculty of Pharmacy, Charles University, Hradec Králové, Czech Republic
Lancet fluke (Dicrocoelium dendriticum) is one of the frequent parasites of small ruminants (sheep, mouflons, goat, etc.). Lancet fluke infection (dicrocoeliosis) troubles animals and causes economic losses for breeders in many regions. Therapy and prophylaxis of animals, using anthelmintics, remain the most important means in the struggle against helminthoses, including dicrocoeliosis. The biotransformation of anthelmintics by helminth has been little investigated, so far, although the helminth biotransformation enzymes may, to a certain extent, protect these organisms against the toxic effects of anthelmintics. The present study was focused on the carbonyl-reducing enzymes activities in lancet fluke. For this purpose, in vitro (subcellular fractions of fluke homogenate) as well as ex vivo (live flukes cultivated in flasks with medium) experiments were used. From among the anthelmintics, flubendazole and mebendazole were chosen because carbonyl reduction represents their deactivation. In addition, several model carbonyl-bearing substrates were included in the study. Results showed that lancet fluke enzymes were able to effectively metabolize flubendazole as well as mebendazole via carbonyl reduction. The structure of both metabolites was confirmed by using LC-MS. In vitro, most of the model substrates (e.g., metyrapone, daunorubicin, and DL-glyceraldehyde) were reduced only in cytosol-like fractions, with the exception of oracin, which was reduced also in microsome-like fractions. In cytosol, high specific activities of acenaphthenol dehydrogenase were found. All specific activities of carbonyl-reducing enzymes toward substrates used were relatively high, similar to the activities found in mammals. These results prove that lancet fluke is able to effectively reduce xenobiotic aldehydes or ketones. By this way, carbonyl-reducing enzymes may protect lancet fluke against the toxic effect of drugs with the carbonyl group. Improving of our knowledge about drug biotransformation and drug-metabolizing enzymes in parasitic helminths could improve the pharmacotherapy of helminthoses, including dicrocoeliosis. (This project was supported by the Czech Science Foundation, grant no. 524/07/0611 and by Zentiva (Zentiva, Praha, Czech Republic).)
162. Study of Activity Distribution and Selective Inhibition of Human Esterases
Sònia Sentellas, Daniel Servera, Francisco Jiménez, Miquel Salvà, and Joan Albertí
Department of Pharmacokinetics and Drug Metabolism, Laboratorios Almirall, S.A., Barcelona, Spain
Esterases have an important role in the human metabolism of a wide variety of exogenous and endogenous compounds. Regarding the disposition of drugs, esterases participate in the activation of ester prodrugs as well as drug inactivation. The tissue distribution of esterases and the identification of the enzymes involved in the hydrolysis of a new chemical entity (NCE) are highly relevant in order to understand the biotransformation of this NCE. Extrahepatic hydrolysis may be, then, important following some routes of exposure to xenobiotic esters. Regarding enzyme identification, different strategies can be followed, including the use of selective inhibitors. In this communication, two studies will be presented. On one hand, activities of the most important human esterases, namely, acetylcholinesterase (AChE), butyrylcholenesterase (BChE), carboxylesterase (CbE), paraoxonase, and arylesterase, were evaluated in hepatic and pulmonary subcellular fractions and blood (i.e., plasma and erythrocytes). Results showed that cholinesterases had the highest activity in blood (AChE in erythrocytes and BChE in plasma); paraoxonase was also present, mainly in plasma. In contrast, the highest arylesterase activity was observed in liver microsomes. Finally, the CbE activity was similar in all the tested tissues. The second part of this communication will be focused on the evaluation of the selectivity of some of the most widely used esterase inhibitors. The compound 1,5-bis(4-allyldimethylammoniumphenyl)pentan-3-one (BW284c51), which was thought to be an AChE selective inhibitor, was demonstrated not to be so selective for AChE, showing some inhibitory effect for BChE. In contrast, the selectivity of tetraisopropyl pyrophosphoramide (Iso-OMPA) for BChE was confirmed. Finally, PCMB and HgCl2 were evaluated as selective inhibitors of paraoxonase and arylesterase, but some effects on cholinesterases were also observed.
163. HIF-1alpha Polymorphisms: Interethnic Variability
Ana L. Ribeiro and Vera Ribeiro
CBME–University of Algarve, Faro, Portugal
Hypoxia-inducible factor-1alpha (HIF-1alpha) is a transcription factor that plays a crucial role in the cellular response to hypoxia. The C1772T (P582S) and G1790A (A588T) polymorphisms, within the oxygen-dependent degradation domain of HIF-1alpha protein, seem to be important in the oxygen regulation of the HIF-1alpha protein, influencing the progression of some hypoxic solid tumors. Despite the numerous reports about the influence of these single-nucleotide polymorphisms (SNPs) on cancer incidence and progression, there are no published data concerning the interethnic variability of these polymorphisms. Here, we investigated the SNPs, C1772T and G1790A, in four distinct populations from Portugal, Mozambique, Colombia, and Guinea-Bissau. The allelic frequency of the 1772T allele was 0.122 in Portugal, 0.151 in Colombia, 0.246 in Mozambique, and 0.08 in Guinea-Bissau. Statistically significant differences were observed when comparing the Portuguese population with the Mozambican one (P = 0.020) and the populations from Mozambique and Guinea (P < 0.0001). The Mozambican population had an allelic frequency of 0.006 for the 1790A allele, which was not detected among the other studied populations. In conclusion, there is an ethnicity-related variation in the frequency of C1772T and G1790A polymorphisms of the HIF-1alpha gene that should be relevant in the context of tumor aggressiveness and progression.
164. Genetic Polymorphisms of N-Acetyltransferase 2 (NAT2) in the Hausa, Ibo, and Yoruba Populations of Nigeria
Benjamin U. Ebeshi,1 Oluseye O. Bolaji,1 Alice Matimba,2 and Collen M. Masimirembwa2
1Pharmaceutical Chemistry, Faculty of Pharmacy, Obafemi Awolowo University, Ile-Ife, Nigeria 2African Institute of Biomedical Science and Technology, Harare, Zimbabwe
The human arylamine N-acetyltransferase 2 (NAT2) enzyme encoded by the NAT2 gene is responsible for the acetylation of several clinically useful drugs, such as isoniazid and sulphonamides. The polymorphism in the NAT2 gene is responsible for the marked interindividual and interethnic variations in the metabolic activity as well as susceptibility to drug-induced carcinogenesis. While the molecular basis of such variations is known for many populations, such data are limited on Nigerians. The aim of this study was to determine the allele frequencies of the NAT2 gene in the Nigerian populations in comparison with frequencies in the other ethnic groups. The single-nucleotide transitions, 481 C>T (NAT2*5), 590 G>A (NAT2*6), 857 G>A (NAT2*7), and 191 G>A (NAT2*14), which constitutes the NAT2 alleles, were genotyped in 300 unrelated healthy subjects from the three major Nigerian ethnic populations: Hausa (N = 98), Ibo (N = 101), and Yoruba (N = 101), using polymerase chain reaction/restriction fragment length polymorphism techniques in accordance with the methods of Abe et al. and Delomene et al. The allele frequencies for NAT2 predict slow acetylation frequencies of 66% in Hausa, 72% in Ibo, and 71% in Yoruba. There was no significant difference in the NAT2 alleles in the three populations studied. Mean frequencies of the NAT2 alleles were 30% (NAT2*4), 29% (NAT2*5), 30% (NAT2*6), 3% (NAT2*7), and 7% (NAT2*14). The mean frequency of NAT2 slow acetylation alleles in the Nigerian population was 70%, which is higher than values reported for other African populations, Caucasians, and Orientals. This preliminary investigation indicates that the NAT2 slow allele frequencies may not reflect absolute values in terms of NAT2 slow genotype, as heterozogosity (e.g., NAT2*4/NAT2*5 and NAT2*4/NAT2*6) that are responsible for fast acetylation are not accounted for. The genotype status of NAT2 shows that the Nigerian population may be prone to adverse effects of drug substrates metabolized by NAT2, in comparison to some other African, Caucasian, and Oriental populations.
165. Association of Catechol-O-methyltransferase (COMT) SNPs with Phenotypic Traits in Affective Disorders
Neslihan Aygun Kocabas,1 Carole Faghel,1 Mara Barreto,1 Sylvie Linotte,2 Daniel Souery,2 Pierre Oswald,2 Siegfried Kasper,3 Joseph Zohar,4 Julian Mendlewicz,2 and Isabelle Massat1
1Fonds de la Recherche Scientifique (FNRS), Department of Experimental Neurology, Free University of Brussels (ULB), Brussels, Belgium, 2Fonds de la Recherche Scientifique (FNRS), Free University of Brussels (ULB), Brussels, Belgium,3Department of Psychiatry and Psychotherapy, Medical University of Vienna, Vienna, Austria 4Department of Psychiatry, Chaim Sheba Medical Center, Tel-Hashomer, Israel
Affective disorders (ADs) are considered as heterozygous and complex disorders that result from both genetic and environmental influences. It is predicted to be the second leading cause of death and disability worldwide by 2020. The pursuit of pharmacogenetics in psychiatry can help predict response to treatment or severe adverse events by characterizing individual genetic differences underlying susceptibility to AD and the response to therapeutics. Catechol-O-methyltransferase (COMT) has been suggested to be involved in the pathogenesis as well as the pharmacological treatment of AD because of one of the major degradative pathways of the catecholamine neurotransmitters and catechol drugs. Although up to 50 SNPs were observed in the hCOMT gene, it has been more focused on the common Val158Met polymorphism (rs4680), generating alleles encoding high- and low-activity forms of the enzyme, when there could be less common SNPs that actually contribute to disease. In this aspect, we carried out this study to define the functional impact of COMT SNPs and haplotypes in susceptibility of AD and on clinical phenotypes of AD. We assessed clinical phenotypes, such as treatment outcome phenotype (e.g., response, remission, and resistance to antidepressant treatment), melancholia, anxiety, and suicidality in 587 patients with DSM-IV major depression. The patients were retrospectively characterized for clinical response to adequate antidepressant treatment administered at least 4 weeks, as measured by the Hamilton Depression (HAM-D-17) score. We analyzed the patients for rs2075507, rs737865, rs6269, rs4633, rs4818, rs4680, rs165599, and compared to controls (n = 215). Therefore, after extraction of DNA, all SNP regions were separately amplified and ABI Prism SNaPshot Multiplex Reactions were performed on purified samples (Applied Biosystems, Foster City, California, USA). After postextention treatment, all samples were genotyped by using an ABI 3130 Genetic Analyzer. None of the seven SNPs, including the rs4680, was significantly associated with AF; however, we found associations for rs4633 and rs2075507, rs6269, rs4680, and rs165599 with remission and suicidality, respectively (P < 0.05). Haplotype analyses with different SNP combinations revealed significance in patients, compared to controls, and in patients stratified by clinical phenotypes.
166. DNA Repair Gene XRCC1 Arg399Gln Polymorphism Is Associated with Increased Risk of Acute Lymphoblastic Leukemia in Turkish Children
Tugba Boyunegmez Tumer, Duygu Yilmaz, Cihan Tanrikut, Gulen Ulusoy, and Emel Arinç
Biochemistry Graduate Programme and Department of Biological Sciences, Middle East Technical University, Ankara, Turkey
It is well known that genes functioning in DNA repair pathways play a crucial role in maintaining genomic integrity as well as protection against mutations that may cause cancer. Polymorphisms of DNA repair genes impair the DNA repair capacity and may lead to genetic instability and carcinogenesis. X-ray repair cross-complimenting group 1 (XRCC1) is one of the most important genes functioning in the base excision repair pathway, and a number of studies suggest that polymorphic forms of XRCC1 have a role in susceptibility to various kinds of cancer including breast, prostate, lung, and bladder cancers. However, there are limited and contradictory data on the association between the XRCC1 Arg399Gln variant allele and childhood acute lymphoblastic leukemia (ALL). In this pilot study, we investigated the possible association of the XRCC1 Arg399Gln allele with the risk of incidence of childhood ALL in the Turkish population. The genotypes were determined with the use of PCR/RFLP techniques on 195 healthy controls and 168 ALL patients. The heterozygous (Arg/Gln) and homozygous mutant (Gln/Gln) genotypes were more common in the ALL patients than the controls (OR: 1.7; 95% CI: 1.1–2.5), resulting in a defective allele (399Gln) frequency of 40% in patients and 30% in controls (P = 0.01). In addition, we observed that homozygosity for the variant allele may be an important factor for risk of incidence of childhood ALL, since we found that individuals with the homozygous mutant genotype had a 2.1-fold higher risk (OR: 2.1; 95% CI: 1.1–4.1) of devoloping childhood ALL than those with the wild-type genotype. In conclusion, the present study suggested the involvment of XRCC1 codon 399 polymorphisms in the genetic predisposition to ALL among Turkish children.
167. Effect of SLCO1B1 Polymorphism on Plasma Bile Acid Concentrations in Humans
Xiaoqiang Xiang, Yi Han, Mikko Neuvonen, Marja K. Pasanen, Annikka Kalliokoski, Janne T. Backman, Jouko Laitila, Pertti J. Neuvonen, and Mikko Niemi
Department of Clinical Pharmacology, University of Helsinki and Helsinki University Central Hospital, Helsinki, Finland
Organic anion-transporting polypeptide 1B1 (OATP1B1, encoded by SLCO1B1) is a sinusoidal influx transporter of human hepatocytes. Our aim was to characterize the role of OATP1B1 in the hepatic uptake of bile acids in vivo. Fasting blood samples were drawn from 24 healthy volunteers with the SLCO1B1 c.388AA-c.521TT (*1A/*1A) genotype, 8 with the c.388GG-c.521TT (*1B/*1B) genotype, 24 with the c.521TC genotype, and 9 with the c.521CC genotype. Plasma concentrations of 15 endogenous bile acids were determined by liquid chromatography coupled with tandem mass spectrometry. The concentrations of ursodeoxycholic acid, glycoursodeoxycholic acid, chenodeoxycholic acid, and glycochenodeoxycholic acid were about 50–240% higher in individuals with the SLCO1B1 c.521CC, c.521TC, or c.388AA-c.521TT genotype than in those with the c.388GG-c.521TT genotype (P < 0.05), with the largest differences seen between the c.521CC and c.388GG-c.521TT individuals. Similar trends were also observed in the concentrations of tauroursodeoxycholic acid and taurochenodeoxycholic acid. The cholic acid concentration was about 30% higher in subjects with the c.521CC or c.388AA-c.521TT genotype than in those with the c.388GG-c.521TT genotype (P < 0.05), but its conjugates remained unaffected by the genotype. Thus, the SLCO1B1 polymorphism considerably affects the disposition of several endogenous bile acids, indicating that OATP1B1 plays an important role in the hepatic uptake of bile acids in vivo in humans.
168. Ethnic Differences in the Frequency of Polymorphisms in Influx Transporters SLC10A1, SLC22A1, SLCO1B3, and SLCO1B1
Vera Dias and Vera Ribeiro
Center for Molecular and Structural Biomedicine, Faro, Portugal
Influx transporters play a key role in the disposition and efficacy of many drugs, such as statins, as well as in the control of endogenous mediators, such as cholesterol, bile acids, or vasodilators. The genes coding for these transporters are polymorphic, leading to interindividual variability in disease risk, as well as in the risk of adverse events or treatment failure. The aim of this work was to study selected genetic polymorphisms in four influx transporters, in individuals from distinct ethnic/geographic backgrounds, from Portugal, Mozambique, and Colombia. PCR-RFLP genotyping methods were developed for a total of 13 SNPs in SLC10A1, SLC22A1, SLCO1B3, and SLCO1B1. The variants, C800T in SLC10A1 and C217T in SLCO1B1, were not detected in any population studied. High frequencies were determined for SLC22A1 (T4215C) and SLC22A1 (C83G), SLC10A1 (A2192G), and SLCO1B3 (G699A), with a similar distribution in individuals from the three populations. G1564T (SLCO1B3) was present in a low frequency in the three different ethnic groups. Significant differences were observed for A2587G (SLC10A1), C480G (SLC22A1), A388G, C463A and T521C (SLCO1B1), and T334G (SLCO1B3). This is, to our knowledge, the first description of pharmacogenetic variability in influx transporters in Portuguese Caucasian, native Africans, or Latin Americans.
169. Functional Characterization of CYP1A2 Genetic Polymorphism in Beagle Dogs
Yin Hu,1 Anna W,2 Helen D,1 Dennis H,2 Hugh S,3 and Janet H1
Departments of 1DMPK, 2Disease Biology, Local Discovery CNS and Pain, and 3Disease Biology, AstraZeneca R&D–Sodertalje, Sodertalje, Sweden
There are large differences between individuals in the way they respond to medications in man. The genetic polymorphism of cytochrome P450s is known to be an important factor determinate of such differences. Similarly to humans, there are also remarkable individual variations in exposure of drug in dog plasma after drug administration. Genetic polymorphism could be a potential cause for the variability in drug exposure in dogs. We have investigated genetic polymorphism of CYP1A2 in 24 beagle dogs. Genotype was evaluated via the screening of SNPs (single-nucleotide polymorphisms) in the CYP1A2 gene. The CYP1A2 enzyme activity from different genotypes of the dogs was evaluated by incubating the microsomes with its specific substrate (7-ethoxyresorufin for CYP1A2 /1A1) measuring the formation of metabolite over time. Quantification of P450 protein was performed by Western blot. The CYP1A2 gene was highly polymorphic. There is not only a stop-codon at R372 created by a C1179T resulting in a null CYP1A2 enzyme activity, but also many other SNPs with varying CYP1A2 activity. The discovery of genetic polymorphism in dog P450s will have an impact on drug discovery and development in the future.
170. Genetic Analysis of Thiopurine Methyltransferase Polymorphism in Jordanian Population
Nancy Hakooz,1 Tawfiq Arafat,2 Debbie Payne,3 William Ollier,3 Julie Andrews,4 and Bill Newman5
1Biopharmaceutics and Clinical Pharmacy, University of Jordan, Amman, Jordan, 2Jordan Center for Pharmaceutical Research, Amman, Jordan, 3Center for Integrated Genomic Medical Research (CIGMR), University of Manchester, Manchester, UK, 4School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester, UK 5Department of Clinical Genetics (SM2), St Mary’s Hospital, Manchester, UK
Thiopurine methyltransferase (TPMT) catalyses the S-methylation of thiopurine drugs, such as 6-mercaptopurine, 6-thioguanine, and azathiopurine. TPMT polymorphisms are the major determinants of interindividual differences in the severe toxicity or efficacy of 6-mercaptopurine. Several mutations in the TPMT gene have been identified, which correlate with a low-activity phenotype. Four variant alleles, TPMT*2, TPMT*3A, TPMT*3B, and TPMT*3C, are responsible for over 80% of low or undetectable enzyme activity. The allelic frequency of TPMT variants has been established in many populations. The frequencies of three (TPMT*2, TPMT*3A, and TPMT*3C) variants were investigated in 173 Jordanian healthy males (18–45 years of age). Single-nucleotide polymorphisms (SNPs) were genotyped by using the Sequenom® MassARRAY technology (Sequenom, Inc., San Diego, California, USA). The Iplex™ assay (Sequenom, Inc., San Diego, California, USA) was followed, according to manufacturers’ instructions, using approx 20 ng of DNA. TPMT*3A and TPMT*3C were the only deficiency alleles detected in the Jordanian population with an allele frequency of 1.18 and 0.59%, respectively. These allele frequencies are found at a lower frequency than in the European Caucasian population. This study provides the first analysis of TPMT mutant allele frequency in a sample of the Jordanian population and indicates that TPMT*3A is the most common allele in Jordanian subjects.
171. Genetic Polymorphisms of Superoxide Dismutases And Risk for Asbestosis
Alenka Franko,1 Metoda Dodič-Fikfak,1 Niko Arneric1, and Vita Dolzan2
1Clinical Institute of Occupational Medicine, University Medical Centre Ljubljana, Ljubljana, Slovenia, 2Institute of Biochemistry, Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia
Asbestosis is among the most frequent diseases caused by asbestos exposure. Asbestos induces the production of reactive oxygen species (ROS) that are known to be involved in the pathogenesis of asbestosis. Superoxide dismutases (SODs) represent the primary defense system against ROS. The genes coding for manganese superoxide dismutase (SOD2) and extracellular superoxide dismutase (SOD3) are polymorphic. The aim of this study was to investigate whether SOD2 Ala -9Val and SOD3 Arg213Gly polymorphisms represent a risk for asbestosis in workers exposed to asbestos. In total, 262 cases with asbestosis and 265 controls with no asbestos disease were genotyped for SOD2 Ala -9Val and SOD3 Arg213Gly polymorphisms, using a real-time PCR assay. The cumulative asbestos exposure and the pack-years of smoking were calculated for each case and control. Asbestosis risk was assessed by using logistic regression analysis. The OR of asbestosis was 1.50 (95% CI: 1.01–2.24) for the SOD2 -9Ala/Ala genotype versus combined Ala/Val and Val/Val genotypes, and 1.63 (95% CI: 0.62–4.27) for the SOD3 Arg/Gly, compared to Arg/Arg genotype. Asbestosis risk for the SOD2-9Ala/Ala genotype did not change considerably after adjustment by gender, age, smoking, and cumulative asbestos exposure. Gender, age, and smoking did not influence asbestosis risk for the SOD3 Arg/Gly genotype either, but the involvement of cumulative asbestos exposure changed the OR from 1.63 (95% CI: 0.62–4.27) to 2.07 (95% CI: 0.72–5.94), suggesting a possible confounding effect. Our key finding was that SOD2-9Ala/Ala genotype increases asbestosis risk. This result as well as our previous findings that genetic polymorphisms of glutathione S-transferases T1 and P1 also influence asbestosis risk, suggest that in addition to asbestos exposure, genetic factors may have a significant influence on the development of asbestosis.
172. Genotyping and Phenotyping of Individual Variants of Butyrylcholinesterase in Drug Development
Clive J. Brealey,1 Neil Evans,2 Roberta Goodall,2 Navjeet Jolly,1 Christopher J. Logan,3 Tracy Pinel,4 Anne-Marie Sims,4 Dominic Surry,1 Rachael White,1 and Suzy H. Rigby1
1Clinical Pharmacology and DMPK, AstraZeneca R&D Charnwood, Loughborough, UK, 2Clinical Biochemistry, Southmead Hospital North Bristol NHS Trust, Bristol, UK, 3Clinical Pharmacology and DMPK, AstraZeneca R&D Alderley Park, Manchester, UK 4Research and Development Genetics, AstraZeneca, Manchester, UK
Butyrylcholinesterase (BChE; EC 3.1.1.8) is a drug-metabolizing enzyme in plasma known to be responsible for the metabolism of several xenobiotics, including cocaine, heroin, organophosphate, and carbamate pesticides. Pharmacogenetic and phenotypic variations of the enzyme have been studied in detail—largely because individuals with low BChE activity can display prolonged apnea and paralysis following administration of the muscle relaxants, suxamethonium and mivacurium (Goodall, 2006). In addition, BChE has been used in drug design strategies for pro- and antedrugs (e.g., bambuterol; Svensson and Tunek, 1988) given its very high turnover numbers with these drug substrates and ability to contribute to very rapid metabolic conversion to the active drug or inactive metabolite. Clearly, there is a need to investigate the extent to which pharmacogenetic differences in BChE in patients will contribute to variations in exposure. For example, the affinities for bambuterol vary with the serum BChE variants (U, A, F, K, and S) (Kovaric and Simeon-Rudolf, 2004). In this study, we have assembled data from a panel of healthy volunteers and from individuals with known phenotypic variations in BChE. Enzyme activities and genotypes were determined and these observations correlated with the metabolic turnover of a novel antedrug development candidate. There was a wide range of BChE activity (not detected: 8,252 U/L), and this correlated well with the metabolic turnover of the antedrug. Genotyping of individuals allowed the separation of the range of activity with genotype and indicated that, for the low to intermediate range, the antedrug substrate was less affected by a decrease in enzyme activity than was the model substrate, butyrylthiocholine.
References
- Goodall R. (2006). Cholinesterase pharmacogenetics. In: Toxicology of Organophosphate and Carbamate Compounds (pp 187–188). Elsevier.
- Kovarik Z, Simeon-Rudolf V. (2004). Interaction of human butyrylcholinesterase variants and bambuterol and terbutaline, J Enz Inhibit Med Chem 19:113–117.
- Svensson L, Tunek A. (1988). The design and bioactivation of presystemically stable prodrugs. Drug Metab Rev 19:165–194.
173. Oxidative Stress and Response to Acute Antipsychotic Treatment
Matej Kastelic,1 Jure Koprivsek,2 Blanka Kores Plesnicar,3 and Vita Dolzan1
1Institute of Biochemistry, Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia, 2Department of Psychiatry, Teaching Hospital Maribor, Maribor, Slovenia, 3Psychiatric Clinic Ljubljana, Ljubljana, Slovenia
It has been suggested that genetic polymorphisms modifying oxidative stress may influence the response to antipsychotic treatment. In the present study, we investigated the influence of polymorphisms in the genes coding for catalase (CAT), manganese superoxide dismutase (MnSOD), and cytochrome P450 17-hydroxylase (CYP17) on the response to acute antipsychotic treatment in 74 patients acutely treated with haloperidol or risperidone either for the first episode of schizophrenia spectrum disorders or for the psychotic episode after tapering their maintenance treatment. CAT C-262T, MnSOD Ala-9Val, and T-C transition in the promoter region of the CYP17 (A2 allele) were genotyped by TaqMan assay and sequence-specific PCR (Applied Biosystems, Carlsbad, California, USA). Psychopathological symptoms were assessed with BPRS and CGI twice: 8–12 and 36–40 days after the first dose of antipsychotic. Adverse events were assessed with the according rating scales: EPS, BARS, and AIMS. The distribution of CAT 262 CC, CT, and TT genotypes was 0.384, 0.507, and 0.110, respectively, the distribution of MnSOD Ala-9Val genotypes was ValVal (0.338), ValAla (0.568), and AlaAla (0.095), and the distribution of CYP17 TT, TC, and CC genotypes was 0.243, 0.527, and 0.230, respectively. Drug type and high dosages of treatment were associated with higher BPRS and CGI baseline scores as well as with higher final BPRS scores. Akathisia (BARS) was dependent on drug dosage, extrapyramidal symptoms (EPS) were more severe in patients treated with haloperidol and at a high dosage, and tardive dyskinesia (AIMS) was dependent on illness duration and was more severe in patients treated with haloperidol and high dosages. When controlled for age, BMI, illness duration, drug type, and dosage, we found a strong correlation between MnSOD Ala-9Val polymorphism and AIMS (P < 0.000) and BARS (P = 0.001) scores, while association with EPS was marginally significant (P = 0.055). We found no association between CAT or CYP17 variants and extrapyramidal adverse events. Also, none of the investigated polymorphisms influenced the efficacy of antipsychotic treatment. In conclusion, our results support the contribution of MnSOD polymorphism to the development of EPS upon acute antipsychotic treatment.
174. The Influence of hOGG1 and XRCC1 Single-Nucleotide Polymorphisms on DNA Repair Capacity
Nina Erculj, Maja Kamin Zadel, and Vita Dolzan
University of Ljubljana, Faculty of Medicine, Institute of Biochemistry, Ljubljana, Slovenia
Genetic polymorphisms in DNA repair genes may result in reduced DNA repair capacity (DRC). The aim of our study was to determine the frequencies of common functional single-nucleotide polymorphisms in specific base-excision DNA repair (BER) genes in a healthy Slovenian population and evaluate their influence on DRC established by comet assay. In total, 174 unrelated healthy subjects were genotyped for hOGG1 S326C, XRCC1 Q399R, and R194W polymorphisms by a real-time TaqMan assay (Applied Biosystems, Carlsbad, California, USA). The influence of selected BER polymorphisms on the percentage of comet tail DNA (TD) was determined in 26 subjects, in which the comet assay had been previously performed. The genotype frequencies for the hOGG1 S326C polymorphism were 0.661 for Ser/Ser, 0.286 for Ser/Cys, and 0.054 for Cys/Cys. For the XRCC1 Q399R polymorphism, the genotype frequencies were 0.129 for Gln/Gln, 0.415 for Gln/Arg, and 0.456 for Arg/Arg. The frequency distribution of the XRCC1 R194W polymorphism was 0.889 for Arg/Arg, 0.088 for Arg/Trp, and 0.023 for Trp/Trp. In the subgroup of 26 subjects, the percentage of TD was significantly increased among individuals with hOGG1 Ser/Cys heterozygous variant genotype, as compared to the Ser/Ser wild-type genotype (8.36 ± 3.69 vs. 6.79 ± 1.39; P = 0.03). No significant associations between Q399R and R194W polymorphisms of XRCC1 and percentage of TD were found. Our results suggest that DRC might be modulated by the hOGG1 S326C polymorphism.
175. Use of Modeling to Predict Oral Clearance of Drugs which Undergo Metabolism by Enzymes Expressing Genetic Variants
Maurice Dickins,1 Ruth Hyland,1 Geoff Johnston,2 Barry Jones,1 and Paul Morgan1
Departments of 1Pharmacokinetics, Dynamics and Metabolism and 2Molecular Medicine, Pfizer Global R&D, Sandwich, Kent, UK
The ability to predict pharmacokinetic variability in early drug development is important in order to understand potential impact on efficacy and safety. Variability in human pharmacokinetics is evident when a significant proportion of clearance occurs via enzymes, which have functional genetic variants. While such variability may be of no clinical consequence if the therapeutic index of the drug is large enough, there are examples where such variability has significant consequences for drug efficacy and/or safety and thus drug labeling (e.g., clopidogrel, irinotecan, and warfarin). Understanding the clinical significance or not of genetic variants in drug metabolism and clearance requires a detailed understanding of the genetic variants, their functional impact on clinical pharmacokinetics, and the resulting impact on the drug’s efficacy and/or safety profile. The potential impact of any genetic variants will not be clear until patient studies, of sufficient numbers, are completed to properly ascertain any impact on drug efficacy and/or safety. Therefore, early drug development would be facilitated through the ability to model the potential impact of genetic variation in drug metabolic clearance in order to assess the need for actual clinical pharmacogenetic studies. The Simcyp modeling platform has been shown to enable the prediction of human clearance of S-warfarin and the effect of the CYP2C9 genotypes (Simcyp, Sheffield, UK). Using Simcyp software (version 8.0), genotype-specific human clearance has been predicted for a number of compounds to show the utility of such modeling approaches to drug development and labeling considerations.
176. Full-Scan Data Acquisition for Rapid Quantitative and Qualitative Analysis using the Exactive LC-MS High-Resolution Mass Spectrometer
Lester Taylor,1 Yingying Huang,2 and Kristi Akervik1
1Thermo Fisher Scientific, San Jose, California, USA, 2Thermo Fisher Scientific, San Jose, CA, USA
Introduction: Current approaches to discovery-stage drug metabolism studies (i.e., pharmacokinetics, microsomal stability, etc.) have focused on the use of targeted analysis (MRM)-based approaches for quantitative analysis. This necessitates the optimization of parameters, such as Q1 and Q3 m/z values, collision energy, and interface voltages. These studies only detect the specified compound and information about other components, such as metabolites, is lost. The ability to do full-scan acquisition for quantitation eliminates the need for compound optimization while enabling the detection of metabolites and other nondrug-related endogenous components.
Methods: Samples from a range of studies (e.g., microsomal stability or pharmacokinetics) were analyzed by using the Exactive nonhybrid Orbitrap LC-MS (Thermo Scientific, Waltham, MA 02454). The sample introduction used UHPLC and fast generic gradients to enable rapid cycle times and high-throughput analysis. The mass spectrometer consisted of a compact single-stage Orbitrap mass analyzer capable of operating at scan speeds compatible with UHPLC peak widths while maintaining characteristic Orbitrap mass accuracy and resolution.
Preliminary Data: Initially, microsomal stability samples were analyzed. Preliminary results indicate that the instrument provides sufficient quantitative capability together with the ability to identify and confirm metabolites. In addition, structure elucidation of metabolites was facilitated by the collection of high-resolution accurate mass fragmentation data. The use of extracted ion chromatograms, using narrow m/z windows, provides selectivity comparable to MRM analysis. Sample sets were further processed to show that metabolites were present in the plasma samples. Further results will be presented that demonstrate that the quantitative-qualitative workflow is possible with this new benchtop single-stage mass spectrometer.
Novel Aspect: A benchtop single-stage UHPLC-Orbitrap system is used for combined high-throughput qualitative and quantitative analysis.
177. Pharmacokinetics of Oxycodone in Extensive and Poor Metabolizers of CYP2D6 during Cotreatment with Placebo or Quinidine
Andrea Sirhan-Daneau, Véronique Michaud, Maude St-Onge, and Jacques Turgeon
Faculty of Pharmacy, Université de Montréal, Montreal, Quebec, Canada
Background/Aims: Oxycodone is an opioid agonist largely prescribed for the treatment of moderate to severe pain. However, a wide interindividual variability is observed in the response to oxycodone. Drug metabolism studies have indicated that oxymorphone is an active metabolite formed by CYP2D6. Hence, we hypothesized that variability in analgesic efficacy to oxycodone could be explained by intersubject variability in the metabolic clearance of the drug.
Methods: Eleven healthy volunteers (8 extensive metabolizers and 3 poor metabolizers of CYP2D6) received a single oral dose of oxycodone (15 mg) on two occasions: once alone and once during cotreatment with quinidine (60 mg) 2 hours prior to, and 10 hours after, the administration of oxycodone. Serial urine samples were collected between 0 and 24 hours to characterize the excretion of oxycodone and noroxycodone.
Results: Percent urinary excretion of oxycodone represented 5.5 ± 1.7% (3.3–8.33%) of the dose after placebo and 6.3 ± 1.6% (3.3–8.6%) after quinidine. Urinary excretion of noroxycodone was 15.7 ± 4.3% (10.7–24.5%) after placebo and increased to 22.2 ± 6.0% (10.1–29.6%) after quinidine. In the extensive metabolizer subgroup, urinary excretion of noroxycodone increased in all subjects from 14.8 ± 3.3% (11.7–18.7%) to 24.7 ± 3.7% (18.3–29.6%) after quinidine (P < 0.003; paired t-test).
Conclusions: Our results indicate that CYP2D6 has a minor role in the overall clearance of oxycodone, with CYP2D6-dependent metabolic pathways representing about 10% of its clearance. These values are similar to those reported for codeine. A decrease in CYP2D6 activity is likely to cause minimal changes in the pharmacokinetics of oxycodone in plasma. This may nevertheless translate into significant changes in the overall analgesic efficacy of the drug due to the formation of the active metabolite, oxymorphone. We pursue additional studies to characterize the contribution of oxymorphone to the overall analgesic properties of oxycodone.
178. Turnover of Nonesterified Fatty Acids in Rats after Multiple Intravenous Infusions of Nicotinic Acid
Christine Isaksson,1 Lambertus A. Peletier,2 and Johan Gabrielsson3
1Department of Pharmacology, University of Gothenburg, Institute of Neuroscience and Physiology, Gothenburg, Sweden, 2Mathematical Institute, Leiden University, RA Leiden, The Netherlands, 3Discovery DMPK and Bioanalytic Chemistry, AstraZeneca R&D Mölndal, Mölndal, Sweden
This investigation demonstrates the PK/PD relationships of nicotinic acid (NiAc)-induced changes in nonesterified fatty acid (NEFA) plasma concentrations following multiple intravenous infusions of NiAc to rats at different dosing rates and durations. Serial arterial blood samples were taken for the evaluation of NiAc and NEFA concentrations. The PK model was fitted to plasma concentration-time data of NiAc. The PK parameters were kept as constants during the regression of NEFA data. A pharmacodynamic feedback model, with NiAc-induced inhibition of turnover rate of NEFA, was used to model the NEFA concentration-time data. An inhibitory moderator, resembling the homeostatic feedback mechanisms, was incorporated to capture tolerance and rebound phenomena seen in NEFA concentrations. The NEFA response variable acted linearly on the production of the moderator. The turnover of the moderator was then fed back inversely on production of response. Turnover rate (kin), fractional turnover rate (kout), and rate constant responsible for tolerance development (ktol) represent system parameters. The drug parameters were potency and sigmoidicity factor (n). Compartmental analysis adequately described the NiAc plasma concentrations. Clearance and volume of distribution of NiAc were found to be approximately 100 mL min−1kg−1 and 0.3 l kg−1, respectively, resulting in an effective half-life of around 2 minutes. NEFA concentrations were significantly decreased in a dose-dependent manner following NiAc administration. Tolerance was seen in NEFA concentrations, particularly for the extended NiAc exposure periods. Following termination of the drug infusion, a significant rebound was seen in the NEFA concentrations. The model mimicked the NEFA concentration-time data well and resulted in parameter estimates with good precision. R0 was approximately 0.5 mmol l−1, t1/2, kout was less than 5 minutes, and the t1/2, ktol was greater than 10 minutes. The potency (IC50) was 30–50 nmol l−1 and the sigmoidicity factor (n) was 1–4. This model elucidates the rate processes governing NEFA turnover after different rates and routes of NiAc dosing. The model will serve as a tool for analyzing and simulating drug-induced changes in plasma NEFA concentrations after treatment with NiAc analogs.
179. Transient Changes in the Expression Pattern of Genes Involved in Bile Acid Metabolism During the Early Stages of Rat Liver Regeneration
Jorge Correia1 and Vera Ribeiro2
1Center for Molecular and Structural Biomedicine, University of Algarve, Faro, Portugal, 2CBME, University of Algarve, Faro, Portugal
Liver regeneration after partial hepatectomy (PH) is a very complex, well-orchestrated phenomenon. Bile acids (BAs) have been shown to play an important role in hepatic regeneration, and their levels are tightly regulated by complex regulatory networks. The farnesoid X receptor (FXR) is activated by physiological levels of BAs and regulates the expression of genes involved in BA synthesis, transport, and detoxification. In this study, changes in the expression patterns of selected genes involved in BA metabolism were investigated, by RT-real-time quantitative PCR, during the early stages of rat liver regeneration following PH. The most significant changes in the expression pattern occur 1 and 22 hours after the PH, with a significant increase in the expression of total FXR, together with a decrease of the mRNA levels of LXR and the isoforms, FXRα1 and FXRα3, which suggest an involvement of these nuclear receptors in the regulation of hepatocyte cell cycle entry and progression. Surprisingly, the expression of SHP and CYP7A1 changed independently of FXR levels, suggesting the involvement of other factors in this complex regulatory network. Taken together, the results obtained suggest that both FXR and LXR play important roles in the coordination of hepatic regeneration, especially in the initiation phase.
180. Stereoselective Kinetics of R- and S-Enantiomers of the Citrus Flavonoid Hesperetin In Vitro
Walter Brand,1 Jia Shao,1 Maarit J. Rein,2 Denis Barron,2 Gary Williamson,2 Peter J. Van Bladeren,2 and Ivonne M. C. M. Rietjens3
1Division of Toxicology, Wageningen University, Wageningen, The Netherlands, 2Nestlé Research Center, Lausanne, Switzerland, 3Division of Toxicology, Wageningen University, Wageningen, The Netherlands
The flavanone, hesperetin, is the aglycone of the rutinoside, hesperidin, the major flavonoid present in sweet oranges. Hesperidin is hydrolyzed in the intestine, and its aglycone is metabolized into glucuronide and sulfate conjugates in the intestinal cells. The transport of these metabolites back into the lumen by ATP-binding cassette (ABC) transporters is an important factor in the limited bioavailability of hesperetin. In contrast with most other flavonoid classes, flavanones contain a chiral center in their basic molecular structure, and therefore, hesperetin exists as an R- and S-enantiomer. Although 2S-hesperidin glycoside is the predominant form in nature, most studies have been performed with the commercially available racemates of hesperidin or hesperetin. We developed an HPLC method, using α1-acid glycoprotein (AGP) as chiral selector in order to separate R- and S-hesperetin, which we identified with a hydrolyzed sample from citrus fruit, on an analytical and semipreparative scale. In subsequent studies, we investigated differences in intestinal metabolism for the separated hesperetin enantiomers in vitro. To this end, apparent kinetics for the conversion of S- and R-hesperetin by UDP-glucuronosyl transferases (UGTs) and sulfotransferases (SULTs) were studied by using microsomal and cytosolic incubations containing fractions from human small intestinal cells. Results obtained demonstrate that hesperetin was glucuronidated to form 7-O-glucuronide and 3’-O-glucuronide metabolites, and sulfonated to form predominantly the 3’-O-sulfate with some 7-O-sulfate conjugates: identified on the basis of HPLC retention times, DAD-spectra and authentic standards, and/or LC-MS/MS and 1H-NMR analysis. The apparent catalytic efficiency (Vmax/Km) of glucuronidation of S-hesperetin was significantly higher, compared to that for R-hesperetin, but equal efficiency was demonstrated for sulfonation. The results demonstrate that differences between the enantiomeric forms should be taken into account when studying biokinetics of chiral flavonoids, and that studies obtained with a racemic mixture may not represent an adequate model system when, as for hesperetin, one of the two stereoisomers predominates in nature.
181. Characterization of Recombinant Breast Cancer Resistance Protein in a Novel Method to Quantify Multidrug Resistance Transporters in Human Tissue Samples
Theodora Tucker,1 Katherine S. Fenner,2 and Michael Coughtrie1
1Center for Oncology and Molecular Medicine, Division of Medical Sciences, University of Dundee, Dundee, UK, 2Pharmacokinetics, Dynamic and Metabolism, Pfizer Global Research and Development, Sandwich, UK
The ATP-binding cassette (ABC) transporter breast cancer resistance protein (BCRP) transports a broad range of xenobiotics, metabolites, and endogenous compounds and is thus important for drug clearance and multidrug resistance. Owing to their complex and hydrophobic nature, ABC transporters are difficult to purify for use as standards. We use a novel approach employing recombinant S-tagged BCRP. The addition of S-protein to S-tag fusion protein results in reconstituted ribonuclease (RNase S) activity, allowing the quantification of the recombinant protein, which is used to create standards in order to immunoquantify the endogenous protein by immunoblotting. For such experiments, it is necessary to confirm that the recombinant protein behaves in the same way as the endogenous protein. Recombinant BCRP with either C-terminal (S-tag.BCRP(C)), N-terminal ((N)S-tag.BCRP), or no S-tag (BCRP) was expressed transiently in HeLa and MDCK cells, and inducibly in HEK293 cells via the FlpInTRex system. We used RT-PCR and immunblotting to investigate the expression and degradation of recombinant BCRP, immunofluorescence to determine localization, and three assays to ascertain function: MTT assay to determine survival of cells following 24-hour exposure to the BCRP substrate, mitoxantrone; FACS to ascertain mitoxantrone accrual in cells following 1-hour exposure; and real-time efflux of another fluorescent BCRP substrate, Hoescht 33342. Untagged recombinant BCRP has high expression, is functional, and localizes to the plasma membrane, as does (N)S-tag.BCRP. However, BCRP.S-tag(C) is much lower in expression, appears to be retained in the endoplasmic reticulum, and extrudes neither H33342 nor mitoxantrone. Proteasome inhibition via MG132 and lysosome inhibition by BMA1 increased the levels of S-tagged and untagged BCRP, but indicated that BCRP.S-tag(C) may be proteasomally degraded to a greater extent than N-terminal or untagged BCRP. Crucially, (N)S-tag.BCRP exhibits S-tag activity, whereas BCRP.S-tag(C) does not, though RT-PCR and immunoblotting with anti-S-tag indicates S-tag is present. Thus, the location of the S-tag can have a considerable effect on the behavior of BCRP, and these results have significant implications for the study of ABC transporters in recombinant systems.
182. Utility of Caco-2 monolayers for OATP2B1-mediated Drug Interaction Profiling
Zhi-Wei Ye, Patrick Augustijns, and Pieter Annaert
Lab for Pharmacotechnology and Biopharmacy, Katholieke Universiteit Leuven, Leuven, Belgium
Transporter proteins are playing a clinically relevant role in drug absorption, distribution, and elimination. OATP2B1 is an important member of the organic anion-transporting polypeptides (OATP) family and is implicated in the intestinal and hepatic disposition of endo- and xenobiotics. In vitro estimation of the contribution of specific transporter isoforms is useful for the prediction of the hepatic drug uptake and transporter-mediated drug interactions. OATP2Bl is the only OATP isoform exhibiting substantial expression in the apical membrane of Caco-2 monolayers, where it contributes predominantly to the uptake of estrone-3-sulfate. The aim of this work was to develop a method for detecting OATP2B1-mediated interaction potential by using 3H-estrone-3-sulfate (ES) as a substrate. It has been shown that OATP2B1 plays a predominant role in ES accumulation in Caco-2 monolayers. The HIV protease inhibitors (PIs) were used as model interacting drugs. Caco-2 monolayers showed the highest transport activity for ES after 2 weeks of culture. The uptake of ES in Caco-2 cells exhibited biphasic saturation kinetics with Km values of 2.9 ± 0.1 and 2.2 ± 0.5 mM. Uptake was sodium independent and was inhibited by different diagnostic OATP inhibitors, including rifampicin, rifamycin, and bromosulfophthalein. Among eight tested HIV PIs [amprenavir (1–100 μM), atazanavir (1–100 μM), darunavir (1–100 μM), iopinavir (0.1–20 μM), nelfinavir (0.1–20 μM), indinavir (1–100 μM), ritonavir (0.1–50 μM), and saquinavir (0.1–50 μM)], the latter three showed the most potent inhibition of ES uptake in Caco-2 cells, with IC50 values of 7.8 ± 1.3, 10.0 ± 2.4, and 12.1 ± 1.9 μM, respectively. Interference with ES accumulation in commonly available Caco-2 monolayers appears a simple in vitro approach to study interaction potential with the human 2B1-isoform of the OATP family of transporters.
183. Generation and Functional Characterization of Abcc2;Abcc3;Abcg2-/−mice
Maria L.H. Vlaming,1 Anita van Esch,1 Zeliha Pala,2 Olaf van Tellingen,1 Koen van de Wetering,1 and Alfred H. Schinkel1
1Division of Molecular Biology, The Netherlands Cancer Institute, Amsterdam, The Netherlands, 2Department of Pharmacology, Istanbul University, Istanbul, Turkey
The multidrug transporters, ABCC2 (MRP2), ABCC3 (MRP3), and ABCG2 (BCRP), are involved in the elimination of potentially toxic compounds from the body and have a substantial overlap in substrate specificity. Whereas ABCC2 and ABCG2 are expressed at apical membranes of hepatocytes and epithelial cells of small intestine and kidney, pumping their substrates into bile, feces, and urine, ABCC3 is expressed at basolateral membranes of these cells and transports its substrates toward the blood. To unravel the possibly overlapping physiological and pharmacological functions of Abcc2, Abcc3, and Abcg2 in vivo, we generated and characterized Abcc2;Abcc3-/-, Abcc2;Abcg2-/-, Abcc3;Abcg2-/-, and Abcc2;Abcc3;Abcg2-/- mice, which are all viable and fertile. Combined with previously generated single knockouts, these strains form a complete panel of mouse models to study the functions of these three ABC transporters in vivo. We used the set of mice to investigate the relative impacts of Abcc2, Abcc3, and Abcg2 on the pharmacokinetics of their shared substrate methotrexate and its main toxic metabolite, 7-hydroxymethotrexate after intravenous administration of methotrexate (50 mg/kg). In contrast to the single and double knockout mice, the plasma levels of methotrexate and 7-hydroxymethotrexate in the Abcc2;Abcc3;Abcg2-/- mice 120 minutes after administration were significantly higher than in wild-type mice (4- and 28-fold increased, respectively). Liver levels of methotrexate and 7-hydroxymethotrexate in Abcc2;Abcc3;Abcg2-/- mice were 7- and even 90-fold increased at minutes after administration, respectively, showing important and overlapping roles for Abcc2, Abcc3, and Abcg2 in limiting liver accumulation of these compounds. We conclude that Abcc2, Abcc3, and Abcg2 are necessary for the fast elimination of methotrexate and 7-hydroxymethotrexate, and that they can clearly compensate for absence of each other. The generated mouse models will provide useful tools to study the relative roles of Abcc2, Abcc3, and Abcg2 in the pharmacokinetics of a wide range of drugs and their metabolites.
184. Comparison of Three Assay Systems utilizing a Common Probe Substrate for Studying P-gp using a Selected Set of Compounds
Peter Szeremy, Pal Akos, Dora Mehn, Peter Krajcsi, and Krisztina Heredi-Szabo
R&D, Solvo Biotechnology, Szeged, Hungary
Aim: The aim of this work was to develop a set of assays, utilizing CalceinAM as the common probe substrate for the investigation of P-gp-mediated drug-drug interactions.
Methods: A dye efflux assay, utilizing P-gp overexpressing human cells and CalceinAM, a substrate of the transporter was used to study the inhibitory properties of the selected compound set. Two membrane-based assays were optimized in order to use CalceinAM: in the ATPase assay as activator molecule and as probe substrate in the vesicular transport assay. The compound set, comprising 22 molecules, was chosen based on two papers (Polli et al., 2006; Rautio et al., 2006), including both high- and low-permeability compounds and also P-gp interactors and noninteractors. Results: Even though CalceinAM is a high-permeability compound, it was successfully applied as a probe in vesicular transport studies. For the 22 compounds, IC50 values were calculated in all three assay systems. The compounds were grouped based on their passive permeabilities and the IC50 values compared within each group. A good correlation was observed for compounds with high passive permeability, whereas the correlation was worse for low-to-intermediate permeability compounds. The main differences were observed in the case of the dye efflux cellular assay, where the test compound needs to cross the plasma membrane first to interact with the transporter. The results were also compared with published IC50 values obtained in monolayer assays (Polli et al., 2006; Rautio et al., 2006).
Conclusions: A set of assays were optimized that combined with passive permeability data could predict a test compound’s interaction with P-gp in more complex assay systems. The main strength of this approach is the use of a common probe substrate in all assays.
References
- Polli JW, Wring SA, Humphreys JE, Huang L, Morgan JB, Webster LO, et al. (2001). Rational use of in vitro P-glycoprotein assays in drug discovery. J Pharmacol Exp Ther 299:620–628.
- Rautio J, Humphreys JE, Webster LO, Balakrishnan A, Keogh JP, Kunta JR, et al. (2006). In vitro P-glycoprotein inhibition assays for assessment of clinical drug interaction potential of new drug candidates: a recommendation for probe substrates. Drug Metab Dispos 34:786–792.
185. Drug-Drug Interaction Studies with Caco-2 Cells according to FDA Draft Guidelines
Daniele Pezzetta, Manuela Pesenti, Luisella Vignati, Anna Moscone, and Pietro Grossi
DMPK and ART–Preclinical Profiling, Accelera Srl, Nerviano, Milan, Italy
The FDA issued, in 2006, its Guidance for Industry: Drug Interaction Studies (FDA, 2006), suggesting a number of assays to investigate if a compound is a substrate for drug transporters, with special focus on the P-glycoprotein (Pgp). According to the FDA guidance, the assays are replicated in the presence of specific inhibitors to confirm any preliminary evidence. The compound is also assayed to assess its potential inhibition of transport systems, including, when appropriate, the evaluation of its IC50 value. Validated methods are required for all these studies. In this poster, we present an application of the Caco-2 cell model, according to the FDA guidelines, with digoxin as the reference Pgp substrate. Quantitative analyses were performed by validated LC-MS/MS methods. The final results demonstrate the ruggedness of the model and its suitability for investigating transport-mediated drug-drug interactions following the FDA guidance.
Reference
- FDA. (2006, September). Guidance for Industry: Drug Interaction Studies—Study Design, Data Analysis, and Implications for Dosing and Labeling. Rockville, Maryland, USA: FDA.
186. Effect of Membrane Cholesterol on BSEP/Bsep Activity—Species Specificity Studies for Substrates and Inhibitors
Emese Kis, Eniko Ioja, Tunde Nagy, Krisztina Heredi-Szabo, and Peter Krajcsi
Research and Development, SOLVO Biotechnology, Szeged, Hungary
The efflux transporter responsible for the canalicular elimination of bile salts from the hepatocytes is the bile salt export pump (BSEP, ABCB11). Absence or inhibition of this transporter leads to bile salt retention in the hepatocyte and, in turn, can lead to cholestatic liver disease. We expressed the BSEP/Bsep protein from three species (human, rat, and mouse) in a baculovirus-infected Sf9 system. Vesicles prepared from these cells were used to evaluate bile salt transport of four conjugated bile salts. As the Sf9 system contains less membrane cholesterol than the liver canalicular membrane, the effect of added cholesterol on the kinetics of BSEP/Bsep-mediated bile salt transport was also investigated. Cholesterol treatment increased the Vmax values in all species, with the most pronounced effect observed in the case of the rat transporter. In contrast, Km values, with the exception of GCDC, remained largely unchanged. The species-specific bile salt transport inhibition potentials of three compounds known to cause clinical cholestasis were investigated in vesicles containing BSEP/Bsep. Troglitazone and glibenclamide inhibited the BSEP/Bsep-mediated transport of different bile salts with similar affinities, whereas the potential of cyclosporine A to inhibit bile salt transport showed species- and bile salt–specific variations. In conclusion, the cholesterol-loaded Sf9 vesicles overexpressing BSEP/Bsep appears to be a useful system for the identification of potential cholestatic compounds and can also be used for the investigation of species specificity. We observed greater differences in IC50 values for inhibitors than in Km values for substrates between species.
187. Evaluation of Species Differences in the Expression of Drug Transporters
Erik Koenig,1 Jesse Gray,1 Cindy Q. Xia,2 Hua Yang,2 and Mi-Sook Kim2
Departments of 1Molecular Technology and 2Drug Metabolism and Pharmacokinetics, Millennium Pharmaceuticals, Inc.,, Cambridge, Massachusetts, USA
Drugs and their metabolites are usually eliminated from the body via urine or bile or, sometimes, both. The extent of biliary excretion of xenobiotics varies widely among species. In general, mice, rats, and dogs are good biliary excreters, while rabbits, guinea pigs, monkeys, and humans are relatively poor biliary excreters. The underlying mechanism for the species differences is at present unclear. Species differences in hepatic blood flow and bile flow do not seem to correlate with the biliary excretion of compounds (1, 2). Thus it is difficult to predict the biliary excretion of drugs in humans from animal data. This study was conducted to address potential roles of drug transporters in species-dependent excretion of drugs and their metabolites. To assess the expression across species, mRNA levels of drug transporters including multidrug resistance 1 (MDR1/Mdr1), five multidrug resistance-associated protein (MRP/Mrp 1, 2, 3, 4 and 5) and breast cancer resistance protein (BCRP/Bcrp) were evaluated in human, monkey, dog and rat liver and kidney. Sequence homology evaluations were conducted to enable a cross-species RT-PCR evaluation. A universal RT-PCR assay primer pair and probe was designed to conserved regions for each gene in each non-human species. Sufficient homology (monkey avg. 96%, dog avg. 86%, rat avg. 79%) was obtained in all transporters evaluated excluding MPR4/Mrp4. RNA for each species and tissue was isolated from 6 unique biological replicates, with a balanced selection for male and female donors; no gender differences were observed for genes evaluated. ddCt values were calculated and the following observations were found in relation to human expression. Notably, hepatic expression levels of MRP3/Mrp3 in human and monkey are significantly higher than those in rat and dog. Hepatic BCRP mRNA levels were highest in monkey among species evaluated. In kidney, mRNA levels of Mrp2 and Mrp3 were highest in dog and mRNA levels of BCRP/Bcrp were highest in rat.
188. Evaluation of the Transporter-mediated Hepatic Uptake in Fresh Rat Hepatocytes
Yoshiyuki Yabe, Aleksandra Galetin, and J. Brian Houston
School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester, UK
Transporter-mediated uptake process is known to be involved in the hepatic uptake of several classes of drugs and can represent the rate-limiting step in the overall hepatic clearance. In the present study, the hepatic uptake of selected HMG-CoA reductase inhibitors, pravastatin, rosuvastatin, and atorvastatin, was investigated by using freshly isolated rat hepatocytes over a concentration range of 0.01–100 μM. In order to evaluate the contribution of transporter-mediated active uptake to the overall clearance, studies were performed in rat hepatocytes in suspension, using [3H]-labeled compounds at 37ºC. Aliquots were taken at the designated time points (30–90 seconds), cells and media were separated by centrifugation through the silicone oil layer, and the radioactivity in the cells and media was determined by a liquid scintillation counter. Clearance via passive diffusion (Pdiff), maximum uptake rate (Vmax), and Michaelis constant (Km) were estimated from the initial uptake rate data by using WinNonlin (Pharmsight, St. Louis, Missouri, USA). In addition, the relative importance of the active hepatic uptake, in comparison to the passive process, was estimated over the range of concentrations investigated. Time-dependent increase in the cell-to-media concentration ratio was observed for all the statins investigated, ranging from 80 to 1,700 in the case of pravastatin and atorvastatin, respectively, at 0.01 μM and at the 90-second time point. Substrate concentration-dependent decrease in the hepatic uptake clearance was observed, confirming the involvement of the saturable uptake mechanism. The active uptake clearance (CLuptake) showed the following rank order: atorvastatin>rosuvastatin>pravastatin and was >30-fold greater than the passive component for the statins investigated. Assessment of the contribution of the passive permeability and active uptake, using rat hepatocytes, indicated that at low concentrations (~0.1 μM), the active process contributes >85% to the overall uptake of all the statins investigated. In conclusion, this strategy is useful for evaluating the contribution of active uptake process via transporters to the overall hepatic clearance.
189. Importance of Drug Efflux Transporters and Physical-Chemical Properties of Drugs for Transplacental Pharmacokinetics
Lenka Cygalova, Martina Ceckova, and Frantisek Staud
Department of Pharmacology and Toxicology, Faculty of Pharmacy, Hradec Kralove, Czech Republic
Aim: P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) are ABC (ATP-binding cassettes) drug efflux transporters with overlapping substrate specificity as well as tissue distribution. In placenta, they have been shown to control the transplacental pharmacokinetics of their substrates. In our study, we evaluated the role of placental P-gp and BCRP by using dually perfused rat placenta and specific substrates for P-gp (rhodamine 123; Rho123) and BCRP (Glybenclamide; GLB) as well as a common substrate for both transporters (BODIPY FL Prazosin; BP).
Methods: Rat term placenta was dually perfused either in an open-circuit system or with fetal perfusate recirculation. To study maternal-to-fetal (MF) and fetal-to-maternal (FM) clearances, substrates were added to the maternal or fetal reservoirs, respectively, and their concentrations were measured in the fetal effluent. To study the FM ratio at steady state, both maternal and fetal sides were infused with equal concentrations of substrate and fetal perfusate was recirculated for 60 minutes.
Results: For all substrates, MF clearances were significantly lower than those in the opposite direction. However, BP, compared to Rho123 and GLB, showed the lowest asymmetry between FM and MF clearances (4.0, 11.0, and 11.6, respectively). Similarly, the highest ratio of FM concentrations 60 minutes after the beginning of fetal perfusate recirculation was found for BP (0.47), in comparison to Rho123 (0.32) and GLB (0.20).
Conclusions: Our results show that both P-gp and Bcrp are able to hinder the transport of their substrates from mother to fetus and return them back to the mother. Surprisingly, BP, despite being transported by both P-gp and BCRP, was the least affected of all substrates. We explain these findings by the high lipophilicity of BP, limiting the effect of active transporters. (This work was supported by a grant of Charles University, no. 119007 C 2007.)
190. In Silico and In Vitro Modeling of Hepatocyte Drug Transport Processes: Control Coefficients for the Disposition of the ABCB1 Substrate Rhodamine-123
Samantha Forster,1 Steven R. Hood,2 and Nick Plant3
1Center for Toxicology, University of Surrey, Guildford, UK, 2Mechanism and Extrapolation Technologies, DMPK, GlaxoSmithKline R&D, Ware, UK, 3Center for Toxicology, University of Surrey, Guildford, UK
Drug transporters are increasingly recognized, alongside phase I and II metabolic enzymes, as important drivers in the pharmaco- and toxicokinetic characteristics of therapeutic agents. The aim of this research is to generate an in silico model of cellular response to stimuli, using a whole-cell approach to investigate system dynamics and test the hypothesis that transporters have a central role in determining drug disposition. An initial step for in silico model development is the generation of the base structure: We utilize CellDesigner™, a graphical front-end for the SBML programming language, allowing us to create a foundation for potential interactions (reactions) between drugs and cellular components (species). We have created one such model, based upon the life cycle of the fluorescent P-gp (ABCB1) substrate, rhodamine-123 (R-123), which is currently being populated with quantitative and kinetic data for each species and reaction. Kinetics of R-123 efflux were obtained by using MDCK II cells overexpressing human ABCB1, while native MDCK II cells were used as controls for inherent transport activity and passive diffusion. Background levels of transport are low, thus making this a useful system for obtaining kinetics for human ABCB1 transport. Also, 4°C conditions used to determine the intracellular accumulation of R-123 by passive diffusion showed that cellular entry was limited due to either altered membrane fluidity or the necessity for active uptake. The latter was indicated by the inhibition of R-123 efflux from MDCK II ABCB1 cells without higher loading in the presence of the ABCB1 inhibitor, quinidine. Uptake studies, using recombinant CHO cell lines expressing various OATPs, will be utilized to investigate this further. Rates of R-123 demethylation to its R-110 metabolite are also being determined, along with the percentage of R-123 protein binding and mitochondrial sequestration. Future work will involve the examination of human primary hepatocytes to improve human relevance and allow us to predict drug disposition in primary hepatocytes and extrapolate drug disposition between various in vitro systems.
191. Inhibition of Hepatobiliary Transporters Is a Potential Mechanism of NSAID Hepatotoxicity
Simone Stahl, Jane Barber, and Gerry Kenna
Molecular Toxicology, AstraZeneca, Macclesfield, Cheshire, UK
Drug-induced liver injury (DILI) is a common adverse reaction caused by nonsteroidal anti-inflammatory drugs (NSAIDs), one of the most frequently used medications worldwide. It appears that the underlying mechanisms are complex and include mitochondrial injury, protein-reactive metabolites, genetic factors, and inhibition of cyclo-oxygenase 2(1). In addition, since NSAID-induced hepatotoxicity often presents as intrahepatic cholestasis, it is conceivable that inhibition of the bile salt export pump (Bsep) and/or other biliary efflux transporters could also be involved(2). In the present study, we have investigated whether NSAIDs affect Bsep and multidrug-restistance associated protein 2 (Mrp2) activity in vitro. Transporter inhibition was analyzed in primary rat hepatocytes cultured in sandwich configuration, utilizing the selective fluorescent probe substrates, cholyllysylfluorescein (CLF) for Bsep and carboxydichlorofluorescein (CDF) for Mrp2. Inhibition of biliary transporters was also studied in membrane vesicles derived from Sf21 cells transfected with rat Bsep, human BSEP, or rat Mrp2, using [3H]-taurocholate and CDF as substrates, respectively. In cultured hepatocytes, the potent inhibition of CLF efflux (apparent IC50, 30–60 μM) was observed with the hepatotoxic NSAIDs, sulindac, diclofenac, and bromfenac. In contrast, ibuprofen, naproxen, and salicylic acid, which only rarely cause liver injury in man, did not significantly inhibit CLF efflux (apparent IC50, >200 μM). A good correlation between the inhibition of CLF efflux in rat hepatocytes and inhibition of [3H]-taurocholate uptake into Bsep/BSEP vesicles was observed. Further, sulindac, diclofenac, and bromfenac significantly inhibited Mrp2-mediated CDF uptake into vesicles (apparent IC50, <200 μM), whereas the minimally hepatotoxic NSAIDs did not (apparent IC50, >1,000 μM). The sulindac-sulfide metabolite showed 10-fold more potent Bsep/BSEP inhibition than sulindac or sulindac-sulfone, suggesting that transporter inhibition by metabolites may be an additional factor contributing to the toxicity observed in patients. Our results demonstrate that the inhibition of Bsep/BSEP may contribute to DILI caused by some NSAIDs. Further, inhibition of Mrp2 may confer a greater risk of DILI than BSEP inhibition alone.
192. Short-Chain Ubiquitination Is Associated with the Degradation Rate of a Cell-Surface–Resident Bile Salt Export Pump (BSEP/ABCB11)
Hisamitsu Hayashi and Yuichi Sugiyama
Laboratory of Molecular Pharmacokinetics, Department of Medical Pharmaceutics, Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan
The reduced expression of the bile salt export pump (BSEP/ABCB11) at the canalicular membrane is associated with cholestasis-induced hepatotoxicity due to the accumulation of bile acids in hepatocytes. We previously demonstrated that 4-phenylbutyrate (4PBA) treatment, an FDA-approved drug for urea cycle disorders, induces the cell-surface expression of BSEP by prolonging the degradation rate of cell-surface–resident BSEP (Hayashi and Sugiyama, 2007). Conversely, BSEP mutations, E297G and D482G, found in progressive familial intrahepatic cholestasis type 2 (PFIC2), reduced it by shortening the degradation rate of cell-surface–resident BSEP (Hayashi and Sugiyama, 2007). Accordingly, to help the development of the medical treatment of cholestasis, the present study investigated the underlying mechanism by which 4PBA- and PFIC2-type mutations affect the BSEP degradation from the cell surface, focusing on short-chain ubiquitination. In Madin-Darby canine kidney II (MDCK II) cells expressing BSEP and rat canalicular membrane vesicles, the molecular weight of the mature form of BSEP/Bsep shifted from 170 to 190 kDa following ubiquitin modification (molecular weight: 8 kDa) (Hayashi and Sugiyama, 2009). Ubiquitination susceptibility of BSEP/Bsep was reduced in vitro and in vivo by 4PBA treatment and, conversely, was enhanced by BSEP mutations E297G and D482G (Hayashi and Sugiyama, 2009). Moreover, biotin-labeling studies using MDCK II cells demonstrated that the degradation of cell-surface–resident chimeric protein fusing ubiquitin to BSEP was faster than that of BSEP itself (Hayashi and Sugiyama, 2009). In conclusion, BSEP/Bsep is modified with two to three ubiquitins, and its ubiquitination is modulated by 4PBA treatment and PFIC2-type mutations. Modulation of short-chain ubiquitination can regulate the change in the degradation rate of cell-surface–resident BSEP by 4PBA treatment and PFIC2-type mutations.
References
- Hayashi H, Sugiyama Y. (2007). 4-phenylbutyrate enhances the cell surface expression and the transport capacity of wild-type and mutated bile salt export pumps. Hepatology 45:1506–1516.
- Hayashi H, Sugiyama Y. (2009). Short-chain ubiquitination is associated with the degradation rate of a cell-surface–resident bile salt export pump (BSEP/ABCB11). Mol Pharmacol 75:143–150.(This project was supported by the Ministry of Education of the Czech Republic, project no. MSM 0021620822.)