1. Drug Transporters in the New Drug Discovery and Development
Yuichi Sugiyama
Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan
Drug transporters play key roles in drug absorption, distribution, and excretion. The information on the functional characteristics of drug transporters provides important information to allow improvements in drug delivery or drug design by targeting specific transporter proteins. The use of transporter function offers the possibility of delivering a drug to the target organ, avoiding distribution to other organs (thereby reducing the chance of toxic side effects), controlling the elimination process, and/or improving oral bioavailability. Recently, many studies on genetic polymorphisms in drug transporters and transporter-mediated drug-drug interactions have been published, and these are part of mechanisms of interindividual difference in drug response. It is important to predict such interindividual difference at an early stage of drug development. OATP1B1 is responsible for the hepatic uptake of many organic anions. We have recently investigated the effect of the genetic polymorphism of OATP1B1, especially the *1b allele, whose frequency was very high in all ethnicities, on the pharmacokinetics of three anionic drugs, pravastatin, valsartan, and temocapril, in the Japanese population and found that OATP1B1*1b is one of the determinant factors governing the interindividual variability in pharmacokinetics. We also established a physiologically based pharmacokinetic (PBPK) model that includes the transporter-mediated membrane transport processes and investigated the effect of changes in transporter function on the pharmacokinetics and, ultimately, the pharmacological and/or toxicological effects.
References
- Giacomini, K.M., Sugiyama, Y. (2005). Membrane transporters and drug response. In: Brunton, L., Lazo, J.S., Parker, K.L. (Eds.), Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th ed. (pp 41–70). New York: McGraw-Hill Professional.
- Kusuhara, H., Sugiyama, Y. (2006). Coordination of uptake and efflux transporters in drug disposition. In: Test, B., Kramer, S., Wunderli-Allenspach, H., Folkers, G. (Eds.), Pharmacokinetic Profiling in Drug Research (pp 105–117). Weinheim, Germany: Wiley-Vch.
- Maeda, K., Sugiyama, Y. (2007). In vitro–in vivo scale-up of drug transport activities. In: You, G., Morris, M.E. (Eds.), Drug Transporters (pp 557–588). Hoboken, New Jersey, USA: John Wiley & Sons.
2. Organic Cation Transporters, OCT and MATE: From Drug Discovery to Clinical Applications
Ken-ichi Inui
Department of Pharmacy, Kyoto University Hospital, Kyoto, Japan
Over 10 years after the identification of the organic cation transporter 1 (OCT1/SLC22A1) (Grundemann et al., 1994), the molecular cloning of H+/organic cation antiporter MATE1 (multidrug and toxin extrusion, SLC47A1) was done successfully (Otsuka et al., 2005). After that, the study fields of organic cation transport systems in the kidney and liver were expanded. Among each transporter family, we found the kidney-specific members, OCT2 (SLC22A2; Okuda et al., 1996) and MATE2-K (SLC47A2; Masuda et al., 2006). Although the substrate specificity of liver type OCT1 and kidney type OCT2 were similar, some drugs are preferentially recognized by OCT2, and their major excretion route was urine. Most substrates of OCT1 and OCT2 were transported by MATE1 and MATE2-K, but cisplatin was not transported by MATE transporters. In addition, oxaliplatin was selectively transported by MATE2-K, not by MATE1, in the human kidney, and therefore, the function of MATE2-K in the kidney is responsible for the low nephrotoxicity of oxaliplatin (Yokoo et al., 2007). Recently, we established the MDCK cells expressing the basolateral OCT and apical MATE1 (Sato et al., 2008). Using this monolayer, we can examine the screening of wide variety of chemicals for the interaction with organic cation transport system. The clinical study with the human renal tissues (Ogasawara et al., 2008), the genetic structures affecting the kidney specific expression of OCT2 (Aoki et al., 2008), and transcriptional regulation of MATE1 and MATE2 (Kajiwara et al., 2007), were elucidated. These results indicate the usefulness of molecular information of OCT and MATE transporters for drug discovery/development and clinical situation, including the personalized dosage adjustment.
References
- Aoki, et al. (2008). AJP 295:F165.
- Grundemann, et al. (1994). Nature 372:549.
- Kajiwara, et al. (2007). AJP 293:F1564.
- Masuda, et al. (2006). JASN 17:2127.
- Ogasawara, et al. (2008). JHG 53:607.
- Okuda, et al. (1996). BBRC 224:500.
- Otsuka, et al. (2005). PNAS 102:17923.
- Sato, et al. (2008). Biochem Pharmacol 76:894.
- Yokoo, et al. (2007). Biochem Pharmacol 74:477.
3. Role of Intestinal and Hepatic OATPs in Drug Absorption and Disposition
Ikumi Tamai and Yoshiyuki Shirasaka
Membrane Transport and Biopharmaceutics, Faculty of Pharmacy, Kanazawa University, Institute of Medical, Pharmaceutical and Health Sciences, Kanazawa, Japan
Organic anion transporting polypeptides (OATPs) are expressed in tissues such as liver and small intestine and are thought to be closely involved in drug absorption and disposition. In liver, plural OATPs, including OATP1B1, 1B3, and 2B1 in humans and Oatp1a1, 1a4, and 1b2 in rats, are expressed at the sinusoidal membrane. Since they exhibit similar, but different, functional characteristics, it is not easy to molecularly correlate them between humans and animals. In the case of beta-lactam antibiotics, we found that Oatp1a4 in rats and OATP1B3 and 1B1 in humans contribute mainly to hepatic uptake of the antibiotic derivatives, by the results of kinetic parameters, relative factor method, selective inhibitors, and in vivo PK parameters. Such information is useful for the extrapolation of animal studies to humans and to understand species similarity of hepatic disposition. Coadministration of grapefruit juice affects the intestinal absorption of the beta-blocker, talinolol, by showing an increase of plasma concentration in rats but a decrease in humans. We demonstrated that talinolol is a substrate of Oatp1a5 in rats and OATP1A2 and OATP2B1 in humans that have been shown to be expressed in intestine. Since talinolol is also a substrate of P-gp and is not subject to metabolism, a contribution of intestinal influx (OATP) and efflux (P-gp) transporters was suggested as the cause of such species differences. Naringin, a major constituent of grapefruit juice, inhibited the Oatp1a5-mediated transport of talinolol. Naringin inhibited rat P-gp at the concentration present in regular grapefruit juice, but did not in human P-gp. Such a differential effect of naringin on rat and human P-gps could explain the distinct grapefruit-juice effect on the intestinal absorption of talinolol. Since other beta-blockers, such as acebutolol and celiprolol, and other drugs, such as fexofenadine and statins, are also shown as substrates of Oatp1a5, OATPs must be key transporters for intestinal absorption of drugs. So, intestinal transporters OATPs as well as P-gp are useful, important targets for the improvement and prediction of intestinal absorption of drugs.
4. Natural Plant-derived Compounds Interact with Membrane Transporters and Channels
Varanuj Chatsudthipong
Department of Physiology, Faculty of Science, Mahidol University, Bangkok, Thailand
Stevioside, an abundant component of Stevia rebaudiana leaf, has become well known for its intense sweetness (250–300 times sweeter than sucrose) and is used as a noncaloric sweetener in several countries. A number of studies have suggested that, besides sweetness, stevioside, along with related compounds, may also offer therapeutic benefits, as they have antihyperglycemic, -hypertensive, -inflammatory, -tumor, -diarrheal, diuretic, and immunomodulatory actions. In addition, it is of interest to note that steviol and, possibly, stevioside, to a lesser extent, interact with both organic anion and cation transport systems in several experimental models. Stevioside and steviol inhibit transepithelial transport of PAH in isolated perfused rabbit renal proximal tubule. However, only steviol is an effective inhibitor of hOAT1- and hOAT3-mediated transport expressed in Xenopus oocytes, renal S2 cells, and mouse renal cortical slices (Chatsudthipong and Jutabha, 2001; Srimaroeng et al., 2005; Srimaroeng et al., 2005). Similarly, it was found that steviol, but not stevioside, inhibits organic cation uptake in CHO-K1 cells stably expressing rabbit rbOCT1 and rbOCT2 (Chatsudthipong et al., 2003). In intact renal tubules, both stevioside and steviol are able to inhibit cation uptake, but the inhibitory effect of steviol is greater than that of stevioside. A recent study in a human intestinal T84 cell line showing that steviol and its analog (dihydroisosteviol), but not stevioside, reversibly inhibit cAMP-activated chloride secretion (Pariwat et al., 2008). Electrophysiological and enzymatic analysis indicated that dihydroisosteviol directly targets CFTR. The compound is specific to chloride secretion stimulated by cAMP, but not by calcium. An in vivo mouse model of cholera showed that intestinal fluid secretion stimulated by Vibrio cholera enterotoxin is markedly reduced by dihydroisosteviol. Also, intraperitoneal administration of steviol is effective in reducing intestinal fluid secretion in this mouse model (Muanprasat, C., unpublished observation). These later findings represent a novel option in the development of new antidiarrheal CFTR inhibitors.
References
- Chatsudthipong, Jutabha. (2001). J Pharmacol Exp Ther
- Chatsudthipong, et al. (2003). FASEB J
- Pariwat, et al. (2008). J Pharmacol Exp Ther
- Srimaroengi, et al. (2005). J Pharmacol Exp Ther
- Srimaroengi, et al. (2005). Pharm Res
5. Role of Human Copper Transporter, HCTR1, in Cisplatin Resistance
Im-Sook Song
Department of Pharmacology and PharmacoGenomics Research Center, Inje University College of Medicine, Busan, South Korea
Platinum-based antitumor agents have been effective in the treatments of many human malignancies, but the ultimate success of treatment is often compromised by the development of drug resistance. The copper transporter, Ctr1, the major Cu influx transporter, has been convincingly demonstrated to transport cisplatin and its analogs, carboplatin and oxaliplatin. Evidence also suggests that the two Cu efflux transporters, ATP7A and ATP7B, regulate the efflux of cisplatin. The observations that these copper transporters are also able to transport Pt drugs are intriguing, because conventional thinking of the inorganic physiologic nature of cisplatin and copper is quite different. Hence, understanding the underlying mechanistic aspects of these transporters is exquisitely important. While mechanisms by which hCtr1, ATP7A, and ATP7B transport copper ions have been studied, to some good extent, very little is known about the mechanisms by which these transporters shuffle platinum-based antitumor agents. Previous studies have demonstrated that treating cultured cells with cisplatin (CDDP) upregulated the expression of glutathione (GSH) and its de novo rate-limiting enzyme, glutamate-cysteine ligase (GCL), which consists of a catalytic (GCLC) and a modifier (GCLM) subunit. It has also been shown that many CDDP-resistant cell lines exhibit high levels of GCLC/GCLM and GSH. Because the GSH system is the major intracellular regulator of redox conditions that serve as an important detoxification cytoprotector, these results have been taken into consideration that elevated levels of GCL/GSH are responsible for the CDDP resistance. In contrast to this context, we demonstrated here that overexpression of GSH by transfection with an expression plasmid containing the GCLC cDNA conferred sensitization to CDDP through upregulation of human copper transporter (hCtr) 1, which is also a transporter for CDDP. Depleting GSH levels in these transfected cells reversed CDDP sensitivity with concomitant reduction of hCtr1 expression. Although rates of copper transport were also upregulated in the transfected cells, these cells exhibited biochemical signature of copper deficiency, suggesting that GSH functions as an intracellular copper-chelator and that overexpression of GSH can alter copper metabolism. More important, our results reveal a new role of GSH in the regulation of CDDP sensitivity. Overproduction of GSH depletes the bioavailable copper pool, leading to upregulation of hCtr1 and sensitization of CDDP transport and cell killing. These findings also have important implications, in that modulation of the intracellular copper pool may be a novel strategy for improving chemotherapeutic efficacy of platinum-based antitumor agents.
6. The ICH S2 Revision and New Movement of the Micronucleus Assay
Makoto Hayashi
Biosafety Research Center, Foods, Drugs and Pesticides (BSRC), Iwata, Shizuoka, Japan
The S2A (1995) and S2B (1997) guidelines describe a basic test battery of in vitro and in vivo genotoxicity tests to support the safety of drugs for human use. This three-test battery is a bacterial mutagenicity assay, a mammalian cell assay (either tk mutations in mouse lymphoma cells, or in vitro assay for metaphase chromosome aberrations) and an in vivo assay for cytogenetic damage, typically a test for micronuclei in erythrocytes of rodents. The major motivation prompting revisions to the guidelines was the high frequency of positive results found in the in vitro mammalian cell assays, which were considered not relevant under in vivo conditions, because they included many that were weakly positive, associated with considerable toxicity, and/or only seen at high concentrations, and the other tests in the battery were negative. This led to a great deal of follow-up testing, including additional animal testing to assess whether there was any genotoxic risk. We also take into account the 3Rs concept into the revision of the guidance. The expert working group considered options, such as developing a better guidance on weight-of-evidence and interpretation of results. We also will merge two S2 guidances (A and B) into one guidance (S2(R1)). We proposed two options that were equally acceptable: 1) the bacterial gene mutation assay and one of three assays using mammalian cells (metaphase chromosomal aberration assay, mouse lymphoma TK assay, and in vitro micronucleus assay), followed by one or two in vivo assay(s), depending on the result of the in vitro assays, and 2) the bacterial gene mutation assay and two in vivo assays using different tissues. The in vivo assays should be incorporated into a repeat-dose toxicological study, if possible. Additionally, several revisions were made, including issues related to the in vivo micronucleus assay. Based on the ICH and other international movements of the genotoxicity assay, the rodent micronucleus assay is also moving to the novel stage. For example, the application of the flow-cytometry technique, an omission of concurrent positive control, and the use of tissues other than bone marrow are now considering for the regulatory acceptance. It is, however, more important that the movement to integrate the micronucleus assay into the repeat-dose toxicological study be made and also to combine the assay with other assays, such as the comet assay, taking into account the 3Rs concept.
7. From Barakol Experiences to Novel Strategies of Safer Drugs
Pornpen Pramyothin
Department of Pharmacology and Physiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
Barakol is a major constituent of Senna siamea Lam. Irwin and Barneby (syn. Cassia siamea Lam.), (Caesalpiniaceae), from which young leaves and flowers were used as folk medicine for centuries to treat insomnia, diabetes, hypertension, asthma, and constipation. Water and ethanolic extracts of young S. siamea leaves were confirmed to induce decreased locomotor activity in rats, while in humans, a sedative effect was observed without toxicity. In 1997, an S. siamea tablet (using barakol as a standardization marker) was approved and became commercially available in Thailand as an alternative treatment of anxiety and insomnia. Concurrently, anxiolytic effect of barakol was demonstrated in rats, using the elevated plus maze test. Barakol was also found to behave like a dopamine agonist in rat striatal slices by inhibiting endogenous dopamine release without a change in dopamine uptake. Nonetheless, the attribution of S. siamea central nervous system activities to the presence of barakol is still controversial due to some conflicting results from different laboratories. Four years after the launch of the S. siamea tablet and the gaining popularity in producing natural sleep, incidence of hepatitis in patients treated with the S. siamea tablet was first reported. The mechanism of liver dysfunction associated with S. siamea is not well understood. Chronic toxicity of S. siamea in rats showed hepatocyte degeneration and necrosis after feeding for 6 months with the appearance of fatty liver and increased relative liver weight at 2, 000 mg/kg/day of S. siamea, which was 100-fold higher than the human therapeutic dose and equivalent to approximately 25 mg/kg/day of barakol. Acute and subacute hepatotoxicity of barakol (100– 300 mg/kg) in mice and rats indicated changes in transaminase activities and reduced total bilirubin. All toxicity studies exhibited a dose-dependent manner. Clinical evaluation in 12 healthy Thai volunteers receiving 2–4 tablets of S. siamea (approximately equivalent to 0.3–0.6 mg/kg/day of barakol) for 3–180 days found 8 subjects were admitted with anorexia and jaundice. There was no relationship between the degree of symptom and dosage/duration of S. siamea intake. Based on the clinical evidence, the Food and Drug Administration of Thailand had issued an initial safety warning and later totally removed products containing S. siamea from the market. There is continuing debate whether the hepatotoxicity of S. siamea is due to idiosyncrasy or the major constituent, barakol, or other unexpected constituents. Since few anxiolytic agents are able to induce natural sleep like S. siamea, it is challenging to clarify the anxiolytic activity, safety, and other stability issue related to the drug development of barakol in order to maximize the use of S. siamea.
8. Vanilloid Receptor 1 (VR1) Ligands as the Next Generation of Analgesic for Neuropathic Pain
Jeewoo Lee
Laboratory of Medicinal Chemistry, College of Pharmacy, Seoul National University, Seoul, South Korea
The TRPV1 (Vanilloid receptor 1 or VR1) is a member of the transient receptor potential (TRP) superfamily. Since TRPV1 functions as a nonselective cation channel with high Ca2+ permeability, its activation by endogenous ligands leads to an increase in intracellular Ca2+ that results in excitation of primary sensory neurons and, ultimately, in the central perception of pain. The involvement of this receptor in both pathological and physiological conditions suggests that the blocking of this receptor activation, by desensitization or antagonism, would have considerable therapeutic utility. The therapeutic advantage of TRPV1 antagonism over agonism is that it lacks the initial excitatory effect preceding the desensitization, which represents a limiting toxicity for the systemic application of the agonists. We have previously reported that isosteric replacement of the phenolic hydroxyl group in potent TRPV1 agonists with the alkylsulfonamido group provided a series of compounds that were effective antagonists to the action of capsaicin on rat TRPV1 heterologously expressed in Chinese hamster ovary (CHO) cells Lee et al., 2003). Further optimization provided novel chiral N-(2-benzyl-3-pivaloyloxypropyl) 2-[4-(methylsulfonylamino)phenyl]propionamide analogs, which were characterized as highly potent, stereospecific rTRPV1 antagonists (Ryu et al., 2008). Their high binding affinities and potent antagonisms are comparable, or more potent than, those of 5-iodoRTX under the same assay conditions. On the basis of our findings, a series of TRPV1 antagonists having 4-(methylsulfonylamino)phenyl as an A-region and f -methyl amide as a B-region have been investigated, and then KMJ-372, N-(4-tert-butylbenzyl)-2-[3-fluoro-4-(methylsulfonylamino) phenyl]propionamide, was selected for further examination as a preclinical candidate. KMJ-372 showed a high binding affinity and potent antagonism against capsaicin, pH, heat, and NADA in rat and human TRPV1 and displayed excellent analgesic profiles in inflammatory, neuropathic, and visceral pain models. Its pharmacokinetics and metabolism studies were also examined.
References
- Lee, J., Lee, J., Kang, M., Shin, M-Y., Kim, J-M., Kang, S-U., et al. (2003). N-(3-acyloxy-2-benzylpropyl)-N’-[4-(methylsulfonylamino)benzyl]thiourea analogues: novel potent and high affinity antagonist and partial antagonists of the vanilloid receptor. J Med Chem 46:3116–3126.
- Ryu, H., Jin, M-K., Kang, S-U., Kim, S. Y., Kang, D. W., Lee, J., et al. (2008). Stereospecific high-affinity TRPV1 antagonists: chiral N-(2-benzyl-3-pivaloyloxypropyl) 2-[4-(methylsulfonylamino) phenyl]propionamide analogues. J Med Chem 51:57–67.
9. Proteomics Approach to Find New Biomarkers for Genotoxicity
Takayoshi Suzuki
Division of Cellular and Gene Therapy Products, National Institute of Health Sciences, Tokyo, Japan
We started our trial to find new biomarkers for genotoxicity using nano-LC-MS/MS (DiNa-QstarXL) system. Urine samples form mouse treated ip with genotoxic carcinogens (quinoline, 4-dimethylaminoazobenzene, N-nitrosomorpholine and 2,4-diamonotoluene) or non-genotoxic chemicals (clofibrate, 1,4-dichlorobenzene, 1-naphtylisothiocyanate, glycine), were obtained from the Collaborative Study Group on Toxicogenomics in JEMS/MMS in which differential gene expression was studied by DNA microarray (GeneChip, Affymetrix, San Diego, CA, USA) and TaqMan®, Applied Biosystems, Foster city, CA, USA. Low Density Array. The urine was collected compulsorily at the sacrifice (4 and 48 hours after the treatment). The 5 mL of urine was digested by trypsin and 20 ng of peptides were injected to the online nano-LC MS/MS analysis. All the peptide signals were visualized on three-dimensional map, using Pep3D (freely available from SPC/ISB) and mzMore (originally developing) software. With a sensitive nono LC-MS system, a few milliliters of urine is sufficient to overview the peptidome. By using a 2-hour gradient, we could observe more than a thousand peptide signals. Several peptides changed their intensity both within and between the treatments. Quinoline and N-nitrosomorpholine treatment showed an alteration of six peptide signals in common, but no changes in nongenotoxic chemicals. We tried focusing our identification of these peptides and identified one of them as a novel member of major urinary protein. The high;y abundant proteins found in the mouse urine sample was the major restriction in loading more amounts of peptide sample to cover low abundant peptides. When human urine samples were analyzed, more numbers of peptides were observed without any major urinary proteins, which can increase the chance to detect relevant biomarkers. For more precise quantification of peptides, we used a stable isotope-tagged method (cICAT). The recent introduction of the higher resolution mass spectrometry, the LTQ Orbitrap™, Thermo Fisher Scientific, greatly improved the coverage and identification of low abundant proteins. The future direction of sensitive and automated proteome analysis with or without isotope labeling is being discussed. Proteomic approach could be a promising new field of mutation research, which is readily applicable for exposed human samples.
10. Role of Metabolism in Arsenic-induced Toxicity: Identification and Quantification of Arsenic Metabolites in Tissues and Excreta
Blakely M. Adair1 and Michael F. Hughes2
1Chemical Science and Technology Lab/ Analytical Chemistry Division, National Institute of Standards and Technology, Charleston, South Carolina, USA
2Office of Research and Development/ National Health and Environmental Effects Research Laboratory, US EPA, Research Triangle Park, North Carolina, USA
Arsenic is a known toxicant and carcinogen. Methylation of inorganic arsenic was once thought to be a detoxification mechanism because of the rapid excretion and relatively lower toxicity of the pentavalent organic arsenical metabolites. Advances in analytical chemistry have allowed the identification and quantification of trivalent organic arsenical metabolites that have proven to be potent toxicants. Thus, it appears that the metabolism of arsenic involves toxic activation via the reduction of penta- to trivalent arsenicals and detoxification via oxidative methylation of tri- to pentavalent arsenicals. Multiple analytical techniques are necessary for the accurate identification and quantification of arsenic metabolites. Our research group has used hydride generation-atomic absorption spectrometry with cryogenic trapping to detect trivalent arsenicals in urine and pentavalent arsenicals in urine and tissues, such as liver, lung, and bladder, of arsenic-treated rodents. In addition, High-performance liquid chromatography (HPLC)-inductively coupled plasma-mass spectrometry and HPLC-electrospray ionization-mass spectrometry have been used to quantify and identify thiolated arsenicals in urine, respectively. Mouse and rat metabolism studies with oral administration of arsenicals revealed that the metabolites produced differed by methylation and oxidation state of the arsenicals administered. Metabolites detected in mice following monomethyl arsenic exposure include dimethyl and trimethyl arsenic, with ease of methylation being greater for trivalent monomethyl arsenic. Mice metabolized dimethyl arsenic to produce trimethyl and thiolated arsenicals in urine. In the rat, following a 14-day drinking-water exposure to pentavalent dimethyl arsenic, metabolites included trimethyl and thiolated arsenicals in urine and trimethylarsenic in tissues. Following a similar exposure of rats to arsenate, metabolites in urine included dimethyl, trimethyl, and thiolated arsenicals, and dimethyl and trimethyl arsenicals were detected in tissues. The acute and chronic toxicities of this metalloid may be from one or more of these metabolites of inorganic arsenic. Therefore, it is critical to link results from metabolism and health-effect studies to determine the mode of action for arsenic toxicity.
Disclaimer
This abstract does not necessarily reflect EPA policy.
11. Role of Transporters in Arsenic Disposition and Accumulation
Jin-Ding Huang
Department of Pharmacology, Natl Cheng Kung University Medical College, Tainan, Taiwan
In the study of arsenic exposure and accumulation, it is important to identify the molecular mechanism of arsenic disposition. Although a passive diffusion pathway cannot be totally excluded, the role of influx transporters is gradually revealed. Phosphate transporters can translocate pentavalent arsenate. Aquaglyceroporins 7 and 9 uptake trivalent arsenite. Recently, we knocked down aquaglyceroporin 3 by RNA interference and found a decreased uptake of aresenite in cancer cell lines. Further, we transfected OATP-C into HEK293 cells and found an increased uptake of arsenate, arsenite, but not MMA, and DMA. The knowledge of arsenic efflux transporter is also limited. Arsenite bound to thiol groups of cellular proteins. Inside the cell, there are 10 mM of reduced glutathione. Glutathione liberates arsenite from cellular proteins in the form of ATG (arsenic triglutathione). ATG is methylated by ATG methyl transferase to form MADG (methyl arsenic diglutathione). ATG and its metabolites are not permeable to the cell membrane. It seems that glutathione-conjugate transporters are responsible for the disposition of these arsenic conjugates. In the rat bile, only ATG and MADG (methyl arsenic diglutathione) can be detected. The organic anion transporter, MRP2, is responsible for the biliary excretion of ATG and MADG at the canalicular membrane. Arsenite, arsenate, and MMA are detected after hydrolysis and oxidation. In addition, we found that ATG is also transported by OATP-C in transfected cells.
12. Mode(s) of Action of Arsenic-induced Tumors and Toxicity
Kirk Thomas Kitchin and Kathleen Wallace
Department of Environmental Carcinogenesis, US EPA, Research Triangle Park, North Carolina, USA
Although arsenic has long been known for its toxicity and carcinogenicity, it is unknown exactly how these adverse health effects occur. Three of the biggest unresolved issues in arsenic exposures and adverse health effects are 1) what is (are) the mode of action (MOA)(s) of arsenic-induced carcinogenicity and toxicity, 2) which arsenic species is (are) the cause of adverse health effects, and 3) for carcinogenicity, does arsenic act in a genotoxic or nongenotoxic mode at low concentrations? Three probable MOAs linking arsenic exposures and adverse health effects are 1) binding to sulfhydryl groups of proteins, 2) ROS generation and oxidative stress, and 3) DNA methylation changes. Altered DNA methylation is a recently proposed MOA for arsenic carcinogenicity, which is supported by less experimental evidence than the other two major proposed MOAs. Radioactive arsenic was used to determine the Kd, Bmax, and association and dissociation rates for arsenite binding to some cysteine containing synthesized peptides and proteins. Some of the many cellular targets of trivalent arsenical binding include tubulin, DNA repair enzymes, PARP-1, thioredoxin reductase, estrogen receptor–alpha, and Keap-1. The lifetimes of unidentate (<1 second), bidentate (1–2 minutes), and tridentate (1–2 hours) binding suggest that only bidentate and tridentate binding to arsenite are biologically important in causing adverse health effects. Substantial information (i.e., human, animal, cellular, in vitro systems) suggests the participation of reactive oxygen species and oxidative stress in arsenic carcinogenesis. For example, there are large amounts of data showing arsenic-induced increases in 8-OHdG in many different experimental systems. Regardless of what key events are components of a MOA, it is quite likely that either the accumulation of mutations, cell proliferation, or increased estrogen action (via DNA methylation) in tissues are the final drivers of carcinogenesis. Several of these proposed MOAs may act either together or sequentially in the five different organs in which arsenic causes human cancer (e.g., lung, skin, urinary bladder, kidney, and liver).
Disclaimer
This abstract does not necessarily reflect EPA policy.
13. Arsenic and Immune System: The Involvement of Cholinergic Lymphatic Receptors
Jutamaad Satayavivad and Tawit Suriyo
Laboratory of Pharmacology, Chulabhorn Research Institute, and Center of Environmental Health, Toxicology and Management of Chemicals, Mahidol University, Bangkok, Thailand
The immunotoxic effects of arsenic have recently received a lot of attention, since the integrity of the immune system is required for an adequate immunosurveillance against tumor cells and response to infection. Arsenic, in the micromolar range (μM), can inhibit lymphocyte proliferation and interleukin-2 (IL-2) secretion in vitro (Conde et al., 2007). However, the molecular mechanisms of these effects have not yet been fully elucidated. A recent report indicates the existence of a cholinergic system in lymphocytes that is independent of cholinergic nerves and is involved in the regulation of immune functions. Correspondingly, both T- and B-lymphocytes express functional cholinoreceptors involved in the regulation of development and activation of these cells (Fujii et al., 2008). Recently, we found that methylmercury can alter the lymphatic muscarinic receptors signaling pathway leading to apoptosis and is also involved in the inhibition of IL-2 production (Suriyo et al., 2008). Arsenic has been reported to affect neuronal cholinergic system (Rodriguez et al., 2003). Our previous study has demonstrated that arsenic can decrease response of cholinoreceptors to acetylcholine in heart, trachea, and aortic ring. This prompts us to consider the involvement of the cholinergic lymphatic system in the immunotoxicity of arsenic. The sensitivity of immune cells to arsenic toxicity was investigated. To this aim, the cytotoxic effects of sodium arsenite (NaAsO2) were tested on immune cells, neuronal cells, and lung cells represented, respectively, by MOLT-3, SHSY5Y, and A549 cell lines by using XTT and MTT assays. The results showed that MOLT-3 cells are more sensitive to arsenic-induced cell death, when compared to SHSY5Y and A549 cells. The LD50 of 24-hour exposure to sodium arsenite were 8, 15, and 150 μM in MOLT-3, SHSY5Y, and A549, respectively. More interestingly, it has been found that sodium arsenite can cause the alteration of cholinergic lymphatic receptors. After 24 hours of exposure to 1–15 μM of sodium arsenite in MOLT-3 cells, the expression of the α7 nicotinic receptor (α7 nAChR) increased dose dependently, while the expression of muscarinic receptor subtype 3 (M3 mAChR) had shown the opposite effect, as measured by the Western blot technique. Our preliminary results suggest that immune cells are very sensitive to arsenic, and the alteration of cholinergic lymphatic system by arsenic may be one of the mechanisms of its immunotoxicity.
References
- Conde, P., Acosta-Saavedra, L.C., Goytia-Acevedo, R.C., Calderon-Aranda, E.S. (2007). Sodium arsenite-induced inhibition of cell proliferation is related to inhibition of IL-2 mRNA expression in mouse-activated T-cells. Arch Toxicol 81:251–259.
- Fujii, T., Takada-Takatori, Y., Kawashima, K. (2008). Basic and clinical aspects of non-neuronal acetylcholine: expression of an independent, nonneuronal cholinergic system in lymphocytes and its clinical significance in immunotherapy. J Pharmacol Sci 106:186–192.
- Rodriguez, V. M., Jimenez-Capdeville, M. E., Giordano, M. (2003). The effects of arsenic exposure on the nervous system. Toxicol Lett 145:1–18.
- Suriyo, T., Thiantanawat, A., Chaiyaroj, S.C., Parkpian, P., Satayavivad, J. (2008). Involvement of the lymphocytic muscarinic acetylcholine receptor in methylmercury-induced c-Fos expression and apoptosis in human leukemic T-cells. J Toxicol Environ Health A 71:1109–1123.
14. UDP-Glucuronosyltransferase and Drug Glucuronidation-Linking Structure, Function, and Kinetics
John O. Miners
Department of Clinical Pharmacology, Flinders University, Adelaide, Australia
The UDP-glucuronosyltransferases (UGTs) comprise a superfamily of enzymes that catalyze the covalent linkage of glucuronic acid to a typically hydrophobic substrate bearing a suitable acceptor functional group. Nineteen functional human UGT proteins have been identified, to date, and these have been classified into families and subfamilies, based on sequence identity. The individual enzymes exhibit distinct, but overlapping, substrate selectivities. In particular, most UGTs have the capacity to glucuronidate small planar phenols and aliphatic alcohols, but increasing complexity of the aglycone confers selectivity due to steric, hydrophobic, and polar interactions. In the absence of an X-ray crystal structure for an entire UGT protein, structure-function relationships have been inferred largely from chimeragenesis and site-directed mutagenesis. The presence of a near conserved N-terminal domain histidine plays a pivotal role as the catalytic base in the glucuronidation of phenols and aliphatic alcohols. Substitution of this histidine with proline provides the capacity for UGT1A subfamily enzymes to convert tertiary amine substrates to the corresponding quaternary ammonium-linked glucuronide, as occurs with UGT1A4. The complex kinetic behavior observed for many of the human enzymes is thought to arise from homodimerization, which is a feature of most, if not all, UGT proteins. Notably, data with UGT2B7 are consistent with the existence of two catalytic sites for each substrate within the active site, along with multiple effector sites. The existence of these sites gives rise to positive homo- and heterotropic cooperativity and, consequently, complex kinetic interactions between alternate substrates of this enzyme. Understanding the substrate profile of the human UGTs with respect to both xenobiotics and endogenous compounds has further provided important insights into the modulation of enzyme activity in vitro by membrane long-chain unsaturated fatty acids. In turn, this has allowed the development of experimental paradigms that accurately predict key kinetic parameters (i.e., intrinsic and hepatic clearances) for glucuronidated drugs in vivo.
15. Toxicogenetic Approach to Understand Chemical-induced Liver Toxicity
Masaaki Miyata1 and Yasushi Yamazoe2
1Graduate School of Pharmaceutical Sciences, Tohoku Univiversity Faculty of Pharmaceutical Sciences, Sendai, Japan
2Division of Drug Metabolism and Molecular Toxicology, Tohoku University, Graduate School of Pharmaceutical Sciences, Sendai, Japan
Currently, various combination approaches are introduced to understand the underlying mechanisms of chemical-induced toxicities. In the expression analyses, drug-metabolizing enzymes are often detected as prominent markers, because of their striking changes, depending on the biological conditions. These changes are possible to reflect certain pathophysiological states, but have not been clearly linked with mechanisms of the toxicities. Lithocholic acid is a naturally occurring hydrophobic bile acid and known to induce liver cholestasis. Unlike other bile acids, this bile acid is biotransformed in the body by enzymatic oxidation and conjugation in ways similar to xenobiotics and drugs. Lithocholate also behaves as receptor ligands. These data prompted us to investigate lithocholate-induced liver damage as an endogenous-model of drug-induced toxicity. Feeding of lithocholic acid in C57BL6 mice induced perturbation of lipid metabolism and, later, resulted in the appearance of liver disorders. In parallel with liver-toxicity parameters, mRNA levels of various enzymes, such as CYP, SULT, AKR, and transporters of both ABCs and SLCs, are altered. Functional proteins associated with lipid import and export are also changed. Certain changes may be linked with specific nuclear receptor signals. Some of the changes are also connected with a shift of energy metabolisms in the liver. Increased loss of phospholipids to cope with the increased excretion demand of hydrophobic bile acids to bile resulted in the shortage of fatty acid and phospholipids in liver of lithocholate-treated mice. These changes are expected to affect the expression levels of drug-metabolizing enzymes associated with fatty acids and glycerides. In fact, various liver CYPs are found to alter in response to lipid levels. These changes occurred in proceed with the alteration of peroxisomal and mitochondrial proteins for energy metabolisms. These data suggest the quantification of drug-metabolizing enzymes, which function to reverse the excess condition of lipids, as early markers of liver disorders.
16. Metabolic Activation of Capsaicin and Induction of NADPH:Quinone Oxidoreductase: Capsaicin-derived ROS Overproduction Induces HO-1 Expression and Provides Protection against Cytotoxicity of Peroxynitrite in HEPG2 cells
Young-Nam Cha
Pharmacology, Inha University, School of Medicine, Incheon, South Korea
Capsaicin (trans-8-methyl-N-vanillyl-6-nonenamide) is a major pungent catechol-ester ingredient in red pepper and is reported to have cytoprotective properties. However, the mechanisms underlying its cytoprotective effects remain largely unresolved. In the present study, capsaicin was found to induce the expression of heme oxygenase-1 (HO-1), the rate-limiting enzyme that catalyzes oxidative degradation of free-heme into carbonmonoxide (CO) gas and antioxidant biliverdin/bilirubin. Capsaicin increased the production of reactive oxygen species (ROS) in HepG2 cells, induced transient increase in the phosphorylation of AKT, promoted nuclear translocation of Nrf2 and enhanced its binding to ARE, and increased HO-1 expression. However, prior exposure of HepG2 cells to N-acetylcysteine, a membrane-permeable cysteine antioxidant, blocked not only the capsaicin-derived ROS production, nuclear translocation of Nrf2, and its ARE binding, but also abolished HO-1 induction. In HepG2 cells treated with capsaicin, while the HO-1 expression was induced, the activity of NADPH:quinone oxidoreductase (NQO) was inhibited, and also, the NQO-1 expression was decreased. Dicoumarol, a well-known inhibitor of NQO activity, produced an identical result with capsaicin and showed that while HO-1 was induced, the NQO-1 expression was diminished. We hypothesize that the O-demethylated metabolite of capsaicin (a catechol) generated in HepG2 cells undergoes further redox-cycling metabolism to generate a reactive capsaicin semiquinone radical that binds covalently to NQO, inhibits its activity, and destroys newly induced NQO-1 protein and leads to the overproduction of ROS. This ROS, in turn, would trigger the phosphorylation of AKT, activation, and nuclear translocation of Nrf2 as well as its binding to ARE, finally stimulating the HO-1 expression as a delayed adaptive cytoprotective response. As a result, the HepG2 cells pretreated with capsaicin could survive better against the cytotoxicity of a peroxynitrite challenge.
17. UGT Enzymes: Drug Interactions, Protein-Protein Interactions and Regulation
Tsuyoshi Yokoi, Ryoichi Fujiwara, Hiroyuki Yamanaka, Akiko Nakamura, Eriko Higashi, Tomohito Matsui, Miki Katoh, and Miki Nakajima
Kanazawa University, Faculty of Pharmaceutical Sciences, Kanazawa, Japan
Human UDP-glucuronosyltransferases (UGTs) play a key role in the metabolism of endogenous and exogenous compounds. Human UGTs are now extensively studied in terms of their biochemical and clinical aspects. In this presentation, our recent studies on 1) drug interactions, 2) protein-protein interactions, and 3) the regulation of human UGTs will be introduced and discussed (<Ref 1 citing here>). Hyperbilirunemia, induced by tranilast, an oral antiallergic agent, was suspected during clinical trials. We elucidated the mechanism that would be responsible for the inhibition by tranilast and the 4-demethyltranilast of the bilirubin glucuronosyltransrerase activity, as well as the UGT1A1 genotype (<Ref 1 citing here>). We demonstrated that UGT substrates with high turnover rates might confuse the identification of the UGT isoform responsible for the glucuronidation of drugs, when such compounds were used as inhibitors for the activities in human liver microsomes, owing to the production of UDP and/or glucuronide or reduction of the inhibitor per se (<Ref 2 citing here>). Protein-protein interactions between human UGT1A1, UGT1A4, and UGT1A6 were investigated by using double expression systems in HEK293 cells. These UGTs interacted with each other, possibly by heterodimerization, and their effects on enzymatic activities were complex, depending on the isoforms and substrates (<Ref 2 citing here>). Studies to investigate the key amino-acid residues responsible for the differences in substrate specificity of human UGTs will be presented (<Ref 3 citing here>). UGT2B7 catalyzes the glucuronidation of morphine and steroid hormones. The molecular mechanisms of the inducible expression of UGT2B7 were investigated. As a result, UGT2B7 was found to be transcriptionally regulated by NF-E2 p45-related factor 2 (Nrf2), but the mechanism is hindered by polymorphisms in the promoter region of UGT2B7*2. The allele-specific mechanism may cause variability in the glucuronidation in response to oxidative stress.
References
- (2009). Drug Metab Pharmacokinet In press.
- (2009). Drug Metab Dispos 37:41–46.
- (2008). Drug Metab Dispos 36:361–367.
- (2008). Pharmacogenet Genom 18:709–720.
- (2007). Drug Metab Dispos 35:1781–1787.
- (2007). Drug Metab Dispos 35:583–589.
18. Drug-metabolizing Enzymes in Thalassemia Patients
Veerapol Kukongviriyapan
Department of Pharmacology, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand
Thalassemia remains a significant health problem in many regions of the world. The diseases are characterized by mutations of the alpha- and beta-globin genes, resulting in decreased or absent production of the normal globin chains. Hemoglobin E-beta thalassemia, the most common form of thalassemia throughout South Asian and Southeast Asian populations, exhibits a wide range of clinical manifestation varying from mild to severe, blood transfusion-dependent thalassemia major. Iron overload is implicated in many complications found in thalassemia major, including liver fibrosis, cardiovascular diseases, and endocrine dysfunction. These pathological features may affect the pharmacokinetics of drugs used in thalassemia. There are very few studies in pharmacokinetics and drug-metabolizing enzymes in thalassemia patients. Assessment of CYP enzymes in patients, using probe compounds, revealed that there is no suppression of many CYP activities. On the other hand, metabolic probes (i.e., antipyrine, caffeine, chlorzoxazone, cortisol, and paracetamol), if not all of them, have showed increases in CYP or phase II enzyme activities in thalassemia patients. Alteration in drug-metabolizing enzyme activity and clearance of the compounds may be associated with various pathophysiological changes in thalassemia, such as hyperdynamic state of cardiovascular system secondary to chronic anemia, deficiency of growth and thyroid hormone, chronic oxidative stress, and others. The modification of pharmacokinetics of drugs used in thalassemia may be of clinical significance.
19. Diversity of Phytochemicals of Vegetables and Herbs in Thai Cookings and Their Potential Role in Health Promotion and Cancer Prevention
Maitree Suttajit
School of Medical Science, Naresuan University at Phayao, Phayao, Thailand
Indigenous medicinal plants are numerous and commonly used by the natives in Southeast Asia and are rich in phytochemicals, including antioxidative compounds. These edible herbs are not only used in only traditional medicine, but they are also used as flavor ingredients in daily food cooking and healthy tea. Thai foods have a reputation as traditional cuisine due to their tasty flavor and contents of several kinds of local vegetables, spices, and medicinal plants, such as lemon grass, curcumin, galangal, peppers, bitter melon, garlic, onion, lemon, kaffir, and so on. They also have healthy, delicious properties that depend on the different kinds of mixed vegetables and plants used in the cooking. Some famous, well-known Thai foods are, namely, Tomyum kung (sour and spicy herbal soup), Gang Keawwan (green and sweet curry with coconut milk), Somtum (sweet and sour papaya salad), etc. As statically compared to other countries, there is a significantly lower incidence of cancers at all sites, including gastrointestinal cancer and chronic diseases, and among Thais, we propose that these Thai traditional foods may have played some important roles for health promotion and cancer prevention. The antioxidative ingredients contained in such cooking herbs.were studied. It was found that there is high antoxidant activity and polyphenol content in several Thai dietary indigenous vegetables and medicinal herbs. The group of the tested plants and herbs with high polyphenolic substances always had strong antioxidative and antiglycation action. Those polyphenols and other phytochemicals were identified as flavanone, flavonol, flavone, flavanonol, catechin, anthocyanin, and anthocyanidin. Several known polyphenolic antioxidants exhibit anti-inflammation, antimicrobial activity, antidiarrheatic activity, antidiabetes, and antiapoptosis. Such various types of polyphenolic compounds might be beneficial as potent cancer chemopreventive agents. Their anticancer properties will be reviewed and discussed in the presentation.
20. Induction of Antioxidant and Detoxifying Enzymes by Dietary Phytochemicals with Chemopreventive and Cytoprotective Potential
Young-Joon Surh, Hye-Kyung Na, Joydeb Kumar Kundu, Jeong-Sang Lee, and Jun-Wan
Shin College of Pharmacy, Seoul National University, Seoul, South Korea
Induction of phase-2 detoxifying or antioxidant genes represents an important cellular defense in response to oxidative and electrophilic insults. A wide array of dietary phytochemicals have been reported to induce the expression of enzymes involved in both cellular antioxidant defenses and elimination/inactivation of electrophilic carcinogens. Induction of such cytoprotective enzymes by edible phytochemicals largely accounts for their cancer chemopreventive and chemoprotective activities. Nuclear transcription factor erythroid 2p45 (NF-E2)-related factor 2 (Nrf2) plays a crucial role in regulating expression of diverse phase-2 detoxifying/antioxidant enzymes, such as NAD(P)H:quinone oxidoreductase-1, heme oxygenase-1, glutamate cysteine ligase, glutathione S-transferase, glutathione peroxidase, thioredoxin, etc. Under physiologic conditions, Nrf2 is normally sequestered in the cytoplasm as an inactive complex with the repressor Kelch-like ECH-associated protein 1 (Keap1). The release of Nrf2 from its repressor and subsequent nuclear translocation are, most likely, to be achieved by alterations in the structure of Keap1. Nrf2, once migrated to the nucleus, forms a heterodimer with another protein, such as small Maf, which, in turn, binds to the antioxidant response elements (ARE) or electrophile response elements (EpRE), located in the promoter region of genes encoding various antioxidant and phase-2 detoxifying enzymes. Some edible chemopreventive and cytoprotective agents target Keap1 by oxidizing or directly modifying one or more of its specific cysteine thiols, thereby facilitating the dissociation of Nrf2 from Keap1 and nuclear translocation. In addition, the phosphorylation of specific serine residues present in Nrf2 by distinct upstream kinases may also facilitate the nuclear localization of Nrf2.
Acknowledgment
This work was supported by the NRL Grant from the Ministry of Science and Technology, Republic of Korea.
References
- Kundu, J.K., Na, H.-K., Surh, Y.-J. (2008). Intracellular signaling molecules as targets of selected dietary chemopreventive agents. In: Surh Y.-J., Dong, Z., Cadenas, E., Packer, L. (Eds.), Dietary Modulation of Cell Signaling Pathways (pp 1–44). Boca Raton, Florida, USA: CRC Press.
- Lee, J.-S., Surh, Y.-J. (2005). Nrf2 as a novel molecular target for chemoprevention. Cancer Lett 224:171–184.
- Na, H.-K., Kim, E.-H., Jung, J.H., Lee, H.H., Hyun, J.W., Surh, Y.-J. (2008). (-)-epigallocatechin gallate induces Nrf2-mediated antioxidant enzyme expression via activation of PI3K and ERK in human mammary epithelial cells. Arch Biochem Biophys 476:171–177.
- Na, H.-K., Surh, Y.-J. (2006). Transcriptional regulation via cysteine thiol modification: a novel molecular strategy for chemoprevention and cytoprotection. Mol Carcinog 45:368–380.
- Surh, Y.-J. (2003). Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer 3:768–780.
- Surh, Y.-J., Kundu, J.-K., Na, H.-K. (2008). Nrf2 as a master redox switch in turning on the cellular signaling Involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med 74:1526–1539.
- Surh, Y.-J., Kundu, J.K., Na, H.-K., Lee, J.S. (2005). Redox sensitive transcription factors as prime targets for chemoprevention with anti-inflammatory and antioxidative phytochemicals. J Nutr 135:2993S–3001S.
- Surh, Y.-J., Na, H.-K. (2008). NF-kB and Nrf2 as prime molecular targets for chemoprevention and cytoprotection with anti-inflammatory and antioxidant phytochemicals. Genes Nutr 2:313–317. 21.
21. Dietary Modulation to Prevent Cancer and Malformation in Mice
Taisei Nomura
Nomura Project, National Institute of Biomedical Innovation, Ibaraki, Osaka, Japan
Various bioactive substances in food can prevent cancer, malformation, and mutation in mice. Error-prone repair inhibitors, caffeine, and some methylxanthines inhibit chemically induced tumors and/or malformations in mice (Nomura, 1976, 1983). However, results were agent dependent (e.g., these substances enhanced X-ray and alkylating agent-induced defects). Vitamins (vitamins A, D, retinoic acid, nicotinamide, etc.) inhibited malformations and tumors in mice (Nomura et al., 1983), though a higher dose of retinoic acid was teratogenic. Pyran, BCG, and activated macrophages inhibited X-ray and urethane-induced or spontaneously developed malformations as well as tumors, probably by eliminating outliers (i.e., preteratogenic and -tumorigenic altered cells) and replacing them with normal cells (Nomura et al., 1990). Immunosurveilance, at later processes, seems nonspecific and more effective. Recently, we found that active hexose correlated compound (AHCC) from editable mushroom prevents radiation-induced malformation and leukemia in mice. AHCC (0.2 mg/g bw) was injected intraperitoneally to N5 mice on days 3.5 and 5.5 of pregnancy, and mice were irradiated with 1.4 Gy of 137Cs gamma-rays on the day 9.5. Congenital malformations were significantly inhibited by AHCC pretreatments (7.1 vs. 24.8% in AHCC untreated group; P < 0.01). 137Cs gamma-ray–induced leukemia in C57BL/6J mice was also suppressed by continuous drinking of AHCC (2%)-contained water until 24 months of age (P < 0.05). Further, AHCC pretreatment significantly suppressed numbers of tumor nodules/mouse (4.3 vs. 39.3 in untreated group; P < 0.01) after intraperitoneal injection of human mesothelioma cell line JMN-1B into SCID mice.
Acknowledgment
This work was supported by MEXT Japan and Amino Up Chemicals Research Fund.
References
- Nomura, T. (1976). Diminution of tumorigenesis initiated by 4-nitroquinoline-1-oxide by post-treatment with caffeine in mice. Nature 260:547–549.
- Nomura, T. (1983). Comparative inhibiting effects of methylxanthines of urethan-induced tumors, malformations, and presumed somatic mutations in mice. Cancer Res 43:1342–1346.
- Nomura, T., et al. (1983). Antiteratogenic effects of tumor inhibitors, caffeine, antipain, and retinoic acid in mice. Cancer Res 43:5156–5162.
- Nomura, T., Hata, S., Kusafuka, T. (1990). Suppression of developmental anomalies by maternal macrophages in mice. J Exp Med 172:1325–1330.
22. Recent Advances in Styryl-lactones-induced Apoptosis
Salmaan H. Inayat-Hussain
Environmental Health Program and UKM Medical Molecular Biology Institute, Faculty of Allied Health Sciences, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia
The lactone and stilbene moieties are important components of an increasing number of compounds that have potential therapeutic roles, especially for anticancer therapy. Since the significance of these moieties were recognized, a myriad of structurally related or different analogs have been investigated and tested against various pharmacological modalities, which may provide novel targets in therapies, such as in conditions where insufficient apoptosis leads to the formation of cancer. Recently, there have been a number of studies directed at the search for novel anticancer compounds incorporating lactone and/or stilbene moieties. Alternatively, replacement of one of these benzene rings in the stilbene with a six-membered ring lactone gives rise to the styryl-lactones. We and others have recently demonstrated the in vitro cytotoxic and apoptogenic effects of these styryl-lactones, such as altholactone and goniothalamin (GTN), which are especially effective in killing different tumor cell lines, including leukemia cells. The mechanisms by which GTN induces apoptosis are currently unclear and have, therefore, been investigated by using T-cell lines. In this study, the potential role of several genes, which were stably transfected into T-cell lines and which regulate apoptosis in different ways, on goniothalamin-induced apoptosis, was examined. Further, the role of the apical caspase-2 and mitochondria in GTN treatment confirmed the role of an intrinsic pathway of apoptosis. These data provide insight into the mechanisms of GTN-induced apoptosis, which may have important implications in the development of novel anticancer drugs from styryl-lactones.
23. Transcriptional Control of UDP Glucuronosyltransferases
Peter Ian Mackenzie, Dong-Gui Hu, Robyn Meech, and Dione A. Gardner-Stephen
Department of Clinical Pharmacology, Flinders University School of Medicine, Adelaide, Australia
Several constitutive, tissue-specific, and ligand-activated transcription factors are important regulators of UDP glucuronosyltransferase (UGT) gene expression. Identifying those transcription factors that regulate the spatial and temporal expression of a specific UGT gene is important for strategies to manipulate UGT expression in the therapeutic setting. In this presentation, recent advances in defining the factors that control UGT gene expression will be discussed. The liver and gastrointestinal tract are major organs involved in the elimination of lipophilic chemicals by glucuronidation. We have previously shown that hepatocyte nuclear factor (HNF)1α regulates most UGT genes in the liver, in cooperation with other factors, such as HNF4α, Octamer transcription factor 1, and pre-B-cell homeobox 2. HNF1α also regulates gastrointestinal UGTs, together with the caudal-related homeodomain protein 2. However, little is known about how UGT genes are regulated in other organs, such as the breast and prostate, especially as the latter two steroid-responsive tissues do not contain HNF1α. We focussed on the breast and have used the breast cancer cell line, MCF-7, to identify UGTs responsive to estrogen and to elucidate the mechanisms underlying this responsiveness. Quantitative polymerase chain reaction demonstrated that only UGT2B15 and 2B17 expression was upregulated 10-fold or more by β-estradiol in MCF-7 cells. Using gene-reporter assays, the response to β-estradiol was shown to be mediated by the proximal 500 bp of the UGT2B15 or 2B17 gene promoter. Using the UGT2B15 proximal promoter, three estrogen-response half-sites and 2 AP1 sites necessary for activation by β-estradiol were identified by site-directed mutagenesis. Gel-shift and CHIP assays, and Si-RNA knockdown experiments, showed that the AP1 factors, c-Jun and Fra2, and the estrogen receptor participated in a cooperative manner to mediate the β-estradiol activation of UGT2B15. Interestingly, despite the presence of 2 AP1-binding sites, UGT2B15 and 2B17 RNA levels were not altered by the phorbol ester, PMA. As UGT2B15 and 2B17 are the main enzymes that glucuronidate testosterone and dihydrotestosterone, control of their expression by β-estradiol may be of importance in modulating the response of the breast to circulating androgens.
24. Novel Transcriptional Activation of the CYP3A4 Gene
Kiyoshi Nagata
Department of Environmental and Health Science, Tohoku Pharmaceutical University, Sendai, Japan
Cytochrome P450 3A4 (CYP3A4) plays a very important role in the metabolism of over 50% of all clinically prescribed drugs. Changes in CYP3A4 expression can affect drug metabolism, altering the therapeutic or toxicological response to a drug. CYP3A4 is induced by a number of drugs through the activation of pregnane × receptor (PXR). PXR has been shown to form a heterodimer with retinoid × receptor α (RXRα), and to bind to the distal nuclear receptor-binding element 1 (dNR1), and an everted repeat separated by six nucleotides in the proximal promoter (prER6) of the CYP3A4 gene. In this presentation, a new rifampicin- and clotrimazole-responsive region, located at −7.6 kb upstream from the transcription initiation site, has been identified by using reporter assays in HepG2 cells. This region contains a cluster of possible nuclear receptor-binding half-sites, AG(G/T)TCA-like sequence. Of these putative half-sites, we focused on six half-sites, called α-η half-sites. Introduction of a mutation into either α- or β-half-site of the CYP3A4 reporter gene almost completely diminished the rifampicin-induced transcription. HepG2-based transactivation assays, using the reporter-gene constructs with or without mutations in the PXR binding element(s), demonstrated that this DR4 motif is essential for the transcriptional activation not only by rifampicin, but also by various human PXR activators, including clotrimazole. However, the introduction of a mutation into either γ-half-site of the CYP3A4 reporter gene decreased the clotrimazole-induced transcription, but not the rifampicin-induced transcription. In addition, reporter assays performed in human hepatocytes and mice with adenoviruses expressing luciferase derived from various CYP3A4 reporter genes, and that expressing human PXR, supported the results of experiments in HepG2 cells. These findings suggest that the newly identified responsive region of the CYP3A4 gene plays an essential role for the xenobiotic induction of CYP3A4.
25. Interindividual Variation of Transcription Factors Regulating DMPK Genes
Sang Seop Lee
Pharmacology and Pharmacogenomics Research Center, Inje University College of Medicine, Busan, South Korea
Drug-metabolizing enzymes and drug transporters play an essential role in the absorption, distribution, metabolism, and elimination of clinical drugs, nutrients, and toxicants. The importance of these molecules is confirmed by the therapeutic failure or adverse drug reactions, in some patients, due to altered pharmacokinetics of clinical drugs with narrow therapeutic ranges. In addition, these molecules may be involved in serious drug-drug interactions. As an effort to understand the mechanism underlying the interindividual variation of drug disposition, genetic and environmental factors influencing the expression or function of the drug-metabolizing enzymes and drug transporters have been extensively explored in recent decades. Among them, genetic polymorphism of drug- metabolizing enzymes and transporters has generated much interest with the finding of functional single-nucleotide polymorphisms within those genes. Besides these molecules, nuclear receptors also modulate drug disposition by regulating the expression of drug-metabolizing enzymes and drug transporters. Among many xenobiotic receptors, pregnane × receptor, constitutive and rostane receptor, and hepatocyte nuclear factors have been most well characterized, since these receptors regulate the expression of important enzymes involved in drug disposition and are affected by various environmental factors. In this presentation, the functional characterization of genetic variations of nuclear receptors will be addressed. And, interindividual diversity in nuclear receptor expression will be also discussed.
26. Nonsteroidal Anti-Inflammatory Drugs and the Advancing Tide of Toxicity
Kathleen M. Knights
Clinical Pharmacology, Flinders University, Adelaide, Australia
Nonselective nonsteroidal anti-inflammatory drugs (nsNSAIDs) are among the most widely used drugs in the world for their analgesic, antipyretic, and anti-inflammatory effects. Adverse effects of nsNSAIDs were noted in the early 1900s, with reports, at that time, of renal and gastrointestinal toxicity. The mechanism of renal and gastrointestinal toxicity has been well described and is attributed to the inhibition of cyclooxygenases (COX-1 and COX-2) and, hence, reduced synthesis of renal vasodilatory prostanoids and gastrointestinal mucosal protective prostaglandins. In addition, nsNSAIDs reduce platelet reactivity and increase blood pressure in normotensive individuals. The development of selective COX-2 inhibitors, such as celecoxib and rofecoxib, reduced gastrointestinal toxicity, but adverse renal and cardiovascular effects persisted. It has been postulated that the selective inhibition of prostacyclin production by COX-2 inhibitors results in a shift in the thromboxane A2/prostacyclin balance that augments the response to thrombotic and hypertensive stimuli. Indeed, the increased incidence of myocardial infarction with rofecoxib resulted in its withdrawal in 2004. However, all nsNSAIDs inhibit COX-2, and current epidemiological evidence is inconclusive regarding the risk of cardiovascular events with nsNSAID exposure. This presentation will provide an overview of the cardiovascular renal toxicity of nsNSAIDs and results of recent in vitro/in vivo studies using diclofenac, ibuprofen, and naproxen, with a focus on their cardiovascular toxicity.
27. Molecular and Genomic Approach for Understanding the Toxicological Mechanisms of ROS-inducing Agents in Human Cells Using Three-Dimensional (3D) Cell Culture System
Young R. Seo
Department of Pharmacology, Department of Biomedical Science, School of Medicine, Kyung Hee University, Seoul, South Korea
Toxicogenomics has been defined as a study of the response of a genome to hazardous substances, using omics technologies, such as genomic-scale mRNA expression called transcriptomics, cell- and tissue-wide protein expression called proteomics, and metabolite profiling called metabolomics, in combination with the conventional method of toxicology. This technology has provided a number of advantages to understand the mechanism of toxicity of chemicals, drugs, environmental agents, and stressors, as well as to identify the biomarkers of toxicity in a variety of biosystems. Here, we utilized the toxicogenomics in an in vivo mimic culture system for the estimation of toxicity in the radiotherapeutic process. First, the sensitivity to ROS-inducing gent ionizing radiation (IR) to H1299 human cancer cells was measured in in vivo mimic three-dimensional (3D) tissue culture systems. Further, the global analysis of gene-expression profiles was carried out by using DNA microarray for characterizing the state of the cells in response to IR. Remarkably, 449 expressed genes were upregulated and 196 genes were downregulated after the treatment. About 20% of genes, including Bcl-2 and Cullin 1 in the 2-fold-upregulated group, were closely related with apoptosis. Genes involved in DNA metabolism, cell-cycle regulation, cytoskeleton organization, and biogenesis were downregulated under 2-fold in response to the ROS-inducing agent, IR. We suggested that the application of toxicogenomics in 3D human cell culture system might give an insight for minimizing the toxicity of various therapeutic processes in the clinic.
28. Adaptive Cellular Survival Response to Oxidative and Inflammatory Stresses
Young-Joon Surh
National Research Laboratory of Molecular Carcinogenesis and Chemoprevention, College of Pharmacy, Seoul National University, Seoul, Korea
In an almost every moment, living organisms are subjected to diverse types of stress, both from external and internal sources. While excessive stress leads to necrotic or apoptotic death, moderate amounts of noxious stimuli may render the cells adaptive to subsequent stress. Such adaptive response to stress normally accompanies de novo synthesis of proteins via the activation of distinct stress-responsive signaling. One of the key signaling molecules involved in cellular adaptation or tolerance to a wide array of stressful conditions is NF-κB. Our previous studies have revealed that NF-κB plays a pivotal role in Bcl-2-mediated resistance to oxidative PC12 cell death through the augmentation of cellular antioxidant capacity. Induction of phase-2 detoxifying or antioxidant genes represents an important cellular defence in response to oxidative and electrophilic insults. Nrf2 plays a crucial role in regulating phase-2 detoxifying/antioxidant gene induction. This transcription factor is sequestered in the cytoplasm as an inactive complex with the inhibitory protein, Keap1. Upon activation, Nrf2 binds to antioxidant responsive element (ARE) sites, leading to the coordinated upregulation of downstream target genes that boost cellular antioxidant potential. Many dietary phytonutrients can induce ARE-driven antioxidant/phase-2 detoxifying gene expression, thereby fortifying cellular defense against oxidative insult. Cysteine thiols present in various transcription factors, and their regulators function as redox sensors in the fine-tuning of transcriptional regulation of many genes essential for maintaining cellular homeostasis. Thus, oxidation or covalent modification of thiol groups present in the above redox-sensitive transcription factors and their regulating molecules can provide a unique strategy for molecular target-based chemoprevention and cytoprotection.
29. Evaluation of Anti-Inflammatory Agents from Natural Origin: Problem of Efficacy Study
Krongtong Yoovathaworn
Department of Pharmacology and Multidisciplinary Unit, Faculty of Science, Mahidol University, Bangkok, Thailand
Reports on the effectiveness of herbal antiinflammatory drugs for the clinical use and availability of safe drugs in the market is in no way comparable to the increase in the number of promising anti-inflammatory compounds of herbal origin. Test systems available make it easy to demonstrate such activity. Rat paw edema as well as arachidonic acid (AA)-induced and phorbol myristate acetate (PMA)-induced mouse ear edema models are often used as screening tests. Assessments for myeloperoxidase (MPO), a marker of neutrophil infiltration and TNF-α presence in the blood have been employed to show the anti-inflammatory action. The in vitro methods include the determination of PGE2 and LTB4 in the activated macrophage and the inhibitory effect on PLA2. The expression of known proinflammatory marker genes, such as IL-1, iNOS, and COX2, in cells such as LPS-stimulated mouse macrophages are now available. The data of biological availablilty and pharmacological activity, with no obvious toxicity after a prolonged treatment in animal, are studied for agents intended to be used systemically. Topical drugs are tested for irritation and sensitizing potential. However, to demonstrate clinical efficacy in humans with a particular disease or condition, such as trama, arthritis, and muscle or joint pain, the use of the double-blinded, crossover design, in comparison with the standard drug, is nearly imposible. Variations in the severity and pathophysiology of inflammation make the scoring of the anti-inflammatory effect, and most are subjective, difficult, and less reliable. Slow dermal absorption and hence the delayed onset of action resulted in a drop out of subjects or requesting for comedication. Even a convenient path is to choose herbs used by people for centuries; and without known adverse effects, the results were, sometimes, disappointing, due to the difference in the form of preparation, the method of administration, and the patient’s condition. At present, most of the studies reporting anti-inflammatory activity from herbs usually concluded with “further investigation to fully identify the biologically active ingredients and to define the underlying molecular mechanisms of anti-inflammatory effect is required” or “detailed preclinical pharmacology and toxicology studies as well as well control clinical studies are needed to further establish the agent of interest as a promising candidate for human clinical use.”
30. Discovery of Biomarkers for Gene Expression and Drug Toxicity and Efficacy
Frank J. Gonzalez, Chi Chen, Andrew D. Patterson, Kristopher W. Krausz, and Jeffrey R. Idle
Laboratory of Metabolism, National Cancer Institute, Bethesda, Maryland, USA
Metabolomics is the study of changes in the population of small molecules in cells, tissues, and biological fluids, such as serum and urine. Metabolomics, using 1H-NMR as a platform, has gained prominence and has been embraced by the pharmaceutical industry as a means to predict the potential toxic effects of drugs, using animal models. For example, a chemical is administered to rodents and urine and serum examined for the presence of metabolites that reflect organ-specific toxicities. Metabolomics can also be used in human epidemiology studies and clinical trails to assess drug efficacy and toxicity. In the past few years, mass spectrometry, coupled with liquid (LC-MS) or gas chromatography (GC-MS), is gaining increased use as a platform for metabolomics due to lower equipment and running costs and more widespread equipment availability, as compared to 1H-NMR. MS-based platforms also yield higher sensitivity and increased throughput than 1H-NMR. However, in contrast to 1H-NMR that gives the structure of each metabolite, time-of-flight MS yields the exact molecular mass and the structure must be determined by database searching and additional experimentation culminating in the comparison of the metabolite in the biological fluid with chemical standards. A major breakthrough in metabolomics platforms was development of ultra-performance liquid chromatography coupled with quadrupole-time-of-flight mass spectrometry (UPLC-QTOFMS) that yields high-resolution metabolite separation and exact mass determination of compounds in biological fluids and extracts. The UPLC-QTOFMS data are deconvoluted by various multivariate data analysis (MDA) software, such as principal components analysis (PCA), partial least squares data analysis (PLSDA), and Random Forests, Salford systems, San Diego, CA, USA, to find differences between samples. To study xenobiotic metabolism and toxicity, test compounds are administered to mice, and serum and urine examined for the presence of the parent compound and its metabolites that are not found in untreated mice. Metabolites derived from the drug as well as endogenous metabolites that reflect a drugs biological activity or toxicity can be resolved. These biomarkers can then be validated for use as sentinels for exposure to toxic agents. Metabolomics is also being used to search for biomarkers for various diseases, such as cancer, and metabolic disorders, including diabetes. This can be accomplished by the use of disease-inducing bioassays in mice and by directly studying, both restrospectively and prospectively, human patients and controls. Biomarkers can be developed for use in early disease detection and for determining the efficacy/toxicity of therapy. Metabolomics can be of value in human genetics and response to drug therapy. By the use of mouse genetic models, metabolomics can be used to find biomarkers associated the expression of enzymes and transcription factors, such as xenobiotic receptors. By use of the mouse model, acetaminophen metabolism and toxicity was studied by using metabolomics. Mice are administered the drug and urine and serum collected at various times after dosing and subjected to analysis by UPLC-QTOFMS, followed by MDA. Metabolites derived from acetaminophen and metabolites unrelated to the drug are identified; the latter can be used to determine mechanism of hepatocytes damage by acetaminophen. Data derived from this study will be presented and discussed.
31. CYP2D6 Genomic Structure and Sequence Characterization in Thai Revealed Such Complexity that Argue against Universal Use of Current Commercial Gene Chip Platform
Payiarat Suwannasri,1 Pornpen Pramyothin,2 Anunchai Assawamakin,3 and Chanin Limwongse3
1Department of Pharmacology and Physiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
2Department of Pharmacology and Physiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
3Molecular Genetics Unit, Department of Research and Development, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand
CYP2D6 is a major drug-metabolizing enzyme in the liver responsible for pharmacokinetic alteration of many clinically important medications. We undertook a study to determine the genetic epidemiology of polymorphic variants of CYP2D6 in Thailand by the combination of long-range nested polymerase chain reaction (PCR) and automated sequencing in 665 Thai subjects. We compared the spectrum of polymorphism found to those reported in the reference database for Caucasian, African-American, and other Asian populations. In a small cohort of the sample, an additional functional study was done, using biopsied liver tissue, to compare predicted enzyme activity with the corresponding genotype. We found unforeseen complexity in the genomic structure of CYP2D6 in Thai, as compared with those in Western countries. Copy-number variation resulting from gene duplication was found in 35% and ranged from 1 to 5 copies. Each of these copies may, in addition, harbor different combinations of single-nucleotide polymorphisms, thus making the attempt to give correct nomenclature to each allele and haplotype a very daunting task. Moreover, new allele resulting from exon 9 gene conversion was found prominently among Thai samples (>42.5%). Gene deletion was found in 10% or less, similar to those reported from other populations. From our data, we concluded that the current commercial CYP2D6 microchip platform cannot dissect this unexpected complexity and is more than likely to be inadequate for the correct genotyping of CYP2D6 allele, at least in our population. From our sample cohort undergoing a functional assay, we found a correlation between genotype and phenotype in those predicted to be ultrarapid and poor metabolizers, whereas a correlation was not straightforward in the intermediate metabolizer group, possibly due to the copy-number variations of CYP2D6 alleles. Using standard labor-intensive, long-range nested PCR thus, from our data, still offers superior and more reliable results than those derived from a commercially available CYP2D6 genotyping microchip.
32. Abstract Not Available at Time of Publication
33. How Close Are We to the Pharmacogenomics-based Personalized Pharmacotherapy?
Jae Gook Shin
Department of Pharmacology and Clinical Pharmacology, Pharmacogenomics Research Center, Inje University College of Medicine and Busan Paik Hospital, Busan, South Korea
Medical doctors have long been asked to solve the issue of why the same drug causes different drug responses among different patients. All clinicians well know about the environmental factors influencing drug response and they usually consider these factors for the optimum dosage selection of the given patients (i.e., body weight, age, renal and hepatic dysfunction, drug interactions, etc.). Pharmacogenetics and pharmacogenomics added lots of new information to understand the genetic factors influencing the drug responses of individual patients during the last 30 years. This information became increasingly recognized and applied to the genetic screening for the personalized pharmacotherapy for the existing medicines, although it is limited to the a few classes of therapeutic drugs, so far. The pharmacogenetics and pharmacogenomics is also likely to become a core important principle in drug discovery and development to identify optimal patient populations to be treated by a particular drug. In addition, the molecular diagnostics for screening target genetic variants are already approved to be applied for patient care (e.g., AmplicChip™ CYP450, Roche Diagnostic corp Indianapolis, IN, USA). It is, therefore, not surprising that some of the genetic information is already applied to the medical practice in some hospitals, and more than 10% of drugs listed in the Physician’s Desk Reference include the pharmacogenetic information, at the moment. Moreover, ethnic-sensitive drugs are approved on the market, and currently, we know some drugs can be effective only to the given ethnic patient populations (e.g., gefitinib is effective for Asian lung cancer patients). However, it is still very limited in the way that genetic tests for personalized pharmacotherapy have become popular in medical practice, and it is expected to take over 15–20 years, at minimum, for patients’ genetic information to become a major factor in choosing the best pharmacotherapy of an individual patient. It seems to be clear that we are already living in the era of personalized medicine, although a limited number of prescription drugs are available, so far, and further development of technology, information, and training of expert in the field of pharmacogenomics will accelerate for us to solve the problem of individual variation of drug responses in the near future.
34. Research and Development of Chili and Chili Products: From Market to Development
Nuntavan Bunyapraphatsara
Pharmacognosy, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
In Thailand, several studies on herbal medicine have been carried out, but only a few projects can be implemented. This failure is due to the rational basing on scientist interest and expertise. To achieve implementation, the researchers have to consider market trend and feasibility. Research and development of chili and a chili product project is a good example. The project started with the selection of product type, and the main criteria were market size and competitiveness. Feed additive was selected because the local market was big enough to start a business. At present, it is necessary to find a new growth promoter to replace the antibiotic growth promoter (AGP), having been banned owing to a drug-resistant problem in humans through food chain and contact. This trend will open an opportunity for new products to enter the feed-additive market. The second step is selection of plants, based on the following criteria: availability of raw material, cost of raw material, biological activity and safety data, and chili was selected for this research and development program. The analysis of previous reports indicates that several studies, including preparation of extract, product development, test in chicken, laying hen, and swine, are required. Not only the studies on the efficacy and safety of feed additive product, but it is also necessary to do research and development of good quality of extract with competitive prize. Therefore, our research group carried out research on the cultivation of chili, which will cover the search for variety with high capsaicin content, improving yield by cultivation technology. To compete with the world market, prize and quality are important. One approach is to expand the demand, which will result in low cost of production. Therefore, we have carried out the research and development of several products, such as analgesic cream, health food products, pesticide, and antisleep spray. All these research projects led us to the implementation of chili project research results in animal husbandry, as well as the cultivation of high-quality chili.
35. Meeting the Challenges of Studying Chinese Medicine: Quality, Pharmacology, and Clinical Issues
Chun-Tao Che
School of Chinese Medicine, The Chinese University of Hong Kong, Hong Kong, China
Modern studies on Chinese medicine (CM) present many challenges, which can be tackled through a multidisciplinary approach, interfacing the traditional knowledge, biological, chemical, and pharmaceutical sciences, as well as clinical expertise. In this presentation, the scenario of studying a multi-item herbal formula for the treatment of gastrointestinal disorder is described. The work is part of an international collaboration to establish the pharmacological potential, safety, and preliminary therapeutic effect of an herbal prescription for the treatment of irritable bowel syndrome (IBS). The study includes the following aspects of investigations: 1) Based on the CM diagnosis of IBS patients (predominantly the diarrhea type), a Chinese herbal prescription was formulated for treating the major CM syndromes identified in these patients; 2) authentication, standardization, and quality assurance of herbal materials was carried out toward the goal of providing a well-characterized herbal mixture for use in the animal studies and human clinical trials. Quality analysis of individual herbs and the combined medicinal formula was performed by using conventional and chromatographic approaches, including liquid chromatography–mass spectrometry. HPLC fingerprints were established for each herb and the herbal mixture; 3) biological evaluation of the herbal formula was performed by using in vitro bioassays (sensorimotor-modulation, receptor expression, and binding) and a stress-induced visceral hyperalgesia animal model (the neonatal maternal separation rat model); 4) preclinical toxicology of the herbal formula was studied by using acute, subchronic, chronic toxicity, and mutagenicity tests; and 5) a dose-escalation study was carried out on IBS patients. Multidisciplinary and interinstitutional collaboration was emphasized in the present work, and this was shown to be a most desirable and effective approach for the study of Chinese herbal medicine.
Acknowledgments
This project was partially supported by the National Center for Complementary and Alternative Medicine, National Institutes of Health (1 U19 AT003266-01; Bethesda, Maryland, USA). Contents are solely the responsibility of the author and do not represent the official views of NCCAM. Collaborators at the University of Maryland School of Medicine, the Chinese University of Hong Kong, the University of Illinois College of Pharmacy, and the University of Western Sydney are acknowledged for their contributions to the work described in this presentation.
36. Importance of Standardization in Botanical Drug Development in Korea
Jinwoong Kim
College of Pharmacy, Seoul National University, Seoul, South Korea
A new botanical drug entity was developed from the standardized extract of Scrophularia buergeriana roots that could be used in therapeutics for Alzheimer’s disease. In animal models, this botanical drug shows both cognitive-enhancing and antioxidant activities. It significantly improves the performance of the passive avoidance test in scopolamine- or β-amyloid-induced memory-impaired mice in single and prolonged administration for 15 days. Further, this drug entity reduces the short- and long-term memory deficits significantly in the Morris water-maize test in single and prolonged administration for 15 days. Standardization of the S. buergeriana extract was achieved by qualitative and quantitative analysis of its active constituents, such as cinnamic acid, p-methoxycinnamic acid, harpagoside, and 8-O-(E-p-methoxycinnamoyl)harpagide. It was found that the quality of this botanical drug depends on the origin of plant materials, manufacturing process of the herbal preparation, and the properties of the final product. This presentation will address the importance of quality assurance and standardization in the development of a botanical drug.
37. Natural Products as Lead Structures in Drug Discovery
Ji-Wang Chern
Research and Development, National Taiwan University, Taipei, Taiwan
Studies on natural products play an important role in drug discovery and development in the last century. It has led to a unique contribution to make new pharmaceuticals, either as the actual drug or as lead compounds for drug development. Today, with the use of modern screening and isolation techniques, the potential for the discovery of new drug from natural products drugs or novel lead compounds has been significantly enhanced. Our approach, using natural products as lead structures, such as doridosine, curcumine, camptothecin, and tryptanthrin, has successfully led to the discovery of novel, potent bioactive compounds. This talk will be focused on the structural modification of natural products such as lead-tryptanthrin as a DNA triplex binding agent on the basis of rational design of the molecular level. Tryptanthrin (1), indolo[2,1-b]quinazolin-6,12-dione, and its derivatives were reported to show antimicrobial activity and inhibitory activity against tuberculosis. The planar tetracyclic system is common for some antitumor drugs, such as camptothecin and ellipticine. In continuing our studies of quinazoline derivatives, we have designed and synthesized a series of tryptanthrin derivatives as potential anticancer agents, and some of them showed better cytotoxicity than tryptanthrin itself in many cancer cell lines. Importantly, 1 and 2 were found to be reversal agents of some anticancer drugs with different mechanisms. We integrated an alkylamine side chain onto tryptanthrin to form 3, in which indolo[2,1-b]quinazolinone can interact with DNA through p-p stacking and hydrogen bonds, and the alkylamine side chain offers binding with DNA via electrostatic interactions. Both computer modeling and thermal denaturation experiments revealed that 3 stabilized not only duplex DNA, but also triplex DNA. This structurally new planar skeleton ring system has never been reported previously, and it is not only a new series of heterocycles worthwhile to develop, but is also expected to have potential in anticancer activity.
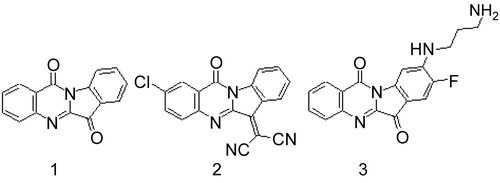
49. Role of OATP Transporters for Hepatic Uptake and Biliary Excretion of Beta-Lactam Antibiotics in Rats
Yuta Shibue,1 Yasushi Morohashi,1 Taiki Shimada,2 Choujin You,1 Masanori Nakakariya,2 Yoshiyuki Shirasaka,1 Takeo Nakanishi,1 and Ikumi Tamai1
1Faculty of Pharmacy, Institute of Medical, Pharmaceutical and Health Sciences, Kanazawa University, Kanazawa, Japan
2Faculty of Pharmaceutical Sciences, Tokyo University of Science, Noda, Japan
Introduction: Organic anion transporting polypeptides (OATPs) are involved in the hepatic uptake of various xenobiotics, including beta-lactam antibiotics. Oatp1a1, Oatp1a4, and Oatp1b2 are expressed at the basolateral membranes of rat hepatocytes. Among them, we have recently shown that nafcillin, a high biliary excretion type of beta-lactam antibiotic, was mainly transported by rat liver via Oatp1a4 in vitro (Nakakariya et al., 2008a, 2008b). In the present study, we pursued generality of the importance of Oatps, especially Oatp1a4, for the hepatic disposition of beta-lactam antibiotics in rats.
Methods: In vitro transport of beta-lactam antibiotics was evaluated by using Xenopus oocytes expressing each Oatp molecule, isolated rat hepatocytes, and sandwich-cultured rat hepatocytes (SCRH). PK parameters were determined by in vivo study performed in rats. In all experiments, beta-lactam antibiotics were quantified by high-performance liquid chromatography.
Results and Disucussion: Nafcillin uptake by isolated rat hepatocytes was inhibited by rifampicin at concentrations up to 10 μM, where the inhibitory effect seemed to be exclusive to Oatp1a4 because of the differece in IC50 values between Oatp1a4 (IC50 = 3.53 μM) and other Oatps, Oatp1a1 (IC50 = 323 μM) or Oatp1b2 (IC50 = 32.6 μM). Nafcillin (10 mg/kg) was intravenously administered and PK parameters were compared with or without rifampicin at a dose of 20 mg/kg that could attain plasma concentration enough for the inhibition of Oatp1a4. Plasma concentration of nafcillin at 4 hours after coadministration with rifampicin was 8.5 times higher than that of nafcillin alone. Biliary excretion clearance and Kpliver were significantly decreased by the coadministration of rifampicin. In SCRH, similar results were obtained with those by in vivo study. For other beta-lactam antibiotic derivatives, similar, but not identical, results were obtained. These results suggest that Oatps, in particular Oatp1a4, play an important role in the biliary excretion of beta-lactam antibiotics. Further study is currently underway to identify the contribution of other Oatps to hepatic dispotition of beta-lactam antibiotics.
References
- Nakakariya M., et al., (2008a). Identification and species similarity of OATP transporters responsible for hepatic uptake of beta-lactam antibiotics. Drug Metab Pharmacokinet 23:347–355.
- Nakakariya, M., et al. (2008b). Predominant contribution of rat organic anion transporting polypeptide-2 (Oatp2) to hepatic uptake of beta-lactam antibiotics. Pharm Res 25:578–585.
50. Phase II Metabolism of Cannabinoids by Human Hepatic and Extrahepatic UDP-glucuronosyltransferase (UGTS) Depends on Upstream Processing by Cytochrome P450S 2C9 and 3A4
Anna Mazur,1 Cheryl F. Lichti,2 Stacie Bratton,3 Anna Gallus-Zawada,3 Moshe Finel,4 Grover P. Miller,5 Jeffery H. Moran,2 and Anna Radominska-Pandya6
1Biochemistry and Molecular Biology, University of Arkansas for Medical Sciences, Little Rock, Arkansas, USA
2Division of Health, AR Public Health Laboratory, Little Rock, Arkansas, USA
3Department of Biochemical and Molecular Biology, University of Arkansas for Medical Sciences, Little Rock, Arkansas, USA
4Center for Drug Research (CDR), Faculty of Pharmacy, University of Helsinki, Helsinki, Finland
5Department of Biochemical and Molecular Biology, University of Arkansas for Medical Sciences, Little Rock, Arkansas, USA
6Department of Biochemical and Molecular Biology, University of Arkansas for Medical Sciences, Little Rock, Arkansas, USA
Tetrahydrocannabinol (Δ9-THC), the primary psychoactive ingredient in marijuana, is subject to cytochrome P450 oxidation and subsequent UDP-glucuronosyltransferase (UGT)-dependent glucuronidation. Many studies have shown that CYP2C9 and CYP3A4 are the primary phase I enzymes responsible for cytochrome P450–dependent oxidations, but little work has been done characterizing phase II metabolic pathways. The purpose of this study was to test the hypothesis that there are specific human UGTs responsible for classical cannabinoid metabolism. The activities of 12 human recombinant UGTs toward classical cannabinoids [cannabinol (CBN), cannabidiol (CBD), (-)-Δ8-THC, (-)-Δ9-THC, (±)-11-hydroxy-Δ9-THC (11-OH-THC), and (-)-11-nor-9-carboxy-Δ9-THC (COOH-THC)] were evaluated by using HPLC-MS/MS and labeling assays. Despite activity by UGT1A1, -1A3, -1A8, -1A9, -1A10, and -2B7 toward CBN, CBD, 11-OH-THC, and COOH-THC, only selected UGTs demonstrate sufficient activity for further characterization of steady-state kinetics. CBN was the most recognized substrate, as evidenced by activities from hepatic UGT1A9 and extrahepatic UGT1A7, UGT1A8, and UGT1A10. These results may reflect the the introduction of an aromatic ring to Δ9-THC leading to favorable π stacking with phenylalanines in the UGT active site. Similarly, oxidation of Δ9-THC to 11-OH-THC results in UGT1A19 and UGT1A10 activity toward the cannabinoid. Further oxidation to COOH-THC, surprisingly, leads to a loss in metabolism by UGT1A9 and UGT1A10, while creating a substrate recognized by UGT1A1 and UGT1A3. The resulting glucuronide of COOH-THC is the main metabolite found in urine, and thus these hepatic enzymes play a critical role in the metabolic clearance of cannabinoids. Taken together, phase II metabolism of cannabinoids depends on upstream processing, including enzymes such as CYP2C9 and CYP3A4.
Acknowledgments
This work was funded by the National Institutes of Health (Bethesda, Maryland, USA; grant nos. DK60109 and GM075893 to AR-P and U90/CCU616974-07 to JM) and the Academy of Finland (project 210933 to MF).
51. OATP-mediated Intestinal Absorption of HMG-COA Reductase Inhibitors in Rats
Kensuke Suzuki, Yoshiyuki Shirasaka, Takeo Nakanishi, and Ikumi Tamai
Faculty of Pharmacy, Institute of Medical, Pharmaceutical and Health Sciences, Kanazawa University, Kanazawa, Japan
Purpose: Pravastatin and pitavastatin are effective HMG-CoA reductase inhibitors after oral administration despite their hydrophilic natures at physiological pHs with their intermediate bioavailability from about 20 to 60%. Previously, we have shown that intestinal apical uptakes of these inhibitors were carrier-mediated using brush-border membrane vesicles. More recently we demonstrated that human OATP2B1 (OATP-B) plays a role in the intestinal uptake of pravastatin (Tamai et al., 1995; Kobayashi et al., 2003; Nozawa et al., 2004). For pitavastatin, it is known that human OATPs (OATP1B1, OATP1B3 and OATP2B1) are involved in hepatic uptake, while contribution of intestinal OATPs for pitavastatin remains unclear. The purpose of the present study is to clarify impact of OATPs on the intestinal absorption of pravastatin and pitavastatin by evaluating one of rat orthologues, Oatp1a5 (Oatp3), which is expressed at the apical membranes of enterocytes in rats and is thought to be important for drug absorption (Shirasaki et al., 2009).
Methods: In vitro transport was measured using Xenopus oocytes expressing Oatp1a5. Rat intestinal absorption was evaluated by in situ closed loop method. These drugs were quantitated by LC/MS/MS.
Results and Discussion: Uptakes of pravastatin and pitavastatin by Xenopus oocytes expressing Oatp1a5 were significantly increased, demonstrating that both pravastatin and pitavastatin are substrates of Oatp1a5. Uptake of pravastatin by Oatp1a5 was saturable, and kinetic analysis indicated an involvement of two components (Km1 = 0.915 μM; Km2 =177 μM). Uptake of pitavastatin by Oatp1a5 was saturable (Km = 2.40 μM) and was pH-dependent with higher activity at acidic pH, which is preferable at upper part of small intestine. By in situ closed loop study, intestinal absorption of pravastatin was decreased by naringin, a typical Oatp1a5 inhibitor. Our findings indicate that Oatp1a5 at least in part contributes to the intestinal absorption of these HMG-CoA reducatase inhibitors in rats.
References
- Kobayashi, D., et al. (2003). J Pharmacol Exp Ther 306:703–708.
- Nozawa, T., et al. (2004). J Pharmacol Exp Ther 308:438–445.
- Shirasaka, Y., et al. (2009). Pharm Res In press.
- Tamai, I., et al. (1995). Pharm Res 12:1727–1732.
52. Functional Role of ST1B Subfamily of Cytosolic Sulfotransferase to Form Natriuretic Xanthurenic Acid Sulfate
Laddawan Senggunprai, Kouichi Yoshinari, Miki Shimada, and Yasushi Yamazoe
Division of Drug Metabolism and Molecular Toxicology, Graduate School of Pharmaceutical Sciences, Tohoku University, Sendai, Japan
Natriuretic substances are a group of molecules affecting sodium homeostasis in the body. Recently, xanthurenic acid (XA; 4,8-dihydroxyquinoline-2-carboxylic acid) derivatives [i.e., XA 8-O-β-D-glucoside and XA 8-O-sulfate (XA sulfate)] isolated from human urine have been shown their natriuresis effects. XA is a product upon tryptophan degradation via the kynurenine pathway. In the present study, we have investigated the sulfation of XA in mouse tissues to assess the contribution of specific sulfotransferases (STs) to the metabolism. Cytosols from tissues of both sexes of C57BL/6N mice (i.e., liver, stomach, jejunum, colon, and kidney) were capable of forming XA sulfate, with various Km values. Jejunum cytosol showed the highest affinity and exhibited the lowest Km value. The kinetic analyses with recombinant mouse (m) STs showed the lowest Km value for mSult1b1, and the value was comparable with that for jejunum cytosol. The highest expression of mSult1b1 in small intestine was confirmed at the mRNA and protein levels. mSult1b1 is thus suggested as a major enzyme responsible for XA sulfation in jejunum. Human (h) SULT1B1 and rat Sult1b1 also mediated XA sulfation efficiently. Thus, XA is likely to be an endogenous substrate for ST1B members. In contrast to XA, kynurenic acid (KYNA; 4-oxo-1H-quinoline-2-carboxylic acid), another tryptophan metabolite having antagonistic effects on glutamate receptors, exhibited potent selective inhibitory effects on mSult1b1 activities with the IC50 values in a low micromolar range. KYNA also exerted the inhibitory activity toward both hSULT1A1 and hSULT1B1 with fair potency. Taken together, these results indicate the functional role of ST1B subfamily of ST in XA sulfate formation in the body and demonstrate another distinct aspect of KYNA as an endogenous inhibitor of STs.
53. LC-MS/MS Assessment of Phase I Hydroxylation and Phase II Conjugation to the Warfarin Metabolome in Human Urine
Drew R. Jones, Shane Z. Sullivan, Anna Radominska-Pandya, Jeffery H. Moran, and Grover P. Miller
Biochemistry and Molecular Biology, University of Arkansas for Medical Sciences, Little Rock, Arkansas, USA
As a step toward exploring a metabolomics approach to personalized warfarin (Coumadin) therapy, we developed a liquid chromatography–tandem mass spectrometry (LC-MS/MS) method capable of quantitating specific enantiomeric (R and S) contributions of the following: warfarin (WAR), phase I hydroxywarfarins (OH-WAR), phase II glucuronides (-GLUC), and phase II sulfates (-SULF). The two-step analytical approach measures 36 unique warfarin metabolites, as confirmed by quality-control samples. Our evaluation of 3 commercially available human urine samples revealed the significance of specific metabolites among patients and the corresponding enzymatic capacity to generate these compounds, including the first report of direct WAR conjugation. The major P450 metabolite was (S)-7-OH-WAR, indicating the significance of CYP2C9 in WAR metabolism, although other CYP2C enzymes also contributed to the clearance of this isomer. (R)-WAR hydroxylation to OH-WARs was more diverse among the patients, as reflected in varying contributions of CYP1A2 and multiple CYP2C enzymes. There was wide variation in the glucuronidation and sulfonation of WAR and the OH-WARs with respect to the compounds and patients. 7-OH-WAR was primarily excreted as a glucuronide in F84 but was excreted equally as sulfate and as free OH-WAR in F85. 8-OH-WAR was primarily sulfonated in F84 and F85 but was not significantly conjugated in F87. For all patients, UGT1A1 was likely responsible for 6-OH-WAR-GLUC production, although UGT1A10 likely contributed in 1 patient. 7-OH-WAR-GLUC levels reflected contributions from, potentially, five different UGT1A enzymes. 7-OH-WAR was also extensively sulfonated (29–30%) with other OH-WARs undergoing sulfonation to varying degrees. Altogether, sulfonated compounds represented between 27 and 42% of all metabolites. In the absence of further study, it is not possible to assign sulfonated metabolites to specific enzymes. For all patients, WAR, 4’-OH-WAR, 8-OH-WAR, and their corresponding glucuronides and sulfates were each minor metabolites, but collectively represent between 12–24% of all metabolites. These data suggest that both phase I and II reactions contribute to the generation of excretable products in human urine, thereby generating complex metabolic networks that require further clinical study to elucidate their significance.
54. Investigation of the Role of Thiazolidinedione Ring of Troglitazone in Inducing Hepatotoxicity in Humans
Sudipta Saha,1 Lee Sun New,1 Ho Han Kiat,2 Wai Keung Chui,1 and Eric Chun Yong Chan1
1Department of Pharmacy, Faculty of Science, Natioanal University of singapore, Singapore, Singapore
2Institute of Medical Biology, Singapore, Singapore
Troglitazone (TGZ) is an orally active hypoglycemic agent that is used for the treatment of noninsulin-dependent diabetes mellitus (NIDDM). It had been associated with severe drug-induced liver failure, which resulted in its withdrawal from the market in 2002. While the mechanism of its toxicity remains unknown, it has been postulated that the formation of toxic reactive metabolites may play an important role in the hepatotoxicity of TGZ. The purpose of this study was to investigate the role of sulfur moiety of thiazolidinedione nucleus in inducing liver toxicity via the formation of reactive metabolites. An analog of TGZ, trosuccinimide (TSN), was synthesized chemically where the sulfur moiety of thiazolidinedione ring was replaced by a methylene group. Both compounds were incubated with human liver microsomes containing glutathione and normal human hepatocytes (THLE2 cell line) to profile reactive metabolites using ultra-performance liquid chromatography tandem mass spectrometry (UPLC/MS/MS). Two reactive metabolite-glutathione conjugates of TGZ were identified during the profiling experiments, of which one was related to the sulfur moiety of the thiazolidinedione ring. No reactive metabolite of TSN was detected in both microsomes and hepatocytes. MTT assays were performed by using THLE2 hepatocytes to measure the levels of toxicity of TGZ and TSN in vitro. Glutathione assays were further performed to measure the oxidative stress produced during incubation of these two drugs with hepatocytes. Peroxisome proliferator activated receptor gamma (PPARã) binding activity was measured finally to calculate the binding affinities of both TGZ and TSN to PPAR-alpha ligand. Our results indicated collectively that TSN (EC50 = 138.5 ± 7.3 μM) was less toxic than TGZ (EC50 = 27.2 ± 4.8 μM) in the THLE2 hepatocytes. As both compounds were shown to bind to the PPAR-alpha ligand, the substitution of the thiazolidinedione moiety may be beneficial from a drug-design perspective. In conclusion, our study indicates that the thiazolidinedione ring may be partially responsible for liver toxicity of TGZ in humans.
55. Prevention of Free Fatty Acid–induced Hepatic Lipotoxicity by S-Allyl Cysteine through AMPK Pathways
Yong Pil Hwang, Jae Ho Choi, Hyo Jeong Yun, and Hye Gwang Jeong
BK21 Project Team, Department of Pharmacy, Chosun University, Gwangju, South Korea
Lipid accumulation in nonadipose tissues leads to cell dysfunction and apoptosis, a phenomenon known as lipotoxicity. It is well-accepted that free fatty acid (FFA)-induced lipotoxicity plays a pivotal role in the pathogenesis of nonalcoholic fatty liver disease (NAFLD). Inhibition of FFA-associated hepatic toxicity represents a potential therapeutic strategy. Aged garlic extract (AGE) possesses multiple biological activities. In this study, we evaluated the protective effect of S-allyl cysteine (SAC), one of the organosulfur compounds of AGE, against FFA-induced lipotoxicity in in vitro HepG2 (human liver cell line) NAFLD models. Pretreatment of HepG2 cells with SAC significantly reduced the FFA-induced generation of ROS, caspase-3 activation, and subsequent cell death. Also, SAC inhibits cholesterol and triglyceride synthesis in a similar manner to the AMP-activated protein kinase (AMPK) activator, metformin. Significant increase in AMPK phosphorylation was observed when the cells were incubated with SAC. Activation of AMPK was also demonstrated by measuring the phosphorylation of acetyl-CoA carboxylase, a substrate of AMPK, correlated with a subsequent increase in fatty acid oxidation. The results indicated that by reducing triglyceride and cholesterol accumulation, SAC could protect the liver from NAFLD through the activation of AMPK. Therefore, SAC might have the therapeutic potential for preventing or treating NAFLD.
56. Fluorescence-based Screening of Pharmaceutical Reactive Metabolites Using Glutathione Immobilized 96-Well Microsomal Assay
Xiang Ma1 and Eric Chun Yong Chan2
1Department of Chemical and Biomolecular Engineering, National University of Singapore, Singapore, Singapore
2Department of Pharmacy, Faculty of Science, Natioanal University of Singapore, Singapore, Singapore
The screening and structural characterization of pharmaceutical reactive metabolites (RMs) has increasingly become an integral part of the ADMET-guided lead optimization process in drug discovery. While sensitive and selective methods for profiling RM are available based on mass spectrometry technologies, these methods are generally low-throughput and are reserved for lead compounds that have progressed further in the research pipeline. In this study, a fluorescence-based method in screening pharmaceutical RMs, using the glutathione (GSH)-immobilized 96-well mouse liver microsomal (MLM) assay, was developed and validated. A poly(2-hydroxyethylmethacrylate) (pHEMA) polymeric membrane was coated on a 96-well plate to provide a functional support for GSH immobilization. In order to sustain the nucleophilic activity of the thiol-trapping group of GSH after immobilization, oxidized GSH (GSSG) was conjugated on a cyanogen bromide (CNBr)-activated pHEMA surface. The immobilized GSH was regenerated after the reduction of GSSG by using D,L-dithiothreitol (DTT). X-ray photoelectron spectroscopy and Ellman’s assay were applied to validate the chemistry while the N-(iodoacetylaminoethyl)-5-naphthylamine-1-sulfonic acid-based fluorescence assay was used to optimize the multiple steps of GSH immobilization. The performance of the 96-well assay was further cross-validated using N-acetyl-p-benzo-quinone imine (NAPQI), a RM of acetaminophen (APAP), and an in vitro MLM assay of APAP. Optimum conditions were achieved for pHEMA loading, CNBr activation of pHEMA, GSSG coupling, and reduction. The detection limit of the assay for NAPQI was 500 nM with good specificity. In vitro MLM assay of APAP demonstrated positive result of 20% RM trapping efficiency, when calculated as a percentage, based on the total amount of immobilized GSH. In conclusion, a fluorescence-based GSH-immobilized 96-well platform for the screening of RM by using MLM was successfully developed and validated. Our novel RM screening method holds great potential for the high-throughput and cost-effective toxicity screening of hit compounds during early stages of drug discovery and development.
57. Abstract Withdrawn
58. Ginsenoside Rb1 Induces Heme Oxygenase-1 via the PI3K and Nrf2 Pathway to Protect Human Dopaminergic Neurons from 6-Hydroxydopamine-derived Oxidative Stress
Yong Pil Hwang and Hye Gwang Jeong
BK21 Project Team, Department of Pharmacy, Research Center for Proteineous Materials, Chosun University, Gwangju, South Korea
In the present study, we investigated the mechanisms of ginsenoside Rb1 protection of neuronal cells from cell death induced by the Parkinson’s disease–related neurotoxin, 6-hydroxydopamine (6-OHDA). Pretreatment of SH-SY5Y cells with ginsenoside Rb1 significantly reduced the 6-OHDA-induced generation of ROS, caspase-3 activation, and subsequent cell death. Ginsenoside Rb1 also upregulated heme oxygenase-1 (HO-1) expression, which conferred neuroprotection against 6-OHDA-induced oxidative injury. Moreover, ginsenoside Rb1 induced PI3K activation, which is involved in the induction of Nrf2, HO-1 expression, and neuroprotection. These results suggest that regulation of the antioxidant enzyme HO-1 via the PI3K- and Nrf2-signaling pathways controls the intracellular levels of ROS.
59. Stability-Indicating Assay Method for Determination of Sultamicillin Tosilate in Pharmaceutical Formulations by HPTLC
Krishnamurthy Bhat,1 Parul Sinha,1 Prashant B. Musmade,1 and N. Udupa2
1Pharmaceutical Quality Assurance, Manipal College of Pharmaceutical Sciences, Manipal, India
2Pharmaceutical Management, Manipal College of Pharmaceutical Sciences, Manipal, India
A sensitive, selective, precise, and stability indicating the high-performance thin-layer chromatographic method of analysis of sultamicillin tosilate tablets was developed and validated. The method employed HPTLC precoated silicagel (60GF254) plates as the stationary phase and chloroform: methanol: ammonia at a 8:2:0.2 (v/v) ratio as the mobile phase. Densitometric analysis of sultamicillin tosilate was carried out in the absorbance mode at 215 nm. This system gave compact spots for sultamicillin tosilate (Rf value, 0.32 ± 0.05). The method was validated for specificity, precision, accuracy, linearity range, and robustness. The linear regression analysis data for the calibration plots showed a good linear relationship over the concentration range of 500–3,500 ng per spot with the regression equation Y = 1.6742 × −60.617 and regression coefficient of (r2) 0.9986, The method precision was determine as inter- and intraday precision and found to be 1.23 and 1.06% as coefficient of variation, respectively. The present method was applied for the estimation of sultamicillin tosilate in bulk drug and dosage forms. Sultamicillin was subjected to acid and alkali hydrolysis, oxidation, dry-heat treatment, and photo degradation to prove the stability, indicating power of the developed method. The present method demonstrated that all the degradation products formed under stress testing were well resolved from the parent peak, and peak purity of the sultamicillin proves that no there is not any interference at the Rf of sultamicillin. Due to its simplicity and accuracy, the method can be useful for routine quality-control testing and stability analysis of samples.
60. Fast and Reliable Determination of Amlodipine and Irbesartan in Beagle Dog Plasma Using Monolithic Silica Rod Liquid Chromatography
Wen Jun,1 Wei Hua,1 Xie Rui,1 Pan Yaju,1 Fan Guorong,1 Wu Yutian,1 and Hu Zhuohan2
1Department of Pharmaceutical Analysis, School of Pharmacy, Second Military Medical University, Shanghai Key Laboratory for Pharmaceutical Metabolite Research, Shanghai, China
2Research Institute for Liver Diseases (Shanghai) Co. Ltd., Shanghai, China
A fast, reliable isocratic reversed-phase HPLC method with UV detection, using internal standards, has been developed and validated for the simultaneous determination of the calcium-channel antagonist, amlodipine, and the angiotensin-II antagonist, irbesartan, in beagle dog plasma. A simultaneous solid-phase extraction with a mixed-mode column was used to extract amlodipine, irbesartan, and two internal standards, verapamil and telmisartan, from the beagle dog plasma. The separation was performed on a Chromolith RP-18e column (4.6 × 100 mm), using the mobile phase of acetonitrile-methnal (0.02 M) sodium dihydrogen phosphate adjusted to pH 3.0 (25:20:55; v/v/v) to achieve an entire separation of the two analytes and two internal standards in less than 6.5 minutes. The wavelength was set at 238 nm. The assay enables the measurement of amlodipine and irbesartan with a minimum quantification limit of 5 and 10 ng•mL−1, with good linearity (r > 0.999) over the linear range of 5–500 and 10–5,000 ng•mL−1, respectively. The within- and between-run precision for these two analytes was less than 7.9% and accuracy ranged from 96.3 to 105.1% for quality-control samples. The proposed assay for amlodipine and irbesartan in beagle dog plasma is fast, sensitive, and reliable, and thus, well suited for the pharmacokinetic study in beagle dogs after coadministration of amlodipine and irbesartan and routine therapeutic monitoring of resistant hypertensive patients. It can be predicted that the use of monolithic silica rod chromatography will substantially shorten the turnaround time in the therapeutic drug-monitoring laboratory.
61. Determination of Faropenem in Human Plasma by Online Solid-Phase Extraction Coupled to High-Performance Liquid Chromatography with UV Detection and its Applications to a Pharmacokinetics Study
Xie Rui,1 Wen Jun,1 Wei Hua,1 Fan Guorong,1 Wu Yutian,1 and Hu Zhuohan2
1Department of Pharmaceutical Analysis, School of Pharmacy, Second Military Medical University, Shanghai Key Laboratory for Pharmaceutical Metabolite Research, Shanghai, China
2Research Institute for Liver Diseases (Shanghai) Co. Ltd., Shanghai, China
Sample preparation is a crucial step in the analysis of biological samples. When a large number of samples from clinical studies were to be analyzed, manual procedures became relatively tedious, time-consuming, and sample-consuming, which highly limits their applications in pharmacokinetics study. High-thoughput sample preparation protocols are required. Here, we developed an online SPE column switching HPLC-UV method for the determination of faropenem in human plasma, using gatifloxacin as an internal standard. Analyses were performed isocratically with two reversed-phase columns connected by a column switch. After perciptation by 6% perchloric acid, the plasma sample was injected for online SPE column switching HPLC-UV analysis. The analyte was retained on the extraction column (Lichrospher, 25 μm, 4.6 mm, Agitent technologies, Foster city, CA, USA) with the loading solvent (20 mM NaH2PO4 adjusted to pH 3.5). Most matrix materials were removed from the column to waste. After 0.5 minutes of washing, the value switched to another position so that the target analytes could be eluted in the back-flush mode by the mobile phase (acetoniteile 20 mM NaH2PO4 adjusted pH 3.5, 16:84; v/v) at a flow rate of 1.5 mL/min, and then separated on the analytical column (Ultimate™ XB-C18, 5 μm, 50 mm° A’ 4.6 mm). The complete cycle of the online SPE preconcentration and HPLC separation of the analytes was 5 minutes. The method was linear over the studied range (20¨C5000 ng/mL), with r2 > 0.99. The intra- and interassay precision rates were in the ranges 0.6¨C3.9 and 1.5¨C2.9%, respectively.The intra- and interassay accuracy rates were >90%. The absolute recoveries were approximately 93 (20 ng/mL), 105 (50 ng/mL), 102 (500 ng/mL), and 104% (4,000 ng/mL). The limits lower of quantification were 20 ng/mL. Also, the method was successfully utilized to quantify faropenem in human plasma to support the clinical pharmacokinetics study.
62. SPE-Liquid/Liquid Extraction Assay for Analysis of CYP3A Activity: Applications to Drug Metabolism of Cancer Patients
Hong Zhang, Yu Fang, and Ying Li
Clinical Pharmacology, Tongji University, Tongji Hospital, Shanghai, China
A highly sensitive SPE–liquid/liquid extraction method has been developed for the analysis of CYP3A (the ratio of urinary 6â-hydroxycortisol, i.e., metabolite, and cortisol used as an indicator for the activity of CYP3A) in urine of cancer patients in the clinical setting. Sample test solutions were separated by reserved-phase high performance chromatography (RPLC), using a C18 analytical column with acetonitrile (A) and 0.5% ammonium sulphate solution (B) as the mobile phase at a flow rate of 1.0 mL min−1. From 0 to 2 minutes, the mobile phase ratio of A/B was kept as 10/90 (v/v), then an increase in A/B to 35/65 (v/v) in gradient within the time range of 10 minutes, keeping A/B at 35/65 (v/v) in the following 10 minutes, but would then be back to the initial mobile phase ratio gradiently within 3 minutes. Eluted compounds were detected and quantitated at 240 nm by an ultraviolet diode-array detector (DAD). The method was validated for linearity, accuracy (i.e., recovery from urine), repeatability (within- and between-day precision), specificity, sensitivity, and stability. Compared with the original cortisol method for 6alpha-hydroxycortisol/cortisol ratio measurement, this method is rapid, simple, accurate, and reproducible, and is especially suitable for monitoring CYP3A activity of cancer patients in the clinical setting.
63. Simultaneous Glucuronidation of Native and Oxidized Estrogens and Warfarin by Human Recombinant UGT1A10: Potential for Drug-Drug Interactions
Anna Radominska-Pandya,1 Anna Mazur,2 Stacie Bratton,1 Moshe Finel,3 Jeffery H. Moran,4 and Grover P. Miller5
1Department of Biochemical and Molecular Biology, University of Arkansas for Medical Sciences, Little Rock, Arkansas, USA
2Biochemistry and Molecular Biology, University of Arkansas for Medical Sciences, Little Rock, Arkansas, USA
3Center for Drug Research (CDR), Faculty of Pharmacy, University of Helsinki, Helsinki, Finland
4Division of Health, AR Public Health Laboratory, Little Rock, Arkansas, USA
5Department of Biochemical and Molecular Biology, University of Arkansas for Medical Sciences, Little Rock, Arkansas, USA
Human UGT1A10 glucuronidates physiologically important endogenous substrates, such as estrogens, and a variety of drugs, including hydroxylated warfarin. It is hypothesized that patients receiving both warfarin and estrogen therapy can suffer from altered metabolism and disposition, with both compounds resulting in alterations of its status in the body and drug effectiveness. Here, the effects of selected estrogens on UGT1A10 glucuronidation of hydroxylated warfarin derivatives (and of warfarins on estrogens) were investigated. Comparisons of detailed inhibition analyses (IC50 and Ki values), were made for each pair of substrates. Initial experiments using E2 as a marker substrate show that 7- and 8-OH-warfarin inhibit turnover through different mechanisms. The kinetic profile for E2 in the presence of 7-OH-War demonstrates substrate inhibition, whereby the observed rate increases and then decreases with E2 concentration. By contrast, 8-OH-War results in a hyperbolic curve reflecting an increase in the apparent Km, as expected for a simple competition model. In complementary experiments, the use of a saturating E2 fails to effectively inhibit glucuronidation of either 7- or 8-OH-War. These contrasting inhibitory results for the marker reactions clearly indicate that OH-Wars and E2 bind to distinct catalytic sites. Nevertheless, the occupancy of the OH-War site allosterically alters E2 reaction kinetics. The nonhyperbolic kinetic profile for 7-OH-War reflects this complex interaction between substrates on respective activities. Rather than act as a simple competitor, 8-OH-War suppresses E2 binding. These different mechanisms presumably reflect the differences in location of the hydroxyl group. These studies are still in progress and findings are undergoing further analysis to elucidate the mechanism of action, which could have significant clinical and pharmacological applications for designing UGT inhibitors.
Acknowledgment
This work was supported by the National Institutes of Health (NIH; Bethesda, Maryland, USA; grant nos.DK60109 and GM075893 to AR-P).
64. Interindividual Difference in CYP2A6 Genotypes as Determined with Restrictional Fragment Length Polymorphism in Relation to GSTM1 and GSTT1 Genes Among the Patients with Prostate Carcinoma
Davut Alptekin,1 Esra Ozturk,2 Muzeyyen Izmirli,3 Sevil Zencir,4 Yildirim Bayazit,5 and Zeki Topcu6
1Department of Medical Biology, Cukurova University, Faculty of Medicine, Adana, Turkey
2Department of Biology, Ege University Faculty of Sciences, Izmir, Turkey
3Deaprtment of Medical Biology, Cukurova University, Faculty of Medicine, Adana, Turkey
4Department of Biochemistry, Ege University Faculty of Sciences, Izmir, Turkey
5Department of Urology, Cukurova University, Faculty of Medicine, Adana, Turkey
6Department of Pharmaceutical Biotechnology, Ege University Faculty of Pharmacy, Izmir, Turkey
Cytochrome P450 (CYP) is a heme-containing enzyme superfamily metabolizing a wide variety of xenobiotics, including drugs and carcinogens (van Schaik, 2008). Among the CYP isoforms, the CYP2A6 is characteristic of its catalytic properties to nitrosamines (Topcu et al., 2002). Previous studies reported a novel deletion-type mutant of the CYP2A6 gene, CYP2A6*4C, as one of the major polymorphic genes in Japanese lacking CYP2A6 activity (Topcu et al., 2002; Miyamoto et al., 1999). In this study, we intended to clarify whether CYP2A6 genetic polymorphism could be related to individual susceptibility to prostate cancer by employing restrictional fragment-length polymorphism in polymerase chain reaction–amplified DNA samples obtained from the blood samples of individuals with or without prostate carcinoma. Because of the known involvement of phase II enzymes in detoxification of reactive oxygen species, genotyping was extended to cover determination of glutathione-S-transferase enzymes, GSTM1 and GSTT1. A total of 25 cancer subjects, with an average age of 65.8 ± 9.0 years (range, 50–87) showed 56 and 40% null genotypes for GSTM1 and GSTT1, respectively, with no estimated homozygote genotype of CYP2A6*4C/*4C. Our results are discussed in relation to possible roles for these particular enzymes in therapy and protection of prostate tissue from the xenobiotics.
References
- Miyamoto, M., Umetsu, Y., Dosaka-Akita, H., Sawamura, Y., Yokota, J., Kunitoh, H., et al. (1999). CYP2A6 gene deletion reduces susceptibility to lung cancer. Biochem Biophys Res Commun 261:658–660.
- Topcu, Z., Chiba, I., Fujieda, M., Shibata, T., Ariyoshi, N., Yamazaki, H., et al. (2002). CYP2A6 gene deletion reduces oral cancer risk in betel quid chewers in Sri Lanka. Carcinogenesis 23:595–598.
- van Schaik, RH. (2008). CYP450 pharmacogenetics for personalizing cancer therapy. Drug Resist Updat 11:77–98.
65. Human CYP4A11 and CYP4A22 Genetic Polymorphism: Identification and Characterization by Molecular Modeling of Natural Allelic Variants
Dany Chevalier
Christian Lacks Lino Cardenas, Amaury Farce, Nicolas Renault, Michel Lhermitte, Philippe Chavatte, and Franck Broly Toxicologie, Faculté de Pharmacie de Lille, Lille, France
The CYP4A subfamily is expressed in liver and kidney and is involved in the metabolism of arachidonic acid into 20-hydroxyeicosatetranoic acid (20-HETE), a metabolite with renovascular and tubular functions (Lasker et al., 2000). A deficiency in the renal formation of 20-HETE has been linked to the development of hypertension (Gainer et al., 2005). The CYPs, 4A11 and 4A22, are the only members of the human CYP4A gene subfamily (drnelson.utmem.edu/CytochromeP450.html www.drnelson.utmem.edu). A genetic variant of CYP4A11 (8610T>C, Phe434Ser), resulting in decreased synthesis of 20-HETE, has been associated with essential hypertension (Gainer et al., 2005; Mayer et al., 2005). The second member, the CYP4A22 gene, was recently identified and exhibits a 95% sequence identity to the CYP4A11 gene (Bellamine et al., 2003). In the present study, we report the first systematic investigation of polymorphisms in the promoter and coding regions, of both CYPs 4A11 and 4A22 genes, in DNA samples from 48 French Caucasians, using single-strand conformational polymorphism analysis of polymerase chain reaction products. For the CYP4A11, two polymorphisms were identified in the promoter region and eight in the coding regions, including the known functional variant (8610T>C, Phe434Ser) and two novel missense mutations. For the CYP4A22, three polymorphisms were identified in the promoter region and 13 in the coding regions, including seven missense mutations. In order to better understand the functional significance of these natural variants, we built the three-dimensional (3D) models of CYP4A11 and CYP4A22 by using the known CYP3A4 3D structure (Yano et al, 2004). Interactive docking studies with both substrates and inhibitors of these enzymes indicate key residue interactions with the putative active-site regions of each natural variant investigated. These data confirmed that CYPs 4A11 and 4A22 genes are highly polymorphic and suggested that some of these variants may be functionally relevant.
66. Differences in CYP2A13 Expression Level in Various Types of Human Lung Cancer
Tatsuki Fukami,1 Miki Nakajima,1 Yoh Zen,2 Isao Matsumoto,3 Makoto Oda,3 and Tsuyoshi Yokoi1
1Department of Drug Metabolism and Toxicology, Kanazawa University, Kanazawa, Japan
2Division of Pathology, Kanazawa University Hospital, Kanazawa, Japan
3Department of General and Cardiothoracic Surgery, Kanazawa University School of Medicine, Japan
CYP2A13 is the most efficient enzyme in the metabolism of nicotine and metabolic activation of tobacco-specific nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), with a substrate specificity similar to that of CYP2A6 because of the high amino-acid identity between them (93.5%). Previously, we found that CYP2A13 also catalyzes the metabolism of 4-aminobiphenyl, naphthalene, styrene, and toluene that are included in tobacco smoke (Nakajima et al., 2006;Fukami et al., 2008). Former studies using RT-PCR analysis have revealed that CYP2A6 is predominantly expressed in liver, whereas CYP2A13 is predominantly expressed in the respiratory tract, such as lung and trachea. In this study, we investigated the CYP2A13 expression at the protein level in various types of lung cancer. First, we prepared a mouse monoclonal antibody against human CYP2A13. Immunoblot analysis revealed that this antibody specifically reacted with a recombinant CYP2A13, but not with a recombinant CYP2A6. We performed an immunohistochemical analysis using this antibody to determine the expression level of CYP2A13 protein in each of 10 squamous-cell carcinoma, adenocarcinoma, large-cell carcinoma, and small-cell carcinoma specimens. Staining in lung cancer cells was much higher than in normal cells. Strong staining was observed in 5 adenocarcinoma, 3 large cell carcinoma, and 2 squamous cell carcinoma specimens. In contrast, only 4 small-cell carcinoma specimens showed marginal staining. In conclusion, we found that CYP2A13 is highly expressed in non-small-cell lung carcinoma, especially in adenocarcinoma. It has been reported that NNK induces adenocarcinoma. Therefore, the high expression of CYP2A13 in adenocarcinoma might be one of the causes of the NNK-dependent carcinogenesis.
References
- Fukami, T., Katoh, M., Yamazaki, H., Yokoi, T., Nakajima, M. (2008). Human cytochrome P450 2A13 efficiently metabolizes chemicals in air pollutions: naphthalene, styrene, and toluene. Chem Res Toxicol 21:720–725.
- Nakajima, M., Itoh, M., Sakai, H., Fukami, T., Katoh, M., Yamazaki, H., et al. (2006). CYP2A13 expressed in human bladder metabolically activates 4-aminobiohenyl. Int J Cancer 119:2520–2526.
67. Protective Effect of Platycodi Radix on Acute Ethanol-induced Hepatotoxicity in Mice
Tilak Khanal,1 Jae Ho Choi,1 Young Chul Chung,2 and Hye Gwang Jeong1
1BK21 Project Team, Department of Pharmacy, Chosun University, Gwangju, South Korea
2Division of Food Science, International University of Korea, Jinju, South Korea
The protective effects of a Platycodi radix (Changkil: CK), the root of Platycodon grandiflorum A. DC (Campanulaceae) against alcoholic steatosis in liver injury induced by acute ethanol administration were investigated. Pretreatment with CK prior to the administration of ethanol significantly prevented the increased serum alanine aminotransferase activity, hepatic TNF-alpha level, hepatic lipid peroxidation, and hepatic triglyceride level in a dose-dependent manner. CK prevented ethanol-induced steatosis and necrosis, as indicated by liver histopathological studies. CK protected against ethanol-induced depletion of hepatic glutathione levels. The concurrent administration of CK efficaciously abrogated the CYP2E1 induction and CYP2E1-dependents hydroxylation of aniline, as compared to the individual treatment at higher doses. These findings suggest that CK may prevent ethanol-induced acute liver injury, possibly through its ability to block CYP2El-mediated ethanol bioactivation and its free radical scavenging effects.
68. Comparison of CYP3A4 and CYP3A5 in Drug Metabolism and Drug Interactions
Shuanmei Li,1 Duan Liu,1 Juanli Zhu,2 Yan Cui,2 Ruimin Zhou,2 and Chao Chen1
1School of Life Science, Northwest University, Xi’an, China
2Shaanxi Lifegen Co., Ltd., National Engineering Research for Miniaturized Detection Systems, Xi’an, China
CYP3A4 and 3A5 are regarded as predominant functional forms of human CYP3A in liver and intestine. They are involved in oxidation, peroxidation, and reduction of approximately 50% of commonly used drugs. The substrates metabolized by CYP3A4 and 3A5 are generally overlapped, but the kinetic parameters are usually different. In addition, the content ratios of CYP3A4 to CYP3A5 in different individuals are variable and thus may result in different drug responses. In the purpose of analyzing the differences between these two CYP3A enzymes, CYP3A4 and 3A5 were expressed in Saccharomyces cerevisiae and characterized by two metabolic reactions, dibenzylfluorescein (DBF)-O-dealkylation and nifedipine oxidization. Further, 19 chemical compounds were 2-fold serial diluted and added to the two reactions for IC50 determination. Both substrate affinities for CYP3A5 were lower than that for CYP3A4 (with 2- to 3-fold higher Km values than CYP3A4). Intrinsic clearance (CLint) of DBF and nifedipine by CYP3A5 were 0.20 and 0.06 μL/min/pmol, respectively. The CLint were 2- and 3-fold lower than that of CYP3A4. (P < 0.05, n ≥3). In drug-interaction studies, ketoconazole strongly inhibited both reactions catalyzed by CYP3A4. IC50 values to DBF-O-dealkylation and nifedipine oxidization are 0.012 and 0.036 μM, respectively. However, CYP3A5 was less inhibited by ketoconazole in those two reactions, for which IC50 values are 0.040 and 0.049 μM. To the other 18 compounds, except for which IC50 values were unavailable in the concentration range we tested, IC50 values of those compounds to CYP3A5 were, on average, 4-fold higher than that of CYP3A4, no matter which probe substrate was tested. The results indicate that CYP3A5 generally has a lower capacity in drug metabolism and was less inhibited by compounds other than CYP3A4.
69. Effect of Albumin on In Vitro Omeprazole Kinetics in Human Liver Microsomes
Nitsupa Wattanachai,1 David J. Elliot,2 Wichittra Tassaneeyakul,1 and John O. Miners2
1Department of Pharmacology, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand
2Department of Clinical Pharmacology, Flinders University and Medical Centre, Adelaide, Australia
Omeprazole (OMP) is metabolized by CYP2C19 and CYP3A4 to 5-hydroxyomeprazole and omeprazole sulfone, respectively. It has been demonstrated that polyunsaturated fatty acids (PUFAs) are substrates and potent inhibitors of CYP2C19. We hypothesise that the addition of bovine serum albumin (BSA) to incubations of human liver microsomes (HLMs) will sequester PUFAs that are released from the membranes of HLM during the course of incubation, therefore, providing true estimates of kinetic parameters. This study investigated the effect of BSA on the kinetics of OMP 5-hydroxylation and sulfoxidation by microsomes from 5 human livers. In the absence of BSA, 5-hydroxyomeprazole and omeprazole sulfone formation followed two enzyme Michaelis-Menten kinetics. The mean apparent (± SD) kinetic parameters for OMP 5-hydroxylation were: Km1 = 8.63 ± 6.83 μM, Vmax1 = 97.3 ± 46.7 pmol/min.mg and CLint1 = 13.8 ± 8.39 μL/min.mg; Km2 = 156 ± 123 μM, Vmax2 = 239 ± 77.4 pmol/min.mg and CLint2 = 2.34 ± 1.47 μL/min.mg. The mean apparent (± SD) kinetic parameters for OMP sulfoxidation were: Km1 = 16.5 ± 11.5 μM, Vmax1 = 63.0 ± 60.2 pmol/min.mg and CLint1 = 3.16 ± 1.50 μL/min.mg; Km2 = 195 ± 105 μM, Vmax2 = 240 ± 116 pmol/min.mg and CLint2 = 1.38 ± 0.54 μL/min.mg. In the presence of 2% BSA, 5-hydroxyomeprazole formation also followed two enzyme Michaelis-Menten kinetics, whereas omeprazole sulfone formation followed a single-enzyme Michaelis-Menten model. In the presence of 2% BSA, the Km1 and Vmax1 for OMP 5-hydroxylation were significantly reduced (2- to 4-fold), and consequently, CLint1 was increased (2-fold). Hence, the addition of BSA to the reaction incubations had a minor effect on the CYP2C19 catalyzed high-affinity component (Km1 and Vmax1) of OMP 5-hydroxylation, whereas the effect of BSA on OMP sulfoxidation was less clear. Further investigations of the effect of BSA on OMP 5-hydroxylation and sulfoxidation by recombinant CYP2C19 and CYP3A4 are required to clarify the role of BSA on in vitro OMP kinetics.
70. Twenty-Year Retrospective Analysis of CYP Activity Levels in Microsomes Isolated from Human Donor Livers
Charles L. Crespi,1 Sweta Parikh,2 Z. E. Barter,3 Teri Bordonaro,2 Edward Francis,2 and Christopher J. Patten1
1R&D, BD Biosciences, Woburn, Massachusetts, USA
2BD Biosciences, Woburn, Massachusetts, USA
3University of Sheffield and Simcyp Ltd, Sheffield, United Kingdom
Human donor livers, originating from U.S.-based organ procurement organizations, have been the basis for the preparation of key reagents used for the characterization of drugs and drug candidates. Over time, the practices for liver transplantation have evolved, which has indirectly affected the nature of organs that are unused for transplantation and are made available for research use. We have prepared microsomes from, and characterized the enzyme activity levels of, CYP1A2 (phenacetin O-deethylase), CYP2B6 ((S)-mephenytoin N demethylase), CYP2C9 (diclofenac 4-hydoxylase), CYP2C19((S)-mephenytoin 4’-hydoxylase), CYP2D6 (bufuralol 1’-hydrocylase), and CYP3A (testosterone 6f”-hydoxylase) in over 300 donor livers, procured from 1985 to the present, and conducted detailed statistical analyses of activity distributions and trends. We have found the following: 1) CYP2B6 and CYP2C19 show the highest variability among donors (CV 150–175%) CYP2C9 (CV 47%) shows the lowest variability, with CYP1A2, CYP2D6, and CYP3A4 (CV 82–85%) being intermediate. (Note: the CV values above are based on an assumption of normality); 2) the age of donors did not change significantly with year of donation. The median age for females was 52, while for males it was 50; 3) the gender distribution was 60% male and 40% female; 4) the gender difference was observed in CYP3A4. On average, females had 34% higher activity. While the average activity for CYP2C19 was 48% higher in females, this difference was not statistically significant; and 5) no significant change in any enzyme activity as a function of year of donation. This result indicates that liver samples obtained over a large period of time are suitable for creation of pools, and that all historical data can be used to predict the properties of such pools. Monte Carlo analyses for a 50-donor pool predicted an average CV for these 6 enzyme activities of 13%. The actual CV observed for four pools prepared in a manufacturing context was 12%. The implications of these observations on the design and performance of reagents derived from human liver will be discussed.
71. Interindividual Variation and Gender Difference of Ginsenoside Rb1 Biotransformation by Human Intestinal Microflora
Weng-Im Leong, Ru Yan, and Yitao Wang
Institute of Chinese Medical Sciences, University of Macau, Macao SAR, China
Objectives: Ginsenoside Rb1 is one of the major bioactive components of Panax ginseng and P. notoginseng. In past decades, the biotransformation of Rb1 by intestinal microflora was extensively reported. However, quantitative characterization of Rb1 metabolism by human microflora is lacking and effect of gender is unclear. The present study aimed to quantitatively determine Rb1 metabolism by human microflora and to characterize gender difference, if there is any.
Methods: Five independent experiments, including microflora samples from 50 healthy volunteers from both genders, were conducted by incubating Rb1 with microflora samples pooled from 10 individuals. Incubates were collected at 0, 2, 4, 12, 18, 24, 36, 48, and 72 hours, twice extracted with butanol before high-performance liquid chromatography (HPLC) analyses. The time course of Rb1 depletion and Rd, F2, and compound K generation were determined by using an HPLC-UV method. In experiments to delineate the effect of gender, Rb1 was incubated with microflora samples from 21 healthy volunteers (10 females plus 11 males) and incubates collected at 18 hours were compared.
Results: Metabolic patterns of Rb1 in 5 independent experiments were similar: Rb1 decreased rapidly with less than 20% of Rb1 remaining intact at 12 hours. Gensenoside Rd, F2, and compound K appeared in incubates, in turn with Rd, reaching its maximum at 12 hours (represented ∼50% of Rb1), followed by F2 at relatively low levels. Compound K peaked at 36 hours, then kept at a constant level for another 36 hours. A greater than 10-fold interindividual variation in Rb1 biotransformation was observed. Females exhibited the tendency to catalyze Rb1 metabolism at a higher rate. More interestingly, the metabolic pathway of Rb1 by males is the same as previously reported. However, females showed one additional pathway, in which two unknown peaks in addition to the three major metabolites were observed, in some samples.
Conclusions: Gensenoside Rb1 biotransformation was quantitatively characterized, for the first time. The results indicated a great interindividual variation. Females exhibited a higher metabolic capacity and an additional pathway that was absent in males.
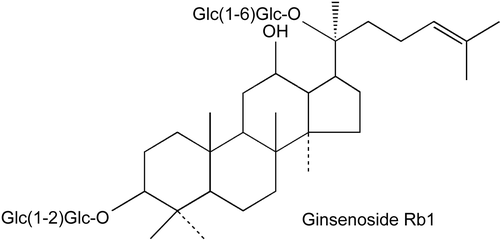
72. Hepatobiliary Transport of PDE5 Inhibitors in the Isolated Perfused Rat Liver
Ching-Ling Cheng,1 Bo-Guang Chen,2 and Chen-Hsi Chou2
1Department of Pharmacy, Chia-Nan University of Pharmacy and Science, Tainan, Taiwan
2Institute of Clinical Pharmacy, National Cheng Kung University, College of Medicine, Tainan, Taiwan
PDE5 inhibitors are used for the treatment of male erectile dysfunction and pulmonary arterial hypertension. The structures of PDE5 inhibitors are similar to cGMP and they are mainly metabolized by CYP3A isozyme in the liver. Currently, the interaction studies of PDE5 inhibitors are primarily focused on metabolic aspects, with little or no address on drug transporters. However, it has been suggested that P-gp, in addition to CYP3A, was responsible for the increase of concentrations of PDE5 inhibitors when concomitant with ritonavir in clinical settings. Therefore, it is of great interest to investigate the role of drug transporters on the hepatobiliary disposition of PDE5 inhibitors. In this study, the hepatic extraction, disposition kinetics, and biliary excretion of sildenafil, tadalafil, and vardenafil were examined by using the isolated in situ perfused rat liver. The liver was perfused in a single-pass mode with protein-free Krebs bicarbonate medium at a flow rate of 20 mL/min. During constant infusion with 1-mg/L PDE5 inhibitors, the hepatic uptake approached equilibrium at about 40, 60, and 80 minutes for tadalafil, sildenafil, and vardenafil, respectively. Sildenafil was highly extracted by the liver with a clearance (19.96 mL/min) that approached the flow rate. In contrast, tadalafil was only moderately extracted and had the greatest steady-state availability (0.8) among the three drugs. All the PDE5 inhibitors showed active biliary secretion as the ratios of drug concentration in bile to that in the outflow perfusate were 18, 155, and 1,930, respectively, for tadalafil, vardenafil, and sildenafil. Using sildenafil as the model drug, the role of transporters on the hepatobiliary disposition of PDE5 inhibitors was further investigated in the presence of known transporter inhibitors, such as cyclosporine A, rifampicin, and probenecid.
73. Primaquine Affects Imatinib Pharmacokinetics and Enhances Liver, Kidney, and Brain Penetration
Jason H.K. Law, Grace Chay, Elaine Kan, Gian Wan Soo, Shin Yee Tan, Wei Yin Lim, Nadeem I. Bukhari, and Ignacio Segarra
Department of Pharmaceutical Technology, International Medical University, Kuala Lumpur, Malaysia
Background: Imatinib, a c-KIT and BCR-ABL tyrosine kinases selective inhibitor, has been approved for chronic myelogenous leukemia and gastrointestinal stromal tumors. Studies suggest further therapeutic activity, including autoimmune nephritis, glioblastoma, renal-cell carcinoma, and other neoplasms. Imatinib is well tolerated, CYP3A4 metabolized, highly bound to alpha-1-acid glycoprotein, and a P-gp substrate. Primaquine is an antimalarial agent that binds extensively to plasma protein alpha-1-acid glycoprotein. Therefore, it was investigated whether the concurrent administration of primaquine enhances the tissue distribution to expand imatinib’s therapeutic applications.
Experimental Design: Primaquine (12.5 mg/kg) was administered orally to male ICR mice (9–12 weeks old), followed 20 minutes later by 50 mg/kg imatinib PO (study group, n = 45). The control group (n = 43) was given only imatinib PO at 50 mg/kg. Imatinib concentration in plasma, kidney, liver, and brain samples at prescheduled time points (2, 5, 10, 20, and 40 minutes and 1, 2, 4, 6, and 10 hours following imatinib administration) was measured by using a validated HPLC method. Pharmacokinetic parameters were estimated by using a noncompartmental technique.
Results and Discussion: Primaquine coadministration increased 1.6-fold plasma AUC0→∞ (control: 21.49 μg·h/mL; study group: 34.24 μg·h/mL), CMAX decreased 24% (4.65 ± 0.95 μg/mL), TMAX halved (20 minutes), and kel decreased (0.293 h−1 control; 0.160 h−1 study group) resulting in longer t1/2 (control: 2.4 hours; study group: 4.3 hours) and longer MRT (3.6 hours for control and 7.3 hours for study group). Cl/F decreased 37%, but VSS/F increased 28%, suggesting increased tissue penetration. Kidney and liver AUC0→∞ increased from 107.57 to 182.18 μg·h/mL and from 49.19 to 134.04 μg·h/mL, respectively. Brain penetration increased: control AUC0-last was 0.72 ± 0.15 and 2.54 ± 0.25 μg·h/mL (P < 0.01, based on Bailer’s method for sparse sampling) and CMAX increased 2.7-fold.
Conclusions: Primaquine coadministration increased imatinib plasma, kidney, liver, and brain exposure, probably due to alpha-1-acid glycoprotein imatinib displacement. The increased brain penetration may be a new option to treat glioblastoma. However, potential renal and liver toxicity may warrant monitoring of their physiological functions.
74. Effect of Ketoconazole on the Bioavailability, Pharmacokinetics, and Tissue Distribution of Imatinib after Oral Coadministration to Mice
Gian Wan Soo, Shin Yee Tan, Wei Yin Lim, Grace Chay, Elaine Kan, Jason H. K. Law, Nadeem I. Bukhari, and Ignacio Segarra
Department of Pharmaceutical Technology, International Medical University, Kuala Lumpur, Malaysia
Background: Imatinib is currently approved for the treatment of chronic myelogenous leukemia and gastrointestinal stromal tumors. As a selective tyrosine kinase inhibitor, imatinib also shows therapeutic potential in glioblastoma, autoimmune nephritis, and other neoplasms. In mice, imatinib has 27% bioavailability and is eliminated via CYP3A4. It is also a P-gp substrate.
Objectives: To assess changes in imatinib bioavailability and tissue distribution after coadministration with ketoconazole, a P-glycoprotein substrate and CYP3A4 inhibitor antifungal agent commonly used by cancer patients was used.
Study Design: Male ICR mice were given oral (50 mg/kg) imatinib alone (control group; n = 43) or 15 minutes after a 50-mg/kg ketoconazole dose (study group; n = 51) orally. Imatinib plasma, brain, kidney, and liver concentrations at predetermined time points (2, 5, 10, 20, and 40 minutes and 1, 2, 4, 6, and 10 hours postimatinib administration) were measured by using a validated HPLC assay. Noncompartmental pharmacokinetic analysis, aided by Bailer’s method for sparse sampling, was performed.
Results: Following ketoconazole coadministration, the plasma, kidney, and liver disposition profile presented double peaks. In the control group, plasma imatinib AUC0→last was 20.72 ± 0.74 μg/mL. After ketoconazole coadministration, plasma AUC0→last increased 57% to 32.48 ± 1.56 μg/mL (P < 0.01) (AUC0→∞ increased 65%), CMAX increased 2-fold (11.61 ± 3.44 μg/mL), TMAX was halved (20 minutes), but elimination t1/2 remained similar (2.5 hours), suggesting that this ketoconazole dose did not affect imatinib elimination. Cl/F and VSS/F decreased 40 and 34%, respectively. Ketoconazole coadministration increased imatinib tissue AUC0→last 28% in kidney and 29% in liver. Liver AUC0→∞ increased proportionally (54%), but it could not be calculated for kidney. Liver and kidney TMAX was earlier (20 minutes), liver CMAX remained similar (16.00 ± 1.72 for control vs. 18.59 ± 4.71 μg/g in study group) but doubled in kidney (20.48 ± 9.93 μg/g). Brain penetration remained negligible. Conclusions: Ketoconazole increased imatinib bioavailability with subsequent kidney and liver increased exposure, but failed to improve brain penetration. Careful liver and renal-function monitoring is recommended when ketoconazole is used concurrently, as renal and hepatic toxicity associated to imatinib have been previously observed.
75. Cytotoxic Effect of Solid Lipid Nanoparticles Containing Doxorubicin on Adriamycin-Resistant Breast Cancer Cells
Hoo-Kyun Choi, Jung Woo Kim, and Keon Wook Kang
College of Pharmacy, Chosun University, Gwangju, South Korea
The failure of chemotherapy in cancer patient is often due to the development of multidrug resistance. Multidrug resistance is mainly due to the overexpression of p-glycoprotein. Properly designed nanoparticles containing anticancer agents could achieve passive tumor targeting, through either avoiding p-glycoprotein–mediated efflux or enhanced permeability and retention (EPR) effect of tumor tissue. In the present study, we developed doxorubicin (Dox)-loaded solid lipid nanoparticles (SLN-Dox) using biocompatible compounds and assessed in vitro hemolytic effect and examined its in vivo effects on the drug retention and apoptosis intensity in p-glycoprotein–overexpressed MCF-7/ADR cells, a representative Dox-resistant breast cancer cell line. Our SLN did not show hemolytic activity in human erythrocytes. In comparison to Dox, SLN-Dox efficiently enhanced apoptotic cell death through the higher accumulation of Dox in MCF-7/ADR cells. Thereby, SLN-Dox could be proposed one of useful therapeutic approaches to overcome the chemoresistance of adriamycin-resistant breast cancer.
76. Modulation of NF Kappa B Activity by a Sri Lankan Herbal Remedy during Early Chemical Hepatocarcinogenesis
Prasanna B. Galhena,1 M. Ira Thabrew,2 Kamani Tennekoon,2 and Mayuri G. Thammitiyagodage3
1Department of Biochemistry and Clinical Chemistry, Faculty of Medicine, University of Kelaniya, Ragama, Sri Lanka
2Institute of Biochemistry, Molecular Biology, and Biotechnology, University of Colombo, Colombo 3, Sri Lanka
3Animal Section, Medical Research Institute, Colombo 8, Sri Lanka
NF kappa B protein, which is closely associated with tumor progression, participates in the transcription of several cellular genes, which are involved in proliferation and inflammation. The aim of the present investigation was to determine the effect of an antihepatocarcinogenic herbal decoction comprised of Nigella sativa, Smilax glabra, and Hemidesmus indicus on the activation of NF kappa B protein during early hepatocarcinogenesis in mice induced by diethylnitrosamine (DEN). Five-week-old, male C3H mice (n = 16) were used for the experiment under standard laboratory conditions. Animals were randomly divided in to four equal groups. Animals in groups 1 and 2 were orally administered distilled water and the decoction (at a dose of 10 g/kg body weight/day), respectively, for a period of 4 weeks. Animals in groups 3 and 4 were administered DEN at a dose of 20 g/kg body weight, as a single intraperitoneal injection. Subsequently, they were orally administered with distilled water and the decoction as in groups 1 and 2, respectively. At the end of the week 4, immunohistochemistry was performed in liver sections fixed in 10% formaldehyde, using rabbit anti-p65 antibody. Primary antibody binding was localized by goat antirabbit IgG antibody HRP and visualized with a goat ABC staining kit. A significant nuclear translocation of hepatic NF kappa B protein was observed in animals induced with DEN confirming the persistent activation of NF kappa B during early chemical carcinogenesis. However, a decline in the nuclear translocation of the protein was observed in the animals treated with the decoction subsequent to DEN induction. This observation was further supported by a significant cytosolic localization of the protein in the animals orally treated with the decoction, when compared with the distilled water control. Inhibition of NF kappa B transcriptional protein may be a mechanism by which the anticarcinogenic effect of the decoction comprised of N. sativa, S. glabra, and H. indicus is mediated.
77. MDR1 Regulation by Ginsenoside Rd
Keon Wook Kang and Yuba R. Pokharel
College of Pharmacy, Chosun University, Gwangju, South Korea
MCF-7/ADR cells, a doxorubicin-resistant human breast cancer cell line, acquire resistance to several chemotherapeutic agents, such as anthracylines and taxol, which has been attributed to overexpression of the multidrug resistance 1 (MDR1) gene. The present study was designed to clarify whether ginsenosides affect the expression of the MDR1 gene in MCF-7/ADR cells. Ginsenoside Rd, Re, Rb1, and Rg1 (100 μg/mL) decreased the MDR1 protein levels in MCF-7/ADR cells. Especially, ginsenoside Rd resulted in the concentration-dependently inhibited MDR1 protein expression without cytotoxicity. However, MDR1 mRNA levels were not decreased by ginsenoside Rd and nuclear levels of key transcriptional factors for MDR1 gene expression, hypoxia inducible factor-1ƒñ, CCAAT-enhancer binding proteinƒ”, Forkhead box-containing protein, O subfamily 1, and Y-box binding protein-1 were also not affected. Reporter-gene analyses showed that ginsenoside Rd did not cause decreases in the transcriptional activity of MDR1 gene and pregnane × receptor reporter. The protein stability of MDR1 is known to be dependent on the ubiquitin-dependent protein degradation. We further found that ginsenosides Rd increased ubiquitination of MDR1 protein. Moreover, doxorubicin-resistance in MCF-7/ADR cells was reversed by ginsenosides Rd treatment. These results propose that ginseng administration with other anticancer agents may be useful for the treatment of chemotherapy-resistant breast cancer through the downregulation of MDR1 protein.
78. Study on the Effective Constituents in Xiexin Decoction for Anti-Inflammation
Bing-liang Ma and Yue-ming Ma
College of Chinese Material Medica, Shanghai University of Traditional Chinese Medicine, Shanghai, China
Aim: The aim of this study was to ascertain the effective constituents in Xiexin decoction (XXD) for anti-inflammation and the interactions of these constituents at the pharmacodynamic level.
Methods: XXD was prepared and qualified by high-performance liquid chromatography (HPLC). Rats were administered oral XXD 1 hour before intraperitoneal (i.p.) lipopolysaccharide (LPS; 3.2 mg/kg); venous serum samples were then collected from the eyehole. Nitric oxide (NO) production was quantified by the Griess reaction, and anthraquinones, flavonoids, and alkaloids were quantified by HPLC. Correlations between NO production and the serum concentration of these constituents were calculated. RAW 264.7 cells were stimulated with LPS (0.25 ìg/mL) and one or more XXD constituents; cell viability and NO production was quantified.
Results: XXD significantly decreased NO production in vivo, which correlated well with rhein, baicalin, berberine, emodin, and aloe-emodin. All the typical constituents of XXD, with the exception of physcion and chrysophanol, dose dependently inhibited NO production in LPS-stimulated Raw 264.7 cells. However, since serum concentrations of some constituents were all very low, only rhein, baicalin, and berberine were adopted in an orthogonal designed study to further ascertain the most important anti-inflammatory constituents in XXD and the interactions of these constituents in vitro. The results showed that rhein was the most powerful constituent, followed by baicalin, then berberine. No synergy was found among these constituents.
Conclusions: Rhein was the most effective anti-inflammatory constituent in XXD, followed by baicalin; no synergy was found among these constituents at the pharmacodynamic level in vitro.
79. Imatinib Pharmacokinetics in Mice after Coadministration with Pain Management Drugs Ibuprofen and Paracetamol Does Not Show Apparent Drug-Drug Interaction
Wei Yin Lim, Grace Chay, Elaine Kan, Shin Yee Tan, Jason H. K. Law, Gian Wan Soo, Nadeem I. Bukhari, and Ignacio Segarra
Department of Pharmaceutical Technology, International Medical University, Kuala Lumpur, Malaysia
Background and Purpose: Paracetamol and ibuprofen are commonly used drugs for pain relief in chronic myeloid leukemia (CML) and gastrointestinal stromal tumors (GIST) treated with imatinib, a molecularly targeted anticancer agent, which presents good pharmacokinetics, is eliminated via CYP3A4 metabolism, and is a substrate of P-glycoprotein. This study investigated whether a pharmacokinetic drug-drug interaction exists between imatinib and both analgesic drugs in a preclinical mouse model.
Experimental Approach: Male ICR mice (9–12 weeks) were orally administered either imatinib 50 mg/kg alone (control group, n = 43) or in combination (15 minutes apart) with 150 mg/kg paracetamol (study group 1; n = 41) or 30 mg/kg ibuprofen (study group 2; n = 40) orally. Mice were euthanized at predetermined time points (2, 5, 10, 20, and 40 minutes and 1, 2, 4, 6, and 10 hours) and plasma imatinib concentration quantified, using a validated HPLC assay. Noncompartmental pharmacokinetic analysis was performed, and the bioequivalence criterion (80–125%) was used to assess the occurrence of drug interaction.
Results and Discussion: In the control group, imatinib AUC0→∞ was 21.49 μg·h/mL, CMAX was 6.12 ± 1.96 μg/mL, TMAX was 40 minutes, and the elimination t1/2 was 2.4 hours, Cl/F was 2.3 L/h/kg, and VSS/F was 8.3 L/kg. Following paracetamol coadministration, AUC0→∞ increased 12% (24.06 μg·h/mL), CMAX was lower: 4.52 ± 3.11 μg/mL, but was not statistically different, which might be due to variability, TMAX was unchanged, and t1/2 increased (3.1 hours). Cl/F slightly decreased (2.1 L/h/kg) and VSS/F increased to 9.9 L/kg. Ibuprofen coadministration resulted in 8% higher AUC0→∞ (23.17 μg·h/mL), 21% higher CMAX (7.39 ± 0.98 μg/mL), similar TMAX, and double t1/2 (4.5 hours). Cl/F was unchanged and VSS/F increased to 12.2 L/kg.
Conclusions: Main plasma pharmacokinetic parameters remained within the bioequivalent range. However, after paracetamol or ibuprofen coadministration, imatinib VSS/F increased 19 and 46%, respectively, suggesting larger tissue distribution and a likely interaction at tissue distribution level. Since severe hepatoxicity has been reported with paracetamol coadministration, liver function monitoring may be necessary, although no interaction seems apparent in plasma. (Note: WYL and GC contributed equally).
80. Metronidazole Affects the Tissue Distribution of Imatinib but Does Not Change Its Plasma Pharmacokinetics in Mice
Shin Yee Tan, Gian Wan Soo, Grace Chay, Jason H. K. Law, Wei Yin Lim, Elaine Kan, Nadeem I. Bukhari, and Ignacio Segarra
Department of Pharmaceutical Technology, International Medical University, Kuala Lumpur, Malaysia
Background: Imatinib is a chemotherapeutic agent that selectively inhibits the c-KIT and the BCR-ABL tyrosine kinases, currently approved for chronic myelogenous leukemia (CML) and gastrointestinal stromal tumors (GIST) treatment. In humans, imatinib presents favorable pharmacokinetics and undergoes CYP3A4-mediated metabolism. It is also a P-gp substrate. In GIST treatment, imatinib is used as a neoadjuvant prior to surgery and continued as adjuvant therapy with metronidazole, a broad-spectrum antibiotic, as prophylaxis and treatment against postoperative infections. Thus, it is hypothesized that metronidazole concurrent administration may affect imatinib pharmacokinetics and tissue distribution.
Methods: Male ICR mice (9–12 weeks) were given 50 mg/kg imatinib orally (control group; n = 43). The study group (n = 51) was given 40 mg/kg of metronidazole orally 15 minutes prior to 50 mg/kg imatinib PO. Mice were euthanized at predetermined time points (2, 5, 10, 20, and 40 minutes and 1, 2, 4, 6, and 10 hours after imatinib administration); plasma, brain, liver, and kidney collected and imatinib concentrations measured by HPLC. Non-compartmental pharmacokinetic parameters were estimated and Bailer’s method for destructive sampling used to aid AUC comparison between the two groups. Results: In plasma, coadministration caused lower CMAX (6.12 ± 1.96 μg/mL for control vs. 3.82 ± 1.80 μg/mL for study group) and 50% shorter TMAX. No differences between both groups were found for AUC0-last (20.72 ± 0.74 μg·h/mL vs. 18.00 ± 1.43 μg·h/mL, respectively), elimination half-life (2.4 hours) or Cl/F. However, tissue exposure was largely affected. The control group showed AUC0-t tissue-to-plasma ratios for kidney and liver of 2.28 and 2.15, respectively, which increased to 5.32 and 3.90, respectively, after coadministration. Brain penetration in the control group was minimal (AUC0-last = 1.81 ± 0.25 μg·h/g) but increased ∼20-fold to 40.57 ± 5.03 μg·h/g in the study group. Conclusions: Metronidazole coadministration results in a drug-drug interaction at tissue level that is not perceptible in plasma. The increased brain exposure may open possibilities for the treatment of glioma. Conversely, the larger tissue penetration may advise renal and hepatic function monitoring due to associated toxicity.
81. Apoptotic and Necrotic Cell Percentages in Kidney Cells from Rats Exposed to Magnetic Field
Mustafa Emre,1 Salih Cetiner,2 Sevil Zencir,3 and Zeki Topcu4
1Department of Biophysics, Cukurova University Faculty of Medicine, Adana, Turkey
2Central Laboratory, Cukurova University Faculty of Medicine, Adana, Turkey
3Department of Biochemistry, Ege University Faculty of Sciences, Izmir, Turkey
4Department of Pharmaceutical Biotechnology, Ege University Faculty of Pharmacy, Izmir, Turkey
Apoptosis has a central role in many physiological processes, and apoptotic cell death can be distinquished from other forms of cell death by a number of morphological and biochemical characteristics. The aim of this study was to investigate the influence of magnetic field on cell death. The percentage of apoptotic and necrotic cells of the kidney extracts from female Wistar rats were determined by the flow-cytometry method. There was not a statistically significant difference between alive and apoptotic cell percents among the experimental and control groups. Our results were further confirmed by the analyses of DNA fragmentation by conventional agarose gel electrophoresis. However necrotic cell percent of the group exposed to magnetic field was significantly lower than that of either unexposed or sham control groups (P < 0.05), which suggests a possible relationship between exposure to magnetic field and cell death.
82. Validation of CYP2C8 Induction Assays in Primary Cultures of Human Hepatocytes Using RT-PCR and Enzyme Activity
George Zhang, Yujia Weng, Erica Liu, Charles L. Crespi, and David M. Stresser
BD Gentest Contract Research Services, BD Biosciences Discovery Labware, Woburn, Massachusetts, USA
Cytochrome P450 (CYP)2C8 metabolizes a number of important drugs and endogenous substances. The isoform is inducible, and the potential for drug-drug interactions due to CYP2C8 induction has been recently highlighted in the U.S. Food and Drug Administration (FDA) draft guidance. We have validated real-time reverse-transcriptase polymerase chain reaction (RT-PCR) and enzyme activity assays in hepatocytes to assess CYP2C8 induction potential. Hepatocytes were cultured in 24-well collagen I–coated plates and treated with solvent vehicle control (dimethyl sulfoxide; DMSO) or 20 μM of rifampicin (RIF) with a daily media change and replenishment of the inducer for 3 days. The PCR amplification efficiency was similar for both CYP2C8-actin and was found to be greater than 93% in two-β and the housekeeping gene donor livers. Treatment with RIF caused a 2.3- and 7.8-fold induction of CYP2C8 mRNA in 2 donors. The CYP2C8 catalytic activity was determined in situ by measuring N-desethyamodiaquine (DEAQ) formation from the corresponding probe substrate, amodiaquine (AQ). Incubation of 100 μM of AQ (near saturating) with hepatocytes treated with DMSO or RIF resulted in a linear formation of DEAQ up to 30 minutes for 3 donors. In general, substrate dependence experiments revealed hyperbolic behavior for up to 175 μM and apparent substrate inhibition from 175 to 250 μM. The Km values were not significantly affected by treatment but were found to be 22, 31, and 153 μM among 3 donors. These data suggest Km values are donor-dependent, ranging up to 7-fold in this limited donor set. Treatment of hepatocytes with 20 μM of RIF caused a 4.9-, 4-, and 2.6-fold induction of CYP2C8 activity in these 3 donors. In conclusion, these results validate the real-time RT-PCR and in situ catalytic enzyme assays for assessment of CYP2C8 induction potential in plated human hepatocytes in this laboratory.
83. Protein Kinase C Inhibitors Prevent Alterations in BKCA Current and Myofilament Ca2+-Sensitivity in Vascular Smooth Muscle Cells at Radiation-Evoked Arterial Hypertension
Igor V. Kizub and Anatoliy I. Soloviev
Experimental Therapeutics, Institute of Pharmacology and Toxicology of the AMS of Ukraine, Kiev, Ukraine
Ionizing radiation increases vascular responsiveness and leads to arterial hypertension, but the mechanisms remain unclear. We have supposed that protein kinase C (PKC) inhibitors may prevent changes in large-conductance Ca2+-activated K+ channels (BKCa) activity and myofilament Ca2+ sensitivity in vascular smooth muscle cells (VSMCs), which both may contribute to arterial hypertension. The study was performed on single isolated rat aorta VSMCs using patch-clamp techniques in whole-cell mode and chemically (β-escin) permeabilized aorta VSMCs using vascular tone measurement, from healthy and whole-body irradiated (source Co60, dose 6 Gy) animals taken on 9 (IR9) and 30 days postirradiation (IR30). Whole-cell outward K+ current in VSMCs from IR9 and IR30 was decreased. Ca2+-activated K+ current was separated pharmacologically by using 1 μM of apamin, 1 μM of charybdotoxin, and 500 nM of paxilline, showing in rat aorta VSMCs BKCa current is the main component of K+ current. Selective BKCa current inhibitor, paxilline (500 nM), depressed reduced K+ current in IR9 and had no effect on this current in IR30 VSMCs. PKC inhibitor, chelerythrine (100 nM), renewal K+ current was reduced by the action of ionizing irradiation in IR9 and IR30 aorta VSMCs. In permeabilized VSMCs from irradiated animals, myofilaments Ca2+ sensitivity was significantly increased. The value of pCa50 (-log [Ca2+] 50% effective), which reflects shifting in the pCa-tension relationship curve, was lower in IR9 vs. control. Chelerythrine (1 μM) and staurosporine (0.1 μM), other PKC inhibitors, had no effect on Ca2+ sensitivity in healthy tissue, but significantly decreased Ca2+ sensitivity in tissue from IR9 and IR30. PKC activator, phorbol dibutyrate (10 μM), significantly increased Ca2+ sensitivity in control tissue but was without effect in irradiated vascular rings. Our data indicate that ionizing radiation inhibits outward K+ current in rat aorta VSMCs manly via PKC-mediated inhibition of BKCa current, as well as PKC, contributes to radiation-evoked myofilament Ca2+ sensitization. The inhibitors of PKC prevent these changes, which can be involved to vascular tone elevation, and subsequent arterial hypertension development under ionizing radiation impact.
84. Inhibition- and Mechanism-based Inactivation of Human CYP3A4 BY Phyllanthus amarus
Theerada Taesotikul,1 Aimon Somanabandhu,2 Vichien Navinpipatana,3 Sirimas Kanjanawart,4 Wongwiwat Tassaneeyakul,5 and Wichittra Tassaneeyakul1
1Department of Pharmacology, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand
2Department of Pharmacognosy, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
3Queen Sirikit Hospital, Sattahip, Chonburi, Thailand
4Department of Pharmacology, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand
5Department of Toxicology, Faculty of Pharmaceutical Sciences, Khon Kaen University, Khon Kaen, Thailand
Phyllanthus amarus (P. amarus) has long been used as a medicinal plant in several countries around the world, including Thailand, for the treatment of jaundice, hepatitis, diabetes, and other diseases. It has been reported that alcoholic and aqueous extract of P. amarus could inhibit certain drug-metabolizing enzymes both in vivo as well as in vitro. Since CYP3A4 is the major enzyme that responsible for the metabolism of a number of drugs, therefore, the inhibitory effects of P. amarus extractions and its two major lignans, phyllanthin and hypophyllanthin, were determined in the present study by using human hepatic microsomal testosterone 6α-hydroxylase as a selective CYP3A4 marker. The ethanolic and aqueous extracts of P. amarus inhibited CYP3A4 activity in a dose-dependent manner with IC50 values of 1.26 ± 2.29 μg/mL and 65.57 ± 4.93 μg/mL, respectively. Further, the soxhlet extractions of P. amarus by hexane, dichloromethane, and methanol showed IC50 values of 0.39 ± 0.15 g/mL, 11.6 ± 4.3 g/mL, and >100 g/mL, respectively. Phyllanthin and hypophyllanthin, which are extracted from the hexane fraction, showed time-dependent inhibition on CYP3A4. Mechanism-based inhibition kinetics of these lignans were also determined.
85. Expression and Functional Analysis of Human Cytochrome P450 2C19 Polymorphic Alleles
Hui-Juan Wang,1 Yi-Wen Gao,1 Ping-Ping Ma,1 Juan-Li Zhu,2 and Chao Chen2
1College of Life Science, Northwest University, Xi’an, China
2National Engineering Research Center for Miniaturized Detection System, Shanxi Lifegen Co. Ltd., Xi’an, China
Aim: Construct a Saccharomyces cerevisiae expressing system and develop an enzymatic detection method for human cytochrome P450 2C19(CYP2C19) polymorphic genes.
Methods: The prototype cDNA of CYP 2C19 was obtained by RT-PCR from human liver tissue and cloned into the pYES2/CT vector for galactose-inducible expression in yeast. The cloned cDNA was subsequently used as a template to introduce polymorphisms by site-directed mutagenesis and were cloned into the same vector. Transformed yeasts produced large quantities of microsome-bound 2C19 enzymes, as determined by Western analysis. The isolated microsomes were used to measure the kinetic constants of 2C19 enzymes in real-time assays, using a fluorogenic substrate, CEC (3-cynao-7-ethoxycoumarin). The inhibition of CYP 2C19 prototype enzyme by a known inhibitor drug (tranylcypromine) was tested by serial titration of drugs in the fluorogenic assays.
Results: Five cDNA of CYP2C19 corresponding to 2C19*1A (prototype) and variant alleles of 2C19*1B (991A>G, I331V), 2C19*6 (395G>A, R132Q), 2C19*9 (431G>A, R144H), and 2C19*13 (1228C>T, R410C) have been constructed for expression in yeast. The results showed that all enzymes possess robust activity with the exception of 2C19*6; the 2C19*6 enzyme failed to show activity even at the highest enzyme concentration tested (3 mg/mL). The Km value of four catalytically active enzymes showed little difference, which were all close to 20 μmol/L. The Vmax value of 2C19*1B and 2C19*13 enzymes were very similar to 2C19*1A, but the Vmax value of 2C19*9 enzyme was 10-fold lower than that of 2C19*1A. Then, the inhibition testing of 2C19*1A enzyme testified that tranylcypromine had specific inhibition on CYP2C19.
Conclusions: The prototypical and four variant forms of human cytochrome P450 2C19 have been expressed in functional form in yeast. The recombinant enzymes show robust activity in real-time fluorogenic assays, and the enzyme activity was specifically inhibited by a known inhibitor of 2C19 enzyme. This work demonstrates the feasibility of large-scale analysis of CYP2C19 polymorphic enzymes in drug mtebabolism and drug-drug interaction study.
86. Expression Profile of Genes Involved in 5-Fluorouracil Metabolic Pathway as Predictive Parameters for 5-Fluorouracil Response in Cholangiocarcinoma
Jariya Chaiyagool,1 Chariya Hahnvajanawong,1 Banchob Sripa,2 Vajarapong Bhudhisawasdi,3 Narong Khuntikeo,3 Ake Pugkhem,3 Siri Chau-in,3 and Wichittra Tassaneeyakul4
1Microbiology, Khon Kaen University, Khon Kaen, Thailand
2Pathology, Khon Kaen University, Khon Kaen, Thailand
3Surgery, Khon Kaen University, Khon Kaen, Thailand
4Department of Pharmacology, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand
Cholangiocarcinoma (CCA), a common bile duct tumor, accounts for approximately 15% of liver cancers worldwide. The highest incidence of CCA has been reported in Laos and Northeastern Thailand. At present, 5-fluorouracil (5-FU) is the most common drug used for the palliative treatment of CCA. The aim of this study was to determine the relationship between the gene-expression profile of the four enzymes in the metabolic pathway of 5-FU, that is, orotate phosphoribosyltransferase (OPRT), dihydropyrimidine dehydrogenase (DPD), thymidylate synthetase (TS), and thymidylate phosphorylase (TP) and an in vitro sensitivity to 5-FU of CCA. Fresh tumor specimens from 25 CCA patients were used for the determination of 5-FU sensitivity, using the histoculture drug-response assay. The mRNA levels of the target genes were quantified by using a TaqMan• (Taqman, Applied Biosystems, Foster city, CA, USA) gene-expression assay. Significant interindividual difference in the expression levels were noticed (i.e., 182-fold for DPD, 67-fold for TP, 34-fold for OPRT, and 25-fold for TS). Marked difference in the response to 5-FU of CCA tissues were also observed with an inhibition index of 5-FU that ranged from 0 to 95%. The mean values of TS, TP, and DPD mRNA levels obtained from the CCA tissues which well responded to 5-FU were not significantly different from those of the poor response group. However, the mean value of OPRT mRNA level in the 5-FU-responsive tissues was significantly higher than those in the poor responsive group. OPRT mRNA level appears to be a good marker for the prediction of 5-FU response in CCA. However, the usefulness of OPRT as a predictive marker for 5-FU response needs to be further verified in vivo.
87. Houttuynia Cordata Water Extracts Suppresses Anaphylactic Reaction and IgE-mediated Allergic Response
Eun Hee Han, Jin Hee Park, and Hye Gwang Jeong
BK21 Project Team, Department of Pharmacy, Chosun University, Gwangju, South Korea
Houttuynia cordata has been used as a traditional medicine in Korea and is known to have antioxidant, anticancer, and anti-allergic activities. The precise effect of H. cordata, however, remains unknown. In this study, we investigated the effects of H. cordata water extract (HCWE) on passive cutaneous anaphylaxis (PCA) in mice and on IgE-mediated antiallergic effect in rat mast RBL-2H3 cells. Oral administration of HCWE inhibited IgE-mediated systemic PCA reaction in mice. HCWE also reduced antigen (dinitrophenyl (DNP)-BSA) induced release of ƒ”-hexosaminidase, histamine, and reactive oxygen species in IgE-sensitized RBL-2H3 cells. In addition, HCWE inhibited the antigen-induced IL-4 and TNF-a production and expression in IgE-sensitized RBL-2H3 cells. HCWE inhibited antigen-induced activation of NF-kB, regulator of cytokines, and degradation of IkB-a. Further, antigen induced phosphorylation of Akt and MAP kinases (ERK1/2 and JNK1/2, except p38 MAP kinase) were inhibited by HCWE. Taken together, in vivo/in vitro antiallergic effect of HCWE suggests possible therapeutic applications of this agent for inflammatory allergic diseases by the inhibition of cytokines in mast cells.
88. Inhibitory Mechanism of Saponins Derived from Roots of Platycodon grandiflorum on Anaphylactic Reaction and IGE-mediated Allergic Response in Mast Cells
Eun Hee Han,1 Jin Hee Park,1 Young Chul Chung,2 Hoo Kyun Choi,1 and Hye Gwang Jeong1
1BK 21 Project Team, Department of Pharmacy, College of Pharmacy, Chosun University, Gwangju, South Korea
2Division of Food Science, International University of Korea, Jinju, South Korea
The purpose of this study was to investigate the protective effects of saponins isolated from the root of Platycodi radix (Changkil saponins: CKS) antiallergic effects in mice and mast cells. Oral administration of CKS inhibited the dinitrophenyl (DNP)-IgE antibody-induced systemic PCA reaction in mice. CKS reduced the ƒ”-hexosaminidase and histamine release from anti-DNP-IgE-sensitized RBL-2H3 cells. In addition, CKS inhibited the IgE antibody-induced increases in IL-4 and TNF-α production and expression in RBL-2H3 cells. In order to explore the inhibitory mechanism of CKS in PCA and mast cell degranulation, we examined the activation of intracellular signaling molecules. CKS suppressed DNP-IgE antibody-induced Syk phosphorylation. Further downstream, CKS also inhibited the phosphorylation of Akt and MAP kinases. Taken together, the in vivo/in vitro antiallergic effects of CKS suggest possible therapeutic applications for this agent in allergic diseases through the inhibition of inflammatory cytokines and Syk-dependent signaling cascades.
89. Tetrabromobisphenol A Induces Cyclooxygenase-2 Gene Expression–mediated PI3-Kinase/Akt/MAPK Signaling Pathways in Murine Macrophages
Eun Hee Han, Jin Hee Park, and Hye Gwang Jeong
BK21 Project Team, Department of Pharmacy, Chosun University, Gwangju, South Korea
Tetrabromobisphenol A (2,2-bis-(3,5-dibromo-4-hydroxyphenyl)propane; TBBPA), a halogenated derivative of bisphenol A, is widely used throughout the world as a flame retardant in numerous products. TBBPA was developed as a relatively nontoxic flame retardant. Cyclooxygenase-2 (COX-2) acts as a link between inflammation and carcinogenesis through its involvement in tumor promotion. In the present study, we examined the effect of TBBPA on COX-2 gene expression and analyzed the molecular mechanism of its activity in murine RAW 264.7 macrophages. Exposure to TBBPA markedly enhanced the production of prostaglandin E2 (PGE2), a major COX-2 metabolite, in macrophages. Further, TBBPA dose dependently increased the levels of COX-2 protein and mRNA. Phosphatidylinositol 3 (PI3)-kinase, its downstream signaling molecule, Akt, and mitogen-activated protein kinases (MAPK) were significantly activated by TBBPA. These results demonstrate that TBBPA induced COX-2 expression through the PI3-K/Akt/ERK, JNK, and p38 MAP kinase pathways. These findings provide further insight into the signal-transduction pathways involved in the carcinogenic effects of TBBPA.
90. Bergamottin Suppresses PMA-induced MMP-9 Expression by Blocking the Nuclear Factor-kB Activation via MAPK Signaling Pathways in HT1080 Human Fibrosarcoma Cells
Yong Pil Hwang, Jae Ho Choi, and Hye Gwang Jeong
BK21 Project Team, Department of Pharmacy, Chosun University, Gwangju, South Korea
Matrix metalloproteinase-9 (MMP-9) plays an important role in the invasion and metastasis of cancer cells. In this study, we examined the inhibitory effect of bergamottin from Citrus paradis on phorbol myristate acetate (PMA)-induced MMP-9 expression in HT1080 human fibrosarcoma cells. Bergamottin significantly suppressed PMA-induced MMP-9 and membrane-type 1 (MT1)-MMP expression and the matrigel invasion. However, bergamottin had no effect on expression of tissue inhibitor of metalloproteinases (TIMP)-1 and TIMP-2, the major endogenous inhibitors of MMPs. Further, as evidenced by the MMP-9 promoter, bergamottin specifically inhibited MMP-9 gene expression by blocking PMA-stimulated activation of NF-kB. In addition, bergamottin suppressed PMA-induced phosphorylation of JNK and p38 mitogen-activated protein kinase (MAPK), upstream factors involved in NF-kB, whereas the phosphorylation of extracellular signal-regulated kinase (ERK) was not affected by bergamottin, suggesting that the primary target of bergamottin for suppression of the NF-kB induction is present in upstream of JNK or p38 MAPK signaling pathway. These results suggest that bergamottin represents a potential antimetastatic agent=suppressing cancer-cell invasion through the specific inhibition of NF-kB–dependent MMP-9 gene expression.
91. N-Acetylglucosamine Inhibits UVB-induced MMP-1 AND -13 Expressions in Human Skin Fibroblasts
Yong Pil Hwang, Jae Ho Choi, Hyo Jeong Yun, and Hye Gwang Jeong
BK21 Project Team, Department of Pharmacy, Chosun University, Gwangju, South Korea
Ultraviolet (UV) B irradiation induces the production of matrix metalloproteinases (MMPs) by activating cellular signaling transduction pathways, which are responsible for the degradation or synthesis inhibition of collagenous extracellular matrix in connective tissues, causing skin photoaging. Using the human skin fibroblast (HS68) cell line in the present study, we investigated the inhibitory effects of N-acetylglucosamine (NAG) on MMP-1 and -13 expressions by various in vitro experiments and elucidated the pathways of inhibition. Treatment with NAG decreased the UVB-induced generation of reactive oxygen species (ROS). In addition, pretreatment with NAG inhibited UVB-induced MMP-1 and -13 expressions in a dose-dependent manner. Moreover, NAG reduced UVB-induced AP-1 transcriptional activity. NAG inhibited UVB-induced expression of c-Jun and c-Fos. Extracellular signal regulated kinase (ERK) activation was markedly inhibited by treatment with NAG. These results suggested that NAG can inhibit UVB-induced MMP-1 and -13 expressions might be mediated through the AP-1/ERK inactivation and inhibition of ROS formation in HS68 cells. In conclusion, the results presented that NAG may be useful in the prevention of UVB-induced photoaging.
92. N-Acetylglucosamine Protects from UVB Irradiation-induced Degradation of Hyaluronan and against UVB-Damage in Human Skin Fibroblasts
Yong Pil Hwang, Jae Ho Choi, Hyo Jeong Yun, and Hye Gwang Jeong
BK21 Project Team, Department of Pharmacy, Chosun University, Gwangju, South Korea
Solar ultraviolet (UV) B radiation damages human skin, affecting skin tone and resiliency, leading to premature aging (photoaging). Hyaluronan (HA), a major component of the cutaneous extracellular matrix, is involved in tissue repair. Human skin is exposed to and damaged by UVB-irradiation. Here, we investigate the effect of NAG on HA metabolism in human skin fibroblasts during acute UVB-induced damage. Pretreatment of human skin fibroblasts with NAG significantly reduced UVB-induced generation of reactive oxygen species (ROS) and subsequent cell death. Also, NAG protects HA from UVB light-induced degradation in a concentration-dependent manner. Pretreatment with NAG attenuated UVB-reduced hyaluronan synthase- 2 expression. In contrast, pretreatment with NAG inhibited UVB-induced hyaluronidase- 1 expression. In addition, NAG prevents the UVB-suppressed procollagen protein expression. These findings indicate that NAG may exert a protective effect against UVB irradiation-induced damage in the skin.
93. Inhibition of Lipid Synthesis through Activation of AMP-activated Protein Kinase by Saponins Derived from Roots of Platycodon grandiflorum
Hye Jin Park and Hye Gwang Jeong
BK21 Project Team, Department of Pharmacy, Chosun University, Gwangju, South Korea
Platycodon grandiflorum is a traditional oriental herbal medicine that is known for its immunostimulatory and antitumor effects. The present studies were performed to determine the extent to which the effect of saponin fraction (CKS) derived from roots of P. grandiflorum on hepatocellular lipids accumulation in insulin-resistant states in human HepG2 hepatocytes. CKS elevates expression of FAS and lipid accumulation. In addition, CKS increased Sirt1 and AMP-activated protein kinase (AMPK) activity in hepatocytes exposed to high glucose. In addition, CKS increased phosphorylation of acetyl coenzyme A carboxylase (ACC), an AMPK downstream effector. These effects of CKS are largely abolished by inhibition of Sirt1 activity, suggesting the stimulation of AMPK. In conclusion, AMPK signaling by CKS, which Sirt1 induces, may have potential therapeutic implications for dyslipidemia and accelerated atherosclerosis in diabetes and metabolism-related disease.
94. Mechanism of Puerarin-induced Endothelial Nitric Oxide Synthase through Estrogen Receptor Alpha-Dependent of PI3K/AKT and AMP-Activated Protein Kinase
Hien Tran Thi, Hyung Gyun Kim, and Hye Gwang Jeong
BK21 Project Team, Department of Pharmacy, Chosun University, Gwangju, South Korea
Nitric oxide (NO), produced by endothelial nitric oxide synthase (eNOS), represents an antithrombotic and antiatherosclerotic principle in the vasculature. In the present study, we demonstrated that puerarin, an isoflavonoid from Pueraria radix increased activation of eNOS in human endothelial ECV 304 cells. Puerarin increased production of NO in cells. Moreover, puerarin increased the activation of Akt/PKB, eNOS, and AMPK leading to increase NO production in cells. Inhibitors of Akt/PKB (LY294002) and ER-alpha (ICI 182,780) failed to suppress the puerarin-induced eNOS phosphorylation and Akt/PKB. Taken together, these results indicate that puerarin stimulates eNOS phosphorylation and NO production via activated PI3K/Akt requires estrogen receptor and AMP-activated protein kinase.
95. Metformin Inhibits P-Glycoprotein Expression in Breast Cancer Cell MCF-7/ADR via Inhibition of NF-kB Activation
Hien Tran Thi, Hyung Gyun Kim, and Hye Gwang Jeong
BK21 Project Team, Department of Pharmacy, Chosun University, Gwangju, South Korea
P-glycoprotein (P-gp) accounts for the most intrinsic and acquired cancer multidrug resistance (MDR). To inhibit the expression of P-gp is one of the effective ways to reverse cancer drug resistance. In the present study, we investigated the effects of metformin (1,1-dimethylbiguanide hydrochloride) on the expression of P-gp in MCF-7/adriamycin (MCF-7/adr), a human breast MDR cancer cell line. Metformin treatment significantly increases in intracellular accumulation of fluorescent P-gp substrate rhodamine-123, indicating that metformin treatment leads to reversal of the MDR phenotype resulting from an increased accumulation of anticancer drugs by inhibiting the drug-efflux function of P-gp. In addition, the activity of MDR1 was suppressed by metformin. Further, metformin also dose dependently inhibited TNF-α-induced NF-kB activation and IkB kinase activity. Taken together, these results suggest that the metformin can be a new adjuvant agent for human breast cancer chemotherapy and molecular mechanism of reversing MDR1 of MCF-7/adr cells by metformin via inhibition of NF-kB activation.
96. The Antimetastatic Effects of Aqueous Extract Isolated from Prunella vulgaris by Modulating Expressions of MMP-9
Jae Ho Choi and Hye Gwang Jeong
BK21 Project Team, Department of Pharmacy, Chosun University, Gwangju, South Korea
Matrix metalloproteinases (MMPs) play important roles in tumor metastasis. In this study, we showed that the effects of aqueous extract isolated from Prunella vulgaris (PVAE) on the experimental lung metastasis induced by B16F10 melanoma cells in C57BL/6 mice. The antimetastatic properties of PVAE were investigated by evaluating MMP-9 production in HT-1080 cells. In a B16F10 melanoma lung metastasis experiment, PVAE showed a great inhibition effect on the number of lung metastatic colonization in a dose-dependent manner. PVAE was reduced by the phorbol 12-myristate 13-acetate-induced MMP-9 gelatinolytic activity and expression in a dose-dependent manner, without changing tissue inhibitors of the metalloproteinase-1 level. Further, PVAE suppressed PMA-induced MMP-9 luciferase activity by inhibiting the function of NF-kB. Using in vitro wound-healing assays, we found that cell migration was also significantly inhibited by PVAE. These results suggest that the antimetastatic effects of PVAE are mediated by the suppression of MMP-9 gelatinolytic activity and its expression by the inhibition of NF-kB.
97. Regulation of WNT/ß-Catenin Signaling in Colon Cancer Cells by Caffeic Acid Phenethyl Ester
Guk Heui Jo,1 Chun Yeon Choi,1 Won Hee Jang,1 Dae-Hyun Seog,1 Yeong-Hong Park,1 Jungsug Gwak,2 Sangtaek Oh,2 and Sung Su Yea1
1Department of Biochemistry, College of Medicine, Inje University, Busan, South Korea
2Department of Pharmacology, College of Medicine, Inje University, Busan, South Korea
The Wnt/ß-catenin signaling pathway is known to be associated with the development of colon cancer. Caffeic acid phenethyl ester (CAPE) is an active component of propolis with many pharmacological properties, including immunomodulation and antitumor activity. In the present study, we investigated the antitumor effect of CAPE associated with the Wnt/ß-catenin signaling pathway. To screen the effect of CAPE on the Wnt/ß-catenin pathway, we used the HEK293 cells transfected with the TOPFlash reporter and the human Frizzled-1 expression plasmid. Wnt3a-conditioned medium (Wnt3a CM) induced TOPFlash reporter activity, which was markedly inhibited by CAPE in a dose-dependent manner. To investigate the effect of CAPE on the intracellular ß-catenin level, we performed Western blot analysis. The ß-catenin level in cytosolic and nuclear fractions was increased by incubation with Wnt3a CM, which was downregulated by CAPE. However, the effect of CAPE on the reduction of ß-catenin was abrogated by the addition of MG-132, which blocks proteasome-mediated protein degradation. CAPE also inhibited ß-catenin response transcription (CRT) and the level of ß-catenin in LiCl-induced HEK293 reporter cells. To test the effect of CAPE on colon cancer cells, CRT and the level of ß-catenin was measured in HCT116 and SW480 cells transfected with TOPFlash. Treatment with CAPE resulted in a decrease of CRT and downregulation of ß-catenin level. Collectively, these results suggest that CAPE inhibits Wnt/ß-catenin signaling in colon cancer cells, and that this effect is mediated, at least in part, through a degradation of the intracellular ß-catenin.
Acknowledgment
This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MEST) (no. R01-2007-000-20669-0).
98. Kahweol Blocks STAT3 Phosphorylation and Induces Apoptosis in Human Lung Adenocarcinoma A549 Cells
Hyung Gyun Kimand Hye Gwang Jeong
BK21 Project Team, Department of Pharmacy, Chosun University, Gwangju, South Korea
Kahweol, the coffee-specific diterpene, has been reported to have anticarcinogenic properties. Animal data support such a chemopreventive effect of coffee. However, the precise underlying protective mechanisms are poorly understood. In this study, we have investigated the effect of kahweol on proliferation and apoptosis in human lung adenocarcinoma A549 cells. In cell viability and cell proliferation assays, kahweol exhibited antiproliferative and proapoptotic effects on A549 cells in a time- and dose-dependent manner. Kahweol considerably inhibited the expression of Bcl-2 but increased that of Bax; it also stimulated the cleavage of caspase-3 and PARP (poly ADP-ribose polymerase). In addition, kahweol-induced apoptosis was confirmed by TUNEL assay. Further, kahweol inhibited dose-dependent STAT3 phosphoryation. Wild-type STAT3 completely blocked the apoptotic effect of kahweol. These findings suggest that kahweol induced apoptosis via downregulation of the STAT3 signaling pathway.
99. Antioxidant and Anti-Inflammatory Potential of Arctic Seaweeds: Activity Screening and Mechanism Identification
Sung Su Yea,1 Chun Yeon Choi,1 Guk Heui Jo,1 and Youngwan Seo2
1Department of Biochemistry, College of Medicine, Inje University, Busan, South Korea
2Division of Marine Environment and Bioscience, Korea Maritime University, Busan, South Korea
We had screened useful pharmacological materials from 20 Arctic seaweeds and identified that Alaria esculenta has antioxidant and anti-inflammatory activity. A. esculenta, a brown seaweed known as dabberlocks or winged kelp, is a traditional food along the coasts of the far north Atlantic Ocean. In the present study, we investigated the effect of A. esculenta extract (AEE) on nitric oxide (NO) production and its mode of action in activated macrophages. Lipopolysaccharide (LPS) induced NO production in RAW264.7 macrophages, which was markedly inhibited by AEE in a dose-dependent manner. AEE also decreased the expression of inducible nitric oxide synthase (iNOS) protein and mRNA in LPS-stimulated RAW264.7 cells. To characterize further the inhibitory mechanisms of AEE, we examined the transcriptional activities of NF-kB. AEE inhibited NF-kB-dependent transcriptional activity by decreasing the nuclear translocation of p65, which is a component of NF-kB. In addition, we examined the effect of AEE on the expression of heme oxygenase-1 (HO-1), which has been known to be a cytoprotective and anti-inflammatory enzyme as well as a negative regulator for iNOS expression. AEE induced the expression HO-1 mRNA and protein in RAW264.7 macrophages. AEE also activated upstream signaling of HO-1 gene expression, such as phosphorylation of AKT. Collectively, these results suggest that A. esculenta has antioxidant and anti-inflammatory activity, which is mediated, at least in part, through downregulation of iNOS expression and induction of HO-1 expression.
Acknowledgment
This work was supported by the Korea Science and Engineering Foundation(KOSEF) grant funded by the Korea government (MEST) (no. R01-2007-000-20669-0).
100. CAR-mediated Transcriptional Activation of Human CYP1A1 and CYP1A2 Genes
Kouichi Yoshinari, Noriaki Yoda and Yasushi Yamazoe
Graduate School of Pharmaceutical Sciences, Tohoku University, Sendai, Japan
CYP1A subfamily of enzymes is induced by polycyclic aromatic hydrocarbons via the Ah receptor (AhR). Phenobarbital is also capable of inducing CYP1A2 independent of AhR, although its precise mechanism remains unclear. Because constitutive androstane receptor (CAR) is primarily responsible for the phenobarbital induction of CYP2B/2C/3A enzymes, we have examined whether CAR is also involved in the transcriptional activation of CYP1A subfamily genes. Human CYP1A1 and CYP1A2 genes are located in a head-to-head orientation and share a ∼23 kb of the 5′-flanking region. Dual reporter plasmids containing the whole or partially deleted 5′-flanking region between two different reporter genes, firefly luciferase and secreted alkaline phosphatase, were used to simultaneously evaluate transcriptional activation of the genes. Reporter assays in HepG2 cells with or without human CAR expression plasmid and human CAR activator 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime demonstrated that CAR mediated the transcriptional activation of both CYP1A1 and CYP1A2 genes through the promoter regions from −461 to −554 and from −18,089 to −21,975 of the CYP1A1 gene. The former overlaps with the region essential for the AhR-mediated transcription of both genes. Several nuclear receptor binding motifs (AG(G/T)TCA-like sequences) in this region were mutated independently for reporter assays. The results indicated that one motif with a perfect AGGTCA sequence is crucial for the CAR-mediated transcriptional activation of both genes. In gel-shift assays, CAR/retinoid × receptor heterodimer bound to an everted repeat separated by eight nucleotides (ER8) containing the AGGTCA motif, located at around −520 of the CYP1A1 gene. Reporter assays using a construct lacking the ER8 confirmed the important role of this motif for the CAR-mediated transcriptional activation of the CYP1A1 and CYP1A2 genes. Interestingly, the ER8 motif is well conserved among human, mouse and rat genes in its sequence and relative position to the CYP1A1 transcriptional start site. These results suggest that CAR activates transcription of human CYP1A1 and CYP1A2 genes through its binding as a heterodimer with retinoid × receptor to the ER8 motif located in the proximal promoter of the CYP1A1 gene.
101. Proteomic Profiling of Bezafibrate-treated Primary Cultures of Human Hepatocytes
Magalie Alvergnas,1 Alain Rouleau,2 Bruno Heyd,3 Georges Mantion,4 Lysiane Richert,5 and Hélène Martin1
1Laboratoire de Toxicologie Cellulaire, EA 4267, IFR 133, UFR des Sciences Médicales et Pharmaceutiques/Plateforme protéomique CLIPP Besançon-Dijon, Besançon, France
2Plateforme Protéomique CLIPP Besançon-Dijon, Besancon, France
3Centre de Transplantation Hépatique, IFR 133, Service de Chirurgie Viscérale et Digestive, Besançon, France
4Centre de Transplantation Hépatique, IFR 133, Service de Chirurgie Viscérale et Digestive, Besançon, France
5Laboratoire de Toxicologie Cellulaire, EA 4267, IFR 133, UFR des Sciences Médicales et Pharmaceutiques/Kaly-Cell, Besançon, France
Bezafibrate (BezaF), a lipid-lowering fibrate, belongs to a group of structurally very diverse compounds designated as peroxisome proliferators, which elicit pathological changes, including proliferation of peroxisomes, cell proliferation, hepatomegaly, and the development of carcinomas in rodents. In contrast, human hepatocytes do not exhibit these changes. The aim of the present study was to generate proteomic cartography of human hepatocytes treated with BezaF (0 and 250 μM), in the presence of N-acetylcysteine (Nac; 0 and 1 mM), known for its antioxidant properties and its capacity to modulate redox status in hepatocyte cultures. After using an adequate lysis buffer for the extraction of hepatic proteins, we injected samples into the two-dimensional (2D) chromatography system, the ProteomeLab PF2D (Beckman Coulter: Fullerton, CA, USA), to separate proteins from human hepatocytes. In this system, proteins were fractionated by isoelectric points (pI) in a pH gradient (pH 8.5 to 4) in the first dimension, and each of these pI protein fraction was further separated by hydrophobicity using nonporous silica reverse-phase chromatography in the second dimension. Then, 2D maps of nontreated and treated samples were generated and were compared using Mapping Tools software (DeltaVue and MultiVue; Beckman Coulter). Modifications of protein expression after BezaF treatment were evidenced and were shown to be dependent on the cellular redox status. Protein fractions after the second dimension of separation were stored until mass spectrometry analysis, to identify the modulated proteins.
102. Cryopreserved Human Hepatocytes for the Prediction of Metabolic Clearance and Drug-Drug Interactions in Humans; An Update
Eliane Alexandre,1 Dumrongsak Pekthong,2 Audrey Baze,1 Celine Parmentier,1 Philippe Bachellier,3 Georges Mantion,4 Bruno Heyd,5 Nicola Hewitt,1 and Lysiane Richert6
1KaLy-Cell, Illkirch, France
2Narusuan University, Phitsanulok, Thailand
3Hôpital de Hautepierre, Centre de Chirurgie Viscérale et de Transplantation, Strasbourg, France
4Centre de Transplantation Hépatique, Service de Chirurgie Viscérale et Digestive, Besançon, France
5Centre de Transplantation Hépatique, Service de Chirurgie Viscérale et Digestive, Besançon, France
6UFR des Sciences Médicales et Pharmaceutiques, Laboratoire de Toxicologie Cellulaire, EA 4267, IFR 133 / Kaly-Cell, Besançon, France
Cultured human hepatocytes are standard in vitro tools for clearance studies;however, they, like microsomes, tend to underpredict in vivo clearances (Jouin et al., 2006). Recently, we found that the addition of serum, combined with mixing of human hepatocytes in a 96-well plate mixed by rotation at 900 rpm, resulted in an about a 2-fold increase in the CLint, in vitro of midazolam, diclofenac, and bufuralol, compared to parallel incubations in primary culture (Simon et al., 2009), improving prediction of in vivo CLHEP. The CLint, in vitro of midazolam, diclofenac, and bufuralol were determined in 6 different batches of cryopreserved human hepatocytes. These compounds were also incubated with a pool of the same 6 donors in a single incubation. The average CLint, in vitro for midazolam, diclofenac, and bufuralol measured in hepatocytes as single donors was the same as the CLint, in vitro in the pooled incubation. This was further confirmed with additional sets of 6 different batches of cryopreserved human hepatocytes. Cultured human hepatocytes are also standard in vitro tools for induction studies. We recently found that cryopreserved, plateable human hepatocytes are suitable to assess induction potential of drugs (Abadie et al., 2009). We compared the basal activities of a set of cryopreserved, plateable human hepatocytes with their fresh counterparts and confirmed that the activities were maintained after cryopreservation. In addition, the induction responses of cryopreserved plateable human hepatocytes to prototypical inducers were in the range of our historical data on responses obtained with freshly isolated cultured human hepatocytes. In conclusion, in vitro clearance determined in fresh or cryopreserved human hepatocytes rotated at 900 rpm in 96-well plates improves prediction of in vivo CLHEP and in vitro assessment of the induction potential of drugs can be performed with either freshly isolated or plateable cultured human hepatocytes.
References
- Abadie, C., et al. (2009). Toxicol In Vitro (submitted).
- Jouin, D., et al. (2006). Eur J Pharmac Biopharmac 63:347–355.
- Simon, S., et al. (2009). 10th European HUG Meeting, Copenhagen, Denmark.
103. Abstract Withdrawn
104. Evaluation of the High-Throughput Method for the Quantification of Trophoblast Drug Transport with Differentiating Human Choriocarcinoma JEG-3 Cells
Kenji Ikeda,1 Naoki Utoguchi,2 Michiaki Myotoku,1 and Yoshihiko Hirotani1
1Laboratory of Clinical Pharmaceutics, Faculty of Pharmacy, Osaka Ohtani University, Osaka, Japan
2Department of Biopharmaceutics, School of Pharmaceutical Sciences, Teikyo University, Kanagawa, Japan
Placental drug transport is mainly regulated by the trophoblast layers. The human choriocarcinoma cell line, JEG-3, has been used as a model for studying placental drug transport. A problem associated with this model is that the drugs are transported through the intercellular junctions. The purposes of this study were to evaluate the placental drug transport model associated with the tight junctions that form during the differentiation of choriocarcinoma JEG-3 cells and to quantify the drug transport in a high-throughput manner by using the 96-well culture plate. For the differentiation of JEG-3 cells, several differentiating reagents and some media were used. The tight junction-forming ability was estimated by measuring transepithelial electrical resistance values (TEERs). The mRNA expression levels of the not-expressed choriocarcinoma 1 (NECC1) gene were mainly estimated as a signal for differentiation. In order to confirm the expression profile of transporters in the differentiated cells as well as that in the normal trophoblast cells, the mRNA expression levels of multidrug resistance–associated proteins (MRPs) and breast cancer resistance protein (BCRP) were determined by using our previous multiplex analysis method (Ikeda et al., 2008). TEERs were the highest in a mixed medium (D/F) containing Dulbecco’s modified Eagle’s and Ham’s F-12 medium in the ratio of 1:1. NECC1 expression was induced under all the differentiating conditions. BCRP expression was the highest in the D/F medium. JEG-3 cells differentiating in the D/F medium were used as a subsequent model. The permeability coefficient (P) of valproic acid, a high transport drug in the placenta, was more than twice that of fluorescein, an intercellular transport reagent. The P-values of fluorescein found by using the 24- and 96-well methods were 3.28 and 2.27 (x 10−6 cm/sec), respectively. We suggested that this D/F-differentiating JEG-3 cells model would be beneficial for the high-throughput study of drug transport across the trophoblast in vitro.
Reference
- Ikeda, K., et al. (2008). Characterization of multidrug resistance-associated protein mRNAs expression profiles in Caco-2 and HT-1080 cell lines induced by methotrexate. Pharmazie 63:883–889.
105. Synergism of Colistin and Rifampicin in the Treatment of Nosocomial Infections with Pandrug-Resistant Nonfermenters: An Observational Study
Chiranjay Mukhopadhyay, N. Nagalakshmi, Kiran Chawla, Gunjan Dutta, and Indira Bairy
Microbiology, Kasturba Medical College, Manipal, India
The increased incidence of nosocomial infections by multi- and pan-drug-resistant (MDR and PDR) nonfermenters, such as Pseudomonas aeruginosa and Acinetobacter baumannii, creates demand on the use of some older antibiotics, such as colistin and rifampicin. We have conducted the present observational study to evaluate the efficacy of colistin as a monotherapy and colistin in combination with rifampicin in the treatment of nosocomial infections with MDR and PDR nonfermenters. Routine biochemical and antibiotic sensitivity tests (CLSI 2006) were done for P. aeruginosa (n = 2339) and A. baumannii (n = 1037). MDR strains were checked for PDR status by determining the minimum inhibitory concentration (MIC) against meropenem and colistin sulphate by the agar dilution method (CLSI 2006). A two-dimensional, two-agent (colistin and rifampicin) broth microdilution checkerboard was done to evaluate the synergism of the antibiotics against PDR strains. There were 46 MDR isolates (31 P. aeruginosa and 15 A. baumannii), of which 29 strains (24 P. aeruginosa and 5 A. baumannii) were detected to be PDR (resistant to colistin also). There was no significant effect of the synergism of colistin and rifampicin as tested against 15 PDR strains, so far. The study is ongoing. Clinical cure was observed in 5 cases with infection with PDR strains, where colistin was used in 1 case. Comorbid conditions might have contributed to high fatality (82.7%) in patients with infection with PDR strains. Although colistin and colistin+rifampicin are not adequately tested clinically, the in vitro findings of their effect on PDR strains are not very encouraging, so far. For MDR strains, colistin alone or along with rifampicin is the “last resort” and should be used with caution. Resistance should be monitored globally and locally.
106. Proinflammatory Cytokines Induced Cellular Oxidative Stress and Suppression of NAD(P)H:Quinone Oxidoreductase 1
Benjaporn Buranrat,1 Auemduan Prawan,1 Veerapol Kukongviriyapan,1 and Upa Kukongviriyapan2
1Department of Pharmacology, Faculty of Medicine, and Liver Fluke and Cholangiocarcinoma Research Center, Khon Kaen University, Khon Kaen, Thailand
2Department of Physiology, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand
Proinflammatory cytokines can induce cellular stress with consequences of alteration of drug-metabolizing enzyme activities. NAD(P)H:quinone oxidoreductase 1 (NQO1) is a xenobiotic metabolizing enzyme that plays important roles in biotransformation of quinone compounds and protects cells against cytotxicity, mutagenicity, and carcinogenicity. Expression and activity of NQO1 is activated by several conditions. However, the effect of inflammation on NQO1 in biliary cells is uncertain. We treated two human cholangiocarcinoma (KKU-100, KKU-OCA17 cells) and normal liver Hela Chang cells with cytokine combinations (interferon-γ, interleukin-1β, and tumor necrosis factor-α) for 48 hours. NQO1 activity and NQO1 mRNA expression were assayed by spectrophotometry and semiquantitative polymerase chain reaction, respectively. The basal activity of NQO1 is very high in KKU-100 and KKU-OCA17 and very low in Hela Chang cells. Inflammatory cytokines significantly decreased NQO1 activity in all cell lines. In addition, NQO1 mRNA expression was apparently suppressed by cytokine combination, especially in KKU-OCA17, with a time-dependent manner. The mechanisms of inflammation to suppressed NQO1 activity were explored. Cellular redox status, as measured as reduced glutathione (GSH), was relatively high in CCA cells and remarkably reduced after treatment with cytokine combination. Inflammatory cytokines increased the nitric oxide production approximately 40, 10, and 2 times in KKU-100, KKU-OCA17, and Hela Chang cells, respectively. Moreover, treatment with nitric oxide donor S-nitroso-glutathione induced reduction of the ratios of GSH and GSSG in KKU-100 and KKU-OCA17, but not Hela Chang, cells. Similarly, treatment of S-nitroso-glutathione suppressed NQO1 activity in KKU-100 and KKU-OCA17 but increased NQO1 activity in Hela Chang cells. Our findings show that inflammation induces oxidative stress and subsequently suppresses NQO1, and that may be implicated in inflammation-associated cancer risk.
Acknowledgments
This work was supported by the Thailand Research Fund (TRF), National Science and Technology Development Agency (NSTDA), and Grant-in-aid from Khon Kaen University. BB is supported by a scholarship from the Commission on Higher Education, the Ministry of Education.
107. Toxic Effect and Neuroendocrine Mechanism of Caffeine on Fatal Development in Pregnant Rats
Gai Liang, Jr., Dan Xu, Jr., Wenwen He, Benjian Zhang, and Hui Wang
Department of Pharmacology, Basic Medical School of Wuhan University, Wuhan, China
Caffeine is the most widely consumed xenobiotic in pregnancy, but it has not been consistently documented whether caffeine intake is a risk factor for intrauterine growth retardation (IUGR). Steroidogenic acute regulatory protein (StAR) and cholesterol side-chain cleavage enzyme (P450scc) are key enzymes in the steroidogenesis of the adrenal, which is important for fetal development. The purpose of the present study was to investigate the effect and possible neuroendocrine mechanism of pregnant caffeine exposure on fetal development in rats. Rat dams of caffeine groups were treated with caffeine at the doses of 20, 60, and 180 mg.kg−1.d−1 intragastrically from gestational day (GD) 12 to GD 20. Control groups were treated with tdistilled water of equal volume. The fetal body weights, body and tail lengths, and placenta weights were recorded. Blood samples of fetuses were obtained by decapitation. Blood of all fetuses of one dam were combined. Plasma corticosterone and adrenocorticotrophin (ACTH) levels were measured by ELISA and radioimmunoassay kits, respectively. The mRNA expressions of StAR/P450scc in fetal adrenals were detected by real-time RT-PCR. The results showed that, compared with the control group, caffeine caused significantly dose-dependent decrease in pups’ body weights, placenta weights, and body and tail lengths at GD 20 (P < 0.01). The incidence rate of IUGR in caffeine groups were 26.7, 29.6, and 44.7%, obviously higher than the control group (P < 0.01). Meanwhile, StAR and P450scc mRNA levels in fetal adrenal of caffeine groups were decreased about 10-fold, compared with control (P < 0.05), but the levels of plasma corticosterone were significantly dose-dependent increased (P < 0.01), and the level of plasma ACTH at dose of 180 mg.kg−1.d−1 was obviously higher than the control group (P < 0.01). These results suggested that pregnant caffeine exposure can significantly suppress fetal growth and development and, eventually, result in IUGR. The possible mechanism is that caffeine intake during pregnancy inhibits the steroidogenesis of fetal adrenals and leads to fetal HPA dysfunction and IUGR.
Acknowledgment
This work was supported by the grants from the China Nature Science fund (30830112,30672566, and 30800931).
108. Trigonostemon reidioides Extracts Induced Apoptosis in Mouse P19 Embryonic Carcinoma Cells
Punnee Nusuetrong,1 Boonrat Chantong,2 Pornpen Pramyothin,3 and Duangdeun Meksuriyen4
1Department of Physiology, Faculty of Medicine, Srinakharinwirot University, Bangkok, Thailand
2Faculty of Veterinary Sciences, Mahidol University, Nakornpathom, Thailand
3Department of Pharmacology and Physiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
4Department of Biochemistry and Microbiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
Trigonostemon reidioides Craib (Euphorbiaceae) is a Thai traditional medicine used for the treatment of drug addiction, asthma, food poisoning, constipation, and snake bites (Tempeam et al., 2005). Until now, it has no evidence of the cytotoxicity of T. reidioides in mouse P19 embryonic carcinoma cells, a cell model to evaluate teratogenicity of toxic substances, such as valproic acid Kultima et al., 2004). The aim of this study was to examine whether reactive oxygen species (ROS) was involved in T. reidioides extract-induced apoptotic signal in P19 cells. The cell viability was assessed by MTT reduction assay. Treatment of ethanolic and aqueous extracts of T. reidioides (0.0005–0.5 mg/mL) inhibited cell proliferation in a concentration-dependent manner, with estimated IC50 values of 0.001 and 0.01 mg/mL, respectively. The apoptotic cell numbers increased in a concentration-dependent manner after treatment of T. reidioides extracts ranging from 0.001–0.1 mg/mL for 24 hours, measured by flow-cytometric staining with propidium iodide. Staining with 2,7-dichlorodihydrofluorescein diacetate demonstrated that intracellular ROS were enhanced after treatment of both extracts. These findings suggested that the inhibition of cell proliferation as well as the induction of apoptosis in P19 cells by the extracts may be through the generation of ROS. In addition, the apoptotic induction of the ethanolic extract is more potent than of the aqueous extract. Thus, our findings suggested that the treatment of T. reidioides should not be used by addicted pregnant women.
References
- Kultima, K., Nyström, A.M., Scholz, B., Gustafson, A.L., Dencker, L., Stigson, M. (2004). Valproic acid teratogenicity: a toxicogenomics approach. Environ Health Perspect 112:1225–1235.
- Tempeam, A., Thasana, N., Pavaro, C., Chuakul, W., Siripong, P., Ruchirawat, S. (2005). A new cytotoxic daphnane diterpenoid, rediocide G, from Trigonostemon reidioides. Chem Pharm Bull (Tokyo) 53:1321–1323.
109. Curcumin Inhibits Cholangiocarcinoma Cell Growth by Modulation of the Cellular Redox Status
Bunliang Suphim,1 Auemduan Prawan,1 Upa Kukongviriyapan,2 and Veerapol Kukongviriyapan1
1Department of Pharmacology, Khon Kaen University, Faculty of Medicine, Liver Fluke and Cholangiocarcinoma Research Center, Khon Kaen, Thailand
2Department of Physiology, Khon Kaen University, Faculty of Medicine, Khon Kaen, Thailand
Curcumin, a major yellow pigment and active component of turmeric (Curcuma longa Linn.), exhibits antioxidant activity and chemoprevention. In this study, we investigated the effect of curcumin on induction of cell killing on cholangiocarcinoma cells, which are derived from the malignant tumors usually resistant to most chemotherapeutic agents. KKU-M214 cells, a cholangiocarcinoma cell line (CCA), were treated with curcumin (1–100 μM) for 0–48 hours. The cell viability was determined under a fluorescence microscope by using acridine orange and ethidium bromide staining. The glutathione was assayed by the enzymatic method. The protein expressions of transcription factor, nuclear factor-kappaB (NF-κB), cyclin D1, and Bcl-xL were determined, at varied time periods, by Western immunoblotting. Curcumin treatment resulted in an antiproliferative effect on KKU-M214 cells at low concentrations (1–3 μM), whereas induction of apoptotic and necrotic cell death were observed at higher concentrations (10–100 μM). The translocation of NF-κB into the nuclear compartment was unchanged during the course of treatment, whereas Bcl-xL and cyclin D1 were decreased during the same period. Cellular redox ratios of GSH and GSSG were initially increased and markedly decreased afterward. The changes in redox status were correlated with induction of apoptosis and necrotic cell death. It is suggested that curcumin inhibits cell growth and induces apoptosis of CCA cells, probably via modulation of the cellular redox status. In conclusion, curcumin demonstrates a possible therapeutic potential in cholangiocarcinoma.
Acknowledgment
This work was supported by Grant-in-aid from Khon Kaen University and Thailand Research Fund (TRF).
110. Suppression Mechanism by Polycyclic Aroma Carbons in Malondialdehyde-modifided Low-Density Lipoprotein-induced Cell Growth
Hiroyuki Suzuki,1 Takeshi Kumagai,1 and Kiyoshi Nagata2
1Pharmacy, Tohoku Pharmaceutical University, Sendai, Japan
2Tohoku Pharmaceutical University, Sendai, Japan
Polycyclic aromatic carbons (PAHs) are common environmental pollutants. Exposure to air pollutants with PAHs has been suspected to be associated with the occurrence of pulmonary diseases. Benzo[a]pyrene (BaP), a carcinogenic model compound of PAHs found in cigarette smoke and air pollutants, has been extensively studied. BaP is a potent inducer for CYP1A1, which plays important roles in the genotoxicity of BaP. Metabolic activation of BaP by CYP1A1 is required to cause its toxic, mutagenic, and carcinogenic effects. Further, PAHs have also been reported to induce atherosclerosis.Generally, pathogenesis of atherosclerosis has been accelerated through malondialdehyde-modifided low-density lipoprotein (MDA-LDL) and oxidized LDL. MDA-LDL was reported to be involved in atherogenesis causing smooth muscle cell proliferation, which may be generated by the uptake of MDA-LDL by macrophages via scavenger receptors. However, the association between PAHs and MDA-LDL, which have also been reported to induce atherosclerosis, has not been established. Recently, we found that PAHs suppressed MDA-LDL-induced human acute monocyte leukemia suspension cell line (THP-1 cell) growth. In this study, we investigated the suppression mechanism by PAHs in MDA-LDL-induced THP-1 cell growth. Cotreatment with BaP or 3-methylchoranthrene decreased the MDA-LDL-induced THP-1 cell growth, but, benzo[e]pyrene or pyrene, that is not a ligand for the arylhydrocarbon receptor (AhR), did not decrease. The suppression of THP-1 cell growth was clearly restored by the addition of α-naphtoflavone, which is a partial antagonist to AhR. Cotreatment with MDA-LDL and BaP markedly induced the expression of CYP1A1 mRNA. These results predicted that activated BaP caused DNA damage and, consequently, increased tumor suppressor protein p53. As a result, the proliferation inhibitor protein, p21, might be induced. As expected, cotreatment with BaP and MDA-LDL significantly induced expressions of p53 and p21. These findings strongly suggest that the MDA-LDL-induced THP-1 cell growth is suppressed by the induction of p21 through DNA damage caused by activated metabolites of PAHs. In this presentation, the interplay between PAHs and MDA-LDL, which are risk factors for atherosclerosis, are discussed.
111. Placental Dysfunctions in Intrauterine Growth Retardation-induced by Smoking and Alcohol Drinking and Effect of Sodium Ferulate
Youe Yan,Yan LI, and Hui Wang
Department of Pharmacology, Basic Medical College of Wuhan University, Wuhan, China
The aim of this study was to explore the placental dysfunction mechanism of intrauterine growth retardation (IUGR) induced by smoking and alcohol drinking, and to validate the effect of sodium ferulate (SF) on it. Pregnant rats were divided into four groups: normal control group, IUGR control group, and SF (25, 50 mg•kg−1) groups. The IUGR model was produced by passive smoking and alcohol drinking. SF was given by intragastric administration. Fetal developmental parameters (body weight, body length, etc.) were measured. Placental weight and pathological changes were examined. The contents of malondialdehyde (MDA) and glutathione (GSH), and the activities of superoxide dismutase (SOD) and catalase (Cat) in placentas were assayed. Mdr1a and mdr1b levels of the placenta were examined by reverse-transcriptase polymerase chain reaction. Results showed that intrauterine tobacco and alcohol exposure had resulted in significant reduced postnatal body and organ weights (P < 0.01, P < 0.05). Placental weights in the IUGR control group were lower than those in the normal one (P < 0.01). Obvious damages occurred in placenta of the IUGR control group, such as interstitial and endovascular hemorrhage, villi edema, and swollen. Further, the contents of MDA and GSH were higher in the IUGR control group than those in the normal one (P < 0.05). Meanwhile, compared to the normal control group, the activities of SOD and Cat were lower in the IUGR control group (P < 0.01, P < 0.05). In addition, the mdr1a level remarkably decreased (P < 0.01), and the level of mdr1b remained stable in the IUGR control group. By SF treatment, fetal developmental parameters and placental weight were near to those in the normal control. The pathological changes were returned to or near normal. SF significantly decreased MDA and GSH contents, and increased SOD and Cat activities, compared to the IUGR control group (P < 0.05). SF induced mdr1a expression (P < 0.01) and was unchanged in the level of mdr1b, compared to the IUGR control group. Our study indicated IUGR may be related to placental dysfunctions by smoking and alcohol drinking. SF may be effective for treating IUGR, in which its mechanism may be due to enhanced antioxidative and transport functions of the placentas.
Acknowledgment
This work was supported by grants from the National Nature Science Foundation of China (nos. 30830112 and 30672566).
112. Metabolic Activation of Benzodiazepines by CYP3A4
Katsuhiko Mizuno, Miki Katoh, Hirotoshi Okumura, Nao Nakagawa, Miki Nakajima, and Tsuyoshi Yokoi
Department of Drug Metabolism and Toxicology, Kanazawa University, Kanazawa, Japan
Cytochrome P450 (CYP) 3A4 is the predominant isoform in liver and metabolizes more than 50% of the clinical drugs commonly used. However, CYP3A4 is also responsible for the metabolic activation of drugs leading to liver injury. An in vitro cell-based assay made by combining recombinant CYP3A4 with HepG2 cells was applied in the present study to screen for hepatotoxicity by drugs. Benzodiazepines are widely used for anxiety as hypnotics and sedatives, but some of them induce liver injury in humans. To clarify whether benzodiazepines are metabolically activated, 14 benzodiazepines were investigated for their cytotoxic effects on HepG2 cells treated with recombinant CYP3A4. By exposure to 100 μM of flunitrazepam, nimetazepam, or nitrazepam, the cell viability in the presence of CYP3A4 decreased more than 25%, as compared with that of the control. In contrast, in the case of other benzodiazepines, the changes in the cell viability between CYP3A4 and control supersomes were less than 10%. These results suggested that nitrobenzodiazepines, such as flunitrazepam, nimetazepam, and nitrazepam, were metabolically activated by CYP3A4, which resulted in cytotoxicity. To identify the reactive metabolite, the glutathione adducts of flunitrazepam and nimetazepam were investigated by liquid chromatography-tandem mass spectrometry. The structural analysis for the glutathione adducts of flunitrazepam indicated that a nitrogen atom in the side chain of flunitrazepam was conjugated with the thiol of glutathione. Therefore, the presence of a nitro group in the side chain of benzodiazepines may play a crucial rule in the metabolic activation by CYP3A4. The present study suggested that metabolic activation by CYP3A4 was one of the mechanisms of liver injury by nitrobenzodiazepines. The present assay system was useful to detect metabolic activation by CYPs and would be beneficial to predict drug-induced cytotoxicity in preclinical drug development.
Reference
- Mizuno, K., Katoh, M., Okumura, H., Nakagawa, N., Negishi, T., Hashizume, T., et al. (2009). Mitabolic activation of benzodiazepines by CYP3A4. Drug Metab Dispos In press.
113. Abstract Withdrawn
114. Intelligent Workflows (MSM) for Metabolite Screening and Characterization Using an LTQ Orbitrap
Yingying Huang,1 Ji Ma,2 Jae Schwartz,1 Robert Cho,2 Yan Chen,1 and Tim Calson2
1Thermo Fisher Scientific, San Jose, California, USA
2Department of PKDM, Amgen, South San Francisco, California, USA
An integral part of drug discovery and development is the identification of drug metabolites that indicate an intrinsic pharmacokinetic mechanism, pharmacological activity, or specific toxicity. In this work, we describe the use of MSM, which utilizes multiple collision cells, dissociation methods, scan modes, mass analyzers, and detectors to perform intelligent metabolite identification experiments. A modified LTQ Orbitrap XL (Orbitrap, Thermo Fisher Scientific, Waltham, MA, USA) with an HCD (higher-energy collisional dissociation) collision cell was used. An isolation mass window up to 600 amu for HCD scans was enabled. In this experiment, a high-resolution full scan was acquired, followed by a high-resolution HCD MS/MS of all incoming ions within that 600-amu window. In parallel, the linear ion trap (LIT) acquired data-dependent MSn spectra. Accela ultra-performance liquid chromatography (UPLC) and a Hypersil Gold C18 column (Hypersil column, Thermo Fisher Scientific, Waltham, MA, USA) (1 × 100 mm, 1.9 μm) with a 20-minute gradient was used. Next, 10 and 1 μM of rat and human hepatocytes incubation samples from verapamil and haloperidol were analyzed. The performance of MSM was also evaluated by using in vivo (1 mg/kg intravenously) rat urine, plasma, and bile samples. The HCD cell can excite a large number of precursors simultaneously, and fragmentation information for all ions within the 600-amu window was acquired. By comparing the data from HCD with the full-scan MS, it is possible to mimic conventional neutral loss scanning, precursor ion scanning, and multiple reaction monitoring experiments. This was achieved by data mining, using accurate mass and high resolution. Once potential metabolites were identified through using such comparisons, the parallel data-dependent CID MSn data allowed an unambiguous structure interpretation. Using m/z 165.0910, a diagnostic fragment ion of verapamil, for example, an accurate mass-precursor ion analysis of a 10-μM rat hepatocytes sample led to the identification of more than 16 putative metabolites. Definitive structural elucidation of these metabolites was achieved by analysis of the data-dependent LIT CID MS2 and MS3 data that were collected in parallel. Analysis of 1-μM rat hepatocytes sample, using the same approach, also led to the identification of all phase I and II metabolites.
115. Aromatic Hydroxylation and Catechol Formation: a Novel Metabolic Pathway of the Mycotoxin, Zearalenone, and the Growth Promotor, Zeranol
Manfred Metzler, Erika Pfeiffer, Andreas Hildebrand, and Georg Damm
Chemistry and Biosciences, Karlsruhe Institute of Technology (KIT), Karlsruhe, Germany
The Fusarium toxin, zearalenone (ZEN), and its semisynthetic congener, zeranol (alpha-zearalanol; ZAL), which is used as a growth promotor in cattle, are macrocyclic resorcylic acid lactones with considerable estrogenic activity. Although both compounds have two positions for metabolic hydroxylation at the aromatic ring, no such metabolites have been reported, to date. In order to probe for aromatic hydroxylation of ZEN and ZAL, both compounds were labeled with deuterium at the aromatic ring. When 13,15-D2-ZEN or 13,15-D2-ZAL were incubated with human liver microsomes, two of the major metabolites were products of monohydroxylation, which, according to LC-MS and GC-MS analysis, had lost one deuterium. This is clear evidence for the formation of the 13-hydroxy- and 15-hydroxy-metabolites of ZEN and ZAL. As the parent ZEN and ZAL carry hydroxyl groups at C-14 and C-16, metabolic hydroxylation at C-13 or C-15 leads to catechols. In order to confirm their catechol structure, the novel metabolites were isolated by high-performance liquid chromatography (HPLC) and incubated with catechol-O-methyl transferase in the presence of S-adenosyl methionine. In agreement with the proposed structures, each of the catechol metabolites of ZEN and ZAL gave rise to two monomethylated products. Further structural confirmation of the catechol metabolites of ZEN was obtained when reference compounds were synthesized: Nitration of ZEN yielded two mononitro-ZENs, which were purified by preparative HPLC. Analysis by nuclear magnetic resonance clearly showed that one compound was 13-nitro-ZEN and the other one was 15-nitro-ZEN. Subsequent reduction to the respective amines, oxidation to quinone imides, hydrolysis to quinines, and reduction of the quinones gave rise to 13-hydroxy-ZEN and 15-hydroxy-ZEN, which were identical with the catechol metabolites of ZEN in LC-MS and GC-MS analysis. This is the first study to show that catechol formation represents the major pathway in the oxidative metabolism of ZEN and ZAL in human liver microsomes in vitro. The toxicological sequelae of this pathway and its in vivo relevance should now be studied.
Acknowledgment
This work was supported by Deutsche Forschungsgemeinschaft (Grant ME 574/32-1) and “Food and Health” of KIT.
116. Pharmaceutical Metabolite Profiling Using Quadrupole/ Ion Mobility Spectrometry/ Time-of-Flight Mass Spectrometry
Eric Chun Yong Chan,1 Lee Sun New,1 Chun Wei Yap,1 and Lin Tang Goh2
1Department of Pharmacy, National University of Singapore, Singapore, Singapore
2Waters Asia Limited, Singapore, Singapore
Metabolite profiling plays an increasingly important role in drug development as a large proportion of drug candidates failed due to poor pharmacokinetic properties (e.g., poor metabolic stability) and toxicity (e.g., reactive metabolite). Recently, ion mobility spectrometry time-of-flight mass spectrometry (IMS/TOFMS) had been explored by several research groups for the analysis of pharmaceuticals and excipients. However, the use of this technique in metabolite profiling has not been reported. In this work, the use of hybrid quadrupole ion mobility spectrometry time-of-flight mass spectrometry (Q/IMS/TOFMS) in the metabolite profiling of leflunomide (LEF) and acetaminophen (APAP) is presented. The IMS drift times (Td) of the drugs and their metabolites were determined in the IMS/TOFMS experiments and correlated with their exact monoisotopic masses and other in silico generated structural properties, such as Connolly molecular area (CMA), Connolly-solvent excluded volume (CSEV), principal moments of inertia along the X, Y, and Z Cartesian coordinates (MI-X, MI-Y, and MI-Z), inverse mobility, and collision cross-section (CCS). The correlation of Td with these parameters were presented and discussed. IMS/TOF tandem mass spectrometry experiments (MS2 and MS3) were also performed on N-acetyl-p-benzoquinoneimine glutathione (NAPQI-GSH) adduct derived from the in vitro microsomal metabolism of APAP. As a comparison, similar experiments were also performed by using the hybrid triple quadrupole linear ion trap mass spectrometer (QTRAPMS) and quadrupole time-of-flight mass spectrometer (QTOFMS). Our results indicated that the isobaric and structurally similar metabolites, such as LEF and A77-1726 or M1 and M2, were not resolved by using IMS. This inherent challenge of IMS in resolving isobaric and structurally related ions needs to be further explored and improved. The successful IMS/TOF MS2 and MS3 experiments performed on NAPQI-GSH were also presented. The ability to perform the MS3 experiment effectively on all product ions in a single analysis demonstrated the potential applicability of the IMS/TOF MS3 method in pharmaceutical metabolite profiling.
117. In Vitro Metabolism of Alicyclic Ketoxime to Ketone by Rabbit Liver Preparations
Hideo Kurebayashi
Division of Pharmacological/Biological Safety Research Center, NIHS, Tokyo, Japan
N-oxidation is an important route for in vitro and in vivo metabolism of primary and secondary amines. N-oxidation of aromatic primary amines and amides, such as 2-naphthylamine, are known as the metabolic activation of ultimate carcinogens. In spite of numerous publications describing the aromatic amines, there is very little information on the hydroxylation of aliphatic amines. Cyclohexylhydroxylamine (CHHYA) was detected in urine following cyclamate administration to monkeys, dogs, and men. CHHYA arose from an incubation mixture of cyclohexylamine with rabbit liver microsomes. Phenylacetone oxime was detected at incubation of N-hydroxylamphetamine with a hepatic 9,000-g supernatant fraction (S9) following incubation of amphetamine. Then, it is likely that cyclohexanone oxime is formed from CHHYA. Phenylacetone was detected at an incubation mixture of phenylacetone oxime with S9. It is likely that cyclohexanone is formed from cyclohexanone oxime, but no tissue site has yet been identified for these reactions. The oxime of alicyclic ketone was incubated with the hepatic preparations of cytosol, microsomes, and a 9,000-g supernatant fraction (S9) for 30 minutes. The ether extraction from the incubation was injected onto the gas-chromatograph for quantification. The stability of alicyclic ketoxime depends upon structure. Although the autodecay of alicyclic ketoxime to alicyclic ketone occurred, in some degree, the rate of enzymatic ketone formation from the oxime was in the descending order: the oximes of cycloheptanone, cyclohexanone, cyclooctanone, and cyclopentanone. The rate of enzymatic ketone formation by microsomal fraction was faster with NADPH or NADH, and under anaerobic conditions. The rate of enzymatic ketone formation by cytosol fraction was faster with 2-hydroxypyrimidine, and under anaerobic conditions. The mechanism of ketone formation from the alicyclic ketoxime seemed to be the enzymatic reduction by both fractions.
118. Detrimental Effect of Prenatal Exposure to Cyclophosphamide on Ovarian Folliculogenesis in Charles Foster Rat
Biswabina Ray and Bhagath Kumar
Anatomy, Kasturba Medical College, Manipal, India
Aim: pollutants such as petroleum fumes, insecticides, pesticides, plastics, and insulators have adverse effects on human ovary, leading to infertility. The metabolically active intermediate of cyclophosphamide (i.e., acrolein and phosphoramide mustard) are present in those pollutants. Cyclophosphamide is a known teratogen, the effect of which on developing organs indirectly gives a clue to the hindering effect of its metabolites, hence the environmental pollutants mentioned above. We hypothesize that cyclophosphamide hinders the embryogenesis of female gonad by interfering with ovarian folliculogenesis. The aim of this study was to observe the effect of cyclophosphamide on ovarian folliculogenesis in the embryonal stage of the rat.
Methods: In this experiment, pregnant rats (n = 12) were randomly assigned into two groups: the control (n = 6) and cyclophosphamide treatment groups (n = 6). The animals in the cyclophosphamide treatment group were injected with cyclophosphamide intraperitoneally from day 10 of gestation, at 2 mg/kg of body weight per dose. The pregnant rats were sacrificed on gestation day 20 and we collected the fetus. The collected fetuses were processed for sectioning and stained with hematoxyline and eosin for microscopical observation of the ovary.
Results: A meshwork-like appearance of mesenchyme with a lesser number of somatic cells and an absence of most of the germ cells in the ovarian follicles were found in the treated fetus. Nonavailability of primordial germ cells stopped the interaction between primordial germ cells and somatic supporting cells, leading to nonproliferation and degeneration of somatic cells and fluid-filled vacant spaces in between the meshwork -like arrangement of mesenchymal cells.
Conclusions: We conclude that cyclophosphamide exposure prevents folliculogenesis causing anovulation and resulting in infertility. The same detrimental effect might be seen on human fertility with environmental pollutants, which are also metabolites of the drug.
119. Intestinal Transport and Glucuronidation Studies on Flavonoid Scutellarein in Caco-2 Cell Monolayer and Human Intestine/Liver Microsomes
Yazhi Wang, Yitao Wang, and Ying Zheng
Institute of Chinese Medical Sciences, University of Macau, Macau SAR, China
Scutellarin (SG), which belongs to flavonoid glycosides, is a well-known Chinese medicine to treat cardiovascular disease. Previous studies suggested that SG needs to be hydrolyzed into its aglycone scutellarein (S) by bacterial enzymes before being absorbed in vivo. A new monoglucuronidation conjugate (iso-scutellarin) was found in human plasma after the oral administration of SG. To better elucidate above in-vivo findings, the present study aimed to elucidate the intestinal transport mechanism as well as the role of glucuronidation of S in both intestine and liver in vitro. The bi-directional transports of S were studied in the Caco-2 cell monolayer and quantified by HPLC/UV. Glucuronidation was investigated by incubation S with human liver microsome (HLM) and human intestine microsome (HIM). The generated metabolites were identified by LC/MS/MS and quantified by HPLC/UV. Metabolic kinetics parameters, including Km and Vmax, were obtained by fitting the data to the typical Michaelis-Menten equation. S has a high apparent permeability coefficient (Papp) from both directions without significant efflux [1.27 ± 0.0915 × 10−5 cm/s (AP to BL); 1.58 ± 0.0932 × 10−5 cm/s (BL to AP)], suggesting that S is mainly transported via the transcellular pathway by passive diffusion. A new metabolite, which has the same molecular weight but different retention time as that of SG, was detected from the bi-direction transport studies on the Caco-2 monolayer. It was preferred to be transported into the apical side. The major glucuronidation metabolite in HLM and HIM was identified as the SG. The Vmax and the Km of glucuronidation of scutellarein in HLM is 0.8471 ± 0.05099 nmol/min/mg and 6.098 ± 1.556 μM, respectively. In summary, S could be readily passed across the Caco-2 cell monolayer by passive diffusion and extensively metabolized into the monoglucuronidation conjugate in human intestine and liver.
Acknowledgment
This project was supported by the Macao Science and Technology Development Fund (008/2007/A1).
120. Potential Targeting Region for the Specific Inhibition of Human Monomeric Carbonyl Reductases of the Short-Chain Dehydrogenase/Reductase Superfamily
Takeshi Miura, Toru Nishinaka, and Tomoyuki Terada
Laboratory of Biochemistry, Faculty of Pharmacy, Osaka Ohtani University, Tondabayashi, Osaka, Japan
Human monomeric carbonyl reductases 1 (CBR1, SDR21C1) and 3 (CBR3, SDR21C2) are phase I enzymes, which catalyze the reduction of many endogenous and xenobiotic carbonyl compounds by using NADPH as a coenzyme. Althouh the two enzymes are highly homologous (79% homology and 72% identity) in their amino-acid sequences, reductase activities of CBR1 are much higher than those of CBR3. CBR1 physically metabolizes daunorubicin, and the metabolite is believed to cause congestive heart failure. CBR1-specific inhibitors would serve as useful adjuncts to daunorubicin therapy by protecting against heart failure and enhancing the antitumor activity of daunorubicin. In the present study, we investigated the structural origin of the functional differences between CBR1 and CBR3, and on the basis of these differences, we searched CBR1-specific inhibitors. First, we carried out a Harr plot analysis of the amino-acid sequences of CBR1 and CBR3 in order to identify the structural origin of the functional differences. The results showed that the amino-acid sequence identity between the two enzymes was low at the region near catalytic residue Ser-139 (N-terminal low-identity region; LirN) and the substrate-binding region (C-terminal low-identity region; LirC). Kinetic studies of several chimeric enzymes have revealed that LirC is responsible for the unique enzymatic properties of CBR. Thus, this region appears to be a potential target for specific enzymatic inhibition. Flavonoids are reported to inhibit the catalytic activity of CBR1 by binding to the substrate-binding loop and substrate-binding cleft and may thus include a candidate CBR1-specific inhibitor that does not inhibit CBR3. Because clinically applied flavonoids are known to be safe for humans, we tested two such flavonoids, namely, ipriflavone and flavoxate, for their inhibitory effect against CBR1 in vitro. The results revealed that both flavonoids weakly inhibited CBR1. In conclusion, the substrate-binding region of CBR is responsible for the functional differences between CBR1 and CBR3, and flavonoids that bind to this region may serve as CBR1-specific inhibitors.
121. pH-Dependence of Mouse and Human Flavin-containing Monooxygenase 5
Meike Motika, Xueying Zheng, Jun Zhang, Kirstin Riedler, and John R. Cashman
Human BioMolecular Research Institute, San Diego, California, USA
Microsomal NADPH-dependent flavin-containing monooxygenases (FMOs) are a family of drug-metabolizing enzymes that catalyze the oxygenation of a wide variety of nucleophilic xenobiotics, drugs, and endogenous chemicals. In humans, five isozymes (i.e., FMO1–FMO5) have been described. FMO5 is a prominent FMO form, but, to date, only a few substrates are known. This is surprising, because FMO5 expresses a functional enzyme in high yield in several mammalian tissues, including humans, and has the highest mRNA levels of all FMOs in the main drug-metabolizing organ, the liver. A comparison of FMOs showed the pH-dependent activity profile of FMO5 differed significantly from that of other FMO enzyme forms. The optimum pH range of FMO1 and 3 was between pH 8 and 10, respectively. For FMO5, the prototypical substrate 10-(N,N-dimethylamino octyl)-2-trifluoromethyl)phenothiazine (8-DPT) N-oxygenation activity continued to increase from pH 7 to 11. The objective of this study was to examine the pH dependence of FMO5 to gain insight into the mechanism of action of FMO5 enzymes. Functional recombinant mouse and human FMO5 (i.e., mFMO5 and hFMO5) were expressed as maltose-binding fusion proteins (MBP-mFMO5 and MBP-hFMO5) from Escherichia coli, purified with affinity chromatography and examined with 8-DPT for their N-oxygenation functional activity at different pH values. Human FMO5 showed a broader range and greater functional activity from pH 6 to 11, compared to mFMO5. Mouse FMO5 lost almost all functional activity at pH 6, while hFMO5 maintained normal enzyme activity. This led to the preliminary conclusion that hFMO5 was more stable under slightly acidic conditions. In order to identify the region of amino-acid residues involved in the effects of pH on hFMO5 and mFMO5 functional enzyme activity, pH-studies in the range of pH 6 through 11 were done with chimeras of recombinant mouse and human MBP-FMO5 fusion proteins and variants of mouse and human MBP-FMO5. The results of these studies will help explain the mechanism of FMO function.
Acknowledgment
This study was supported by NIH grant DK59618.
122. Functional Consequences of OATP1B3 SNPs in Korean Population
Jung-Byung Chae, Kyung-Ha Yu, Kyeong-Ryoon Lee, Young-Sun Choi, Dae-Duk Kim, Chang-Koo Shim, and Suk-Jae Chung
College of Pharmacy, Seoul National University, Seoul, South Korea
The human organic anion transporting polypeptide, 1B3 (OATP1B3, also known as OATP8), is expressed specifically in the liver and is thought to be responsible for transporting a variety of endo-/enxo-genous chemicals into the liver. However, the pharmacogenetic significance for the anion transporter has not been fully delineated in the literature. Therefore, the objectives of the study were to screen OATP1B3 single-nucleotide polymorphisms (SNPs) in the Korean population and to determine the functional consequences. SNPs of the anion transporter were first scanned in samples from 48 Korean subjects by sequencing. Seven SNPs of OATP1B3 were found to be located in the exon region. A statistically significant linkage disequilibrium was found among certain variants. In particular, two novel SNPs were newly identified in the present study. To determine whether the variant forms have the functional relevance, OATP1B3 was cloned from the human liver mRNA library, and the variant genes were generated by using site-directed mutagenesis. OATP1B3 wild type and variants were expressed in Xenopus laevis oocytes and HEK293/FRT cells and the functional activities were studied by using estrone-3-sulfate, digoxin, and methotrexate as substrates. The transport study revealed that certain variants exhibited markedly reduced uptake of the substrates, indicating that the functional consequence of these SNPs is substrate specific. The newly identified SNP exhibited no difference in the function. In the promoter region, eight SNPs were apparently located. In addition, statically significant linkage disequilibrium was found among certain variants. In particular, two novel SNPs were newly identified in the present study. To determine the functional relevance of the variant forms, the promoter region of OATP1B3 was cloned from the human genomic DNA library, and the variant genes were generated by using site-directed mutagenesis. The promoter region of OATP1B3 wild type/variants were transfected into HepG2 cells and the functional activities were determined by using the reporter gene assay. The study revealed that a newly identified SNP exhibited markedly reduced expression of the reporter gene. Therefore, these observations indicate that certain variants of OATP1B3 have functional consequence and the polymorphism is clinically relevant.
123. Influence of PXR Haplotype Variants on Paclitaxel and Docetaxel Pharmacokinetics and Pharmacodynamics in Asian Cancer Patients
Balram Chowbay,1 Edwin Sandanaraj,1 Suman Lal,1 Wang Zhenping,1 Lim Wan Teck,2 Ang Cher Siang,2 Edmund Lee,3 and Tan Eng Huat2
1Clinical Pharmacology Laboratory, Division of Medical Sciences, National Cancer Center, Singapore, Singapore
2Department of Medical Oncology, National Cancer Center, Singapore, Singapore
3Department of Pharmacology, National University of Singapore, Singapore, Singapore
Background: Paclitaxel is primarily metabolized by CYP3A4 and CYP2C8, while docetaxel is mainly metabolized by CYP3A4/5. Both paclitaxel and docetaxel are effluxed by ABCB1. CYP3A4/5, CYP2C8, and ABCB1 are also downstream targets of the pregnane × receptor (PXR) gene. The objective of this exploratory study was to investigate the influence of PXR genetic variants on the pharmacokinetics of paclitaxel and docetaxel in Asian cancer patients.
Materials and Methods: A total of 25 Asian cancer patients receiving intravenous infusions of paclitaxel either as a weekly (80 mg/m2; N = 11) or three weekly (170 mg/m2; N = 14) dosage regimens or a weekly docetaxel regimen of 30 mg/m2 (N = 46) were recruited. Pharmacogenetic and pharmacokinetic data were available for all the patients. Paclitaxel and docetaxel pharmacokinetic parameters were estimated by using noncompartmental analysis (WinNonlin) and the Mann-Whitney U test was used to assess genotypic-phenotypic correlations.
Results: Two main PXR haplotype groups were identified: the PXR*1B and non-PXR*1B haplotype groups. The PXR*1B haplotype group was tagged by the IVS6-17C>T and 2654T>C SNPs. Patients harboring the PXR*1B haplotypic constitution had significantly lower clearance [CL/dose (mL*h−1*mg−1); median: 94.0; range: 45.3–207.2] and significantly higher exposure levels of paclitaxel [AUC0-∞/dose (hr*μg*mL−1); median: 52.8; range: 35.5–89.0], compared to patients belonging to the non-PXR*1B haplotype group [CL/dose (mLh−1*mg−1); median: 229.50; range: 65.5–624.3; P = 0.03) and AUC0-∞/dose (hr*μg*mL−1); median: 27.2; range: 12.5–53.9; P = 0.007), respectively]. Patients carrying the PXR*1B haplotype group also had significantly higher Cmax levels of paclitaxel [Cmax/dose (μg*mL−1); median: 17.6; range: 12.1–40.9], compared to patients belonging to the non-PXR*1B [Cmax/dose (μg*mL−1); median: 11.03; range: 1.3–22.5; P = 0.03] haplotype group. Pharmacodynamic analysis revealed that patients carrying the PXR*1B haplotypic constitution had 2.1- and 1.7-fold lower absolute neutrophil counts and platelet counts, when compared to the patients bearing the non-PXR*1B haplotypic constitution. No significant genotypic effects were found between PXR haplotypes and docetaxel pharmacokinetics.
Conclusions: This exploratory study suggests that PXR haplotype constitution may be important in influencing interindividual variations in the disposition of paclitaxel but not docetaxel.
124. Carbamazepine—the Most Common Cause of Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis and an Association with HLA-B*1502 in a Thai Population
Thawinee Jantararoungtong,1 Somsak Tiamkao,2 Suda Vannaprasaht,3 Kasemsin Pavakul,4 Narong Auvichayapat,5 Charoen Choonhakarn,6 and Wichittra Tassaneeyakul1
1Department of Pharmacology, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand
2Department of Medicine, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand
3Department of Pharmacology, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand
4Khon Kaen Hospital, Khon Kaen, Thailand
5Department of Paediatrics, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand
6Department of Medicine, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand
Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are potentially life-threatening drug-induced cutaneous reactions. The drugs commonly implicated as the cause of these severe reactions vary, depending on host factors and the drug prescription pattern in that particular area. The aim of the present study was to explore the epidemiology of SJS and TEN as well as an association between carbamazepine-induced SJS/TEN and HLA-B*1502 in a Thai population. The case records of all patients with a clinical diagnosis of SJS or TEN admitted in Srinagarind Hospital and Khon Kaen Hospital, Thailand (between 1995 and 2008) were studied in detail regarding the drugs implicated as the cause, the management, and the clinical outcome. A total of 216 cases of SJS and 54 cases of TEN were identified. The mean age of the patients was 38.67 ± 20.45 years. About 77.04% of cases were possibly caused by drugs. The culprit drugs were anticonvulsants (35.58%), antibiotics (35.10%), antigout (9.61%), antituberculosis (7.21%), and nonsteroidal anti-inflammatory drugs (5.29%). In addition, the common drugs implicated as the cause of SJS/TEN was carbamazepine (20.67%), sulfonamides (19.23%), and allopurinol (9.62%). The time from the first exposure to the culprit drugs to the onset of SJS and TEN was 9.66 ± 10.01 days. Length of hospital stay was 9.13 ± 9.24 days (n = 140). The mortality rate of SJS was 1.36% (2/147) and TEN was 2.04% (3/147). In addition, the strong association between carbamazepine-induced SJS/TEN and HLA-B*1502 in a Thai population was noted.
125. Polymorphisms in the Promoter Region of UGT1A9 Gene in Thais
Porntipa Korprasertthaworn1, Krongtong Yoovathaworn,2 and Wandee Udomuksorn3
1Department of Pharmacology, Faculty of Science, Mahidol University, Bangkok, Thailand
2Department of Pharmacology and Multidisciplinary Unit, Faculty of Science, Mahidol University, Bangkok, Thailand
3Department of Pharmacology, Faculty of Science, Prince of Songkla University, Songkhla, Thailand
The human UDP-glucuronosyltransferase, UGT1A9, catalyzes glucuronidation of various endobiotics and xenobiotics. Genetic polymorphisms of UGT1A9 can influence detoxifying capacities and have considerable effect on the metabolisms of numerous drugs. The purpose of this study was to investigate the polymorphisms in the 5’-flanking region of UGT1A9 gene in the Thai population. Genomic DNA from 93 healthy unrelated volunteers was amplified by tpolymerase chain reaction (PCR), and DNA sequencing was performed to determine mutations. There was a novel single- nucleotide polymorphism (SNP) in the UGT1A9 promoter region, heterozygous −688A>C, with a frequency of 0.2473. In addition, three known polymorphisms were found: −440T>C, −331C>T, and one base insertion of thymidine resulting in −118A(T)10AT (UGT1A9*1b), with frequencies of 0.9785, 0.9677, and 0.5323, respectively. In conclusion, this is the first study to demonstrate the genetic variations in the promoter region of the UGT1A9 gene in the Thai population.
126. Genetic Polymorphism of NAD(P)H-Quinone Oxidoreductase-1 in Thais and Its Association with Cholangiocarcinoma
Pornsin Zeekpudsa, Auemduan Prawan, Veerapol Kukongviriyapan, and Vajarabhongsa Bhudhisawasdi
Department of Pharmacology, Faculty of Medicine, Liver Fluke and Cholangiocarcinoma Research Center, Khon Kaen University, Khon Kaen, Thailand
NAD(P)H-quinone oxidoreductase-1 (NQO1) is a detoxifying/antioxidant enzyme that plays a critical role in cellular defense against reactive oxygen species and toxic quinone derivatives, which, in turn, confer cytoprotection, inhibition of mutation, and carcinogenesis. The gene coding for NQO1 has a genetic polymorphism (C→T) at nucleotide position 609 (amino-acid codon 187) of the NQO1 cDNA. This mutation has been associated with a decreased enzymatic activity and increased risk of chemically induced cytoxicity and susceptibility to various forms of cancer. However, the role of NQO1 polymorphism in relation to carcinogenesis of cholangiocarcinoma (CCA), the most common liver cancer in the Northeast of Thailand, is unknown. In the present study, we genotyped the NQO1 C609T polymorphism by PCR-RFLP in 210 CCA patients and 189 healthy control subjects matched for age, sex, and ethnicity. Among 189 Northeastern Thai healthy controls investigated, the NQO1 609T was present in 44% and the frequency distributions of C/C, C/T, and T/T genotype were 32, 53, and 15%, respectively. T-allele frequency in the CCA patients was comparable with the healthy controls (P = 0.4). In an analysis for the association of the NQO1 genotype and survival time of the CCA patients at the time of diagnosis, the variant NQO1 genotypes (C/T and T/T) were not associated with lower survival time, when compared with the C/C genotype (P = 0.092). Our findings suggested that the NQO1 C609T polymorphism may not directly represent a genetic risk factor for CCA; however, the present study cannot exclude NQO1 as a possible modifier for CCA development. Further study in a larger population and biological function of NQO1 gene is required to verify the role of NQO1 in CCA.
127. CNS Penetrant or Not? Pitfalls with In Vivo Determination Using Brain to Blood Ratio
Ziqiang (Zack) Cheng,1 Jinqiang Zhang,1 Rongxia Liu,1 Yiwen Wu,1 Zhen Ge,1 Yi Li,1 Zong-ping Zhang,1 Yu Yang,1 Yan Chen,1 Hong Lu,1 Hans Hu,2 Barry Wang,2 Jason Meng,2 Raymond Zhao,2 and Eric Yang1
1Drug Metabolism and Pharmacokinetics, GlaxoSmithKline, Shanghai, China
2Neuroinflammation Discovery Performance Unit, GlaxoSmithKline, Shanghai, China
Purpose: The total brain-to-blood ratio (B/B) at a single time point obtained from in vivo experiments has traditionally been used to determine whether a molecule is a central nervous system (CNS) penetrant. Despite the effectiveness for early screening, the B/B determination has several caveats: 1) B/B at single time point is a snapshot; 2) brain concentration is more important than the ratio for PK/PD correlation; 3) the unbound exerts drug’s intended pharmacological activity; 4) brain penetration in the animal diseased model may be different from naïve animals. In this poster, we propose an integrated, tiered approach to address these caveats.
Methods and Results: Full blood and brain profiles of five GSK compounds were obtained after single oral dose administration. B/B at eight separate time points and AUC ratio were determined. The B/B increased over time, and half-lives in blood and brain were similar for the drugs tested. B/B and CSF concentration at steady state for two compounds were determined after intravenous (i.v.) bolus administration, followed by i.v. infusion to rats. Rat B/B at steady state was similar to the AUC ratio in mice, and CSF concentrations were in line with unbound concentrations in blood and brain. A single oral dose of two GSK compounds was given to EAE mice, the disease model for multiple schlerosis, at the peak of the disease. Blood and brain were collected 4 hours postdose and analyzed. Brain penetration was found to be similar between EAE and naïve mice.
Conclusions: The integrated, tiered approach should be applied to investigate drug delivery across the blood-brain barrier. We recommend to initially determine B/B at selected time points (2, 4, and 8 hours) during PK screening. Brain-to-blood AUC ratio, brain PK, and B/B at steady state should be assessed for the precandidates, along with some estimation on unbound drug concentration in brain.
128. Pharmacokinetic Study of Borneol and Menthol in Traditional Chinese Medicine
Xin He,1 Xuefang Xu,1 Yang Wang,1 Xinbing Zhu,2 Zhijuan Chen,1 Shuizhen Lin,1 Qiang Li,1 and Changxiao Liu1
1Department of Chinese Pharmacy, Tianjin Key Laboratory of Chemical and Analysis of Chinese Materia Medica, Tianjin University of Traditional Chinese Medicine, Tianjin, China
2The 2nd Teaching Hospital, Tianjin University of Traditional Chinese Medicine, Tianjin, China
Both borneol and menthol are bioactive substances derived from traditional Chinese medicine. In order to understand the pharmacokinetics of borneol and menthol in Qingyan dropping pills, a rapid, sensitive, and simple gas chromatographic method with flame ionization detection was developed for the simultaneous determination of borneol and menthol in rat plasma. Sample preparations were carried out by liquid-liquid extraction with an internal standard solution in naphthalene. The analytes and internal standard (naphthalene) were well-separated on an HP-1 capillary column. The standard curves were linear over a wide concentration rang of 5.0–50.0 ng/μL (r = 0.9963) and 8.7–62.2 ng/μL (r = 0.9994) for both borneol and menthol in plasma, respectively. The pharmacokinetic parameters were computed by the software program, DAS ver 1.0. The results showed that the open one-compartment model was fitted to both borneol and menthol in rat plasma. The method described was successfully applied to the pharmacokinetic study of rat plasma after the oral administration of Qingyan dropping pills.
Acknowledgments
This project was supported by the Tianjin Science and Technology Foundation of China (no. 07JCYBJC18800), the National Natural Science Foundation of China (no. 30772778), and the National Administration of Chinese Traditional Medicine of China (no. 20080751).
129. Pharmacokinetic and Pharmacodynamic Analysis of Distigmine Bromide, Acetylcholinesterase Inhibitor
Yoshihiko Ito, Aya Kato, Taketsugu Harada, Kazumi Fushimi, and Shizuo Yamada
Department of Pharmacokinetics and Pharmacodynamics and Global COE Program, School of Pharmaceutical Sciences, University of Shizuoka, Shizuoka, Japan
Distigmine bromide (distigmine), a reversible, long-acting carbamate cholinesterase inhibitor, is commonly used for the treatment of urine discharge disorders in the underactive bladder. The aim of this study was to clarify the relationship between the inhibitory effect of distigmine on blood acetylcholinesterase (AChE) activity and its plasma concentration. Distigmine (0.3, 1.0, and 3.0 mg/kg) was orally administered to male Sprague-Dawley (SD) rats, and blood samples were collected at 0, 3, 10, and 30 minutes and 1, 1.5, 2.5, 4, 6, and 12 hours after administration. The plasma concentrations of distigmine were determined by liquid chromatography/tandem mass spectrometry. The blood AChE activity was measured by the dithiobisnitrobenzoic acid (DTNB) method. The plasma concentration of distigmine peaked 0.5 hours after oral administration at doses of 0.3, 1.0, and 3.0 mg/kg and decreased gradually. The PK of distigmine was best described by a one-compartment PK model. On the other hand, the maximum inhibition of blood AChE activity was observed 3 hours after the distigmine administration and lasted until 12 hours. The plasma concentration and AChE inhibition by these doses of distigmine were dose-related. Thus, a profound counterclockwise hysteresis loop was observed for the relationship between the plasma concentration and blood AChE inhibition of distigmine at each oral dose. Such delay between the plasma concentration and pharmacological effect of this drug was significantly minimized by the sigmoidal Emax pharmacodynamic model with values for Ke0 of 0.470–0.428 ng/mL in each oral dose. In conclusion, it has been shown that the concentration-effect relationship of distigmine could be described satisfactorily by the sigmoidal Emax pharmacodynamic model approach linked to the plasma concentration by a first-order process.
130. Determination of the Primary Physicochemical Properties for the Delivery of Drugs to the Brain after the Nasal Administration
Kyeong-Ryoon Lee, Mi-Hwa Kim, Jung-Byung Chae, Jun-Hyeng Son, Dae-Duk Kim, Chang-Koo Shim, and Suk-Jae Chung
College of Pharmacy, Seoul National University, Seoul, South Korea
Recently, it was shown that intranasal administration, compared with other administration routes, of certain drugs led to an enhanced transport to the brain. However, in the literature, physicochemical characteristics of drugs, necessary for an enhanced delivery from the nose to the brain, has not been systemically studied. Therefore, the objective of this study was to determine the primary physicochemical properties of drugs for the delivery to the brain after nasal administration. Therefore, 17 model drugs, with varying physicochemical properties, were used: The drugs were administrated to rats at the dose of 8∼ 16 mg/kg (20 μL/rat) to each nostril for 2 minutes. Plasma samples were collected up to 15 minutes, and the brain samples were collected immediately after the last plasma sampling. HPLC-UV, fluorescence, and mass spectroscopy were used to determine the concentration of the model drugs in the plasma and brain samples. The pharmacokinetic parameters (e.g., AUClast, Xbrain, and CLuptake,brain) were calculated and compared with those obtained after corresponding intravenous administrations. Among the drugs studied, only five (approximately 29.4% of the drugs studied) drugs showed enhanced delivery to the brain after nasal administration, indicating that certain specificity exists for an efficient nose-to-brain transport, and that the five model drugs are primarily transported to the brain via the olfactory pathway. Multiple linear regression analysis indicates that the physicochemical properties, such as molecular weight, partition coefficient, and log D, are the primary determining factors in the prediction of brain transport after nasal administration of the model drugs. Since efficient delivery of drugs to the brain is essential for optimal pharmacotherapy for drugs targeted to the brain, the present study may be practically relevant, particularly in the targeted delivery of drugs to the brain after nasal application.
131. Muscarinic Receptor Binding and Pharmacokinetics of a Novel Anticholinergic Agent, Imidafenacin, for the Treatment of Overactive Bladder
Shizuo Yamada, Masanao Seki, and Yoshihiko Ito
Department of Pharmacokinetics and Pharmacodynamics and Global COE Program, School of Pharmaceutical Sciences, University of Shizuoka, Shizuoka, Japan
A novel antimuscarinic agent, imidafenacin, is currently developed for the therapy of overactive bladder (OAB). It is reported that imidafenacin exhibited pharmacological selectivity for the bladder over the salivary gland, and also, high doses of imidafenacin had little pharmacological effect on the central nervous system (CNS). With regard to the pharmacokinetics, the serum concentration of imidafenacin peaked 0.2 hours after oral administration of this drug (0.1–2.0 mg/kg) in rats and declined rapidly. The present study was undertaken to characterize muscarinic receptor binding in the target (bladder) and nontarget (salivary gland, heart, colon, lung, and cerebral cortex) tissues in relation to the pharmacokinetics after the oral administration of imidafenacin. Following the oral administration of imidafenacin (0.5, 2.0 mg/kg), there was significant muscarinic receptor binding measured by the radioreceptor binding assay, using [N-methyl-3H]scopolamine ([3H]NMS), in rat tissues, except the cerebral cortex. The receptor binding activity was characterized by significantly longer lasting binding in the bladder than other tissues. Even a high oral dose of imidafenacin (10 mg/kg) exerted little binding activity of brain muscarinic receptors, being consistent with little CNS effect of this drug. The present study, with the pharmacokinetic data on the extremely higher concentration of imidafenacin in the bladder than in the serum of rats, suggests that imidafenacin orally administered distributes more preferentially to the bladder than other tissues of rats after the intestinal absorption and subsequently binds to the muscarinic receptors. Thus, it is likely that a long-lasting receptor binding in the bladder after oral administration of imidafenacin underlies the pharmacological selectivity of this drug.
132. Food-Drug Interaction Involving the Ferulic Acid/H+ Transporter
Shirou Itagaki, Yoko Kobayashi, Yoshitaka Saito, Masaki Kobayashi, Takeshi Hirano, and Ken Iseki
Faculty of Pharmaceutical Sciences, Hokkaido University, Sapporo, Japan
Dietary polyphenols are thought to be beneficial to human health by exerting various biological effects, such as free-radical scavenging, metal chelation, modulation of enzymatic activity, and alteration of signal-transduction pathways. Ferulic acid is a phenolic compound that is a ubiquitous plant constituent. Ferulic acid exhibits a wide range of therapeutic effects against various diseases, including cancer, cold, flu, and influenza. Moreover, ferulic acid is commercially prepared and used as a component of functional foods. Self-medication with functional foods and over-the-counter (OTC) drugs is an economical choice of treatment for common self-limiting illnesses, including pain. Nonsteroidal anti-inflammatory drugs (NSAIDs) are among the most useful medicines for pain relief. Functional foods and OTC drugs are usually taken orally. Several transporters contribute to the absorption of administered compounds, and interactions involving transporters may affect their therapeutic efficacy. Intestinal uptake of ferulic acid is associated with a monocarboxylate transport system. We focused on this transporter and investigated the interaction of ferulic acid and NSAIDs. Our results showed that monocarboxylate-type NSAIDs interact with ferulic acid and suggested that changing monocarboxylate-type NSAIDs to drugs that are absorbed mainly via other transporters is an effective way for preventing reduced therapeutic response of ferulic acid. The results obtained in this study provide important information for counseling support in the OTC market.
Acknowledgment
This work was supported, in part, by The Agricultural Chemical Research Foundation and OTC Self-Medication Promotion Foundation.
133. The Involvement of Mrp2/MRP2 in the Species Different Excretion Route of Benzylpenicillin between Rat and Human
Min-Koo Choi,1 Hyunmi Kim,1 Su-Jeong Yim,1 Yong-Hae Han,2 Chang-Koo Shim,3 and Im-Sook Song1
1Department of Pharmacology and PharmacoGenomics Research Center, Inje University College of Medicine, Busan, South Korea
2Department of MAP/MS F13-07, Bristol-Myers Squibb, Princeton, New Jersey, USA
3National Research Laboratory of Transporters Targeted Drug Design, College of Pharmacy, Seoul National University, Seoul, South Korea
The purpose of this study was to investigate the involvement of rat Mrp2 and human MRP2 in benzylpenicillin (BPC) transport by using canalicular liver plasma membrane (cLPM) vesicles isolated from Sprague-Dawley or Easai hyperbilirubinemic (EHBR) rats and MDCKII cells overexpressing MRP2. The ATP-dependent uptake of BPC and estradiol-17b-D-glucuronide (E217bG), a representative substrate for Mrp2, into EHBR-cLPM vesicles was decreased relative to that seen with control-cLPM vesicles, which may reflect the absence of Mrp2 in the EHBR. The ATP-dependent uptake of taurocholate, which is not a substrate for Mrp2, was similar in both control and in EHBR-cLPM vesicles. The concentration dependence of ATP-dependent BPC uptake was reflected in a Km of 44.0 μM and a Vmax of 508.4 pmol/mg/min. Additional inhibition studies using E217bG and methotrexate as representative substrates for Mrp2/MRP2 demonstrated the involvement of rat Mrp2, but not human MRP2, in BPC efflux. BPC appears not to be a substrate for, or inhibitor of, other human efflux transporters, such as MDR1, MRP1, MRP3, or BCRP. In conclusion, rat Mrp2, but not human MRP2, plays an important role in ATP-dependent BPC uptake in the bile canalicular membrane, which may explain why biliary excretion of BPC is high in the rat but negligible in humans.
134. Identification and Functional Characterization of Novel Mammalian Riboflavin Transporter RFT1
Atsushi Yonezawa, Satohiro Masuda, Toshiya Katsura, and Ken-ichi Inui
Department of Pharmacy, Kyoto University Hospital, Kyoto, Japan
Riboflavin, a water-soluble vitamin (B2), is essential for normal cellular function. Although riboflavin transporters were thought to play an important role in the maintenance of riboflavin homeostasis, a mammalian riboflavin transporter has yet to be identified. In the present study, we searched for novel transporters using our rat kidney mRNA expression database (Horiba et al., 2004), and SOSUI program, which can predict the number of transmembrane domains from amino-acid sequence. We found a predicted 10-transmembrane protein, GPR172B, which we renamed as RFT1, as a candidate novel transporter. Human RFT1 consists of 448 amino acids, which does not show the similarity to known ABC and SLC transporters. BLAST search of the GenBank database identified the several homologous of RFT1 from the mammalian, fish, and amphibian species. Real-time polymerase chain reaction revealed that RFT1 mRNA was expressed strongly in the placenta and small intestine and was detected in all tissues examined. In addition, RFT1 was also expressed in HEK293 and Caco-2 cells. HEK293 cells transfected with green fluorescent protein-tagged RFT1 exhibited a fluorescent signal in the plasma membrane. By overexpression in the system with HEK293 cells, a screening of 26 compounds was carried out. However, we could not identify the substrate of RFT1. We tried to search for the substrate by using siRNA targeting RFT1. The uptake of [3H]riboflavin by HEK293 cells was significantly decreased by siRNA for RFT1. In addition, the transfection of siRNA significantly decreased the uptake of [3H]riboflavin by Caco-2 cells. With some modifications of uptake condition, [3H]riboflavin uptake was apparently increased by the overexpression of RFT1. Functional analyses were carried out by using HEK293 cells natively expressing RFT1. Riboflavin transport by HEK293 cells was Na+-, membrane potential- and pH-independent. Kinetic analyses demonstrated that the Michaelis-Menten constant for the uptake by HEK293 was 28.1 nM. These results suggested that RFT1 is a novel mammalian riboflavin transporter, which could play a role in the riboflavin absorption.
Reference
- Horiba, N., et al. (2004). Kidney Int 66:29–45.
135. Effect of Thai Plant Extracts on P-Glycoprotein Function and Cell Viability in HepG2 and Paclitaxel-Resistant HepG2 Cells
Ryoko Yumoto,1 Masashi Kawami,1 Junya Nagai,1 Denpong Patanasethanont,2 Bung-orn Sripanidkulchai,2 Varaporn Junyaprasert,3 Noppamas Soonthornchareonnon,3 and Mikihisa Takano1
1Graduate School of Biomedical Sciences, Hiroshima University, Hiroshima, Japan
2Khon Kaen University, Khon Kaen, Thailand
3Mahidol University, Bangkok, Thailand
Resistance of cancer cells to anticancer drugs is a serious problem in cancer chemotherapy. P-glycoprotein (P-gp; ABCB1) is well known to confer multidrug resistance to cells by extruding various anticancer drugs. Agents such as immunosuppressants were reported to reverse P-gp-mediated multidrug resistance in cancer cells. However, because most of them are P-gp substrates and have pharmacological effects, their clinical use is limited. We previously found that ethanol extracts and constituents of Kaempferia parviflora, which has been used in Thai traditional medicine, showed a P-gp inhibitory effect (Patanasethanont et al., 2007). In this study, we examined the effect of ethanol extracts of Thai plants on P-gp function and cell viability in HepG2 and paclitaxel-resistant HepG2 (PR-HepG2) cells. PR-HepG2 cells were established from HepG2 cells, a human hepatoma cell line, by long-term treatment of cells with paclitaxel (PTX), and found to show a higher P-gp expression level than HepG2 cells (unpublished observation). We first examined the effect of extracts on the cellular uptake of rhodamine-123 (Rho123), a typical P-gp substrate. Ethanol extracts from Cratoxylum formosum, Aganosma marginata (KP008), Microcos tomentosa, and Ancistrocladus tectorius enhanced Rho123 uptake (i.e., inhibited P-gp). KP008 enhanced Rho123 uptake for 1 hour in a concentration-dependent manner in PR-HepG2 cells. This effect would not be due to its cytotoxicity, because 1 hour of incubation with KP008 did not affect cell viability, though cytotoxicity of KP008 was observed after 48 hours of incubation. In addition, KP008 potentiated PTX-induced cytotoxicity in PR-HepG2 cells. Interestingly, however, the effect of various extracts on the uptake of two P-gp substrates, Rho123 and paclitaxel, was not necessarily comparable. For example, extracts from Polyalthia evecta and Micromelum minutum enhanced PTX, but not Rho123, uptake. Though further studies are needed, these results indicate that some extracts of Thai plants and their constituents may be useful as chemosensitizers of multidrug resistance in cancer cells.
Reference
- Patanasethanont, D., et al. (2007). Effects of Kaempferia parviflora extracts and their flavone constituents on P-glycoprotein function. J Pharm Sci 96:223–233.
136. Determination of Atorvastatin in Human Plasma and the Application to Pharmacokinetics Study
Pattarawit Rukthong and Korbtham
Sathirakul Pharmaceutics, Mahidol University, Bangkok, Thailand
Wide interindividual and interethnic variability exists in the plasma concentrations of the cholesterol-lowering drugs, HMG-CoA reductase inhibitors (statins), in their efficacy and risk of adverse effects. Determination of atorvastatin in human plasma, using LC MS/MS, has been widely accepted as an effective method, owing to its practical applicability in routine drug analysis in pharmacokinetics and pharmacogenetics study. Nevertheless, the method sensitivity can be compromised, once limited sample volume is available. This problem is typically encountered in pharmacokinetics studies, from which a small volume of plasma samples either from humans or animals can be obtained. Drug extraction from human plasma was performed with the aid of reversed-phase C18 solid-phase microextraction. The detection was accomplished by triple quandruple mass spectrometer interfaced operated in electrospray positive ion mode. Quantitation was achieved by using multiple reaction monitoring (MRM) precursor-production transitions at m/z 559.2 and 440.1 for atorvastatin and m/z 412.2 and 224.2 for fluvastatin (internal standard). Linearity was found within the atorvastatin concentration range of 0.2–80 ng/mL. The limit of detection was found to be 0.05 ng/mL. The lower limit of quantification was 0.2 ng/mL, with a relative standard deviation (%CV) of less than 12%. Acceptable precision and accuracy were obtained for the concentrations within the calibration curve range. The average recovery at low, medium, and high atorvastatin concentrations from spiked plasma:quality control samples were 84.16 ± 7.99, 96.82 ± 6.14, and 102.84 ± 6.73%, respectively. The need of plasma volume less than 250 μL for each sample made it possible to decrease sample preparation time. The method was successfully validated and proved appropriate for the analysis of atorvastatin in human plasma and can be applied for further planned study to determine the relationship between transporeter gene polymorphism and pharmacokinetics of atorvastatin in the Thai healthy volunteer.
137. Determination of Paeonol O-Demethylation Activities In Vitro by Ultra-Performance Liquid Chromatography with MS Detection (UPLC-MS)
Hui-Xin Liu and Ling Yang
Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China
O-demethylation of paeonol has been implicated as an important mechanism of paeonol elimination. To characterize paeonol metabolism by O-demethylation, a rapid, specific ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS) method was developed for the qualitative and quantitative determination of paeonol O-demethylation activity in human liver microsomes (HLMs). A HLMs incubation system with paeonol resulted in the formation of a single product peak. Following the purification, this peak was confirmed as O-demethylated metabolite of paeonol (2,4-dihydroxyacetophenone; DHA) by MS and NMR. The method was validated for the determination of DHA with respect to specificity, linearity, detection limit, recovery, stability, precision, and accuracy. The chromatographic separation was achieved on a UPLC BEH C18 column, Waters, Millford, MA, USA (50 mm × 2.1 mm i.d., 1.7 μm), with a phase of acetonitrile-water (ratio, 30:70). A selective ion reaction (SIR) monitor was specific for paeonol, DHA, and I.S. The method was linear over the concentration range of 0.5–100 μM for DHA in spiked HLMs. Acceptable precision and accuracy were obtained for concentrations over the standard curve range. DHA were stable at 4°C for at least 72 hours in spiked liver microsomes samples. The method was successfully used to determine the kinetics of DHA activities toward paeonol in liver microsomes from different species.
138. Heavy Metal Contents of the Rhizomes in Thai Zingiberaceous Plants
Chutima Limmatvapirat,1 Komas Sang-Uthai,1 Juree Charoenteeraboon,2 and Thawatchai Phaechamud3
1Pharmaceutical Chemistry, Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
2Biopharmacy, Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
3Pharmaceutical Technology, Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
The aim of this study was to determine the concentration levels of 11 heavy metals (Al, Cr, Mn, Fe, Ni, Cu, Zn, As, Cd, Hg, and Pb) in rhizomes of Thai herbal plants in the family, Zingiberaceae. Eleven kinds of samples were collected in the Nakhon Pathom province. One gram of each dried sample was mixed with nitric acid and digested until the solution was clear. Reagent blanks were also analyzed in parallel in all steps. All prepared samples and blanks were analyzed immediately by inductively coupled plasma-mass spectrometry (ICP-MS). It was found that epidermal peel of Alpinia galanga Sw. (galangal) contained the highest Al and Mn contents, 1040.918 and 1464.072 mg/kg, respectively. The epidermal peel of Curcuma mangga Val. and .Zijp (white curcuma) showed the highest Cr, Ni, and Cu contents, 16.731, 36.807, and 17.114 mg/kg, respectively, while that of Curcuma xanthorrhiza Roxb. (Wan-chak- mot-luk) showed the highest Fe content, 444.5872 mg/kg. The epidermal peel and ground tissue of all samples contained Cd and Hg contents lower than safe limits, 0.3 and 0.5 mg/kg, respectively. Both epidermal peel and ground tissue of white curcuma, Zingiber cassumunar Roxb. (Plai), and C. aeruginosa Roxb. (Wan-ma-ha-mek) contained As, Cd, Hg, Pb, and Zn contents lower than the permissible limits, 4, 0.3, 0.5, 10, and 100 mg/kg, respectively. However, the epidermal peel of galangal, Z. officinale (L.) Sw. (ginger), C. longa Linn. (turmeric), and Boesenbergia pandurata (Roxb.) Holtt. (Kra-chai-luang) showed As and Zn contents higher than safe limits, 4, and 100 mg/kg, respectively. Zinc contents in ground tissue of Wan-chak-mot-luk, Kra-chai-luang, C. zedoaria (Berg.) Roscoe (Khomin-io), and Kaempferia parviflora Wall. Ex Baker (Kra-chai-dum) were higher than the safe limit (100 mg/kg). Further, the epidermal peel of Khomin-io and the ground tissue of C. aromatica Salisb. (Wan-nang-khum) contained Pb contents higher than the safe limit (10 mg/kg). These overlimits of As, Pb, and Zn contents in some herbal plants in the family, Zingiberaceae, suggested that the quality control of heavy metal contamination should be concerned.
139. Method Development and Validation for Determination of 3,4-Methylenedioxymethamphetamine, Methylenedioxyamphetamine, and Methamphetamine in Human Urine Using GC-FID
Benjamas Janchawee,1 Apichai Phonchai,2 Sathaporn Prutipanlai,1 and Sittipoom Thainchaiwattana3
1Department of Pharmacology, Faculty of Science, Prince of Songkla University, Songkhla, Thailand
2Forensic Science Program, Faculty of Science, Prince of Songkla University, Songkhla, Thailand
3Regional Forensic Science Division 4, Songkhla, Thailand
A gas chromatography with a flame ionization detection (GC-FID) method was developed and validated for the simultaneous determination of 3,4-methylenedioxymethamphetamine (MDMA), methylenedioxyamphetamine (MDA), and methamphetamine (MA) in human urine. Alkalinized (pH 12) samples were prepared by solid-phase extraction, using an Oasis® HLB cartridge (Oasis catridge, Waters, Milford, MA, USA). The cartridge was washed with a 5% methanol-water mixture containing 2% ammonium hydroxide and eluted with a 70% methanol-water mixture containing 2% acetic acid. One microliter of the reconstituted sample was injected into the GC system, using a CP-Sil 24 CB WCOT fused silica capillary column (30 m × 0.32 mm i.d.; 0.25-μm film thickness). Flow rate of the carrier gas (N2) was 2.6 mL min−1. Flow rates of both fuel gas (H2) and make-up gas (N2) were 30 mL min−1. Oxidant gas (O2) flow rate was 300 mL min−1. Temperatures of the injector and the detector were 290 and 300°C, respectively. Column temperature was initially 80°C (0 minutes) and increased with a ramp rate of 20°C min−1 to a final temperature of 270°C, where it was held for 1 minute. The analytes were eluted at 4.2, 6.8, 7.1, and 9.1 minutes for MA, MDA, MDMA, and DPA (internal standard), respectively. Calibration curves were linear over the concentration range of 1–20 μg mL−1 for MDMA (r = 1.0000), MDA (r = 0.9971), and MA (r = 0.9998). The intra- and interday precisions (%RSD) were ranged from 2.97 to 17.31% for MDMA, 6.28 to 16.54% for MDA, and 6.86 to 17.18% for MA, respectively. The accuracy (%DEV) ranged from (–) 16.68 to (+) 4.28% for MDMA, (–) 18.10 to (+) 14.94% for MDA, and (–) 18.47 to (+) 0.60 for MA, respectively. The extraction recoveries ranged from 84.17 to 95.01% for MDMA, 92.56 to 107.60% for MDA, and 80.35 to 90.74% for MA. The lower limit of quantification (LLOQ) of all analytes was 1 μg mL−1.
140. Physicochemical Characteristics Related to Decomposition of Quercetin
Saengrawee Sutthiparinyanont,1 Aroonsri Priprem,1 and Malyn Chulasiri2
1Pharmacy, Khon Kaen University, Khon Kaen, Thailand
2Reseach and Development, S&J International Enterprises PCL, Bangkok, Thailand
Quercetin (MW 338.3 g/mol), a flavonol compound, is generally found in fruits, vegetables, flowers, botanicals, and beverages. It is a potent antioxidant that could be potentially used as for antiaging and photoprotection. Its physicochemical properties in relevance to stability in aqueous environments provide better product development, particularly for external use. Various techniques were used to elaborate its properties and interactions with solvents at 25°C. Its crystallographic structure by XRD suggests a crystalline form of initial and stored quercetin dihydrate powder. NMR showed that C3-OH is involved in intramolecular bonding; also, FT-IR spectrophotometry revealed the attacking at C3-OH of quercetin in storage aqueous conditions, toward protocatechuic acid (MW 154.1 g/mol), when it was accelerated by light and elevated temperature, as confirmed by UV spectrophotometry. Thermal analysis by DSC and TGA indicates physicochemical changes of quercetin with three major steps of phase-transition temperature at about 116, 320, and 350°C. Its zero-order decomposition at 25°C was also shown by a series of stability profiles of a range of solvents and the effect of some additives. In the aqueous system, quercetin presents as almost insoluble in water (0.2 and 0.7 μg/mL at 25 and 37°C), and the solubility was a little bit increased when adding phosphate ions (1.1 and 4.3 μg/mL at 25 and 37°C), which also enhanced protocatechuic acid production and reduced activation energy (Ea). Solubilities and decomposition profiles in some alcohols and polyols, polymers, surfactants, and albumin will be discussed. Partition coefficient, log koctanol/water of quercetin of 2.48 at 25°C suggests a concentration-dependent permeability. It could be concluded that quercetin is a hydrophobic compound with planar structure, but its affinity with certain aqueous moieties could be induced via C3-OH, which is the most active site and is prone to oxidative degradation and complex formation of this compound.
141. Preliminary Evaluation of GC-MS for the determination of Terpinen-4-Ol in Cutaneous Microdialysis Samples
Kotchaphan Chooluck,1 Kittisak Sripha,2 Hartmut Derendorf,3 and Korbtham Sathirakul4
1Department of Pharmacy, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
2Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
3Department of Pharmaceutics, College of Pharmacy, University of Florida, Gainesville, Florida, USA
4Department of Pharmacy, Mahidol University, Bangkok, Thailand
Terpinen-4-ol (T4) is the main active ingredient in Phlai oil, obtained from steam distillation of the rhizome of Zingiber cassumunar Roxb., which was commonly used as topical anti-inflammatory herbal medicine. The aim of this work was to develop an analytical method for the assessment T4 in cutaneous microdialysis samples by means of gas chromatography-mass spectrometry (GC-MS). In order to confirm a suitable method for intended use of the procedure employed for this determination, our developed analytical method needs to be firstlvalidated. The calibration curve demonstrated a linear relationship between the peak-area ratio of T4 and methyl salicylate (MeS), which was used as an internal standard, over a range of T4 concentrations (0.36–1.79 ppm). The intra- and interday precisions at all concentrations tested were less than 1.5 and 4.0% RSD, respectively. The recoveries of T4 were in the range of 101.22–111.44%. The analyte was shown to be stable in working standard solutions after 40 hours at room temperature and in standard stock solutions after 3 days at −20°C without a relevant loss of signal. The limit of detection and quantification were 0.0294 and 0.0883 ppm, respectively. According to the results, our analytical method met the requirement of method validation followed by the International Conference on Harmonisation guideline. However, based on this preliminary evaluation, further method testing of this developed approach in the conditions of cutaneous microdialysis should be performed. Thereafter, it can be applied to the determination of T4 in topical formulations by using a microdialysis model for the dermatopharmacokinetic study. This study will be Thailand’s first pharmacokinetic study of Thai herbal medicine.
142. Solid-Phase Extraction and Method Validation of Mitragynine in Urine by HPLC Technique
Sathaporn Prutipanlai,1 Orchuma Botpiboon,2 Benjamas Janchawee,1 and Niwat Keawpradub3
1Department of Pharmacology, Faculty of Science, Prince of Songkla University, Songkhla, Thailand
2Forensic Science Program, Faculty of Science, Prince of Songkla University, Songkhla, Thailand
3Department of Pharmacognosy and pharmaceutical Botany, Faculty of Pharmaceutical Sciences, Prince of Songkla University, Songkhla, Thailand
Mitragynine is an indole alkaloid extracted from the leaves of Mitragyna speciosa Korth. (Rubiaceae). The mitragynine level in serum is developed, but its level in urine was not conducted. Therefore, an extraction procedure for mitragynine from urine was implemented in this study. The analyses were extraction from rat urine by solid-phase extraction on an Oasis® HLB SPE column (Oasis, Waters, Milford, MA, USA) and quantitative analysis by high-performance liquid chromatography with ultraviolet detection. The separation system consisted of a C18 column heated to 35°C, a methanol-water (80:20, v/v) mobile phase, and a flow rate of 0.8 mL/min. Mitragynine, with a retention time of 9.2 minutes, was well resolved from any interference in rat urine. The calibration curve was linear from 0.1 to 10 μg/mL (r = 0.9992). The method of mitragynine extraction from urine, using HLB®, shows good validation parameters with a rate of recovery rate over 94%. The intra- and interday precision rates of the method were 0.67–3.28% RSD and 0.74–3.49% RSD, respectively. The accuracy ranged from −2.69 to +13.72% DEV. The lower limit of quantification was 0.1 μg/mL.
143. Validated HPLC for Analysis of Flavonoid Content in Siamese Neem Young Flowers
Worarat Chaisawangwong and Wandee
Gritsanapan Pharmacognosy, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
The Siamese neem tree (Azadirachta indica A. Juss. var. siamensis Valeton) is a medicinal plant found in every part of Thailand. The young flower is consumed as a bitter tonic vegetable. Moreover, it has been used as an element tonic and for the treatment of fever. The flower extract was reported to exhibit in vitro free radical– scavenging activity and can inhibit lipid peroxidation of the bronchogenic cancer cell line. Active compounds in the flowers are flavonoids, such as rutin and quercetin. To evaluate the quantity of active components in Siamese neem young flowers, a high-performance liquid chromatographic (HPLC) method was developed for the assessment of two bioactive flavonoids: rutin and quercetin in the aqueous extract of this plant. A Hypersil® BDS C18 column (Hypersil column, Thermo Fisher Scientific, Waltham, MA, USA) (250 × 4.6 mm, 5-μm particle size) was used. The elution was carried out with isocratic solvent systems with a flow rate of 1 mL/min at an ambient temperature. The mobile phase consisted of 0.5% formic acid in methanol:water (3:7) (A) and methanol (B). Quantitaitve analysis was programmed at A:B (8.5:1.5). The wavelength of the UV-vis detector was set at 254 nm. Separation of the two compounds achieved by the proposed method showed good linearity in the range of 15.62–500 and 0.49–31.25 μg/mL for rutin and quercetin, respectively. The correlation coefficient was higher than 0.999. Intra- and interday precisions showed relative standard deviation (%RSD) less than 2%. Accuracy of the method was determined by a recovery study conducted at three different levels, and the average recoveries of rutin and quercetin were 101.97 and 99.05%, respectively. The LOD and LOQ of rutin were found to be 0.6518 and 1.9752 μg/mL, while of quercetin were 0.0357 and 0.1081 μg/mL, respectively. This proposed HPLC method was sensitive, accurate, and precise for quantitative analysis of flavonoid content in the extracts of Siamese neem flowers. The method should be useful for routine analysis of flavonoids in the flower raw materials, extracts, and their preparations of this plant.
144. Comparison of In Vitro Drug Release and In Vitro Skin Permeation of Q10-loaded NLC
Veerawat Teeranachaideekul,1 Varaporn Junyaprasert,1 Prapaporn Boonme,2 Rainer Muller,3 and Eliana Souto4
1Department of Pharmacy, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
2Department of Pharmaceutical Technology, Faculty of Pharmaceutical Sciences, Prince of Songkla University, Thailand, Songkla, Thailand
3Department of Pharmaceutical Technology, Biotechnology and Quality Management, Free University of Berlin, Berlin, Germany
4Department of Pharmaceutical Technology, Faculty of Health Sciences, Fernando Pessoa University, Portugal, Porto, Portugal
Even though several advantages of solid lipid nanoparticles (SLNs), such as protection of the incorporated sensitive drug molecules due to the solid matrix around drug molecules, have been shown, some drawbacks of SLNs have been reported, for example, low drug payload, drug expulsion during storage, and unpredictability of gelation phenomenon. The concept of SLN with the less ordered structure of the lipids matrix within solid particles’ so-called nanostructured lipid carriers (NLCs), therefore, has been introduced around the beginning of the millennium. NLCs can be produced by mixing various different lipid molecules, that is, blending solid lipid with liquid lipid (oils). In a previous study, the localization of oil molecule was elucidated by means of 1H-NMR. The aim of this study was to evaluate the effect on oil content on the localization of oil molecule within the lipid nanoparticles. Cetyl palmitate (CP) and medium-chain triglycerides (MCTs) were chosen as solid lipid and liquid lipid (oil) of NLCs, respectively. Q10-loaded NLCs were prepared with different ratios of CP and MCT by the high-pressure homogenization (HPH) technique. The 1H-NMR of Q10-loaded NLC was compared to that of Q10-loaded nanoemulsions (NEs). It was found that Q10-loaded NLCs showed a broader signal between 0.9 and 2.5 ppm, in comparison to that of Q10-loaded NE, depending on the MCT loading content. This indicates that MCTs are strongly restricted to and/or incorporated into the solid matrix of CP, especially Q10-loaded NLCs containing the low ratios of MCT and CP.
145. Evaluation of Intestinal Absorption Kinetics after Oral Administration Using Positron Emission Tomography (PET)
Tadayuki Takashima,1 Makoto Kataoka,2 Shunichi Kitajima,3 Machiko Murai,1 Hiroyuki Ou,2 Yasuhiro Wada,1 Emi Hayashinaka,1 Yilong Cui,1 Shinji Yamashita,2 and Yasuyoshi Watanabe1
1Molecular Probe Dynamics Laboratory, RIKEN Center for Molecular Imaging Science, Hyogo, Japan
2Fuculty of Pharmaceutical Sciences, Setsunan University, Osaka, Japan
3GMJ Inc., Tokyo, Japan
It is important to evaluate the rate-limiting steps in the intestinal absorption process following oral administration in vivo. In vivo intestinal absorption studies, so far, were mainly evaluated by blood pharmacokinetics, in situ intestinal perfusion data, or tissue concentration data based on single time point study, which made it difficult to evaluate each absorption process at the same time. Positron emission tomography (PET) is a useful tool for in vivo intestinal absorption study, since it has higher sensitivity and spatial-temporal resolution to evaluate the gastrointestinal transit and absorption elements following oral administration of a radiolabeled drug continuously. We performed a quantitative PET study for the evaluation of the intestinal absorption kinetics in normal Sprague-Dawley (SD) rats by using 2-[18F]fluoro-2-deoxy-D-glucose ([18F]FDG) as a functional PET probe for hexose absorption. In addition, the effect of anesthesia on the gastrointestinal transit and absorption was also evaluated by using the PET image analysis. After the oral administration of [18F]FDG, a 90-minute PET scan was conducted as well as continuous blood sampling from the femoral vein, and distribution of the radioactivity in the gastrointestinal region was analyzed. The gastrointestinal transit and absorption rates were calculated based on the movement and elimination of the radioactivity in the gastrointestinal tract, respectively. The gastric emptying rate of [18F]FDG in conscious or isoflurane anesthetized rats were 0.36 and 0.01 min−1, respectively. In addition, the elimination rate of [18F]FDG in the intestine were 0.08 and 0.01 min−1 in the conscious and the anesthetized rats, respectively, showing gastric empty is the rate-limiting step in the anesthetized rats. Meanwhile, blood pharmacokinetic data showed that Tmax after the oral administration of [18F]FDG in conscious rats were around 20 minutes, whereas Tmax were more than 45 minutes in the anesthetized rats, which supported PET image results. Therefore, PET image results help elucidate the rate-limiting steps in the intestinal absorption process, and we suggest that PET study is useful for the analysis of intestinal absorption kinetics in vivo.
146. MRI diagnosis and Treatment of Multiple Drug Delivery Development
Hyo Jeong Kim,1 Sun Hang Cho,1 Byung Jin Kim,1 Seo yung Jeong,2 Soon Hong Yuk,1 Sung Joo Hwang,3 Byung Kook Kwak,4 Youn Woong Choi,5 Hyo Jeong Kim,2 Su Ji Kim,1 Byung Gu Min,6 and Dae Chul Ha7
1Biomaterials Reseach Center, Korea Research Institute of Chemical Technology, Daejeon, South Korea
2Practical Pharmacy, College of Pharmaceutical Science, Kyunghee University, Seoul, Republic of Korea, Seoul, South Korea
3National Research Laboratory of Pharmaceutical Technology, College of Pharmacy Chungnam National University, Daejeon, South Korea 4College of Medicine, Chung-Ang University, Seoul, Republic of Korea, Seoul, South Korea
5Managing Director, Korea United Pharm. Inc., Seoul, Republic of Korea, Seoul, South Korea
6Korea United Pharm. Inc., Seoul, Republic of Korea, Seoul, South Korea
7Pharmaceutical R&D Center, Korea United Pharm. Inc., Seoul, Republic of Korea, Dae Jeon, South Korea
Magnetic resonance imaging (MRI) is a noninvasive, usually painless, medical test that helps physicians diagnose and treat medical conditions. MRI contrast agents have been used specifically to improve the image contrast between normal and pathological tissues. In this study, multifunctional nanoparticles were developed for the application of them to an imaging-combined therapy system. Poly(amino acid)s have been used as medical biomaterials and drug-delivery carriers because of their water solubility, nontoxicity, biodegradability, and ease of modification for favorable bioavailability. As the multifunctional nanoparticle, PSI-USPIO was prepared and then a hydrophobic anticancer drug, doxorubicin, was loaded into the PSI-USPIO. Physical properties of PSI-USPIO-DOX nanoparticles were characterized by ELS, TEM, and ICP. The intracellular uptake of PSI-USPIO-DOX to B16F10 murine melanoma cells was verified by fluorescence-activated cell sorter (FACS) and conforcal laser microscopy.The anticancer activity was verified with the B16F10 murine melanoma-bearing mice model by monitoring tumor growth pattern and also MRI of liver was observed with the VX2 hepatoma-bearing rabbit model by using a 3.0T clinical magnetic resonance image (MRI) system. PSI-USPIO-DOX may be used as a multifunctional drug-delivery carrier for imaging and therapeutic agents.
147. Self-Emulsifying Drug-Delivery Systems (SMEDDSS) for Bioavailability Enhancement of MASTIC
Suji Kim,1 Byung Jin Kim,1 Yang No Yoon,1 Hyo Jeong Kim,1 Soon Hong Yuk,1 Sun Hang Cho,1 Sung Joo Hwang,2 Suji Kim,2 Young Sig Gil,3 and Sang-Young Jeong3
1Biomaterials Research Center, Korea Research Institute of Chemical Technology, Deajeon, South Korea
2College of Pharmacy, National Research Laboratory of Pharmaceutical Technology, Chungnam National University, Daejeon, South Korea
3RNB Center/Life Science Laboratory, Kolmar Korea Co., Ltd., Chungcheongnam, South Korea
The bioavailability enhancement of a poorly water-soluble compound could be achieved from the enhancement of gastrointestinal absorption by using a self-microemulsifying drug-delivery system (SMEDDS), because SMEDDS could microemlsify the drug easily with the aid of weak gastrointestinal movement. In this study, we designed a SMEDDS to enhance the solubility and bioavailability of natural product MASTIC having poor water solubility and an inhibitory effect to Helicobacter pylori in the digestive organs. To prepare SMEDDS for MASTIC, we investigated the solubility of MASTIC by using various oils and co-/surfactants through visual assessment and the stability was evaluated by GC-MS. As the components for MASTIC formulation, based on its solubility, glyceryl monooleate (Peceol®), polyoxy 40 hydrogenated caster oil (Cremophor CO 40®), and diethylene glycol monoethyl ether (Cabitol®), were used as an oil, a surfactant and a cosurfactant, at the mixture ratio of 40/25/35% (w/w), respectively. We measured dispersion time for microemulsifying of self-microemulsified MASTIC in pH 1.2 and 6.8 buffer and pH 6.5 DI water with 100 rpm stirring at 37.5°C. The stability of oil/water (O/W) microemulsion was conformed by monitoring particle size in their buffer through visual assessment and using ELS (surface zeta potential and particle size analyzer) measurement. The formulation had approximately 100 nm of particle size and excellent stability in aqueous media over 3 months. Also, we confirmed that the self-microemulsified MASTIC had the spherical shape the particle had and high concentration of MASTIC, using FE-SEM and GC-MS. From these results, it is expected that the SMEDDS for MASTIC capsularized in hard or soft capsules could be used for the enhancement of bioavailability and eradication of H. pylori. In vitro and in vivo experiments of the SMEDDS of MASTIC on eradication of H. pylori will be carried out in a further study.
148. Prediction of Steady-State Serum Carbamazepine Concentrations of Thai Epileptic Patients by a Modified Jiao’s Population Pharmacokinetic Model
Manat Pongchaidecha,1 Chompoonuch Werawattanachai,2 Somchai Towanabut,3 and Arkhom Arayawichanont4
1Department of Pharmaceutics (Clinical Pharmacy), Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
2Department of Pharmacy, Prasimahabhodi Psychiatric Hospital, Ubonratchatani, Thailand
3Neurological Information Center, Prasart Neurological Institute, Bangkok, Thailand
4Department of Medicine, Sappasithiprasong Hospital, Ubonratchatani, Thailand
The objective of this research was to compare bias and precision of the prediction of steady-state serum carbamazepine concentrations of modified Graves, Reith, Chan, Jiao 2003, and Jiao 2004 population models. The models were modified based on prospective observational data from 131 psychiatric patients. The selection of modified models were performed according to their mean errors and root mean square errors, with the lowest of these values in a particular model indicating the best modified model. Additional prospective data obtain from 40 subjects were used for validating the best modified model and conventional models. The best model for Thai epileptic patients was the modified Jiao 2004 model that estimated the clearance based on the following equations: CL/F (L/hour) = 0.165 × dose (mg/day) 0.41 × total body weight (kg) 0.11 × 1.25 valproic × 1.18 phenytoin × 1.27 phenobarb. The modified Jiao 2004 model produced the lowest mean error, 0.02 (95% CI: −0.3, 0.40) and root mean square error, 2.13 (95% CI: 1.83, 2.38). The estimated parameter was used for predicting the trough concentration of carbamazepine via the following formulation: Cssmin (mcg/mL) = dose (mg/day)/CL (L/day). Mean error and root mean square error of the modified Jiao 2004 model and other models were compared (test data: n = 40). Mean error and root mean square error of the modified Jiao 2004 model were −0.96 (95%CI: −1.67, −0.26) and 2.39 (95%CI: 1.80, 2.85). Mean error of the modified Jiao 2004 was lower than the Reith, Jiao 2003, and Jiao 2004 models with significance at P = 0.004, P < 0.001, and P < 0.001, respectively, but not different from the Graves and Chan models. Root mean square error of the modified Jiao 2004 model was also lowest (2.39) and different from the Reith, Chan, Jiao 2003, and Jiao 2004 models at P = 0.034, 0.002, 0.005, and 0.001, respectively, but not different from the Graves model. In conclusion, the findings suggest that the best population model is the modified Jiao 2004 model.
149. Comparative Role of Glutathione S-Transferases Polymorphisms Between Liver and Bladder Cancer in Thais
Chonlada Viratroumanee,1 Kannika Pradabkaew,1 Pornpen Pramyothin,1 Chanin Limwongse,2 Payiarat Suwannasri,1 and Anunchai Assawamakin2
1Department of Pharmacology and Physiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
2Division of Molecular Genetics, Department of Research and Development, Faculty of Medicine, Siriraj Hospital, Mahidol University, Bangkok, Thailand
Glutathione-S-transferases (GSTs) are multigene family encoding enzymes that play a major role in phase II biotransformation of drugs and numerous carcinogenic compounds. Among eight major classes of GSTs, three isozymes, GSTM1, GSTT1, and GSTP1, are considered to contribute mainly on toxic compound metabolism and reportedly are associated with susceptibility to various types of cancer, which varied in different populations. Cigarette smoking and certain occupational exposure with cyclic chemicals are the main risk factors for bladder cancer, while liver is well known for bearing important detoxification process of both endogenous and exogenous compounds, including carcinogens. Both types of cancer are influenced by multifactors, including genetics. This study aimed to investigate the role of GSTM1, GSTT1, and GSTP1 polymorphisms in bladder and liver cancer risk in Thais. GSTM1 and GSTT1 were genotyped by multiplex PCR and used denaturing high performance liquid chromatography (DHPLC) to differentiate between GSTM1 hemizygous deletion and wild type. GSTP1 was identified by polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP) method. Null alleles of GSTM1 and GSTT1 present no association with the risk of neither bladder nor liver cancer. However, GSTP1 null genotype apparently plays a major contribution to the decreased risk of both types of cancer. GSTP1 (Ile/Val) individuals showed the protective effect to bladder cancer, compared to wild type [OR = 0.53 (0.33–0.84); P = 0.006), similar to liver cancer [OR = 0.57 (0.36–0.90); P = 0.0174). For double-gene interactions, GSTP1 combined with GSTT1 was surprisingly found to reduce risk only with liver cancer. Additionally, in liver cancer, the protective effect of GSTP1 is also shown in any combinations of GSTP1 (Ile/Val or Val/Val) with other two genes. In summary, this study demonstrates the protective role of GSTP1 (Ile/Val) to liver and bladder cancer risk in Thais.
150. Cytochrome P450 3A4 Enhances UDP-Glucuronosyltransferase 1A9-catalyzed Glucuronidation of SN-38, an Active Metabolite of Irinotecan
Yuji Ishii,1 Yuki Iwamoto,1 Toshiya Oizaki,1 Arief Nurrochmad,1 Yoshio Nishimura,1 Shinichi Ikushiro,2 Futoshi Taura,1 Satoshi Morimoto,1 Kiyoshi Nagata,3 Yasushi Yamazoe,4 Peter I. Mackenzie,5 and Hideyuki Yamada6
1Graduate School of Pharmaceutical Sciences, Kyushu University, Fukuoka, Japan
2Toyama Prefectural University, Toyama, Japan
3Tohoku Pharmaceutical University, Sendai, Japan
4Division of Drug Metabolism and Molecular Toxicology, Graduate school of Pharmaceutical Sciences, Tohoku University, Sendai, Japan 5Department of Clinical Pharmacology, Flinders University, SA, Australia
6Graduate School of Pharmaceutical Sciences, Kyushu University, Fukuoka, Japan
Glucuronidation is one of the major pathways to eliminate SN-38, an active metabolite of irinotecan. Although UDP-glucuronosyltransferase 1A1 (UGT1A1) has been considered to primarily contribute to this metabolism, UGT1A9 is also known to catalyze SN-38 glucuronidation. We have previously demonstrated that cytochrome P450 3A4 (CYP3A4) alters the regioselectivity of UGT2B7-catalyzed morphine glucuronidation (Takeda et al., 2005). Further, the J-helix or surrounding region of CYP3A4 is suggested to be involved in the interaction with UGT2B7 (Takeda et al., 2009). Another group has shown that CYP3A4 interacts with members of the UGT1A subfamily as well as with UGT2B7. However, the effects of CYP3A4 on UGT1A1/9-catalyzed SN-38 glucuronidation have not been elucidated. To address this issue, we investigated whether hepatic UGT1A1/9-catalyzed SN-38 glucuronidation is altered by simultaneous expression of CYP3A4. When CYP3A4 was coexpressed with UGT1A1 or 1A9 in Sf-9 cells, their activities were significantly elevated, compared with the corresponding UGT single-expression system. The enhancement in SN-38 conjugation by CYP3A4 was much higher for UGT1A9 than for UGT1A1. The Kms for the UGT1A1/9-mediated reaction remained unchanged, even with coexpressed CYP3A4. In contrast, the Vmax values for both UGTs were increased by coexpression with CYP3A4, although the effect on UGT1A1 catalysis was not large. Thus, the intrinsic clearance (Vmax/Km) was increased 7-fold by dual expression, compared with UGT1A9 single expression. These results suggest that CYP3A4 interacts with UGT1A9 to markedly affect the efficiency of detoxification of SN-38. It has been reported that hepatic CYP3A4 levels vary by 40-fold. This work suggests that the CYP3A4-dependent facilitation of SN-38 glucuronidation is, at least partially, a factor affecting an individual’s capacity to eliminate SN-38.
References
- Takeda, S., et al. (2005). Modulation of UDP glucuronosyltransferase function by cytochrome P450: evidence for the alteration of UGT2B7-catalyzed glucuronidation of morphine by CYP3A4. Mol Pharmacol 67:665–672.
- Takeda, S., et al. (2009). Mol Pharmacol In press.
151. Effects of Curcuma comosa Extracts on Phase II Drug-Metabolizing Enzymes in Rat Livers
Neeranart Jiwapornkupt,1 Nuansri Niwattisaiwong,1 Laddawal Phivthong-ngam,2 Khemchat Apipalakul,3 Pawinee Piyachaturawat,4 and Somsong Lawanprasert1
1Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
2Faculty of Medicine, Srinakharinwirot University, Bangkok, Thailand
3Faculty of Dentistry, Chulalongkorn University, Bangkok, Thailand
4Faculty of Sciences, Mahidol University, Bangkok, Thailand
Drug-drug interaction study is one of the processes required during the drug research and development process. Induction and/or inhibition of phase I and/or phase II enzymes are always involved. The objective of this study was to investigate effects of Curcuma comosa hexane and ethanolic extracts on phase II drug-metabolizing enzymes involved in drug metabolism, such as UDP-glucuronosyltransferase (UDPGT), sulfotransferase (SULT), glutathione S-transferase (GST), and NAD(P)H quinoneoxidoreductase (NQOR) in rat livers. Fifty male Wistar rats were randomly divided into five groups of 10 rats each. Rats in the control group were given corn oil at 1 mL/kg/day, whereas rats in groups 2 and 3 received C. comosa hexane extract (which was dissolved in corn oil) orally at dosages of 250 and 500 mg/kg/day. Rats in groups 4 and 5 received C. comosa ethanolic extract orally at dosages of 250 and 500 mg/kg/day. At the end of the extract administration (30 days), rats were anesthesized. Microsomes and cytosols were prepared from the livers for enzyme-activity assays. The results showed that C. comosa hexane extracts, at the dosages of 250 and 500 mg/kg/day, significantly increased UDPGT activity, whereas NQOR activity was significantly increased when C. comosa hexane extract at 500 mg/kg/day was given. Both extracts did not affect SULT and GST activities. These results indicated that coadministration of C. comosa hexane extract with some drugs that are detoxified by UDPGT and/or NQOR may affect the level of those drugs. In addition, an increase of UDPGT and NQOR activities by C. comosa indicated a potential benefit of this plant for a decrease in risks of chemical-induced carcinogenesis and/or mutagenesis.
Acknowledgment
This study was financially supported by the National Research Council of Thailand—The Comprehensive Research Project for Indigenous Plants, 2006.
152. Multiple Effects of L-Ascorbic Acid on the Conjugation Reactions in the Human Colon Carcinoma Caco-2 Cells
Hiroomi Tamura, Hidetaka Tsuruta, Toshiaki Yagishita, and Mikiko Shimizu
Graduate School of Pharmaceutical Sciences, Keio University, Tokyo, Japan
Vitamin C is an essential dietary vitamin and is used in large doses either on its own or in multivitamin preparations. Although vitamin C has been reported to have little clinical relevance in the metabolism and bioavailability of drugs that are predominantly metabolized by cytochrome P450s (CYPs), several studies have demonstrated effects of ascorbic acid on conjugation reactions catalyzed by UDP-glucuronosyl transferases (UGTs) or sulfotransferases (SULTs). However, precise molecular mechanisms underlying the effects of ascorbic acid on the phase II reactions have not yet been fully documented. To elucidate the molecular mechanism of the effect of vitamin C on the conjugation reactions in the gastrointestinal tracts, we investigated the effects of L-ascorbic acid (AA) on the conjugation of 1-naphthol in Caco-2 cells, a model of human intestinal cells.
Methods: Caco-2 cells were incubated with 200 μM of 1-naphthol (NA) plus AA at various concentrations for 24 hours, and the conjugates of NA were measured by high-performance liquid chromatography. Expression of UGT and SULT genes was determined by reverse-transcriptase polymerase chain reaction (RT-PCR) or real-time PCR. Assays of UGT and SULT activities were performed according to the methods reported previously.
Results and Discussion: By AA treatment of Caco-2 cells for 24 hours, AA inhibited the accumulation of both glucuronyl- and sulfoconjugates in Caco-2 cells in a dose-dependent manner with IC50 values of 28 and 42 mM, respectively. However, AA did not show any significant inhibition of the cytosolic phenol SULT activity, and it exhibited weak inhibition of the microsomal UGT activity. On the other hand, AA induced the expression of UGT1A genes, whereas it reduced the SULT1A gene expression. These results indicate that AA might affect conjugation reactions in the gastrointestinal tract via multiple effects on both enzymatic activities and gene expression of UGTs and SULTs.
153. Roles of N-Terminal Domain Histidine and Proline Residues in the Substrate Selectivities of Human UDP-Glucuronosyltransferase (UGT) 1A1, 1A6, 1A9, 2B7, and 2B10
Oranun Kerdpin,1 Peter I. Mackenzie,2 Kushari Bowalgaha,2 Moshe Finel,3 and John O. Miners2
1Department of Pharmacy Practice, Faculty of Pharmaceutical Sciences, Naresuan University, Phitsanulok, Thailand
2Department of Clinical Pharmacology, Flinders University, Adelaide, Australia
3Faculty of Pharmacy, University of Helsinki, Helsinki, Finland
An N-terminal domain histidine (corresponding to position 39 of UGT1A1) is conserved in all UGT1A and UGT2B subfamily proteins, except UGT1A4 (Pro-40) and UGT2B10 (Leu-34). Apart from UGT1A4 and UGT2B10, all UGT1A and UGT2B xenobiotic metabolizing enzymes glucuronidate the planar phenols 4-methylumbelliferone (4MU) and 1-naphthol (1NP). However, only UGT1A4 glucuronidates the tertiary amines, lamotrigine (LTG) and trifluoperazine (TFP), while UGT2B10 is known to N-glucuronidate cotinine. The conserved N-terminal domain histidine of UGT1A1, UGT1A6, UGT1A9, and UGT2B7 was mutated to proline and the leucine-40 of UGT2B10 was substituted with histidine. Wild-type and mutant proteins were expressed HEK293 cells and the capacity of each enzyme to glucuronidate 4MU, 1NP, LTG, and TFP was characterized. Expression levels of UGT1A1(H39P), UGT1A6(H38P), and UGT1A9(H37P) in HEK293 cells were comparable to UGT1A4, and all His→Pro mutants glucuronidated LTG. Km values for LTG glucuronidation by UGT1A1(H39P) and UGT1A9(H37P) were 774 and 3812 μM, respectively, compared to 1579 μM for UGT1A4. UGT1A1(H39P) also glucuronidated TFP with a Vmax/Km value comparable to UGT 1A4, but UGT2B7(H35P) lacked activity toward all substrates studied here. Unlike the wild-type enzyme, UGT2B10(L34H) glucuronidated 4MU and 1NP with respective Km values of 260 and 118 μM. The data indicate that the presence of proline instead of the conserved N-terminal domain histidine of UGT1A subfamily proteins favors the N-glucuronidation of tertiary amines over the metabolism of planar phenols, such as 4MU and 1NP. In contrast, the observation that UGT2B10(L34H) glucuronidated 4MU and 1NP confirms an important role for the conserved N-terminal domain histidine in the glucuronidation of planar phenols and related compounds.
154. UGT1A6 and UGT1A9 Involving in the Metabolism of Daphnetin by Human Liver Microsomes
Si-Cheng Liang, Hui-Xin Liu, Guang-Bo Ge, and Ling Yang
Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China
Daphnetin (7, 8-dihydroxycoumarin), one of the coumarin derivatives, is the major bioactive constituent from Daphne odora (Thymelaeaceae). Previous studies indicated that daphnetin has anticoagulant activity like other coumarin analogs and other functions, including anti-inflammatory, arthritis, and rheumatic activities. Though daphnetin has been generally used and been found with good pharmacological applications, its metabolites and metabolic pathway in humans remain unknown. In the present study, the in vitro metabolic studies were carried out. The results showed that no metabolite was formed in NADPH-dependent metabolic system with human liver microsomes (HLMs). In contrast, two metabolites (M-1 and M-2) can be formed rapidly in the presence of UDPGA with HLMs and were identified to be monoglucuronides of daphnetin by liquid chromatography/mass spectrometry (LC/MS). Further study revealed that UGT1A6 and UGT1A9 was primarily involved in the formation of M-1 and M-2 by using a range of specific UGT inhibitors, recombinant UGTs, and correlation analysis. In human liver microsomes, the Km values of M-1 and M-2 are 111.0 + 6.2 iM and 93.2 + 5.0 ìM, respectively, and the Vmax values are 19.0 + 0.4 imol/min/mg and 16.3 + 0.4 imol/min/mg, respectively. The metabolic pathway and kinetic properties of daphnetin indicated that daphnetin can be cleared by UGTs and its UGT metabolites may paly an important role in some pharmacological activities in vivo.
155. Circadian Rhythm of Cytochrome P4502E1 and its Effect on Disposition Kinetics of Chlorzoxazone in Rats
Phisit Khemawoot,1 Kousuke Nishino,2 Sai Yoshimichi,3 and Ken-ichi Miyamoto3
1Department of Immunology and Medicine, AFRIMS, Bangkok, Thailand
2Department of Clinical Pharmacy, Graduate School of Natural Science and Technology, Kanazawa, Japan
3Department of Hospital Pharmacy, Faculty of Medicine, Kanazawa, Japan
The aim of this report is to study the circadian rhythm of cytochrome P4502E1 (CYP2E1) and its effect on the disposition kinetics of chlorzoxazone in male Wistar rats. The rats were housed under a 12-hour light-dark cycle (lights from 9:00 a.m. to 9:00 p.m.) with food and water ad libitum for 3 months. It was found that the expression of microsomal CYP2E1 mRNA in the liver during the dark phase was significantly lower than during the light phase, whereas the content of CYP2E1 protein and its hydroxylation activity were significantly higher. Therefore, chlorzoxazone (20 mg/kg) was intravenously administered at 12:00 p.m. (light-phase group) or 12:00 a.m. (dark-phase group) to determine the effect on the disposition kinetics. The value of the area under the plasma concentration-time curve from 0 to 8 hours (AUC0-8 h) of chlorzoxazone showed no significant difference between the two groups. However, the value of chlorzoxazone half-life in plasma of the light-phase group was significant longer than the dark-phase group. The AUC0-8 h of 6-hydroxychlorzoxazone, a metabolite formed from chlorzoxazone mainly by CYP2E1, was significantly higher in the dark than in the light phase. In conclusion, microsomal CYP2E1 shows a substantial circadian variation in rats, and this was associated with a decrease of chlorzoxazone half-life and an increase of 6-hydroxychlorzoxazone production. Therefore, the temporal variations of therapeutic response and toxicological effects may have to be taken into consideration for other xenobiotics that are predominantly metabolized by CYP2E1, particularly those with a short half-life.
156. Effect of Pinocembrin on Xenobiotic-Metabolizing Enzymes in Rat Liver
Charatda Punvittayagul,1 Suphachai Charoensin,1 Sirinya Taya,1 Wilart Pompimon,2 and Rawiwan Wongpoomchai1
1Department of Biochemistry, Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand
2Department of Chemistry, Faculty of Science, Lampang Rajabhat University, Lampang, Thailand
Pinocembrin (5,7-dihydroxyflavanone) is one of the flavanones found in rhizome of fingerroot (Boesenbergia pandurata) or Kra-chai in Thai. Several lines have shown some pharmacological and biochemical functions, including in vitro antimutagenicity. Our previous study demonstrated that pinocembrin had no toxicity in male rat. This study aimed to investigate the effect of pinocembrin on phase I and II xenobiotic-metabolizing enzymes in rat liver. Male wistar rats were divided into four groups: Group 1 was a vehicle control, while groups 2–4 were intragastrically fed with various doses of pinocembrin (1, 10, and 100 mg/kg b.w., respectively) for 7 days. All animals were sacrificed at day 8 and livers were analyzed for enzyme activities and protein expressions. In the present study, pinocembrin slightly increased protein expressions of CYP2E1, CYP3A1, and NADPH:cytochrome P450 reductase in a dose- response manner. However, it did not affect heme oxygenase and glutathione-S-transferase activities. These results demonstrated that short-term oral administration of pinocembrin had no affect on both phase I and II xenobiotic metabolizing enzymes in rat liver.
157. Effect of Tea Polyphenols on the Content of Cytochrome P450 and B5 along with Expression of CYP2E1 and CYP1A2 in Male Mice
Xia Chen, Guozhu Han, Jinyong Peng, Kexin Liu, Qin Zhou, and Huijun Sun
College of Pharmacy, Dalian Medical University, Dalian, China
Aim: The aim of this study was to investigate the beneficial effect of tea ployphenols (TPs) on the hepatic production of cytochrome P450 (CYP450) and b5 along with the expression of CYP2E1 and CYP1A2 in mice.
Methods: Kunming male mice were intraperitoneally injected with TPs (25, 50, and 100 mg/kg/day) for 2 days. Normal saline (NS) and chloramphenicol were used as negative and positive controls separatively. The livers were removed and microsomes were isolated, and then, the contents of CYP450 and b5 were measured by UV-spectrophotometry. To examine the expression of CYP2E1 and CYP1A2, male mice were pretreated with TPs (100, 200, and 400 mg/kg/day) for 5 days. NS was used as the negative control. CYP2E1 and CYP1A2 protein and mRNA expression levels in the liver were evaluated by Western blotting, immunohistochemical staining and reverse-transcriptase polymerase chain reaction.
Results: The contents of hepatic CYP450 and b5 in TP-treated groups (100, 200, and 400 mg/(kg¨Bd)) were dose-dependently decreased. The contents of CYP450 in the negative control, low, medium, and high TP groups and positive control groups were 0.354 ± 0.024, 0.279 ± 0.014, 0.204 ± 0.016, 0.130 ± 0.016, and 0.147 ± 0.019 separatively. The contents of b5 in these groups were 0.160 ± 0.011, 0.108 ± 0.004, 0.077 ± 0.006, 0.048 ± 0.006, and 0.061 ± 0.009 differently. TPs at the doses of 200 and 400 mg/kg could significantly reduced the expression of both CYP2E1 and CYP1A2 at gene and protein levels, compared with the NS group. The immunohistochemical study showed that both CYP450 and CYP1A2 positively stained cells in TPs (200 and 400 mg/(kg¨Bd)) groups were obviously less than that in the NS group.
Conclusions: TPs could suppress both CYP450 and b5 in a dose-dependent manner. Also, TPs could reduce CYP450 isoform CYP2E1 and CYP1A2 expression dose dependently. These results might be very important for drug interaction when TPs are used in combination with other drugs.
158. Effects of Curcuma comosa Ethanolic Extract on Hepatic Cytochrome P450 Activities
Chonthicha Kittichanun,1 Laddawal Phivthong-ngam,2 Nuansri Niwattisaiwong,1 Khemchat Apipalakul,3 Pawinee Piyachaturawat,4 and Somsong Lawanprasert1
1Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
2Faculty of Medicine, Srinakharinwirot University, Bangkok, Thailand
3Faculty of Dentistry, Chulalongkorn University, Bangkok, Thailand
4Faculty of Sciences, Mahidol University, Bangkok, Thailand
Curcuma comosa Roxb. (Zingiberaceae) is an indigenous plant of Thailand. Its rhizome has been widely used in Thai traditional medicine for the treatment of abnormal uterine symptoms. The purpose of this study was to investigate the effect of C. comosa ethanolic extract on hepatic cytochrome P450 (CYP) in rats. Thirty male Wistar rats were randomly divided into three groups. Rats were given orally C. comosa ethanolic extract at doses of 250 and 500 mg/kg/day, once-daily for 30 days. Corn oil was administered to rats in the control group in the same manner. At the end of treatment, rats were sacrified and liver microsomes were prepared. The results showed that C. comosa ethanolic extract, at 250 mg/kg/day, caused a significant increase of total CYP contents and the activity of CYP1A1. Also, the extract caused a dose-dependent increase of CYP2B1/2B2 activities, whereas CYPs1A2, 2E1, and 3A activities were not modulated. These results indicated the possibilities of C. comosa ethanolic extract regarding drug-drug interactions and the increased risk of toxicity, mutagenesis, and/or carcinogenesis of drugs or compounds that are metabolized or bioactivated via CYP1A1 and CYP2B1/2B2.
Acknowledgment
This study was financially supported by the National Research Council of Thailand–The Comprehensive Research Project for Indigenous Plants, 2005.
159. Effects of the Standard Extract of Centella asiatica (ECa233) on Rat Hepatic Cytochrome P450
Kornphimol Kulthong1, Mayuree H. Tantisira2, Songpol Chivapat3, Nuansri Niwattisaiwong2, Khemchat Apipalakul4, and Somsong Lawanprasert2, 5
1National Nanotechnology Center, NSTDA, Phathumthani, Thailand
2Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
3Medicinal Plant Research Institute, Ministry of Public Health, Nontaburi, Thailand
4Faculty of Dentistry, Chulalongkorn University, Bangkok, Thailand
Centella asiatica (L.) Urban (Umbelliferae) has been used traditionally in various diseases. The extracts of this plant and its active compounds possess several pharmacological effects. The aim of this study was to examine the subchronic effects of the standard extract of C. asiatica (ECa233) on hepatic xenobiotic/drug-metabolizing enzymes, particularly cytochrome P450 (CYP), such as CYP1A1, CYP1A2, CYP2B1/2B2, CYP2E1, and CYP3A in rats. Both male and female Wistar rats were randomly divided into four groups of 10 rats each. Rats in the treatment groups were given orally ECa233 at 10, 100, and 1, 000 mg/kg/day, whereas rats in the control group were given distilled water for 90 consecutive days. At the end of treatment, rats were sacrificed and liver microsomes were prepared for enzyme activity assay. The results showed that ECa233 did not affect total CYP content, as well as the activities of CYP1A1, CYP1A2, CYP2B1/2B2, CYP2E1, and CYP3A, in both male and female rats. Inhibitory effect of ECa233 was examined in vitro, using rat liver microsomes. ECa233 caused a concentration-dependent decrease of the activity of CYP2B1/2B2 (BROD: IC50 = 523 μg/mL, PROD: IC50 = 563 μg/mL) and a very slight effect on CYP1A2 (MROD: IC50 > 1,000 μg/mL). ECa233 had no inhibitory effect on the activities of CYP1A1, CYP2E1, and CYP 3A at concentrations up to 2,000 μg/mL. The inhibitory effect of ECa233 on CYP2B1/2B2 indicated a possibly beneficial effect of the compound regarding the protection of chemical-induced carcinogenesis, but the concern was regarding the possibilities of drug-drug interaction with the medicines metabolized by these CYPs.
160. Enhanced Bacterial Expression of Several Mammalian Cytochrome P450s by Codon Optimization and Molecular Chaperones
Zhong-Liu Wu,1 Zhi-Gang Zhang,1 F. Peter Guengerich,2 Yan Liu,1 and Xiao-Qiong Pei1
1Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu, China
2Department of Biochemistry, Center in Molecular Toxicology, Vanderbilt University School of Medicine, Nashville, Tennessee, USA
Drug metabolism and toxicity research demand a substantial amount of active mammalian cytochrome P450 enzymes. Bacterial expression systems have proven to be a valuable tool for that purpose because of ease of use, low cost, and high protein production. In order to achieve active expression, various techniques have been developed. Codon optimization and molecular chaperon are two of those recently introduced ones (Yun et al., 2006; Wu et al., 2006a, 2006b). Their effects on the active expression of mammalian cytochrome P450s 2B1 (rat), 2S1 (human), 2U1 (human), 2W1 (human), and 27C1 (human) were investigated. Codon-optimized cDNAs of P450s 2B1, 2W1, and 27C1 were synthesized by using a PCR-based oligonucluotide assembly designed online (http://helixweb.nih.gov/dnaworks/). N-terminal-contiguous rare codons of P450s 2S1 and 2U1 were optimized by site-directed mutagenesis to suit the codon bias of Escherichia coli. The results showed positive effects for these two techniques on the expression of active P450s in general, albeit to different extents. With codon optimization, the expression level (nmol/L) increased 22-fold for P450 27C1 (1,300 vs. 60), 3.6-fold for P450 2U1 (180 vs. 50), and 2.1-fold for P450 2W1 (1,800 vs. 840) but remained unchanged for P450s 2B1 and 2S1. Coexpression of a molecular chaperone, GroES/EL, brought the expression level up 14-fold for P450 27C1 (1,300 vs. 90) and 3- to 5-fold for P450s 2B1, 2S1, and 2W1 (with or without codon optimization). Taken together, the results suggest that the general enhancement effect of the two techniques, plus the use of other available candidate chaperones, could be extended to the optimal expression of other mammalian P450s.
Acknowledgments
This work was supported, in part, by the 100 Talents Program of the Chinese Academy of Sciences, the Provincial Sci and Tech Foundation for Young Scholars of Sichuan, China (08ZQ026-023) and the National Natural Science Foundation of China (20802073/B020104 to Z-LW; and USPHS R37 CA90426 and P30 ES000267 to FPG).
References
- Yun, C.H., et al. (2006). Curr Drug Metab 7:411–429.
- Wu, Z.L., et al. (2006a). Arch Biochem Biophys 445:138–146.
- Wu, Z.L., et al. (2006b). Mol Pharmacol 69:2007–2014.
161. Identification of CYP1A2 as the Principal Enzyme Catalyzing Paeonol O-Demethylation in Human Liver Microsomes
Hui-Xin Liu and Ling Yang
Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China
Paeonol, the primary active component of a traditional Chinese medicine, Moutan Cortex, has a wide range of pharmacological activities. As compared to the extensive research of the pharmacological activities of paeonol, few studies have dealed with its metabolism and pharmacokinetics. Several phase II metabolites and the only one phase I metabolite of paeonol (the O-demethylated metabolite of paeonol, resacetophenone) has been detected in studies with rats. As one of the major metabolites, resacetophenone reached a maximum concentration approximately 20 minutes after dosing. However, no information is available, to date, about the phase I metabolites and which isozyme of phase I metabolic enzyme is involved in this reaction in humans. In present study, we elucidated the O-demethylation pathway of paeonol and identified one O-demethylated metabolite (resacetophenone) in HLMs by comparing the tandem mass spectra and the chromatographic retention time with that of the standard compound. A kinetic study showed that paeonol O-demethylation by HLMs followed Michaelis-Menten kinetics. The kinetics parameters values of HLMs for paeonol O-demethylation were Km = 24.1 ± 1.3 M and Vmax = 577.7 ± 10.8 pmol/min/mg protein. Among recombinant CYP isozymes examined in the present study, only CYP1A2 and CYP2A6 isozymes exhibited paeonol O-demethylation activity. However, it is noteworthy that in 11 individual human liver microsomes, the activities of paeonol O-demethylation were significantly correlated with the activities toward phenacetin, a proposed CYP1A2-selective probe substrate, but not with the activities toward coumarin, a proposed CYP2A6-selective probe substrate. In addition, the extensive inhibition in HLMs obtained with furafylline (CYP1A2-selective inhibitor) provide further evidence of CYP1A2 involvement. In combination, we demonstrate that CYP1A2 and CYP2A6 are involved in the paeonol O-demethylation, and that CYP1A2 plays a major role in paeonol O-demethylation in HLMs. The idenficiation of CYP1A2 as being responsible for paeonol O-demethylation will greatly improve future investigations of CYP1A2 interindividual differences associated with paeonol clinical trials and the magnitude of drug-drug interactions.
162. In Vitro Antimalarial Interactions between Mefloquine and Cytochrome P450 Inhibitors
Raewadee Wisedpanichkij,1 Wanna Chaijaroenkul,1 Piyanan Sangsuwan,2 Jintana Tantisawat,2 Kanyarat Boonprasert,1 and Kesara Na-Bangchang1
1Pharmacology and Toxicology Unit, Graduate Program in Biomedical Sciences, Pathum Thani, Thailand
2Department of Medical Technology, Faculty of Allied Health Sciences, Pathum Thani, Thailand
The treatment and control of malaria is becoming increasingly difficult due to the resistance of Plasmodium falciparum strains’ resistance to commonly used antimalarials. Combination therapy is currently the strategy for combating multidrug-resistant falciparum malaria, through exploiting phamacodynamic synergistic effect and delaying the emergence of drug resistance. The objective of the present study was to investigate antimalarial activity of inhibitors of cytochrome P450 (CYP) enzyme, including their interactions with the antimalarial mefloquine against chloroquine-resistant (K1) and chloroquine-sensitive (3D7) P. falciparum clones in vitro. Results showed IC50 (drug concentration, which produces 50% schizont maturation inhibition) values [mean (range)] of mefloquine against K1 and 3D7 clones to be 8.6 (8.0–9.3) and 12.1 (10.5–13.8) nM, respectively]. The corresponding values for the IC50 values of quinidine were 32.2 (31.9–32.5) and 28.7 (28.4–29.0) nM and for ketoconazole were 3.9 (3.7–4.1) and 4.8 (4.6–5.1) nM, respectively. Analysis of isobologram revealed a trend of decreasing of fraction IC50 (FIC), which indicates synergistics of either quinidine or ketoconazole with meloquine for both chloroquine-resistant and chloroquine-sensitive clones.
163. Influence of Genetic and Environmental Factors on Cytochrome P450-mediated Nicotine Metabolism in Thai Population
Wiratchanee Mahavorasirikul, Rungnapa Apinan, Soisungwan Satarug, Wichittra Tassaneeyakul, and Kesara Na-Bangchang
Graduate Program in Biomedical Sciences, Faculty of Allied Health Sciences, Thammasat University, Pathumthani, Thailand
The influence of genetic and environmental factors, particularly cadmium exposure, on cytochrome P450–mediated nicotine metabolism (CYP2A6) was investigated in a total of 196 Thai subjects (94 males, 101 females), using nicotine as a probe substrate. Subjects were given 2 mg of nicotine gum (Nicorette™, GlaxoSmithKline, Middlesex, UK) to chew for 30 minutes. The pattern of urinary excretion of cotinine (metabolite of nicotine) over 2 hours (logarithmically transformed) was normally distributed. Individuals with levels in the ranges of 0.01–2.40, 2.19–14.83, and 14.93–94.99 μg/2 hours were categorized as poor metabolizers (PM), intermediate metabolizers (IM), and extensive metabolizers (EM), respectively. The majority (61%) of subjects were classified as IM, whereas 24 and 15% were classified as EM and PM, respectively. Factors influencing CYP2A6-mediated nicotine metabolism included cadmium body burden, gender, age, and smoking status. Age, gender, iron stores, residential areas of exposure, types of food consumption, and smoking status were found to significantly influence cadmium body burden. A weak positive, but significant, correlation was observed between total amounts of urinary cadmium excretion (ng/2 hours) and age, and total cotinine (μg/2 hours) over 2 hours. Males excreted cotinine at a higher rate than females. In individuals classified as EM, mean total urinary excretion of cotinine in smokers (males) was significantly higher than nonsmokers (males and females). There was no significant correlation between smoking dependency and CYP2A6 genotypes (CYP2A6*1A/*1B, CYP2A6*1B/*1B, CYP2A6*1B/*9, CYP2A6*1A/*1A, CYP2A6*1A/*4C, CYP2A6*1A/*4C, CYP2A6*1B/*4C, CYP2A6*4C/*4C, CYP2A6*1A/*7, CYP2A6*1A/*8, CYP2A6*1A/*9), CYP2A6*1A/*10, CYP2A6*1B/*7, CYP2A6*1B/*8, CYP2A6*1B/*10, CYP2A6*4C/*9, CYP2A6*7/*9, CYP2A6*7/*10). However, a significant association was observed between smoking dependency and CYP2A6 phenotypes, where the proportions of smokers who smoked more than five cigarettes/day was found to be higher in EM, compared with IM and PM.
164. Modification of the Plasma Disposition Profile of Imatinib through Protein Binding and Cytochrome P450 Inhibition
Elaine Kan, Jason H.K. Law, Gian Wan Soo, Wei Yin Lim, Grace Chay, Shin Yee Tan, Nadeem I. Bukhari, and Ignacio Segarra
Department of Pharmaceutical Technology, International Medical University, Kuala Lumpur, Malaysia
Background: Imatinib inhibits the Bcr-Abl, c-kit, and PDGFR tyrosine kinases and it is approved for the treatment of chronic myeloid leukaemia and gastrointestinal stromal tumors. It has been shown to inhibit the growth of glioblastoma cells in vitro and in vivo. Imatinib binds to α-1-acid glycoprotein, is metabolized by CYP3A4 and CYP3A5 isoenzymes in the intestinal wall and liver, and is a substrate of efflux transporters located at the apical membrane of intestinal epithelial cells and the blood-brain barrier. As a result of α-1-acid glycoprotein, the influence of efflux transporters, and CYP-mediated metabolism, the bioavailability and pharmacokinetics of imatinib is affected. Purpose: The aim of this study was to investigate the combined effect of ketoconazole, a CYP3A4 and p-glycoprotein inhibitor, and primaquine, which binds to α-1-acid glycoprotein, on the disposition profile of imatinib.
Experimental Design: Male ICR mice (9–12 weeks) were dosed with imatinib (50 mg/kg) orally (control group, n = 43). The study group (n = 48) was given primaquine (12.5 mg/kg) 20 minutes and ketoconazole (50 mg/kg) 15 minutes before imatinib administration (50 mg/kg). All drugs were given orally. Plasma imatinib concentration was quantified at predetermined time points (2, 5, 10, 20, and 40 minutes and 1, 2, 4, 6, and 10 hours) after imatinib administration, using a validated high-performance liquid chromatography method. Pharmacokinetic parameters were calculated by using noncompartmental methods.
Results and Discussion: Coadministration of ketoconazole and primaquine with imatinib increased imatinib CMAX by 35% (8.27 ± 1.5 μg/mL) and AUC0→∞ by 31% (28.33 μg·h/mL). TMAX was unaffected (40 minutes) and elimination t1/2 was 1.5 hours (control: 2.4 hours). Cl/F and VSS/F decreased 23% to 1.7 L/h/kg and 20% to 6.6 L/kg, respectively. In addition, imatinib plasma disposition profile, when administered in combination with ketoconazole and primaquine, showed a characteristic plateau, which lasted up to 6 hours.
Conclusions: The combined effects of primaquine and ketoconazole on α-1-acid glycoprotein binding, P-glycoprotein, and CYP-mediated metabolism inhibition increases imatinib bioavailability, affects Cl and VSS, and configures to imatinib a distinct, sustained release–like disposition profile.
165. Using Traditional Chinese Medicine Meridine Guide Drug as a New Drug Delivery Method for Targeting: a Hypothesis and Proof
Ruizhi Zhao,1 Shaojun Liu,1 Shirui Mao,2 Yanjun Wang,1 and Yang Wang3
1Second Affiliated Clinical College, Guangzhou University of Traditional Chinese Medicine, Guangzhou, China
2School of Pharmacy, Shenyang Pharmaceutical University, Shen Yang, China
3College of Traditional Chinese Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, China
Background and Purpose: In traditional Chinese medicine, a meridian guide drug (MGD) is usually used to enhance the effect of the active drug and decrease its side effects. The function of the MGD is similar to that of the modern target delivery system. We hypothesize that the MGD could be used as a new method for achiving target delivery. In order to prove this hypothesis, we chose resveratrol as a model drug and investigated the effect of an MGD–vinegar-processing Radix bupleuri (VPRB) on the tissue distribution of resveratrol in mice.
Experimental Approach: Animals were divided into four groups randomly: the resveratrol group as the control and resveratrol coadministered with three different doses of VPRB. Concentrations of resveratrol in different tissues were determined by high-performance liquid chromatography and the target effeciency was evaluated by RUE and RTE.
Key Results: Compared to the control group, medium-dose VPRB enhanced the targeting efficiency of resveratrol significantly, and the RUE and RTE were 1.79 and 46.9%, respectively. Meanwhile, it considerably reduced the distribution of resveratrol in lung and blood, with the RUE and RTE in blood 1.1 and −22.6% and were 0.88 and −55.0% in lung, respectively. VPRB reduced the Cmax of resveratrol in almost all the tissues except for liver, especially in heart and kidney, with the extent between 26 and 61%, which is beneficial to reduce side effects.
Conclusions and Implications: This article demonstrated medium-dose VPRB could enhance the liver targeting of resveratrol, implying that MGD may be a potential way for achieving target therapy.
166. In Vitro Study on Anti-Inflammatory Activity of Thai Mulberry Extracts
Suchitra Thongpraditchote,1 Omboon Luanratana,2 Kwanchai Rattanamanee,3 Narongchai Pongpan,2 and Yuvadee Wongkrajang1
1Department of Physiology, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
2Department of Pharmacognosy, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
3Department of Pharmacy Practice, Faculty of Pharmaceutical Sciences, Naresuan University, Pitsanulok, Thailand
The anti-inflammatory effect of the crude extracts from different parts of Thai Mulberry was determined. The stem bark, stem wood, root bark, and root wood were extracted by soxhlet extraction, using hexane, dichloromethane, and methanol as the extraction solvent. All extracts were tested in vitro for their inhibition on interleukin-1 beta (IL-1β) production and their promotion of IL-10 production. All methanolic extracts inhibited the production of IL-1β at a dose of 0.1 mg/mL. The extract from stem wood showed the best activity (58.2%), followed by root wood (57.9%), root bark (45.6%), and stem bark (36.1%). The stimulation of anti-inflammatory cytokine, IL-10, was also determined in this study. The percentage of stimulation of IL-10 was 58, 67.3, and 30% for the stem wood, the root bark, and the root wood extracts at a dose of 0.01 mg/mL, respectively. These results indicated the anti-inflammatory activity of Thai Mulberry extracts from the stem wood and the root. These findings support the traditional use of Mulberry stem and root in the treatment of inflammation.
Acknowledgment
This research project was supported by Mahidol University.
167. Antibacterial Activity of Seed Coats of Certain Thai Tamarind Cultivars
Piyanuch Wongprapairoj, Waleewan Eaknai, Vimolmas Lipipun, and Sunanta Pongsamart
Department of Biochemistry and Microbiology, Chulalongkorn University, Bangkok, Thailand
Tamarind seed coats have been reported as containing phenolic antioxidants, such as 2-hydroxy-30, 40-dihydroxyacetophenone, methyl 3,4-dihydroxybenzoate, 3,4-dihydroxyphenyl acetate, and epicatechin. The objective of the present study was to preliminarily assess the antibacterial potential of seed coat extracts from certain Thai tamarind cultivars. Three tamarind cultivars from the Nakhonratchasima (Korat) province, including Tamarindus indica “Priao” (TI-P/K), “Srichomphu” (TI-SP/K), and “Sithong-nak” (TI-STN/K), were collected, tamarind seed coats were extracted with 70% ethanol followed by chloroform, and then the aqueous phase was extracted with ethyl acetate and dried. The seed coat extracts were estimated for antibacterial activity against the representative bacteria; gram-positive, Staphylococcus aureus ATCC 6538P, and gram-negative, Escherichia coli ATCC 25922 by both the microdilution susceptibility test and drop-plate technique were used for colony counts. Gentamycin sulfate was used as the positive control. The results showed that seed coat extracts of the three tamarind cultivars inhibited bacterial growth at concentrations 1.56–6.25 mg/mL. MICs and MBCs of seed coat extracts were determined. S.aureus was more susceptible to the seed coat extracts than E.coli. Tamarind seed coats seemed to be economically important as a good food source containing antioxidants and antibacterial agents for nutracenticals.
168. Antioxidant Activities of Thai Tamarind Seed Coat Extracted by Two Different Solvent Extraction Methods
Oranuch Nakchat and Sunanta
Pongsamart Biochemistry and Microbiology, Chulalongkorn University, Bangkok, Thailand
Tamarind seeds have been widely used in pharmaceuticals and used to be a coffee adulterant widely in Thailand. Tamarind seed coats have also been found recently to be a good source of antioxidants. The present study was done to compare antioxidant activities in tamarind seed coat extracts from two different solvent extraction methods. Seed coats of Tamarindus indica “Priao-Yak” were extracted with ethyl acetate (method 1), and 70% ethanol followed by partition with chloroform, and finally with ethyl acetate (method 2). Total phenolic contents were determined by Folin-Ciocalteu assay, and antioxidant activities were estimated by various methods, including DPPH. scavenging, OH radical scavenging, iron chelating, reducing power, NO radical scavenging, ABTS.+scavenging and lipid peroxidation assay. BHA and Trolox were used as the positive control. The results showed that tamarind seed coat extract from method 1 possesses significantly higher phenolic contents (74.21 ± 0.77 g GAE/100 g dry extract) than that of the extract from method 2 (52.17 ± 1.45 g GAE/100 g dry extract). Antioxidant activities of extracts from the two methods were pretty high, compared to positive controls. The extract of method 2 exhibited antioxidant activities with DPPH. scavenging, OH radical scavenging, reducing power, as well as the iron chelating activity test that were stronger than that of the extract of method 1. But, antioxidant activities of the extract of method 2 with NO scavenging, ABTS.+ scavenging, as well as the lipid peroxidation inhibition test were lower than that of the extract of method 1. Different extraction methods were effected with the components as well as antioxidant activities in tamarind seed coat extracts.
169. Antioxidants of Seed Coat Extracts of Thai Tamarind Cultivars from Nakhonratchasima Province
Waleewan Eaknai, Piyanuch Wongprapairoj, Maneewan Suksomtip, and Sunanta
Pongsamart Department of Biochemistry and Microbiology, Chulalongkorn University, Bangkok, Thailand
Antioxidants in Tamarind seed coats have been widely studied recently in Thai tamarind cultivars. Tamarind seems to be a good source of food containing high levels of antioxidants. Value-added products from waste of seed coats of Thai tamarind cultivars were investigated. Seed coat extracts of tamarind cultivars collected from the Nakhonratchasima (Korat) province were examined for contents of total phenol and proanthocyanidin together with antioxidant activities, and DPPH radical scavenging activity with lipid peroxidation inhibition and reducing power assays were evaluated. Seed coat extracts tested comprised total phenolic content equivalent to GAE 42–51 g/100 g dry extract, while proantocyanidins measured absorbance at 550 nm were 0.83–0.95 per mg dry extract. Antioxidant activity with the DPPH radical scavenging assay exhibited EC50 value ranges of 119–160 μg/mL, compared to 109 μg/mL of BHA and 102 μg/mL of vitamin C; the reducing power assay exhibited EC50 value ranges of 108–131 μg/mL, compared to 96 μg/mL of BHA and 45 μg/mL of vitamin C; and the lipid peroxidation inhibition assay showed EC50 value ranges of 70–81 μg/mL, compared to 15 μg/mL of BHA and 314 μg/mL of vitamin C, respectively. Seed coats of three tamarind cultivars from the Nakhonratchasima province seemed to be good sources of food with high antioxidants, compared to vitamin C.
170. Antioxidative and Genotoxicity Assessment of Cissus quadrangularis L. Extracts
Tarat Sapsrithong,1 Weeraya Kaewprem,1 Punnee Nusuetrong,2 Thitima Pengsuparp,1 and Duangdeun Meksuriyen1
1Department of Biochemistry and Microbiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
2Department of Physiology, Faculty of Medicine, Srinakharinwirot University, Bangkok, Thailand
Cissus quadrangularis L. (CQ) is widely used in Thai traditional medicine as an over-the-counter drug for the treatment of hemorrhoids. Based on scientific evidence, CQ contains antioxidant agents (i.e., quercetin and resveratrol) that might lead to the prevention of blood vessel damage or to strengthen them. According to the World Health Organization guideline for the safe use of herbs, the quality, efficacy, and safety of CQ extracts were evaluated in the present study. To establish the quality of CQ extracts, TLC densitometric analysis was performed. Quercetin was found in ethanolic CQ extracts by exhibiting an Rf value equal to standard quercetin (Rf 0.52) in a solvent system of toluene-ethyl acetate-formic acid (6:4:1, v/v/v). The highest yield of quercetin was found in the ethanolic extracts, while quercetin was not detected in either 40% ethanol or aqueous extracts. The ethanolic CQ extracts were then performed with soxhlet and reflux apparatus, giving the extraction yield of about 27.8% and 29.2% of dried plant material and quercetin content at 1.82 ± 0.09 and 1.46 ± 0.06 μg/mg of crude extracts, respectively. To determine the antioxidative activity of the ethanolic CQ extracts, DPPH scavenging assay was assessed and showed that the CQ extracts by soxhlet and reflux extraction had a 50% effective concentration (EC50) at 0.33 and 0.44 mg/mL, respectively, while quercetin itself exhibited an EC50 at 3.30 μM. To evaluate the cytotoxic potential of the CQ extracts, an MTT reduction assay was determined by using the human umbilical vein endothelial cell line (ECV304). No cytotoxicity on ECV304 cells was preliminary observed after a 24-hour exposure to the CQ extracts at a concentration of 1 mg/mL. Besides the antioxidant property, quercetin was also reported as a well-known genotoxicant. To confirm the safety of use during long-term use of CQ containing quercetin, a genotoxicity assessment of the CQ extracts in ECV304 cells was, therefore, investigated. A comet assay for the evaluation of the genotoxic potential of CQ extracts at a low noncytotoxic concentration range is now underdeveloped.
171. Anxiolytic Effect of Michelia alba DC. Oil
Sarunya Laovitthayanggoon
Pharmaceutical and Natural Products Department, Thailand Institute of Scientific and Technological Research, Pathumthani, Thailand
The pharmacological study of Michelia alba DC. oil in mice via the inhalation route was investigated by using open-field, elevated plus maze (EPM) and the pentobarbital-induced sleeping time assay. Six male mice were inhalated with 10% of the M. alba DC. oil in sterile water for 1 hour before the experiment started. The results showed that treated mice exhibited significantly changed behavior and emotion in the open-field trial. After pentobarbital (40 mg/kg body weight) injection, all treated mice slept longer than the control group. Moreover, their having spendt time in the open arms during the EPM test were found to be a prolonged period. These results indicated that M. alba DC. oil played a key role as an anxiolytic and had hypnotic effects.
172. Application of Uniform Design for Preparation of Icariin Solid Lipid Nanoparticles and Study of Transdermal Drug Release Mechanism of it In Vitro
Qian Wang, Yang Wang, Xiaoli Liu, Wendi Wang, and Xiaojuan Zhang
College of Traditional Chinese Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, China
Aim: The aim of this study was to optimize the formulation for the preparation of the Icariin solid lipid nanoparticles (Ica-SLN) and to study the transdermal drug-release mechanism of it in vitro.
Method: The optimal preparation was selected by the U9 (94) uniform design. Ica-SLN was prepared by the high-temperature melting and cool solidification method. The comprehensive score of the entrapment efficiency, ratio of loading drug, diameter, and zeta potential was used as an index. The experiment on percutaneous absorption of Ica-SLN in vitro was taken by using Franz diffusion cells and rat abdomen skin, and the Ica-SLN was prepared according to the optimal formulation. We took Ica water solution, which was the same concentration with the SLN as the control, to study the transdermal drug-release mechanism of the Ica-SLN in vitro and to study the influence of pH value and concentration on the permeability of this drug.
Results: The entrapment efficiency and the ratio of loading drug of Ica-SLN was 73.50 ± 4.74 and 2.44 ± 0.16%, respectively. The average diameter was 108.3 ± 2.71 nm and the zeta potential was −46.76 ± 3.46) mV. Compared whit the Ica water solution, Ica-SLN could significantly increase the accumulative penetration amount of Ica. The permeation of Ica-SLN was in accord with the following equation: Q = 27.66t1/2 – 19.09 (r = 0.9963). Concentration had a remarkable effect on the accumulative penetration amount of Ica-SLN (P < 0.01), while the pH value did not show any obvious effect (P > 0.05).
Conclusions: The application of the uniform design is useful to achieve high entrapment efficiency and ratio of loading, and the diameter is of great satisfaction. As a drug-loading system, the SLN could obviously improve the permeability of Ica.
173. Chitosan Microencapsulated Capsules for Colonic Drug Delivery: Cytotoxicity and Drug Uptake Enhancement
Kampanart Huanbutta,1 Jurairat Nunthanid,1 and Lee-Yong Lim2
1Faculty of Pharmacy, Silpakorn University, Nakorn Pathom, Thailand
2Pharmacy, School of Biomedical, Biomolecular and Chemical Sciences, The University of Western Australia, Perth, Australia
The purpose of the study was to evaluate the cytotoxicity of microcapsules (MC) using the MTT assay and to investigate drug-uptake enhancement of chitosan (CS) microcores containing diclofenac sodium (DS) in comparison with DS solution. Chitosan microcores containing DS were prepared from CS (Molecular weight, 45 kDa; 87% degree of deacetylation) at ratios of DS:CS, 1:2, and 1:4. Chitosan MC were prepared by coating CS microcores with Eudragit®; S100 (ED) (Eudragit, PharmaPolymers, Piscutaway, NJ, USA) by the desolvation method, using sodium sulfate as a desolvating agent at ratios of DS:CS:ED, 1:2:6, and 1:4:6, respectively. This system was expected to pass through the stomach and small intestine to deliver the drug to the colon. Cytoxicity of MC to Caco-2 cells was determined by the inhibition of mitochondrial dehydrogenase (MTT). Caco-2 cells were seeded into 96-well microtiter plates. After a 48-hour incubation, growth medium was replaced with fresh medium containing 0.01–0.1 g/mL of MC. The cells were incubated for 5 hours, then MTT was added and the cells were incubated for an additional 2 hours. The medium was removed and the formazan crystals were dissolved. The optical density was measured by using a microplate reader. Moreover, the drug-uptake enhancement of DS-CS microcores or DS solution in Caco-2 cells was also evaluated. The cells were incubated with the test samples up to 4 hours. The cell lysates were measured for DS amount and protein content by using the plate reader. Viability results of Caco-2 cells showed that the MC tested in our present study in the range of 0.01–1.0 mg/mL did not show any toxicity. Further, drug-uptake enhancement of DS-CS microcores evaluated through Caco-2 cells showed that the microcores exhibited the higher ability of enhancement over the DS solution, and the ratio of 1:2 can encourage the drug into the cells better than the ratio of 1:4. It might be due to mucoadhesiveness as well as loosening of tight junctions, according to interaction on epithelium cells of CS, that enhances the uptake and permeation of drugs.
174. Chronic Treatment with Tocotrienol, AN Isoform of Vitamin E, Prevents Intracerebroventricular Streptozotocin-induced Cognitive Impairment and Oxidative-Nitrosative Stress in Rats
Vinod Tiwari, Anurag Kuhad, Mahendra Bishnoi, and Kanwaljit Chopra
Pharmacology Division, University Institute of Pharmaceutical Sciences (UIPS), Chandigarh, India
Introduction: Oxidative stress has been implicated in neurodegenerative disorders, including Alzheimer’s disease (AD). Intracerebroventricular (ICV) streptozotocin (STZ) has been shown to cause cognitive impairment, which is associated with increased oxidative stress in the brain of rats. Vitamin E is a potent free radical scavenger and antioxidant. In the present study, we investigated the effect of both the isoforms of vitamin E, α-tocopherol (100 mg/kg per orally; p.o.) and tocotrienol (50 and 100 mg/kg p.o.) against ICV STZ-induced cognitive impairment and oxidative-nitrosative stress in rats.
Materials and Methods: Adult male Wistar rats were injected with ICV STZ (3 mg/kg) bilaterally on days 1 and 3. α-tocopherol and tocotrienol were administered chronically to rats for 21 days starting from day 1 of STZ injection. The learning and memory behavior was assessed by using the Morris water maze and elevated plus maze on days 15, 16, 17, 18, 19, and 20. The rats were sacrificed on day 21 and whole-brain homogenate was used for the estimation of parameters of oxidative stress, including malondialdehyde, reduced glutathione, superoxide dismutase and catalase, nitrite levels, and acetylcholinesterase (AChE) activity.
Results: α-tocopherol as well as tocotrienol showed significantly less cognitive impairment in both the behavioral paradigms, but the effect was more potent with tocotrienol. Both isoforms of vitamin E effectively attenuated the reduction in glutathione and catalase and reduced the malonaldehyde, nitrite as well as cholinesterase activity in the brains of ICV STZ rats in a dose-dependent manner.
Conclusions: The study demonstrates the effectiveness of vitamin E isoforms, with tocotrienol being more potent in preventing the cognitive deficits as well as the oxidative and nitrosative stress caused by ICV STZ in rats and suggests its potential in the treatment of neurodegenerative diseases such as AD.
175. Comparison of Anti-Acne Inducing Bacteria Activity and α-Mangostin Content of Mangosteen Fruit Rind Extracts Prepared Using Different Solvents
Werayut Pothitirat,1 Mullika Traidej Chomnawang,2 and Wandee Gritsanapan1
1Department of Pharmacognosy, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
2Department of Microbiology, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
Mangosteen (Garcinia mangostana Linn.) of the family Guttiferae is one of the important exported fruits of Thailand and known as the “Queen of Fruit.” The fruit rind of this plant has been used in Thai traditional medicine for the treatment of skin infections, wounds, diarrhea, and dysentery. The extract of the fruit rind has demonstrated various biological effects, especially antimicrobial activity. It contains a high amount of xanthones, especially α-mangostin, which is a major constituent and is responsible for antibacterial activity. The purpose of this study is to compare antiacne-inducing bacteria activity and α-mangostin content of mangosteen fruit rind extracts, which were prepared by using different solvents (i.e., hexane, dichloromethane, ethanol, and water). All extracts were tested for antibacterial activities against bacteria-inducing acne, including Propionibacterium acnes and Staphylococcus epidermidis, using the broth microdilution method. The dichloromethane extract exhibited the strongest antibacterial effect with MIC values for both bacterial species at 3.91 μg/mL, while MBC values against P. acnes and S. epidermidis were 3.91 and 15.63 μg/mL, respectively. TLC autobiography indicated that α-mangostin in all extracts, except the water extract, was a major active component against both P. acnes and S. epidermidis. By high-performance liquid chromatography, the dichloromethane extract yielded the highest content (46.21%; w/w) of α-mangostin, followed by ethanol extract (18.03%; w/w), hexane extract (17.21%; w/w), and water extract (0.54%; w/w), respectively. These findings suggest that the dichloromethane extract of G. mangostana fruit rind should be a potential source of natural antiacne-inducing bacteria for the further development of antiacne pharmaceutical preparations.
176. Cyclooxygenase-2 Assay and Molecular Modeling Study of Artifacts from Prasaplai, a Thai Traditional Medicine
Prasan Tangyuenyongwatana,1 Nipa Jongkon,2 Chak Sangma,2 and Wandee Gritsanapan1
1Department of Pharmacognosy, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
2Department of Chemistry, Faculty of Science, Kasetsart University, Bangkok, Thailand
The rhizome of Zingiber cassumunar Roxb. (Zingiberaceae) and the seed of Nigella sativa L. (Ranunculaceae) are components in Prasaplai, a Thai traditional medicine used for relieving dysmenorrhea and adjusting the menstrual cycle. The rhizome of Z. cassumunar contains (E)-4-(3,4-dimethoxyphenyl)but-3-en-1-ol (compound D) as a major active anti-inflammatory component, while the seed of N. sativa contains fatty acids, which are linoleic, oleic, and palmetic acids. The powdered mixture of these two plants produced three artificial fatty acid esters, which are (E)-4-(3,4-dimethoxy-phenyl)but-3-en-1-yl linoleate (1), (E)-4-(3,4-dimethoxy-phenyl)but-3-en-1-yl oleate (2), and (E)-4-(3,4-dimethoxy-phenyl)but-3-en-1-yl palmitate (3) after storage. The objectives of this study were to examine the cyclooxygenase-2 (COX-2)-inhibitory activity of these artificial fatty acid esters in vitro and to predict their bindings with COX-2 by using molecular docking and comparing them to compound D. The immortalized mouse PGH-1 and PGH-2 null cells were used as the COX-1 and COX-2 deficient cell lines, respectively, to investigate the inhibition activities and selectivity of these compounds. From the experimental results, all three artifacts showed inhibition potencies less than 50% and suggested that they were inactive compounds, which corresponded to the poor binding results from molecular docking with the AutoDock 3.0.5 program(AutoDock, Scripper Research Institute, Lusolla, CA, USA). These results suggest that these artifacts might act as prodrugs of compound D, which were not active directly to COX-,2 and they would be hydrolyzed after absorption in the intestinal tract and release the parent active anti-inflammatory compound (compound D).
177. Determination of Three Components in Sonneratia caseolaris L. by High-Performance Liquid Chromatography
Chutima Limmatvapirat,1 Sarawut Sukjindasathein,1 Juree Charoenteeraboon,2 and Thawatchai Phaechamud3
1Pharmaceutical Chemistry, Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
2Biopharmacy, Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
3Pharmaceutical Technology, Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
Sonneratia caseolaris L., which belongs to the plant family Sonneratiaceae, mainly grows in intertidal forests of Thailand. The methanol extract was rich in polyphenols. The phytochemical studies on S. caseolaris L. have revealed the presence of gallic acid, luteolin, and luteolin-7-O-glucoside. Two flavonoids, luteolin, and luteolin-7-O-glucoside, isolated from this plant, showed anti-inflammatory, antioxidant, and hepatoprotective activities. The development of a quality-control method is an essential issue for effective clinical study of this plant. However, there has been no report on controlling the quality of S. caseolaris L. extract and the simultaneous determination of its multiple constituents. The purpose of this study was to develop a simple, rapid high-performance liquid chromatography and ultraviolet spectrophotometric (HPLC-UV) method for the simultaneous determination of the three active compounds (gallic acid, luteolin, and luteolin-7-O-glucoside) to evaluate the quality of this medicinal plant. In order to obtain an optimal extraction procedure, 70% methanol showed the highest extraction efficiency. The optimal extraction time was 15 minutes. The HPLC separation was performed on a RP-18 semipreparative column, Waters, Milford, MA, USA (250 mm × 4.6 mm i.d.; 10 μm) with a gradient of acetonitrile and 0.1% (v/v) aqueous phosphoric acid, at a flow rate of 5.0 mL/min, detected at 280 nm. The regression equations showed the good linear relationships between the peak area of each component and their concentration. This assay procedure was successfully performed to the determination of three compounds in extracts of sepals, seeds, and stamens. The developed assay method was rapid and could be readily utilized as a guideline for the quality control of S. caseolaris L. extracts.
178. Effect of Gastric pH on Calcium Availability of Various Calcium Salts
Yossanan Weerapol, Kamonrak Cheewatanakornkool, and Pornsak Sriamornsak
Department of Pharmaceutical Technology and Pharmaceutical Biopolymer Group (PBiG), Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
Calcium absorption does not depend only on the amount of the calcium present in the products, but also on other factors, such as solubility and ionization. The objective of this study was to compare calcium release and ionization from various salts of calcium supplement tablets (i.e., calcium gluconate, calcium lactate, calcium lactate gluconate, calcium acetate, calcium hydrogen phosphate, and calcium carbonate). The effect of pH of test fluid on calcium availability was also studied. Compressed tablets of different calcium salts were prepared by direct compression. Calcium release from tablets was tested in simulated gastric fluid without pepsin (SGF; pH 1.2) or fed state-simulated gastric fluid (FSSGF;, pH 4.8), using USP dissolution apparatus. The ionized calcium concentration in test fluid was determined by calcium ion selective electrode. The results demonstrated that most of the formulations released calcium within 1 hour. Tablets made of higher solubility calcium salts demonstrated a faster calcium release. The tablets containing calcium hydrogen phosphate showed the slowest calcium release, followed by calcium carbonate, in SGF. The calcium release from calcium hydrogen phosphate and calcium carbonate tablets in gastric fluid with higher pH (FSSGF) showed a slower calcium release than in SGF. The results suggested that most of the calcium salts could be used as calcium supplements. However, the use of calcium hydrogen phosphate and calcium carbonate required an acidic environment in order to be dissolved in the gastrointestinal tract.
179. Evaluation of Antioxidant Activity of Ginger (Zingiber officinale, ROSCOE) Extract
Petchompoo Siriphan,1 Chaiyo Chaichantipyuth,2 and Suchada Chutimaworaphan1
1Department of Pharmacy, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
2Department of Pharmacognosy, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
Ginger (Zingiber officinale, Roscoe) is widely used in traditional medicine, with beneficial effects reported in numerous diseases. Gingerols, the main pungent principles of ginger, have antioxidative activity. In this study, the antioxidative activity of hydroalcoholic, acetone, and isolated [6]-gingerol extracts were evaluated for their H-donating and hydroxyl radical activities, using the 1,1-diphenyl-2-picrylhydrazyl (DPPH) and the hydroxyl radical-scavenging assay, and compared this with standard antioxidants, such as α-tocopherol, ascorbic acid, and quercetin. The coarse powder of the rhizome was extracted with solvents of different polarities by maceration and then partitioned. All extracts exhibited good antioxidant activity with concentration-dependent quenching of DPPH and hydroxyl radical. Maximum inhibitory activities noticed were 50–93 and 60–85%, respectively. The DPPH-scavenging activity of acetone and isolated [6]-gingerol extracts were lower than α-tocopherol and ascorbic acid, but standard [6]-gingerol activity was greater than these reference standards. Regarding hydroxyl radical-scavenging activity, the acetone, isolated [6]-gingerol extract, and standard [6]-gingerol exhibited identical activities to protect deoxyribose degradation and higher than reference standard quercetin. Hydroalcoholic extract had the lowest activities in both tests. It was observed that the oxidative activity of isolated [6]-gingerol were not much higher than the crude acetone extract. It was found that acetone extract of ginger comparatively exhibited potent free radical–scavenging activities. These results suggested that the extract from ginger locally grown in Thailand showed potential oxidative activity that is capable in the formulation of pharmaceutical and cosmetic products.
180. Factors Affecting of Cannabinoids Content of Hemp (Cannabis sativa L.) for Characteristic Guidelines and Forensic Applications
Surapol Natakankitkul,1 Prapatsorn Tipparat,2 Pipop Chamnivikaipong,3 Sirot Chutiwat,4 and Maitree Suttajit5
1Pharmaceutical Science, Faculty of Pharmacy, Chiang Mai, Thailand
2Regional Medical Science Center, Chiang Mai (RMSC), Chiang Mai, Thailand
3Institute of Survey and Narcotic Herbs, Northern Office the Narcotic Control Broad, Chiang Mai, Thailand
4Queen Sirikit Botanic Garden, Botanical Garden Organization, Chiang Mai, Thailand
5School of Sciences and Technology, Naresuan University, Phayao, Thailand
Cannabis has been used as a psychoactive drug and mostly as a source of fiber and seed for centuries. Although hemp cultivation was primarily prohibited due to its narcotic effect as marijuana or the drug cannabis, it has recently been rediscovered to be a potentially important crop for world fiber and seed production. We have studied and analyzed three major cannabinoids: delta-9-tetrahydrocannabinol (THC), cannabidiol CBD, and cannabinol CBN by gas chromatography in hemp grown in the north of Thailand. Both THC and CBD were related to stage of plant development, which is highest in the flowering stage. The range of THC and CBD contents at flowering were 1.032–1.220 and 0.324–0.407% (w/w) by dry weight, respectively, with the CBD:THC ratio of 0.31–0.4. The CBN content was found to be less than 0.005% (w/w) by dry weight, whereas mean THC and CBD contents in marijuana (three seized samples) were 2.926 and 0.092% (w/w), respectively, giving a CBD:THC ratio of only 0.02. The plant height was significantly (P < 0.05) correlated to diameter of stem, location (i.e., elevation of growing area), and stage of plant development. Data suggest that the other environmental factors, plant growth and development factors, and species of cannabis plant may influence the THC and CBD contents. Using cannabinoid composition, particularly the CBD:THC ratio, will be as possible tools in marker-assisted characteristic guidelines in hemp, which are useful for breeding and forensic applications.
181. Gastric Ulcer Protective Effect of YA-hom in Rats
Wisuda Suvitayavat, Pathawee Intayoong, and Suwan Thirawarapan
Department of Physiology, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
Gastric ulcer can be induced by several factors, including an imbalance between aggressive and defensive factors, such as increasing gastric secretion and decreasing gastric mucus secretion. Ya-hom, a Thai traditional medicine, has been shown to inhibit gastric secretion and stimulate gastric mucus secretion, which supports its use for gastric ulcer protection and treatment. However, the effect of Ya-hom on gastric ulcer protection has not been reported. Thus, this study aimed to assess the gastric ulcer protective activity of Ya-hom. The gastric lesion protective effect of Ya-hom was evaluated by oral administration of Ya-hom (1, 2, and 4 g/kg) before induction of gastric ulcer by hydrochloric acid (0.6 M HCl, 6 mL/kg), aspirin (ASP; 200 mg/kg), and water immersion restraint stress (WIR; 16 + 2°C), in comparison to cimetidine (0.1 g/kg). Four, 6, and 5 hours after HCl-, ASP-, and WIR-gastric ulcer induction, respectively, the rats were sacrificed for the determination of gastric ulcer. The results showed that Ya-hom (1, 2, and 4 g/kg) inhibited HCl-, ASP-, and WIR-induced gastric ulcer in a dose-dependent manner with the maximum inhibition of 93.4, 54.5, and 61.8%, respectively. Cimetidine inhibited HCl-, ASP-, and WIR-induced gastric ulcer with the inhibition of 78.4, 76.3, and 48.0%, respectively. These data indicate that Ya-hom can protect gastric ulcer induced by acid, aspirin, and stress.
182. Herbal Denture-Cleansing Solution
Sumarn Saraya,1 Jiranee Puttikulbovorn,2 and Rungravi Temsiririrkkul3
1Department of Microbiology, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
2Regional Medical Sciences Center, Suratthani, Thailand
3Departrment of Pharmaceutical Botany, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
Candida albicans is regarded as the pathogenesis of candidosis-associated denture stomatitis. This research was done to develop and evaluate the use of herbal denture-cleansing solution for the treatment of Candida adherence on denture materials. The antifungal properties of medicinal plants were determined by the agar diffusion method. The four plant species that inhibited the growth of Candida albicans ATCC 1032 were Alpinia galanga (L) Willd., Punica granatum L, Terminalia chebula Retz., and Lawsonia inermis L. and showed antifungal activities in the minimum concentrations (MICs) of 6.25, 6.25, 12.5, and 12.5 mg/mL, respectively. The evaluation of the anticandidal effect of herbal denture-cleansing formulations by time-kill assay demonstrated that the herbal denture-eleansing formulation A, which contained the crude extract of A. galangal (L) Willd., exhibited antifungal activity 2 times stronger than formulation B. Formulation B contained the crude extract of A. galangal (L) Willd. combined with P. granatum L. A serial MIC of formulation A showed that concentrations of 16MIC, 8MIC, and 4MIC inhibited fungal cells within 60, 90, and 180 minutes, respectively. The same MIC concentrations of formulation B showed antifungal activity at concentrations of 16MIC, 8MIC, and 4MIC within 120, 180, and 240 minutes. In vitro anticandidal assessment of herbal denture-cleansing formulation to Candida adherence on acrylic resin material was studied. The results showed that concentrations of 16MIC, 8MIC, and 4MIC of herbal denture-cleansing formulation A inhibited C. albicans within 3, 5, and 60 minutes, while the formulation B at the concentrations of 16MIC and 8MIC inhibited the fungal cells on the surface of acrylic resins within 15 and 30 minutes. Consequently, the result of the physical stability testing showed that all serials of MIC of the herbal denture-cleansing formulation were not stable after 6 months.
183. Investigation of Modified Shellac as a Potential Material for Enhanced Enteric Drug Delivery of Xenobiotic Agents
Danuch Panchapornpon,1 Jurairat Nunthanid,1 Manee Luangtana-Anan,1 Chutima Limmatvapirat,2 Satit Puttipipatkhachorn,3 and Sontaya Limmatvapirat1
1Department of Pharmaceutical Technology, Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
2Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
3Department of Industrial Pharmacy, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
Xenobiotic agents that are susceptible to degradation in the stomach due to the acidic environment or gastric enzymes need to be stabilized by enteric film coating. Shellac was one of the enteric polymers that showed prominent acid and enzyme resistance and could be used for coating. However, native shellac showed major disadvantages, including lower solubility at intestinal pH and instability. The purpose of this study was to improve the properties of shellac by structure modification. Native shellac was partially esterified with various cyclic anhydrides, including trimellitic, succinic, and phthalic anhydrides by dry media reaction. The formation of shellac esters was monitored by instrumental analysis, including DSC, FTIR, and NMR spectroscopy. The shellac esters were then comparatively characterized for physicochemical properties, mechanical properties, and stability profiles. The DSC results suggested the utilization of cyclic anhydrides in the reaction, as indicated by decreasing the enthalpy of endothermic peaks. The presence of new FTIR peaks confirmed the formation of shellac esters. Shellac succinate was easily formed at 60°C, while those of shellac phthalate and shellac trimellitate occurred at higher temperatures. The level of esterification should be related with the molecular size of cyclic anhydrides, as suggested by the lowest molecular weight of succinic anhydride. In addition, shellac esters demonstrated better solubility than native shellac. Both shellac succinate and shellac phthalate were dissolved in the range of intestinal pH (5.8–6.7), while the gastric protection was still in the acceptance level. The mechanical properties of film were changed after esterification. Percent elongation of films prepared from shellac esters was increased, as compared to films prepared from native shellac. The result suggested the greater flexibility of shellac esters. Shellac esters also demonstrated better stability profiles. The polymerization, which was the major cause of instability, did not significantly occur in shellac esters. In conclusion, the findings could give the solution for the problems of native shellac and gade it more potential as an alternative polymer for enteric drug delivery.
184. Molecular Cloning of Anti-HIV Protein-Encoding Gene from Canna indica L
Chanpen Wiwat and Apanchanid Thepouyporn
Microbiology, Mahidol University, Bangkok, Thailand
Various parts of Canna indica L. is widely used in traditional medicine as a diaphoretic and diuretic in fevers and dropsy. Its decoction showed antitumor activity in rats, weak cytotoxic activity in cell culture, and antibacterial activity. Previous study of the water extract of C. indica L. rhizomes showed HIV-1 reverse-transcriptase inhibitory activity (IC50 22.56 μg/mL). Two purified proteins from C. indica L., namely 31 and 14 kDa, showed significant HIV-1 RT inhibition with IC50 values of 17.41 and 19.25 μg/mL, respectively. The aims of this research work were to confirm the anti-HIV activity of C. indica L. Tat/Rev MC99 virusΔproteins by the syncytium reduction assay, using a defective and Tat/Rev transfected 1A2 cell line, and to clone and express the anti-HIV-encoding gene. The results showed that the purified proteins from roots, rhizomes, and leaves gave positive results of the syncytium inhibition assay with EC50 values of 7, 12.2, and 2.8 μg, respectively. The N-termini of purified 14- and 31-kDa proteins were determined, and the amino-acid residues were shown to be as follows: FN(d)VLLGGNGGVLAIV and NPSPLQNYTVAN(A)NXS, respectively. The specific primers were designed based on these N- terminal sequences and used for amplification of the specific genes. The mRNAs extracted from various parts of the plant were reverse transcribed to cDNAs. Subsequently, they were used as a template for gene amplification by polymerase chain reaction. Molecular cloning of the specific genes is in progress.
185. Phytochemical Screening, Antioxidant, and Antimutagenic Activities of Polygonum odoratum Extracts
Methin Phadungkit1 and Thidarat Somdee2
1Pharmaceutical Sciences, Faculty of Pharmacy, Mahasarakham, Thailand
2Faculty of Public Health, Mahasarakham University, Mahasarakham, Thailand
Studies showed that strong antioxidant and antimutagenicity activities of plant extract possess great potential as functional foods for cancer prevention. The aims of the current study were to evaluate antioxidant and antimutagenic activities and to study the chemical constituents of Polygonum odoratum extracts. Antioxidant activity was determined by the ability of each extract to scavenge the free radical, 1,1-diphenyl-2-picrylhydrazyl (DPPH). Antimutagenic activity was evaluated by using the Ames test in Salmonella typhimurium (TA 98 and TA 100 strains). The results showed that the herbal extract showed high antioxidant activity with the EC50 value at concentrations of 12.71 ± 0.28 mcrg/mL. The extract showed strong antimutagenicity activity against the S. typhimurium TA 100 strain, with percentage inhibition values ranging from 64.48 ± 11.57 to 76.59 ± 12.76% but showed weak activity against the S. typhimurium TA 98 strain. In the present study, flavonoids and condensed tannins from herbal exteacts were proposed to be antioxidant and antimutagenic agents, respectively. In conclusion, the apparent antioxidant and antimutagenic activities of the herbal extracts further suggests their potential usefulness in cancer prevention.
186. Preparation and Evaluation of Antimicrobial Nanoemulsion Containing Herbal Extracts
Sirikarn Pengon,1 Chutima Limmatvapirat,2 and Sontaya Limmatvapirat1
1Department of Pharmaceutical Technology, Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
2Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
Some herbal extracts, especially volatile oils from plants, demonstrate interesting antimicrobial activity. However, most of them have a limited application due to water immiscibility and incompatibility with body fluids. The purpose of the study was to solve the problems by conversion of volatile oils into biocompatible oils in water (o/w) nanoemulsion. The nanoemulsion was prepared by blending oil components with the water phase and subjected it to a high-pressure homogenizer. Formulation factors and process variables were evaluated by monitoringparticle sizes, particle charges of oil droplets, and other physical stabilities. The optimized formula was also evaluated for antimicrobial activity. The results demonstrated that molecular structure and polarity of volatile oils affected physical stability. Some formulas containing clove oil showed cracking of emulsion after storage. Physical stability was improved by increasing the percentage of surfactants (e.g., Cremophore® and Tween®). The emulsion containing 3–10% (w/w) of surfactants did not show any evidence of creaming or cracking. As percentage of Cremophore increased, the particle sizes of oil droplets were decreased. These particle sizes were reduced to less than 100 nanometers in emulsion containing 10% (w/w) of Cremophore. The increasing number of surfactants in the o/w interface and the decreasing surface tension of emulsions should be a possible explanation. The process variables, including pressure and number of feed cycles, also affected the particle sizes of oil droplets. The particle sizes were rapidly decreased from 2,500 to 500 nm after increasing the pressure to 10,000 psi for 1 cycle. The selected nanoemulsion formula was to determine the antimicrobial activity against Staphylococcus aureus. This nanoemulsion demonstrated almost 100% killing at the level of 10-fold dilution. The possible mechanism might be involved in the disruption of bacterial cell wall. In conclusion, the study demonstrated the crucial variables that determined the properties of nanoemulsions. The knowledge gained could support the formulation of stable, effective antimicrobial nanoemulsions in the near future.
187. Screening of Cytotoxic Activity of Thai Medicinal Plants against Human Cholangiocarcinoma Cells In Vitro
Wiratchanee Mahavorasirikul,1 Wanna Chaijaroenkul,1 Arunporn Itharat,2 and Kesara Na-Bangchang1
1Graduate Program in Biomedical Sciences, Faculty of Allied Health Sciences, Thammasat University, Pathumthani, Thailand
2Applied Thai Traditional Medicine Center, Faculty of Medicine, Thammasart University, Pathumthani, Thailand
The study aimed to investigate cytotoxic activities of crude ethanol extracts of a total of 30 selected Thai medicinal plants against the human cholangiocarcinoma cell line in vitro. The cytotoxic effect was evaluated by using the MTT assay. Six ethanolic extracts from Zingiber officinale (rhizome), Piper chaba (fruits), Atractylodes lancea (rhizome), Kaempferia galanga (rhizome), Mesua ferrea (flower), and Prasapoayai regimen exhibited promising activity with 50% inhibitory concentrations (IC50) of 40.74, 34.26, 24.09, 37.36, 48.28, and 44.12 μg/mL, respectively. Further studies to investigate for their activities in animal model of cholangiocarcinoma, including mechanisms of action, together with identification of chemical structures of active principles, are required.
188. Skin Irritation and Skin Sensitization Tests of the Mixed Extracts
Sareeya Reungpatthanaphong
Pharmaceutical and Natural Products Department, Thailand Institute of Scientific and Technological Research, Pathumthani, Thailand
According to Thai tradition medicine, the rhizome of Zingiber montanum (Koenig) link ex Dietr (Syn. Z. cassumunar Roxb; in Thai “Phlai”; Fam. Zingiberaceae) was therapeutically used as an anti-inflammatory, analgesic, etc. Also, Mulberry (Morus alba L.; in Thai “Mon”; Fam. Moraceae) leaves, which are widely used in Oriental medicine, were scientically proved as having antioxidant, melanocyte inhibition, and whitening agent. A mixture of the extracts of “Phlai” and Mulberry (PM) is proposed to be used as a skin vitality promoter and a whitening agent. In this research article, the skin irritation and the skin sensitization were conducted, following the Test Guidelines No. 404; Acute Dermal Irritation/Corrosion and No. 406; Skin Sensitization of the OECD Guidelines for the testing of chemicals in the rabbits and the guinea pigs, respectively. The results demonstrated that the PM did not show any toxicity signs.
189. The Inhibitory Potential of Thai Mango Seed Kernel Extract against Methicillin-Resistant Staphylococcus aureus
Pimsumon Jiamboonsri,1 Pimolpan Pithayanukul,1 Mullika Traidej Chomnawang,2 and Rapepol Bavovada3
1Department of Pharmacy, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
2Department of Microbiology, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
3Department of Pharmaceutical Botany, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
Plant extracts are a valuable source of novel antibacterials in combating pathogenic isolates of methicillin-resistant Staphylococcus aureus (MRSA), a global nosocomial problem. Mangos (Mangifera indica L.), which belong to the family Anacardiaceae, grow in tropical and subtropical regions, and their components are commonly used in folk medicine to produce a wide variety of remedies. Among the edible portions, the extract from Thai mango seed kernel (MSKE) (Mangifera indica L. c.v. “Fahlun”) has been shown to have a relatively high phenolic content and to exhibit antityrosinase and antihepatotoxic activities (Nithitanakool et al., 2009a, 2009b). The aim of this study was to assess in vitro antibacterial susceptibility against Staphylococcus aureus and MRSA of the MSKE and its phenolic principles (gallic acid, methyl gallate, and pentagalloylglucose). The MSKE and its principles were screened for inhibitory zone, and the minimum inhibitory concentration (MIC)/the minimum bactericidal concentration (MBC) were determined by serial dilution with a standardized microdilution broth methodology. Ten clinical isolates of MRSA and a standard control strain (ATCC 25923) were cultured and exposed to the plant extract. Vancomycin was used as a positive control agent. The MSKE and its principles showed promising anti-MRSA activities and inhibited the growth of MRSA continually for at least 24 hours at 2- and 4-MIC concentrations. The standard control strain demonstrated similar results.
References
- Nithitanakool, S., Pithayanukul, P., Bavovada, R. (2009a). Antioxidant and hepatoprotective activities of Thai mango seed kernel extract. Plant Med in press.
- Nithitanakool, S., Pithayanukul, P., Bavovada, R., Saparpakorn, P. (2009b). Molecular docking studies and anti-tyrosinase activity of Thai mango seed kernel extract. Molecules 14:257–265.
190. Tocotrienol Attenuates Diabetic Neuropathic Pain: Involvement of Oxidative-Nitrosative Stress and Inflammatory Cascade Pathway
Anurag Kuhad and Kanwaljit Chopra
University Institute of Pharmaceutical Sciences, Panjab University, Chnadigarh, India
Diabetic neuropathic pain, an important microvascular complication in diabetes mellitus, is recognized as one of the most difficult types of pain to treat. The development of tolerance, inadequate relief, and potential toxicity of classical antinociceptives warrant the investigation of the newer agents to relieve this pain. Reactive oxygen/nitrogen species, cytokines, and apoptosis are implicated in the pathogenesis of diabetic neuropathy. The aim of the present study was to explore the effect of tocotrienol on thermal and mechanical hyperalgesia, allodynia, oxidative-nitrosative stress, inflammation, and apoptosis in streptozotocin-induced experimental diabetes. Diabetic rats developed neuropathy, which was evident from a marked hyperalgesia and allodynia associated with enhanced nitrosative stress, release of inflammatory mediators (TNF-α, IL-1α, and TGF-1α), and caspase 3. Chronic treatment with tocotrienol (25, 50, and 100 mg/kg body weight; p.o.) for 4 weeks starting from the week 4 of streptozotocin injection significantly attenuated behavioral, biochemical, and molecular changes associated with diabetic neuropathy. Moreover, diabetic rats treated with the insulin-tocotrienol combination produced a more pronounced beneficial effect, as compared to their per se groups. The major finding of the study is that insulin alone corrected the hyperglycemia and partially reversed the pain response in diabetic rats. However, the combination with tocotrienol not only attenuated the diabetic condition, but also reversed neuropathic pain through the modulation of oxidative-nitrosative stress, inflammatory cytokine release, and caspase-3 in the diabetic rats, and thus it may find clinical application to treat neuropathic pain in diabetic patients.
191. In Vitro Assessment of Cytochrome P450 Inhibition by Cilostazol and Its Metabolites
Min-Jung Kim,1 Ji-Hyun Mun,2 and Soo Kyung Bae3
1Department of Pharmacology, Inje University College of Medicine, Busanjin-gu, South Korea
2Department of Pharmacology, Inje University, Busan, South Korea
3Department of Pharmacology and PharmacoGenomics Research Center, Inje University College of Medicine, Busan, South Korea
Aim: The aim of this study was to assess the potential for drug-drug interactions between cilostazol and substrate of cytochrome P450 isozymes.
Methods: Using pooled human liver microsomes, the in vitro inhibitory effects of cilostazole and its metabolites on CYP1A2 (phenacetin O-deethylation), CYP2A6 (coumarin 7-hydroxylation), CYP2B6 (bupropion 6-hydroxylation), CYP2C8 (paclitaxel 6α-hydroxylation), CYP2C9 (tolbutamide 4-hydroxylation), CYP2CI9 (S-mephenytoin 4’-hydroxylation), CYP2D6 (dextromethorphan O-demethylation), CYP2E1 (chlorzoxazone 6-hydroxylation), and CYP3A (midazolam 1’-hydroxylation/ testosterone 6α-hydroxylation) activities were examined. To judge the mechanism-based inhibitor of them, after preincubation for 30 minutes, interaction in the presence of NADPH was also examined. After incubation, formed respective metabolites were analyzed by liquid chromatography/tandem mass spectrometry (LC-MS/MS).
Results: No significant reversible inhibition of nine CYP activities were observed for 0.1, 1, and 10 μM of cilostazol and its metabolites. However, the inactivation of CYP3A by cilostazol was NADPH-, time- and concentration dependent, and was characterized by Kinact values of 0.25 min−1, and K1 values of 25.9 μM, respectively. The kinact/KI ratio (inactivation efficiency), which is used to evaluate the potential clinical impact of mechanism-based inhibition, for cilostazol on inhibition of CYP3A activities is lower than that of CYP3A inactivation by clarithromycin or diltiazem used as positive controls.
192. Inhibitory Effects of Dibenzocyclooctadiene lignans from Fructus schisandrae on CYP2C8-mediated Paclitaxel Metabolism in Human Liver Microsomes
Yan-Yan Zhang and Ling Yang
Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China
Dibenzocyclooctadiene lignans are the principle active ingredients of Fructus Schisandrae, which is extensively used as an antitussive, sedative, and tonic agent in China, Korea, Japan, and Russia. According to the Chinese Pharmacopoeia 2005 edition, these lignans are recognized as the major constituents in this Chinese herb medicine. Recently, Schisandra extract and some purified lignans have been found to enhance the efficacy of paclitaxel against multidrug-resistance tumor cell lines Huang et al., 2008). In the present study, an in vitro study was undertaken to evaluate the influence of seven major Schisandra lingans on CYP2C8, which is the primary metabolic enzyme for paclitaxel. Among all these components, gomisin G (IC50 = 4.3 ± 0.7 μM) exhibited the most potent inhibitory effect on CYP2C8-dependent paclitaxel 6α-hydroxylation in a noncompetitive manner, with an apparent Ki value of 4.1 μM. Gomisin A (IC50 = 8.7 ± 0.8 μM, Ki = 8.6 μM) and pregomisin (IC50 = 9.3 ± 0.8 μM, Ki = 8.0 μM) were also found to potently inhibit CYP2C8 in a noncompetitive and competitive manner, respectively. However, schizandrol A (IC50 > 100 μM), schizandrin A (IC50 = 31.8 ± 1.7 μM), B (IC50 = 12.6 ± 1.9 μM), and C (IC50 > 100 μM) were considered less likely to cause drug-drug interactions via the inhibition of CYP2C8 activity. Since paclitaxel holds a complex pharmacological and toxicological profile with a narrow therapeutic window (Mielke et al., 2005), special attention should be taken on the alteration of paclitaxel pharmacokinetic behavior, when coadministered with the corresponding dibenzocyclooctadiene lignans or the crude extract of Fructus Schisandrae.
References
- Huang, M., Jin, J., Sun, H., Liu, G.T. (2008). Reversal of P-glycoprotein-mediated multidrug resistance of cancer cells by five schizandrins isolated from the Chinese herb, Fructus schizandrae. Cancer Chemother Pharmacol 62:1015–1026.
- Mielke, S., Sparreboom, A., Steinberg, S.M., Gelderblom, H., Unger, C., Behringer, D., et al. (2005). Association of paclitaxel pharmacokinetics with the development of peripheral neuropathy in patients with advanced cancer. Clin Cancer Res 2005 11:4843–4850.
193. Abstract Withdrawn
194. The Evaluation of Pharmacokinetic Drug Interaction Potential OF 9 Herbal Medicines Marketed IN Korea
Soo-Jin Park,1 Eun Jeong Choi,1 Im-Sook Song,1 Ji-Hong Shon,2 and Jae-Gook Shin3
1Department of Pharmacology and PharmacoGenomics Research Center, Inje University College of Medicine, Busan, South Korea
2Department of Pharmacology and Clinical Pharmacology, Pharmacogenomics Research Center, Inje University College of Medicine and Busan Paik Hospital, Busan, South Korea
3Department of Pharmacology and Clinical Pharmacology, Pharmacogenomics Research Center, Inje University College of Medicine and Busan Paik Hospital, Busan, South Korea
The primary objective of this study was to establish basic in vitro herb-drug interaction information. For this, the inhibition and induction potential of nine herbal medicines (HMs) marketed in Korea were investigated on drug-metabolizing enzymes, drug transporters, and nuclear receptors. To investigate the inhibitory potential of ethanol extract of HMs, nine CYP and three UGT isoform specific probes were incubated with human liver microsomes in the absence and presence of HMs and preincubation experiment were performed for CYPs to test mechanism-based inactivation. In addition, the regulation of expression of target genes in human hepatocytes were evaluated after exposure to nine HMs for 24 hours. Analysis was performed by real-time polymerase chain reaction in the presence of a TaqMan® (Taqman, Applied Biosystems, Foste city, CA, USA) probe. In the inhibition test, Scutellaria baicalensis Georgi (Hwanggeum) inhibited CYP1A2 activity with an IC50 value of 2.16 μg/mL. The inhibition of the CYP2C8 and CYP2C9 activities by Hwanggeum (50%) was dependent on the preincubation process, suggesting that Hwanggeum may be a mechanism-based inactivator of CYP2C8 and CYP2C9. Hwanggeum also strongly inhibited UGT1A1 activity with an IC50 value of 13.4 μg/mL. In human hepatocytes, treatment with nine HMs also resulted in significant modulation in target gene expression. Desmodium styracifolium (Osbeck.) Merr. and Hwanggeum showed more than 1.5-fold induction on several target genes. Gardenia jasminoides Ellis (Chija) appeared to be one of the most noticeable regulators of 12 target genes. Interestingly, the pattern of regulation had been shown to be changed by concentration of Chija. Especially, Hwanggeum showed an inhibition/induction effect on UGT1A1, depending on conditions of the experiment. These results indicated that some of the ethanol extract of the HMs used in Korea can significantly induce or inhibit drug-metabolizing enzymes, transporter, and nuclear receptor, which could cause herb-drug interactions. Consequently, it would be expected that both the acute and chronic herb administration may have some roles in the pharmacokinetic results of coadministered drug, although it remains to further evaluate the clinical relevance of these effects.
195. Abstract Withdrawn
196. Effect of Aqueous Extract of Cleistocalyx nervosum on Antioxidant and Detoxifying Enzymes in Wistar Rat
Sirinya Taya, Charatda Punvittayagul, Wanida Inbut, and Rawiwan Wongpoomchai
Department of Biochemistry, Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand
Cleistocalyx nervosum var. paniala (Thai name “Ma-kiang”) is a local fruit in the northern region of Thailand. High amounts of polyphenols in C. nervosum showed in vitro antioxidant and antitumor activities. The present study was designed to determine the effect of aqueous extract of C. nervosum on antioxidant and detoxifying enzymes in rat liver. Male wistar rats, weighing about 190 g, were divided into three groups. Group 1 was represented as a vehicle control. Groups 2 and 3 were received 100 and 500 mg/kg body weight of aqueous extract via intragastrium for 4 weeks, 5 times per week. The effect of C. nervosum on oxidative stress and antioxidant and detoxifying systems were evaluated. C. nervosum induced glutathione levels and the activities of glutathione peroxidase, heme oxygenase, and glutathione-S-transferase and did not affect TBARS formation in rat liver. In conclusion, the aqueous extract of C. nervosum enhanced some hepatic antioxidant and detoxifying enzymes in male rat.
197. Inhibitory Effects of Caffeic Acid and Its Amide Derivatives on Human Cytochrome P4501A2
Churdsak Jaikang,1 Kanokporn Niwatananun,2 Siripun Narongchai,3 Paitoon Narongchai,3 and Chaiyavat Chaiyasut1
1Department of Pharmaceutical Sciences, Faculty of Pharmacy, Chiang Mai University, Chiang Mai, Thailand
2Department of Pharmaceutical Care, Faculty of Pharmacy, Chiang Mai University, Chiang Mai, Thailand
3Department of Forensic Medicine, Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand
Caffeic acid (CAF) and its analogs are found in fruits, vegetables, and coffee beans. Caffeic amide analogs possess antioxidant activity and free radical–scavenging potential. The effects of caffeic acid and its amide derivatives on CYP1A2 activity in human liver microsomes, using phenacetin as a probe substrate, were examined. Ethyl 1-(3′,4′-dihydroxyphenyl) propen amide (EDPA), octyl 1-(3′,4′-dihydroxyphenyl) propen amide (ODPA), phenylmethyl 1-(3′,4′-dihydroxyphenyl) propen amide (PMDPA), and phenethyl 1-(3′,4′-dihydroxyphenyl) propen amide (PEDPA) were synthesized, using PyBOP as a coupling agent. The formation of the CYP-specific metabolite following coincubation with CAF or its amide derivatives were determined to establish Ki values for this enzyme. The inhibition of CAF and its amide derivatives (EDPA, ODPA, PMDPA, and PEDPA) on CYP1A2, according to Dixon and Cornish-Bowden plots, were uncompetitive. The inhibitory potency of CAF and its amide derivatives on phenacetin O-deethylation was comparable with Ki values of 1.34, 0.68, 0.55, 1.77, and 0.53 nM, respectively. The results indicate that CAF and its amide derivatives exhibit an uncompetitive mode of inhibition on CYP1A2 activity in human liver microsomes. This may lead to important implications in the prevention of CYP1A2-mediated chemical carcinogenesis.
198. Investigating the Molecular Mechanism of Lapatinib-induced Liver Toxicity
Eric Chun Yong Chan,1 Jing Wen Oh,1 Lee Sun New,1 and Han Kiat Ho2
1Department of Pharmacy, Faculty of Science, Natioanal University of singapore, Singapore, Singapore
2Institute of Medical Biology, Sinagpore, Singapore
Lapatinib, the first FDA-approved oral dual inhibitor of the ErbB-1 and ErbB-2 tyrosine kinases, has demonstrated immense potential in metastatic breast cancer that has failed standard therapy. However, the release of a black-box warning for lapatinib-related hepatoxicity threatened its success. Structurally, lapatinib has the propensity to generate toxicologically relevant quinoneimine-reactive metabolites upon O-dealkylation and oxidation. Therefore, we aimed to investigate lapatinib’s potential in reactive metabolite generation to enhance our understanding of the molecular mechanism of its hepatotoxicity. In vitro microsomal metabolic stability assay was performed to assess lapatinib’s susceptibility to biotransformation and its stability over time. Using reduced glutathione (GSH) as an oxidative stress biomarker, GSH-reactive metabolite profiling and GSH consumption experiments demonstrated that lapatinib did not yield GSH-binding reactive metabolites directly and indirectly, respectively. Subsequently, an in vitro mechanism-based inactivation study was performed to investigate the lapatinib-based inactivation of CYP3A4 by using both human liver microsomes (HLMs) and recombinant CYP3A4 isozyme (rCYP3A4). All quantitative analyses were performed by using UPLC/MS/MS. Finally, the in vitro toxicity of lapatinib was compared with other quinazolinamine analogs (erlotinib and gefitinib) by quantification of the ATP in culture through luciferase-generated luminescent signal, which directly correlates with THLE-2 cell viability. Our results showed, for the first time, that lapatinib inactivated the testosterone-6α-hydroxylase activity of both rCYP3A4 and HLM in a mechanism-based inactivation approach, exhibiting values of Ki of 2.679 μM and Kinact 0.0212 min−1. On the other hand, lapatinib did not inhibit cell proliferation more significantly than erlotinib and gefitinib at similar doses in our in vitro cell assay.
199. Human Hepatocyte Nuclear Factor 4α Is Regulated by miR-24
Shingo Takagi, Miki Nakajima, Katsuhiko Kida, Tatsuki Fukami, and Tsuyoshi Yokoi
Graduate School of Medical Science, Kanazawa University, Kanazawa, Japan
Hepatocyte nuclear factor (HNF) 4α is a key transcription factor regulating the expression levels of various drug-metabolizing enzymes and transporters. MicroRNAs (miRNAs) are small noncoding RNAs that regulate gene expression by translational repression or mRNA degradation. We identified several potential miR-24 recognition elements (MRE24) in the coding region as well as the 3′-untranslated region (3′-UTR) of human HNF4α mRNA. By luciferase analyses in HEK293 cells, it was demonstrated that the reporter activity of plasmid containing the HNF4α coding region downstream of the luciferase gene was significantly decreased by the overexpression of miR-24. In contrast, the reporter activity of plasmid containing the MRE24 in the 3’-UTR was not affected. Exogenously expressed HNF4α protein in HEK293 cells was significantly decreased by the overexpression of miR-24, when an expression plasmid excluding the 3’-UTR was transfected. These results suggest that the MRE24s in the coding region, but not in the 3′-UTR, are functional for the negative regulation. Endogenous HNF4α protein was significantly decreased (27% of control) by the overexpression of miR-24 in HepG2 cells. Under this condition, HNF4α mRNA level was also significantly decreased (20% of control). These results suggest that the human HNF4α is repressed by miR-24 through the mRNA degradation, rather than the translational repression. We also found that treatment of HepG2 cells with the protein kinase C activator, phorbol 12-myristate 13-acetate, significantly increased (8-fold) the expression level of the precursor, miR-24. The induction was diminished by the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) kinase (MEK) inhibitor, U0126, and the p38 inhibitor, SB202190. These results suggest that the ERK and p38 pathways are involved in the induction of miR-24 expression. In conclusion, we found that miR-24, of which expression is regulated by MAPK, negatively regulates human HNF4α expression.
200. Oxidative Stress Is Required for the Complete Activation of the Nrf2 Signaling Mechanism
Azman Abdullah,1 Neil Kitteringham,2 Christopher E. Goldring,2 Rosalind Jenkins,2 Jane Hamlett,2 and B. Kevin Park2
1Department of Pharmacology, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia
2Department of Pharmacology and Therapeutics, The University of Liverpool, Liverpool, United Kingdom
The ability to control protein expression levels in cells provides many advantages. For example, quantitative and temporal information on the role of a heterologous gene in normal cellular physiology and following exposure to xenobiotics can be obtained. A human hepatoma-derived cell line (HepG2) incorporating a second-generation doxycycline-inducible gene expression system (rtTA2S-M2) has been developed by Goldring et al. (2006). Nrf2 (nuclear factor erythroid 2 p45-related factor 2) is a transcription factor that responds to cellular stress. The rtTA2S-M2, controlling the Nrf2 gene, was studied in the HepG2 cell line. Stable clones transfected with the Nrf2 transgene showed high levels of functional expression occurred in a time- and dose-dependent manner, and they also exhibited stable inducible expression (Goldring et al., 2006). The functionality and effectiveness of this Nrf2 clone in mediating an actual response toward chemical insult was then tested. The clone was incubated with doxycycline at the dose and incubation period that would cause maximal Nrf2 induction, and at the same time, it was also subjected to concurrent diethyl maleate (DEM) treatment at a concentration and incubation period that would allow maximal Nrf2 translocation into the nucleus. The proteome of control and treated Nrf2 clones were then analyzed by using the two-dimensional gel electrophoresis (2-DE) proteomics method. The results showed that there were not many differences between the controls and DEM-treated samples in terms of 2-DE protein expression changes. It was, therefore, suggested that the cultured transgenic cells might lack certain Nrf2 binding partners or had lost some metabolic capabilities and signaling pathways that are inherent in primary liver cells. It could also be that cells need to undergo oxidative stress as well as an increase in Nrf2 levels in order to elicit a full Nrf2 response.
Reference
- Goldring, C.E., et al. (2006). Development of a transactivator in hepatoma cells that allows expression of phase I, phase II, and chemical defense genes. Am J Physiol Cell Physiol 290:C104–C115.
201. Possible Role of Heme Oxygenase-1 and Prostaglandins in Pathogenesis of Cerebral Malaria: Induction of Heme Oxygenase-1 by Prostaglandin D2 and Metabolite by Human Astrocyte CCF-STTG1 Cells In Vitro
Jiraporn Kuesap and Kesara Na-Bangchang
Pharmacology and Toxicology Unit, Graduate Program in Biomedical Sciences, Faculty of Allied Sciences, Thammasat University, Pathumthani, Thailand
Astrocytes are the most abundant cells in the central nervous system that play a role in maintaining the blood-brain-barrier and in neural injury, including cerebral malaria, a severe complication of Plasmodium falciparum. Prostaglandin (PG) D2 is abundantly produced in the brain and regulates sleep response. Moreover, PGD2 is a potential factor derived from Plasmodium falciparum within erythrocytes. Heme oxygenase-1 (HO-1) catalyzes enzymes in the heme breakdown process to release iron, carbon monoxide, and biliverdin/bilirubin and may influence iron supply to the falciparum parasites. Here, we showed that treatment of human astrocyte cell, CCF-STTG1, with PGD2 significantly increased the expression levels of HO-1 mRNA. Western blot analysis showed that PGD2 treatment increased the level of HO-1 protein in a dose- and time-dependent manner. Thus, PGD2 may be involved in the pathogenesis of cerebral malaria by inducing HO-1 expression in malaria patients.
202. Sauchinone as a Novel AMPK-activating Candidate Treats Iron-Overload Liver Disease
Young Woo Kim,1 Sung Min Lee,1 Sang Mi Shin,1 Janie S. Brooks,1 Sang Chan Kim,2 and Sang Geon Kim1
1College of Pharmacy, Seoul National University, Seoul, South Korea
2College of Oriental Medicine, Daegu Haany University, Daegu, South Korea
Iron overload disorders, such as hepatic siderosis and hemochromatosis, cause hepatocyte injury and inflammation by oxidative stress, which may lead to liver fibrosis and hepatocellular carcinoma. Presently, no pharmacological therapy is available for these disorders. Previously, we reported that sauchinone (a lignan from Saururus chinensis) has anti-inflammatory activity. This study investigated the in vivo and in vitro efficacy of sauchinone as a novel candidate for iron-overload liver disease and explored the underlying mechanism. As an effort to establish an in vivo model, iron accumulation and liver histopathology were assessed in mice injected with phenylhydrazine (hemolytic agent). Phenylhydrazine treatments promoted iron accumulation and ferritin expression in the liver, causing hepatocyte death and inflammation with an increase in plasma levels of arachidonic acid (AA), a proapoptotic inflammatory fatty acid. In the absence of interference of iron accumulation or AA production, sauchinone remarkably attenuated liver injury and inflammation and activated AMPK in mice. In hepatocyte models, iron and AA synergistically increased cytotoxicity, as indicated by increases in reactive oxygen species and mitochondrial permeability transition. Sauchinone prevented the ability of iron+AA to induce mitochondrial dysfunction and apoptosis. Sauchinone treatment results in LKB1 activation in hepatocytes, which, in turn, leads to the activation of AMPK: These events contribute to cell survival. Evidence of these cytoprotective effects is shown by the the reversal of sauchinone’s restoration of mitochondrial membrane potential by either overexpression of a dominant negative mutant of AMPK alpha or compound C treatment. Conclusions: Our results demonstrate that sauchinone protects the liver from toxicity induced by iron accumulation that may be mediated by LKB1-dependent AMPK activation.
203. Specification of the Binding and Interaction Features: Effect on CYP2A6 Inhibition
Chunzhi Ai and Ling Yang
Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China
It is well known that the inhibition of cytochorme P450 2A6 (CYP2A6), a nicotine-metabolizing enzyme, plays a significant role in decreasing cigarette consumption, smoking intensity, and the concurrency of tobacco-dependent diseases. A computational investigation has been carried out on P450 2A6 and its naphthalene inhibitors to explore the crucial molecular features contributing to binding specificity. The molecular bioactive orientation was obtained by docking (FlexX) these compounds into the active site of the enzyme. Also, the density function theory (DFT) method was further used to optimize the structures, with subsequent optimizations by the molecular lipophilic potential (MLP) and molecular electrostatic potential (MEP) analysis. The results reveal that MLP and MEP methods are of significant value to describe the binding process and interactions between CYP2A6 and its inhibitors. An increase trend was observed for the pIC50s values with the ascendance of the minimal MLPs. And the minimal MEPs appear to locate at the hydrogen-bond-acceptor substitutes. Consequently, the differences between the MLP and MEP, together with the highest occupied molecular orbital (HOMO) energy, are the key factors in determining the inhibitory effects of the series of compounds on CYP2A6. This investigation will be helpful to provide a better understanding of the interaction between CYP2A6 and the ligand, which will aid in the rational design of novel potent inhibitors of the enzyme.
204. In Vitro Skin Permeation and Stability Studies of Quercetin
Aroonsri Priprem,1 Saengrawee Sutthiparinyanont,1 and Malyn Chulasiri2
1Pharmacy, Khon Kaen University, Khon Kaen, Thailand
2Pharmacy, Mahidol University, Bangkok, Thailand
Quercetin is one of the plant flavonoids that is potentially beneficial for the skin. It is hypothesized that skin permeation should deliver quercetin to bypass its metabolism via diffusion at a subtoxic range. Shed snake skin and human skin, as the barrier membranes, were mounted between donor and receptor compartments of Franz diffusion cells, with quercetin solution as the donor and phosphate buffer (pH 7.4) or albumin solution as the receptor solutions. An initial phase (0–1 hour) with a permeation rate of 0.0010 μg/square cm•min of quercetin was found when using shed snake skin into phosphate buffer (pH 7.4) at 37°C. Followed this initial phase, decomposition of quercetin was observed due to catalysis from phosphate ions. The decomposition rate of quercetin in phosphate buffer was found to be 5 times higher than that in water. Saturated concentration of quercetin in water and phosphate buffer, investigated by using UV spectrophotometry, was 0.2 and 1.1 μg/mL, whereas its log partition coefficient (log Ko/w) was 2.5 at 25°C. Thermal analysis by TGA and DSC indicates first-phase transition at 90° and decomposition temperature at 320–350°C. Decomposition of quercetin in an aqueous environment could affect its active antioxidant structure and activity. Quercetin decomposition in an aqueous environment profoundly increased with an increase in pH and temperature. Using albumin solutions as the receptor, the permeation rate of quercetin was in line with the results using water and phosphate buffer, while its permeation coefficient was not significantly different when using shed snake and human skin as the barriers (P < 0.05). However, a permeation rate when using shed snake skin was 4 times higher than that using human skin. This leads to conclude that the permeation of quercetin through the shed snake and human skin occurred via passive diffusion, and after permeation, it is subjected to rapid chemical decomposition in the presence of phosphate ions but complex formation in the presence of albumin.
205. Mitragynine and Methanol Extract of Kratom Leaf (Mitragyna speciosa Korth.) Inhibited Compound Nerve Action Potential
Somsmorn Chittrakarn,1 Niwat Keawpradub,2 Kitja Sawangjaroen,1 Supaporn Kansenalak,1 and Benjamas Janchawee1
1Pharmacology, Prince of Songkla University, Hat-Yai, Thailand
2Pharmacognosy and Pharmaceutical Botany, Prince of Songkla University, Hat-Yai, Thailand
Kratom (Mitragyna speciosa Korth.) is a Thai medicinal plant known to produce narcotic-like actions. Native people also use it to treat muscle ache and fatigue. The effects of mitragynine and a methanolic extract of kratom leaves were investigated on an isolated sciatic nerve preparation. The rat was anesthetized by intraperitoneal injection of sodium pentobarbital in the concentration of 50 mg/kg body weight. Both the left and right sciatic nerves were dissected out and placed in a three-compartment chamber, in which bipolar stimulating and recording platinum electrodes were placed under the nerve. The central compartment had a volume of 1 mL containing Tyrode’s solution or test drugs. The nerve was stimulated with a supramaximal voltage at a frequency of 0.6 Hz and duration of 0.04 msec by a Grass S88 stimulator, Grass Simulator, Grass Technologies, West Warwick, RI, USA via an SIU5 stimulus isolation unit. The compound action potentials were displayed on an oscilloscope via an AM502 Differential Amplifier for recording nerve conduction (ms), amplitude (mV), and duration (ms). Kratom extract in the concentrations of 10– 40 mg/mL and mitragynine (2 mg/mL) blocked the compound action potential. Mitragynine, present at 2 mg/mL, caused a gradual decrease of the nerve conduction, amplitude, and duration of the compound action potential. High concentrations of mitragynine did not produce a complete blockage of compound action potential. The mechanism of action of kratom extract or mitragynine should be further investigated on the neuromuscular junction.
206. Arsenic-induced VEGF Expression in Human Lung Adenocarcinoma A549 Cells: Involvement of Transcription Factors, HIF-1 α and β-catenin
Sumitra Suntararuks,1 Zarni Min,2 Piyajit Watcharasit,1 and Jutamaad Satayavivad1
1Laboratory of Pharmacology, Chulabhorn Research Institute, Bangkok, Thailand
2Graduate Program in Environmental Toxicology, Chulabhorn Graduate Institute, Bangkok, Thailand
Arsenic is a potent human carcinogen; however, the mechanism underlining arsenic carcinogenesis is not well understood. Arsenic-stimulated expression of vascular endothelial growth factor (VEGF), which is a key signaling molecule in angiogenesis, a crucial process for carcinogenesis and tumor progression, has been suggested to play an important role in arsenic carcinogenesis (Kamat et al., 2005; Soucy et al., 2004). It is well established that hypoxia-inducible factor-1α (HIF-1α) is a major regulator for VEGF expression; however, the role of HIF-1α in VEGF upregulation by arsenic remains inconclusive. Kamat et al. reported that chronic exposure of arsenic caused VEGF upregulation through a mechanism involving HIF-1α (Kamat et al., 2005), while Soucy et al. showed that inhibition of HIF-1α by siRNA did not alter arsenic ability to stimulate VEGF (Soucy et al., 2004). Recently, α-catenin has been shown to be involved in VEGF transcriptional regulation (Skurk et al., 2005). This present study, therefore, investigated whether HIF-1α and α-catenin are involved in arsenic-induced VEGF in human lung adenocarcinoma A549 cells. Arsenic caused an increase in VEGF mRNA level, as measured by reverse-transcriptase polymerase chain reaction in a dose-dependent manner. Determination of HIF-1α by immunoblot analysis revealed that nuclear accumulation of HIF-1α increased following arsenic treatment. We also measured the level of α-catenin in the nucleus and found that arsenic treatment did not alter α-catenin levels in the nucleus. Taken together, these results suggested that arsenic-induced VEGF may involve transactivation of HIF-1α but not α-catenin.
References
- Kamat, C.D., Green, D.E., Curilla, S., Warnke, L., Hamilton, J.W., Sturup, S., et al. (2005). Role of HIF signaling on tumorigenesis in response to chronic low-dose arsenic administration. Toxicol Sci 86:248–257.
- Skurk, C., Maatz, H., Rocnik, E., Bialik, A., Force, T., Walsh, K. (2005). Glycogen-synthase kinase3beta/beta-catenin axis promotes angiogenesis through activation of vascular endothelial growth factor signaling in endothelial cells. Circ Res 96:308–318.
- Soucy, N.V., Klei, L.R., Mayka, D.D., Barchowsky, A. (2004). Signaling pathways for arsenic-stimulated vascular endothelial growth factor-A expression in primary vascular smooth muscle cells. Chem Res Toxicol 17:555–563.
207. Characterization of Houttuynin Adducts with Phenylalanine and Lysine in Insulin B Chain
Zhipeng Deng, Xiaoyan Chen,and Dafang Zhong
Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China
Houttuynia cordata injection was once widely used in the clinical setting for the treatment of fever, cold, inflammation, diarrhea, and other infection diseases in China, but was withdrawn from the market in 2006 due to serious hypersensitivity reactions occasionally. Houttuynin (decanoyl acetaldehyde) is the major antibacterial constituent in the injection. Houttuynin has an α-dicarbonyl group in its structure, which may covalently bind to critical proteins in vivo. In this study, we used the oxidized B chain of insulin as a model peptide to investigate the binding of houttuynin to proteins. To identify the modification site and structures of adducts generated in the houttuynin-modified insulin B chain, both the houttuynin-pretreated and untreated peptides were digested with V8 protease and Pronase E, then the resulting peptides and amino acids were subjected to LC/MSn analysis. The adducts were found mainly to occur at N-terminal phenylalanine, which afforded 1:1 and 2:1 houttuynin- phenylalanine adducts with the loss of one and three water molecules, respectively. The concentration of the former was higher than the latter. Their structures were characterized as Schiff’s base adduct and pyridinium adduct, respectively, on the basis of the chemical and MSn evidence. In addition, a small amount of houttuynin-lysine adducts was also identified, but the concentration of pyridinium-type adduct was far higher than the Schiff’s base adduct. In conclusion, houttuynin adducted more readily with the N-terminal α-NH2 group presented in the peptides or proteins than the primary amine group under physiological conditions. The protein adducts are speculated to be responsible for the adverse reactions induced by Houttuynia cordata injection.
208. CYP2E1 Distribution in Rat Liver Coincides with Site of Acute Liver Injury but Not the Lobe-Specific Response to Administration of the Cholangiocarcinogenic Agent, Furan
Kevin Hickling,1 Jonathan Hitchcock,2 John Evans,3 Tim Hammond,4 and Kevin Chipman5
1Safety Assessment, Astrazeneca, Loughborough, United Kingdom
2Molecular Toxicology Drug Safety Research and Development, Pfizer Ltd., Kent, United Kingdom
3Safety Assessment, AstraZeneca, Cheshire, United Kingdom
4AstraZeneca, Cheshire, United Kingdom
5Professor of Cell Toxicology and Head of School, School of Biosciences, The University of Birmingham, Birmingham, United Kingdom
Furan is an industrial solvent and byproduct of certain food-cooking processes. Exposure to furan is of concern, as it is a potent liver carcinogen in rat with inconclusive evidence of genotoxicity. The toxic properties of furan are attributed to the formation of the reactive metabolite, cis-2-butene-1,4-dial via CYP2E1 metabolism. We have investigated the potential for furan to induce initial hepatotoxicity, oxidative stress, and DNA damage in the rat and how this leads to lobe-specific cholangiofibrosis and eventual cholangiocarcinoma. Furan was administered daily to rats orally via gavage at 30 mg/kg body weight and livers were analyzed at time points between 8 hours and 30 days after treatment. After a single dose, furan induced an initial centrilobular hepatocyte necrosis. There was a marked association between the sites of injury and location of CYP2E1 protein in hepatocytes. Indicators of DNA oxidation (8-oxo-dG), markers of cell apoptosis, and cell-cycle arrest in hepatocytes also correlated with these sites. However, the extent of initial injury was significantly increased in severity in the right and caudate lobes, which did not correlate with the density of CYP2E1 distribution. From day 3, there was significant repair by hepatocyte replication, in spite of continued furan administration, but CYP2E1 expression was significantly reduced in these newly formed cells at all time points and was confined to sporadic individual hepatocytes in centrilobular areas. Reduced expression may, therefore, confer a significant survival advantage to hepatocytes during continued furan exposure. Cholangiofibrosis was observed by day 12 arising from the biliary system of the initially more severely injured caudate and right liver lobes. These areas again showed no recovery in CYP2E1 expression, compared to naïve liver, indicating that, apart from the first exposure to furan, CYP2E1 metabolism of furan is of only minor significance to the genesis of cholangiofibrosis and its progression to cholangiocarcinoma in the rat.
209. Effect of Thunbergia laurifolia on Rat Splenocyte Detected by FTIR
Chatkul Techakitiroj,1 Ratana Sindhuphak,2 Songpol Chivapat,3 Teeradech Suramana,2 Nikorn Dusitsin,2 and Palarp Sinhaseni4
1Department of Microbiology and Biochemistry, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
2Institute of Health Research, Chulalongkorn University, Bangkok, Thailand
3Medicinal Plant Research Institute, Department of Medical Sciences, Ministry of Public Health, Nonthaburi, Thailand
4Department of Pharmacology and Physiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
FTIR spectroscopy is a well-established technique for the study of protein structural modifications, which provide new findings that reveal important information. In this study, we applied FTIR to study spleen of rat exposed to water extract of Thunbergia laurifolia in conjunction with serum biochemicals. It was found that FTIR spectra at amide I in spleen of rats exposed water extract of T. laurifolia significantly shifted, as compared to control rats. This finding is in agreement with bilirubin-protective effects against nitrosative stress, since ferric-nitrosyl heme oxygenase complex was possibly reflected in the bond vibration detected by FTIR 5 cm-1, characterized by lower and broader complex. These patterns allow a greater degree of ligand conformational freedom resulting in changes in amide I, the vibration of the amino-acid side chain inside the protein. We proposed that the action of T. laurifolia through heme oxygenase may be an adaptive response to the dopamine-like action of the water extract of T. laurifolia. The mechanistic understanding of signalings through nitrosative stress in the modulation of rat adaptive response should be further investigated to explain the widely known antidote effects of Thunbergia laurifolia.
210. Effects of Multiwalled Carbon Nanotubes on Human B Lymphocytes
Rawiwan Maniratanachote,1 Tina Thurnherr,2 Peter Wick,2 and Harald Krug2
1National Nanotechnology Center, National Science and Technology Development Agency, Pathumthani, Thailand
2Laboratory of Materials-Biology Interactions, EMPA-Swiss Federal Laboratories of Materials Testing and Research, St. Gallen, Switzerland
Multiwalled carbon nanotubes (MWCNTs) represent a very promising class of carbon nanomaterials with potential use in many technological and medical fields. They consist of several coaxial graphite cylinders that form tubes with diameters of 10–30 nm. Because of their unique properties and promising advantages, MWCNTs are used in various applications and are, therefore, produced in large quantities. However, there is increasing concern that carbon nanotubes might exhibit toxic effects similar to those of other fibrous, biopersistent particles, such as asbestos. Several studies using rodent models revealed that carbon nanotubes have the potential to cause inflammation and formation of granuloma in the lung. In addition, carbon nanotubes can internalize into the cells and may interact with organelles, thereby producing specific pathologies in various organs. The immune system is an important defense mechanism against environmental insults and thus might be affected by the exposure to these novel nanomaterials. We, therefore, studied the effects of three industrially produced MWCNTs on human B lymphocytes. Asbestos was used as a reference material. Various biological responses and toxicological end-points were thoroughly investigated. Generally, all MWCNTs tested showed a low toxicity on B lymphocytes. There were some differences in toxicity and intracellular responses among the three MWCNTs, suggesting differences in the quality and constituents of these materials. In contrast to MWCNTs, which induced some oxidative stress responses, asbestos showed no toxicity toward these cells.
211. Hepatitis B Virus × Inhibits Phase II Detoxifying Enzyme Expression through Repression of C/EBPβ Activity Il
Je Cho, Sung Hwan Ki, S. Carroll Brooks, III, and Sang Geon Kim
College of Pharmacy, Seoul National University, Seoul, South Korea
A genome-wide in silico screening rendered the genes of phase II enzymes in the rat genome whose promoters contain the putative DNA elements interacting with CCAAT/enhancer binding protein (C/EBP) and NF-E2-related factor (Nrf2). The hepatitis B virus × (HBx) protein modulates the transactivation and/or the repression of genes regulated by some bZIP transcription factors. This study investigated the effects of HBx on the induction of phase II enzymes with the aim of elucidating the role of HBx interaction with C/EBPβ or Nrf2 bZIP transcription factors. Immunoblot and reporter gene analyses revealed that HBx interfered with the constitutive and inducible GSTA2 transactivation promoted by oltipraz (C/EBPβ activator), but not that by tert-butylhydroquinone (t-BHQ, Nrf2 activator). Moreover, HBx transfection completely inhibited GSTA2 reporter gene activity induced by C/EBPβ, but failed to inhibit that by Nrf2. Gel-shift assays identified that HBx inhibited the increase in C/EBPβ–DNA complex formation by oltipraz, but not the increase in Nrf2–DNA complex by t-BHQ. Immunoprecipitation and immunoblot assays verified the direct interaction between HBx and C/EBPβ. Moreover, chromatin immunoprecipitation assays confirmed HBx inhibition of C/EBPβ binding to its binding site in the GSTA2 gene promoter. HBx repressed the induction of other phase II enzymes, including GSTP, UDP-glucuronyltransferase 1A, microsomal epoxide hydrolase, GSTM1, GSTM2, and γ-glutamylcysteine synthase. These results demonstrate that HBx inhibits the induction of phase II detoxifying enzymes, which is mediated by its interaction with C/EBPβ, but not Nrf2, substantiating the specific role of HBx in phase II detoxifying capacity.
212. Influence of Gold Nanoparticles on Intracellular Organelles and Cell Viability in Hela Cells
Oraya Kamnerdsin,1 Veerawat Korkiatsakul,1 Kasama Rakphetmanee,1 Rojrit Rojanathanes,2 Chintana Chirathaworn,3 and Amornpun Sereemaspun4
1Department of Anatomy, Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand
2Department of Chemistry, Faculty of Science Chulalongkorn University, Bangkok, Thailand
3Department of Microbiology, Faculty of Medicine Chulalongkorn University, Bangkok, Thailand
4Department of Anatomy, Faculty of Medicine Chulalongkorn University, Bangkok, Thailand
Gold nanoparticles have been widely used in medicine as drug delivery and biosensor for many years. However, the cytotoxicity of gold nanoparticles has been conflicted. In this study, we aimed to examine the in vitro cell biological response and intracellular localization of the xenobiotic nanoparticles. Citrate-stabilized gold nanoparticles were synthesized and tested with HeLa cells, the human cervical cancer cell line, at concentrations of 10, 50, and 100 ug/mL, in order to investigate the cytotoxicity and morphological change, using the MTT assay and LM and TEM, respectively. The results indicate that cell viability was decreased in 100 ug/mL citrate/gold nanoparticle-treated groups in day 3 after gold nanoparticle incubation. The existence of gold nanoparticles in HeLa cells was found. The mitochondria, rER, and nuclear membrane alteration were also observed. In conclusion, this study suggests that gold nanoparticles are capable of decreasing cell viability and intracellular organelles could be altered structurally and functionally. The possible exposure risk of gold nanoparticles should be of concern.
213. NADP(H) Quinone Oxidoreductase 1 and Caspase-2: Key Orchestrators in Goniothalamin-induced Coronary Artery Smooth Muscle Cell Apoptosis?
Chan Kok Meng,1 Nor Fadilah Rajab,1 Laily bin Din,1 David Siegel,2 David Ross,2 and Salmaan H. Inayat-Hussain1
1Environmental Health Program and UKM Medical Molecular Biology Institute, Faculty of Allied Health Sciences, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia
2Department of Pharmaceut Sciences, University of Colorado Health Sciences Center, Denver, Colorado, USA
The activation of p53 has been shown to inhibit in-stent stenosis (ISS) by abrogating coronary artery smooth muscle cell (CASMC) proliferation as well as facilitating apoptosis. Therefore, pharmacotherapeutic agents that induce p53 may play a potential role in preventing this iatrogenic disease. Goniothalamin (GN), a styryllactone derived from Goniothalamus andersonii, has been shown to induce apoptosis in various cell lines, including CASMCs. We have recently demonstrated that GN induces DNA damage leading to CASMC apoptosis. In this study, we have further investigated the molecular mechanisms of GN-induced apoptosis. Our results demonstrate that DNA damage induction by GN occurs via pathways dependent of reactive oxygen species and independent of topoisomerase II inhibition. As a result of DNA damage, p53 was elevated in CASMCs as early as 2 hours after GN treatment. However, in contrast to previous findings, NQO1 protein expression and activity did not correlate with p53 elevation in GN-treated CASMCs. In agreement with the role of p53-induced apoptosis, GN-treated CASMCs resulted in a time-dependent increase in caspase-2 activity. Subsequently, caspase-9 and -3 activity were simultaneously activated. The apical caspase-8 in the receptor mediated apoptosis was not activated in the execution of CASMCs. In addition, GN-treated Jurkat caspase-8 null cells demonstrated no significant difference in cytotoxicity, as compare to wild type, which further corroborated the insignificant role of caspase-8 in our study. These data provide insight into the mechanisms of GN-induced apoptosis, which may have important implications to be developed as a drug for the treatment of ISS.
214. Pinostrobin Attenuates Diethylnitrosamine-induced Rat Hepatocarcinogenesis through Some Xenobiotic-metabolizing Enzymes
Rawiwan Wongpoomchai,1 Suphachai Charoensin,1 Umnat Mevatee,2 and Wilart Pompimon3
1Department of Biochemistry, Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand
2Department of Anatomy, Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand
3Department of Chemistry, Faculty of Science, Lampang Rajabhat University, Lampang, Thailand
Pinostrobin (5-hydroxy-7-methoxyflavanone) is found in Boesenbergia pandurata (fingerroot or krachai). Many previous studies suggested that pinostrobin possessed pharmacological and biochemical properties. It had no mutagenic effect on both bacteria and rat liver. The chemopreventive effect of pinostrobin, using a rat liver micronucleus assay, and possible inhibitory mechanism were investigated. Male Wistar rats were divided into four groups and intraperitoneum was injected by 30 mg/kg body weight of diethylnitrosamine (DEN), a rodent hepatocarcinogen, at days 1 and 4 of the experiment. Groups 1–3 received 10– 50 mg/kg body weight of pinostrobin by gavage feeding for 12 days, before the first DEN administration for 6 days. Group 4 was fed with 5% Tween-80 as a vehicle. The treatment of 10– 50 mg/kg of pinostrobin reduced micronucleated hepatocyte formation in a dose-dependent manner in DEN-initiated rats. To clarify the chemopreventive mechanism of pinostrobin in DEN-initiated rat hepatocarcinogenesis, the expression and activity of some phase I and II xenobiotic-metabolizing enzymes were investigated. The level of hepatic CYP2E1 protein, a major enzyme activating DEN to its DNA-reactive form, in rats administered 50 mg/kg of pinostrobin, was significantly decreased, compared with control rats. While others, such as NADPH:P450 reductase, heme oxygenase-1, and glutathione S-transferase (GST) isozyme A and P, were unaffected. Interestingly, GST activity was significantly enhanced at the same dose of CYP2E1 reduction. From these results, pinostrobin possibly inhibited DEN-induced micronucleated hepatocyte formation by decreasing the CYP2E1 expression and inducing GST activity in the liver. It was suggested that pinostrobin from Thai fingerroot may exert a chemopreventive effect on an early stage of DEN-induced rat hepatocarcinogenesis.
215. Protective Effect of Quercetin on Hydrogen Peroxide–induced TJ Disruption
Somrudee Chuenkitiyanon,1 Thitima Pengsuparp,2 and Suree Jianmongkol3
1Interdisciplinary Program in Pharmacology, Graduate School, Chulalongkorn University, Bangkok, Thailand
2Biochemistry and Microbiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
3Pharmacology and Physiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
Oxidative stress has been an underlying cause of endothelium dysfunction and a loss in vascular protective barrier against xenobiotics. Several studies showed that oxidative stress affected the expression and distribution of tight junction proteins, leading to tight junction disruption and increase in permeability. The purposes of this study were to investigate the effect of quercetin, a known antioxidant flavonoid, on the hydrogen peroxide induced–disruption of tight junction. ECV304 cells were pretreated with quercetin (10 μM) for 30 minutes prior to the addition of hydrogen peroxide (100 μM). After the incubation period of 4 hours, the permeability was determined by following phenol red transport. In addition, the expression and localization of tight junction proteins were determined with Western blot and immunofluorescent techniques. In this study, exposure of hydrogen peroxide at 100 μM for 4 hours markedly increased the paracellular permeability and decreased TEER value without an observable effect on cell viability. Pretreatment with quercetin prevented hydrogen peroxide–induced hyperpermeability as well as a loss of TEER value. The disruption of occludin and ZO-1 at the cell border was also prevented in the quercetin pretreatment group. In addition, pretreatment with quercetin could prevent the hydrogen peroxide–induced reduction of expression of these two tight junction proteins, significantly. In conclusion, quercetin could preserve the barrier integrity of ECV304 upon challenge with hydrogen peroxide. Our findings suggested that quercetin was able to prevent the loss of expression of tight junction protein as well as its assembly.
216. Rosiglitazone, PPARgamma Agonist, Stimulates Paracellular Transport in Primary Inner Medullary Collecting Duct Cells
Sunhapas Soodvilai,1 Zhanjun Jia,2 and Tianxin Yang2
1Department of Physiology, Faculty of Science, Mahidol University, Bangkok, Thailand
2Internal Medicine, University of Utah and Veterans Affairs Medical Center, Salt Lake City, Utah, USA
The involvement of peroxisome proliferator-activated receptor gamma (PPAR-gamma) in mediating thiazolidinedione-induced fluid retention has been reported. However, the mechanism of increased fluid reabsorption in the distal nephron in response to the PPAR-gamma agonist is still controversial. We determined the mechanisms of rosiglitazone (RGZ)-stimulated ion transport by using the electrophysiological studies on primary cultures of inner medullary collecting duct (IMCD) cells. Exposure of IMCD cells monolayers to RGZ, amiloride-sensitive short-circuit current (Isc),an index of ENaC activity, was unchanged at 24 hours but was significantly decreased at 48 hours, corresponding to parallel inhibition of mRNA expressions of alpha-, beta-, and gamma-ENaC. Despite ENaC inhibition, the transepithelial resistance (TER) was significantly reduced by 50% of control (P < 0.01) at 24 hours and by 70% of control at 48 hous (P < 0.01) after being treated with RGZ, suggesting that an alterative route contributed for an increased of ion transport. Therefore, we examined whether RGZ affected paracellular Na+ and Cl− transport. RGZ treated IMCD cell monolayers exhibited increases of the paracellular Cl− transport by 230% of control, to a lesser extent, the paracellular Na+ transport (50% of control) suggesting increased permeability to Cl− over Na+. Taking advantage of the availability of the conditional CD PPAR-gamma KO mice, we established PPAR-gamma deficient IMCD cell cultures to address the specificity of RGZ in increasing paracellular ion transport. The mutant cells were largely resistant to RGZ-induced changes in the paracellular Na+ or Cl− permeability seen in the wild-type cells. Together, our data suggest that PPAR-gamma activation by PPAR-gamma agonist stimulates paracellular ion transport and inhibited ENaC in primary IMCD cells.
217. Synergist Effect of Hydrogen Peroxide with Ethanol on U937 in Cell Death
Sirikul Dangmanee and Wisit Tangkeangsirisin
Biopharmaceutical Sciences, Silpakorn University, Nakornpathom, Thailand
Hydrogen peroxide, one of the most reactive oxidants in the gas phase of cigarette smoke, induces cell death in several cell types by enhancing oxidative stress. On the other hand, ethanol-induced oxidative stress promotes macrophage apoptosis, which frequently occurs in advanced human atherosclerotic plaques. This study was done to prove whether hydrogen peroxide cotreatment with ethanol in human monocytic leukemic cell line U937 causes cell death in a synergistic fashion. We found that ethanol alone did not affect cell death. However, hydrogen peroxide induced apoptosis in a dose-dependent manner. Cotreatment of ethanol and hydrogen peroxide proved to be synergistic in inducing apoptosis and the IC50 value of cotreatment was smaller, compared to hydrogen peroxide alone. We conclude that ethanol, in concomitance with hydrogen peroxide, induces apoptosis synergistically in U937. These results support that cigarette smoking with alcohol drinking is linked to high risk for atherosclerosis.
218. The Possible Induced Genotoxicity in Dry-Cleaning Egyptian Workers Exposed to Tetrachloroethylene
Ashraf M. Emara
Tanta Poison Center, Clinical Toxicology, Faculty of Medicine, Tanta, Egypt
Environmental exposures that increase the rate of damage above background levels increase the potential for unrepaired lesions to become permanent mutations. This concept has led to the use of oxidative DNA damage in the assessment of occupational and environmental exposures to chemicals that are capable of inducing an oxidative stress. The objective of the present study was to determine the possible tetrachloroethylene-induced genotoxicity in Egyptian dry-cleaning smokers and nonsmoking workers. The study was carried out on 80 adult male workers. Subjects designated as controls (n = 40) were healthy persons with no occupational exposure to tetrachloroethylene frequency-matched to exposed tetrachloroethylene dry-cleaning subjects (n = 40) by age and lifestyle. The following parameters were measured blood tetrachloroethylene, lymphocyte hydroxydeoxyguanosine (8-OHdG), urinary 8-OHdG, and chromosomal aberrations and damage. The average level of tetrachloroethylene in air was found to be ≤140 ppm for all workers. The present study revealed that tetrachloroethylene induced oxidative DNA damage in Egyptian dry-cleaning workers that was detectable as increased lymphocyte 8-OHdG and urinary excretion of 8-OHdG in both smokers and nonsmokers. This is more evidenced by the increase in the percentage of chromatid breaks in tetrachloroethylene-exposed workers. It could be concluded that tetrachloroethylene induces oxidative DNA damage in Egyptian dry-cleaning workers. So, Egyptian dry-cleaning workers are at high risk to diseases that result from oxidative damage of DNA, including cancer. It is recommended to reduce their exposure working hours or to reduce the concentration of tetrachloroethylene in their working environment and to increase the use of exogenous antioxidant with the withdrawal of cigarette smoking to reduce the tendency of oxidative DNA damage.
219. Vasorelaxation Effects of Glutathione in the Isolated Rat Aorta Model
Nattaya Chaothanaphat, Prasan Dhumma-upakorn, and Suree Jianmongkol Chulalongkorn
University, Bangkok, Thailand
This study investigated the influence of endothelium on the vasorelaxant activity of GSH, using in the in vitro model of isolated rat aorta. The vascular tensions upon treatment with GSH in the endothelium-intact and -denuded aortic rings were compared in various conditions. Our results showed that GSH was able to suppress the responsiveness of aortic vessels toward the cumulative addition of PE (1 nM–10 μM). The presence of endothelium had no markedly influence on the extent of GSH-inhibition. In PE-precontracted aortic rings, GSH (2–8 mM) significantly induced vasorelaxation in a concentration-dependent manner. The presence of endothelium also affected the characteristic of GSH-induced relaxation profiles, in addition to the magnitude of the decreased tension. Treatment of GSH (at the concentration up to 5mM) caused a loss of vascular tensions at a higher degree in the endothelium-intact aortic rings than in the endothelium-denuded preparations. GSH-induced vasorelaxation of endothelium-intact rings was inhibited by glibenclamide (3 μM), methylene blue (10 μM), and L-NAME (10 μM). In contrast, only glibenclamide could inhibit the vasorelaxation effect of GSH in the denuded preparations. Further, the endothelium-dependent vasorelaxation effect of GSH was abolished in Ca2+-free medium. Taken together, our findings suggested that GSH decreased vascular tone through the activation of Ca2+-dependent NO-cGMP pathway in the endothelium along with its hyperpolarizing actions in both endothelium and smooth muscle cells.
220. Metabolic Profiling and Cytochrome P450 Reaction Phenotyping of Schizandrin
Yun-Feng Cao,1 Jun Yin,1 and Ling Yang2
1School of Traditional Chinese Medicine, Shenyang Pharmaceutical University, Shenyang, China
2Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China
Schizandrin, one of the major lingans isolated from Schisandra fruits, has been shown to produce potent neuroleptic and anticonvulsive effects in animals. In Chinese Pharmacopoeia, this lingan is recognized as a phytochemical marker for the quality control of Fructus Schisandrae. The present study aimed to profile the phase I metabolites of schizandrin and identify the human cytochrome P450 (CYP) isoforms involved. After schizandrin was incubated with human liver microsomes (HLMs) and a NADPH-generating system, three main metabolites (namely, M-1, M-2, and M-3) were isolated by high-performance liquid chromatography, and their structures were identified to be 7,8-dihydroxy-schizandrin, 7,8-dihydroxy-2-demethyl-schizandrin, and 7,8-dihydroxy-3-demethyl-schizandrin by LC-MS and 1H-NMR, respectively. A combination of correlation analysis, chemical inhibition studies, assays with recombinant CYPs, and enzyme kinetics indicated that all these metabolites were generated predominantly by CYP3A4. Rat and minipig liver microsomes metabolized schizandrin in a similar way to HLMs. The Km values ranged from 23.4 to 58.0 μM, and the Vm values ranged from 1,168 to 11,310 pmol/min/mg for M-1 in different species. All these data demonstrated that minipig and rat could be surrogate models for human in schizandrin pharmacokinetic studies. Better knowledge of schizandrin’s metabolic pathway could provide vital information for understanding the pharmacological and toxicological properties of Fructus Schisandrae.
221. Metabolic Profiling of [3H]20(S)-Protopanaxadiol in Rat Bile
Jian Meng, Lizhi Zhao, Xiaoyan Chen, Youhong Hu, and Dafang Zhong
Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China
20(S)-protopanaxadiol (PPD) is an antidepressant candidate under preclinical development. In this study, [3H]PPD was prepared and used in metabolic profiling in rat bile. By reduction of the PPD dioxide with NaB3H4 in N,N-dimethyl acetamide, [3H]PPD was selectively labeled with a radiochemical purity of 98.6%. Off-line liquid scintillation counting was conducted for total radio activities in urine and bile. The metabolic profiles were determined by high-performance liquid chromatography (HPLC) with dynamic on-line radio flow detection (AIM Research Inc., Hockessin, USA) coupled with ion-trap mass spectrometry. Samples were analyzed by using a Zorbax SB-C18 column (Zorbax, Agilent Technologies, Santa Clava, CA, USA) by gradient elution. After a single oral administration of [3H] PPD to male Sprague-Dawley rats at a dose of 160 μCi/ 100 mg/kg, radioactivity excreted into the bile was 32.5% of the dose up to 48 hours after administration, while the radioactivity excreted into the urine was 1.54% during the same period, indicating bile is the major excretion route. Radiochromatograms of the bile collected from bile duct–cannulated rats over the 3–6-hour interval showed massive metabolite peaks with retention time ranging from 8.0 to 16 minutes, accounting for >95% of the billiary radioactivity, while the [3H]PPD peak at 27.1 minutes, accounting for only 1%. The results indicated that PPD was extensively metabolized into polar molecules and excreted by bile. HPLC-MSn analysis revealed that these metabolites included three isomers of hydroxy-PPD-glucuronides, 3 isomers of hydroxy-PPD-sulfates, and 3 isomers of dihydroxy-PPD-sulfates, which were all formed by hydroxylation and then conjugation. These conjugates were major metabolites of PPD in rat bile, and their exact structures will be further investigated. Surprisingly, no direct conjugation products of PPD were observed, although three hydroxyl groups exist in the structure of PPD.
222. Metabolism of Evodiamine by Human Cytochrome P450 Enzymes
Zhong-Ze Fang and Ling Yang
Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China
Evodiamine is the main active alkaloid of Evodia rutaecarpa and has been demonstrated to have many pharmacological activities. In the present study, the phase I metabolites of evodiamine were profiled, and the cytochrome P450 isoforms involved were identified. Human liver microsomal incubation of evodiamine in the presence of NADPH resulted in the formation of five major metabolites (M-1, M-2, M-3, M-4, and M-5). Four major metabolites (M-1, M-2, M-3, and M-5) were identified to be monohydroxylated evodiamine, and one metabolite (M-4) was identified to be N-demethylated evodiamine by liquid chromatography/mass spectrometry (LC/MS). CYP3A4 and CYP1A2 were identified to be the isoforms primarily involved in the formation of M-1, M-2, M-3, M-4, and M-5 in studies with specific P450 inhibitors, recombinant P450s, and correlation analysis. In human liver microsomes, the Km values ranged from 4.7 to 19.7 μM, and the Vm values ranged from 86.3 to 447.5 pmol/min/mg for M-1, M-2, M-3, M-4, and M-5. Clarification of the metabolite profile and kinetic properties of evodiamine by human P450s provide important information relevant to the pharmacology and toxicology properties of evodiamine and E. rutaecarpa.
223. A Comparison between Microdosing and Normal Dose Metabolic Profiles
Carmai Seto,1 Takeo Sakuma,1 Jinsong Ni,2 Fred Ouyang,3 Devin Welty,3 Van Dinh,3 Gabriella Szekely-Klepser,3 Andrew Acheampong,3 and Vince C. Gao4
1MDS Analytical Technologies, Concord, Ontario, Canada
2Allergan, Irvine, California, USA
3Allergan, Irvine, California, USA
4Life Technologies Asia Pacific, Wanchi, Hong Kong
Introduction: Microdosing provides a new approach for early assessment of human pharmacokinetics. Microdosing studies are designed to evaluate pharmacokinetics or imaging of specific targets without inducing pharmacologic effects. Microdosing studies may not predict the pharmacokinetic behavior of the drug at clinical doses, since it is not known whether or not dose linearity is maintained between a microdose and a clinical dose. Nonlinearity of pharmacokinetics could be induced when absorption is dose dependent or when metabolism or elimination becomes saturated. The work presented investigated if there is a dose dependency of metabolism between a microdose and normal dose in rat.
Methods: PK Studies: Atorvastatin, methimazole, ofloxacin, omeprazole, and tamoxifen were administered to jugular vein–cannulated male Sprague-Dawley rats via oral gavage. The compounds were administered at two different doses at 1.67 μg/kg (microdose) and 5.0 mg/kg (normal dose). LC/MS/MS Analysis: Multiple reaction-monitoring, information-dependant acquisition (MRM-IDA) was used to detect and characterized metabolites in plasma and urine. The analyses were conducted by using a QTRAP® 5500 (QTRAP, Applied Biosystems, Foster city, CA, USA) LC/MS/MS system with automated method creation, using LightSight™ software. (LightSight, Applied Biosystems, Foster city, CA, USA) Preliminary Data: The type and the number of metabolites detected and confirmed in the plasma and urine from the microdose and the normal dose were compared. This was to determine if there is dose dependency of the metabolic processes of the compounds. Metabolites were detected and characaterized for atorvastatin, methimazole, ofloxacin, omeprazole, and tamoxifen were first identified in the plasma and urine collected from the rats given the normal dose of each compound. Then, a predicted MRM-IDA method was created for each compound with the information obtained from the normal dose and a biotransformation set that contained an extensive list of possible metabolites. These MRM-IDA methods were used to characterize and confirm the circulating metabolites in the plasma and urine collected from the microdose. The MRM scan mode allowed for the detection of the metabolites and the full-scan MS/MS sensitivity of the linear ion trap allowed for the characterization and confirmation of these metabolites.
224. Albumin Effect on In Vitro Codeine (COD) Glucuronidation: Improving the Prediction of Hepatic Intrinsic Clearance
Pritsana Raungrut,1 Benjamas Janchawee,2 Verawan Uchaipichat,3 David J. Elliot,4 and John O. Miners4
1Biomedical Sciences, Prince of Songkla University, Hat Yai, Thailand
2Pharmacology, Prince of Songkla University, Hat Yai, Thailand
3Pharmaceutical Practice, Khon Kaen University, Khon Kaen, Thailand
4Clinical Pharmacology, Flinders University and Flinders Medical Center, SA, Australia
COD is metabolized in vivo to a major metabolite (C6G) via glucuronidation. The screening experiment for COD glucuronidation activity was performed in 14 different UGTs (viz. UGT 1A1, 1A3, 1A4, 1A5, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B10, 2B15, and 2B17) and kinetic parameters for COD glucuronidation was investigated by human liver microsome (HLM) and recombinant UGT2B7. These experiments were performed in the absence and presence of 2% bovine serum albumin (BSA). Recombinant UGTs were screened for COD glucuronidation activity at three COD concentration (0.5, 2, and 10 mM). The results showed that only UGT2B4 and UGT2B7 formed the C6G metabolite; however, UGT2B4 only affected at highest concentration in the absence of BSA and at 2 and 10 mM in the presence of BSA. In contrast, UGT2B7 exhibited a concentration-dependent activity both in the absence and presence of BSA. Rates of C6G formation by HLM was best fitted by the single-enzyme Michaelis-Menten model with a mean apparent Km = 2.32 ± 0.33 mM, and Vmax = 573 ± 113 pmol/min.mg. The kinetics of COD glucuronidation changed to substrate inhibition (Ksi = 11.44 ± 1.28 mM). The addition of BSA resulted in a significant reduction of the mean Km value (to 0.29 ± 0.02 mM), without significantly affecting Vmax (541.60 ± 97.82 pmol/min.mg). In contrast to HLM, recombinant UGT2B7 was best described with sigmoidal kinetic both of the absence and presence of BSA. The addition of BSA affected to reduce the apparent Km from 1.09 to 0.33 mM and it did not significantly affect to Vmax and Hill coefficient (n). When considered in the term of intrinsic clearance (CLint or CLmax), the BSA increased the CLint in HLM 7.5-fold (from 0.26 ± 0.05 to 1.95 ± 0.39 uL/min.mg) and the CLmax in recombinant UGT2B7 3.44-fold (from 0.027 to 0.093 uL/min.mg). Inclusion of BSA in in vitro experiments decreases the apparent Km for COD glucuronidation and hence increases CLint (or CLmax) and improves the prediction of hepatic intrinsic clearance.
225. Effect of Leech Lime Juice on Cytochrome P450 3A4 and P-Glycoprotein Activities
Nusara Piyapolrungroj,1 Uthai Sotanaphun,2 and Panadda Phattanawasin3
1Department of Biopharmacy, Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
2Department of Pharmacognosy, Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
3Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
The purpose of this study was to investigate the effects of leech lime juice on the cytochrome P450 3A4 (CYP3A4) activity and the function of P-glycoprotein (P-gp). Crude extract was prepared from squeezed juice of leech lime (Citrus hystrix), using adsorption chromatography on Amberlite, Rohn & Hans, XAD-16 and then extracted with dichloromethane. The active compounds, 6′,7′-dihydroxybergamottin and oxypeucedanin hydrate, were isolated from the dichloromethane extract by Silica gel 60 PF254. Their abilities to inhibit CYP3A4 were examined in vitro by utilizing the CYP3A4/BFC screening kit (Gentest™ BD Biosciences, San Jose, CA, USA) and testosterone 6β-hydroxylation reaction. To elucidate the effects on P-gp function, the accumulation of calcein-AM into LLC-PK1 and LLC-GA5-Col300, as well as bidirectional transport of daunorubicin across Caco-2 cell monolayer, were examined. The results revealed that both 6′,7′-dihydroxybergamottin and oxypeucedanin hydrate inhibited CYP3A4 activity with IC50 values of 8.50 and 26.36 μM, respectively. The elongation of the preincubation period of both active compounds with CYP3A4 led to the enhancement of the CYP3A4 inhibition, suggesting an involvement of a mechanism-based inhibition. The accumulation of calcein-AM into LLC-PK1, compared with that into LLC-GA5-Col300, substantiated that only 6′,7′-dihydroxybergamottin, not oxypeucedanin hydrate, inhibited P-gp function in a concentration-dependent manner. In summary, leech lime juice is composed of furanocoumarin components that inhibit both CYP3A4 and P-gp activity. Consequently, consuming leech lime juice with drugs may cause juice-drug interaction, if these drugs are CYP3A4 and/or P-gp substrates.
226. Full-Scan Data Acquisition for Rapid Quantitative and Qualitative Analysis Using the Exactive LC-MS High-Resolution Mass Spectrometer
Lester Taylor,1 Yingying Huang,2 and Kristi Akervik1
1Thermo Fisher Scientific, San Jose, California, USA
2ThermoFisher Scientific, San Jose, California, USA
Introduction: Current approaches to discovery-stage drug-metabolism studies (i.e., pharmacokinetics, microsomal stability, etc.) have focused on the use of targeted analysis (MRM)-based approaches for quantitative analysis. This necessitates the optimization of parameters, such as Q1 and Q3 m/z values, collision energy, and interface voltages. These studies only detect the specified compound, and information about other components, such as metabolites, is lost. The ability to do full-scan acquisition for quantitation eliminates the need for compound optimization while enabling the detection of metabolites and other non-drug-related endogenous components.
Method: Samples from a range of studies (i.e., microsomal stability, pharmacokinetics) were analyzed by using the exactive nonhybrid Orbitrap LC-MS(Orbitrap, Thermo Fisher Scientific, Waltham, MA, USA). Sample introduction used ultra-high-performance liquid chromatography (UHPLC) and fast generic gradients to enable rapid cycle times and high-throughput analysis. The mass spectrometer consisted of a compact single-stage Orbitrap mass analyzer capable of operating at scan speeds compatible with UHPLC peak widths while maintaining characteristic Orbitrap mass accuracy and resolution.
Preliminary Data: Initially, microsomal stability samples were analyzed. Preliminary results indicate that the instrument provides sufficient quantitative capability together with the ability to identify and confirm metabolites. In addition, structure elucidation of metabolites was facilitated by the collection of high-resolution accurate mass-fragmentation data. The use of extracted ion chromatograms, using narrow m/z windows, provides selectivity comparable to MRM analysis. Sample sets were further processed to show that metabolites were present in the plasma samples. Further results will be presented that demonstrate that the quantitative-qualitative workflow is possible with this new bench-top single-stage mass spectrometer.
Novel Aspect: A bench-top single-stage UHPLC-Orbitrap system is used for combined high-throughput qualitative and quantitative analysis
227. Identification of Metabolites in Feces from Rats with Cinobufagin
Xiao chi Ma II, Ke-xin Liu, Bao-jing Zhang, Jing Ning, Yan Tian, and Sa Deng
School of Pharmaceutical Sciences, Dalian Medical University, Dalian, China
Cinobufagin (1) is one of the major bufadienolides with antitumor activity and potent cytotoxicity in the traditional Chinese medicine Chan-Su (Ma et al., 2008). However, it also can adversely affect the myocardium and the most life-threatening toxicity includes ventricular ectopy and hyperkalemia (Arena and Drew, 1985). Therefore, it is significant to study in vivo metabolites of 1 for evaluating its pharmacological effects and safety. In the present study, the in vivo metabolites of 1 in rats were studied. Eight metabolites were isolated from feces of rats, and their structures were identified as deacetycinobufagin (2), 3-ketodeacetycinobufagin (3), 3-epi-deacetycinobufagin (4), 12b-hydroxyl- 3-epi-deacetycinobufagin (5), 5b-hydroxyl-3-epi-deacetycinobufagin (6), 1a, 5b- dihydroxyl-3-epi-deacetycinobufagin (7), 1a-hydroxyl-3-epi-deacetycinobufagin (8), and 9a, 12a-dihydroxyl-3-epi-deacetycinobufagin (9), respectively, on the basis of widely spectroscopic studies, including 2D-NMR (). Among them, metabolites 5–9 are new compounds. In addition, most of them are the derivates of metabolite 4, and it is also a major metabolite of 1 in serum and liver of rat after oral administration. The metabolites 5–9 were detected in bile of rats with large amounts.
Figure 1. The in vivo metabolites of cinobufagin in rats.
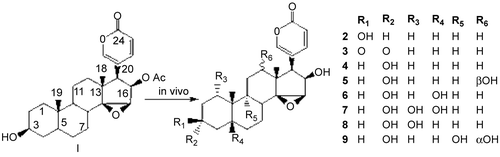
References
- Arena, J.M., Drew R.H. (1985). Poisoning: Toxicology, Symptoms, Treatments, 5th ed.(p 562). Springfield, Illinois, USA: Charles C. Thomas.
- Ma, X.C., Xin, X.L., Liu, K.X., Han, J., Guo D.A. (2008). Microbial transformation of cinobufagin by Syncephalastrum racemosum. J Nat Prod 71:1268–1270.
228. In Vitro Metabolic Study of Boc5 in human tissues and Animal Liver Microsomes
Guang-Bo Ge, Si-Cheng Liang, Yong Liu, and Ling Yang
Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China
As the first nonpeptidic agonist of glucagon-like peptide 1 receptor, Boc5 can invoke sustained glycemic control and weight loss in diabetic C57BL/6J mice and has become a promising hit compound for antidiabetic drug. Boc5 displays good efficiency via intravenous administration but loss efficiency via oral administration, thus its ADME properties should be studied. In the present study, the in vitro investigation on the metabolic stability of Boc5 was first reported. Both phase I and II metabolic studies were carried out by using a series of tissues and microsomes from human and three experimental animals, including pig, rat, and C57BL/6J mice. Significant species difference in metabolism of Boc5 was presented; Boc5 cannot be metabolized by human plasma, liver and intestine microsome, as well as rat and pig liver microsomes. In contrast, Boc5 can be rapidly metabolized by C57BL/6J mice liver microsome (∼50% conversion at 1 hour), and three metabolites (M-1, M-2, and M-3) were detected by using liquid chromatography/mass spectrometry (LC/MS). M-1 was confirmed as 2-thenoic acid, and M-3 and M-2 were assigned as two metabolites after the hydrolysis of one and two thenoic acids from Boc5, respectively, based on the retention times and mass spectra by comparison with standards. These results revealed that the hydrolysis of thiophene carboxylic ester bond was the major metabolic pathway of Boc5, indicating esterases invlolved in this biotransformation. Further studies have shown that carboxylesterase in C57BL/6J mice liver display the key role in rapid hydrolysis of Boc5 by using a range of inhibitors. All of these results were very useful to develop the next-generation nonpeptidic agonists and to choose suitable experimental animals for whole tests.
229. Candidate Gene Analysis of Warfarin Doses in Korean Patients
Kyoung-Woo Je,1 Ho-Sook Kim,1 Yin-Jin Jang,2 Jae-Gook Shin,3 and Sang Seop Lee2
1Pharmacology, Pharmacogenomics Research Center, Inje University College of Medicine, Busan, South Korea
2Pharmacology, Pharmacogenomics Research Center, Busan, South Korea
3Department of Pharmacology and Clinical Pharmacology, Pharmacogenomics Research Center, Inje University College of Medicine and Busan Paik Hospital, Busan, South Korea
Warfarin is a commonly prescribed anticoagulant that is difficult to use because of the wide interindividual variation in dose requirements, the narrow therapeutic range, and the risk of serious bleeding. In order to find genetic factors contributing to the interindividual variation of warfarin doses, functions of genetic variations in cytochrome P450 2C9 (CYP2C9), a major drug-metabolizing enzyme for warfarin oxidation, and vitamin K epoxide reductase complex 1 (VKORC1), a molecular target for warfarin, have been extensively addressed. The genetic studies have revealed that common genetic variations in these genes, together with biological/environmental factors, can explain interindividual variation of warfarin doses by up to 50–60%. For the revelation of additional genetic factors affecting warfarin doses, genetic association studies on several candidate genes involved in pharmacokinetics and pharmacodynamics of warfarin have been performed. In the present study, the effect of common genetic variations, in some candidate genes on warfarin doses, were investigated in the patients with heart-valve replacement. Based on the linkage disequilibrium properties in each gene, a number of candidate SNPs were selected in genes, including ApoE, GGCX, CALU, NQO1, PROS1, etc., and genotyped in 265 patients by the TaqMan®, Applied Biosystems, Foster city, CA, USA real-time polymerase chain reaction method. Logistic regression analysis showed that there were no genetic variations with strong predictive power for warfarin doses, such as CYP2C9 or VKORC1. However, the genetic variations in CALU and PROS1 were associated with warfarin doses at the significance level of P < 0.05. According to the regression model, these genetic variations contribute to the 1–2% of warfarin dose variations. Further clinical studies with a larger number of patients will reveal the clinical relevance of these genetic variations.
230. CYP1A1 Msp1 Polymorphism Frequency in Thai Population
Sutira Lerdtragool, Dawan Shimphu, and Saisiri Mirasena
Department of Biochemistry, Faculty of Medical Science, Naresuan University, Phitsanulok, Thailand
Hepatic CYPs are the major enzyme system involved in the phase I metabolism and toxicity of many drugs and xenobiotics. CYP1A1 is primarily an extrahepatic enzyme that catalyzes the first step in the metabolism of many substrates, such as polycyclic aromatic hydrocarbon (PAHs), benzo[a]pyrene (B[a]P) in cigarette smoke, steroids with a phenol ring, combustion products from meats and fats, and caffeine. Interindividual variation in drug or xenobiotic metabolism can occur as a result of specific CYP genetic polymorphisms. The CYP1A1 3801 T>C (Msp1) polymorphism has been shown to associate with xenobiotic-induced diseases, such as lung cancer, stroke, therapeutic failure, and adverse effects of drugs. The aim of the present study was to determine the frequency of 3801 T>C polymorphism in CYP1A1 gene in Thai populations. CYP1A1 3801 T>C (Msp1) allele was genotyped in 510 unrelated healthy individuals by using a real-time polymerase chain reaction assay in peripheral blood DNA. The observed CYP1A1 Msp1 allelic frequency was 0.52 in the total samples. The genotype distribution was in Hardy-Weinberg equilibrium. The allelic frequency of CYP1A1* (Msp1) allele in Thais (0.52) was much higher than in Caucasians (0.094) and other Asians (0.36). The data obtained indicated that the CYP1A1 Msp1 polymorphism is common in Thai populations, which, therefore, may be useful as a reference for future studies of an influence of CYP1A1 Msp1 polymorphism on cancer susceptibility, chemotherapy evaluation, and adverse drug reactions.
231. CYP2D6 Copy Number Variation: the First and Only Example of CYP Genes
Payiarat Suwannasri,1 Pornpen Pramyothin,1 and Chanin Limwongse2
1Department of Pharmacology and Physiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
2Research and Development, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand
Cytochrome P450 2D6 (CYP2D6) has been extensively studied for its high occurrence of polymorphisms and genotype-phenotype correlation. Although various detection techniques have been developed to solve its complexity, there is still a conflict of genotype, in some cases. Copy number variations (CNVs) were then discovered from an attempt to unravel CYP2D6 gene rearrangement. This is the first example among CYP genes that presents whole gene deletion, in tandem duplication and multiplication. To improve the accuracy of genotyping, single-nucleotide polymorphisms (SNPs) should be determined in parallel with copy number detection. Semiquantitative denaturing hig- performance liquid chromatography (DHPLC) analysis was thus developed to identify relative CYP2D6 copy numbers along with the single-base extension (SBE) technique for 10 SNPs detection. Blood samples of 284 Thai healthy subjects (control) and 135 liver cancer patients (case) were collected for genotyping and analyzing for gene-disease association. CNVs ranging from zero to 5 copies of CYP2D6 were detected in the Thai population and surprisingly found as high as 38.87% in control and even higher in some cases, up to 50.37%. Individuals presenting four copy numbers are significantly higher in cases, when compared to control (P = 0.000). For SNP frequency analysis, those having deletion or duplication were excluded in order to prevent the mixed-up genotype. Among 10 SNPs, three (C100T, C1039T, and C2850T) were significantly shown to affect liver cancer risk (P < 0.05). In contrast, it was not found likewise when including those having CNVs in SNP analysis. This example obviously shows the importance of CNV awareness in CYP2D6 genotyping and also demonstrates the detection technique of CNV screening in CYP gene.
232. Cytidine Deaminase c.79A>C Polymorphism Affected Optimal Dose Delivery of Gemcitabine
Ekkaphon Metharom,1 Peter Galettis,2 Susan Manners,2 Maria Jelinek,2 Paul de Souza,2 and Matthew Links2
1Cancer Chemotherapy and Pharmacology Group, St. George and Sutherland Hospitals, Sydney, Australia
2St. George and Sutherland Hospitals, Cancer Chemotherapy and Pharmacology Group, Sydney, Australia
Gemcitabine (GEM) is a nucleoside analog with anticancer activity against many human tumors. Deoxycytidine kinase (dCK) is the rate-limiting enzyme for GEM activation to its active metabolite, gemcitabine triphosphate (GEM-TP). Cytidine deaminase (CDA) is the enzyme responsible for the metabolic clearance of gemcitabine. Single-nucleotide polymorphisms (SNPs) have been reported in genes involved in GEM metabolism and catabolism. Controversy exists over the optimal dosing of GEM. We performed a randomized, crossover, phase II clinical trial to study the effect of standard infusion (30 minutes) and prolonged infusion (100 minutes) on intracellular GEM pharmacokinetics. An attempt to find a single optimal dose rate is problematical because of interpatient variability in factors determining the GEM threshold. We hypothesized that polymorphisms in genes involved in gemcitabine pathway, particularly the CDA c.79A>C may influence the optimal dosing regimen in individual patients. Three SNPs (c.79A>C, c.208G>A, and c.435T>C) in the CDA gene were genotyped in 32 patients participated in the clinical trial. Other factors, including infusion rate and the week in which the drug is administered, affected GEM-TP accumulation. The relationship between CDA SNPs, leukocytes GEM-TP accumulation, and GEM infusion rate were examined by using repeated measure analysis of variance. The univariate model demonstrated a trend for increase accumulation of GEM-TP with the variant c.79 SNP; however, this trend was not statistically significant. The multivariate model demonstrated an interaction between c.79 genotype, infusion rate, and week of administration that affected GEM-TP accumulation (P = 0.01). Patients who carried variant c.79 accumulated higher GEM-TP than those carrying wild-type c.79 when receiving GEM as a 100-minute infusion and when the threshold for GEM-TP accumulation was increased (week 2). These findings demonstrate the importance of considering both pharmacokinetics and pharmacogenetics in optimizing gemcitabine accumulation. This represents a classical interaction between genes and environment; and provides strong support for the inclusion of CDA genotype in an individualized dosing strategy.
233. Development of Genotyping Method for Warfarin Therapy
Yin-Jin Jang,1 Woo-Young Kim,2 Jae Gook Shin,3 and Sang Seop Lee1
1Pharmacology, Pharmacogenomics Research Center, Busan, South Korea
2Department of Pharmacology and Pharmacogenomics Research Center, Inje University College of Medicine, Busan, South Korea
3Department of Pharmacology and Clinical Pharmacology, Pharmacogenomics Research Center, Inje University College of Medicine and Busan Paik Hospital, Busan, South Korea
Due to the risk of bleeding or stroke by unintended PK/PD of warfarin, the prediction of warfarin doses is one of the serious questions in the field of personalized medicine. Since 2002, the genetic association studies of CYP2C9 and VKORC1 have revealed that the common genetic variation of these genes is one of the major determinants of safe, efficient warfarin doses. Now, it is well agreed that the use of pharmacogenetic information is clinically relevant. U.S. FDA recommends genetic tests for warfarin therapy and has approved at least four in vitro diagnostics for warfarin thearpy. Most of these IVD focus on the detection of CYP2C9*2 and *3 in addition to the determination of VKORC1 haplotypes. Our previous study showed that functionally defective CYP2C9*13 and *14 alleles are observed in Korean patients. In the present study, the multiplex pyrosequencing method was developed to analyze CYP2C9*3, *13, and *14 at the same time. The newly developed genotyping method genotyped all of the three CYP2C9 alleles without ambiquity, and the results were completely concordant with genotyping results of direct sequencing. We also developed the duplex pyrosequencing method for VKORC1 haplotype determination for Asians. VKORC1 genotype results were also verified by direct sequencing. Our pyrosequencing method provides a rapid, accurate, and economic genetic test for warfarin therapy and is especially proper to Asian patients.
234. Genetic Polymorphisms of Human ABCC3 in Different Ethnic Populations and Their Functional Assessment
Tomohiro Sasaki, Yuriko Ryokai, Takeshi Hirota, Shun Higuchi, and Ichiro Ieiri
Department of Pharmaceutical Sciences, Kyushu University, Fukuoka, Japan
The multidrug resistance–associated protein 3 (MRP3; ABCC3) is a member of the ATP-binding cassette transporters and expressed at several tissues, including adrenal grand, liver, small intestine, colon, kidney, and pancreas in humans. Remarkable changes in the pharmacokinetic profiles of clinically important drugs, such as methotrexate and fexofenadine, were observed in ABCC3 gene knock-out mice, suggesting MRP3 has a significant role in the disposition of these drugs. mRNA and protein expression levels of MRP3 in human tissues show large variabilities among individuals by 86- and 84- fold, respectively. It is reported that −211C>T in the 5’-falnking region of ABCC3 altered mRNA expression levels significantly in the human liver. However, it is a not fully elucidated ethnic difference of these variations and the contribution of other mutations or haplotype structures on the expression of ABCC3. In the present study, we investigated genetic variations of ABCC3 gene in three ethnic groups (Caucasian, American-African, and Japanese, 96 in each) and assessed their functional impacts by reporter gene assay. ABCC3 sequences, including all 31 exons, their surrounding introns, and approximately 2 kb of the 5’-flanking region, was determined by PCR-SSCP and -RFLP methods, followed by DNA sequencing. The haplotypes and their frequencies were calculated by ARLEQUIN ver. 3.1 software, Arlequin. We found 59 genetic variations, including 17 novel ones. Although the frequencies of variants in the coding region were relatively low (maximum allele frequency was 5%), some variants observed in the 5’-flanking region were found at high frequency. The frequencies of each variants and haplotype structures indicated interethnic differences. Functional impacts of genetic variations in the 5’-flanking region by reporter gene assay will be presented in the poster.
235. Regulation of Expression of ABCB1 Gene in Human Placenta by Noncoding MicroRNAs
Takeshi Hirota,1 Yuuki Eto,2 Rumi Tanaka,2 Shun Higuchi,2 and Ichiro Ieiri2
1Clinical Pharmacokinetics, Graduate School of Pharmaceutical Sciences, Kyushu University, Fukuoka, Japan
2Clinical Pharmacokinetics, Graduate School of Pharmaceutical Sciences, Kyushu University, Fukioka, Japan
ABCB1 (P-glycoprotein, P-gp) plays a large role in the distribution and elimination of many clinically important drugs. To date, the contribution of the ABCB1 gene polymorphisms on large interindividual differences in ABCB1 gene expression is still controversial among studies. Some studies suggest that changes in stability of mRNA or in the translational efficiency due to certain ABCB1 polymorphisms are likely reasons for the large variations. In contrast to genetic polymorphisms, recently, post-transcriptional control of gene expression by noncoding small RNA (microRNA, miRNA) is becoming of interest. After recognizing the 3’-untranslated region (UTR) of the target mRNA, miRNA causes translational repression or mRNA cleavage. We hypothesize that miRNA plays an important role for large interindividual differences in ABCB1 expression. We identified near-perfect matching sequences in the 3’-UTR of human ABCB1 gene with following miRNAs, using the miRNA registry: miR-27a and miR-374a. Since polymorphisms in the 3’-UTR affect the gene expression by interfering with miRNAs function, we initially investigated genetic polymorphisms in the 3’-UTR of the ABCB1 gene. We found the following two genetic variations: +187G>C and +193A>G. The frequency of +193A>G was 0.28 in Japanese (n = 30), and this variation was located on the predicted target sequence of miR-374a. To determine the effect of miRNAs and +193A>G on the regulation of ABCB1 gene, wild-type and mutant 3’-UTR were cloned into luciferase reporter vectors. We analyzed whether the reporter activity of the plasmid containing the wild or mutant 3’-UTR was reduced by cotransfection of each precursor miRNA. Exogenously expressed miR-27a and miR-374a could decrease the luciferase activity, which was independent on the polymorphism in the 3’-UTR. The expression levels of miR-27a, miR-374a, and ABCB1 protein were evaluated by using human placental samples. Significant negative correlations were observed between the expression levels of two miRNAs and ABCB1 protein. Our findings suggest that the expression of miR-27a and miR-374a contributed to the interindividual variability in ABCB1 gene expression, leading to variations in P-glycoprotein function.
236. UGT1A6 Genotype-related Pharmacokinetics of Propranolol in Healthy Volunteers: Preliminary DATA
Jeeranut Tankanitlert,1 Praveena Yamanont,2 L. Michael G. Limenta,3 and Noppawan P. Morales3
1Pharmacology, Phramongkutklao College of Medicine, Bangkok, Thailand
2Pharmacology, Faculty of Science Mahidol University, Bangkok, Thailand
3Department of Pharmacology, Faculty of Science, Mahidol University, Bangkok, Thailand
Background: Propranolol is a nonselective beta-adrenergic receptor-blocking drug widely use for the treatment of cardiovascular diseases. The drug was metabolized by oxidation, N-dealkylation, and glucuronidation pathway. The oxidation product and active metabolite was conjugated with glucuronide. The glucuronidation reaction is activated by UDP-glucuronosyltransferase (UGT1A) to terminate its action; we, therefore, explore UGT1A6 genotype-related pharmacokinetics of propranolol.
Methods: To elucidate the pharmacokinetics of propranolol in normal subjects, blood samples were collected and DNA was extracted. The UGT1A6 genotypings were performed by using the restriction fragment-length polymorphism (RFLP) methods. Nine normal subjects were grouped according to UGT1A6 genotypes (wild-type, heterozygous, and homozygous group). Blood samples were collected before and 15, 30, 60, 90, 150, 210, 330, and 450 minutes after a single oral dose of 40 mg of propranolol. We followed propranolol level using HPLC.
Results: The maximum concentration (Cmax), area under concentration-time curve (AUC0->), half-life (t1/2), and volume of distribution (Vd) were higher, while elimination rate (ke) and clearance were lower in the wild-type group than those of the heterozygous and homozygous groups (P < 0.005). The results seem to explain how the UGT1A6 polymorphisms may affect the pharmacokinetics of propranolol. Additional data from this ongoing study are required to confirm these preliminary observations.
237. Antidiabetic Effect of the Water Extract of Mitragyna speciosa Korth. Leaves
Supaporn Kansenalak,1 Somsmorn Chittrakarn,1 Niwat Keawpradub,2 Ekkasit Kumarnsit,3 Kitja Sawangjaroen,1 and Juntipa Purintrapiban4
1Department of Pharmacology, Faculty of Science, Prince of Songkla University, Hat-Yai, Thailand
2Department of Pharmacognosy and Pharmaceutical Botany, Faculty of Pharmaceutical Sciences, Prince of Songkla University, Hat-Yai, Thailand
3Department of Physiology, Faculty of Science, Prince of Songkla University, Songkhla, Thailand
4Department of Biomedical Science, Faculty of Medicine, Prince of Songkla University, Songkhla, Thailand
Mitragyna speciosa Korth., called kratom in the Thai language, is known locally in southern Thailand. It is in the Rubiaceae family. A red-veined variety of kratom is claimed to have strong biological activities and is widely used by Thai and Malay villagers as a folklore medicine for diarrhea, muscle pain, diabetes mellitus, and hypertension. A chronic metabolic disorder, diabetes mellitus is characterized by hyperglycemia. Various complications develop as a consequence of the metabolic derangement in diabetes, and its complications constitute a major health problem in modern societies. The aim of this work was to investigate the hypoglycemic effect of the water extract of kratom leaves on streptozotocin-induced diabetic rats and enhancement of glucose transport in L8 muscle cells. The water extract of M. speciosa was prepared from dried powder by the freeze-dried technique. Male Wistar rats (200–220 g) bred in Center Animal House, Prince of Songkla University, were used. The rats were injected with streptozotocin (65 mg/kg, intraperitoneally). Since streptozotocin was capable of producing total hyperglycemia (48 hours) with moderate blood glucose over 150 mg/dL, the rats were used in this experiment. The diabetic rats were pretreated with the various doses (50, 100, 200, and 400 mg/kg) of the water extract (WE) and glibenclamide (0.6 mg/kg) for 7 days. Fasting blood was collected from the orbital plexus for glucose assay. The results showed that the WE at 50, 100, 200, and 400 mg/kg pretreated for 7 days were caused by the dose-dependent hypoglycemic effect. Further, it was suggested to activate 2-deoxyglucose transport in a dose- and time-dependent manner. It was, therefore, supposed that the water extract might exert antidiabetic activity via insulin-like action on the stimulation of glucose transport in L8 muscle cells.
238. Comparative Pharmacokinetics of Cephradine in Diabetic and Normal Rats
Guo-zhu Han, Jun Liang, Yilan Wang, Ting Fu, and Kexin Liu
College of Pharmacy, Dalian Medical University, Dalian, China
Objective: The aim of this study was to study effects of diabetes mellitus (DM) on pharmacokinetics of cephradine (CED) by comparing the difference in pharmacokinetic behaviors of CED between diabetic and normal rats.
Methods: DM was induced in male rats by a single intravenous (i.v.) injection of alloxan; rats whose blood glucose was over 16 mmol/L were taken as the DM group. The rats were divided into the DM and normal control (CTL) groups, which were subdivided into low- (90-mg/kg) and high-dose (180-mg/kg) subgroups. CED was administered by i.v. or per oral (p.o.) routes. Plasma samples were analyzed by reverse-phase high-performance liquid chromatography method. Chromatographic separation was achieved on a Kromasil C18 column, Kromasil, Bohur, Sweden with the mobile phase consisting of 0.025 mol/l KH2PO4-MeOH-CH3CN (87:6:7), delivered at 1.0 mL/min; the UV detector was set at 261 nm. The peak area ratio of CED to cephalexin (CEX) as internal standard vs. concentration of CED was used to construct the calibration curve. Then, 50-uL aliquots of TCA-deproteinized plasma samples were injected into chromatograph.
Results: In i.v. experiment, DM group, when compared with the CTL group, presented a significantly shortened t1/2 b and MRT as well as increased CL, as reflected by t1/2 b 84–91 vs. 116–120 minutes, MRT 61–70 vs. 103–119 minutes; CL 23–25 vs. 18–19 mL/min/kg (P < 0.05); in the p.o. experiment, a markedly shorter t1/2 K and tmax as well as greater CL and Cmax in the DM group than in the CTL group were found; meanwhile, DM rats suffered from remarkably increased kidney weight (KW) and KW/BW ratio relative to CTL rats.
Conclusions: DM pathological status can speed up the elimination of CED from body of rats; the compensatory hypertrophy and thereby hyperfunction of kidneys in early-stage diabetics may explain, in part at least, this accelerated elimination.
239. Development and Determination of Fentanyl Using High-Performance Liquid Chromatography with Ultraviolet Detection in Bypass Graft Patients
Pitinuch Piochai,1 Korbtham Satirakul,1 and Siriluk Chumnanvej2
1Pharmaceutical Technology, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand
2Department of Anaestheties, Phramongkutklao Hospital, Bangkok, Thailand
A reversed-phase high-performance liquid chromatography method for the determination of plasma concentrations of the narcotic analgesics: Fentanyl using diazepam as the internal standard is presented. This method has been developed, with sufficient sensitivity to allow analysis of patient plasma samples generated following administration of a clinically relevant dose. Chromatographic separations were achieved with a C18, Thermo Fisher Scientific, Waltham, MA, USA 25 cm × 4.6 mm i.d. column and was monitored at 210 nm. The assay was linear over the clinically relevant plasma range of 20–200 ng mL ∼ for fentanyl and has the sensitivity and specificity necessary to determine plasma concentrations. Inter- and intraday precision (RSD) did not exceed ±15% in these ranges. Although less sensitive than liquid chromatography-mass spectrometry (LC-MS), reliable measurements of fentanyl can be performed with this HPLC-UV system. The assay procedure was utilized for pharmacokinetic studies of plasma concentrations in subjects receiving fentanyl during and after cardiac surgery (bypass graft). This will allow better elucidation of pharmacokinetic variables in this population.
240. Development and Validation of a Sensitive and Rapid Nonaqueous LC-ESI-MS/MS Method for Measurement of Diosgenin in the Plasma of Normal and Hyperlipidemic Rats: a Comparative Study
Lina Xu III, Yuetao Liu, Xu Han, Jinyong Peng, Xinqiang Tang, and Kexin Liu
College of Pharmacy, Dalian Medical University, Dalian, China
In this study, a sensitive, specific electrospray ionization (ESI) liquid chromatography–tandem mass spectrometry (LC-MS/MS) method was developed to detect diosgenin in the plasma of normal and hyperlipidemic rats for pharmacokinetic study. Diosgenin was extracted from plasma by using the solvent system composed of n-hexane-ethyl acetate (9:1, v/v), and the compound sarsasapogenin was used as the internal standard (IS). The separation was performed on an Elite Kromasil C18, Kromasil, Bohur, Sweden (150 2.1 mm i.d., 5 μm) by isocratic elution in 3.0 minutes, and pure methanol was used as the mobile phase with a flow rate of 0.5 mL/min. With multiple reaction-monitoring modes, linear calibration curves were obtained in the range of 10–1,500 ng/mL (r ≥ 0.9979), and the limit of quantification was 10 ng/mL. The intra- and interassay variabilities were within 7.74%, and the accuracies were between −5.33 and 1.50%. The extraction recoveries at low, medium, and high levels in two groups were all more than 74.59%. Diosgenin in rat plasma was stable over three freeze-thaw cycles, at ambient temperature for 8 hours and 30 days storage in a freezer at −70°C. The assay was successfully applied to study the pharmacokinetic parameters in normal and hyperlipidemic rats after an oral administration of diosgenin. As a result, the plasma half-life (t1/2) was 5.81 ± 1.15 hours and the AUC0-t was 4,530.59 ± 953.16 ng h/mL in normal rats, compared with 8.35 ± 2.42 hours and 23,598.63 ± 7,145.58 ng h/mL for hyperlipidemic rats. The maximum plasma concentrations (Cmax) at the levels of 274.13 ± 73.43 and 881.76 ± 252.84 ng/mL were achieved at tmax of 5.81 ± 1.15 and 9.69 ± 1.90 hours for normal and hyperlipidemic groups, respectively. In a word, a significantly different pharmacokinetics of diosgenin was existed between normal and hyperlipidemic rats, which would be benefit for the clinical use of diosgenin and some related natural products.
241. Influence of CYP3A5 and MDR1 (ABCB1) Polymorphisms on the Pharmacokinetics of Tacrolimus in Patients with Rheumatoid Arthritis and Lupus Nephritis
Tomonori Nakamura,1,Yuko Okada,1 Keiju Hiromura,2 Yoshihisa Nojima,2 Katsunori Nakamura,1 and Koujirou Yamamoto1
1Department of Clinical Pharmacology, Gunma University Graduate School of Medicine, Maebashi, Japan
2Department of Medicine and Clinical Science, Gunma University Graduate School of Medicine, Maebashi, Japan
Aims: Tacrolimus (TAC) is a substrate for cytochrome P450 (CYP) 3A5 and P-glycoprotein encoded by CYP3A5 and MDR1 (ABCB1), respectively, having multiple single-nucleotide polymorphisms. In this study, we genotyped CYP3A5 A6986G (CYP3A5*3), MDR1 G2677(A/T), and C3435T polymorphisms and investigated the effect of these polymorphisms on the pharmacokinetics of TAC in patients with rheumatoid arthritis (RA) and lupus nephritis (LN).
Methods: Thirty-six consecutive recipients (RA, n = 20; LN, n = 16) were enrolled in this study. The patients received oral TAC at a dose ≤ 3 mg once-daily after the evening meal. Whole-blood concentrations of TAC 12 hours after administration were measured by semiautomated microparticle enzyme immunoassay. The polymerase chain reaction–restriction fragment length polymorphism and direct sequence method were used for genotyping the CYP3A5 and MDR1 polymorphisms, respectively.
Results: The dose-adjusted 12-hour TAC level (ng/mL per mg/kg) were significantly lower in the patients with CYP3A5*1 than CYP3A5*3/*3 (Kruskal-Wallis test, P = 0.0007). The MDR1 polymorphism was not associated with any pharmacokinetic parameters.
Conclusions: RA and LN patients with the CYP3A5*1 allele required a higher daily TAC dose, compared with those with the CYP3A5*3/*3 genotype to maintain the target 12-hour concentration, suggesting that this polymorphism is useful for determining the appropriate dose of TAC.
242. Pharmacokinetic Study of the Main Antioxidant Active Components of Tea Polyphenols in Rats Following a Single-Dose IV Administration
Guo-zhu Han,Ting Fu, Jun Liang, and Kexin Liu
College of Pharmacy, Dalian Medical University, Dalian, China
Tea polyphenols (TPs) has been shown to exhibit potent free-radical–scavenging activities and extensive health-promoting and therapeutic effects. In the present study, pharmacokinetics of the main antioxidant active components, EGCG and ECG, of TPs extracted from green tea were investigated in rats after a single-dose intravenous (i.v.) administration of a TP injection. The EGCG and ECG concentrations in rat plasma were simultaneously determined by means of a specific, accurate HPLC-UV assay developed in our lab. This assay took advantage of a reversed-phase C18 column and gradient mobile phase consisting of 0.1% citric acid+CH3CN and a gradient flow rate of eluent as well as a detection wavelength of 280 nm. Groups of rats were administered i.v. at doses of 50, 100, and 150 mg/kg. The plasma samples collected postdose were subjected to a liquid-liquid extraction with EtoAc prior to HPLC analysis and quantified by peak-area internal method. The plasma concentration vs. time profiles showed EGCG and ECG decayed from plasma in a biexponential fashion and could be described by the two-compartment model and the first-order kinetics. EGCG and ECG eliminated from the body rapidly with t1/2 of 112–145 and 46–61 minutes, CL of 0.034–0.044 L.(kg.min)−1 and 0.010–0.015 L.(kg.min)−1, and distributed widely with Vd of 6.28–7.9 and 0.9–1.22 L.(kg.min)−1. The two compounds follow linear kinetics over the range of the doses studied. Compared with ECG, EGCG has longer t1/2 and larger Vd, indicating the different pharmacokinetic behaviors between the two.
243. Population Pharmacokinetics of Cilostazol in Relation to Genetic Polymorphisms of CYP3A, CYP2C19, and MDR1 Gene
Hee-Doo Yoo,1 Sun-Ae Park,1 Hea-Young Cho,2 and Yong-Bok Lee1
1College of Pharmacy, Chonnam National University, Gwangju, South Korea
2Pharmacological Research Department, NITR, KFDA, Seoul, South Korea
The aim of this study was to investigate the influence of genetic polymorphisms in CYP3A, CYP2C19, and MDR1 gene on population pharmacokinetics of cilostazol in healthy subjects. This study was a single-dose, randomized, two-way, open-label, crossover, and dose escalation design. A total of 104 healthy Korean volunteers was administered orally as a 50- or 100-mg single dose. The serum concentrations of cilostazol were measured for up to 48 hours, using a validated high-performance liquid chromatography method. All subjects were also determined for CYP3A (CYP3A5*3), CYP2C19 (CYP2C19*2 and CYP2C19*3), and MDR1 (C1236T, G2677T/A, and C3435T) by polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP). The influences of gene polymorphisms on the variability of pharmacokinetic parameters were evaluated by using a nonlinear mixed effects modeling (NONMEM) method. The two-compartment model with first-order absorption and lag time well described the cilostazol serum concentrations. CYP3A5*3 genotype group had a statistically significant (P < 0.05) influence on the systemic clearance (CL/F). In regard to CYP2C19, statistically significant differences (P < 0.05) were observed among the three groups [extensive metabolizers (EMs), intermediate metabolizers (IMs), and poor metabolizers (PMs)] for the systemic clearance (CL/F). Also, the combination of CYP3A5*3 and CYP2C19 is associated with statistically significant differences (P < 0.05) in the systemic clearance (CL/F). The interindividual coefficients of variation (CV) in CL/F were 24.17% and Vc/F was 66.71%. However, no significant differences were found between MDR1 genotype groups and cilostazol pharmacokinetic parameters. The present study represents that CYP3A5 and CYP2C19 partly explain the substantial interindividual variability in the metabolism of cilostazol, whereas MDR1 seems not to be involved in the disposition of cilostazol.
244. The Effect of Palm Fruit Fiber (Borassus flabellifer) on Streptozotocin-induced Diabetic Rats
Wandee Udomuksorn,1 Supaporn Kansenalak,1 and Uraporn Vongvatcharanon2
1Department of Pharmacology, Faculty of Science, Prince of Songkla University, Songkhla, Thailand
2Department of Anatomy, Faculty of Science, Prince of Songkla University, Songkhla, Thailand
Palmyra palm or Borassus flabellifer is widely found in Asian country. It was used as the source of sugar, sometimes call “sugar palm,” food, furnitures, etc. The fruit fiber of this palm has been used as a folk remedy for diabetics in the Thai temple. There are some people who have used it as an aqueous extract for diabetic control; finally, all of them do not need to take oral antidiabetic drugs anymore. But, no scientific evidence occurred. So, this study aimed to characterize the effect of aqueous extract of palm fruit fiber (PFF) on blood sugar and pancreas cells in diabetic rats. Male Sprague-Dawley (170–200 g) rats were used and divided into three groups (n = 8), group 1 control, group 2 aqueous extract of PFF 2 g/kg, oral OD for 14 days, and group 3 glibenclamide 600 μg/kg, oral OD for 14 days. All groups were treated with streptozotocin (60 mg/kg, intraperitoneally) to induce diabetics. Before and after treatment, animals were anesthesized and the blood was collected via orbital plexus puncture for blood sugar level. The pancreas were removed and fixed in 10% formaldehyde to process the pancreas cells for histopathological examination and 4 levels of the severity score: 0–3 were classified by using the number of Islet of Langerhan and % of beta cell damage. The levels of blood sugar of all groups at day 0 were 75 ± 4.5, 104 ± 4.3, and 94 ± 2.9 mg/dL, respectively, and were scored as 0 (normal). At day 3, the levels of blood sugar were 134 ± 1.8, 133 ± 2.6, and 127 ± 3.8 mg/dL, respectively, and were scored as 3 (severe). At day 7, the levels of blood sugar of the PFF group were significantly decreased to 105.4 ± 1.2 mg/mL (P < 0.01, when compared with day 3 in the same group) and were scored as 1 (mild). The lowering of blood sugar level showed at day 14 and was scored as 2 (moderate) for the glibenclamide group. The fact that the aqueous extract of PFF (2 g/kg, orally) reduces blood sugar level may due to recovering the pancreas beta cells.
245. In Vitro–In Vivo Relationship for Hepatic Uptake Clearance
Masahiro Suzuki, Yoshiyuki Akiyama, Hiroshi Komura, and Motohiro Kogayu
Drug Metabolism and Pharmacokinetics Laboratories, CPRI, Japan Tobacco Inc., Osaka, Japan
The hepatic uptake process is one of the determinant factors of total clearance for drugs actively taken up to the liver. In such drugs, drug interaction based on inhibition of the hepatic uptake process has been reported, and extent of increase of plasma concentration by concomitant drugs is influenced by the contribution of the transporter-mediated uptake clearance to the total uptake clearance. Therefore, it is important to predict the in vivo hepatic uptake clearance and the contribution of transporter from in vitro hepatocyte uptake data. The purpose of the present study was to investigate in vitro–in vivo relationship for the uptake clearance and the contribution of transporters using rats. Nine model compounds, substrates toward anion uptake transporters, were applied in in vitro and in vivo studies. The in vivo hepatic uptake clearances were determined based on the well-stirred model, using reported in vitro kinetic parameters (Km, Vmax, and PSdif), blood unbound fraction, and hepatic blood flow. The predicted values were compared to the in vivo clearances estimated by an in vivo integration plot analysis. A good in vitro–in vivo correlation for uptake clearance was observed with r = 0.93. Further, the involvement of transporters to the total uptake for pravastatin was evaluated from Km, Vmax, and PSdif. The corresponding in vivo data was estimated from the integration plot analysis with and without coadministration of cyclosporine A. The in vitro and in vivo contribution was in the similar range (0.92 and 0.76, respectively). Further investigation will be performed to reveal the in vitro and in vivo relationship in the involvement of transporters using another compound.
246. Anti-Inflammatory in Induced MCF-10A Cells of Quercetin Delivered by Liposomes
Malyn Chulasiri,1 Saengrawee Sutthiparinyanont,2 Aroonsri Priprem,2 Hyun-Hee Lee,3 Hye-Kyung Na,3 and Young-Joon Surh3
1Reseach and Development, S&J International Enterprises PCL, Bangkok, Thailand
2Pharmacy, Khon Kaen University, Khon Kaen, Thailand
3College of Pharmacy, National Research Laboratory of Molecular Carcinogenesis and Chemoprevention, Seoul, South Korea
Quercetin, a dietary bioflavonoid, exerts its anti-inflammatory activity via its broad antioxidant activities. To investigate the permeation of quercetin into cells and to protect quercetin from exposure to aqueous conditions, four formulas of quercetin-encapsulated liposomes were developed and tested with human epithelial MCF-10A cell line. Phospholipid, cholesterol, and Span 60 at a molar ratio of 1:1:1 was used to form liposomes with and without either piperine or polyethylene glycol 400 (PEG) or sodium alginate as the additional ingredients. Overall, the particles of the liposomes were averaged 200 nm in diameter with negative zeta potentials and encapsulation efficiencies of higher than 85%. Using Western blot analysis, pretreatment of MCF-10A cells with these liposomes at 7.5 and 15 μM of quercetin, compared with controls, COX-2 expression induced by TPA was significantly reduced when PEG was the additional ingredient of the quercetin liposomes. Also, quercetin at 7.5 μM was found effective in COX-2 inhibition, provided that it was delivered in a liposomal encapsulation with PEG coating. Electrophoretic mobility shift assay for NF-κB DNA binding activity confirmed the anti-inflammatory activity of quercetin in the nuclei after it was delivered into the cells delivered by this liposome formula. It is concluded that to be able to exert its cellular anti-inflammatory activity, quercetin requires a delivery system.
247. Assessment of Transporter Activities in the Liverchip
Yi-An Bi, Veronica Zelesky, and David Duignan
Pdm, Pfizer, Inc., Groton, Connecticut, USA
Purpose: An emerging bioengineering technology—the 3D LiverChip model developed by Dr. Linda Griffth at MIT—raises new possibilities for the study of complex physiological and pathophysiological processes in vitro. This work was done to characterize transporter-mediated clearance in the LiverChip.
Methods: Human hepatocytes were cultured in LiverChips with consistent medium flow at 0.2 mL/min. The time course of cell culture and various media were examined. Transporter function was assessed with CDF, taurocholate, digoxin, salicylate, and several statins. Comparison of transporter clearance (CLint,app) values in LiverChip and SCHH was also conducted.
Results: The results show that hepatocyte cultures in LiverChip could last 10 days, and the cells in the scaffolds could form tissue-like structures in three dimensions. Metabolic CLint,app for midazolam (CYP3A4) and naloxone (UGT2B7) was 20 and 17 μL/min/mg (N = 2), respectively, and similar to that seen in SCHH. Uptake and efflux transporter function were confirmed in the LiverChip with TC, digoxin, rosuvastatin, fluvastatin, and cerivastatin. Efflux transporter function was also confirmed by incubating estradiol and measuring metabolite in media. For the lipophilic statins—fluvastatin and cerivastatin—which are OATP substrates, but their hepatic clearance is driven by CYP metabolism, CLint,app values were 14 and 3.1 uL/min/mg. In contrast, for the uptake/efflux substrates, TC and digoxin, compounds which are not metabolized and for which uptake clearance drives hepatic clearance, the apparent uptake clearance was 3.1 and 1.1 uL/min/mg, compared to 15.9 and 2.9 in SCHH. Similarly, efflux clearance for these compounds was very low relative to that obtained in SCHH. Overall, the data suggest that the LiverChip contains either leaky tight junctions and/or a significant percentage of open bile canaliculi, which, in turn, leads to underestimations of uptake and efflux clearance.
Conclusions: Overall, LiverChip technology shows the potential to be a useful tool for assessing hepatic disposition. However, more optimization is needed before the LiverChip can be used to measure transporter-mediated activity.
248. CFTR Chloride Channel Inhibition by Tannic Acid: Mechanism and In Vivo Efficacy
Nisa Wongsamitkul,1 Lalida Sirianant,1 Chatchai Muanprasat,2 and Varanuj Chatsudthipong3
1Toxicology Graduate Program, Faculty of Science, Mahidol University, Bangkok, Thailand
2Department of Physiology, Faculty of Science, Mahidol University, Bangkok, Thailand
3Department of Physiology, Faculty of Science, Mahidol University, Bangkok, Thailand
Secretory diarrhea remains a clinical important problem worldwide. At present, either rehydration therapy or antibiotics treatment are the principle management options of this disease. Unfortunately, rehydration therapy cannot reduce the severity of diarrhea and antibiotics resistance occurs frequently. Therefore, several efforts have been made to discover new antidiarrheal medications. Currently, several natural products have been used as sources of pharmaceutical products due to their reasonable cost, accessibility, and ancestral experiences. Tannic acid (penta-m-digalloyl-glucose) is a plant-derived polyphenol that was suggested to have a lot of clinical benefits, including antioxidantive effects, antimutagenicity, and anticarcinogenicity. However, its effect on the cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP-activated chloride channel that is directly involved in the pathogenesis of secretory diarrhea induced by bacterial enterotoxins, is not known. This study was aimed to characterize a novel effect of a tannic acid on CFTR function in fisher rat thyroid (FRT) cells stably expressing human CFTR and in human colonic epithelial cell lines (T84 cells) by measuring apical chloride current and short-circuit current, respectively. In addition, antidiarrheal efficacy of tannic acid was tested in mouse closed loop models of cholera. The data in FRT cells showed that tannic acid inhibited CFTR-mediated apical chloride current in a dose-dependent fashion with an IC50 value of 10 μM. Similarly, tannic acid inhibited CFTR mediated apical Cl− current induced by cpt- cAMP, a cell-permeable cAMP, and apigenin, a direct CFTR activator, with IC50 values of 11 and 22 μM, respectively. The CFTR inhibitory effect of tannic acid was reversible, and tannic acid did not cause cytotoxicity. Short-circuit current measurement in T84 cells also demonstrated effectiveness of tannic acid in inhibiting cAMP-induced chloride secretion. In the mouse closed loop model, tannic acid reduced cholera toxin-induced intestinal fluid secretion by about 71.3% without affecting intestinal fluid absorption. These data indicated that tannic acid targets CFTR and may be clinically useful for development to be a new antidiarrheal therapy.
249. Changes of Renal Drug Transporters in the Metabolic Acidosis and Its Effects on Urinary Drug Excretion
Hideyuki Motohashi, Arong Gaowa, Toshiya Katsura, and Ken-ichi
Inui Department of Pharmacy, Kyoto University Hospital, Kyoto, Japan
The kidney is one of the main organs for drug elimination as well as the liver. In the proximal tubular cells, many transporters play the important roles for secretion and reabsorption of the various drugs. On the other hand, metabolic acidosis is induced by excessive acid production or acid ingestion. Under the acidotic conditions, a number of adaptive changes occur in the kidney. However, changes of drug transporters in the metabolic acidosis and the effects of these changes on urinary drug excretion were not investigated. In this study, we investigated the effects of metabolic acidosis on the renal drug excretions. In addition, expression levels of renal drug transporters under the metabolic acidosis were examined. Wistar/ST rats were given the 1.5% NH4Cl ad libitum for 48 hours. Decreased pH in the blood indicated that the metabolic acidosis was induced in the model rats. To examine renal drug excretion in the model rats, phenolsulfonphthalein (PSP) and metformin were administered by intravenous infusion. In acidosis rats, serum concentration of PSP was significantly higher than that in normal rats and renal clearance of PSP was decreased. On the other hand, creatinine clearance and renal clearance of metformin were not changed in the acidosis rats, compared with normal rats. Further, the results of Western blotting indicated that the expression level of organic anion transporter (OAT) 3 was decreased in the acidosis rats. In contrast to OAT3, OAT1 or organic cation transporter (OCT) 2 were not affected. The immunohistochemical analysis showed that basolateral localization of OAT3 in the proximal tubules was not affected by the induction of acidosis. These results suggested that renal excretion of anionic drug was decreased during metabolic acidosis and that the decrease might be at least partly due to the reduction of OAT3 protein.
250. Characterization of Efflux Transport of Three PDE5 Inhibitors, Sildenafil, Vardenafil, and Udenafil
Im-Sook Song, Jian-wei Deng, Su-Jeong Yim, Young-Lim Choi, and Jae-Gook Shin
Department of Pharmacology and PharmacoGenomics Research Center, Inje University College of Medicine, Busan, South Korea
Purpose: Although efflux transporters are known to modulate the disposition of many clinically important drugs, little information is available for the involvement of efflux transporters in the disposition of PDE5 inhibitors. Thus, we aimed to characterize the efflux transport system of representative PDE5 inhibitors, sildenafil, vardenafil, and udenafil, and to compare the kinetic parameters of these compounds in MDCK cells overexpressing MDR1, BCRP, MRP1, MRP2, and MRP3.
Methods: We measured basal to apical transport as well as apical to basal transport of three PDE5 inhibitors within the concentration range of 1–100 M, using MDR1, BCRP, MRP1, MRP2, and MRP3 overexpressed MDCK cells.
Results: Udenafil and vardenafil showed a much greater basal to apical transport rate than the apical to basal transport rate in MDR1 and BCRP overexpressed cells (MDCKII-MDR1 and MDCKII-BCRP, respectively), while sildenafil showed neither MDR1 nor BCRP mediated efflux transport. Udenafil and sildenafil seems to be pumped out via MRP2 transporter. Udenafil showed a greater basal to apical transport rate than the apical to basal transport rate in MDCKII-MRP1 and MRP3 cells; however, vardenafil and sildenafil showed a similar transport rate in both directions.
Conclusions: Vardenafil and udenafil seem to be substrates for MDR1 and BCRP, whereas sildenafil is not a substrate neither of MDR1 nor of BCRP. Only udenafil of the three PDE5 inhibitors examined is a substrate for MRP1 and MRP3, which may increase the plasma concentration of udenafil. Their different contribution to efflux transporters, such as MDR, BCRP, and MRPs of three PDE5 inhibitors, may affect their different oral bioavailability, different half-life, and duration time, although the clinical relevance or relative contribution of efflux transport systems to pharmacokinetics and drug response need to be further evaluated through in vivo clinical studies.
251. Characterization of the 3-O-Sulfate Conjugate of 17α-Ethinylestradiol as an Inhibitor and Substrate of Human Drug Transporters
Yong-Hae Han, Dennis Busler, Yang Hong, and A. David Rodrigues
Department of MAP/MS F13-07, Bristol-Myers Squibb, Princeton, New Jersey, USA
17α-ethinylestradiol (EE2), a potent synthetic estrogen, is an important component of various oral contraceptive formulations. EE2 is metabolized and is known to undergo extensive (first-pass) 3-O-sulfation in both intestine and liver. Once formed, EE2 3-O-sulfate (EE2-Sulf) can circulate in the blood, serve as a source of free EE2, or be excreted. To date, however, the transporters governing the hepatobiliary and urinary excretion of EE2-Sulf are not known. Therefore, cryopreserved human hepatocytes were incubated with EE2-Sulf and the uptake kinetics assessed. Overall, the results were consistent with a transporter-mediated uptake process characterized by two saturable components (Km1 = 220 nM; Km2 = 15.5 μM). Further transporter phenotyping (chemical inhibitors and heterologously expressed transporters) revealed that EE2-Sulf serves as an OATP1B1, OATP2B1, and OAT3 substrate. For OATP1B1, uptake conformed to two-Km kinetics (87 nM and 148 μM, respectively), whereas a single Km described the kinetics of EE2-Sulf uptake by OATP2B1 (10.7 μM) and OAT3 (21.1 μM). Further, EE2-Sulf also behaved as an inhibitor of OATP1B1 (IC50 of 52 nM), OATP1B3 (IC50 of 10.7 μM), and OATP2B1 (IC50 of 4.2 μM). EE2-Sulf was also studied as a substrate of efflux transporters, employing membrane vesicles expressing MRPs (1, 2, 3, and 4), BSEP, and BCRP. The results showed that EE2-Sulf is a BCRP substrate (Km1 = 2.9; Km2 = 307 μM). All other vesicles did not show ATP-dependent uptake. It is concluded that EE2-Sulf uptake into liver is mediated by OATP2B1 and OATP1B1, followed by secretion into the bile via BCRP. On the other hand, renal elimination of EE2-Sulf likely involves OAT3.
252. Effect of OCT1 C1022T Allele on the Pharmacokinetics of Levosulpiride in Korean Subjects
Sang-No Lee,1 Se-Mi Kim,1 In-Ho Kwon,1 Hea-Young Cho,2 and Yong-Bok Lee1
1College of Pharmacy, Chonnam National University, Gwangju, South Korea
2General Pharmacology Team, National Institute of Toxicological Research, Korea Food and Drug Administration, Seoul, South Korea
OCT1 is one of the major transporters responsible for absorption, distribution, and elimination of clinically important drugs. Levosulpiride is a benzamide derivative selectively inhibiting the dopaminergic D2 receptors both in the central nervous system and gastrointestinal tract. The bioavailability of levosulpiride is only 30% after oral administration with large interindividual variability. The aim of this study was evaluate the effect of OCT1 C1022T allele on the pharmacokinetics of levosulpiride in Korean subjects. In total, 96 healthy male Korean subjects were genotyped for OCT1 C1022T, and the genotypes were identified by PCR-RFLP. Each volunteer received a single oral dose of a levosulpiride tablet (25 mg). Blood samples were collected over 36 hours, and the plasma concentrations of the drug were determined using HPLC. The subjects were classified into three groups for genotypes at position 1022 (CC, CT, and TT): 69 for the CC group, 24 for the CT group, and 3 for the TT group for analysis. The pharmacokinetic parameters of AUCt, AUCinf, Cmax, CL/F, and V/F of levosulpiride differed significantly (P < 0.05) among the three groups (P = 0.004, 0.008, 0.038, 0.009, and 0.002, respectively). Compared with the AUCt, AUCinf and Cmax of levosulpiride among the three different OCT1 C1022T genotype groups, those of mutant type were increased significantly with the AUCt to be 608.80 ± 162.71, 743.89 ± 184.15, and 796.73 ± 88.90 (ng•hour/mL), the AUCinf to be 672.56 ± 184.55, 812.66 ± 195.28, and 874.97 ± 143.84 (ng•hour/mL), and the Cmax to be 51.09 ± 19.89, 58.82 ± 21.46, and 75.09 ± 13.45 (ng/mL), respectively, as the number of OCT1 C1022T alleles increased. But, also CL/F and V/F of mutant type were decreased with the CL/F to be 40.17 ± 11.63, 32.38 ± 6.91, and 29.00 ± 4.58 (L/hour) and the V/F to be 589.09 ± 170.83, 459.38 ± 134.36, and 417.00 ± 63.38 (l). These results showed that genetic polymorphisms of the OCT1 C1022T gene can have an impact on levosulpiride disposition in humans, and that these genetic differences can explain the intersubject variabilities of levosulpiride disposition in humans.
253. Functional Impairment of Basolateral Renal Organic Anion Transporters in Experimental Diabetes in Mice
Anusorn Lungkaphin,1 Chutima Srimaroeng,1 and Varanuj Chatsudthipong2
1Department of Physiology, Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand
2Department of Physiology, Faculty of Science, Mahidol University, Bangkok, Thailand
Aims: Renal clearance of various anionic drugs and endogenous organic anion compounds is mainly mediated by the basolateral organic anion transporters 1 and 3 (OAT1 and OAT3). It has been shown that there are changes of renal transport function and several pharmacokinetic drugs in experimental diabetes mellitus. Therefore, alterations of renal OATs in diabetes mellitus could decrease the elimination or retention of drugs and result in severe nephrotoxicity. This study was conducted to examine the function and expression of renal OATs in diabetic mice induced by streptozotocin.
Methods: Diabetes in mice was induced by streptozotocin (200 mg kg−1 body weight). Mice with blood glucose exceeding 300 mg dL−1 were classified as diabetic. Transport activity was determined by fluorescein accumulation, a substrate of OAT1 and OAT3, into isolated renal proximal tubules by using confocal microscopy. Western blot analysis was quantified from total protein and membrane protein of kidney cortex to determine OAT1 and OAT3 expressions.
Results: The experimental diabetes induced by streptozotozin was confirmed by hyperglycemia, hyperlipidemia, and weight loss. Fluorescein uptake was conducted in 14, 21, 28, and 42 days of diabetic mice. However, fluorescein accumulation was significantly decreased only in 28 and 42 days of diabetes mice. There was also a concomitant reduction in OAT3 membrane expression, but not OAT1.
Conclusions: These results indicate that renal anion transport mediated by basolateral OATs is compromised after prolonged (4 weeks or longer) diabetic condition in mice. The mechanisms causing alterations in renal function is due to the reduction of renal OAT3 expression.
254. H+-coupled Efflux Transport of Erythromycin at the Blood-Placenta Barrier in Rats
Yoshimichi Sai, Noriaki Tanaka, Akinori Takagi, Kaori Ochi, Tomohiro Nishimura, Masatoshi Tomi, Noriko Kose, and Emi Nakashima
Faculty of Pharmacy, Keio University, Tokyo, Japan
Erythromycin (EM) is a macrolide antibiotic that is applicable to pregnant women who suffered chlamydial, syphilis, and mycoplasma infections because of its less toxicity and limited placental transfer. Although EM is a known substrate for human and mouse OAT2, the transport mechanism that limits the placental transfer has not been clarified yet. We have aimed to clarify the transport mechanism of EM at the blood-placenta barrier. The uptake of [14C]EM at the blood-placenta barrier was studied by using a conditionally immortalized syncytiotrophoblast cell line, TR-TBT 18d-1. The [14C]EM uptake by TR-TBT 18d-1 cells took place in a Na+ or Cl−-independent manner. The [14C]EM uptake was saturable, showing a Km value of about 400 μM, which was more than 10 times larger than that by human OAT2. The [14C]EM uptake was slightly inhibited by p-aminohippurate and cimetidine but hardly affected by dehydroepiandrosteronsulphate and probenecid. The saturable uptake was greatly increased according to increment of extracellular pH ranging from 6.4 to 8.4. The pH-dependency was further studied by using rat placental brush-border membrane vesicles (BBMVs). The [14C]EM uptake by BBMVs was enhanced in the presence of outwardly directed H+ gradient. An overshoot phenomenon was observed at pHout8.4 and pHin7.4 condition, suggesting involvement of an H+-coupled exchange mechanism. Efflux transport by TR-TBT 18d-1 cells was assessed by examining the effect of cyclosporin A, verapamil, quinidine, fumitremorgin C, probenecid, and NaN3. None of them affected the transport, but ethylisopropylamirolide, a Na+/H+ exchanger NHE inhibitor, significantly reduced the [14C]EM efflux. The unidirectional fetal-to-maternal clearance (Kfm) of [14C]EM across the placenta was evaluated by rat in situ placenta perfusion. The Kfm value of [14C]EM was significantly greater than that of a diffusional permeability marker [14C]inulin. It was significantly reduced in the presence of 5 mM of EM, suggesting the existence of a saturable efflux transport in in vivo condition. In conclusion, our data suggested that the H+-coupled efflux transport limited the placental transfer of EM to the fetus.
255. Oseltamivir (Tamiflu™) Is a Substrate of PEPT1
Takuo Ogihara,1 Takashi Kano,1 Tamae Wagatsuma,2 Sho Wada,1 Hikaru Yabuuchi,3 Shigeki Emonoto,3 Yoshiyuki Shirasaka,4 Kaori Morimoto,1 Shoko Kobayashi,2 and Ikumi Tamai4
1Department of Pharmacology, Faculty of Pharmacy, Takasaki University of Health and Welfare, Takasaki, Japan
2Department of Food and Life Science, Takasaki University of Health and Welfare, Takasaki, Japan
3GenoMembrane, Inc., Yokohama, Japan
4Faculty of Pharmacy, Institute of Medical, Pharmaceutical, and Health Sciences, Kanazawa University, Kanazawa, Japan
Oseltamivir, an ester-type prodrug of the neuraminidase inhibitor, Ro 64-0802, has been developed for the treatment of A and B strains of the influenza virus, but has neuropsychiatric and other side effects. We previously demonstrated that oseltamivir, but not Ro 64-0802, was a substrate of P-glycoprotein (P-gp). Accordingly, low levels of P-gp activity or drug-drug interactions at P-gp may lead to enhanced brain accumulation of oseltamivir, and this, in turn, may account for the central nervous system effects of oseltamivir observed in some patients (Morimoto et al., 2008). During that research, we noticed that increased plasma concentration and toxicity were observed in fasted infant rats compared with nonfasted ones. Therefore, we speculated that transporters involved in the uptake of food components also take part in the absorption of oseltamivir. In this study, we characterized the transport across intestinal epithelial cells and the absorption of oseltamivir. Uptake by Caco-2 cells (human carcinoma cell line) and HeLa cells transfected with peptide transporter1 (HeLa/PEPT1) was time- and temperature-dependent, and was inhibited by typical PEPT1 inhibitors, such as glycyl-sarcosine (Gly-Sar). The uptakes by Caco-2 cells and HeLa/PEPT1 were saturable, with similar Km values. Oseltamivir absorption in adult rats was greatly reduced by the simultaneous administration of milk, casein, or Gly-Sar. Further, the plasma and brain concentrations of oseltamivir were higher in fasting than in nonfasting rats after oral administration. These results suggest that oseltamivir is a substrate of PEPT1, and that PEPT1 is involved in its intestinal absorption. It may be clinically important that milk peptides inhibit the absorption of oseltamivir, especially in infants who routinely receive milk, because this could result in reduced efficacy, as well as reduced toxicity. Influx transporters, such as PEPT1, may contribute to the absorption of various ester-type prodrugs, not only peptide-mimetic ones.
Reference
- Morimoto, K., Nakakariya, M., Shirasaka, Y., Kakinuma, C., Fujita, T., Tamai, I., et al. (2008). Oseltamivir (Tamiflu) efflux transport at the blood-brain barrier via P-glycoprotein. Drug Metab Dispos 36:6–9.
256. Regulatory Role of Sex Hormones on Organic Cation Transport: In Vivo and In Vitro Studies
Paranee Meetam,1 Chutima Srimaroeng,2 Sunhapas Soodvilai,3 Titiwat Sungkaworn,3 and Varanuj Chatsudthipong3
1Department of Biopharmacy, Faculty of Pharmacy, Silpakorn University, Nakornpathom, Thailand
2Department of Physiology, Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand
3Department of Physiology, Faculty of Science, Mahidol University, Bangkok, Thailand
The renal proximal tubule (RPT) plays a crucial role in the secretion of organic cation (OC). The OC secretory transport process is vectorial which involves the transporters located at both basolateral and apical membranes. This study was performed to investigate the effect of sex hormones on OC transport process using in vivo clearance and isolated mouse RPT. Tetraethylammonium (TEA) was used as a prototypic organic cation. Castration significantly decreased [3H]-TEA uptake in isolated RPT and its renal clearance by about 35.5 and 35.9%, respectively. Supplementation of testosterone to castrated mice was able to restore [3H]-TEA uptake and clearance to normal levels. In contrast, [3H]-TEA uptake and clearance were higher in ovariectomized mice than that of the control by about 47.7 and 35.1%, respectively, and could be restored to normal levels by estrogen treatment. Kinetic analysis of [3H]-TEA uptake suggested that a decrease in transport rate of TEA observed was due to a lower number of transporters in castrated mice, rather than a change in transport affinity, and conversely, an increase in transport rate in ovariectomized mice was due to a higher number of transporters. The mRNA levels of the relevant transporters at basolateral (OCT1, OCT2, and OCT3) and apical (OCTN1, OCTN2, and MATE1) measured by quantitative polymerase chain reaction suggested that OCT2 is the major transporter responsible for TEA transport in our study. The OCT2 mRNA level was significantly reduced after castration. Interestingly, castrated mice also showed a modest increase in organic cation/carnitine transporter 1(OCTN1) mRNA level, indicating that testosterone may also regulate apical OCTN1 expression. In the female group, mRNA levels of transporter at the basolateral membrane were not affected by estrogen, whereas kinetic study suggested the modulating role of estrogen on the number of transporters. The controversial between the kinetics study and mRNA level suggested that the effect of estrogen may affect OC transport by post-transcriptional regulation. Collectively, our findings indicate that testosterone and estrogen act differently in regulating OCT-mediated transport activity in mouse kidney.
257. Substrate-Dependent Effect of Genetic Variant of NTCP on the Uptake of Statins
Im-Sook Song, Wei Pan, Ho-Jung Shin, and Young-Lim Choi
Department of Pharmacology and PharmacoGenomics Research Center, Inje University College of Medicine, Busan, South Korea
Na+-taurocholate cotransporting polypeptide (NTCP, SLC10A1) greatly contributes to the recirculation of bile acids and also contributes to the hepatic intake of drugs. This study was designed to identify common NTCP genetic variants and determine their in vitro functional alteration. The in vitro function of genetic variants of NTCP was evaluated in an Flp-In 293 expression system. Sequence analysis revealed 6 NTCP variants, including a novel nonsynonymous SNP, G190A (A64T, *6), in the NTCP gene with 1% allele frequency in Koreans (n = 150). However, this SNP was not found in the 366 Chinese and the 152 Vietnamese subjects studied. In contrast to NTCP*2 (S267F), NTCP*6 (A64T) lost most of the uptake activity toward both taurocholic acid (TA) and estrone-3-sulfate (ES), when compared with the reference. Km of NTCP*1,*2 and *6 for TA were, respectively, 10.50 ± 2.01, 84.01 ± 4.38, and 46.02 ± 5.56 M, while the Vmax were 307.83 ± 12.68, 9.80 ± 0.26, and 289.77 ± 14.00 pmol/mg pro/min, respectively. Subcellular localization was similar among the reference and the variants of NTCP. The effect of genetic variants of NTCP on the uptake of various statins was further investigated. Among statins investigated in this study, rosuvastatin, pitavastatin, and atorvastatin have a great affinity to NTCP, pravastatin and fluvastatin have a moderate affinity to NTCP, but NTCP did not facilitate the simvastatin uptake. Moreover the effect of genetic variant of NTCP, p.S267F, was substrate dependent. For example, the uptake of rosuvastatin and atorvastatin was significantly increased by the NTCP-S267F variant; however, the uptake of pravastatin, pitavastatin, fluvastatin, and simvastatin was not altered by the NTCP-S267F variant. Considering the uptake of taurocholate by the NTCP-S267F variant was greatly decreased, the effect of NTCP genetic variation on the transport activity for the substrates should be investigated with care, depending on the substrate groups.
258. Abstract Withdrawn
259. Effects of Curcuma comosa Extracts on Choline Acetyltransferase and Acetylcholinesterase in Rat Brain
Sasithorn Lupreechaset,1 Nuansri Niwattisaiwong,2 Somlak Poungshompoo,3 Laddawal Phivthong-ngam,4 and Somsong Lawanprasert2
1Nopparatrajathanee Hospital, Bangkok, Thailand
2Faculty of Phamaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
3Faculty of Veterinary Sciences, Chulalongkorn University, Bangkok, Thailand
4Faculty of Medicine, Srinakharinwirot University, Bangkok, Thailand
Curcuma comosa Roxb., a plant in the family of Zingiberaceae, is one of the well-known medicinal plants in Thailand. Several previous studies reported that C. comosa exhibited estrogenic-like effects. The objective of this study was to investigate the effects of C. comosa hexane and ethanolic extracts on the activities of choline acetyltransferase (ChAT) and acetylcholinesterase (AChE) in rat brain. Forty-five rats were randomly divided into six treatment groups. Rats were administered orally with 1 mL/kg/day of corn oil (control group), 250 and 500 mg/kg/day of C. comosa hexane extract (groups 2 and 3), and 250 and 500 mg/kg/day of C. comosa ethanolic extract (groups 4 and 5) for 30 days. Each group comprised 8 rats. Estradiol valerate was given intramuscularly to 5 rats at a dose of 10 g/kg/day for 7 days, serving as an estrogen reference group. At the end of the treatment, rats were anesthesized, then cervically dislocated. Serum samples were determined for estradiol concentrations. Brain was collected and dissected for cerebral cortex, basal forebrain, and hippocampus. All brain-region homogenates were prepared for an enzyme activity assay. The results showed that both dosages of C. comosa ethanolic extract caused an increase of ChAT activity in cerebral cortex and hippocampus but not in basal forebrain. C. comosa hexane and ethanolic extracts did not affect AChE activities in all three brain regions. Estradiol, given at the dosage regimen in this study, did not affect both ChAT and AChE activities. Even though an increase of serum estradiol level from C. comosa extracts was less than the estradiol group, both extracts caused a significant increase of serum estradiol level, as compared to the control group. These findings demonstrated that C. comosa ethanolic extract might potentially possess a beneficial effect on the cholinergic nervous system as shown by an enhancement effect on the activity of ChAT, an enzyme responsible for brain acetylcholine synthesis.
260. Involvement of MATE1 in the Biliary and Urinary Excretion of Metformin in Mice
Sumito Ito,1 Hiroyuki Kusuhara,1 Yushun Kuroiwa,1 Chunyong Wu,1 Yoshinori Moriyama,2 and Yuichi Sugiyama1
1Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan
2Okayama University Graduate School of Medicine, Density, and Pharmaceutical Science, Okayama, Japan
An antidiabetic drug, metformin, is eliminated predominantly from the kidney as unchanged form. Metformin is an organic cation at physiological pH, and undergoes the tubular secretion in the kidney by the organic cation transport system. Although it has been demonstrated that the renal uptake of metformin is mediated by OCT2/SLC22A2, the luminal efflux mechanism for this remains unknown. Further, the target of metformin is liver mitochondrial respiratory complex 1, and inhibition of mitochondrial respiration results in an inhibition of gluconeogenesis in the liver. OCT1/SLC22A1, a hepatic uptake transporter, is deeply associated with the pharmacological effect/adverse reaction of metformin. The present study focused on the multidrug and toxin extrusion 1 (MATE1/SLC47A1), a proton/organic cation exchanger that is expressed in the liver and kidney. The substrate specificity and membrane localization of this transporter suggests its involvement in the biliary and urinary excretion of cationic drugs. Pyrimethamine (PYR) was found to be a potent inhibitor of MATE1 with Ki values of 0.29 μM for mouse Mate1. In vitro transport study using cDNA transfectants elucidated that inhibition potencies of PYR to the basolateral transporters (Oct1 and Oct2) than Mate1 (the Ki values were 3.6 and 6.0 μM, respectively), indicating that PYR can inhibit selectively luminal efflux mechanisms. PYR (2 μmol/kg, intravenously; i.v.) was given to ddY mice (male, 8–9 weeks) before starting the i.v. infusion of metformin. The plasma, liver, and kidney concentrations, and urinary and biliary excretion rates, were determined under steady-state conditions. Although the plasma concentrations, and biliary and urinary excretion rates, were unchanged, both liver and kidney concentrations of metformin were significantly greater in mice treated with PYR, and the tissue-to-plasma concentration ratios were 3.5- and 3.8-fold greater in mice treated with PYR in the kidney and liver, respectively. Consistent with increased liver concentration, the plasma lactate following metformin treatment was significantly higher in mice treated with PYR. These results suggest that Mate1 mediates the efflux of metformin both in the liver and kidney, and it acts as one of the critical factors for the pharmacological action of metformin.