1. Abstract not available
2. Abstract not available
3. Predicting the human pharmacodynamics of monoclonal antibodies: implications for first in man studies
Stacey J. Tannenbaum1, and Philip Lowe2
1Modeling and Simulation, Novartis Pharmaceuticals Corp., East Hanover, NJ, USA, 08807
2Modeling and Simulation, Novartis Pharma AG, Basel, Switzerland, CH-4056
Although the phases of clinical drug development are well known, it is relatively unappreciated that there are similar phases in preclinical development. “Phase I” the initial, normally Research Discovery driven pharmacology; “Phase II” non-GLP dose range finding, followed by pivotal “Phase III” GLP toxicology. Together with an array of in vitro experiments comparing species, these stages should enable an integrated safety assessment (ISA) prior to entry into man, documenting to investigators (IB, Protocol) and authorities (IND/CTA) evidence that the new pharmaceutic is unlikely to cause harm. Following the lessons learned from TeGenero TGN1412 and subsequent updates to regulatory guidelines, there are aspects peculiar to biotherapeutics, especially those that target the immune system. Two of these are i) target mediated drug disposition, where the binding of the drug to its target quantitatively affects the pharmacokinetics and ii) the ability to quantitate soluble target binding directly by analysing for drug-target complexes. A pharmacokinetic-pharmacodynamically drug-target binding guided process will be exemplified for the aforementioned aspects using both in-house and literature studies. The objective of the process is to deliver a justification of a safe starting dose and therapeutically relevant escalation doses for human studies based on mode of action considerations.
4. Abstract Not Available
5. Structure-function relationships of the cytochrome p450 2c9 drug metabolizing enzyme and impact of genetic variation
Timothy S. Tracy
Experimental and Clinical Pharmacology, University of Minnesota, Minneapolis, MN, USA, 55455
Cytochrome P450 2C9 (CYP2C9) is involved in the metabolism of several drugs of therapeutic importance, including (S)-warfarin, phenytoin, losartan and the NSAIDs. Drugs that are substrates of CYP2C9 typically are weakly acidic due to key binding determinants within the CYP2C9 active site. Crystal structures of CYP2C9 have confirmed results from site-directed mutagenesis studies with respect to the importance of the R108, F110 and F476 residues in substrate binding interactions. The R108 residue facilitates ionic interactions with anionic moieties on the substrate and the phenylalanine residues serve to facilitate pi-stacking interactions. Interestingly, atypical kinetic profiles including substrate inhibition, auto-activation, biphasic kinetics and heteroactivation, suggestive of simultaneous binding of two substrates within the CYP2C9 active site have been observed leading to predictions of a flexible active site that can alter conformation and size (confirmed by crystallographic studies) to accommodate multiple substrate molecules. In the case of heteroactivation, the effector molecule appears to enhance substrate metabolism through effects on the P450 cycle efficiency and alterations in the water network. CYP2C9 also exhibits genetic polymorphisms with the most common amino acid substitutions being the R144C (allele frequency ≈ 8%) and I359L (allele frequency ≈6%) variants. The R144C variant is located outside the enzyme active site but nevertheless results in an approximately 30% reduction in enzyme activity. More significantly, the I359L variant is located within the active site but not thought to be directly involved in substrate binding, yet results in an 80% reduction in enzyme activity. Stoichiometry studies suggest both variants affect P450 cycle efficiency but at different steps. CYP2C9 allelic variants not only affect substrate metabolism rates but can also affect degree of drug interactions, through effects on inhibitor potency and reductions in clearance rates. In summary, CYP2C9 metabolizes several important drugs, exhibits complicated structure-function relationships due to active site plasticity and is subject to genetic polymorphisms that alter both substrate metabolism rate and drug interaction potential. Supported by NIH Grants #GM063215 and GM069753.
6. Cytochrome p450 erad: functional partnerships, molecular interactions and post-translational modifications for proteasomal targeting
Maria Almira Correia
Cellular and Molecular Pharmacology, University of California, San Francisco, San Francisco, CA, USA, 94158
Cytochromes P450 (P450s) are endoplasmic reticulum (ER) membrane-anchored hemoproteins engaged in the metabolism and disposition of endo- and xenobiotics. Because of their pivotal role in cellular drug detoxification, the principal research focus has been on their functional role in drug metabolism (DM) and drug-drug interactions (DDIs). These P450-dependent processes are directly controlled by the cellular content/activity of the enzymes involved. While drug-mediated P450 induction and inhibition are clearly major determinants of clinically relevant DM and DDIs, it is now becoming increasingly evident that drug-mediated P450 inactivation/ stabilization/turnover can also influence these processes, as the grapefruit juice-mediated inactivation/degradation of CYP3A4 amply exemplifies. Assessment of the physiological role of P450 inactivation/stabilization/turnover in DM and DDIs requires a fundamental understanding of the intrinsic cellular/molecular processes. However, relatively little is known about the mechanisms involved in P450 stabilization and turnover. We have therefore focused on elucidating P450 stabilization and turnover at the basic cellular/molecular level by characterizing the cellular proteins and posttranslational modifications involved in this process using various eukaryotic (yeast, rat and human) cell models. Diagnostic chemical probes of ubiquitin (Ub)-dependent 26S proteasomal degradation (UPD) and autophagic-lysosomal degradation (ALD) were also employed for molecular analyses of P450 protein degradation pathways. Our collective findings reveal that in spite of the vast evolutionary diversity, the cellular degradation pathways (involving UPD, ALD or both) for hepatic CYPs 3A, 2B1, 2C11 and 2E1 as P450 prototypes are essentially conserved from Saccharomyces cerevisiae to man. Accordingly, we have found that native or suicidally inactivated hepatic CYPs 3A (3A4/3A5/3A23) subscribe to UPD in a classical ER-associated degradation (ERAD) process. This involves phosphorylation of specific residues to target the CYP3A proteins to an E2/E3 Ub-ligase complex for ubiquitination, followed by the AAA ATPase p97-mediated extraction of the ubiquitinated CYP3A out of the ER-membrane and its delivery to the cytosolic 26S proteasome for degradation. The specific cellular chaperones, kinases and the E2/E3 partnerships involved in this process along with chemicals including clinically relevant drugs that affect P450 turnover in human and rat hepatocytes will be discussed. Supported by NIH Grants GM44037, DK26506, and DK26743.
7. Conformational plasticity and heterogeneity of cytochromes p450 2b and 3a: implications for prediction of drug metabolism
James R. Halpert
Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California, San Diego, La Jolla, CA, USA, 92093-0657
P450 2B enzymes are very versatile catalysts with a broad range of substrates and inhibitors. In addition to marked species differences, the presence of functionally distinct allelic variants of rat P450 2B1 and human P450 2B6 and of highly structurally related but functionally distinct enzymes in rats and rabbits has made P450 2B enzymes an excellent model system for investigating structure-function relationships. X-ray crystal structures of rabbit P450 2B4 represent the greatest diversity of conformations of a single mammalian P450 reported to date. Our central hypothesis is that P450 2B ligand binding affinity and specificity are determined by enzyme plasticity as well as ligand access and binding. Recently, the 2.5 å structure of P450 2B4 in complex with 1-(4-phenyl)benzylimidazole was solved (1). This complex is a structural intermediate to the previous closed 4-(4-chlorophenyl)imidazole and open bifonazole structures (2-3). Comparisons of five 2B4 structures reveal trends in plastic region mobility that may allow predictions of their positions in future structures and hence ligand binding to this flexible enzyme. The central hypothesis in our studies of P450 3A4 is that conformational changes resulting from ligand binding and/or protein-protein interactions play a key role in homotropic and heterotropic cooperativity. Our work has utilized pressure-perturbation spectroscopy, rapid scanning stop flow, and advanced absorbance and fluorescence techniques. Very recent results with P450 3A4 and the allosteric bacterial enzyme P450eryF indicate a three-step “cascade” mechanism of ligand binding (4). Thus, binding of the first substrate molecule to yield the SE complex promotes a second, spectrally silent binding event that causes expulsion of the first substrate molecule to yield the effector complex ES. Binding of an additional molecule triggers the formation of the final binary complex (ESS), where the substrate-induced spin shift is observed. In addition, conformational transitions affect P450-P450 interactions and modulate heterogeneity of the P450 3A4 pool, which consists of multiple and functionally distinguishable conformers that do not interconvert during the time frame of our experiments (5). (Supported by NIH grants ES003619 and GM54995).
References
- Gay, S.C., Sun, L., Maekawa, K., Halpert, J.R., and Stout, C.D. (2009). Crystal structures of cytochrome P450 2B4 in complex with the inhibitor 1-biphenyl-4-methyl-1H-imidazole: ligand induced structural response through alpha-helical respositioning. Biochemistry, in press.
- Scott, E.E., White, M.A., He, Y.A., Johnson, E.F., Stout, C.D., and Halpert, J.R. (2004). Structure of mammalian cytochrome P450 2B4 complexed with 4-(4-chlorophenyl)imidazole at 1.9 å resolution: Insight into the range of P450 conformations and coordination of redox partner binding. J. Biol. Chem. 279:27294–27301.
- Zhao, Y., White, M.A., Muralidhara, B.K., Sun, L., Halpert, J.R., and Stout, C.D. (2006). Structure of microsomal cytochrome P450 2B4 complexed with the antifungal drug bifonazole: Insight into P450 conformational plasticity and membrane interaction. J. Biol. Chem. 281:5973–5981.
- Davydov, D.R., Davydova, N.Y., and Halpert, J.R. (2008). Allosteric transitions in cytochrome P450eryF explored with pressure-perturbation spectroscopy, lifetime FRET, and a novel fluorescent substrate, Fluorol-7GA. Biochemistry 47:11348–11359.
- Davydov, D.R., and Halpert, J.R. (2008). Allosteric P450 mechanisms: multiple binding sites, multiple conformers, or both? Expert Opinion Drug Metab. Toxicol. 4:1523–1535.
8. Diverse functions of microsomal p450 enzymes – insights from various engineered mouse models
Xinxin Ding, Cheng Fang, Xin Zhou, Yuan Wei, Jaime D’Agostino, Fang Xie, Jun Gu, and Qing-Yu Zhang
Wadsworth Center, NYSDOH, Albany, NY, USA, 12201-0509
Engineered mouse models are increasingly utilized for the determination of the roles of P450 enzymes in target tissue metabolic activation and tissue-selective toxicity, including carcinogenicity, of drugs and other xenobiotic compounds. These mouse models are also used to explore the potential biological functions of P450 enzymes in various organ systems, such as the brain and the olfactory chemosensory organ. This presentation will focus on our recent studies using various mouse models that target the cytochrome P450 reductase (CPR), an enzyme required for the function of all microsomal P450 enzymes, as well as mouse models that target selected enzymes in the Cyp2 gene family. Mouse models with tissue-specific Cpr deletion in liver, lung, heart, forebrain, or intestine have been produced, and utilized for drug metabolism and toxicity studies. A forebrain neuron-specific Cpr-null mouse was also found to have interesting biological phenotypes, including an impaired ability to retrieve spatial memory, and a decreased sensitivity to electric shock, accompanied by decreased activities of brain microsomes toward steroid hormone metabolism. Additional mouse models are being studied, including a Cpr-low mouse, which has decreased CPR expression in all tissues; an extrahepatic Cpr-low mouse, which has normal CPR expression in the liver, but decreased CPR expression in all extrahepatic organs; and several P450 knockout mouse models. Studies using the Cpr-low mouse model suggested that a decreased CPR expression can slow the progression of neurodegenerative changes in a transgenic mouse model of Alzheimer disease. Studies on a Cyp2a5-null mouse indicated that, while hepatic CYP2A5 does not play a significant role in systemic clearance of endogenous testosterone, the loss of CYP2A5 is accompanied by significant increases in tissue levels of testosterone in selected extrahepatic organs, such as the lateral nasal gland, a gland important for nasal mucus secretion. These studies illustrate the utility of engineered mouse models for exploration of the in vivo functions of P450 enzymes. (Supported in part by NIH grants ES007642 and CA092596)
9. Abstract not available
10. Therapeutic peptide; exploring charged based detection and ion-ion reaction as alternative quantitation technique
J.C. Yves Le Blanc
Research, MDS Analytical Technologies, Concord, ON, Canada, L4K 4V8
A rapidly growing area in the biopharmaceutical industry is the analysis of therapeutic peptides, typically generated from in vivo studies at the discovery stage. Traditionally, ELISA is used to quantitate these peptides/proteins since it provides sufficient dynamic range, and rapid, high-throughput analyses. However, the development of ELISA assays is time-consuming and is a poor match for rapid drug-discovery screening requirements. Mass spectrometry-based techniques could provide results much faster at the discovery stage if the appropriate sensitivity and selectivity can be achieved. For conventional drug candidates, the preferred approach is MRM-based detection, which provides the required selectivity and sensitivity for rapid analyses. However, the MRM approach is not as routine and simple when analyzing multiply charged (z>>2) peptides and proteins. Experiment optimization can take days to perform, and multiply charged peptides/proteins typically fragment to form a multitude of fragments, diluting the analytical signal and making the analysis less sensitive [Berna and Ackerman, 2009]. It is of great interest to discover alternative mass spectrometry-based techniques that offer selectivity comparable to MRM analyses without their disadvantages. In this work, we report on two approaches that enable selective and quantitative detection of multiply charged peptides/proteins. The first, targeted-Enhanced Multiply Charged (targeted-EMC) scans, relies on the selective discrimination of singly charged noise prior to mass analysis in a hybrid quadrupole linear ion trap (QqLIT). The main advantage of this technique is that no compound specific tuning is required. The second approach relies on controlled charged reduction using ion-parking [McLuckey et al, 2002]. In this method, the multiply charged peptide/protein ions are reduced to lower charge states in vacuum via ion-ion reactions. This process is stopped at a specific charge state based on selective inhibition of the reaction. With these approaches, it was possible to perform selective detection of peptides at levels comparable to MRM analyses. Results for both techniques will be presented, as will a discussion regarding the analytical figures of merits and limitations of these techniques.
11. The emerging role of high-resolution single stage mass spectrometry in quantitative bioanalysis
Richard King
PharmaCadence Analytical Services, LLC, Hatfield, PA, USA, 19440
Over the past two decades, mass spectrometric detection has become perhaps the most widely used of all analytical techniques for studying biological systems. The most often cited advantages of mass spectrometry and particularly liquid chromatography coupled on-line with mass spectrometry are selectivity, sensitivity, and broad applicability. Active research in the area of LC-MS is primarily aimed at improving performance in one or more of these areas. This presentation is focused on the selectivity and sensitivity aspects of modern LC-MS. Currently, the most selective strategy is to use selected reaction monitoring (SRM) with liquid chromatography. This has become the method of choice when implemented on the triple quadrupole instruments, for performing targeted quantitative analysis. While highly effective, the technique requires experimental determination of the fragmentation characteristics of the targeted analyte under a variety of experimental conditions. There are a number of automated strategies for reducing the work required to generate the information necessary to use SRM, however, if single a single stage of MS had the required selectivity, dynamic range, and acquisition speed, the process might be eliminated entirely saving time, effort and ultimately money. Additionally, the means by which SRM is selective also inherently limits the detection of other species, particularly unknown species in the sample. Recent advances in high resolution mass spectrometry that allow for resolution greater than 30,000 with linear dynamic range in excess of 4 orders of magnitude and acquisition rates greater than 10 spectra/second have opened the door for practical application of high resolution, single stage, mass spectrometry to quantitative bioanalysis. Here we present the results from an in-depth investigation of the relative selectivity and sensitivity of the high-resolution MS versus that of SRM. We also examine the potential advantages of using full scan single stage MS for quantitative bioanalysis and the future applications that might be enabled by the technique.
12. Single molecule mircoscopy techniques for the study of antibody trafficking in live cells
Sripad Ram, Prashant Prabhat, Jerry Chao, Raimund J. Ober, and E. Sally Ward
Immunology, University of Texas Southwestern Medical Center, Dallas, TX, USA, 75390
Antibody molecules of the IgG class represent an essential component of humoral immunity. The study of 3D intracellular trafficking pathways of IgGs in live cells is of relevance to understanding their role in mediating immune responses. Current research efforts have mainly focused on studying intracellular trafficking events either on the plasma membrane or deep inside the cell. However, the 3D trafficking itineraries that span from the plasma membrane to the intracellular compartments have not been well studied. The main reason for this is that currently available imaging technologies are not well adapted for imaging rapidly moving intracellular objects in 3D. Recently we developed a microscopy modality called multifocal plane microscopy for 3D imaging and tracking of subcellular objects and single molecules in live cells. This has allowed us to quantitatively track and characterize 3D intracellular events that are otherwise difficult to capture with conventional imaging techniques. We have imaged the 3D intracellular trafficking pathways of IgG and its salvage receptor, FcRn (neonatal Fc receptor), in live endothelial cells. FcRn is distinct from the classical Fcg receptors and is involved in recycling/transporting IgGs and immune complexes at blood-tissue barriers. Our imaging data reveals highly complex trafficking itineraries for IgG and FcRn that span from the plasma membrane to intracellular compartments deep inside the cell. The use of quantum dot labeled IgGs enabled us to study the endocytic and exocytic pathways at the single molecule level for extended periods of time. Our data shows evidence for novel mechanisms of FcRn-mediated IgG exocytosis and endocytosis and provides new insights into the 3D intracellular trafficking pathways of IgGs in cells. The multifocal plane imaging technology is not limited to tracking IgGs and can be used to study the 3D intracellular trafficking of other proteins as well as therapeutic agents such as drug/gene delivery vectors, toxin-fusion protein conjugates, etc. This research was supported in part by the NIH and in part by a postdoctoral fellowship from the National Multiple Sclerosis Society.
13. Role of transcription factors in the regulation of hepatic drug metabolism and transporter genes
Curtis D. Klaassen
Pharmacology, Toxicology, and Therapeutics, The University of Kansas Medical Center, Kansas City, KS, USA, 66160
The elimination of many drugs and other xenobiotics requires transporters for uptake into hepatocytes, phase-I and -II biotransformation to form more water-soluble compounds, as well as transport out of hepatocytes. While it has been known for about a half century that one drug can enhance the biotransformation of other drugs, the molecular mechanisms of induction of the major phase-I enzymes, the Cytochrome P450s (CYPs), were not delineated until recently. The four transcription factors that appear to be most important for the induction of CYPs are Aryl hydrocarbon Receptor (AhR), Constitutive Androstane Receptor (CAR), Pregnane X Receptor (PXR), and Peroxisome-Proliferator Activated Receptor Alpha (PPARα). It was discovered recently that these transcription factors can also induce the expression of uptake transporters, phase-II biotransformation enzymes, as well as efflux transporters. A fifth transcription factor, namely Nuclear factor E2-related factor 2 (Nrf2), although not important for the induction of CYPs, is mechanistically very important for the up-regulation of phase-II enzymes and transporters. In addition, it is now evident that dioxin (TCDD) induction of many phase-II enzymes requires both AhR and Nrf2 transcription factors. Thus, although drug-drug interactions have been recognized for a half century, the molecular mechanisms accounting for such interactions through induction of uptake transporters, phase-I and -II drug metabolizing enzymes, as well as efflux transporters have not been unraveled until recently, which will be covered in this lecture. (Supported by NIH grants ES009649, ES009716, ES013714, and DK081401).
14. The role of shp in bile acid-induced liver toxicity
Li Wang
Medicine and Oncological Sciences, University of Utah School of Medicine, Salt Lake City, UT, USA, 84132
Bile acid homeostasis is controlled by coordinated feedback and feedforward regulation of genes involved in bile acid uptake, efflux, and biosynthesis. Nuclear receptor farnesoid X receptor (FXR, NR1H4) is identified as a primary bile acid receptor and sensor and plays a key role in the transcriptional regulation of bile acid metabolism. Small heterodimer partner (SHP, NR0B2), a FXR target gene and transcriptional repressor, is a crucial component in the negative feedback regulation of bile acid synthesis. Cholestatic liver injury has been shown to be an important stimulus for the development of fibrosis. Cholestasis results in intrahepatic accumulation of cytotoxic bile acids which ultimately leads to fibrosis and cirrhosis, the latter is the advanced stage of fibrosis. Hepatic fibrosis is a scarring process of the liver that includes components of both increased and altered deposition of extracellular matrix (ECM) and reduced breakdown of ECM components. Hepatic stellate cells (HSCs) are the major source of ECM in the liver and proliferation and differentiation of HSCs into myofibroblast-like cells have been associated with the development of liver fibrosis. With the progression of fibrosis induced by bile duct ligation (BDL), alpha smooth muscle actin (α-SMA) is highly expressed by HSCs, which is classically considered as an indicator and marker of cell activation to myofibroblast-like cell. Interestingly, the FXR-/- mice died within a week upon bile acid feeding due to severe liver damage, whereas they exhibited reduced response to BDL-induced liver damage, possibly due to the adaptive overexpression of hepatic ABC transporters in response to cholestasis. In contrast, the SHP-/- mice appeared to be less sensitive to bile acid feeding-induced liver damage, however, they were more prone to BDL-induced cholestasis. Consistently, liver fibrosis induced by BDL was attenuated in SHP-overexpressed transgenic mice. We demonstrated that SHP suppressed gene expression of -SMA in activated HCSs mediated by reducing the transactivation activity of Nrf2 (nuclear factor erythroid-2 related factor 2). Our study identified a novel molecular component, the Nrf2 gene, through which SHP inhibited α-SMA function and liver fibrosis. We conclude that SHP and Nrf2 crosstalk plays an important role in the progression of liver fibrosis induced by cholestatic liver injury.
15. Nuclear receptor regulation of hepatic hydroxysteroid sulfotransferase (SULT2A)
Melissa Ann Runge-Morris
Insitute of Envir Hlth Sci, Wayne State University, Detroit, MI, USA, 48201
Hepatic hydroxysteroid sulfotransferase (SULT2A) catalyzes the sulfonation of endogenous hormones and xenobiotics, selected bile acids and oxysterols. Because of its multi-faceted role in physiology, the precise molecular regulation of SULT2A is being increasingly recognized as an important determinant of xenobiotic detoxication, carcinogen bioactivation, and the ligand-activation of lipid-sensing nuclear receptors. Unlike its rodent counterpart, human hepatic SULT2A1 gene transcription is regulated by the peroxisome proliferator-activated receptor alpha (PPARalpha), a nuclear receptor that is ligand-activated by fibrate xenobiotics and endogenous fatty acids. PPARalpha transactivates human hepatic SULT2A1 transcription through a cis-acting PPAR responsive element located at -5949 to-5929 nucleotides distal from the SULT2A1 transcription start site. Also unlike the rodent SULT2A, human hepatic SULT2A1 gene transcription undergoes dose-dependent suppression in response to pregnane X receptor (PXR) activation by rifampicin. The PXR is a nuclear receptor that is ligand activated by a broad spectrum of steroids, bile acids and xenobiotics, and therefore, represents an important control nexus for a number of diverse target gene pathways. Rifampicin-activated PXR negatively regulates SULT2A1 transcription through interactions with HNF4alpha, a liver-enriched nuclear receptor that is not known to be ligand activated. Higher concentrations of rifampicin induce SULT2A1 gene transcription through a PXR-independent mechanism. In vitro coactivator recruitment analysis demonstrates that the SULT2A1-catalyzed sulfonation of oxysterols antagonizes oxysterol-mediated activation of the liver X receptor (LXR), another important nuclear receptor engaged in the integrated control of lipid metabolism. In systemic diseases that feature either a lipid imbalance or the emergence of dyslipidemias, dynamic disturbances in the population of biologically active lipids in human liver would be expected to titrate the activation of down-stream nuclear receptor pathways. This cascade of events is poised to influence both the expression of lipid metabolism target genes and that of SULT2A, a xenobiotic metabolizing enzyme that plays an added role as a rheostat for endogenous lipid signaling. Supported by NIH Grant ES05823 and NIEHS Center Grant ES06639.
16. Mechanism of FXR regulation of bile acid synthesis
John Y. Chiang
Dept of Integrative Medical Sciences, Northeastern Ohio Univ’s Col of Med and Pharmacy, Rootstown, OH, USA, 44272
Bile acids are physiological detergents that generate bile flow and facilitate intestinal absorption and transport of lipids, nutrients and vitamins. Bile acids also are signaling molecules and inflammatory agents that rapidly activate nuclear receptors and cell signaling pathways that regulate lipid, glucose and energy metabolism. The enterohepatic circulation of bile acids exerts important physiological functions not only in feedback inhibition of bile acid synthesis but also in control of whole body lipid homeostasis. In the liver bile acids activate a nuclear receptor, farnesoid X receptor (FXR) that induces an atypical nuclear receptor small heterodimer partner (SHP), which subsequently inhibits nuclear receptors, liver related homologue-1 (LRH-1) and hepatocyte nuclear factor 4α (HNF4α), and results in inhibiting transcription of the critical regulatory gene in bile acid synthesis, cholesterol 7α-hydroxylase (CYP7A1). In the intestine FXR induces an intestinal hormone, fibroblast growth factor 15 (FGF15) (or FGF19 in human), which activates hepatic FGF receptor 4 (FGFR4) signaling to inhibit bile acid synthesis. However, the mechanism by which FXR/FGF19/FGFR4 signaling inhibits CYP7A1 remains unknown. Chenodeoxycholic acid (CDCA) and an FXR-specific agonist GW4064 strongly induced FGF19 but inhibited CYP7A1 mRNA levels in primary human hepatocytes. FGF19 strongly and rapidly repressed CYP7A1 but not SHP mRNA levels. Kinase inhibition and phosphorylation assays revealed that the MAPK/ERK1/2 pathway played a major role in mediating FGF19 inhibition of CYP7A1. Interestingly, CDCA stimulated tyrosine phosphorylation of the FGFR4 in hepatocytes. FGF19 antibody and siRNA specific to FGFR4 abrogated GW4064 inhibition of CYP7A1. These results suggest that bile acid-activated FXR is able to induce FGF19 in hepatocytes to inhibit CYP7A1 by an autocrine/paracrine mechanism. It may be concluded that the hepatic FGF19/FGFR4/ERK1/2 pathway may inhibit CYP7A1 independent of SHP. During cholestatic liver injury, bile acids may activate the FGF19/FGFR4 signaling pathway to inhibit bile acid synthesis and prevent accumulation of toxic bile acid in human livers. Bile acids and bile acid receptors are therapeutic targets for development of drugs for treatment of cholestatic liver diseases, fatty liver diseases, diabetes, obesity and metabolic syndrome.
17. Abstract not available
18. Abstract not available
19. Abstract not available
20. Abstract not available
21. Mtiochondrial toxicity assessment in the drug discovery safety paradigm
Yvonne Will
Compound Safety Prediction, Pfizer, Groton, CT, USA, 06340
Mitochondria produce almost all the energy in cells, but also chronically expose the cell to cytotoxic free radicals. Many widely prescribed therapeutics undermine mitochondrial function by interfering with DNA replication or expression, and more acutely, by uncoupling or inhibiting oxidative phosphorylation, leading to a variety of organ toxicities such as hepatic, cardiac, muscle, kidney and CNS.
Mitochondrial dysfunction is increasingly implicated in the etiology of drug-induced toxicities.1,2 Members of diverse drug classes directly and acutely undermine mitochondrial function by uncoupling electron transport from ATP production, and/or by inhibiting electron transport.3,4 Retrospective analysis reveals that such impairment contributes to, or in many cases is likely the proximate mediator, of organ toxicity that has forced market withdrawl of drugs (phenformin, troglitazone, cerivastatin), or attracted a Black Box warning from the FDA (pio- and rosiglitazone, tolcapone). In addition, ‘off-target’ mitochondrial impairment is implicated in idiosyncratic adverse drug reactions where organ history and mitochondrial genetics render specific patient populations more susceptible to mitochondrial dysfunction. Here, we will introduce commonly used in vitro methods to study mitochondrial toxicity. Examples include oxygen sensors to quantify mitochondrial respiration in isolated mitochondria and cells, immunocapture of xenobiotic agents that target mitochondrial proteins using isolated mitochondrial proteins and O2 and pH measurements., Examples of each technique will be presented and strengths and limitations of each approach will be discussed. We will discuss where to position these assays within drug development.
References
- Kitamura, S., Sugihara, K., and Ohta, S. (2006) Drug Metab Pharmacokinet 21(2), 83–98 2.
- Dowers, T. S., Rock, D. A., Rock, D. A., Perkins, B. N. S., and Jones, J. P. (2004) Drug Metab. Dispos. 32(3), 328–332 3.
- Alfaro, J. F., and Jones, J. P. (2008) J. Org. Chem. 4. Obach, R. S., and Walsky, R. L. (2005) J Clin Psychopharmacol 25(6), 605–608 5.
- Torres, R. A., Korzekwa, K. R., McMasters, D. R., Fandozzi, C. M., and Jones, J. P. (2007) J. Med. Chem. 50(19), 4642–4647
22. Reactive metabolite target proteomics - quo vadis?
Robert P. Hanzlik1, Yakov Koen1, and Jianwen Fang2
1Medicinal Chemistry, University of Kansas, Lawrence, KS, USA, 66045-7582
2Bioinformatics Core Facility, University of Kansas, Lawrence, KS, 66047
The biological significance of macromolecule modification by reactive electrophiles derives from the works of Karl Landsteiner, James and Elizabeth Miller, and Bernard B. Brodie. The specific association between protein “covalent binding” (CVB) and direct cytotoxicity, first demonstrated in the Brodie lab, has been amply reinforced by myriad examples, and today most practitioners accept CVB as being capable of causing cytotoxicity, although this has never been proven (in the sense of Koch’s Postulates), and counter-examples (CVB without toxicity) are well known. Protein CVB has been investigated from many perspectives in search of biochemical mechanisms of cytotoxicity. Early studies focused on enzymatic bioactivation mechanisms and structure-reactivity-selectivity-toxicity relationships. This approach revealed interesting new chemistry of reactive metabolite formation and disposition but few direct insights into mechanisms of toxicity. MS-based methods accelerated the identification of reactive metabolite target proteins. The Reactive Metabolite Target Protein Database now lists almost 300 individual proteins (ca. 1000 synonyms) targeted by one or more of 28 different protoxins in seven tissues of three species. Protein adduction is surprisingly selective. Relatively few targets are common to multiple reactive metabolites, and seldom have adducted peptides been observed in proteins derived from in-life experiments (as opposed to chemical model systems). Target lists have not revealed conspicuous “Achille’s heels” among target proteins, but “death by a thousand cuts” is not particularly appealing as a mechanism of toxicity. More recently various pathway or systems approaches have been used to analyze lists of target proteins. One that we have used involves analysis of the interacting partners of target proteins. This presentation will review the current “knowns and known unknowns” of protein CVB, as well as illustrate the application of bioinformatic and other approaches to the elucidation of mechanisms of reactive metabolite toxicity. (Supported by NIH grant GM-21784.)
23. Using metabonomic approach to study toxicity mechanisms
Nathalie Priymenko1, Céline Domange2, Cécile Canlet2, Amidou Traore3, Guy Bielicki3, Cécile Keller3, Nicolas Violle4, Julie Pflieger4, Andrée Morel4, Henri Schroeder4, and Alain Paris2
1Umr 1089, Université de Toulouse, ENVT, INRA, F-31027 Toulouse cedex 3, France
2UMR1089, INRA, F-31027 Toulouse cedex 3, France
3QuaPA STIM, INRA Clermont-Ferrand/Theix, F-63122 St Genès Champanelle, France
4Ur Afpa, INRA, ENSAIA, Université de Nancy, F-54506 Vandoeuvre les Nancy Cedex, France
Metabonomics aims to measure the global, dynamic metabolic response of living system to biological stimuli. This approach requires to get a fingerprint analysis of body fluids (urine, plasma, etc.) using various spectroscopic methods, like 1H-NMR or mass spectrometry. Therefore, statistical datamining of spectra is necessary to extract the useful metabolic information. Metabonomics, by monitoring all influencing factors without making any assumption about their respective involvement, can detect properties about new toxicants and some putative related mechanisms of so-induced disruptive events. Metabonomics was used to investigate the toxicity of a plant, Hypochoeris radicata (HR), involved in a neurological horse disease, called Australian stringhalt. First described in 1848, this peripheral neuropathy induced by this plant, only observed in horse, leads to abnormal characteristic movements of hindlimbs and a laryngeal paresis. This affection was chosen as a toxicological model to apply metabonomics on a complex matrix to reveal unknown toxic principles without having any pathophysiological information. Without clinical trouble observed, the urinary fingerprinting of HR-treated mice allowed to reveal the main involved metabolite, scyllo-inositol, which is also found as a biomarker in liver and brain. In order to know whether scyllo-inositol is really a HR intoxication marker, brains of HR-treated mice were analyzed by magnetic resonance imaging, and confirmed the presence of abnormal concentrations of scyllo-inositol. Besides, we carried out behavioural tests demonstrating the HR infraclinical impact. Metabonomics provides a powerful approach to explore retrospectively the markers of a disease risk and to provide some indications of a pathophysiological situation. Several doses of the xenobiotics have to be tested to improve chemometric analysis of biological NMR spectra. Interestingly, this technology is being developed in the pharmaceutical industry for drug screening and for new candidate medicine selection. If genetic and individual effects on the metabolic status can be attenuated in laboratory animals, the challenge is from now on detection of subtle disruptions within the huge adaptable and complex biochemical system of the sick horses.
24. Can idiosyncratic hepatotoxicity be modeled in the rat?
Michael McMillian
Mechanistic Toxicology, Johnson & Johnson Pharmaceutical Research and Development, LLC., Raritan, NJ, USA, 08869
A number of human idiosyncratic hepatotoxicants can be detected by their gene expression patterns in rat liver, despite lacking toxicity by classical, regulatory methodology. Many of these compounds produce robust oxidative stress/ reactive metabolite (OS/RM)-regulated gene expression responses, apparently via Nrf2 activation. Carbamazepine (225 mg/kg by gavage), a model non-toxic compound in rats which produces a high incidence of adverse effects in humans, was chosen based on its OS/RM gene expression responses to test in two possible models of idiosyncratic hepatotoxicity. In a low dose lipopolysaccharide (LPS) model (1 mg/kg iv tail vein two hours prior to carbamazepine), carbamazepine became extremely hepatotoxic in some rats, based on ALT elevations and histopathology, but the response was quite variable. In agreement with published findings, LPS strongly repressed several cytochrome P450 (CYP) and transporter mRNAs and their inductions by carbamazepine. In addition, LPS strongly repressed several OS/RM gene responses to carbamazepine, and some of these genes were differentially affected in responders/ nonresponders for hepatotoxicity. In a dextran sodium sulfate (DSS) model of colonic inflammation (500 mg/ml in drinking water overnight or for four days), where endogenous LPS reportedly enters the systemic circulation, carbamazepine remained non-toxic despite pronounced repressions of CYP and transporter mRNA responses. Surprisingly, DSS potentiated inductions of OS/RM genes in response to carbamazepine. LPS administration after DSS converted carbamazepine to a hepatotoxin in some rats. Differences between these two models of immune activation suggest that OS/RM responsive genes may play a role in the idiosyncratic effects of carbamazepine.
25. In vitro models of the blood-brain barrier to study drug delivery to the brain
Pierre-Olivier Couraud
Institut Cochin, Université Paris Descartes, INSERM U567 / CNRS UMR 8104, Paris, France
Brain microvascular endothelium, which constitutes the blood-brain barrier (BBB), differs from that of peripheral organs by low paracellular permeability due to highly impermeable intercellular tight junctions as well as by active influx transport of nutriments and efflux of xenobiotics. Modeling the BBB is a key issue for understanding the mechanisms of maintenance of BBB integrity and for facilitating drug screening in industrial R&D programs. A number of in vitro BBB models have been proposed for the last ten years: primary cultures of brain endothelial cells of bovine, porcine or murine origin, grown on semi-permeable filters in two-chambers devices in the presence of glial cells, have been shown to recapitulate most of the characteristics of the BBB. In some cases, a correlation between in vitro and in vivo drug permeability across the BBB has been demonstrated, thus validating these in vitro models. Establishment of a human model of the BBB has proven to be a difficult goal: we recently produced and characterized a human brain endothelial cell line hCMEC/D3 which retains in culture a stable endothelial phenotype highly reminiscent of the human BBB. These cells express a variety of tight junction proteins and membrane transporters: we recently investigated the expression of the ABC-transporters and cytochromes P450 expressed by these cells and their regulation by the aryl hydrocarbon receptor (AhR). In addition, we could study the migration of activated lymphocytes across the BBB and elucidate the mechanisms of migration of neural precursor cells into the brain parenchyma in inflammatory situation. In conclusion, in vitro BBB models constitute valuable tools for investigating the biology of the BBB and for studying drug delivery to the brain. In particular, on the basis of the extensive characterization of the hCMEC/D3 cell line provided so far by us and others, we propose this cell line as a unique in vitro model of human BBB.
26. Delivery of drugs to brain: importance of drug transporters and membrane permeability in the disposition of lapatinib
Joseph W. Polli
Preclinical Drug Metabolism and Pharmacokinetics, GlaxoSmithKline, Inc., Research Triangle Park, NC, USA, 27709
The delivery of a new drug candidate to the central nervous system (CNS) can be a significant challenge for a drug discovery project team. CNS penetration of a compound is often poor due to exclusion at the blood-brain barrier (BBB) and/or blood-cerebrospinal fluid (BCSF) barrier. In vitro membrane permeability studies are often completed to rank compounds for CNS penetration. As well, membrane transporters, such as P-glycoprotein and Breast Cancer Resistance Protein, can influence the CNS penetration of a drug. The importance of drug transporters and membrane permeability will be illustrated through the work completed during the development of Lapatinib, a tyrosine kinase inhibitor. Results and use of data from drug transporter in vitro and in vivo assays along with clinical findings will be described. Integration of in vivo and in vitro data along with an understanding of CNS physiology can assist in selecting better CNS penetrating compounds during drug development.
27. Unbound brain concentration determines brain receptor occupancy
Xingrong Liu
Drug Metabolism and Pharmacokinetics, Genentech, South San Francisco, CA, USA, 94080
The objective of the present study was to test the free drug hypothesis by examining the relationship between in vitro binding affinity (KI) and in vivo activity for 18 serotonin (SERT) and dopamine (DAT) transporter inhibitors. The in vivo activity was quantified as the drug concentration occupying 50% of the transporters (OC50). In vivo rat OC50 was determined by autoradiography using [3H]DASB and [3H]WIN3,5428 as the ligands to assess SERT and DAT occupancy, respectively. The unbound brain concentrations were calculated from total brain concentrations and unbound brain fraction which was determined by the brain homogenate method. The in vivo total brain SERT and DAT OC50 (Mean ± SD) was 408 ± 368 and 410 ± 395 fold greater than the KI, respectively. In contrast, the in vivo unbound brain SERT and DAT OC50 was only 3.3 ± 2.1, and 4.1 ± 4.0 fold different from the KI. The in vivo total plasma SERT and DAT OC50 was 44 ± 60 and 53 ± 77 fold greater than the KI, respectively. In contrast, the in vivo unbound plasma SERT and DAT OC50 was only 4.6 ± 5.5, and 4.6 ± 5.1 fold different from the KI. Therefore, prediction of the biophase drug concentration using unbound brain concentration for this data set results in an approximately 100-fold improvement for the accuracy than using the total brain concentration. The prediction of the biophase drug concentration using unbound plasma concentration results in an approximately 10-fold improvement for the accuracy than using the total plasma concentration. This study supports the hypothesis that CNS activity is driven by the unbound and not by the total brain drug concentration. The strategies and experimental approaches to characterize and optimize the unbound brain concentration for CNS compounds in drug discovery and development settings will be presented.
28. Can drug concentration in the cerebrospinal fluid be a surrogate for the unbound concentration of cns drugs in the brain?
Yuichi Sugiyama
Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan, 113-0033
The unbound drug concentration in the brain (Cu, brain) is a critical factor determining pharmacological/adverse effects in the brain and, therefore, its prediction in humans is critical for a proper understanding of the PK-PD relationship of new drugs. Because of the difficulty in measuring it directly in humans, the concentration in the cerebrospinal fluid (CSF) (CCSF) has been used as a surrogate for Cu, brain. However, the factors responsible for a difference in the unbound concentration in the brain and CSF remain obscure. Compounds were administered by continuous infusion to rats and mice. Blood, CSF and brain samples were collected at the end of infusion. Unbound fractions in the brain were determined by using brain slice. In vitro transport experiments with P-gp and Bcrp expressing polarized cells were carried out. Simulation using a three-compartment model (blood, brain and CSF) suggests that the Cu, brain became lower than the CCSF when the efflux clearance across the blood-brain barrier was greater than the other membrane permeability clearances, the efflux clearance from CSF to plasma and bulk flow of CSF. Theoretically, Kp,CSF/brain,u (CCSF/Cu, brain) is one if drugs do not undergo extensive efflux across the blood-brain barrier. Kp,CSF/brain,u of most of Bcrp and/or P-gp substrates were found to be less than one while they were increased to one in Bcrp−/- mice and/or Mdr1a/1b−/- mice. In contrast, the Kp,CSF/brain,u of relatively hydrophilic P-gp substrates was one, although the Kp,brain of these P-gp substrates was significantly increased in Mdr1a/1b−/- mice. Diffusion of these compounds across the ependyma surface may not be negligible compared with the efflux across the blood-brain and CSF barriers. The results of the present study suggest that, even although drugs undergo extensive efflux by P-gp and Bcrp, the CCSF can be a surrogate of the Cu, brain for those drugs with low membrane permeability.
References:
- Enokizono J., Kusuhara H., Ose A., Schinkel A.H. and Sugiyama Y. Quantitative investigation of the role of breast cancer resistance protein (bcrp/abcg2) in limiting brain and testis penetration of xenobiotic compounds. Drug Metab Dispos 36: 995-1002 (2008)
29. Aldehyde oxidases and other molybdenum hydroxylases
Christine Beedham
Clinical Sciences, University of Bradford, Bradford W Yorks, United Kingdom, BD7 1DP
Drug oxidation (and reduction) mediated by the molybdenum containing enzyme, aldehyde oxidase (AO), is often overlooked in comparison to reactions catalyzed by cytochrome P450. However, aldehyde oxidase and the closely related xanthine oxidoreductase (XOR) catalyse the oxidation of a wide range of N-heterocyclic drugs in addition to aldehyde oxidation and reductive reactions. Typically, in vivo conversion of drugs to AO-generated metabolites is rapid and metabolites are excreted directly without conjugation. Consequently, drugs may be quickly inactivated or bioavailability may be reduced due to AO or XOR-catalysed oxidation in liver (AO and XOR) or gut (XOR). It is difficult to express AO and XOR activity in recombinant systems and until recently, information on these enzymes has predominantly come from in vitro animal studies. This has provided valuable data on substrate/inhibitor specificity and possible clinical implications. Many in vitro AO inhibitors have been identified but, to date, there is little clinical indication that inhibitory drug interactions are significant in vivo. In contrast, co-administration of XOR inhibitors can be used to modulate drug therapeutics. Recently, full elucidation of the crystal structure of bovine milk XOR and Desulfovibria gigas AO and determination of their gene structure has expanded our knowledge of these enzymes considerably. AO and XOR each have a single functional gene in human liver (hAOX1 and XOR) but, unlike XOR, the AO gene is not conserved in all species and thus results from animal studies have to be interpreted with caution. Human AO and XOR genes appear to be controlled by complex but differential mechanisms at both transcriptional and post-translational levels. For example, studies in mice have indicated that both enzymes may be induced via the aryl hydrocarbon receptor (AhR) pathway, however, there is no clinical evidence that metabolism of drugs catalyzed by these enzymes is altered in smokers or during exposure to other AhR inducers. There are conflicting reports on the implications of AO polymorphism but some preliminary studies have indicated that human AO single nucleotide polymorphisms (SNPs) can influence drug efficacy whereas XOR regulation and polymorphism may be more important in various physiological and pathophysiological mechanisms.
30. Flavin-Containing monooxygenases
Ronald N. Hines1, David E. Klick1, Sevasti B. Koukouritaki2, Eugene W. Gerner3, Patricia Thompson3, and Frank L. Meyskens Jr.4
1Depts. of Pediatrics and Pharmacology/Toxicology & Children’s Research Institute, Medical College of Wisconsin & Children’s Hospital & Health System, Milwaukee, WI, USA, 53226
2Depts. of Pediatrics & Children’s Research Institute, Medical College of Wisconsin & Children’s Hospital & Health System, Milwaukee, WI, USA, 53226
3Arizona Cancer Center, University of Arizona, Tucson, AZ, USA, 85724
4Chao Family Cancer Center, University of California-Irvine, Irvine, CA, USA, 92668
The flavin-containing monoxygenases (FMOs) oxidatively metabolize numerous toxicants and approximately 2% of clinically relevant drugs. In the human, 5 FMO genes encode functional enzymes (FMO1-5) with FMO1, 2 and 3 being most important for drug and toxicant metabolism. FMO1 is expressed at high levels in the fetal liver, small intestine and kidney while FMO3 is expressed at high levels in the adult liver. FMO2 is primarily a lung-specific enzyme, but its impact is minimized by a premature stop codon common in populations outside of Africa. In the human, FMO1 is expressed at its highest level in the 1st trimester fetal liver, then declines and is silenced a few days after birth. In contrast, FMO3 is essentially absent in the fetal and neonatal liver, but is detectable in most individuals by 1 to 2 years of age. Intermediate expression is observed in individuals between ages 2 and 11. Adult expression is usually seen by age 18. FMO3 promoter analyses revealed the presence of an NFY (position -75 to -59), Pbx2/HOX (position -115 to -103), HNF4α (position -167 to -152), YY1 (position -258 to -248) and C/EBPβ (position -456 to -444) responsive elements. The NFY, HNF4α and C/EBPβ sites appear most important for constitutive expression in the adult while developmental changes in the C/EBPβ LAP:LIP ratio likely are involved in regulating FMO3 developmental expression. Several functional FMO3 variants have been identified. Hypomorphic variants have been associated with increased sulindac chemoprevention efficacy in familial adenomatous polyposis (FAP) patients. However, preliminary analysis of data from a recently completed phase III randomized placebo-controlled trial in which patients with sporadic colorectal polyps received a combination of difluoromethylornithine (DFMO) and sulindac (N=191) or placebo (N=184) do not appear to corroborate these earlier findings. A significant association was observed with decreased colonic mucosa PGE2 levels and the FMO3 G308 variant (rs2266780) (P=0.038) and increased colonic mucosa putrescine levels and the FMO3 K158 variant (rs2266782) (P=0.036). However, no association was observed between the FMO3 variants and risk of adenoma recurrence. These contradictory data may be explained by the recently discovered opposing actions of FMO3. Consistent with earlier studies, hypomorphic FMO3 variants would favor a cyclooxygenase-dependent antitumor effect, but hypermorphic variants would favor an antitumor effect through the ability of sulindac sulfoxide and sulfone to enhance SAT1 expression through PPARγ and reduce cellular polyamine levels via the SLC3A2 transporter.
31. UDP-glucuronosyl transferases
Gerhard M. Gross
Cpd, AstraZeneca, Macclesfield, United Kingdom, SK10 4TG
Symposium 7: Scientific and Regulatory Perspectives on Non-CYP Drug Metabolizing Enzymes and Transporters UDP-glucuronosyl Transferases Gross Gerhard, AstraZeneca R & D, Alderley Park, Macclesfield, UK UPD-glucosyl transferases are a superfamily of endoplasmatic reticulum bounded enzymes which belong to the so called phase II enzymes. They catalyse the attachment of glycolsyl groups (glucuronic acid, glucose and xylose) to an acceptor molecule, the so called aglycone. There is a great variety of functional groups which may function as attachment group e.g. any alcohol, aromatic as well as aliphatic; carboxylic acids, thiols, amines and acidic carbon atoms. Most common reaction is the transfer of a glucuronic acid moiety to an aglykone. This process is called glucuronidation. The group of enzymes catalyzing this reaction is referred to as UGTs. UGTs are divided into two families UGT1 and UGT2. For human 31 genes have been identified expressing UGTs including some pseudogenes. The most important human UGTs are: UGT1A1, UGT1A3, UGT1A4, UGT1A5, UGT1A6 as well as UGTs 1A7, 1A8, 1A9, 1A10; from the UGT1 family and UGT2B4, UGT2B7, UGT2B10, UGT2B11, UGT2B15 and UGT2B17 from the UGT2 family. These enzymes are expressed mainly in the alimentary system, liver, kidney, brain, lung as well as (low level expression) in steroidogenic tissues like breast, prostate and adrenal. They play a significant role in metabolism of many endogenous (bilirubine, bile acids and steroids) and xenobiotic compounds. Glucuronidation converts a substrate/aglykon to a more polar and water soluble form and therefore facilitates its elimination through the kidney via glomerular filtration or active secretion as well as active transport through bile into small intestine. Genetic polymorphism has been reported for quite a few UGTs e.g. UGT1A1 (polymorphisms of this enzyme also relevant for Criegler-Najjar syndrome and Gilberts disease), UGT1A6, UGT1A7, UGT2B4, UGT2B7 and UGT2B15. Functional significance so far seems to be only clearly proven for UGT1A1. Nuclear receptors are able to regulate UGT expression, e.g. UGT1A1 induction through PXR and CAR. Major relevant aspects of UGTs and glucuronides for drug development are basically all factors contributing to safety and efficacy of a drug: drug/drug interactions; impact of genetic polymorphism of UGTs; interplay e.g. metabolic switching of CYP mediated metabolism with UGT metabolism as well as interactions with transporter. Acyl glucuronides are in particular seen as relevant for safety. Case studies will be presented to illustrate and discuss all these points. Regulatory guidance’s, where glucuronides are explicitly mentioned or of relevance are discussed also.
32. Abstract not available
33. Abstract not available
34. A Systematic analysis of predicted phosphorylation sites within the human PXR protein
Jeff L. Staudinger
Pharmacology & Toxicology, University of Kansas, Lawrence, KS, USA, 66045
The pregnane × receptor (PXR, NR1I2) regulates the expression of genes that encode drug metabolizing enzymes and drug transporter proteins in liver and intestine. Understanding the molecular mechanisms that modulate PXR activity is therefore critical for the development of effective therapeutic strategies. Several recent studies have implicated activation of kinase signaling pathways in the regulation of PXR biological activity, though direct evidence and molecular mechanisms are currently lacking. We therefore sought to characterize potential phosphorylation sites within the PXR protein using a rational, comprehensive and systematic site-directed mutagenesis approach to generate phosphomimetic mutations (Ser/Thr → Asp) and phospho-deficient mutations (Ser/Thr → Ala) mutations at eighteen predicted consensus kinase recognition sequences in the human PXR protein. Here, we identify amino acid residues S8, T57, S208, S305, S350, and T408 as being critical for biological activity of the PXR protein. Mutations at positions 57 and 408 abolish ligand-inducible PXR activity. Mutations in the extreme N-terminus and in the PXR ligand-binding domain at positions S8, S305, S350, and T408 decrease the ability of PXR to form heterodimers with RXRa. Mutations at positions S208, S305, S350, and T408 alter PXR-cofactor interactions. Finally, the sub-cellular localization of the PXR protein is profoundly affected by mutations at position T408. These data suggest that PXR activity can potentially be regulated by phosphorylation at specific amino acid residues within several predicted consensus kinase recognition sequences to differentially affect PXR biological activity.
35. Regulation of AhR signaling by PPARβ/δ in skin
Michael G. Borland1, Gary H. Perdew2, Frank J. Gonzalez3, and Jeffrey M. Peters1
1Center for Molecular Toxicology and Carcinogenesis, The Pennsylvania State University, University Park, PA, USA, 16802
2Ctr for Molec Toxicol, Penn State Univ, University Park, PA, 16802
3Lab of Metab/Bldg 37 Rm 3106B, NIH/NCI, Bethesda, MD, 20892
Peroxisome proliferator-activated receptor- β/ δ(PPARβ/δ) is a ligand activated transcription factors that regulates specific target genes that modulate many biological processes. Previous work shows that PPARβ/δ can attenuate skin tumorigenesis. We examined whether this phenotype could be due to differences in the metabolism of the carcinogen. Expression of cytochrome P450 1A1 (CYP1A1) or CYP1B1 mRNA or protein is increased in mouse skin and primary keratinocytes by polycyclic aromatic hydrocarbons (PAH) and surprisingly, these changes are not found in the absence of PPARβ/δ expression. Interestingly, expression of phase II xenobiotic metabolizing enzymes also exhibits differential patterns in response to PAH treatment in the absence of PPARβ/δ expression. This effect does not appear to be due to differences in the relative level of the AhR, ARNT, XAP2 or HSP90. To begin to delineate the mechanism by which PPARβ/δ regulates AhR signaling, ligand binding and the nuclear translocation of AhR were examined, but no difference in these responses were observed between genotypes. Evidence suggests that there is not a functional interaction between the two receptors, although this must be examined in greater detail. The occupancy of the CYP1A1 promoter in response to PAH was examined by chromatin immunoprecipitation, and in the absence of PPARβ/δ expression, reduced AhR occupancy of the CYP1A1 promoter was observed. Stable short-hairpin RNA technology was utilized to generate a human keratinocyte cell line with knockdown of PPARβ/δ to being to examine the human relevance of these changes. Similar to results observed in mouse keratinocytes, PAH-induced expression of CYP1A1 and CYP1B1 is diminished in HaCaT keratinocytes when PPARβ/δ expression is knocked down as compared to controls. Collectively, these findings suggest a novel mechanism in mouse and human skin where PPARβ/δ modulates AhR-dependent signaling and potentially alters the carcinogenic effect induced by PAH, likely through modifying the balance between bioactivation and clearance of PAH. (Supported by CA89607, CA97999, CA124533, ES04869)
36. Nuclear receptors LXR and CAR in the crossroad of drug metabolism and energy metabolism
Wen Xie
Department of Pharmaceutical Sciences, University of Pittsburgh, Pittsburgh, PA, USA, 15261
The liver X receptor (LXR) and constitutive androstane receptor (CAR) are two receptors postulated to have distinct functions. LXR is a sterol sensor that promotes lipogenesis, whereas CAR is a xenosensor that controls xenobiotic responses. Here we show that LXRalpha and CAR are functionally related in vivo. Loss of CAR increased the expression of lipogenic LXR target genes, leading to increased hepatic triglyceride accumulation; whereas activation of CAR inhibited the expression of LXR target genes and LXR ligand-induced lipogenesis. Conversely, loss of LXR alpha and beta increased the basal expression of xenobiotic CAR target genes; whereas activation of LXR inhibited the expression of CAR target genes and sensitized mice to xenobiotic toxicants. The mutual suppression between LXRalpha and CAR was also observed in reporter gene assays. The ligand-free LXRalpha, like its CAR counterpart, exhibited high constitutive activity by recruiting nuclear receptor co-activators. The competition and/or sequestering of co-activators represent a plausible mechanism for the mutual suppression between these two receptors. Our results have revealed dual functions of LXRalpha and CAR in lipogenesis and xenobiotic responses, establishing a unique role of these two receptors in integrating xenobiotic and endobiotic homeostasis.
37. Enhanced predictability of P450-Mediated drug metabolic pathways by integrated quantum mechanics/molecular dynamics computational models
Garold S. Yost
Dept of Pharmacol & Toxicol, Univ of Utah, Salt Lake City, UT, USA, 84112-5820
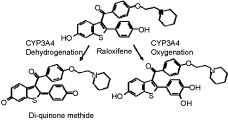
Predictions of metabolic pathways for novel therapeutic agents with structure-based computational models of drug metabolism enzymes have improved significantly when x-ray structures of cytochrome P450 enzymes were available. However, the utility of these static models is quite limited because they cannot accommodate normal solution dynamics, particularly when substrate binding causes conformational changes, which appear to be normal for most P450 enzymes. Therefore, many biochemists use sophisticated computer modeling to derive more precise structures of solution-equilibrated enzymes. To gain insight into the contributions of electronic and thermodynamic factors to substrate-enzyme interactions during P450 catalysis, we used a combination of quantum mechanics and molecular dynamics techniques to refine the x-ray structures of CYP3A4. The selective estrogen receptor modulator drug, raloxifene, was used to evaluate “normal” oxygenated metabolites, and bioactivation to a dehydrogenated electrophilic di-quinone methide intermediate. Docking studies of raloxifene with the x-ray structures (1WOE or 1TQN) of CYP3A4, without the assignment of partial charges to the heme, did not produce any low-energy highly-populated active site conformations, in which the known sites of metabolism for raloxifene were within 5 angstroms of the center of the heme. However, refined docking studies with quantum mechanics-based partial charge assignments to the heme prosthetic group provided conformations that specifically predicted dehydrogenation. The new model was also used to identify amino acid residues that would be expected to control positioning of the substrate in the active site of CYP3A4. In the refined docking studies, the lowest energy and highest populated cluster of the model-predicted enzyme/substrate conformations predicted regiospecific dehydrogenation of the benzothiophene moiety of raloxifene at the 6-hydroxy end of the molecule, to generate the di-quinone methide. Analysis of the model identified phenylalanine 215 as an important contributor to substrate positioning within the active site. Incubations of purified CYP3A4 with raloxifene showed that the dehydrogenated product was the major metabolite, precisely agreeing with the computer-simulated results. To also test the accuracy of the refined docking results, the F215G mutant was produced and tested. LC/MS/MS analysis of incubations of native enzyme and the F215G mutant clearly demonstrated a statistically significant increase in the oxygenation/dehydrogenation ratio. We opine that the loss of dehydrogenation efficiency of the mutant was caused by decreased conformational specificity for the dehydrogenation pathway, due to the loss of steric effects from the phenylalanine and the loss of T-stacking interactions with the aromatic portion of raloxifene. Thus, our integrated QM/MD computational approach provided a more robust model with greater predictive power than methods based on the unrefined static x-ray structures of this important drug metabolism enzyme, and enhanced the predictability of this metabolic pathway. It seems possible that these methods can be optimized to provide much better molecular models of many drug metabolism enzymes that can be valuable in the drug development process. Supported by NIH Grant #GM074249 from the National Institute of General Medical Sciences.
38. Optimization of antimicrobial drug therapies through multiscale computational modeling
Brad Reisfeld1, Arthur N. Mayeno2, and Michael A. Lyons3
1Department of Chemical and Biological Engineering & School of Biomedical Engineering, Colorado State University, Fort Collins, CO, USA, 80523
2Department of Environmental and Radiological Health Sciences, Colorado State University, Fort Collins, CO, USA, 80523
3Department of Chemical and Biological Engineering, Colorado State University, Fort Collins, CO, USA, 80523
The millions of lives saved by antimicrobial agents has been a testament to the important impact of new agents for fighting infectious disease scourges. Dampening this success has been the emergence of a number of drug resistant organisms for which clinicians often have a limited antibacterial armamentarium to draw upon. With tuberculosis (TB) as an example, once the emergence of resistance occurs for first-line agents such as rifampin and isoniazid, the next best agents (even in combination) are much less effective therapies, and therapy is required for many months or years. Thus, a critical challenge in the treatment of drug-resistant infectious diseases is to determine the combination of drugs, and the optimal dose, frequency, and duration for each component in the drug combination to minimize treatment duration, reduce the likelihood of emergence of resistant strains, and enhance tolerability. We hypothesize that computational modeling can be an effective tool in meeting this challenge and in the optimization of antimicrobial drug therapies. To this end, we are developing an integrated, multiscale modeling framework for the optimization of combination drug therapies for TB treatment, using capreomycin, moxifloxicin, linezolid, and ethionamide as the second-line drugs of interest. This framework is based on Bayesian models for population variability, physiologically-based pharmacokinetic models for drug absorption, distribution, metabolism, and excretion, and pharmacodynamic models for the development of TB granulomas, including the effects of the antimicrobial drugs and the immune system. Here, we present preliminary results for a single drug, capreomycin. These results include the predicted pharmacokinetic behavior of the drug, and comparisons of these results to our own experimental data in rodents and those of Le Conte et al. [1]. We also present the time-dependent populations of bacteria, macrophages, and T-cells in the absence and presence of capreomycin and compare these results to those in the literature, e.g. [2,3]. 1. Le Conte P, Le Gallou F, Potel G, Struillou L, Baron D, Drugeon H (1994) Pharmacokinetics, toxicity, and efficacy of liposomal capreomycin in disseminated Mycobacterium avium beige mouse model. Antimicrob Agents Chemother 38:2695-2701. 2. Segovia-Juarez J, Ganguli S, Kirschner D (2004) Identifying control mechanisms of granuloma formation during M. tuberculosis infection using an agent-based model. J Theor Biol 231:357-376. 3. Wigginton J, Kirschner D (2001) A model to predict cell-mediated immune regulatory mechanisms during human infection with Mycobacterium tuberculosis. J Immunol 166:1951–1967.
39. Aldehyde oxidase in drug metabolism: Experimental and theoretical predictions of structure and function
Jeffrey P. Jones and Josh F. Alfaro
Chemistry, Washington State University, Pullman, WA, USA, 99164
At present over 40 drugs, nutritional supplements, and xenobiotics are metabolized to some extent by AOX or XO (1). One method to slow aromatic oxidation by P450 enzymes is incorporation of a nitrogen to make a heteroaromatic ring (2). The electron withdrawing characteristics of nitrogen slow the electrophilic chemistry of the P450 enzymes. Aldehyde oxidase (AOX) and xanthine oxidase (XO) both catalyze the oxidation of heteroaromatic compounds by nucleophilic addition of oxygen from water (3). The net result can lead to a change in the metabolic pathways from P450 mediated electrophilic addition to XO and AO mediated nucleophilic addition by the molybdenum iron-sulfur flavoprotiens, xanthine oxidase (XO) and aldehyde oxidase (AOX) (4).
We will present results comparing both computational and experimental data for XO and AO, omparing and contrasting the mechanism, regioselectivity (5), and substrate specificity of each enzyme. We will also present results from in silico and in vitro site-directed mutants of AOX and XO, and the affect of these mutants on reactivity.
References
- Kitamura, S., Sugihara, K., and Ohta, S. (2006) Drug Metab Pharmacokinet 21(2), 83–98 2.
- Dowers, T. S., Rock, D. A., Rock, D. A., Perkins, B. N. S., and Jones, J. P. (2004) Drug Metab. Dispos. 32(3), 328–332 3.
- Alfaro, J. F., and Jones, J. P. (2008) J. Org. Chem. 4. Obach, R. S., and Walsky, R. L. (2005) J Clin Psychopharmacol 25(6), 605–608 5.
- Torres, R. A., Korzekwa, K. R., McMasters, D. R., Fandozzi, C. M., and Jones, J. P. (2007) J. Med. Chem. 50(19), 4642–4647
40. Accelerating ADME screening: Application of multivariate in silico approaches to discriminate substrate affinity of homologous drug transporters
Peter Swaan
Department of Pharmaceutical Sciences, University of Maryland, Baltimore, MD, USA, 21201
The ability to identify ligands for drug transporters is an important step in drug discovery and development. It can both improve accurate profiling of lead pharmacokinetic properties and assist in the discovery of new chemical entities targeting transporters. In silico approaches, especially pharmacophore-based database screening methods have great potential in improving the throughput of current transporter ligand identification assays, leading to a higher hit rate by focusing in vitro testing to the most promising hits. More recently, multivariate methods (e.g. genetic algorithms, probabilistic neural networks, support vector machines, k-Nearest Neighbors, recursive partitioning, random forest algorithms, self organizing maps, etc.) have been explored as a way to deal with information from many transporters. This presentation will provide an overview of methodology, algorithms and recent applications in the field of transporters, enzymes and nuclear receptors.
41. Developmental issues in the selection of candidate genes for pharmacogenetic studies of pediatric ADRs
J. Steven Leeder
Division of Clinical Pharmacology and Medical Toxicology, Children’s Mercy Hospitals and Clinics, Kansas City, MO, USA, 64108
Untoward events occurring in children have been pivotal for the subsequent development of the Food and Drug Administration and legislation aimed at protecting the public. Clinical experience has revealed that children differ from adults in terms of risk for particular adverse drug reactions (ADRs). For example, delayed maturation of drug metabolizing enzymes may contribute to concentration-dependent toxicities, particularly in newborns where drug biotransformation capacity is most limited. Furthermore, compelling data have been presented indicating that several severe idiosyncratic ADRs, such as aspirin and Reye’s syndrome, valproic acid hepatotoxicity, cefaclor serum-sickness-like reactions, and cutaneous toxicity associated with lamotrigine occur more frequently in children, compared to adults. The mechanisms of toxicity are poorly understood at best and as a result, explanations for the apparent increased risk of these events in children are unknown. Contributing to the problem is the fact that the developmental continuum between birth and adolescence is a very dynamic, complex period of life. Based on available in vitro data and in vivo pediatric pharmacokinetic studies, drug clearance pathways undergo dramatic changes throughout the maturation process. The activities of many enzymes involved in drug biotransformation are absent or very limited at birth, raising the possibility that there may be periods of relatively increased vulnerability to concentration-dependent drug toxicity. On the other hand, in vivo pharmacokinetic and therapeutic drug monitoring data in pediatric patients imply that some drug biotransformation pathways may exceed adult capacity, at least when dosage requirements/drug clearance between children and adults are compared and expressed relative to body weight. This issue is far from resolved but developmentally increased cytochrome P450 (CYP) activities during childhood could conceivably result in increased formation of reactive, potentially toxic metabolites. Furthermore, genetic variation in detoxification pathways (UGTs, SULTs and GSTs, for example) has the potential to contribute to a net increase in reactive metabolite formation and therefore, also represents a significant determinant of risk for the development of idiosyncratic ADRs in young children. To facilitate the development of testable hypotheses concerning genetic and developmental determinants of risk for specific ADRs in children, and to aid in the design of protocols to test those hypotheses, we have applied a systematic approach collect information and aid in the identification and prioritization of candidate genes for developmental pharmacogenetic studies. This approach will be illustrated using specific examples of contemporary pediatric drug safety concern.
42. Abstract not available
43. Abstract not available
44. Metabolomic Mapping of Atypical Effects in Schizophrenia
Rima Kaddurah-Daouk1, Rebecca A. Baillie2 and Joseph McEvoy1
1Psychiatry and Behavioral Sciences, Duke University Medical Center, Durham, NC
2Rosa Pharmaceuticals, Inc, Cupertino, CA
Background: Several atypical antipsychotic drugs induce weight gain and hypertriglyceridemia. To date there has not been a comprehensive evaluation and mapping of global biochemical changes in schizophrenia and upon treatment with antipsychotics. Such mapping could provide novel insights about disease mechanisms and metabolic side effects of therapies used for its treatment. We used a specialized metabolomics platform, lipidomics, that quantifies over 300 polar and non polar lipid metabolites (across 7 lipid classes) to evaluate global lipid changes in schizophrenia and upon treatment with three commonly used atypical antipsychotics.
Methods: Lipid profiles were derived for close to a 100 patients with schizophrenia before and after treatment for 2-3 weeks with either olanzapine, risperidone or aripiprazole. Plasma was isolated after overnight fasting and frozen at -80°C. Immediately prior to analysis, samples were thawed, spiked with internal standards and extracted with chloroform:methanol (2:1, v/v). Individual lipid classes within the extract were separated by preparative chromatography as described by Watkins et al 2007. Isolated lipid classes were trans-esterified in 3 N methanolic HCl in a sealed vial under a nitrogen atmosphere at 100°C for 45 min. The resulting fatty acid methyl esters were extracted with hexane containing 0.05% butylated hydroxytoluene, separated and quantified by a gas chromatograph (Hewlett-Packard model 6890, Wilmington, DE) equipped with a 30-m DB-225MS capillary column (J&W Scientific, Folsom, CA) and a flame-ionization detector. BPRS at baseline and at the time of follow-up blood draws was used for Clinical assessment. For analysis we used ANCOVA (patients pre- and post-treatment) and correlations (BPRS, body weight, waist circumference, and lipid levels) as well as paired samples t-test (patients pre- vs. post-treatment). Significance=P<0.05.
Results: We mapped specific changes for olanzapine, risperidone and aripiprazole within the phosphatidylethanolamine, phosphatidylcholine, and triglyceride lipid classes and highlighted pathways affected by antipsychotics. Arachidonic acid pathway changes specific for risperidone treatment were mapped. Several pretreatment triglyceride, fatty acid and lysophosphatidylcholine metabolites correlated with body wt and waist. Baseline free fatty acid metabolites were most common in predicting change in waist circumference, while triglyceride and diglyceride metabolites were more common when predicting changes in body wt. Full analysis of metabolites correlated with therapeutic benefit is underway.
Discussion: Metabolomics offers unique insights into mechanism of therapeutic benefit and metabolic side effects of drugs. We have started to map metabolic pathways implicated in the metabolic side effects seen with individual antipsychotic drugs. Each antipsychotic drug appears to have selective changes on lipid metabolism but some changes are common to the class of atypical antipsychotics.
45. Clinical drugs in the environment
Bryan W. Brooks
Environmental Science and Biomedical Studies, Baylor University, Waco, TX, USA, 76798 Clinical Drugs in the Environment Bryan W. Brooks, Baylor University
In recent years pharmaceuticals have been identified as classes of “emerging” environmental contaminants. In addition to being introduced to the environment through centralized and decentralized wastewater treatment plant discharges, pharmaceuticals may also be transported to the environment following land application of biosolids and effluents, and livestock husbandry practices in agricultural settings. Whereas the majority of research efforts to date have included standardized ecological toxicity screening and developing analytical methodologies with GC/MSMS, LC/MSMS and (more recently) LC/TOF-MS to support environmental monitoring activities, recent studies are focusing on contaminant source tracking, chemical fate, mechanistic and comparative toxicology, and human health and ecological risk assessment approaches. Because therapeutics often possess physiochemical (e.g., ionizable compounds) and biological (e.g., therapeutics are designed to target specific biomolecules, pathways) properties that differ from many historical environmental contaminants, these compounds are presenting unique challenges to environmental assessment approaches. For example, current efforts are examining how existing models ranging from fugacity-based fate predictions to traditional ecotoxicology assays and risk assessment paradigms may require modifications to predict environmental consequences of pharmaceutical exposure. Our group is specifically examining various “read-across” approaches for pharmaceutical pharmacology and toxicology data from mammals to non-target vertebrates. This presentation will provide current and future perspectives on the state-of-the-science of clinical drugs in the environment, including a summary of research needs to support environmental monitoring, fate, effects and risk assessment activities.
46. Metabolic interactions of environmental chemicals
Andrew D. Wallace, Parikshit C. Das, Richard C.T. Casabar, Yan Cao, and Ernest Hodgson
Dept of Evironmental and Molecular Toxicology, North Carolina State University, Raleigh, NC, USA, 27695-7633
Environmental chemicals can impact human metabolism, but the health risks of environmental chemicals are determined primarily from studies using surrogate animals. Extrapolation from surrogate animals is problematic due to the uncertainties of the relevance to humans and to considerations of human variation and species-specific differences. Thus, the hypothesis for these studies was that metabolic interactions that result from inhibition and/or induction of xenobiotic metabolizing enzymes are probable cause for human health risks. To study interactions based on inhibition, the bioactivation of the organophosphorothioate insecticide chlorpyrifos was investigated using human liver microsomes. Formation of the reactive metabolite chlorpyrifos-oxon (CPO) was found to cause P450 (CYP) inhibition of both xenobiotic and endogenous substrate metabolism. To determine the effects of environmental chemicals on CYP enzyme protein expression, human hepatocytes were treated with the environmental chemicals chlorpyrifos, DEET, endosulfan, and fipronil. Increases in CYP3A4 expression were seen by 100μM chlorpyrifos, 100μM DEET, 10μM endosulfan, and also 1μM fipronil. CYP2B6 protein levels were also found to be significantly elevated by endosulfan at 1μM. To investigate the mechanisms of these increases in CYP protein, HepG2 and COS-1 cells were transiently transfected with the human pregnane X receptor (PXR) or constitutive androstane receptor (CAR) and CYP3A4 or CYP2B6 promoter reporter plasmids. In the presence of hPXR or hCAR, induction of CYP3A4 or CYP2B6 promoter activity was seen by 10μM chlorpyrifos, 0.1μM fipronil, and 10μM endosulfan. Endosulfan (2.5 mg/kg/day) treatment of hPXR-transgenic mice caused a significant reduction of tribromoethanol-induced sleep times of approximately 50%, whereas no significant change in sleep times was observed in PXR-null mice. By the use of human in vitro microsomes and enzymes, human hepatocytes, and a humanized mouse model we have determined the ability of environmental chemicals to cause metabolic interactions based on inhibition or induction. The ability of different environmental chemicals to impact xenobiotic metabolic pathways varies, with some effects occurring at low concentrations that may pose an unacceptable human health risk.
47. PON1, environmental chemicals, and health
Lucio G. Costa, Toby Cole, and Clement E Furlong
Environm. Occup. Health Sciences, University of Washington, Seattle, WA, USA, 98105
Organophosphorus (OP) compounds are one of the major classes of insecticides. Their high acute toxicity to nontarget species is due to inhibition of acetylcholinesterase, with ensuing accumulation of acetylcholine and overstimulation of cholinergic receptors. Metabolism is known to be a major determinant of OP toxicity. An important detoxication pathway for the active oxygen metabolites of certain OPs is represented by the lactonase/A-esterase paraoxonase 1 (PON1), present primarily in liver and in plasma. PON1 null mice are more sensitive than wild-type animals to the acute cholinergic toxicity of diazoxon (DZO) and chlorpyrifos oxon (CPO). In contrast, administration of purified PON1 to rodents provides protection toward the acute toxicity of these compounds. Human PON1 is a polymorphic enzyme; relevant polymorphisms are at position 192 (Q192R) in the catalytic site of the coding region, and at position -108 (T-108C) of the promoter region, which influences the level of PON1 expression. An individual’s “PON1 status” is determined by the Q192R polymorphism and by the enzyme levels. Studies in transgenic humanized mice (expressing either PON1 Q192 or R192 on a knockout background) have shown that the toxicity of CPO is modulated by both the Q192R polymorphism and the level of enzyme, while in case of DZO only enzyme level is of relevance. Interestingly, the toxicity of paraoxon (the OP from which the enzyme’s name derives) is not affected by PON1 in vivo. PON1 levels can be modulated by a number of factors in addition to genetic polymorphisms, age being an important one. PON1 activity is low after birth and increases with age in both rodents and humans. Low PON1 activity during development contributes to the enhanced susceptibility of young animals (and perhaps children) to the acute cholinergic toxicity of OPs. Variants of recombinant PON1, recently expressed in E.coli, have increased catalytic efficiency toward certain substrates, and are promising new tools in antidotal therapy toward OP poisoning. Relevance of PON1 goes beyond its role in metabolizing certain OPs; indeed, PON1 metabolizes oxidized lipids (hence its role in cardiovascular disease), certain drugs or pro-drugs, and quorum sensing factors.
48. Phthalates, phthalate metabolites and human endocrine function
John Meeker
Environmental Health Sciences, University of Michigan, Ann Arbor, MI, USA, 48109
There is concern for adverse human health risks resulting from exposure to environmental endocrine-disrupting compounds (EDCs). Adding to this concern are a number of recent studies of men from several countries that have shown unexplained secular declining trends in semen quality and testosterone levels over the past several decades. Phthalates are a class of compounds that have a wide range of industrial and commercial uses, and are found in many consumer products ranging from fragrances to plastics. This widespread use results in pervasive human exposure through various pathways, and urinary phthalate metabolites can be detected in nearly 100% of individuals in the U.S. general population. Recent findings from human studies on the relationship between phthalate exposure and endocrine-related effects, such as altered hormone levels and semen quality, will be presented, as will new data suggesting a role for individual differences in phthalate metabolism as a modifier in the relationship between phthalate exposure and endocrine function.
49. Contribution of intestinal first-pass metabolism to drug-drug interactions of orally administered drugs
Aleksandra Galetin
School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester, United Kingdom, M13 9PT
There is an increasing interest in the contribution of the small intestine to the extent of observed metabolic drug-drug interactions (DDI), in particular in the case of the most abundant intestinal P450 enzyme, CYP3A4. Intestinal interaction is incorporated in the current DDI prediction models as the ratio of the intestinal availability in the presence and absence of an inhibitor or inducer (FG’ and FG, respectively). The incorporation of intestinal interaction into the DDI prediction strategy has resulted in differential predictions success across studies. Although this approach minimizes the number of false negative predictions, a general DDI over-prediction trend is apparent. This is particularly evident for DDIs with victim drugs with a high intestinal first-pass extraction (>75%), mediated by either extensive intestinal metabolism (e.g., buspirone) or as a result of the proposed interplay of drug metabolism and efflux (e.g., tacrolimus). Considering the multifactorial nature of the DDI prediction models the potential intestinal contribution will be discussed in conjunction with other perpetrator- and victim drug-related properties. Due to the sensitivity of the DDI prediction models to the accuracy of the FG estimates, the current study focuses on different in vitro and in vivo approaches to assess this parameter. Inter-individual variability associated with this parameter will also be addressed. Finally, impact of potential saturation of CYP3A4 and P-glycoprotein in vivo on the estimation of the FG and consequently on the magnitude of DDI will be discussed.
50. Understanding and anticipating drug-drug interactions requires new, interactive model classes that are also mechanism knowledge embodiments
C. Anthony Hunt
Bioengineering and Therapeutic Sciences, University of California, San Francisco, CA, USA, 94143
A dictum of the physicist Richard Feynman was “what I cannot create, I do not understand.” It follows that to begin understanding the consequences of two or more drugs interacting simultaneously with components of complex biological mechanisms, we must build mechanisms that a) exhibit some of the phenomena of interest, b) are extant (actually existing, observable), and initially absent drug interactions. I will describe how we have drawn on a new class of discrete event modeling methods to build extant, abstract yet plausible, hierarchical, spatially organized, semi-modular, biomimetic systems in object-oriented software that are capable of exhibiting emergent phenomena similar to that observed in wet-lab experiments. During simulations, we observe, at multiple levels, the consequences of “drugs” interacting with components. Building these analogue mechanisms is fundamentally different from the traditional, inductive approach of “modeling the data.” In the latter, the mechanisms are all conceptual. Making predictions, especially for new contexts and situations, based on mechanistic concepts that have not been challenged scientifically, is risky at best. I will provide additional arguments for why the study of xenobiotics needs this new class of models. We have constructed and refined liver analogues that are being used to discover, clarify, and challenge plausible, multi-level mechanistic linkages between drug disposition processes and phenotypic phenomena of interest. I will explain how the simultaneous in silico interactions of two or more drugs can be easily explored after having built analogues without drug interactions. Refinement and validation of in silico livers requires cycles of scientific modeling and simulation. The process calls for positing multiple generative hypotheses (using abductive reasoning methods) and selection of the most plausible (using inductive and deductive methods). That activity is followed by in silico experimentation to test and falsify those hypotheses. By following that protocol iteratively, we obtained a rat liver analogue undergoing perfusion in which we simulated drug disposition. We discovered a parameterization for which simulation results mimicked wet-lab results satisfactorily: digoxin and its metabolite in perfusate 1) in the absence of any predose and with a predose of either 2) the uptake inhibitor rifampicin or 3) the efflux inhibitor quinidine. Compounds are represented using objects that identify themselves by carrying physicochemical property information. The concrete nature of these new analogues means that validated descendents will have the potential to evolve into executable, observable representations of what we know (or think we know) about drugs interacting within biological systems: executable biological knowledge embodiments, which have the potential to revolutionize pharmaceutical research.
51. CYP-mediated drug-drug interaction predictions in human using hepatocytes: A paradigm shift?
Chuang Lu
Drug Metabolism and Pharmacokinetics, Millennium Pharmaceuticals, Inc., Cambridge, MA, USA, 02139
The rule of [I]/Ki ratio to predict pharmacokinetic drug-drug interactions (DDI) is the norm set by the FDA. However, this ratio determination has posed a great challenge for the industry due to ambiguity around the determination of [I] value (active concentration at the enzyme’s site) and lab-to-lab variation of Ki value, resulting in less than adequate predictions. A new model was created, and will be described, to circumvent the necessity to measure [I]/Ki value, by using human hepatocytes suspended in human plasma and equating extracellular concentration of the inhibitor in the incubation to that in vivo in human plasma. The resulting data on CYP activity change in the presence of the inhibitor, coupled with quantitative reaction phenotyping data provided the ingredients for improved prediction of DDIs in humans. Applying this method, excellent correlation was observed between predicted and clinically observed AUC changes for 16 marketed compounds with potent CYP3A inhibitor ketoconazole and moderate and multiple CYP inhibitor fluconazole, essentially providing a paradigm shift away from the age old [I]/Ki rule. In addition, clinical DDI study results of an investigational compound with ketoconazole and fluconazole confirmed the preclinical DDI predictions.
52. Metabolism-dependent inhibition (MDI) studies: Ten things worth knowing
Andrew Parkinson
XenoTech, LLC, Lenexa, KS, USA, 66219
This presentation will highlight some Dos and Don’ts, describe some unusual findings and provide examples of system-dependent outcomes with respect to in vitro CYP and UGT inhibition studies. The ten topics to be covered are: 1. In the case of MDI studies, a dilution step can be used to determine KI and kinact values, but it should NOT be incorporated in the design of screening procedures to identify MDI based on IC50 shifts. 2. Why? Because in the absence of MDI (when there is only direct inhibition), IC50 values should be based on the post-dilution concentration of drug candidate, but in the presence of MDI it should be based on the pre-dilution concentration. 3. When a dilution step is incorporated into the study design (to measure KI and kinact), the kinetics of MDI can be altered by inhibitor depletion, which can result in incomplete CYP inhibition. 4. Ultracentrifugation offers many advantages over dialysis and filtration to evaluate the reversibility of MDI. 5. The generally accepted idea that formation of a nitroso metabolite is the mechanism by which tertiary amines form a ~455 nm-absorbing, metabolite inhibitory complex (MIC) with ferrous cytochrome P450 may not be correct. 6. Some oligonucleotides are surprisingly effective inhibitors of microsomal CYP enzymes, especially CYP1A2 and CYP2C8. 7. However, in contrast to the situation with human liver microsomes (HLM), oligonucleotides are weak CYP inhibitors in human hepatocytes. 8. Gemfibrozil provides another example of system-dependent inhibition: It causes weak inhibition of CYP2C8 in NADPH-fortified HLM, but causes potent inhibition in human hepatocytes (as it does in the clinic) due to its conversion to gemfibrozil glucuronide, an irreversible metabolism-dependent inhibitor of CYP2C8. 9. In the case of gemfibrozil, we observe glucuronidation-dependent activation. However, in the case of ezetimibe, we observe glucuronidation-dependent protection, which is why the marked MDI of CYP3A4 observed in NADPH-fortified HLM is not observed in human hepatocytes (or in the clinic). 10. In the case of UGT, the extent of enzyme inhibition by 1-naphthol differs markedly between recombinant UGT enzymes and HLM due to the rapid conjugation of naphthol in HLM and the attendant build-up of UDP, which inhibits UGT activity by competing with UDP-glucuronic acid.
53. Global nuclear occupancy of pxr and co-existence of histone h3k4 di-methylation resulting in activation of drug-processing genes in mouse liver
Yue J. Cui1, Sumedha S. Gunewardena2, and Curtis D. Klaassen3
1Department of Pharmacology, Toxicology and Therapeutics, University of Kansas Medical Center, Kansas City, KS, USA, 66160
2Department of Molecular and Integrative Physiology, University of Kansas Medical Center, Kansas City, KS, USA, 66160
3Pharmacology, Toxicology, and Therapeutics, The University of Kansas Medical Center, Kansas City, KS, USA, 66160
The pregnane X receptor (PXR) is a key regulator of drug metabolism and disposition in liver. Data from this and other laboratories have shown that numerous drug-processing genes (uptake transporters, phase-I and –II drug metabolizing enzymes, and efflux transporters) in mouse liver are up-regulated by PXR activators. However, little is known of the global nuclear occupancy of PXR, or the profiles of direct PXR-target genes in liver. Therefore, the purpose of the present study was to characterize the global binding patterns and target gene profiles of PXR in mouse liver. Eight-week-old male C57BL/6 mice were administered a single dose of the PXR ligand pregnenolone-16α-carbonitrile (PCN) (200mg/kg, i.p. in corn oil) or vehicle, and livers were removed 8h thereafter. Genome-wide ChIP-sequencing analysis revealed constitutive PXR binding sites in control liver, and an increase in the number of PXR binding sites after PCN administration. The most frequent PXR binding motifs were direct repeats 4 and 3. ChIP-on-chip assays of mouse chromosomes 5, 12, and 15 showed that PXR binding did not appear to overlap with DNA methylation or histone H2K27 tri-methylation, which are epigenetic marks for gene suppression; however, PXR binding overlapped with histone H3K4 di-methylation, an epigenetic mark for gene activation. Approximately 600 genes altered by PCN had direct PXR binding sites, and the majority of these alterations (both induction and suppression) appeared to be due to increased PXR-binding. Specifically, increased PXR binding by PCN correlates with the mRNA induction of phase-I enzymes (Cyp3a11, Cyp2b10, Aldh1a1), phase-II enzymes (Gsta1, a4, m1, m2, m3, PAPss2), and transporters (Oatp1a4 and Mrp3), and the induction of these mRNAs was abolished in livers from PCN-treated PXR-null mice. In conclusion, genome-wide profiling of PXR reveals unique DNA binding patterns and co-existence of histone H3K4 di-methylation, resulting in temporal activation of critical genes in drug metabolism and disposition. (Supported by NIH grants ES-09716, ES-09649, ES-013714, DK-081461, RR-021940, RR-016475, and NICHD-02528)
54. Role of CYP2A5 in the metabolism of nicotine and cotinine: insights from a Cyp2a5-null mouse model and a CYP2A6-humanized mouse model
Xin Zhou1, Xiaoliang Zhuo2, Fang Xie3, Kerri Kluetzman4, Yue-Zhong Shu5, William Humphreys6, and Xinxin Ding7
1Laboratory of Molecular Toxicology, Wadsworth Center, Albany, NY, USA, 12201-0509
2Discovery Biotransformation, Bristol-Myers Squibb, Wallingford, CT, USA, 06492
3Laboratory of Molecular Toxicology, Wadsworth Center, NYSDOH, Albany, NY, USA, 12201-0509
4Wadsworth Center, NYSDOH, Albany, NY, USA, 12201-0509
5Pharmaceutical Candidate Optimization, Bristol-Myers Squibb Company, Wallingford, CT, USA, 06492
6Pharmaceutical Candidate Optimization, Biotransformation, Pharmaceutical Research Institute, Bristol-Myers Squibb Company, Princeton, NJ, USA, 08543-4000
7Dept HTME, Wadsworth Ctr, Albany, NY, USA, 12201-0509
Mouse CYP2A5, expressed in liver, kidney, lung, and nasal mucosa, is similar to human CYP2A6 in tissue distribution and substrate specificity. Heterologously expressed CYP2A5 is active in the metabolism of both endogenous substrates, such as testosterone, and xenobiotic compounds, such as nicotine and cotinine. To determine the biological and pharmacological functions of CYP2A5 in vivo, we have generated a Cyp2a5-null mouse. Homozygous Cyp2a5-null mice are viable and fertile; they show no evidence of embryonic lethality or developmental deficits. A CYP2A6-humanized mouse model was also produced, by crossing the Cyp2a5-null mouse with a CYP2A6-transgenic mouse model (which shows a liver-specific CYP2A6 expression). Neither the Cyp2a5-null mouse, nor the CYP2A6(+/-)/Cyp2a5-null mouse, showed any compensatory increases in the expression of other major P450 or UDP-glucuronosyltransferase (UGT) enzymes examined. The Cyp2a5-null mouse, CYP2A6(+/-)/Cyp2a5-null mouse, and WT mouse were then utilized for determinations of the roles of CYP2A5 or CYP2A6 in the metabolism of nicotine and its major circulating metabolite, cotinine. The results indicated that the Cyp2a5-null mouse had decreased systemic clearance of both nicotine and cotinine; for both compounds, substantial increases in plasma half life, and in the area under the concentration-time curve, were observed in the Cyp2a5-null mouse, compared with WT mouse. In contrast, the CYP2A6(+/-)/Cyp2a5-null mouse did not show any increase in the clearance of either nicotine or cotinine in vivo, although it did display increased hepatic microsomal activity toward nicotine C-oxidation in vitro, compared with Cyp2a5-null mouse. Further pharmacokinetics analysis confirmed that the brain levels of nicotine and cotinine are also influenced by the Cyp2a5 deletion. We conclude that CYP2A5 is the major nicotine and cotinine oxidase in the mouse liver. Our findings also support the notion that human CYP2A6 is not as efficient as mouse CYP2A5 in the clearance of nicotine. The Cyp2a5-null mouse should be valuable for studies on the potential role of nicotine as a neuroprotective agent in mouse models. (Supported in part by NIH grant ES-07462)
55. Pharmacokinetic and pharmacodynamic determinants of warfarin anticoagulation response
Inna Y. Gong1, Ute I. Schwarz2, Rommel G. Tirona2, Natalie Crown3, George Dresser3, Alejandro LazoLangner4, GuangYong Zou2, Samantha LaRue5, Nicole Langlois5, Phil Wells5, and Richard B. Kim3
1Physiology and Pharmacology, The University of Western Ontario, London, ON, Canada, N6A3A5
2Physiology and Pharmacology, University of Western Ontario, London, ON, Canada, N6A3A5
3Department of Medicine, Division of Clinical Pharmacology, University of Western Ontario, London, ON, Canada, N6A3A5
4Department of Medicine, Division of Hematology, University of Western Ontario, London, ON, Canada, N6A3A5
5Department of Medicine, Division of Hematology, University of Ottawa, Ottawa, ON, K1Y 4E9
Warfarin is a widely prescribed anticoagulant known for marked interindividual variation in drug requirement and responsiveness. Genetic polymorphisms in genes affecting warfarin metabolism (cytochrome P450 2C9; CYP2C9) and activity (vitamin K epoxide reductase complex 1; VKORC1) together with patient clinical parameters explain only 50% of dose variation. Of the known relevant patient parameters such as age, weight and gender, it was uncertain how these influence pharmacokinetic (PK) and/or pharmacodynamic (PD) responses. Accordingly, we have characterized warfarin PK-PD responses in patients initiating warfarin therapy. Consented patients were enrolled and dosed accordingly to reach therapeutic international normalized ratio range of 2 to 3. Using a developed and validated highly sensitive liquid chromatography-tandem mass spectrometry method, plasma R/S-warfarin levels were measured on treatment days 3, 5 and 8. PK parameters for S-warfarin were determined in 42 patients with pharmacokinetic modeling of plasma concentration profiles. The results demonstrate that after a warfarin loading dose, S-warfarin levels were proportional to weight-normalized dose indicating that volume of distribution relates to body mass. As expected, S-warfarin clearance was lower in CYP2C9 *2 and *3 variant carriers. Therapeutic S-warfarin plasma levels segregated with VKORC1 haplotype indicating that VKORC1-associated dose variation correlates with targeted S-warfarin concentration. Interestingly, clearance of S-warfarin was different between females and males, suggesting gender-dependent differences in the metabolic capacity for warfarin. S-warfarin clearance was not associated with either age or weight, indicating that reduced warfarin metabolism is not the source of lower dose requirement observed in elderly patients. Accordingly, age-dependent PD factors, namely vitamin K status, may be responsible for increased warfarin sensitivity in the elderly. In addition, warfarin dose requirement was significantly related to genetic variation in the vitamin K metabolizing enzyme CYP4F2 (V433M), further confirming the role of vitamin K in warfarin response. In summary, our data reveal important new mechanistic insights regarding warfarin dose variability. Better understanding of warfarin PK-PD interaction will serve as a basis for creating a more predictive dosing algorithm for individualized warfarin therapy.
56. Utility of the unbound cerebrospinal fluid-to-plasma ratio, Kp,uu,CSF, for prediction of brain exposure in rats and humans
Markus Fridén1, Susanne Winiwarter2, Ola Bengtsson3, Gunilla Jerndal2, Ulf Bredberg2, Margareta Hammarlund-Udenaes1, and Madeleine Antonsson2
1Department of Pharmaceutical Biosciences, Division of Pharmacokinetics and Drug Therapy, Uppsala University, Uppsala, Sweden
2Discovery DMPK, AstraZeneca R&D Mölndal, Mölndal, Sweden, 431 83
3AstraZeneca R&D Lund, Lund, Sweden, 431 83
Purpose: This study evaluates the utility of the unbound CSF-to-plasma ratio Kp,uu,CSF as a surrogate measure of the unbound brain-to-plasma ratio Kp,uu,brain in the rat, and the agreement with Kp,uu,CSF determined in humans.
Methods: Forty-three drugs were selected based on the availability of human CSF data and structural diversity. Kp,uu,brain and Kp,uu,CSF were measured in the rat by combining total brain, plasma and CSF concentrations with the unbound fraction in plasma and CSF as well as brain slice estimates of brain tissue binding. Literature reports on total CSF-to-plasma ratios were characterized with regards to the experimental procedures used. Values of human Kp,uu,CSF were calculated with fraction unbound in plasma determined by equilibrium dialysis. The fraction unbound in CSF was calculated from the fraction unbound in plasma and the CSF plasma protein concentration.
Results: The agreement of Kp,uu,CSF and Kp,uu,brain in the rat was generally within three-fold. However, Kp,uu,brain was generally overpredicted for highly effluxed drugs like loperamide, and underpredicted for drugs that were not highly effluxed. Thus, Kp,uu,CSF does not display the same range of values as does Kp,uu,brain. There was some correlation between rat and human Kp,uu,CSF (R2=0.56), but human Kp,uu,CSF was underpredicted by 3-fold on average. This was at least partly related to experimental factors such as patient disease state and the site and timing of CSF sampling.
Conclusion: The results raise concerns as to the utility of CSF sampling for prediction of Kp,uu,brain in particular for highly effluxed drugs. It is suggested that Kp,uu,brain is measured when possible. Although there were many experimental factors to consider for the comparison of rat and human Kp,uu,CSF, the study provides some basic support for the use of the rat as a model in drug discovery for prediction of brain exposure in humans.
57. Impact of drug transport on the tissue distribution of rosuvastatin: Studies in Mrp1 knockout mice
Michael J. Knauer1, and Rommel G. Tirona2
1Physiology and Pharmacology, University of Western Ontario, London, ON, Canada, N6A 5A5
2Physiology and Pharmacology, University of Western Ontario, London, ON, Canada, N6A3A5
The 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, or statins, are important drugs used in the treatment and prevention of cardiovascular disease. Recently, we demonstrated a role for human Organic Anion Transporting Polypeptide (OATP) 2B1 and Multidrug Resistance Associated Proteins (MRP) 1, MRP4 and MRP5 in the uptake and efflux transport as well as toxicity of statins in skeletal muscle cells in vitro. Here, we examined the in vivo and ex vivo role the efflux transporters in regulating the tissue distribution of rosuvastatin in a mouse model of Mrp1 deficiency. Rosuvastatin transport by mouse Mrp1 was assessed in cultured cells overexpressing the transporter. The expression of transporters in mouse tissues was examined by qPCR. Wild-type and Mrp1 knockout (KO) mice were administered [3H] rosuvastatin (1 mg/kg) by tail vein injection. Mice were sacrificed after 6 hrs and the tissue content of [3H] rosuvastatin was examined by liquid scintillation counting. Muscle fibres from wild-type and Mrp1 KO mice were isolated to study [3H] rosuvastatin transport. Rosuvastatin was found to be a transport substrate for mouse Mrp1. The distribution of rosuvastatin in kidney, spleen, brain, heart, and testis was similar in Mrp1 KO mice in comparison to wild-type animals. However, Mrp1 KO animals had a significantly higher liver to plasma ratio of rosuvastatin compared to wild-type animals. Importantly, we observed a lack of difference in skeletal muscle rosuvastatin distribution between Mrp1 KO and wild-type mice. Gene expression analysis demonstrated elevated levels of Mrp2, Mrp4 and Mrp5 in KO mouse tissues which normally express high levels of Mrp1. Tissue distribution of rosuvastatin was not different in tissues of KO animals despite that this drug is an Mrp1 substrate. These findings highlight the interplay between multiple redundant transporters in the tissue pharmacokinetics of rosuvastatin.
58. Impact of saturation of CYP3A4 and P-glycoprotein in vivo on the prediction of intestinal availability
Michael Gertz1, Anthony Harrison2, John Davis2, J. Brian Houston1, and Aleksandra Galetin1
1School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester, United Kingdom, M139PT
2Pfizer Global Research and Development, Sandwich, United Kingdom, CT13 9NJ
CYP3A4 and efflux transporter (P-glycoprotein) interplay in the human small intestine was suggested to contribute to low intestinal availability (FG) of a number of drugs. Previous work has shown that use of the QGut model resulted in FG under-predictions of up to 88% in the case of saquinavir. In the current study, 11 CYP3A4 drugs with high intestinal extraction in-vivo (FG≤0.5) were selected to address potential saturation of CYP3A4 and P-glycoprotein in vivo, as an explanation for observed FG under-predictions. Intrinsic clearances were determined in 9 individual jejunal microsomes, corrected for microsomal binding and intestinal CYP3A4 abundance. In order to address potential saturation, clearance values were also estimated at the respective enterocytic drug concentrations; these were based on the therapeutically used dose and CYP3A4 Km values of the 11 drugs investigated. Out of the drugs in the dataset, impact of P-glycoprotein saturation on FG predictions was assessed for indinavir, saquinavir and tacrolimus using the permeability data from MDCK-MDR1 in the presence and absence of P-glycoprotein inhibitor CP-100356. The analysis indicated that saturation of intestinal metabolism in vivo was not expected for atorvastatin, simvastatin and tacrolimus, in contrast to indinavir, nisoldipine, saquinavir and terfenadine for which the likelihood of saturation was high. For the latter four drugs, a pronounced reduction in clearance at enterocytic concentration (67-99%) was seen in comparison to the estimates obtained under common in vitro conditions. Accounting for CYP3A4 saturation resulted in FG predictions of 6/11 drugs within 1.5-fold of the observed values, a reduction in bias by 26% and no observed FG under-predictions. Incorporating the saturation of efflux processes had minor impact on the predicted FG of indinavir and tacrolimus, in contrast to saquinavir where differential contribution of CYP3A4 and P-glycoprotein saturation was apparent. In the case of indinavir, predicted FG approached 1 due to significantly reduced clearance at enterocytic drug concentration, regardless of permeability data. The implications of these findings on the prediction of FG from in vitro data are discussed.
59. Expression Of Human UGT1A1 in the gastrointestinal tract of humanized ugt1*28 mice plays an important role in bilirubin clearance
Ryoichi Fujiwara, Nghia Nguyen, and Robert H. Tukey
Laboratory of Environmental Toxicology, Department of Chemistry & Biochemistry and Pharmacology, University of California, San Diego, La Jolla, CA, USA, 92093
Bilirubin is an end product of heme catabolism and is metabolized by UDP-glucuronosyltransferase 1A1 (UGT1A1). Gilbert’s syndrome results in congenital hyperbilirubinemia and is associated with a (TA)7 repeat in the TATAA box of the UGT1A1*28 gene. Transgenic mice expressing the human UGT1 locus including the (TA)7 repeat of the UGT1A1*28 allele were crossed into a Ugt1-null background creating humanized UGT1 (hUGT1*28) mice. Knockout of the Ugt1 locus leads to neonatal lethality by 7 days after birth and is linked to excessive bilirubin accumulation in the central nervous system (kernicterus). Expression of the UGT1 locus in hUGT1*28 mice rescues neonatal mice from kernicterus induced lethality, resulting in healthy adult mice with mildly elevated levels of serum bilirubin. Hyperbilirubinemia in adult hUGT1*28 mice is linked to reduced expression of UGT1A1 in liver. In contrast, dramatic hyperbilirubinemia occurs during neonatal development, with peak values being reached at 14 days after birth, followed by a sharp decline over the next 7 days to adult levels. The sharp reduction in serum bilirubin from 14 to 21 days after birth is not associated with liver UGT1A1 expression, but is tightly linked to developmental expression of UGT1A1 in the gastrointestinal tract. To evaluate the role of gastrointestinal UGT1A1 towards bilirubin clearance, neonatal mice were exposed to agents that induced UGT1A1. Treatment of neonatal hUGT1*28 mice with either TCDD or phenobarbital led to increased UGT1A1 expression in both the liver and small intestine and a dramatic lowering of serum bilirubin levels. Arsenic has been shown to regulate several xenobiotic-metabolizing genes. When arsenic was administered orally to neonatal hUGT1*28 mice as bilirubin was accumulating in during development, circulating bilirubin levels were dramatically reduced. Arsenic had no impact on the expression levels of hepatic UGT1A1. However, expression of UGT1A1 in the small intestine was significantly induced. Thus, two lines of evidence support an important role for the gastrointestinal tract in bilirubin metabolism. 1) developmental expression of UGT1A1 and 2) selective induction of UGT1A1 by arsenic.
(This work is supported by NIEHS grant ES010337)
60. Adenovirus delivery of drug transporters to generate a custom polarized epithelial model system
Wendy A. Teft1, Rommel G. Tirona2, and Richard B. Kim3
1Department of Medicine, University of Western Ontario, London, ON, Canada, N6A5A5
2Physiology and Pharmacology, The University of Western Ontario, London, ON, Canada, N6A 5A5
3Division of Clinical Pharmacology, Department of Medicine, The University of Western Ontario, London, ON, Canada, N6A 5A5
The study of the interplay between drug uptake and efflux transporters in vitro remains difficult due to the inherent difficulty associated with generating stable cell lines expressing transporters of interest. Here, we describe a method for targeted transporter expression in a polarized cell line, which may be better reflective of drug transport in organs such as liver, kidney or small intestine. MDCK and HeLa cells were transduced with transporter-encoded Adenovirus to generate transiently expressing cells. Transporter expression was characterized by real-time PCR and confocal microscopy and function was measured by transport assays. Using this system, we tested the bile acid transporter, NTCP and the drug transporter, OATP2B1. Adenoviral delivery of NTCP to cellular monolayers resulted in [3H] taurocholate uptake which was approximately 1800% greater than LacZ transfected control cells at 48 hours post-infection. Additionally, uptake of [3H] estrone sulfate showed a near 300% greater overall uptake in cells infected with OATP2B1-expressing Adenovirus compared to LacZ-transfected control. Cell viability following Adenovirus transduction remained greater than 90%. To measure the vectorial drug transport in the apical-to-basolateral or basolateral-to-apical direction, MDCK cells were grown on cell culture inserts to form a polarized epithelium. Cells were infected with Ad-NTCP or Ad-OATP2B1 and assayed for membrane integrity and transepithelial drug flux. Basolateral-to-apical uptake of [3H] taurocholate by NTCP expressing cells increased from 2 to 10.5 pmol/well over a 4 hour time course, while Ad-LacZ infected cells reached only 1 pmol/well. Polarized cells infected with Ad-OATP2B1 transported [3H] estrone sulfate in the apical-to-basolateral direction with a flux of 25 pmol/well by 4 hours compared to 20 pmol/well in the LacZ control. These results demonstrate that a customized drug transporter expressing model system can be generated using Adenovirus-based system. This system has the potential for simultaneous co-expression of multiple uptake and efflux transporters. Importantly, the interplay of such transporters can be studied so that a more robust in vitro to in vivo prediction of a substrate drug clearance profile can be made.
61. The role of small intestinal P450 enzymes in the protection against systemic exposure of orally administered benzo(a)pyrene
Cheng Fang, and Qing-yu Zhang
Wadsworth Center, NYSDOH, Albany, NY, USA, 12201-0509
Benzo(a)pyrene (BaP), a potent environmental toxicant, is mainly metabolized by CYP1A1. Induction of CYP1A1 by BaP in the liver and small intestine (SI) plays a critical role in the rapid clearance of oral BAP in mice; however, it is not clear which of the two organs play a more important role in controlling systemic bioavailability of orally administered BaP. Here, we utilized an intestinal epithelium-specific cytochrome P450 reductase (CPR)-knockout (IE-Cpr-null) mouse and a liver-specific CPR-knockout (liver-Cpr-null) mouse to determine the roles of liver and SI P450s in BaP metabolism. BaP was given to IE-Cpr-null, liver-Cpr-null, and wild-type (WT) mice, by either oral gavage or intraperitoneal (ip) injection, and blood BaP levels were determined for pharmacokinetic analysis. We found that BaP blood levels (as indicated by Cmax and AUC values) were significantly higher, and BaP clearance was slower, in IE-Cpr-null than in WT mice, after oral BaP treatment. In contrast, BaP clearance was not different between IE-Cpr-null and WT mice, following ip BaP. Furthermore, there was no significant difference between liver-Cpr-null and WT mice in BaP clearance, after either ip or oral administration of BaP. These results indicate that SI P450-mediated first-pass metabolism is a key determinant of the systemic bioavailability of oral BaP. In additional studies, we observed that the differences between WT and IE-Cpr-null mice in systemic bioavailability of oral BaP were even greater in mice pretreated with β-naphthoflavone to induce CYP1A1 expression, compared with mice pretreated with vehicle alone. Moreover, an examination of the time-course of CYP1A1 induction by oral BaP revealed that CYP1A1 protein expression in the SI was already induced by ~3 fold at 2 h following BaP administration, while the induction in other organs analyzed (including liver, lung, and kidney) was not significant until 4 h post-treatment. Taken together, these results point to SI CYP1A1 induction as a critical factor for protection against systemic exposure by orally administered BaP. (Supported in part by NIH grant GM082978)
62. Synergism between CYP2B6 gene polymorphism and pxr-mediated induction
Haishan Li, and Hongbing Wang
Pharmaceutical Sciences, University of Maryland School of Pharmacy, Baltimore, MD, USA, 21201
Cytochrome P450 2B6 (CYP2B6) is a highly polymorphic enzyme involving the metabolism of a number of clinically important drugs. Significant interindividual variation in CYP2B6 gene expression has been attributed to either inherit genetic polymorphisms or chemical-mediated induction through the activation of nuclear receptor CAR or PXR. It was reported, recently, that the -82T>C mutation within the CYP2B6*22 allele, which converts the putative TATA box into a functional CCAAT/ enhancer-binding protein (C/EBP) binding site, enhanced the basal expression levels of CYP2B6 gene. To explore whether the polymorphism-based function change in CYP2B6 promoter could also affect drug-mediated induction of this gene, we have generated CYP2B6 promoter constructs containing various mutants including the -82T>C, and examined their response to PXR activators in cell-based reporter assay. Intriguingly, data obtained from these experiments demonstrated that the luciferase activity in construct of -82T>C was synergistically enhanced in the presence of PXR and its prototypical ligand rifampicin (RIF). Moreover, this synergism was abrogated in the construct containing a mutation that disrupts the -82T>C converted C/EBP binding site. In contrast, cotransfection of C/EBP alpha (C/EBPA) expression vector in the above mentioned reporter assays further increase the luciferase reporter expression in response to RIF treatment. Additionally, coimmunoprecipitation assay showed that PXR interacts with C/EBPA directly in COS-1 cells; and CHIP assay in transfected HepG2 cells demonstrated that treatment with RIF results in increased recruitment of PXR to the PBREM (-1683 to -1733) in -82T>C mutant over the reference constructs. Furthermore, genotyping of a small number (11) of human primary hepatocyte donors found that the liver donor with the highest inducibility to RIF (30 folds) is a heterozygote of -82T>C, while the other 10 donors with an induction range of 3-15 folds by RIF are all wild type homozygotes. Collectively, our results demonstrate a synergistic interaction between gene polymorphism and PXR-mediated induction of CYP2B6, which may contribute to the large individual variations of CYP2B6 expression in humans.
63. Paradoxical reduced lithocholic acid hepatotoxicity in Mrp4-null mice
Yuanyuan Zhang, Jessica Morgan, Yao Wang, and John D. Schuetz
Pharmaceutical Science, St.Jude Children’s Research Hospital, Memphis, TN, USA, 38105
Mrp4 is a member of the multidrug resistance associated gene family that is expressed on the basolateral membrane of hepatocytes and undergoes adaptive up-regulation in response to cholestatic injury or bile acid feeding. We have previously shown that the Mrp4 null mice have an impaired cytoprotective response in obstructive cholestasis (Leggas, 2004; Mennone, 2006). Lithocholic acid (LCA) is a hydrophobic secondary bile acid and hepatotoxic by mechanisms that include alterations in the canalicular membrane, formation of crystalline plugs or altered trafficking of canalicular export pumps. We investigated the hepatotoxicity of LCA in Mrp4-null mice. Female wildtype and Mrp4-null mice were fed either a normal diet or one supplemented with 1% LCA. Regardless of genotype, the LCA-induced loss in body weight was similar for Mrp4-null and wildtype mice. As expected, the total bile acid concentration in the liver was almost two-fold greater in the Mrp4-null mice. Unexpectedly, the serum aminotransferase activities (AST, ALT) were significantly lower in the Mrp4-null compared to the wild type mice. Histopathologic analysis revealed severe multifocal necrosis, fibrosis, and hepatocyte proliferation (consistent with an increased liver size) in wildtype mice fed LCA, with all these parameters being significantly less in Mrp4-null mice. Pathway analysis revealed significant alterations in the nuclear receptor pathway (such as CAR, FXR, PPARα, by Fisher’s Exact Test). Increased LCA-induced damage in the wildtype mice was evidence for enhanced activation of the cytokine-cytokine receptor interaction pathway. Further examination of the microarray data indicated that LCA induced higher collagen and keratin mRNA expression in wildtype vs Mrp4-null. One potential mechanism of protection might be that LCA up-regulates a nuclear receptor pathway in the Mrp4-null mice that minimizes fibrosis and proliferation. While Mrp4 is required for protection against obstructive cholestasis, our data indicate that, unexpectedly, the absence of Mrp4 is protective against liver injury induced by the hydrophobic bile acid LCA.
References
- M. Leggas, M. Adachi, G. Scheffer, D. Sun, P. Wielinga, G. Du, K. Mercer, Y. Zhuang, J. Panetta, B. Johnston, R. Scheper, C. Stewart, and J. Schuetz. Mrp4 Confers Resistance to Topotecan and Protects the Brain from Chemotherapy. Molecular and Cellular Biology 2004; 24: 7612–7621
- A. Mennone, C. Soroka, S. Cai, K. Harry, M. Adachi, L. Hagey, J. Schuetz, and J. Boyer. Mrp4−/- Mice Have an Impaired Cytoprotective Response in Obstructive Cholestasis. Hepatology 2006; 43:1013–1021
64. Identification of aryl hydrocarbon receptor as NOVEL transcriptional regulator of human ABCG2 (BCRP)
Kah Poh Hendrick Tan1, Bernice Wang1, Mingdong Yang1, Haibo Xu1, Paul Boutros2, Kazuhiro Kosuge1, Jane MacCauley1, Alex Wu1, Andrew Chuang1, Patricia Harper1, Douglas Ross3, and Shinya Ito1
1Clinical Pharmacology and Toxicology, Physiology and Experimental Biology Program, Hospital for Sick Children, Toronto, ON, Canada, M5G 1X8
2Pharmacology, University of Toronto, Toronto, ON, Canada, M5S 1A8
3Pathology, University of Maryland School of Medicine, Baltimore, MD, USA, 21201
ATP-binding Cassette G2 (ABCG2) or Breast Cancer Resistance Protein (BCRP) is a cellular efflux pump of xenobiotics and physiologic substrates. Its overexpression contributes to phenotypes of chemotherapeutic drug resistance. Despite its vital role in physiology and pharmacotherapy, the molecular regulation of BCRP remains largely unexplored. Using human colon, liver and mammary cancer cells and human primary hepatocytes and colonocytes, we noticed that exposure of these cells to the Aryl hydrocarbon receptor (AhR) ligands, including tetrachlorodibenzo-p-dioxin (TCDD), significantly induced BCRP gene transcripts. Concomitant increases in BCRP protein and function, as determined by mitoxantrone uptake, were also noted. However, similar treatments to mice or mouse cell lines failed to replicate observations of the human-derived cells, suggesting that this phenomenon is human specific. In silico analysis on 5’-flanking region of the BCRP gene revealed presence of 13 and five putative dioxin-responsive elements (DREs) in human and mouse, respectively, but none of these DREs was conserved between the two species. Knockdown of AhR function via small-interfering RNA or inhibitor dimethoxyflavone significantly attenuated TCDD-induced BCRP expression; whereas ectopic expression of AhR in AhR-deficient cells (MCF7AhR100) restored induction of BCRP gene by TCDD. Luciferase-based gene reporter assays showed that -1.29-kb of the gene promoter of human BCRP was responsive to AhR overexpression and TCDD treatment. Sequential deletion of this promoter construct revealed that four putative DRE(s) residing within -300 bp from the transcription start site may be responsible for gene transactivation. By means of promoter reporter and electrophoretic mobility shift assays, we further examined each of these DREs and found one of the DREs was functional. Existence of AhR binding on regions encompassing this functional DRE in native chromatin cell context was confirmed by chromatin immunoprecipitation analysis. We concluded that human BCRP is directly regulated by the AhR signalling pathway. Our study provides an unprecedented role of AhR in mediating an adaptive response against xenobiotics accumulation by upregulating an important ABC transporter. (Supported by CIHR)
65. Understanding drug failure through pharmacogenetics: Altered cerivastatin in vitro metabolism by CYP2C8 variants found in patients experiencing rhabdomyolysis
Ruediger Kaspera1, Suresh B. Naraharisetti1, Bani Tamraz2, Tariku Sahele3, Pui-Yan Kwok4, Kristin Marciante5, Susan A. Heckbert6, Bruce M. Psaty7, and Rheem A. Totah1
1Department of Medicinal Chemistry, University of Washington, Seattle, WA, USA, 98195
2Cardiovascular Research Institute and Institute for Human Genetics, University of California at San Francisco, San Francisco, CA, USA, 94143
3Department of Medicinal Chemistry, University of Washington, Seattle, WA, USA, 98102
4Cardiovascular Research Institute, Department of Dermatology, and Institute for Human Genetics, University of California at San Francisco, San Francisco, CA, USA, 94143
5Department of Medicine, Cardiovascular Health Research Unit, University of Washington, Seattle, WA, USA, 98101
6a) Department of Epidemiology, Cardiovascular Health Research Unit, b) Center for Health Studies, University of Washington (a) and Group Health (b), Seattle, WA, USA, 98101
7a) Department of Medicine and Department of Epidemiology, Cardiovascular Health Research Unit, b) Center for Health Studies, University of Washington (a) and Group Health (b), Seattle, WA, USA, 98101
Cerivastatin, a HMG-CoA reductase inhibitor, was withdrawn from the market because of the serious adverse effect of myotoxicity and rhabdomyolysis. This drug withdrawal prompted investigations not only related to drug-drug interactions (inhibition of CYP2C8, CYP2C9 and various transporters), but also into genetic factors that may explain variations in the pharmacokinetics of cerivastatin. In this investigation, DNA samples from patients (n = 126) who had rhabdomyolysis after cerivastatin administration were collected, and the CYP2C8 gene was resequenced in search of novel genetic variations that might in part explain the susceptibility of these patients to rhabdomyolysis compared to controls. Sequencing data demonstrated that the patients had a lower minor allele frequency (MAF) of CYP2C8*3 and CYP2C8*4 (0.091 and 0.044) compared with the general Caucasian population (0.150 and 0.075 respectively, Bahadur et al., Biochem Pharmacol, 2002, 64:1579-89). Further, three novel coding region single nucleotide polymorphisms were found (each at an MAF of 0.004). To determine whether any of the CYP2C8 genetic variants influenced the metabolism of cerivastatin, all variant proteins were heterologously expressed in a bacterial expression system and their ability to metabolize cerivastatin was assessed and compared with wild type (WT) protein. Recombinant CYP2C8.3 and CYP2C8.4 displayed an increase in Vmax (up to three fold) while the novel SNPs demonstrated a higher Km but no change in formation clearance (Vmax/Km) compared to WT. Kinetic data from human liver microsomes carrying CYP2C8*3 and *4 mutations demonstrated that carrying one or more of the CYP2C8*3 or *4 alleles was associated with a 2 to 14-fold increase in cerivastatin clearance compared with livers expressing CYP2C8 WT. These results suggest that CYP2C8 genetic variations present in patients who used cerivastatin and had rhabdomyolysis affect cerivastatin metabolism, and that inheriting one or more allele of CYP2C8*3 or CYP2C8*4 might be associated with a lower risk of this complication.
66. Efficient recoveries of drug-related material from rodent eye tissues for metabolite analysis using novel adaptive focused acoustic technology
Caroline J. Sychterz, Michael J. Morris, Kitaw Negash, and May Y. Ho
Drug Metabolism and Pharmacokinetics, GlaxoSmithKline, King of Prussia, PA, USA, 19406
Occasionally during the course of a drug’s development the levels of drug and potential metabolites in specific tissues must be determined; however, when those tissues are fibrous and small (~ 100 mg) efficient sample preparation where homogenization and extraction are required often proves to be a difficult task. Previous attempts to utilize standard homogenization and pulverizing techniques to prepare rodent eyes, such as Waring blenders, probe homogenizers and a mortar and pestle, were either inefficient or labor intensive and technically difficult. In each instance the chances for cross-sample contamination were high as was the time and effort spent by the analyzing scientist during multiple sample preparations. Adaptive focused acoustic (AFA) technology has shown promise in enhancing sample recovery in plant and animal-based samples [Toorchi M, Nouri MZ, Tsumura M and Komatsu S. Acoustic Technology for High-Performance Disruption and Extraction of Plant Proteins. J Proteome Res 7(7):3035-3041 (2008); Wenger MD, DePhillips P and Bracewell DG. A Microscale Yeast Cell Disruption Technique for Integrated Process Development Strategies. Biotechnol Prog 24:606-614 (2008); http://www.covarisinc.com/publications.html (accessed 20.03.2009)]. A novel non-contact sample preparation method was developed for recovering drug-related material from rodent eyes using freeze fracturing with AFA technology for metabolite analysis. Recoveries of [14C]compound-related material in the extracts of homogenized eye tissue were >86%, with spiked control samples exhibiting >93% recovery. Stability samples showed no evidence of compound degradation during sample preparation. In combination with freeze fracturing, AFA is a non-contact technology that can be used to efficiently pulverize rodent eye samples in approximately 2 minutes with minimum cross-sample contamination for multiple sample processing for metabolite analysis.
67. Comparison of human plasma [14C]determinations by low level scintillation counting and Accelerator Mass-Spectrometry (AMS)
Theodore J. Chando, Lisa J. Christopher, Samuel J. Bonacorsi, and Donglu Zhang
Bristol-Myers Squibb Research and Development, Princeton, NJ, USA, 08543
When radiolabeled compounds are dosed in human ADME studies at relatively low specific activities, the LLQ for the determination of total radioactivity in plasma by standard liquid scintillation counting (LSC) can be high compared to a specific assay for parent. A way to eliminate this difference in assay ranges is by accelerator mass-spectrometry (AMS), which has significant sensitivity advantages, but also disadvantages. Since standard LSC is limited by higher backgrounds and counting times are reduced to increase sample throughput, an alternative approach would be to push sensitivity by low-level LSC. This study evaluated the use of low-level method for counting plasma and compared results to standard LSC and AMS. Different cocktails, cocktail volumes/vials, and sample volumes were evaluated to optimize the low-level methodology. Counts were performed for 2 h with refrigerated Packard Tri-Carb Model 3100TR counters equipped with low-level capability and a low-level quench curve. The two cocktails evaluated showed differences in their ability to solubilize human plasma under refrigerated conditions. Standard curves of [14C]BMS-Y were prepared in duplicate from 2500 to 1 dpm with 0.2 and 0.4 mL human plasma and each spiked sample compared with a triplicate blank plasma background. LLQ was ~ 5 dpm/mL, regardless of the plasma volume used. In comparison, regular LSC had an LLQ ~ 185 dpm/mL when 10 min counts were performed, without refrigeration and using a counting threshold of two backgrounds to be considered statistically significant. Although the LLQ of low-level LSC did not approach the LLQ of AMS (≤ 0.1 dpm/mL), it sufficiently reduced the plasma LLQ for radioactivity to allow complete comparison to parent as determined by LC/MS in the BMS-Y clinical trial. When low level counting of BMS-X equivalents was performed on human plasma samples determined by AMS to contain 10-30 dpm/mL, the percent difference ranged from 0.3 to 11.3%. These experiments demonstrate that the two techniques can be used to achieve similar results but have very different costs and convenience.
68. Estimation of steady-state metabolite exposures in humans using a combination of automated liquid handling and LC-MS methods
Nilgün çömezoglu, Nirmala Raghavan, and Ragu Ramanathan
Biotransformation, Bristol-Myers Squibb, Princeton, NJ, USA, 08543
In order to fully understand the efficacy and toxicity profile of a new drug in early development it is important to understand exposure to both parent and metabolites. This has led the pharmaceutical industry to develop new analytical methodologies to obtain a quantitative understanding of circulating human drug metabolites. Ideally, metabolite concentration in human plasma samples could be estimated in the phase I single and multiple ascending dose studies prior to a human ADME study. Quantitative bioanalysis or radioactivity detection is the method of choice to detect and quantify the drug metabolites. When synthetic standards or radiolabeled drug at early stages of drug development are not available, determination of AUC values for metabolites becomes very challenging. What is typically done in this case is that AUC of the parent is obtained using a quantitative bioanalytical method and the AUC of the metabolite is determined using semi quantitative metabolite profiles. The objective of this study was to determine AUC of metabolites of Compound-X in plasma samples obtained from the multiple ascending dose (MAD) study in the absence of standards with the use of a pooled plasma-sample technique in combination with LC-MS. Human plasma (0.5, 1, 2, 4, 8, 24 h) from selected cohorts of an MAD study of Compound-X were pooled using an automated time-proportional pooling approach. Plasma aliquots were extracted with acetonitrile and analyzed by LC-MS. The area response ratio of metabolite to parent was calculated using full scan MS. The MS ratio was compared with the UV response to generate a correction factor assuming equivalent parent and metabolite molar extinction coefficient. A separate validated quantitative LC-MS/MS method was used for determining the AUC of parent drug. AUC contribution of the metabolite was determined using the correction factor and the AUC of the parent drug. It was shown that under steady-state conditions, three metabolites were above 10% of the parent AUC and preclinical species were exposed to major circulating human metabolites under single dose conditions.
69. Validation of an HPLC/MS/MS bioanalytical method for the quantitative analysis of lidocaine from rat and mini-pig plasma
Shane Needham, Derek Laine, Casey Johnson, and Chad Christianson
Alturas Analytics Inc, Moscow, ID, USA, 83843
Lidocaine was one of the first in class amino-amide type local anesthetics. Lidocaine is commonly used to relieve skin irritations and injected during dental and minor surgeries. As lidocaine formulations and delivery techniques are improved, the need to measure systemic levels of lidocaine in plasma is warranted. Previous HPLC or LC/MS/MS assays required run times >10 minutes and/or laborious extraction procedures for the analysis of lidocaine from biological fluids. Here we report on a simple preparation method coupled with LC/MS/MS to provide an accurate and precise assay for the determination of lidocaine from rat and mini-pig plasma. Using the latest Crystal City III Guidelines for Bioanalytical Method Validation, a sensitive HPLC/MS/MS method was developed and validated for the quantitative analysis of lidocaine from rat and mini-pig plasma. The method was validated using a simple acetonitrile precipitation for sample preparation. Across the dynamic range of the assay (10-10,000 ng/mL), the interday accuracy and precision of the method in both species was ±6% and ±10%, respectively. Lidocaine was shown to be stable for at least 20 hours in the plasma of both species on the benchtop and through at least five freeze thaw cycles. Sample extracts from rat and pig plasma were stable for >20 hours when awaiting analysis in the autosampler. No significant matrix interferences were detected during validation in either species. This method has been used for the analysis of >500 samples in support of in-life studies in rat and mini-pig.
70. An analysis of the in vitro metabolism of dextromethorphan by accurate mass spectrometry with stable-isotope labeled metabolite profiling and reaction phenotyping
Joanna E. Barbara, Mark J. Horrigan, Phyllis Yerino, Faraz Kazmi, Paul C. Toren, and Andrew Parkinson
XenoTech, LLC, Lenexa, KS, USA, 66219
Dextromethorphan is of interest as a widely available over-the-counter drug and as an FDA-approved in vitro and in vivo probe substrate for the polymorphically-expressed cytochrome P450 (CYP) enzyme CYP2D6. Mass spectrometric profiling of its major metabolites has been previously published, but structural assignment of less common metabolites has been complicated by the difficulties associated with elucidation of the fragmentation mechanism of the alkaloid ring system. The alkaloid ring system is a structural motif common to opioids (such as codeine, morphine and oxycodone) and information about its fragmentation behavior is useful for improved application of mass spectrometry to analysis of related drugs. Initially, stable-isotope labeled structural analogs for mass spectral structural elucidation were used to facilitate determination of the collisionally-induced dissociation behavior of dextromethorphan. Native and stable-isotope labeled compounds were incubated with NADPH-fortified human liver microsomes for metabolite profiling and enhanced structural elucidation by accurate mass spectrometry. In addition to the expected major metabolites (the O- and N-demethylated metabolites formed mainly by CYP2D6 and CYP3A4/CYP2B6, respectively), several unexpected hydroxylation metabolites of dextromethorphan were detected. Consequently, reaction phenotyping using recombinant CYP enzymes was employed and aligned with the accurate mass spectrometric metabolite profiling for an overview of in vitro dextromethorphan metabolism.
71. Rapid separation of isomeric acylglucuronides using fused-core silica liquid chromatography
Yongying Jiang, Michelle Chang, Tian J. Yang, and David Moore
Dept of Non-Clinical Safety, Hoffmann-La Roche Inc., Nutley, NJ, USA, 07110
Acylglucuronides (AGs) of carboxylic drugs are potential reactive metabolites and have the potential of reacting with proteins both in vitro and in vivo. It has now been generally accepted that reactive AGs are responsible for a number of adverse drug reactions of carboxylic drugs. Therefore, an in vitro assessment of AG reactivity may provide valuable information to predict the potential in vivo toxicity of carboxylic drug candidates. Currently, most in vitro assays to assess AG reactivity use LC separation and MS quantitation of the AG isomers to determine the degradation rate of the primary AG. However, LC separation of these isomers using traditional C18 column often requires a lengthy gradient up to 30 min, posing a significant obstacle for developing a higher throughput AG assay. Fused-core silica particle is a newly developed chromatographic technology that has shown substantial improvement in chromatographic peak efficiencies over fully porous particles and in reducing backpressure over sub 2 mm particles. These advantages have rendered a quick adoption of this technology into the field of bioanalysis. We have found that the LC separation of AG isomers can be significantly improved by using fused-core silica liquid chromatography and developed a rapid LC method of less than 8 min for efficient separation of isomeric AGs using a fused-core C18 column. The method was validated by the separation and quantitation of isomeric AGs of eight nonsteroidal anti-inflammatory drugs (NSAIDs) including tolmetin, zomepirac, diclofenac, suprofen, fenoprofen, ibuprofen, ketoprofen, and furosemide. The resulting rearrangement percentages and the subsequent ranking of the AGs of these NSAIDs from our analysis are consistent with those reported in the literature. The method has been successfully applied to routine AG assays of our carboxylic drug candidates. These results demonstrated that we have developed a highly efficient LC-MS method for the analysis of isomeric AGs and this method can potentially be adapted for the development of higher throughput AG assays.
72. Utilization of fast efficient tandem hybrid instrumentation for identification and comparison of metabolite generation in freshly isolated and cryopreserved human hepatocytes
Cornelia Smith1, Johnie Brown2, Jeffrey D. Miller2, James Ferguson2, Christina Nolan1, Jeanette Hill3, and James Hill4
1Life Technologies, Durham, NC, USA, 27703
2Psm, Applied Biosystems, Framingham, MA, USA, 01701
3Life Technologies, Austin, TX, USA, 78754
4Life Technologies, Austin, TX, USA, 27703
Guidelines for drug development emphasize the identification of metabolic pathways, relevant metabolites and potential for drug-drug interactions for candidates where metabolism is the primary route of elimination. In vitro tools used to assess metabolism include freshly isolated and cryopreserved hepatocytes. Cryopreserved hepatocytes have emerged as a favored model due to commercial availability and convenience for use. Although previous studies have compared CYP activities of fresh and cryopreserved hepatocytes, few have elaborated on the extent that cryopreservation plays on metabolite ID. This study examined the effects of cryopreservation by comparing metabolites generated from phase I and phase II substrate incubations performed in both fresh and cryopreserved cell suspensions processed from the same donors. Incubations proceeded for two hours to allow for optimal metabolite formation using the following compounds: phenacetin, bupropion, paclitaxel, diclofenac, S-mephenytoin, dextromethorphan, testosterone, midazolam, benzydamine and 3-methylindole. A zero timepoint was included for reference. Incubations were terminated by freeze on contact and samples stored at -70°C prior to analyses. All samples were analyzed using a sensitive fast-cycle-time hybrid tandem Qq-LIT mass spectrometer operated in positive ESI mode. Automatic Q1 selection of precursor ions for collision cell fragmentation was employed to allow for fast efficient trap scans of the MS/MS spectra as metabolites eluted. Metabolites were identified and grouped according to class and peak intensity. Statistical analysis was used (t-test) to discern differences in metabolites formed in fresh and cryopreserved hepatocytes. Metabolites were observed in both freshly isolated and cryopreserved hepatocytes (e.g. oxidations, dealkylations, glucuronide conjugations). No metabolites were identified as uniquely belonging to either the fresh or the cryopreserved group, although differences in levels of metabolites were observed. This analytical approach allowed for greater discrimination of the effects of cryopreservation on hepatic drug metabolizing enzymes and could prove particularly useful for metabolic profiling and species comparisons.
73. Software assisted chiral chromatographic method development for the quantitation of four chiral drugs in monkey plasma using LC/MS/MS
Troy Voelker1, Patrick Bennett2, Min Meng2, and Lisa Rhode1
1Analytical, Tandem Labs, Salt Lake City, UT, USA, 84124
2Tandem Labs, Salt Lake City, UT, USA, 84124
Traditional chiral chromatographic separation method development is time consuming even for an experienced chromatographer. This paper describes the application of computer software ACD Lab® to facilitate the development of chiral separation for the quantitation of armodafinil, ramelteon, eszopiclone and dexlansoprazole using LC-MS/MS technology. Assisted by ACD/Chrom Manager and LC Simulator software, the optimal chiral chromatographic development was completed within hours. The baseline chiral separation for all four drugs was achieved less than four minutes. Except dexlansoprazole which is still in the method development stage, armodafinil, ramelteon, eszopiclone were validated under GLP guidance with excellent intra- and inter-day accuracy and precision.
Methods: The LC/MS/MS system was a Sciex API4000 or 5000 under positive ionization mode using turboionspray coupled with a Shimadzu LC-10AD high pressure pump and a CTC PAL autosampler. The SRM transitions for eszopiclone and eszopiclone-D8 (ISTD), armodafinil and modafinil-d5 (ISTD), ramelteon and ramelteon-d3 (ISTD), dexlansoprazole and lansoprazole-13C6 (ISTD) were monitored. ACD/Structure Designer, ChromManager and LC Simulator software (v. 10.0) were utilized for chiral chromatography development. AGP chiral analytical columns, 100x 2 mm or 50x 2 mm, were used to achieve chiral separations. Structure Designer was utilized for the calculation of physical chemical properties, i.e., structure, pKa value and solubility information. ChromManager software was utilized for data editing and processing. LC Simulator software was utilized for LC chromatography simulation and predictions.
Results: Using the ACD Lab software approach, two initial LC/MS/MS chromatographic conditions were acquired in the laboratory using a racemic mixture of the analytes. The same aqueous mobile phase i.e., 10 mM Ammonium acetate, pH unadjusted, and three organic solvents, i.e., isopropyl alcohol, methanol and pentanol, were utilized for all applications. Two injections were performed in the laboratory on a LC/MS/MS system based on semi-random selection of the LC program and composition. Under these conditions, the enantiomeric peaks either completely coeluted or were partially resolved. The Analyst® wiff file from the two injections were then uploaded into the ACD/ChromManager software. After peak editing, i.e., peak picking and labeling, structure and LC condition assignment, these two chromatograms were imported into the ACD/LC Simulator software. The predicted separation conditions were obtained instantly by manipulating the column dimension, flow rate, mobile phase composition within the software. The computational results were then utilized as the guidance for further experimental work in the laboratory. This procedure was repeated until satisfactory chromatography was achieved within a few more injections.
Following the success of the chiral chromatography development, full GLP validations were conducted for the quantitation of armodafinil, ramelteon and eszopiclone. The method validation of dexlansoprazole is in process. The validation demonstrated excellent intra- and inter-day accuracy and precision. In addition, extraction recovery, analyte stability in solution, matrix and processed extracts were conducted and reported per FDA GLP guidelines. These assays have been used to support clinicsample studies.
74. Evaluation of the effect of first-pass metabolism on orally absorbed drugs using the Caco-2 cell line model
Sandra Gagnon1, Alan Bartlett2, and Linh Nguyen1
1Drug Metabolism and Pharmacokinetics, Charles River Preclinical Services Montreal, Senneville, QC, Canada, H9X 3R3
2Laboratory Sciences, Charles River Preclinical Services Montreal, Senneville, QC, Canada, H9X3R3
Caco-2 is a well accepted model for the prediction of drug absorption in humans, and for the assessment of P-glycoprotein-mediated drug-drug interaction. The expression of CYP3A4 and glucuronolsyltransferase (UGT) activity was also reported, but mostly required induction by vitamin D or antioxidant compounds. The inclusion of inducers in the culture medium might interfere or interact with test compounds that utilize the same metabolic and transport pathways in Caco-2 cells. In this study, we show that under regular cell culture conditions (DMEM medium supplemented with L-glutamine, FBS, non-essential aminoacids), Caco-2 cells retained the ability to express the major conjugative Phase II enzymes as well as CYP3A4 and the BCRP2 and P-glycoprotein efflux transporters. Caco-2 cells were seeded on polycarbonate membranes and cultured for up to 3 weeks. Testosterone and 7-hydroxycoumarin were used as probe substrates for directional A-to-B transport, and CYP3A4, UGT and sulfotransferase activities. Digoxin and estrone-3-sulfate were used as markers for P-glycoprotein and BCRP transporter, respectively. Samples collected from the basolateral side were analyzed using appropriate analytical methods (HPLC-UV and LC-MS). Upon differentiation on semi-permeable membranes (ie at least two weeks as suggested by TEER ~250 Ohm-cm2), P-glycoprotein and BCRP efflux transporters were expressed at significant levels as reflected by efflux ratios of ~10 and ~50, respectively. These efflux activities were inhibited by addition of specific inhibitors (verapamil and fumitremorgin C). The expression of CYP3A4 and UGT activities did not appear to be associated with cellular differentiation, as their high activities were detected in the first week post-seeding. Sulfotransferase activity however was detectable only in the second week of culture and represented ~65% of 7-hydroxycoumarin conjugation after the third week. The time-dependent activities were correlated with the data by Western blot analysis. Thus, our Caco-2 clone under controlled culture medium could be utilized to determine the extent of first-pass metabolic effect on drug candidates and thereby providing an indication of their oral bioavailability.
75. Pharmacokinetics and allometric scaling of a TNF nanoformulation
Stephan T. Stern1, Lonnie Myer2, Giulio F. Paciotti2, Lawrence Tamarkin2, and Scott E. McNeil1
1Nanotechnology Characterization Laboratory, Advanced Technology Program, SAIC-Frederick, Inc., NCI-Frederick, Frederick, MD, USA, 21702
2CytImmune Sciences, Inc.,, College Park, MD, USA, 20740
Nanotechnology is increasingly finding application in chemotherapeutic drug delivery, demonstrating enhanced efficacy and diminished toxicity. One challenge for development of nanoformulations is interspecies scaling of pharmacokinetics for estimation of clinical starting dose. The present study demonstrates use of allometric analysis to retrospectively scale the clinical pharmacokinetics of CYT-6091, a nanomedicine consisting of tumor necrosis factor-alpha (TNF) that is covalently bound to 27 nm pegylated gold colloid nanoparticles. Rabbits, rats and oncology patients were treated intravenously with the TNF nanoformulation (11-250 ug/kg TNF-alpha). Blood samples were collected at 6-7 time points over an eight hour period post injection, and analyzed for TNF concentration by an established ELISA. Pharmacokinetic parameters for each resulting blood TNF profile were determined by noncompartmental analysis. Allometric analysis was then used to scale volumes of distribution (V) and clearance (CL) between species, using the power models V=0.64*BW0.79 (R2=0.987) and CL=2.09*BW1.43 (R2=0.957), respectively. CL was found to scale similarly to other macromolecular drugs for power model exponents >1 by Mahmood’s “rule of exponents” (2009), whereby scaling of the brain weight (BrW) product, CL=0.011*BW2.18/BrW (R2=1.000), improved predictability. Brain weight product scaling of V, V=0.002*BW2.03/BrW (R2=0.999), also improved predictability. Funded by NCI Contract No. HHSN261200800001E.
76. Relationship between passive permeability, efflux, and predictability of clearance from in vitro metabolic intrinsic clearance
Liyue Huang1, Loren Berry1, Brett Janosky1, Sindhura Ganga2, April Chen1, Jonathan Roberts1, Adria E. Colletti1, and Min-Hwa Jasmine Lin1
1Dept of PKDM, Amgen Inc, Cambridge, MA, USA, 02142
2Genzyme
In vitro intrinsic metabolic clearance (CLint) is used routinely for compound selection in drug discovery; however, in vitro CLint often under-predicts in vivo clearance (CL). Forty-one proprietary compounds and 16 marketed drugs were selected to determine whether permeability and efflux status could influence the predictability of CL from in vitro CLint obtained from liver microsomal and hepatocyte incubations. For many of the proprietary compounds examined, rat CL was significantly under-predicted using the well-stirred model incorporating both fup and fuinc. Further analysis revealed that the accuracy of the prediction was differentiated by permeability and Pgp- and mBcrp-mediated efflux. For proprietary compounds with passive permeability greater than 5 × 10− 6 cm/s and efflux ratios less than 5 in both MDR1-LLC-PK1 and mBcrp-MDCK cells, CLint provided reasonable prediction. The average fold error (AFE) was 1.8 for rat liver microsomes (RLM) and 2.3 for rat hepatocytes. In contrast, CL was dramatically under-predicted for compounds with passive permeability less than 5 × 10− 6 cm/s; AFEs of 54.4 and 29.2 were observed for RLM and rat hepatocytes, respectively. In vivo CL was also under-predicted for compounds that were good efflux substrates (permeability >5 × 10− 6 cm/s). The AFEs were 7.4 and 8.1 for RLM and rat hepatocytes, respectively. A similar relationship between permeability, efflux status and the predictability of human CL was observed for 16 marketed drugs. These data demonstrate that permeability and efflux status are determinants for the predictability of CL from in vitro metabolic CLint.
77. Assessment of the contribution of transporter-mediated active uptake to the overall hepatic uptake clearance for a range of drugs
Yoshiyuki Yabe, Aleksandra Galetin, and J. Brian
Houston School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester, United Kingdom, M13 9PT
Transporter-mediated uptake process can represent the rate-limiting step in the overall hepatic clearance. In the present study, the hepatic uptake of 11 compounds with differential physicochemical properties was investigated, including five HMG-CoA reductase inhibitors (statins), saquinavir, ritonavir, telmisartan, clarithromycin, repaglinide and fexofenadine. In order to characterize the uptake kinetics and evaluate the contribution of transporter-mediated active uptake to the overall clearance, studies were performed in rat hepatocytes in suspension by oil-spin method over a 0.01-100 μM concentration range. Clearance via passive diffusion (Pdiff), maximum uptake rate (Vmax) and Michaelis constant (Km) were estimated from the initial uptake rate data using WinNonlin. In addition, relative importance of the active hepatic uptake in comparison to the passive process was estimated over the range of concentrations investigated. A wide range of Km values was obtained for the uptake of the compounds in the dataset, with 8/11 drugs showing Km<10 μM. CLactive ranged from 31.7-1500 μL/min/106 cells for pravastatin and atorvastatin, respectively. In the case of statins, CLactive was >13-fold higher than the passive component. Assessment of the contribution of the passive permeability and active uptake indicated that at low concentrations (~0.1 μM) the active process contributes >90% to the overall uptake of all five statins; similar profiles were obtained for telmisartan and fexofenadine. Even though high Pdiff values were obtained for ritonavir and repaglinide, active process contributed predominantly to the uptake at lower substrate concentrations (<1 μM). This is in contrast to saquinavir where high passive permeability limited the impact of transporter-mediated uptake over the range of concentrations. Clarithromycin was mainly taken up via active uptake process with relatively high Km value (68 μM), suggesting that the uptake is the rate-limiting step. Tissue to media unbound concentration ratio (Kp,u) was >10 for all the drugs in the dataset with the exception of saquinavir, ritonavir and repaglinide. Comprehensive analysis of the uptake data of the compounds investigated and the implications of the findings are discussed.
78. Humanized UGT1*28 Mice for Assessing UGT1A1 dependent glucuronidation and Clearance
Young-Sun Yang1, Vincent Peterkin1, Hongliang Cai1, Kathy Hotz1, Nghia Nguyen2, Deirdre La Placa2, Robert H. Tukey2, and Jeffrey C. Stevens1
1Department of Pharmacokinetics, Dynamics, and Metabolism, St Louis Laboratories, PGRD, Chesterfield, MO 63017
2Laboratory of Environmental Toxicology, Departments of Chemistry & Biochemistry and Pharmacology, University of California, San Diego, CA 92093.
Despite the fact that the UGT1A1*28 allelic variant is common in the human population and has been shown to lead to a clinically relevant phenotype in Gilberts Syndrome, there are few tools to assess and predict whether reduced UGT1A1 expression in humans will affect the overall clearance of a new chemical entity. To address this dilemma, a humanized UGT1A1*28 mouse model (hUGT1*28) was generated by introducing the human UGT1 locus expressing the UGT1A1*28 allele into a Ugt1-null background. To evaluate the utility of hUGT1*28 mice for assessing UGT1A1-related glucuronidation and clearance, three compounds were chosen to evaluate pharmacokinetic properties: SN-38 (an exclusive UGT1A1 substrate), Ezetimibe (a partial UGT1A1 substrate), and Naloxone (a UGT2B7 substrate). The well-characterized substrate of UGT1A1, SN-38, displayed the greatest difference in parent drug clearance (≥ 3-fold for each parameter) in hUGT1*28 mice following intravenous administration when compared to Phenobarbital treated hUGT1*28 mice and wild type mice. Moderate changes in pharmacokinetic parameters were observed for Ezetimibe, consistent with the drug displaying partial dependence on UGT1A1 for clearance. In contrast, the clearance of Naloxone was not altered when comparing clearance patterns between hUGT1*28 and wild type mice. The gender differences in UGT1A1 dependent glucuronidation between hUGT1*28 and wild type mice were also investigated in vivo and the preliminary data demonstrated the same rank-order of pharmacokinetic parameters. Additionally, enzyme kinetic parameters assessed for SN-38, Ezetimibe, and Naloxone using liver microsomes prepared from wild type and hUGT1*28 mice showed patterns consistent with the in vivo pharmacokinetic data. Specifically, for SN-38, Vmax decreased 5- and -10-fold in hUGT1*28 mouse liver microsomes compared to microsomes prepared from wild type mice and Phenobarbital treated hUGT1*28 mice, respectively. Overall, the hUGT1*28 mouse model can serve as a tool in pharmacokinetic modeling to investigate the effects of UGT1A1 dependent glucuronidation and clearance of clinically relevant drugs. (Supported by a grant from Pfizer and NIEHS grant ES010337).
79. The molecular rearrangement of sulfotransferase (SULT) 2A1 in the presence of 3′-phosphoadenosine-5′-phosphosulfate (PAPS)
Ian T. Cook1, Thomas S. Leyh2, and Charles N. Falany3
1Department Pharmocology and Toxicology, University of Alabama at Birmingham, Birmingham, AL, USA, 35294
2Department of Biochemistry, Albert Einstein College of Medicine, Bronx, NY
3Department Pharmacology and Toxicology, University of Alabama at Birmingham, Birmingham, AL, USA, 35294
SULT2A1 is an important Phase II conjugation enzyme responsible for the transfer of sulfonate from PAPS to an acceptor compound. The crystal structure of SULT2A1 indicates the region from N230 to D253 is highly dynamic in the absence of PAPS but stabilizes when PAPS is bound. The structural ordering upon PAPS binding results in a 50% reduction in the volume of the active site. This study modeled interactions of dehydroepiandrosterone (DHEA) and raloxifene in the active site of SULT2A1 in the presence and absence of PAPS. The models were then tested by kinetic studies of DHEA and raloxifene sulfation. Computer modeling using the Molecular Operating Environment showed that DHEA bound in a catalytically competent orientation with similar calculated affinities in both the “open” conformation without PAPS bound and the “closed” conformation with PAPS bound. DHEA as a planer hydroxysteroid, was able to bind equally well in both the open and closed conformations. Analysis of the raloxifene binding indicated a significant decrease in the apparent affinity with the closed conformation compared to the open conformation.. In vitro steady state kinetics for SULT2A1 sulfation of DHEA and raloxifene showed that DHEA sulfation involved an apparent random Bi-Bi mechanism, whereas raloxifene sulfation suggested an ordered mechanism with raloxifene binding before PAPS. Initial rate studies indicate that either DHEA or PAPS could bind independently; however, raloxifene could only bind to SULT2A1 in the open confirmation. If PAPS is bound, raloxifene cannot bind in a catalytically competent orientation to the SULT2A1•PAPS complex. In summary, the modeling and kinetic studies support the hypothesis that the structural rearrangement of SULT2A1 caused by the binding of PAPS results in an increased selectivity for planar steroids. Compounds such as raloxifene, can only be sulfated if they bind in the open active site of SULT2A1 in the absence of PAPS.
80. Improvement of in vitro-in vivo extrapolation of udp-glucuronosyltransferase catalyzed metabolic reactions using selective probe substrates and LC-MS quantification of the proteins
Kasja P. Kanebratt1, Johan Lengqvist2, Ermias Melles1, and Pawel Baranczewski1
1Clinical Pharmacology & DMPK, AstraZeneca R&D Södertälje, Södertälje, Sweden, SE-15185
2Molecular Toxicology, AstraZeneca R&D Södertälje, Södertälje, Sweden, SE-15185
The existing in vitro methods underestimate the clearance and magnitude of drug-drug interactions (DDIs) related to metabolic reactions catalyzed by the UDP-glucuronosyltransferases (UGTs), the most important family of phase II enzymes. Therefore, in order to develop a reliable in vitro approach for early estimation of contribution of UGTs to metabolism and DDIs, assays were developed using specific probe substrates for five major, hepatic UGTs. The enzyme kinetic, optimal incubation, and analytical (UPLC-MS/MS based methods) parameters for each reaction using recombinant expressed human enzymes (rh) were estimated for: UGT1A1 and estradiol (Km 5.0 μM), UGT1A4 and lamotrigine (Km 1500 μM), UGT1A6 and 1-naphthol (Km 2.5 μM), UGT1A9 and propofol (Km 36 μM), and finally UGT2B7 and zidovudine (Km 225 μM). The specific reactions were evaluated using different known inhibitors of the UGTs, namely: ketoconazole for UGT1A1 (IC50 2.4 μM), hecogenin for UGT1A4 (IC50 0.30 μM), diclofenac for UGT1A6 (IC50 205 μM), irinotecan for UGT1A9 (IC50 66 μM), and fluconazole for UGT2B7 (IC50 1624 μM). An improvement to the in vitro-in vivo extrapolation (IVIVE) can be made by correcting these results with the amount of UGT proteins in both rhUGTs and human liver microsomes (HLM). A method was developed to quantify UGT protein in these in vitro systems which involves SDS-PAGE separation of rhUGT supersomes or HLM, enzymatic in-gel digestion (trypsine) and absolute LC-MS based quantification of each (hepatic) protein isoform. Isoform-specific peptides from UGT1A1 were verified in both rhUGT supersomes and HLMs. The described combination of analysis of specific probe substrates of hepatic UGTs in rh enzymes and absolute quantification of UGT proteins will enable improved scaling factors, significantly improve IVIVE for UGTs and thus better estimations of the contribution of UGTs to the clearance and DDIs of new drug candidates.
81. Characterization of cysteinylglycine adducts of reactive intermediates of heterocyclic amine generated in vitro
Mary Grubb, Wenying Li, Yanou Yang, and Jonathan Josephs
Biotransformation, Bristol-Myers Squibb Co, Princeton, NJ, USA, 08543
Glutathione is the standard trapping agents for detection of reactive metabolic intermediates, both in vitro and in vivo. However, its usefulness as a trapping agent is limited to soft electrophiles such as Michael acceptors (quinones, quinonimines, and quinonemethides). The usual practice for detecting hard electrophiles like iminium ions is to include potassium cyanide as a trapping agent in microsomal incubations with compounds containing aliphatic cyclic amines, which are likely to form such reactive intermediates. In combination with acidic mobile phases used for LC-MS analysis of incubation mixture, potassium cyanide remaining in these samples presents a hazard to lab occupants. The method presented here provides an alternate safer way to detect reactive intermediates containing hard electrophiles. Nicotine, nefazodone and triprolidine all contain cyclic aliphatic amines, and all form cyanide adducts when incubated with liver microsomes in the presence of potassium cyanide, indicative of an iminium ion intermediate. In this study, the compounds (100 mM) were incubated with human and rat liver microsomes (2 mg/mL) in the presence of 5 mM glutathione or 5 mM cysteinylglycine for 1h at 37°C. Incubates were quenched with acetonitrile and the post-centrifugation supernatant was dried to completion. LC-MS analysis of samples was conducted using an Orbitrap high resolution mass analyser (ThermoFisher). All three drug substrates formed cysteinylglycine adducts in the presence of 5 mM cysteinylglycine, and to a lesser extent with 5 mM glutathione. The adducts feature an imine bond between an alpha carbon of the alicyclic amine and the cysteine nitrogen atom, possibly formed by attack of the cysteine nitrogen on the electrophilic iminium ion, or its aldehyde equivalent. Flutamide is metabolized to a primary alcohol, which could then be oxidized to an aldehyde. A similar incubation conducted with flutamide and either glutathione of cysteinylglycine in the presence of NADPH also yielded a cysteinylglycine adduct. Thus, the presence of cysteinylglycine adducts detected in screening incubations with glutathione may indicate the presence of hard electrophilic reactive intermediates.
82. Separation and glucuronidation of R- and S-hydroxywarfarins by human recombinant UDP-glucuronosyltransferases (UGTs)
Jeffery H. Moran1, Drew R. Jones2, Stacie Bratton3, Anna Gallus-Zawada3, Lindsay M. Pack1, Grover P. Miller4, and Anna Radominska-Pandya3
1Div. of Health, AR Public Health Laboratory, Little Rock, AR, USA, 72201
2Dept of Biochem & Molec Biol, Univ of Arkansas for Med Sci, Little Rock, AR, USA, 72205
3Dept of Biochem & Molec Biol, Univ of Arkansas for Med Sci, Little Rock, AR, USA, 72205-7199
4Dept of Biochem & Molec Biol, Univ of Arkansas for Med Sci, Little Rock, AR, 72205
Coumadin (R-, S-Warfarin [War]) is a challenging drug to accurately dose, both initially and for maintenance, because of its narrow therapeutic range and wide inter-patient variability. Another challenge in this therapy is that Coumadin is typically administered as a racemic mixture. The R and S enantiomers are oxidized by multiple hepatic CYPs at 5 different positions to form 10 different hydroxywarfarin (OHWar) metabolites, which can be conjugated further by UGTs to form 10 different glucuronides. Recently, we have shown that ~30 metabolites of War are generated in vivo and that glucuronidation of racemic OHWar is catalyzed by several UGT isoforms localized in liver and intestine. The goal of the current work is to purify the R and S enantiomers of 6-, 7-, and 8-OH-War and identify the human UGTs involved in their glucuronidation. Semi-preparative HPLC methods have been elaborated and mg quantities of each enantiomer can be efficiently purified. The kinetic and inhibition constants for human recombinant UGTs towards these separated enantiomers have been assessed using HPLC-UV-Vis analysis and product confirmations have been assessed using HPLC-MS/MS methods. The most important observation is that the separated enantiomers of 6-, 7-, and 8-OH-War are glucuronidated in a significantly different manner and compete with each other for the aglycon binding site, as shown by Km and Vmax comparisons. Specifically, extrahepatic UGT1A10 and hepatic 1A1 have significantly higher capacities for (S)-7-OHWar and (R)-7-OHWar glucuronidation, respectively. Further evaluations show that UGT1A10 has little or no activity toward (R)-7-OHWar and the presence of the R enantiomer can significantly inhibit UGT1A10 glucuronidation of (S)-7-OHWar. This is the first demonstration that the R and S enantiomers of OHWars are glucuronidated with significantly different enzymatic affinity and capacity. These results correspond to the metabolic profiles seen in urine from warfarin patients. (NIH-DK60109 to AR-P; Bioterrorism Cooperative Agreement U90/CCU616974-07 and APHL-Fellowship to JHM)
83. Kinetic and structural characterization of the threonine 261 allozyme of human cytosolic sulfotransferase 2A1
Charles N. Falany, Ian T Cook, and Josie L Falany
Pharmacology and Toxicology, University of Alabama at Birmingham, Birmingham, AL, USA, 35294
Sulfotransferase (SULT) 2A1 is involved in the sulfation of dehydroepiandrosterone (DHEA) in the liver and adrenal cortex. In African-Americans, there is a non-synonymous single nucleotide polymorphism (G781A) that occurs at a frequency of 0.133, resulting in the conversion of Ala261 to Thr (T261). Our laboratory has reported that the SULT2A1-T261 allele is associated with a change in the DHEA to DHEA-sulfate ratio in the plasma of young African American men. Previous investigators have suggested that the T261 allozyme expressed in Cos7 cells has decreased activity. The T261 mutation occurs in the putative dimerization domain and has been reported to interfere with dimerization. Our laboratory has investigated the effects of the T261 mutation on DHEA sulfation activity, thermostability and dimerization using both the pure enzyme and liver cytosol. Wildtype (WT) SULT2A1 and the T261 variant cDNAs were subcloned into the pMALc2X vector, expressed in E. coli with a maltose binding protein (MBP) tag and purified by amylose affinity chromatography. The MBP tag was cleaved with Factor Xa and the proteins were chromatographically purified. Sulfation assays with DHEA and 3’-phosphoadenosine-5’-phosphosulfate (PAPS) were performed to evaluate kinetic parameters as well as thermostability. No significant differences were detected in the Kms for DHEA and PAPS or in thermostability between WT SULT2A1 and the T261 allozyme. Sephadex G-100 chromatography performed with both purified WT and T261 mutant SULT2A1 resulted in similar elution profiles, consistent with their elution as dimers. To further confirm these results, liver homogenate prepared from an African American man heterozygous for the T261 allele was resolved by Sephadex G-100 chromatography. DHEA sulfation activity eluted as a dimer. To evaluate the effects of the T261 mutation on SULT2A1 expression, cytosol was prepared from 30 African-American liver specimens, 4 of which possessed one T261 allele. Immunoblot analysis revealed no difference in expression levels between WT/WT and WT/T261 livers. In summary, no differences were observed in activity or expression of WT and T261 SULT2A1 suggesting that the cause for the alteration in DHEA-sulfate formation in African Americans is associated with SULT2A1 activity in the adrenal cortex.
84. Alternative spliced forms of the major phase II conjugating UGT1A gene regulate glucuronidation in native human cells
Judith Bellemare1, Mélanie Rouleau1, Bernard Têtu2, and Chantal Guillemette1
1Pharmacogenomics Laboratory, CHUQ Research Center and Faculty of Pharmacy, Laval University, Quebec, QC, Canada, G1V 4G2
2Department of Pathology, CHUQ Research Center and Faculty of Medicine, Laval University, Quebec, QC, Canada, G1V 4G2
The UDP-glucuronosyltransferase UGT1A gene is a major biotransformation gene involved in the metabolism of a vast array of molecules in the liver and gastrointestinal tract. Recently, we uncovered a new series of alternative splice variant isoforms referred to as isoforms 2 or UGT1A_i2 that use an alternative exon 5 (5b). The function of such mRNAs and the corresponding 45kDa proteins still remains unclear. While devoid of glucuronosyltransferase activity, UGT1A_i2 are widely co-expressed with the enzymatically active and classical UGT1A isoforms (called UGT1A_i1). Here, using a specific antibody directed against exon 5b, we observed abundant signal in human colon tissue samples, predominantly along intestinal crypts. In human cells, UGT1A_i2 proteins were also shown to be expressed in similar sub-cellular compartments as UGT1A_i1 enzymes. Cellular properties of i2 alternative spliced forms were then studied using synthetic short-interfering RNA (siRNA) targeted at exon 5b. Human LOVO and HT-115 colon cell lines demonstrated significant levels of exon 5a and exon 5b mRNAs, and derived microsomal preparations displayed significant enzymatic activities for UGT1A substrates, namely bilirubin and others. In both these cellular models, we observed that siRNA-mediated knockdown of endogenous i2 upregulates cellular glucuronidation activities by 120-170% (p<0.01) for all substrates tested.
Conclusion: Functional data support a dominant-negative function for endogenous exon-5b splice forms of UGT1A, hence potentially affecting in vivo glucuronidation capacity. This new regulatory strategy may ensure an additional mean to modulate cellular response to endo/xeno stimulus.
85. Correlation of bilirubin glucuronidation and estradiol-3-glucuronidation by UGT1A1: studies with model inhibitors
Jin Zhou1, Timothy S. Tracy2, and Rory P. Remmel1
1Medicinal Chemistry, University of Minnesota, Minneapolis, MN, USA, 55455
2Experimental and Clinical Pharmacology, University of Minnesota, Minneapolis, MN, USA, 55455
Bilirubin (BILI) is an end product of heme catabolism, requiring conjugation with glucuronic acid for elimination. The primary enzyme catalyzing BILI glucuronidation is UGT1A1 and insufficient conjugation caused by inhibition of UGT1A1 activity can result in clinical consequences such as jaundice and kernicterus. Thus, it is prudent to screen drug candidates for their ability to inhibit BILI glucuronidation. However, the instability of BILI poses substantial technical challenges assessing effects of drugs on BILI, in vitro. Estradiol (E2)-3-glucuronidation is often used as a probe reaction for UGT1A1. However, E2-3-glucuronidation exhibits a sigmoidal kinetic profile, suggestive of multiple E2 binding sites within the enzyme active site that may confound direct correlations with effects of drugs on BILI. The current work evaluated whether E2-3-glucuronidation can be used as a surrogate to study inhibition on BILI glucuronidation by examining a battery of substrates/inhibitors of UGT1A1 for their effects on both processes. The glucuronidation kinetics of BILI and E2 were carefully characterized with HEK293-expressed UGT1A1. Incubation conditions for BILI kinetics were 0.05 mg/ml of protein for 5 mins and 0.25 mg/ml of protein for 30 mins for E2. Consistent with previous reports, E2-3-glucuronidation displays sigmoidal kinetics (Vmax=115.0 ± 1.72 pmol/min/mg of protein, S50=15.2 ± 0.41μM and n=1.9 + 0.07), whereas BILI glucuronidation exhibits typical hyperbolic kinetics (Vmax=165.0 ± 4.3 pmol/min/mg of protein, Km=0.2 ± 0.018μM). The interactions between E2 and BILI were also studied, with E2 and BILI inhibiting the glucuronidation of each other. Effects of 6 UGT1A1 substrates (ethinylestradiol, 4-hydroxyphenyl hydantoin, 4-methylumbelliferone, 1-naphthol, baicalein, raltegravir) on E2-3-glucuronidation and BILI glucuronidation were studied at substrate concentration equal to S50 or Km. Ethinylestradiol at low concentrations (≤10μM) resulted in mild stimulation of E2-3 glucuronidation, but no such effect on BILI glucuronidation was observed. The IC50 of ethinylestradiol on BILI glucuronidation was 26.6μM and 44.0μM for E2-3-glucuronidation. However, the IC50s of the other 5 compounds on E2-3-glucuronidation and BILI glucuronidation were within 15% of each other, suggesting that E2 may be a good surrogate for studying inhibition of BILI glucuronidation. Additional substrates and inhibitors are being evaluated (Data will be presented in the poster). (Supported by Bristol-Myers Squibb and NIH: # GM063215).
86. Structure-activity study of CYP2B induction in rat liver
Lyudmila F. Gulyaeva1, Vladimir Pustylnyak1, Andrey Yarushkin1, and Nikolay Slynko2
1Molecular carcinogenesis, Institute of MolBiol & Bioph SB RAMS, Novosibirsk, Russia, 630117
2Institute of Cytology and Genetics, Novosibirsk, Russia, 630090
Many xenobiotics are capable of cytochrome P450 induction. In many cases this process occurs via the interaction with specific nuclear receptors (CAR, AhR, PXR). There is a large group of structurally diverse chemicals (PB-like inducers) which activate gene transcription of CYP2B and CYP3A in species- and tissue-specific manner. We have studied induction of these P450s by a specific inducer 2,4,6-triphenyldioxane-1,3 (TPD) and have shown that this compound induces CYP2B in rat liver via a transcriptional mechanism, but not in mouse liver. Moreover, TPD in contrast to phenobarbital specifically activates the Ca++-calmodulin-dependent signal transduction pathway followed by the activation of the CAR receptor. We carry out the synthesis of several analogs of TPD using a reaction with 1,3-diphenylpropane-1,3-diol and corresponding 4-substituted benzaldehydes to verify a hypothesis that minor changes in the inducer structure can cause major changes in induction abilities. The results of this study are presented in the table.
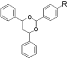
One can see that treatment of rats with TPD and its analog #2 causes the increase of the total P450 content as well as CYP2B protein and mRNA content with enhanced enzyme activity. Another structural analog of # 3 doesn’t produce any induction effect. Thus, minor structural changes in TPD cause dramatical changes in its ability to induce CYP2B. This phenomenon can be explained by various interactions with the CAR receptor or related proteins. Results of this study can shed light on the species-specific effects of PB-like inducers.
This work was supported by the RFBR grant 09-04-00801-à, Russia.
87. Cytochrome P450s catalyze reduction of lipid aldehydes metabolites
Russell A. Prough1, Immaculate Amunom1, Viola Tamasi2, Daniel J. Conklin3, Aruni Bhatnagar3, and Sanjay Srivastava3
1Biochemistry & Molecular Biology, U. Louisville School of Medicine, Louisville, KY, USA, 40292
2Department of Genetics, Cell- and Immunobiology, Semmelweis University, Faculty of Medicine, Budapes, Hungary, H-1445
3Medicine/Cardiology, U. Louisville School of Medicine, Louisville, KY, USA, 40292
Recent studies on the metabolism of lipid-derived aldehydes provided evidence that the cytochromes P450 metabolize lipid peroxidation products, such as the α,β-unsaturated aldehydes, 4-hydroxynonenal (HNE) and related model aldehyde substrates, such as 9-anthracene carboxaldehyde (9-ACA). We observed that reduced products, 1,4-dihydroxynonenol (DHN) and 9-anthracene methanol (9A-MeOH) were produced by P4503A4 and 2B6 with these two aldehyde substrates, respectively; other P450s tested catalyzed only the oxidation reaction. Both monooxygenase and reduction reactions were inhibited by specific inhibitors for the various P450s studied. The products formed under all conditions were confirmed by gas chromatography-mass spectrometry (HNE) or fluorescence spectroscopy (9-ACA). To further characterize the reduction reactions, we performed experiments using argon to establish anaerobic conditions and using carbon monoxide as an inhibitor of P450 function. A glucose-glucose oxidase enzyme system was included in the reaction mixtures to remove trace amounts of oxygen in the reaction mixtures. The monooxygenase reactions forming carboxylic acids of HNE and 9-ACA were potently inhibited by both anaerobiosis (in argon atmosphere) or inclusion of CO atmosphere as anticipated for a P450 catalyzed reaction. However, anaerobiosis had no effect upon the production of the reduced metabolites, DHN and 9-A-MeOH by P4503A4. Surprisingly, metabolism via the reduction pathway was not inhibited in the presence of CO. To assess the possible in vivo significance of these reactions, we performed experiments in primary rat hepatocytes to show that DHN and 4-hydroxynonenoic acid metabolites of HNE (50 μM) are formed during 1 hour of metabolism. We demonstrated that inhibitors of CYP3A and 2B, such as miconazole, inhibited DHN and HNA formation in hepatocytes, implying that these enzymes apparently catalyze both the monooxygenase and reduction reactions in primary hepatocytes. These studies support the work of Walter Levine who demonstrated that these same P450s catalyze oxygen- and CO-insensitive azo reduction reactions in rat liver microsomal fractions. These P450s may limit HNE action due to metabolism in liver and other tissues. Supported in part by USPHS grant 1ES11860.
88. Deoxyschizandrin clearance and its metabolism to schizandrin is mediated by CYP3A4: A new high affinity and turnover enzyme-specific probe
Yun-Feng Cao1, Yan-Yan Zhang2, Guang-Bo Ge2, Jun Yin1, and Ling Yang2
1School of Traditional Chinese Medicine, Shenyang Pharmaceutical University, Shenyang, China, 110016
2Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China, 116023
Deoxyschizandrin (DS), one of the major pharmacological effective lingans isolated from Schisandra fruits, has been shown to provide hepatoprotection, anti oxidation and antitumor activities. The present study aims to investigate the metabolism of DS by cytochrome P450s (CYPs) in human liver microsomes (HLMs). After DS was incubated with HLMs and NADPH-generating system, only one mono-hydroxylated metabolite was detected and was identified as schizandrin by comparing the tandem mass spectra and the chromatographic retention time with that of the standard compound. A combination of correlation analysis, chemical inhibition studies, assays with recombinant CYPs and enzyme kinetics indicated that CYP3A4 was the specific hepatic isoform that cleared DS. The Km and Vmax values for DS hydroxylation were 1.4μM and 16144 pmol/min/pmol for recombinant CYP3A4, and 1.5μM and 39574 pmol/min/mg for HLMs, respectively. Since the in vitro metabolism assay described herein requires a short incubation period (10 min), a relatively low protein concentration (0.05 mg/ml), and low-cost probe and metabolite standards, DS hydroxylation may serve as a alternative high affinity and turnover specific marker reaction for assessing CYP3A4 activity.
89. Substrate-Dependent functional alterations of Seven CYP2C9 variants found in japanese subjects
Keiko Maekawa1, Noriko Harakawa1, Emiko Sugiyama1, Masahiro Tohkin1, Su-Ryang Kim1, Nahoko Kaniwa1, Noriko Katori1, Ryuichi Hasegawa1, Mikihiko Naito1, Haruhiro Okuda1, Kazuki Yasuda2, Kei Kamide3, Toshiyuki Miyata4, Yoshiro Saito1, and Jun-ichi Sawada1
1National Institute of Health Sciences, Tokyo, Japan, 158-8501
2International Medical Center of Japan, Tokyo, Japan, 162-8655
3Osaka University Graduate School of Medicine, Osaka, Japan
4National Cardiovascular Center, Osaka, Japan
Background and purpose: Cytochrome P450 2C9 (CYP2C9) is a polymorphic enzyme responsible for the oxidative metabolism of up to 15% of the drugs that undergo phase I metabolism. Several genetic polymorphisms in CYP2C9 such as *3 (Ile359Leu) are known to affect the metabolism of clinical drugs in a substrate-dependent manner. We have already identified 7 known or novel alleles, *3, *13 (Leu90Pro), *26 (Thr130Arg), *28 (Gln214Leu), *30 (Ala477Thr), *33 (Arg132Gln) and *34 (Arg335Gln), in Japanese populations. This study was aimed to characterize their functional alterations in vitro.
Methods: The wild-type and variant proteins were expressed using baculovirus-insect cells systems, and their protein expression levels were assessed by CO-difference spectra and Western blotting. Catalytic activities of the recombinant microsomes were compared between the wild-type and variants using diclofenac, losartan and glimepiride as substrates.
Results: CYP2C9 holo-protein expression levels were similar among the wild-type (CYP2C9.1) and 6 variants except for CYP2C9.13. A large part of CYP2C9.13 was present in the inactive P420. As compared with CYP2C9.1, all variants except for CYP2C9.34 exhibited substrate-dependent changes in Km, Vmax and intrinsic clearance (Vmax/Km). The Km values were significantly increased in 5 variants (CYP2C9.3, CYP2C9.13, CYP2C9.26, CYP2C9.28 and CYP2C9.30) for diclofenac 4’-hydroxylation and in 2 variants (CYP2C9.13 and CYP2C9.28) for losartan oxidation and in one variant (CYP2C9.13) for glimepiride hydroxylation. For diclofenac 4’-hydroxylation, only 2 variants (CY2C9.13 and CYP2C9.26) exhibited significantly lower Vmax values than CYP2C9.1, whereas, for both losartan oxidation and glimepiride hydroxylation, the Vmax values of all 7 variants were significantly decreased. The intrinsic clearance for losartan oxidation was markedly decreased (> 77%) in 6 variations except for CYP2C9.34. On the other hand, reductions in the intrinsic clearance of glimepiride hydroxylation were >80% in CYP2C9.3, CYP2C9.13, CYP2C9.26 and CYP2C9.33 and 56-75% in CYP2C9.28 and CYP2C9.30.
Conclusion: Careful administrations of losartan and glimepiride would be needed for the patients bearing these variant alleles.
References
- Maekawa, K., Fukushima-Uesaka, H. et al. (2006) Four novel defective alleles and comprehensive haplotype analysis of CYP2C9 in Japanese. Pharmacogenet Genomics 16: 497–514.
- Yin T, Maekawa K. et al. (2008) Genetic variations of CYP2C9 in 724 Japanese individuals and their impact on the antihypertensive effects of losartan. Hypertens Res 31: 1549–1557.
90. Unusual interaction between CYP3A4 and BI-J: Is BI-J a new substrate for the third binding site of CYP3A4?
Arti Mathur, Donald Tweedie, and Yongmei Li
Drug Metabolism & Pharmacokinetics, Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT, USA, 06877-0368
CYP3A4 has been reported to have multiple binding sites (Houston, 2002). Reaction phenotyping studies were performed to determine the human isoform(s) of cytochrome P450 (CYP) responsible for metabolism of BI-J. In the presence of isoform selective chemical inhibitors, the metabolism of BI-J in human liver microsomes (HLM) was shown to be mediated by CYP3A4 (ketoconazole) and CYP2D6 (quinidine). Using recombinant CYP (rCYP) isoforms, CYP3A4 was the enzyme primarily responsible for almost all of the metabolism of BI-J. In incubations with recombinant human CYP3A4, it was also observed that the metabolism of BI-J was inhibited by quinidine. Additional studies confirmed that quinidine did not inhibit the metabolism of testosterone or midazolam by CYP3A4 in HLM. Quinidine has been reported to be an inhibitor of the third binding site of CYP3A4 (Galetin et al., 2002). Therefore, it is proposed that BI-J is a substrate for the third binding site of CYP3A4. Since there are only a few compounds identified to date as substrates of the third binding site of CYP3A4 (e.g. nifedipine, felodipine), the consequences of this finding for drug interactions may be limited. However, these studies illustrate the need to be diligent in conducting reaction phenotyping studies in order to derive the correct conclusions.
91. Characterization of heterologously expressed dog CYP1A2, CYP2A13, CYP2A25, CYP2B11, CYP2C21, CYP2C41, CYP2D15, CYP3A12 and CYP3A26 enzymes
Diansong Zhou, Alban Linnenbach, Rick Luzietti, Catherine Booth-Genthe, and Scott W. Grimm
Clinical Pharmacology & DMPK, AstraZeneca, Wilmington, DE, USA, 19850
The dog is commonly used during preclinical drug development for metabolism, pharmacokinetic, and toxicological testing. However, knowledge of drug metabolizing capability and metabolic specificity of dog CYPs is limited. In this study, nine dog CYPs were co-expressed with dog cytochrome P450 reductase in baculovirus-Sf9 insect cells and enzyme activities towards commonly used human CYP probe compounds were evaluated. Both CYP1A2 and CYP2A13 exhibited phenacetin deethylase activity. CYP1A2 had low affinity (Km, 12.3 μM), high capacity (Vmax, 13.0 pmol/min/pmol) while CYP2A13 showed high affinity (Km, 3.9 μM), low capacity (Vmax, 3.6 pmol/min/pmol). CYP2A13 also catalyzed 7-ethoxycoumarin deethylation more efficiently than CYP2A25 (>200 fold). CYP2B11 catalyzed both R-mephenytoin and bupropion hydroxylation, with Km of 33 and 141 μM respectively. CYP2C21 and CYP2C41 exhibited similar Km values for diclofenac hydroxylase activity, while CYP2C21 demonstrated ~50 fold higher Vmax than CYP2C41. CYP2D15 catalyzed dextromethorphan O-demethylation efficiently with Km of 9 μM. Both CYP3A12 and CYP3A26 hydroxylated testosterone at multiple regiospecific sites. The current study is the first report of dog CYP1A2, CYP2A13 and CYP2A25 expression and characterization using a cDNA-directed expression system. Availability of a battery of dog CYPs will certainly improve our understanding of interspecies differences in P450-mediated drug metabolism.
92. Determination of inter-system extrapolation factors (ISEFs) for scaling recombinant cytochrome P450 data in early reaction phenotyping
Yuan Chen, Liling Liu, Khanh Nugyen, Mario Monshouwer, and Adrian Fretland
Dmpk, Roche Palo Alto LLC, Palo Alto, CA, USA, 94304
Identification of drug metabolizing enzymes is critical in assessing potential drug interactions and human pharmacokinetic variation. Quantitative in vitro phenotyping data can be obtained through studies using chemical inhibitors in human liver microsomes (HLM) or expressed cytochrome P450 (CYP) isoforms. Recombinant human CYPs (rhCYPs) have increasingly been used due to their availability and simplicity; however, translation of this type of data directly to a contribution to metabolism in vivo can be problematic. The ISEF approach was established by incorporating the factors of intrinsic activity, accessory protein expression, and abundance of CYP isoforms into the extrapolation of data from rhCYP system to HLM. In the present study, in-house ISEFs were established for CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 using 11 probe substrates. The ISEFs were determined based on intrinsic clearance (CLint) (CLISEF) and/or metabolite formation enzyme kinetics (VISEF) in rhCYP and HLM. Multiple substrate for CYP2C9, CYP2D6, and CYP3A4 were used because of the relative importance of these isoforms. The data from multiple substrate was in a good agreement. The ISEFs determined using enzyme kinetics of metabolite formation (CYP1A2, 2C9, 2D6, 3A4) were within 2-fold of that determined based on CLint. The validation of ISEF was conducted using ten marketed drugs by comparing the extrapolated data with published data. The major isoforms responsible for the metabolism were identified, and the contribution of the predominant CYPs was similar to that of previously reported data. A further evaluation was conducted using a retrospective analysis of proprietary internal compounds. Phenotyping data extrapolated using the rhCYP-ISEF method were very comparable to that obtained using HLM-based inhibition approach, including identification of the major contributing CYPs, their contribution to total CLint, and scaling rhCYP CLint to a HLM CLint. The ISEF values established in our laboratory provide a convenient early reaction phenotyping method even when complete metabolite information is not available. It is especially useful in situations where the HLM-based inhibition approach is limited due to low turnover compounds.
93. Constitutive androstane receptor (CAR) simultaneously transactivates human CYP1A1 and CYP1A2 genes
Yasushi Yamazoe, and Koichi Yoshinari
Graduate School of Pharmaceutical Sciences, Tohoku University, Aoba-ku, Sendai 980-8578, Japan
Phenobarbital has long been believed to increase CYP1A1 and CYP1A2 levels through a pathway independent of aryl hydrocarbon receptor. In this study, we have investigated the role of constitutive androstane receptor (CAR) in the transactivation of human CYP1A1 and CYP1A2. Because these genes share the 5’-flanking region, reporter assays were performed with dual-reporter constructs, containing the whole or partially deleted promoters between two different reporter genes. The results obtained in HepG2 cells showed that CAR enhanced the transcription of both CYP1A1 and CYP1A2 through the regions from -461 to -554 and from -18089 to -21975 of CYP1A1. With further reporter assays using deleted and mutated constructs and gel shift assays, an ER8-type motif, located at around -520 of CYP1A1, was identified as an CAR-responsive element and a binding motif of CAR/retinoid X receptor a heterodimer. Chromatin immunoprecipitation assays in HepG2 cells transfected with CAR expression plasmid supported the binding of the heterodimer to ER8 in a chromatin context. These results suggest that phenobarbital-type xenobiotics increase CYP1A1 and CYP1A2 levels in human livers through CAR.
94. Generation of the human metabolites of simvastatin and lovastain by bacterial cytochrome P450 BM3 enzymes
Ji-Yeon Kang1, Keon-Hee Kim1, Dong-Hyun Kim1, Heung-Chae Jung2, Jae-Gu Pan2, Taeho Ahn3, and Chul-Ho Yun1
1School of Biological Sciences and Technology and Hormone Research Center, Chonnam National University, Gwang-Ju, South Korea
2Systems Microbiology Research Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon, South Korea
3Department of Biochemistry, College of Veterinary Medicine, Chonnam National University, Gwang-Ju, South Korea
Recently, it was found that the wild-type and mutant forms of cytochrome P450 BM3 (CYP102A1) from Bacillus megaterium oxidize various xenobiotic substrates of human P450 enzymes. Therefore, it was suggested that CYP102A1 can be used to produce large quantities of the metabolites of human P450-catalyzed reactions. Simvastatin and lovastatin, hyperlipidemia or hypercholesterolemia drugs, are oxidized by human P450 3A4 to produce three major metabolites (6b’-OH, 3”-OH, and exomethylene products). In this report, we show that the oxidation of simvastatin and lovastatin was catalyzed by wild-type CYP102A1 and a set of its mutants. One major hydroxylated product (6b’-OH product) and one minor product (exomethylene product) were produced. However, formation of 3”-OH product was not observed. Formation of the metabolites were confirmed by HPLC and GC-MS by comparing the metabolites which were produced by human P450 3A4. These results demonstrate that CYP102A1 mutants can be used to produce human metabolites of simvastatin and lovastatin. Keywords: Simvastatin, Lovastatin, CYP102A1, P450 3A4.
95. Effects of organic solvents on cytochrome P450 probe reactions: Filling the Gap with S-Warfarin and midazolam hydroxylation
Elizabeth A. Connolly1, Larry C. Wienkers2, and Mary F. Paine1
1UNC Eshelman School of Pharmacy, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA, 27599
2Pharmacokinetics and Drug Metabolism, Amgen, Inc., Seattle, WA, USA, 98119
Background: Organic solvents, often required to solubilize xenobiotics, are known to modulate cytochrome P450 (CYP)-mediated reactions, which can confound the interpretation of results from in vitro CYP phenotyping studies. S-warfarin 7-hydroxylation and midazolam 1’-hydroxylation are among the preferred probe substrate reactions for CYP2C9 and CYP3A, respectively. A review by FDA scientists (Yuan et al., Drug Metab Dispos 30:1311-9, 2002) highlighted a lack of information, which remains, regarding solvent effects on these reactions. To rectify this knowledge gap, the effects of six commonly used organic solvents (ACN, MeOH, EtOH, IPA, acetone, DMSO) were evaluated on each hydroxylation reaction in human liver or intestinal microsomes (HLM, HIM). S-warfarin 7-hydroxylation was evaluated in HLM, as CYP2C9 is concentrated in the liver. Midazolam 1’-hydroxylation was evaluated in HIM; although CYP3A is concentrated in both the intestine and liver, solvent effects using HLM have been reported previously.
Methods: Reaction mixtures (n=3) consisting of microsomes (0.1 mg/ml), substrate (4 μM), solvent (0.1%, 1%, 2%), and phosphate buffer (0.1 M, pH 7.4) were initiated with NADPH (1 mM). After 30 (HLM) or 4 (HIM) minutes at 37°C, reactions were quenched with cold MeOH or ACN, respectively. 7-Hydroxywarfarin and 1’-hydroxymidazolam were quantified by HPLC/MS-MS.
Results: Relative to water, from 0.1-2% solvent, the rank order extent of inhibition of S-warfarin 7-hydroxylation was ACN (9-46%) < MeOH (15-76%) ≈ acetone (16-62%) < DMSO (42-90%) ≈ EtOH (46-96%) ≈ IPA (58-98%). The rank order extent of inhibition of midazolam 1’-hydroxylation was ACN (<5-17%) < MeOH (5-22%) ≈ EtOH (<5-25%) ≈ acetone (<5-27%) < IPA (<5-51%) < DMSO (23-73%).
Conclusions: ACN is recommended as the primary organic solvent for the S-warfarin 7-hydroxylation reaction, with MeOH and acetone (0.1%) as secondary solvents. ACN also is recommended as the primary solvent for the midazolam 1′-hydroxylation reaction, with MeOH, EtOH, and acetone (≤2%) as secondary solvents. This information should facilitate the optimization of experimental conditions involving these two preferred CYP probe substrate reactions.
Supported by NIH GM077482
96. Use of high resolution mass spectrometer analysis in early drug discovery cytochrome p450 inhibition/metabolism screens
Peter Germano, Derek Wachtel, Wilmin Bartolini, and Robert Busby
Analytical Pharmacology/DMPK, Ironwood Pharmaceuticals, Cambridge, MA, USA, 02141
Screening new molecular entities (NMEs) for cytochrome P450 inhibition and substrate potential early in drug discovery can lower the risk profile as potential drug candidates move into lead optimization and preclinical development. Most P450 screening assays can be automated to work in a high throughput manner using HPLC-mass spectrometry (LC/MS) to detect the P450 products. In some cases, NMEs with isobaric masses to the monitored assay products will be screened. When this occurs, a new P450 assay will need to be developed. High resolution mass spectrometry can distinguish compounds with the same nominal mass but different exact mass. This technique can provide more specificity than typical quantitative methods that use triple quadrupole mass spectrometers. While triple quadrupole mass spectrometers have high specificity using MRM methods, they can be subject to increased noise from isobaric compounds and endogenous interferences. The approach of using high resolution mass spectra to quantitate also allows for a single LC/MS method to be used for all compounds in a screen. By resolving the mass differences, the interferences can be eliminated and a single, high-throughput LC/MS method can be used.
An automated cassette assay was developed to simultaneously screen NMEs as potential substrates and inhibitors of the cytochrome P450 enzymes in human liver microsomes. Known P450 metabolites of probe substrates, 1′-Hydroxymidazolam (CYP3A4), 1′-Hydroxybufuranol (2D6), 4′-Hydroxydiclofenac (2C9), Resorufin (1A2), 4′-Hydroxymephenytoin (2C19), and 6α-Hydroxypaclitaxel (2C8), were used to measure P450 activity. This high resolution LC/MS procedure allows for the elimination of the potential mass interferences between NMEs and monitored metabolites. As an added benefit, the data from this assay can be further mined and used to identify novel P450 metabolites as an early read on in vitro metabolism for NMEs of interest.
97. CYP2D6 and CYP3A4 inhibitory potential of marketed CNS drugs from LC-MS/MS and fluorescent based assays
Vishwottam Kandikere, Raghavchaudary Palacharla, Ranjithkumar Ponnamaneni, Arunkumar Manoharan, Gopinadh Bhyrapuneni, Koteshwara Mudigonda, and Ramakrishna Nirogi
Discovery Research, Suven Life Sciences Ltd, Hyderabad, India
Drug-drug interactions are a potential cause of severe side effects and can result in early termination of development or restricted use of drugs and there is a need to evaluate the likelihood for such interactions as early as possible in the process of drug development. CYP enzymes are involved in many of the drug-drug interactions and the effect of new chemical entities on CYP enzymes is of major concern (Wilkinson, 2001). Inhibitory potential of NCE’s is most often assessed by human liver microsomes using LC-MS/MS analysis or recombinant CYP’s using fluorescent probe substrates. The current study was initiated mainly to evaluate the CYP2D6 and CYP3A4 inhibitory potential of some of the marketed central nervous system drugs and also to compare the inhibitory potential between the two assays mostly used for evaluation of the CYP inhibition. A panel of xenobiotics covering a wide variety of structural classes has been examined for CYP2D6 and CYP3A4 inhibitory potential. Forty-six drugs including Cholinesterase Inhibitors, Serotonin Reuptake Inhibitors, Benzodiazepines, Anticonvulsants, Antidepressants, Anti Parkinsonism agents, Antipsychotics and Anti migraines were studied. Drugs were screened for their inhibition in human liver microsomes and analysis was performed by LC-MS/MS (Ming et al., 2007). The inhibitory potential of drugs in rCYP’s was determined by micro titer plate assay using fluorescent substrate probes (Crespi et al., 1997). Quinidine and ketoconazole were used as positive controls for CYP2D6 and CYP3A4 respectively. IC50 values were determined by plotting inhibitor concentration versus percentage of remaining activity. The drugs were risk assessed based upon their IC50 values (< 1 μM – High risk; 1- 10 medium risk; and > 10 μM low risk). The results of inhibitory potential of the marketed CNS drugs and the difference in correlations between the LC-MS/MS and fluorogenic assays will be discussed.
References
- Wilkinson G. “Pharmacokinetics” In Goodman & Gilman’s The pharmacological basis of therapeutics 10 th edition, PP 3-29, 2001.
- Ming Yao, Mingshe Zhua, Michael W. Sinz, Hongjian Zhang, W. Griffith Humphreys, A. David Rodrigues, Renke Dai. Development and full validation of six inhibition assays for five major cytochrome P450 enzymes in human liver microsomes using an automated 96-well microplate incubation format and LC-MS/MS analysis. Journal of Pharmaceutical and Biomedical Analysis 44, 211-223:2007.
- Charles L. Crespi, Vaughn P. Miller, and Bruce W. Penman Microtiter plate assays for inhibition of human, drug metabolizing cytochrome P450, Analytical biochemistry 248, 188-190:1997.
98. Effects of 17ß-estradiol (E2) on expression of CYP enzymes in human hepatocytes
Kwi Hye Koh, and Hyunyoung Jeong
Department of Pharmacy Practice, College of Pharmacy, University of Illinois at Chicago, Chicago, IL, USA, 60612
Estrogen, a major female hormone, is known to influence various hepatic functions. One of major hepatic functions is drug metabolism mediated by Cytochrome P450 (CYP) enzymes. To date, the effects of estrogen on hepatic drug metabolism remain unknown. The objectives of this study are (1) to profile expression of genes regulated by estrogen and (2) to characterize effects of estrogen on CYP expression in human liver tissues. We treated 3D-cultured human hepatocyte with vehicle or E2 (100 nM) and extracted RNA for microarray (Affymetrix human Genome U133 plus 2.0 GeneChip). The results showed that E2 increased expression of drug-metabolizing enzymes (CYP2A6, CYP2A7, CYP2B6 and SULT2A1) and nuclear receptors (ESR1, GPRC5B, NR0B2 and GHR) in addition to many other hepatic genes involved in cell death, cellular movement, cell growth and differentiation, and cell cycle. To validate the results of microarray and to investigate effects of E2 on expression of major CYPs in liver, we treated human hepatocytes with vehicle, E2 (100 nM), or positive control drugs (CITCO and rifampin) and determined expression levels of major CYP by quantitative real-time PCR. E2 markedly increased mRNA expression levels of CYP2A6 (15.1-fold), CYP2B6 (10.3-fold) and CYP3A4 (3.1-fold) as compared to vehicle, while decreasing mRNA expression of CYP1A2 (1.5-fold). The expression levels of other CYPs (CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A5) were not affected by E2. The induction of CYP2A6, CYP2B6 and CYP3A4 was E2 concentration-dependent. Expression levels of TFF1 and TFF3, known estrogen-responsive genes, were upregulated by E2 as expected. CITCO (a known CYP2B6 inducer) increased CYP2B6 mRNA levels by 22-fold whereas rifampin (a known CYP3A4 inducer) increased CYP2A6 and CYP3A4 mRNA levels by 15.3- and 60-fold, respectively. In conclusion, this study demonstrates that E2 affects CYP expression in a CYP-pathway dependent manner in human hepatocytes; E2 induced mRNA expression of CYP2A6, CYP2B6 and CYP3A4, while repressing that of CYP1A2.
99. Automation of a multiplexed cell-based assay to measure inhibition and induction of the cytochrome P450 isoform 3A4 by small molecule compounds
Brad Larson1, Peter Banks1, Mary Sobol2, and James J. Cali2
1Applications, BioTek Instruments, Winooski, VT, USA, 05404
2Dept of R & D, Promega Corp, Madison, WI, USA, 53711
The role that CYP3A4 plays in the metabolism of drugs within the body has been well documented. Therefore, it is essential to understand how this P450 isoform is affected by these xenobiotics, to avoid any possible drug-drug interactions. Compounds have been shown to inhibit the activity of CYP3A4, while others have demonstrated the ability to induce the expression of this enzyme, and methods exist to measure each of these phenomena. What is less understood, and can lead to wrong potential interaction assumptions, is how a single drug can both induce gene expression, and also inhibit the activity of the same isoform. Here we demonstrate the automation of a cell-based multiplexed assay which can provide this critical information. Inhibition and induction were measured using a luminogenic, cell-membrane permeable 3A4-specific substrate and a reporter gene assay based on luciferin, respectively. Normalization to cell number was also performed using a fluorescent cell viability assay. DPX-2 cells were used as a model and dosed for 24 hours with variable concentrations of 50 individual compounds. By combining the information gained from this triplexed assay, the full effect of a drug can be seen on the CYP3A4 isoform.
100. Sequential metabolism of the nicotine Δ5′(1′)iminium ion by P450s 2A6 and 2A13
Linda B. von Weymarn, and Sharon E. Murphy
Masonic Cancer Center, University of Minnesota, Minneapolis, MN, USA, 55455
The major metabolite of nicotine, the addictive agent in tobacco, is the nicotine Δ5′(1′)iminium ion. In humans the oxidation of nicotine to the Δ5′(1′)iminium ion is catalyzed primarily by P450 2A6 in the liver, although the extrahepatic enzyme P450 2A13 is a better catalyst of nicotine 5′-oxidation than P450 2A6 in vitro. We have reported that the nicotine Δ5′(1′)iminium ion is a substrate of both enzymes (1). In this study we demonstrated that the primary metabolite of P450 2A6 and 2A13 catalyzed nicotine Δ5′(1′)iminium ion metabolism is cotinine. Interestingly, secondary products of Δ5′(1′)iminium ion metabolism were detected shortly after cotinine was formed by both enzymes. For example, P450 2A13 trans-3’-hydroxycotinine (3OH-Cot) was detected as a product of Δ5′(1′)iminium ion metabolism and only a small amount of 5’-hydroxycotinine (5OH-Cot) was detected. In contrast, 5OH-Cot is the major product of P450 2A13-mediated cotinine metabolism (2). In this study we observed that during P450 2A13-mediated Δ5′(1′)iminium ion the ratio of 3OH-Cot to 5OH-Cot decreases from 5:1 to 1:1 as the concentration of the Δ5′(1′)iminium ion decreases and the concentration of cotinine increases. For P450 2A6-mediated Δ5′(1′)iminium ion metabolism two secondary metabolites were observed; N-(hydroxymethyl)norcotinine and 3OH-Cot. During P450 2A6-mediated cotinine metabolism 5OH-Cot is a more predominant product than 3OH-Cot (2). We hypothesize that this discrepancy in the ratio of products of the secondary metabolism of the Δ5′(1′)iminium ion is due to sequential metabolism of the nicotine Δ5′(1′)iminium ion to 3OH-Cot. The Δ5′(1′)iminium ion is oxidized twice, first to cotinine and then to 3OH-Cot, before leaving the active site. Molecular modeling indicate that nicotine and the nicotine Δ5′(1′)iminium ion binds in the active site of P450s 2A6 and 2A13 in a different orientation than cotinine. The Δ5′(1′)iminium ion binds in an orientation that favors cotinine and subsequent 3OH-Cot formation whereas cotinine binds in an orientation that favors 5OH-Cot formation over 3OH-Cot formation.
References
- von Weymarn, LB, Brown, KM, Murphy, SE. Inactivation of CYP2A6 and CYP2A13 during nicotine metabolism. The Journal of Pharmacology and Experimental Therapeutics, 316:295-303 (2006)
- Brown, KM., von Weymarn, LB., Murphy, SE. Identification of N-(hydroxymethyl) norcotinine as a major product of P450 2A6, but not P450 2A13-catalyzed cotinine metabolism. Chemical Research in Toxicology, 18:1792-1798 (2005)
101. Creation of a genetically engineered protease deficient yeast strain having human Cyp450
Rajeev Soni1, and Praveen Gupta2
1PREMAS BIOTECH PVT LTD, HARYANA, India, 122050
2PREMAS BIOTECH PVT LTD, Haryana, India, 122050
Microsomal cytochrome P450 (CYPs) monooxygenases are mainly expressed in liver and play a crucial role in phase I metabolism of xenobiotics. Here we report the creation of a genetically engineered protease deficient yeast strain having human cytochrome P450 reductase for the heterologous over-expression of cDNA’s encoding six major human Cytochrome P450 isoforms that are responsible for metabolism of more than 90% of drugs and xenobiotics, co-expressed with human NADPH cytochrome P450 reductase. All the recombinant CYPs are composed of the active holo-enzyme as confirmed by reduced CO different spectra with a peak at 450nm. Reductase activity was measured by reduction of cytochrome c and microsomes containing active human CYP450 enzyme was confirmed by measuring their specific activities using their respective specific fluorescence substrates.
102. Structural basis of sequential metabolism revealed by an NMR-based structural model of rabbit cytochrome P450 2B4 with the analgesic amidopyrine and its product didesmethylamidopyrine
Arthur G. Roberts, and James R. Halpert
Skaggs School of Pharmacy, University of California, San Diego, San Diego, CA, USA, 92093-0703
Rabbit cytochrome P450 2B4 (CYP2B4) and the analgesic amidopyrine were used as prototypes of sequential drug metabolism by mammalian cytochromes P450 (CYPs). CYP2B4 metabolizes amidopyrine by two N-demethylation steps to form the product didesmethylamidopyrine. Despite numerous studies of amidopyrine with this enzyme, little is known of the structural basis of sequential metabolism. In order to narrow our gaps in understanding, NMR studies were undertaken to build a structural model of binding of amidopyrine and its product to CYP2B4. Heme-induced relaxation of ligand protons was used to position the ligands in the active site of CYP2B4. Saturation transfer and NOE experiments were used to probe the protein and aqueous environments of the ligands, respectively. This information was combined and then used as constraints for a molecular dynamics simulation of ligands bound to CYP2B4. Implications of the model will be discussed in the context of sequential metabolism by CYPs as a whole and how the technology presented in this study can be applied to other CYPs. (Supported by NIH grant ES003619).
103. Biochemical and structural characterization of mycobacterium tuberculosis CYP124
Jonathan B. Johnston, Petrea M. Kells, Larissa M. Podust, and Paul R. Ortiz de Montellano
Department of Pharmaceutical Chemistry, University of California San Francisco, San Francisco, CA, USA, 94158
Mycobacterium tuberculosis (Mtb) produces a variety of lipids that serve important functions, including modulating the immune response during pathogenesis and contributing to a robust cell wall that is impermeable to many chemical agents. The genome of Mtb encodes for many enzymes involved in lipid biosynthesis and degradation, including twenty P450 enzymes, some of which are potential drug targets. To date only two Mtb P450 enzymes demonstrate catalytic activity. Here we report the first biochemical characterization of Mtb CYP124 (Rv2266) that includes cloning, over-expression and purification. Purified CYP124 displays tight binding to various azole compounds and also binds to a series of lipids that induce concentration-dependent Type-I spin shifts. CYP124 shows robust catalytic activity towards select lipids, importantly with preferential oxidation of the chemically disfavored ω-position. We also report x-ray crystal structures of the ligand-free and substrate-bound CYP124 at 1.4 å and 2.1 å, respectively, which provide the first reported structural insights of this enzyme and its activity. The observed regiospecific catalytic activity reported here suggests roles of CYP124 towards physiologically relevant Mtb substrates and provides a platform for the development of specific inhibitors.
104. Comparison of CYP activities from human liver microsome pools based on weight, gender and age
LaHoma Easterwood, Matt Palmer, Kevin Lehnert, Jason Wright, Lyndee Lowrance, Ammy Santiago, and Jeanette Hill
CellzDirect, Inc., Austin, TX, USA, 78754
Liver microsomes are subcellular fractions containing common drug metabolizing enzymes including cytochrome P450 (CYPs), flavin-monooxygenases (FMOs), carboxylesterases, epoxide hydrolase, UDP-glucuronosyltransferases (UGTs). Because liver microsomes are abundant in enzymes that are involved in the biotransformation of xenobiotics, they serve as a useful biological system for assessing the intrinsic clearance (CLINT) of a particular drug, for identifying which CYPs are involved in the metabolism of a given xenobiotic, and for identifying potential drug interactions associated with inhibition of CYPs or UGTs. Large differences in CYP activity are often observed between individual donors. To determine whether levels of CYP activity correlate to specific donor phenotypes, microsome pools were produced based on weight (normal weight [Body Mass Index (BMI)<25], overweight [BMI =25-30], obese [BMI=30-40] and morbidly obese,[ BMI ≥40]), gender (male or female), and age (18-35 years, 40-60 years, >75 years). CYP activities for each pool were characterized and kinetic parameters (Vmax and Km) were determined for the major drug metabolizing CYP enzymes (CYP1A2, phenacetin O-dealkylation; CYP2A6, coumarin 7-hydroxylation; CYP2B6, bupropion hydroxylation; CYP2C8, paclitaxel 6ahydroxylation; CYP2C9, tolbutamide hydroxylation; CYP2C19, (S)-mephenytoin 4’hydroxylation; CYP2D6, dextromethorphan demethylation; CYP2E1, chlorzoxazone 6hydroxylation; CYP3A4, testosterone 6β-hydroxylation and midazolam 1’-hydroxylation). The results obtained demonstrate that differences in CYP activity correlate with donor phenotypes based on weight, gender and age. These results provide a better understanding of factors that may contribute to individual differences in drug metabolism.
105. Luciferin isopropyl acetal: a new, highly selective and sensitive bioluminogenic CYP3A4 substrate for induction and inhibition assays
James J. Cali1, Poncho Meisenheimer1, Mary Sobol1, Dongping Ma1, Timothy A. Moeller2, Ji Young Lee3, and Scott Heyward2
1Dept of R & D, Promega Corp, Madison, WI, USA, 53711
2Celsis In Vitro Technologies, Baltimore, MD, USA, 21227
3Research and Development, Celsis In Vitro Technologies, Halethorpe, MD, USA, 21230
CYP3A4 induction and inhibition by drugs and other xenobiotics is a significant cause of adverse drug-drug interactions. To predict the potential for these outcomes early in drug discovery, compounds are tested for their capacity to induce or inhibit the conversion of a probe substrate by CYP3A4 to a specific product. We have synthesized a new, highly selective and sensitive CYP3A4 probe substrate, luciferin isopropyl acetal (luciferin-IPA), for use in rapid bioluminescent cell-based and cell-free CYP3A4 assays. With a panel of 21 recombinant human CYP enzymes luciferin-IPA only showed activity with CYP3A enzymes. Selectivity for CYP3A4 was 14 and 137 fold over CYP3A5 and CYP3A7, respectively. CYP3A4 selectivity was confirmed in a rapid, 96-well, intact hepatocyte assay where activity was increased by known CYP3A4 inducers and inhibited by CYP3A4 selective inhibitors. The luciferin-IPA/CYP3A4 hepatocyte assay was easily configured in a multiplex application with a cell viability assay. Cell free luciferin-IPA enzyme assays were sensitive to a wide range of known CYP3A4 inhibitors. The reaction was competitively inhibited by the CYP3A4 probe substrates midazolam, testosterone and nifedipine, suggesting it will detect inhibitors also detected by each of these commonly used probe assays. While luciferin-IPA/CYP3A4 induction and inhibition data was virtually identical to data from conventional CYP3A4 testosterone 6-beta hydroxylation assays, the luminescent assays were simpler, more sensitive and substantially quicker.
106. The role of CYP26A1 in retinoic acid clearance
Jayne E. Thatcher, Alex Zelter, and Nina Isoherranen
Department of Pharmaceutics, University of Washington, Seattle, WA, USA, 98195
Retinoic acid (RA) is a critical signalling molecule that is essential for diverse biological functions. Altered RA metabolism has been linked to several important diseases such as Alzheimer’s disease, type 2 diabetes, obesity and cancer. The family of CYP26 enzymes has been identified as RA hydroxylases in all chordates and these enzymes are believed to be responsible for elimination of RA from cells and whole organisms. The aim of this study was to determine whether CYP26A1 is the major RA hydroxylase in human liver, and to predict using in vitro enzyme kinetic data and CYP expression levels determined by western blotting, the relative importance of CYP26A1, CYP3A4 and CYP2C8 in hepatic clearance of RA. The formation of the major metabolite of RA, 4-OH-RA was measured in incubations with CYP3A4 and CYP2C8 supersomes and using hCYP26A1 membrane preparations from baculovirus infected insect cells. CYP26A1 was found to be over 1000-fold more efficient than CYP2C8 and CYP3A4 in metabolizing RA, with intrinsic clearance values of 760 ml/min/nmolP450, 0.35ml/min/nmolP450 and 0.21ml/min/nmol P450 for CYP26A1, CYP2C8 and CYP3A4, respectively. RA bound CYP26A1 tightly with Km of 21 ± 3.6nM, whereas binding to CYP2C8 and CYP3A4 was weaker with Km-values of 19μM (CYP3A4) and 13 μM (CYP2C8). The Vmax values for the three enzymes were similar, between 3.5 and 4.7pmol/min/pmol P450. Using the kinetic values measured and known expression levels of CYP2C8 and CYP3A4 in human liver, it was predicted that CYP26A1, even if expressed at very low levels, would be the major enzyme eliminating endogenous RA. However, as circulating concentrations of therapeutically administered RA are micromolar, CYP26A1 will be saturated and its relative contribution to the clearance of RA will be diminished. In the absence of CYP26A1 expression, CYP3A4 is predicted to be more important than CYP2C8 in RA clearance due to its higher expression levels in human liver. These data suggest that inhibition of CYP26A1 is necessary to selectively decrease RA clearance, and avoid tolerance to therapy due to auto-induction of RA clearance.
107. CYP3A ER-associated degradation (ERAD) in rat hepatocytes involves p97 AAA ATPase
Poulomi Acharya, Mingxiang Liao, and Maria Almira
Correia Cellular and Molecular Pharmacology, University of California San Francisco, San Francisco, CA, USA, 94158
Cytochromes P450 (P450s) are endoplasmic reticulum (ER) membrane-anchored hemoproteins that metabolize a vast repertoire of endo- and xenobiotics. Hepatic P450 content, determined by synthesis, stabilization and turnover, significantly influences drug metabolism, therapeutics and drug-drug interactions. We sought to characterize the molecular mechanisms and proteins involved in P450 stabilization and turnover. One such protein is the cytosolic p97, an AAA-ATPase that extracts ER-membrane proteins and delivers them for proteasomal degradation. P450s are N-terminally anchored to the ER membrane with their protein bulk readily accessible to the cytosolic proteasome. Thus it is unclear whether they require p97-mediated extraction. However, CYP3A degradation was impaired in Saccharomyces cerevisiae with defective Cdc48 (p97 homolog)1. Moreover, in rat hepatocytes an increased p97 association with ubiquitinated CYP3A was observed which was further enhanced on CYP3A inactivation and/or proteasomal inhibition2. These findings suggested a role for p97 in CYP3A degradation. To examine this, we used RNAi (lentiviral shRNA targeting 3rd and 6th exons) to silence p97 (80% mRNA and 90% protein knockdown relative to a vector control verified by qRT-PCR and immunoblotting respectively) in sandwich-cultured rat hepatocytes. This knockdown was associated with significant (4-fold) increase in both parent and ubiquitinated CYP3A content that was further enhanced upon CYP3A inactivation. These data are the first to reveal that CYP3A ubiquitination precedes p97-mediated membrane extraction and delivery of ubiquitinated CYP3A to 26S proteasome. Thus, p97, an emerging therapeutic target for several cancers, liver and degenerative diseases, functions as a critical chaperone in CYP3A degradation.
Supported by NIH grants DK26506, GM44037, DK26743.
References
- Liao, M., Faouzi, S., Karyakin, A. and Correia, M. A. (2006) Endoplasmic reticulum-associated degradation of cytochrome P450 CYP3A4 in Saccharomyces cerevisiae: further characterization of cellular participants and structural determinants. Mol. Pharmacol. 69, 1897-1904.
- Faouzi, S., Medzihradszky, K.F., Hefner C., Maher J.J. and Correia, M.A. (2007) Characterization of the Physiological Turnover of Native and Inactivated Cytochromes P450 3A in Cultured Rat Hepatocytes: A Role for the Cytosolic AAA ATPase p97? Biochemistry, 46, 7793-7803.
108. Kinetic analysis of cytochrome P450 19A1
Christal D. Sohl, and F. Peter Guengerich
Department of Biochemistry and Center in Molecular Toxicology, Vanderbilt University, Nashville, TN, USA, 37232-0146
Cytochrome P450 19A1 (P450 19A1), or aromatase, catalyzes the conversion of androgens (androstenedione and testosterone) to estrogens (estrone and 17b-estradiol). In both cases, the reaction proceeds sequentially through a 19- hydroxy- and a 19-aldo-containing intermediate before a final aromatization step, requiring three moles each of NADPH and O2. There has been much interest in the characterization of P450 19A1 due to its ability to catalyze a sequential, three-step reaction with an aromatization step, which is rare in nature. Further, aromatase inhibitors have been proven to be effective therapy for estrogen-dependent breast cancer since the 1970’s. Only steady-state kinetic parameters, with a wide range in values, have been measured to date for P450 19A1. The lack of pre-steady state data and variance in Km and Vmax values are probably due to difficulty in both expression and purification and the use of various methods to measure aromatase activity. After assaying various cDNA constructs, bacterial strains, solubilization conditions, and chromatography methods, we have successfully expressed and purified P450 19A1 at moderately high levels in Escherichia coli. These protein preparations consistently yield pure, catalytically active protein with typical reduced-CO difference spectra, the latter of which is a common challenge that at one point led to the questioning of aromatase as a member of the cytochrome P450 family. Using stopped-flow spectroscopy, we are characterizing the pre-steady state parameters of P450 19A1 by measuring binding constants for each ligand in the three-step reaction. By generating single turnover conditions (rapid quench methods), reaction progression curves can be constructed, showing the disappearance of substrate, appearance and disappearance of both intermediates, and the formation of product. These data, in addition to dissociation constants that we have calculated to be in the sub-μM range for androstenedione and the two intermediates, can be fit to a kinetic mechanism. (Supported in part by USPHS R37 CA090426).
109. Regulation of vitamin D receptor and cytochrome P450 3A4 enzyme by microRNAs
Yuzhuo PAN, Wenqing Gao, and Aiming Yu
Pharmaceutic science, University at Buffalo,Suny, Amherst, NY, USA, 14260
Cytochrome P450 3A4 (CYP3A4) metabolizes over 50% of drugs on the market. Transcriptional regulation of CYP3A4 is known to be controlled by a number of nuclear receptors (NR) including vitamin D receptor (VDR/NR1I1), whereas posttranscriptional regulation of CYP3A4 remains elusive. Here we show that small, noncoding microRNAs (miRNAs) may control posttranscriptional and transcriptional regulation of CYP3A4 through their actions on 3’-untranslated region (3’UTR) of CYP3A4 and VDR, respectively. Luciferase reporter assays showed that CYP3A4 3’UTR-luciferase activity was decreased significantly in HEK293 cells after transfected with plasmid expressing miR-27b or mmu-miR-298, whereas the activity was unchanged in cells transfected with miR-122a or miR-328. Disruption of corresponding miRNA response element (MRE) within CYP3A4 3’UTR led to 2- to 3-fold increase in luciferase activity. Immunoblot analyses indicated that CYP3A4 protein was down-regulated over 40% by miR-27b and mmu-miR-298 in LS-180 and PANC1 cells, which was associated with significantly decreased CYP3A4 mRNA levels, as determined by quantitative real-time PCR (qPCR). Likewise, the actions of miR-27b and mmu-miR-298 on VDR 3’UTR were shown by luciferase assays, in which the mmu-miR-298 MRE site is well conserved within the 3’UTR of mouse, rat and human VDRs. Down-regulation of VDR by the two miRNAs was supported by immunoblot and qPCR analyses. Furthermore, Overexpression of miR-27b or mmu-miR-298 resulted in lower cell sensitivity to cyclophosphamide. Together, these findings suggest that CYP3A4 and VDR gene expression may be regulated at the posttranscriptional level.
110. CYP2C9-CYP3A4 protein-protein interactions: Effects on catalysis and mechanisms of interactions
Murali Subramanian, Harrison Tam, Helen Zhang, and Timothy S. Tracy
Experimental and Clinical Pharmacology, University of Minnesota, Minneapolis, MN, USA, 55455
Cytochrome P450s are known to interact with cytochrome P450 reductase (CPR) and cytochrome b5, all membrane bound proteins. In multiple in-vitro systems, P450s also have been observed to interact with other P450s. The current work investigated the effects and mechanisms of interactions between CYP2C9 and CYP3A4 in a reconstituted system. CYP2C9-mediated metabolism of S-naproxen and S-flurbiprofen were inhibited up to 80% by CYP3A4, although Km values were unchanged. The inhibition increased at higher CYP3A4 concentrations whereas increased CPR concentrations reduced the inhibition. The addition of cytochrome b5 only marginally affected the magnitude of inhibition. In contrast, CYP2C9 had no effect on the metabolism of CYP3A4-mediated metabolism of testosterone and midazolam. To explore the mechanisms governing these P450-P450 interactions, dapsone, a CYP2C9 effector that increases P450 cycle efficiency, was co-incubated with the CYP2C9:CYP3A4 mixture and flurbiprofen hydroxylation monitored. Dapsone attenuated the inhibition of CYP2C9 by CYP3A4, suggesting that the CYP2C9:CYP3A4 interaction involves, at least to some extent, effects on cycle efficiency. To assess potential region(s) on the proteins where the interactions occur, incubations were repeated with N-terminus truncated CYP2C9 and full length CYP3A4 and vice-versa. In both instances, the inhibition was fully abolished when at least one of the proteins was a truncated form, unequivocally establishing that the N-terminus hydrophobic membrane-binding regions of CYP2C9 and CYP3A4 were involved in the interaction. Additionally, the CYP2C9:CYP3A4 heteromer complex was isolated by co-immunoprecipitation techniques, confirming the physical interaction of the proteins. In summary, these results demonstrate that the membrane binding domains of CYP2C9 and CYP3A4 proteins interact to form a heteromer complex that inhibits the activity of CYP2C9 by up to 80%, at least in part due to a reduction in P450 catalytic cycle efficiency. These P450-P450 interactions may affect in-vitro in-vivo correlations and predictions of drug clearance.
Acknowledgements
NIH Grant number GM063215
111. Compounds isolated from milk thistle extract differentially modulate human enteric and hepatic CYP3A activity
Scott J. Brantley1, David J. Kroll2, Nicholas H. Oberlies3 and Mary F. Paine4
1UNC Eshelman School of Pharmacy, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA, 27599-7360
2BRITE, North Carolina Central University, Durham, NC
3The University of North Carolina at Greensboro, Greensboro, NC, 27402
4UNC Eshelman School of Pharmacy, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA, 27599
Milk thistle (Silybum marianum) is a top-selling dietary supplement worldwide. Available commonly as oral formulations of an extract, termed silymarin, this inconsistently-regulated product is used frequently to self-treat hepatic disorders. Intravenous formulations are available in Europe and are approved as rescue therapy for deathcap mushroom poisoning. Often marketed as a single compound, silymarin comprises at least seven flavonolignans (silybin A, isosilybin A, silybin B, isosilybin B, silychristin, isosilychristin, silydianin) and one flavonoid (taxifolin). Methods have been developed, for the first time, to isolate and purify gram-scale quantities of each component in silymarin. As such, a rigorous characterization of the modulatory effects of individual silymarin components on drug metabolizing enzymes is now possible. Cytochrome P450 3A (CYP3A) is concentrated in both the small intestine and liver and is highly sensitive to modulation by xenobiotics, which can lead to clinically significant drug interactions. Accordingly, the effect of each component was evaluated on CYP3A-mediated midazolam 1’-hydroxylation using human intestinal and liver microsomes (HIM, HLM). Reaction mixtures (n=3) consisting of HIM or HLM (0.05 mg/ml), midazolam (4 μM), and silymarin or individual component (100 μM) in phosphate buffer (0.1 M, pH 7.4, 3.3 mM MgCl2) were initiated with NADPH (1 mM). After 4 (HIM) or 2 (HLM) minutes at 37°C, reactions were quenched with cold acetonitrile. 1’-Hydroxymidazolam was quantified by HPLC-MS/MS. Relative to control (0.75% methanol), silymarin inhibited activity in HIM by 50%, whereas the individual components inhibited by varying extents, from <5% (isosilybin A) to 72% (silychristin). Similar trends were observed with HLM, but the extents of inhibition were slightly greater than those with HIM; silymarin inhibited activity by 70%, whereas the individual components inhibited activity by 16% (isosilybin A) to 88% (silychristin). Given the batch-to-batch variation in constituent phytochemicals in natural products, the availability of gram-quantities of isolated compounds from silymarin will provide means for standardizing the drug interaction liability of milk thistle products in both the in vitro and clinical settings.
Supported by NIH GM077482
112. Regulation of hepatic cytochrome P450 and cytokine mrnas by citrobacter rodentium infection in cytokine- and cytokine receptor-deficient mice
Beatrice A. Nyagode, Ryan D. Kinloch, and Edward T. Morgan
Pharmacology Department, Emory University, Atlanta, GA, USA, 30322
Following infection with Citrobacter rodentium, the equivalent of enteropathogenic Escherichia coli in humans, murine hepatic cytochrome P450 mRNAs are selectively regulated and several serum pro-inflammatory cytokines are elevated. The role of these cytokines in the in vivo regulation of cytochrome P450s during infection is poorly understood. In this study, female C57BL/6 mice, either wildtype (WT) or deficient in interleukin-6 (IL6−/-), interferon-γ (IFNγ−/-), interleukin-1 receptor (IL1R−/-) or tumor necrosis factor receptor p55 (TNFRp55−/-), were infected orally with C. rodentium and sacrificed 7 days later. Hepatic expression of cytochrome P450 and cytokine mRNAs as well as serum cytokine levels were examined. In all groups, infection affected CYP4A10 and 4A14 the most, down-regulating mRNAs to between 5 and 37% of levels in uninfected animals while CYP2D9 and TNFα mRNAs were induced between 2 to 27-fold. Levels of IL1β mRNA were induced between 5 to 76-fold in all groups except in IFNγ−/- mice. Infection had no effect on CYP3A13 mRNA in all but IL6−/- mice, in which they were reduced to 31% of control levels. CYP3A11 mRNA levels were down-regulated in WT and IL1R−/- to 56 and 9% respectively but were unchanged by infection in the remaining groups. Infection did not affect levels of IL6 mRNAs in WT and IFNγ−/- mice, but caused a 5-fold induction in IL1R−/- mice. The down-regulation of CYP2D22 and 3A25 by infection in WT, IL6−/- and IL1R−/- was abrogated in both IFNγ−/- and TNFRp55−/- mice. In general circulation, the deficiency of one cytokine affected serum levels of one or more of the other cytokines. These results indicate similar involvement of both IFNγ and TNFα in regulation of CYP2D22, 3A11 and 3A25 mRNAs and the involvement of IL6 in the regulation of both CYP3A11 and 3A13 during C. rodentium infection. This work was supported by NIH grant DK072372.
113. Metabolism of leflunomide, a Disease Modifying Anti-Rheumatic Drug (DMARD), by cytochrome P450 BM3 from bacillus megaterium
Seon Ha Park, Dong-Hyun Kim, and Chul-Ho Yun
School of Biological Sciences and Technology, Chonnam National University, Gwangju, South Korea
For drug metabolism, human P450 enzymes expressed in various tissues play an important role where they exclusively metabolize over 80% of clinical drugs. However, due to its low catalytic activities and low stabilities, human P450 enzymes are not suitable for studies on drug efficacy or toxicity, which are significant steps in drug development process. Recent studies have discovered that mutant forms of cytochrome P450 BM3 (CYP102A1) from bacillus megaterium can metabolize various drugs via catalytic reactions similar to that of human P450 enzymes. Unlike human P450 enzymes, CYP102A1 enzymes are fused to their reductase domain, and this fusion of P450 domain and reductase domain makes CYP102A1 enzymes a prospective candidate for industrial enzymes to produce human drug metabolites. Leflunomide, N-[(4-Trifluoromethyl)phenyl]-5-methylisoxazole-4-carboxamide, is a disease-modifying anti- inflammatory agent, widely prescribed to treat advanced rheumatoid arthritis. Metabolism of leflunomide has not been elucidated in detail yet, however, in vitro studies have demonstrated that human P450 1A2 may be the enzyme mainly responsible for leflunomide activation. In this study, we demonstrate that wild-type and a set of CYP102A1 mutants catalyze the hydroxylation reaction that produces the same metabolite as that of human P450 1A2. Leflunomide was metabolized by human P450 1A2 and CYP102A1 wild type and its mutants, which were constructed by site-specific mutagenesis with different mutation sites. The metabolism of leflunomide by human P450 and CYP102A1 was analyzed by high-performance liquid chromatography (HPLC) and its metabolite analysis was performed by liquid chromatography/mass spectrometry (LC/MS). The results suggest that CYP102A1 mutants can be used as an effective tool for the further study on leflunomide metabolism and provide as a precursor to synthesize the derivatives for the development of a new potent dug. Key words: Human P450 1A2, cytochrome P450 BM3 (CYP102A1), leflunomide.
114. Effects of the human CYP3A4*4 (Ile118Val) genetic polymorphism on anandamide metabolism
Matthew J. Pratt-Hyatt, Natasha T. Snider, Haoming Zhang, and Paul F. Hollenberg
Pharmacology, University of Michigan, Ann Arbor, MI, USA, 48109-0600
The endocannabinoid system plays an important role in numerous physiological processes including mood, appetite, and pain-sensation. A critical compound in maintaining cannabinoid tone is the endocannabinoid anandamide (AEA). We have recently shown that AEA is metabolized by several human cytochrome P450s (P450) to form several metabolites; one of which exhibits increased biological activity (Snider et al., A Cytochrome P450-derived Epoxygenated Metabolite of Anandamide is a Potent Cannabinoid Receptor 2-Selective Agonist, Mol. Pharm. 75:965-972, 2009). CYP3A4, one of the major P450s involved in AEA metabolism, produces four metabolites of AEA. One of these metabolites, 5,6 epoxyeicosatrienoic acid ethanolamide (EET-EA), exhibits a much higher affinity than AEA for the cannabinoid 2 receptor (CB-2) and leads to a marked decrease in intracellular cAMP levels in cells expressing CB-2. There are multiple human alleles of CYP3A4 and *4 has been shown to exhibit a significant decrease in activity for cortisol (Wang et al. Ile118Val Genetic Polymorphism of CYP3A4 and Its Effects on Lipid-Lowering Efficacy of Simvastatin in Chinese Hyperlipidemic Patients, Eur J Clin Pharmacol. 60, 843-848, 2005). Using a recombinant version of the mutant allele expressed in E.coli, we determined that in a reconstitution system it produces 10 times less 6-OH-testosterone than wild type 3A4 (WT). We then investigated AEA metabolism by the WT and the *4 variant. The variant produced ten times less 14,15-EET-EA and 11,12-EET-EA and very little 8,9-EET-EA. In addition, it did not produce any significant amount of 5,6-EET-EA, the CB-2 binding metabolite. Besides producing three of the metabolites formed by WT 3A4, the mutant also produced a novel peak corresponding to 19-HETE-EA. The area of this peak is 50 percent of the 11,12-EET-EA peak, the major product formed. These data indicate that individuals expressing this allele may exhibit significant variations in the metabolism of anandamide as well as any compounds resembling anandamide.
115. Identification of ligand-induced conformational dynamics in cytochrome P450 using 2D NMR With the unnatural amino acid 13C-p-Methoxyphenylalanine
Jed N. Lampe, Stephen N Floor, John D Gross, Clinton R Nishida, Michael J Trnka, and Paul R Ortiz de Montellano
Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, CA, USA, 94158
Cytochrome P450 (CYP) enzymes are ubiquitous monooxygenases that are the primary enzymes responsible for the detoxification of xenobiotic compounds in mammals. Although conformational dynamics are thought to play an important role in ligand binding and catalysis by cytochrome P450 enzymes, few techniques exist to examine them in molecular detail. 2D NMR is a useful non-destructive method for obtaining information on ligand-enzyme dynamics, however global isotopic labeling can make assignment of specific protein-ligand interactions difficult. Using a unique isotopic labeling strategy, we have site specifically inserted a 13C-labeled unnatural amino acid residue, 13C-p-methoxyphenylalanine (MeOF), into three different locations in the substrate binding region of CYP119, a thermophilic P450 enzyme. Surprisingly, in all cases the resonance signal from the ligand-free protein is represented by a doublet in the 1H,13C-HSQC spectrum. Upon binding of the high affinity inhibitor ligand 4- phenylimidazole, the signals from the initial ligand-free resonances are reduced in favor of a single new resonance, in the case of the F162MeOF mutant, or two new resonances, in the case of the F153MeOF mutant, suggesting a stable conformational intermediate on the path to the ground state crystal structure. In comparison, the resonance signal shows dramatic line broadening upon binding of the low affinity inhibitor ligand imidazole, suggesting the presence of multiple ligand-bound conformational species in solution. Finally, binding of the high affinity substrate lauric acid shifts the conformational equilibrium to a single species, suggesting direct conversion from the inactive form to the active form of the enzyme. This represents the first direct physical evidence for the ligand-dependent simultaneous existence of multiple P450 conformers in solution. This general approach may be used to further illuminate the role that conformational dynamics plays in the complex enzymatic phenomena exhibited by P450 enzymes.
116. Expression, characterization and functional properties of a selenocysteine substituted cytochrome P450 enzyme
Santhosh Sivaramakrishnan1, Hugues Ouellet1, Yongying Jiang1, Takahiro Hayashi2, Pierre Moënne-Loccoz2, and Paul R. Ortiz de Montellano1
1Department of Pharmaceutical Chemistry, University of California, San Francisco, CA, USA, 94158
2Department of Science and Engineering, Oregon Health & Science University, Beaverton, OR, USA, 97006
Cytochrome P450 enzymes are ubiquitous monooxygenases responsible for the oxidation of various drugs, natural products and xenobiotics. The proximal cysteine thiolate ligand coordinated to the heme iron at the active site is crucial for the biological activity of these proteins. In general, P450-mediated oxygenation is thought to proceed via a reactive ferryl intermediate [Fe(IV)=O] coupled to a heme porphyrin or a protein radical cation, referred to as Cpd I. The exact nature of the reactive species is still ambiguous due to its high reactivity under normal conditions. However, recent QM/MM calculations predicted that replacement of the proximal cysteine thiolate ligand with selenocysteine (SeCys) might increase the life time of the putative Cpd I species, making it detectable.1 Herein we report the expression, characterization and functional properties of SeCys substituted CYP119, a thermophilic cytochrome P450 enzyme. Typically, a change of proximal ligand results in loss of the spectroscopic signature and catalytic properties of the P450 enzyme. However, detailed investigation of SeCys substituted CYP119 using UV-vis, resonance Raman and EPR spectroscopy revealed spectral characteristics comparable to those of the normal cysteine-ligated protein.2 Furthermore, H2O2-mediated shunt oxidation of lauric acid indicated that the seleno mutant retains full catalytic activity. Finally, stopped flow kinetic analysis of the reaction of SeCys mutant with peroxides suggests that the SeCys ligation stabilizes the putative ferryl species compared to wild-type CYP119. These results help elucidate the nature of the reactive species involved in the P450 catalytic cycle and may offer insights into the development of novel catalysts. This work was supported by NIH grants GM25515 and GM74785
References
- Cohen, S.; Kumar, D.; Shaik, S. In silico design of a mutant of cytochrome P450 containing selenocysteine, J. Am. Chem. Soc. 2006, 128, 2649-2653.
- Jiang, Y.; Sivaramakrishnan, S.; Hayashi, T.; Cohen, S.; Moënne-Loccoz, P.; Shaik, S.; Ortiz de Montellano, P. R. Calculated and experimental spin state of seleno cytochrome P450, Submitted to Angewandte Chemie, 2009.
117. CYP2E1 turnover in Saccharomyces cerevisiae and human hepatocytes: Evolutionary conservation of the proteolytic pathways
YongQiang Wang, Poulomi Acharya, and Maria Almira
Correia Cellular & Molecular Pharmacology, University of California, San Francisco, San Francisco, CA, USA, 94158
CYP2E1 exhibits biphasic turnover in the rat liver: Its substrate-complexation results into a stable slow-turn over species (t1/2 ≈ 37 h) degraded largely via autophagic-lysosomal degradation (ALD), whereas its substrate decomplexation results in a fast-turn over species (t1/2 ≈ 7 h) degraded via ubiquitin-dependent proteasomal degradation (UPD) in a typical ERAD process. To molecularly characterize the CYP2E1 degradation pathways, we have heterologously expressed human liver CYP2E1 in S. cerevisiae yeast strains defective in UPD (hrd2-1) and deficient in ALD/vacuolar Pep4p master protease-dependent degradation (pep4D) and corresponding isogenic wild type (WT) strains and its degradation was followed by the stationary-chase method. Cells were harvested at various stages of logarithmic culture growth. Microsomal immunoblotting analyses revealed that a fraction of CYP2E1 is initially stabilized to a significant extent (33%) in the hrd2-1 yeast strain relative to the corresponding WT control, whereas at a later stage (20 h after reaching an OD ≈ 0.8) another fraction is stabilized to a much greater extent (>2-fold) in pep4D-yeast strains. Thus in yeast as in the rat liver, both UPD and ALD function as the major CYP2E1 proteolytic pathways. Because of the controversial role of ubiquitination in CYP2E1 turnover, we also examined CYP2E1 degradation in cultured human hepatocytes after treatment with and without the proteasomal inhibitor MG262 for 6 h. MG262 significantly stabilized both the native (30%) and ubiquitinated CYP2E1 species. This MG262-stabilization of ubiquitinated CYP2E1 was greatly enhanced after suicidal inactivation of the enzyme, consistent with a larger fraction of CYP2E1 being committed to UPD after structural inactivation. These findings reveal that native CYP2E1 is indeed an UPD substrate in yeast as well as rat and human hepatocytes, thereby attesting to the evolutionary conservation of its degradation. Supported by NIH grant GM44037.
118. See abstract 117
119. Clarification of species difference in the cause of non-linear PK profile
Yoko Nagaya, Osamu Takenaka, and Tsutomu Yoshimura
Drug metabolism and pharmacokinetics research, Eisai Co., Ltd., Tsukuba, Japan
New chemical entities often show non-linear pharmacokinetic (PK) profile in experimental animals. There are some considerable reasons for nonlinearity, however, there are few reports focused on species difference in the reasons. Hence, species difference in the cause of nonlinearity was evaluated using ER-429006 as a model compound. The Cmax and AUC increased more than dose proportionally in rats, dogs and monkeys after oral administration. Presumable reasons for nonlinearity were: 1) saturation of hepatic metabolism, 2) saturation of intestinal metabolism, and 3) effect of P-gp on absorption. To assess the possibility of reason 1), kinetic parameters (Km and Vmax) for hepatic metabolism were estimated using liver microsomes of the three species. Comparison between the Km values and unbound plasma concentrations in vivo indicated that saturation of hepatic metabolism would be the cause of nonlinearity in rats and dogs, but not in monkeys. Since ER-429006 was significantly metabolized by monkey intestinal microsomes only, the reason 2) was thought to be important in monkeys. To verify possibility of saturation of intestinal metabolism, effect of ketoconazole on the intestinal metabolism was evaluated in vivo. Oral dose of ketoconazole increased Cmax and AUC of ER-429006 after oral dose in monkeys, but not after intravenous dose of ER-429006. In the case of co-administrating ketoconazole, the dose normalized Cmax and AUC of ER-429006 were almost constant regardless of ER-429006 dose. These results suggest the reason 2) would be cause of nonlinearity in monkeys. Since ER-429006 is a P-gp substrate, the possibility of reason 3) was also assessed. There was no impact of P-gp on the absorption of ER-429006 based on the results of PK profile between wild and mdr1a/1b knock out mice and of the oral absorption estimate by simultaneous measurements of the portal and systemic concentrations in rats.
These results indicated that the main factor of non-linear PK profile is species dependent.
120. Comparison of the formation kinetics of acetaminophen in chimpanzee, cynomolgus monkey, marmoset, rhesus monkey and human liver microsomes
James Grace1, Harvey Wong2, Michael Sinz1, and Kimberley Lentz1
1Metabolism and Pharmacokinetics, Bristol-Myers Squibb Co, Wallingford, CT, USA, 06492
2Dept of Drug Metab & Pharmacokinetics, Genentech Inc, South San Francisco, CA, USA, 94080
Non-human primates are widely used in the pharmaceutical industry for the prediction of human pharmacokinetics and as a preclinical safety model. The metabolism of phenacetin to acetaminophen is widely regarded as a measure of CYP1A2 activity. In order to understand species differences in CYP1A-like activity the formation kinetics of acetaminophen was investigated in vitro using liver microsomes from human and nonhuman primate liver microsomes. The linearity of acetaminophen formation was determined with respect to protein concentration and time, in order to determine optimal incubation conditions for enzyme kinetic analysis. Phenacetin was incubated in the presence of 0.25 mg/mL of pooled microsomes at 37°C over a range of substrate concentrations (2-10,000 μM). After incubation, 100 μL of reaction mixture was quenched with 200 μL of acetonitrile containing the internal standard, altenolol, and samples were analyzed by UPLC-MS-MS. Samples were generated in duplicate and mean results were reported. The kinetics of acetaminophen formation was estimated by nonlinear regression using either the Michaelis-Menton or two-site kinetic models. The models used were selected based on visual inspection of the fit of the data and the goodness of fit criterion. The intrinsic clearance was calculated by Vmax/Km. P450 content was determined by the method of Omura and Sato. The Km for the formation of acetaminophen for cynomolgus monkey was fit to a Michaelis-Menton model while human, rhesus monkey, chimpanzee, and marmoset were fit to a two-site model. Vmax was higher for all non-human primates when compared to humans. In general, the higher Vmax observed in non-human primates is consistent with the higher CYP450 content measured in non-human primate liver microsomes. These results may assist in the understanding of species differences in metabolism and pharmacokinetics observed in non-human primates when compared to humans for compounds that are CYP1A2 substrates.
121. Assessment of the hepatic cytochrome P450 reductase null mouse model: Effect on clearance and exposure of docetaxel, midazolam, nelfinavir and theophylline
Jason W. Boggs1, Edna F. Choo1, Anna Dornier1, Michael Reich2, and Cornelis Hop1
1Dept of Drug Metabolism and Pharmacokinetics, Genentech Inc, South San Francisco, CA, USA, 94080
2In Vivo Studies, Genentech Inc, South San Francisco, CA, USA, 94080
P450 Oxidoreductase (Por) is the essential electron donor for all P450 enzymes and is responsible for the activation of P450 metabolism. The Taconic Hepatic Cytochrome P450 Reductase Null (HRN) Mouse Model possesses a targeted mutation that results in a homozygous genotype that results in liver-specific deletion of the Por gene thereby resulting in a disruption of P450 metabolism in the liver. This model could be useful in assessing new chemical entities (NCE) that could be used as tool compounds for proof of concept studies, e.g., in vivo efficacy, where high hepatic P450 clearance (CL) and low exposure may prevent in vivo evaluation of a NCE. The objective of this set of experiments was to further characterize the HRN mouse using probe drugs metabolized by P450, with moderate to high clearance and to note changes in CL and exposure in HRN mice compared to wild-type (WT) animals. In HRN mice following intravenous (IV) administration of midazolam (2 mg/kg), CL was reduced by 4.5-fold compared to CL in wild-type (WT) mice (8.52 mL/min/kg vs. 38.5 mL.min/kg, respectively). This reduction in CL corresponded to a 6.8-fold increase in half-life of midazolam from 0.182 h to 1.23 h. The oral AUC of midazolam (2 mg/kg) was increased by ~20-fold in HRN mice compared to WT mice (1.75 vs. 0.0843 uM.hr), the greater effect observed with oral dosing suggests that hepatic first pass plays a role in the oral CL of midazolam. A 2-fold and a 5-fold decrease in CL was also observed in HRN mice following IV administration of docetaxel (5 mg/kg) and theophylline (2 mg/kg), respectively, compared to WT mice (docetaxel: 58.4 mL/min/kg vs. 124 mL/min/kg and theophylline: 4.96 mL/min/kg vs. 25.6 mL/min/kg). CL of nelfinavir (5 mg/kg) was very similar in HRN and WT mice following IV dosing (123 mL/min/kg vs. 126 mL/min/kg) which suggests that non-P450 or non-hepatic P450 pathways are involved in nelfinavir’s CL. Further assessment of compensatory changes in HRN mice, e.g, transporter expression may be necessary to characterize the model further. The observations from these experiments demonstrate that changes in CL and exposure are observed in HRN mice compared to WT mice with drugs that are metabolized by P450 metabolism and that the HRN mouse model could potentially be a valuable tool in the process of evaluating in vivo efficacy of tool compounds in drug discovery.
122. Species difference in drug metabolism: CYP2C8 catalyzed formation of unique human metabolites of NVP-LDI133 and NVP-LDI141
Yancy Du, Arpine Vapurcuyan, Helen Gu, Tsu-Han Lin, Jimmy Flarakos, Imad Hanna, and James Mangold
Drug Metabolism and Pharmacokinetics, Novartis Institutes for BioMedical Research, East Hanover, NJ 07936
NVP-LDI133 and NVP-LDI141 were two drug candidates in development for the treatment of Hepatitis C. The compounds are structurally similar consisting of a cyclohexyl-2-quinolino-indole-N-acetamide backbone with either a dimethylthiazole (LDI133) or a methyl pyridine ( LDI141) side chain. The metabolism of LDI133 and LDI141 was compared following incubation with human, monkey, dog, and rat hepatocytes. Both compounds were conjugated to glucuronic acid resulting in the respective acyl-glucronide metabolites in all species. Alternatively, hydroxylation at the cyclohexyl moiety of either compound appeared to be a predominant, and unique metabolic pathway in human hepatocytes. In monkey hepatocyte incubations hydroxylated LDI141, but not LDI133, was present albeit at relatively much lower amounts compared to human hepatocyte incubations. Little (if any) of this type of metabolite was detected following incubation of either compound with rat or dog hepatocytes. In human liver microsomes, a marked inhibition (> 80%) of the formation of the hydroxylated metabolite was observed in the presence of the selective CYP2C8 inhibitor montelukast (1 μM). Moreover, of the seven cDNA-expressed human P450 tested, only CYP2C8 was capable of generating the hydroxylated LDI133 or LDI141. CYP2C8 is known to possess a relatively narrow substrate specificity compared to other human and animal cytochrome P450 enzymes. The results of these studies show that human oxidative biotransformation routes of LDI133 or LDI144 hydroxylation are predominantly catalyzed by CYP2C8. Accordingly, hydroxylation of either compound may be a useful indicator of CYP2C8 activity in various in vitro human test systems. However, the results also suggest that the preclinical development of compounds that are exclusively metabolized by CYP2C8 may not be desirable. The narrow regioselectivity of this P450 relative to other enzymes may result in the formation of human specific metabolite(s). Consequently, the potential toxicity of the resulting metabolites, or their downstream metabolic disposition, may not be sufficiently evaluated in preclinical toxicology studies.
123. Comparative biotransformation and mass balance of radiolabeled BMS-690514 (EVRI), an ErbB/VEGF receptor inhibitor, after oral administration to rats, dogs and humans
Hong Su1, Haizheng Hong1, Steven Zhang1, Blisse. J. Vakkalagadda1, Pamela. L. Clemens1, Alban Allentoff1, Haojun Sun1, Janet Caceres-Cortes2, Ramaswamy. A. Iyer1, W. Griffith Humphreys1, and Lisa J. Christopher1
1Department of Biotransformation-PCO, Bristol-Myers Squibb, Princeton, NJ, USA, 08543
2Pco, Bristol-Myers Squibb, Princeton, NJ, USA, 08543
BMS-690514 (((3R,4R)-4-amino-1-((4-((3-methoxyphenyl)amino)pyrrolo[2,1-f][1,2,4]triazin-5-yl)methyl)-3-piperidinol) is a potent inhibitor of ErbB and VEGF receptor tyrosine kinases that is currently in clinical development for treatment of solid tumor cancers. A series of ADME studies were conducted to determine the routes of excretion and biotransformation of radiolabeled BMS-690514 following administration to rats, dogs and humans. In all 3 species, the majority of the radioactive dose (50-71%) was recovered in the feces after oral administration of [14C]BMS-690514. Urinary excretion was 18-20% of the dose in rats and dogs, and about 34% in humans. The excretion profiles in bile duct-cannulated (BDC) rats and dogs were consistent with intact animals. After oral administration of [14C]BMS-690514 to BDC rats, 83.2 and 16.9% of the dose was recovered in the bile and urine, respectively; similarly, after iv administration of [3H]BMS-690514 to BDC dogs, 59.5% of the dose was recovered in the bile and 15.9% was recovered in urine. For a group of human subjects (n=3), where bile was collected in the 3-8 h interval after an oral dose of [14C]BMS-690514, about 16% of the dose was recovered in the bile. These data suggest that biliary secretion was a major route for the elimination of drug-related radioactivity in all species. The plasma profiles were qualitatively similar in all species; unchanged BMS-690514 was a major drug-related component in the 1-8 h plasma profiles. M6, an O-glucuronide of BMS-690514 was a prominent circulating metabolite in dogs and humans. Other metabolites present in all species included M1 (a hydroxylated metabolite), and secondary metabolites M7 & M9, glucuronide conjugates of hydroxylated and O-demethyl BMS-690514, respectively. The parent compound comprised <11.8, 3.3 and <28% of the dose recovered in the urine and feces from rats, dogs and humans. The primary pathways for the metabolism of BMS-690514 were glucuronidation of the parent molecule, hydroxylation (at several positions), O-demethylation, and glucuronidation of oxidative metabolites. Taken together, the results from these studies suggested that BMS-690514 was well-absorbed and highly metabolized in all 3 species.
124. In Vivo absorption, disposition, metabolism and excretion Of 14C-INCB018424 in non-clinical species
Adam D. Shilling, Frank Nedza, Sharon Diamond, and Swamy Yeleswaram
Drug Metabolism and Biopharmaceutics, Incyte Corporation, Wilmington, DE, USA, 19880
The non-clinical ADME properties were investigated for INCB018424, an inhibitor of the Janus kinase family of protein tyrosine kinases (JAKs). The tissue distribution was determined in Sprague-Dawley (albino) and Long-Evans (pigmented) rats after a single oral dose of 14C-INCB018424 by quantitiative whole-body autoradiography. Separately, the mass balance and metabolism of INCB018424 was determined in CByB6F1 Hybrid mice (Tg.rasH2 non-transgenic littermates), Sprague-Dawley rats and beagle dogs given a single oral (mice, rats and dogs) or IV (rats) dose of 14C-INCB018424. INCB018424-derived radioactivity was widely distributed in a similar pattern to the tissues of albino and pigmented rats. Tmax values were observed within 4 h of dosing with highest concentrations in the gastrointestinal tract, urinary bladder, bile, renal cortex, renal medulla, liver, aorta, adrenal gland, uveal tract and skin (pigmented). Elimination was complete in most tissues except for low levels of radioactivity in the aorta, skin, and liver of albino rats. In rats, the route and extent of elimination was similar after oral and IV administration with 92% and 87% of the dose excreted within 12 h after dosing, respectively. Urine, bile and feces accounted for 49-52%, 37% and 12% of the dose in rats, respectively. Elimination was similarly rapid in dogs with urine and feces accounting for 34-36% and 55-58% of the dose, respectively. In metabolite profiling studies, INCB018424 constituted <3% of the radioactivity in mouse and rat excreta and ~15% in dog, indicating extensive metabolism. In mice and dogs, unchanged drug was the primary component in circulation, while in rats, metabolites predominated. Species differences in metabolite profiles were minor with circulating and excreted metabolites mainly consisting of hydroxylations, ketones and in some cases, subsequent O-glucuronides. There were minimal gender differences in plasma pharmacokinetics for INCB018424 in mice and dog. In rats, however, females had higher circulating levels of INCB018424 compared to males, consistent with in vitro data showing extensive metabolism by male rat specific isozymes, CYP2C11, CYP2C13 and CYP3A2, but not the female specific isozyme, CYP2C12.
125. Metabolic differences between sprague dawley and wistar han male rats
James P. Chovan, Erya Yu, and Steven C. Ring
Dept of Drug Safety & Disp, Cephalon, Inc., West Chester, PA, USA, 19380-4245
While Sprague Dawley (SD) rats are often used for nonclinical safety testing, Wistar Han (WH) rats are also utilized, particularly in certain contract research organizations, sometimes necessitating a mid-program change in the strain being used. The current study was designed to compare the metabolic capabilities of the 2 strains in vitro to enable an assessment of their likely metabolic profiles in vivo. The rates of metabolism of several standard probe substrates for human cytochrome P450 (CYP) enzymes were examined using liver microsomes from male SD and WH rats. While these substrates are not as specific for individual CYP enzymes in rats as in humans and often are metabolized by CYPs in different families of enzymes in the rats, the CYP enzymes associated with each reaction in male SD rats have previously been identified (Xenobiotica 2007;37(5):459-73). Reactions that were faster in SD than in WH liver microsomes were methoxy- and ethoxyresorufin dealkylation (CYP2C6/11 > CYP2A1/2 and CYP2E1), pentoxyresorufin dealkylation (CYP1B1 >> CYP2C6/11), and benzyloxyresorufin dearylation (CYP1B1 >> CYP2C6/11). Reactions that were faster in WH than in SD liver microsomes included triazolam 1-hydroxylation (CYP3A1/2) and lauric acid 12hydroxylation (no CYP identified; recombinant rat CYP4A was not available for testing). Several other reactions were also tested, but showed no material differences between the strains. Overall, the results suggest that there are many metabolic similarities between SD and WH rats, but that the pharmacokinetics and metabolic profiles of test compounds cannot be assumed to be equivalent in the 2 strains.
126. Gender Differences in oral drug exposure in the rat with the gamma-secretase inhibitor BMS-708163
Wendy Joy Clarke1, Donglu Zhang2, Sarah J. Taylor3, Ramaswamy. A. Iyer4, Nirmala Raghavan5, W. Griffith Humphreys4, Kenneth Stephen Santone6, Vinod K. Arora7, Thomas Philip7, Yue-Zhong Shu8, Richard Burrell9, John E. Leet10, Kevin Gillman11, John E. Starrett Jr.11, Richard E. Olson11, Jere E. Meredith12, and Kimberley Lentz13
1Dept of Metabolism and Pharmacokinetics, Bristol-Myers Squibb, Wallingford, CT, USA, 06492-7660
2Bristol-Myers Squibb Research and Development, Princeton, NJ, USA, 08543
3Bioanaytical Research, Bristol-Myers Squibb, Wallingford, CT, USA, 06492-7660
4Department of Biotransformation-PCO, Bristol-Myers Squibb, Princeton, NJ, USA, 08543
5Biotransformation, Bristol-Myers Squibb, Princeton, NJ, USA, 08543
6Dept of Metab & Pharmacok, Bristol-Myers Squibb Co, Wallingford, CT, 6492
7Dept of Discovery Biotransformation, Bristol-Myers Squibb, Wallingford, CT, 6410
8Pharmaceutical Candidate Optimization, Bristol-Myers Squibb Company, Wallingford, CT, USA, 06492
9Chemical Synthesis, Bristol-Myers Squibb, Wallingford, CT, 06492
10Synthesis and Analysis Technology Team, Bristol-Myers Squibb Company, Wallingford, CT, USA, 06492
11Neuroscience Chemistry, Bristol-Myers Squibb, Wallingford, CT, USA, 06492
12Neuroscience Biology, Bristol-Myers Squibb, Wallingford, CT, USA, 06492
13Metabolism and Pharmacokinetics, Bristol-Myers Squibb Co, Wallingford, CT, USA, 06492
The effect of gender on the pharmacokinetics of drugs in rats is well recognized. This work describes a substantial gender difference in oral exposure in Sprague Dawley rats with BMS708163, a potent and selective gamma-secretase inhibitor, which is being developed for the treatment of Alzheimer’s Disease (AD). Oral AUC in female rats was 40-70 fold greater than in male rats at equivalent doses. After an oral predose (100 mg/kg) of 1-Aminobenzotriazole (ABT), a potent nonspecific inhibitor of cytochromes P450, the AUC of BMS708163 was similar between male and female rats, indicating involvement of CYP-mediated metabolism. A similar result was obtained after an IP predose (500 mg/kg) of Troleandomycin (TRO), a CYP3A inhibitor, prior to oral administration of BMS708163. Taken together, these data support the involvement of CYPmediated metabolism in the observed gender differences in male and female rats. Further in vitro and in vivo experiments with [14C]BMS-708163 showed that the metabolic pathways differed between male and female rats. Metabolism of [14C]BMS708163 in male rats was predominantly catalyzed by CYP3A2, to form an oxidative metabolite, M5. In female rats the much slower metabolic reaction of [14C]BMS-708163 appeared to be catalyzed by CYP2C to form M6, an oxadiazole metabolite. These results suggest that the large gender difference in pharmacokinetics of BMS-708163 was caused by differential expression of metabolizing enzymes in male and female rats.
127. Disposition of a gamma-secretase inhibitor 14C-labeled BMS-708163 in mice, rats, rabbits, dogs, and humans. Applications of bile collection in differentiating oxidative versus reductive metabolic pathways
Donglu Zhang1, Mark Ma1, Nirmala Raghavan1, Lifei Wang1, Richard Burrell2, Huidong Gu1, Vinod Arora3, Thomas Philip3, Yue-Zhong Shu4, Ming Zheng5, Randy Dockens5, Wendy Clarke3, Kimberley Lentz3, W. Griffith Humphreys1, and Ramaswamy Iyer1
1Pharmaceutical Candidate Optimization, Bristol-Myers Squibb, Princeton, NJ, USA, 08543
2Chemical Synthesis, Bristol-Myers Squibb, CT, USA
3Pharmaceutical Candidate Optimization, Bristol-Myers Squibb, CT, USA
4Pharmaceutical Candidate Optimization, Bristol-Myers Squibb, CT, USA, 08543
5Discovery Medicines and Clinical Pharmacology, Bristol-Myers Squibb, NJ, USA
BMS708163, a potent and selective gamma-secretase inhibitor, is being developed for the treatment of Alzheimer’s Disease (AD). Following oral administration of 14C-labeled BMS-708163 to mice, rats, rabbits, dogs, and humans, the parent and inactive oxa-diazole metabolite M6 were identified as the predominant circulating components. All species showed good dose recoveries (>85%) with majority of the dose excreted in the feces. There was no parent drug detected in urine, feces, or bile suggesting that the absorbed dose was mainly cleared through metabolism. All the metabolites observed in humans were also detected in animal species, suggesting that the animal species used in toxicology studies were exposed to the same drug-related components as humans. Most of the drug-related components in bile samples were oxidative metabolites. However, fecal samples showed mainly metabolites where the diazole-ring was reduced, suggesting that the biliary metabolites were reduced by GI contents prior to fecal elimination. The oxidative metabolism of BMS-708163 was mediated through cytochrome P450 (CYP3A and 2C). These results showed that bile collection was essential to differentiate role of oxidative versus reductive metabolism in the overall elimination of BMS-708163. In summary, BMS-708163 was cleared mainly through metabolism in all species with oxidation playing a significant role in its elimination.
128. Twenty year, retrospective analysis of CYP activity levels in microsomes isolated from human donor livers
Charles Crespi1, Sweta Parikh2, Zoe E. Barter3, Teri Bordonaro2, Edward Francis2, and Christopher J. Patten4
1Dept of Discovery Labware, BD Biosciences, Woburn, WA, USA, 01801
2BD Biosciences, Woburn, MA, USA, 01801
3Academic Unit of Clinical Pharmacology, University of Sheffield and Simcyp Limited, Sheffield, United Kingdom, S2 4SU
4R & D, BD Biosciences, Woburn, MA, USA, 01801
Human donor livers, originating from US-based Organ Procurement Organizations, have been the basis for the preparation of key reagents used for the characterization of drugs and drug candidates. Over time, the practices for liver transplantation have evolved which has indirectly affected the nature of organs which are unused for transplantation and are made available for research use. We have prepared microsomes from and characterized the enzyme activity levels of CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6 and CYP3A in over 300 donor livers, procured from 1985 to the present and conducted detailed statistical analyses of activity distributions and trends. We have found the following:
CYP2B6 and CYP2C19 show the highest variability among donors (CV 150-175%) CYP2C9 (CV 47%) shows the lowest variability with CYP1A2, CYP2D6 and CYP3A4 (CV 82-85%) being intermediate. (Note: the CV values above are based on an assumption of Normality).
The age of donors did not change significantly with year of donation. The median age for females was 52 while males it was 50. The levels of CYP activity did not vary with donor age for all CYPs except CYP2C19 where a statistically significant decrease in activity with age was observed.
The gender distribution was 63% male and 37% female. The gender difference was observed CYP3A4. On average, females had 34% higher activity. While the average activity for CYP2C19 was 48% higher in females, this difference was not statistically significant.
No significant change in any enzyme activity as a function of year of donation. Therefore, liver samples obtained over a large period of time are suitable for creation and predicting the properties of pools.
Monte Carlo analyses for a 50 donor pool predicted an average CV for these 6 enzyme activities of 13%. The actual CV observed for 4 pools prepared in a manufacturing context was 12%. Monte Carlo analysis on virtual pools comprised of 25 to 200 donors indicates that the donor number needs to be increased to over 120 in order to obtain batch to batch CVs for all enzymes of less than 10%. The implications of these analyses for pool design will be discussed.
129. Interaction of mycobacterium tuberculosis CYP130 with heterocyclic arylamines compounds
Hugues Ouellet1, Larissa M. Podust1, Jens P. von Kries2, and Paul R. Ortiz de Montellano1
1Pharmaceutical Chemistry, UCSF, San Francisco, CA, USA, 94158
2Screening Unit, Leibniz-Institute for Molecular Pharmacology, Berlin, Germany
Tuberculosis remains a leading cause of human mortality. The emergence of strains of Mycobacterium tuberculosis, the causative agent, that are resistant to the major frontline antitubercular drugs increases the urgency for the development of new therapeutic agents. Sequencing of the M. tuberculosis genome revealed the existence of twenty cytochrome P450 enzymes, some of which are potential candidates for drug targeting. CYP130 is absent from the genome of Mycobacterium bovis, suggesting that it might play specific role(s) for the infection of the human host, and thus constitutes a potential candidate for drug targeting. We have shown previously that CYP130 binds azole drugs (econazole, clotrimazole, miconazole and ketoconazole), and crystal structures of ligand-free and econazole-bound protein were determined.1 Since the identification of the in vivo substrate for this P450 enzyme still remains elusive, a high throughput screening study was performed to identify novel molecular substrate/inhibitor scaffolds. This screening assay is based on the detection of ligand induced heme perturbations by type-I or type-II ligands. In this work, we report the results of the high throughput screening of 20,000 compounds, with the identification of heterocyclic arylamines as major hits. We also present a spectroscopic, biochemical and x-ray structure analysis of selected compounds. Remarkably, the topology of the CYP130 active site favors angular coordination of the arylamine group over the orthogonal coordination of azoles. Upon substitution of Gly243 by Ala, the binding mode of azoles and some arylamines reverts from type-II to type-I due to hydrophobic and steric interactions with the alanine side chain. We suggest a role for the conserved Ala(Gly)243-Gly244 motif in the I-helix in modulating both the binding affinity of the axial water ligand and the ligand selectivity of P450 enzymes. This work was supported by NIH RO1 AI07824, GM25515 and GM078553, and BMBF/PTJ grants.
References
- Ouellet H., Podust L. M., Ortiz de Montellano P. R. Mycobacterium tuberculosis CYP130: Crystal structure, biophysical characterization, and interactions with antifungal azole drugs. J. Biol. Chem (2008), 283(8), 5069-5080.
130. Theory vs. realty: Is free drug concentration in adipose equal to free concentration in plasma?
Mei-Mann Hsueh1, Mengmeng Wang2, Bogdan Sleckza1, Celia Dareinzo1, Chris Freeden1, Pamela Abraham1, Saeho Chong1, and Timothy Harper1
1Dept of PCO/MAP, Bristol Myers Squibbs, Pennington, NJ, USA, 8536
2Discovery Pharmacokinetics, Drug Safety and Metabolism, Wyeth Research, Andover, MA, USA, 01810
Purpose: Although it is the free drug concentration in the target organ that is believed to be important in pharmacokinetic and pharmacodynamic (PKPD) modeling (Derendorf and Hochhaus, 1995), it is still a common practice in drug development to collect tissue samples, such as adipose, liver and brain, to determine total drug concentrations in tissues of interest. Under freely diffusible conditions, the concentration of free drug in tissue will be in equilibrium with free drug in plasma. Since the fraction unbound in tissue (fut) = [unbound drug] tissue/[total drug] tissue and the fraction unbound in plasma (fup) = [unbound drug]plasma/[total drug]plasma, the ratio of [total drug]tissue/][total drug]plasma will be equivalent to fup /fut (Fichtl, et al., 1991). In this study, 6 commercial compounds with diverse structures and a wide range of plasma protein binding values (<3% to >99%) were evaluated in vitro and in vivo in an effort to understand the relationship between free drug concentrations in plasma and adipose tissue.
Methods: In vivo - Each compound was administered i.v. at a dose of 5 mg/kg to CD-1 mice. Plasma samples were collected at 3, 10, 30 min, 1, 3, 6, 8, 24 hr after dosing and adipose samples were collected at 1 and 3 hrs after dosing and homogenized. Concentrations of each compound in plasma and adipose homogenate were determined and the PK parameters were estimated. The free adipose and free plasma concentrations were calculated based on total plasma and adipose concentrations determined in vivo, and the fua and fup values determined in vitro. In vitro - Plasma samples were dialyzed against buffer for 4 hours in a 96-well equilibrium dialysis apparatus to determine the fraction unbound in plasma (fup). Homogenized adipose samples were diluted 2-, 5-, 12.5-, 31- and 78-fold and dialyzed against buffer for 4 hr in a 96-well equilibrium dialysis apparatus to determine the fraction unbound in the diluted adipose homogenate (fua-dilution factor) Fraction unbound in undiluted adipose (fua) was then determined by extrapolating from the fua-dilutiong factor vs. dilution factor.
Results and conclusions: The PK parameters of all 6 compounds were consistent with the literature data. For clarithromycin, levofloxacin, and amiodarone, the free concentration in adipose (Adipose*fua) was within 2-fold of the free plasma concentration (plasma*fup). For metaclopramide, gabapentin and meloxicam, the free concentration in adipose was within 3-fold of the free plasma concentration. Overall, the free adipose concentration was roughly equal to free plasma concentration for all the 6 compounds tested, within a 3-fold error range. Assuming the “free-drug hypothesis” holds, these results suggest that determining total drug concentrations in target tissues may be of much less value for PK/PD correlations than determining the free plasma concentration - an important consideration in a resource-limited environment.
References
- B. Fichtl, A.V. Nieciecki and K. Walter, Adv. Drug Res. 20 (1991), pp. 117–165.
- H. Derendorf and G. Hochhaus, (1995) Handbook of Pharmacokinetic/ Pharmacodynamic Correlation. CRC press.
131. Abstract withdrawn
132. Altered pharmacokinetics of iricotecan and its active metabolite, SN-38 in cisplatin-induced acute kidney injury rats
Kensuke Tazoe1, Koji Yokoo2, Yuji Yamakawa2, Akinobu Hamada3, and Hideyuki Saito4
1Department of Clinical Pharmaceutical Sciences, Graduate School of Pharmaceutical Sciences, Kumamoto University, Kumamoto, Japan, 862-0976
2Department of Clinical Pharmaceutical Sciences, Graduate School of Pharmaceutical Sciences, Kumamoto University, Kumamoto, Japan
3Department of Pharmacy, Kumamoto University Hospital, Kumamoto, Japan, 860-8556
4Department of Pharmacy, Kumamoto University Hospital, Kumamoto, Jordan, 860-8556
Irinotecan (CPT-11) is an anticancer drug used in the treatment of various types of cancers. Despite that the urinary excretion rate of CPT-11 is 20-30%, patients with lower creatinine clearance have a higher risk of developing grade 3/4 neutropenia by the treatment with CPT-11. However, the association of renal function with CPT-11 pharmacokinetics remains unknown. The aim of this study was to clarify the details of the alteration of CPT-11 pharmacokinetics in acute kidney injury (AKI) rats induced by cisplatin (CDDP). CPT-11 (10 mg/kg) was intravenously administered to AKI rats made by intraperitoneally administration of 10 mg/kg CDDP 72 h before the CPT-11 treatment. The concentrations of CPT-11 and SN-38 were determined by high-performance liquid chromatography. The plasma concentrations of CPT-11 and SN-38 were markedly elevated in AKI rats compared to those in control rats. In particular, the area under the plasma concentration time curve of CPT-11 and SN-38 increased to 190% and 170%, respectively. The urinary excretion rate of CPT-11 and SN-38 were markedly decreased to 2.5% and 2.0%, respectively. In contrast, the biliary excretion rate of CPT-11, but not of SN-38, was significantly increased. Immunoblotting was performed to determine the protein expression of ABC transporters mediating the excretion of CPT-11 and SN-38. The expressions of P-glycoprotein(P-gp) and breast cancer resistance protein(Bcrp) in the kidney and liver appeared to be elevated by AKI. The expression level of multidrug resistance-associated protein 2 was increased in the kidney and decreased in the liver. It was suggested that the up-regulation of the hepatic P-gp and Bcrp could result in the increased biliary excretion of CPT-11 in AKI rats. In contrast, these efflux transporters might not be involved in renal excretion of CPT-11 and SN-38. CDDP-induced AKI resulted in a decrease in the urinary excretion rate of CPT-11 and SN-38, which is associated in part with the increased plasma concentration of CPT-11 and SN-38.
133. Pharmacokinetics of M118, unfractionated heparin and enoxaparin sodium in normal and 5/6 nepherectomized uremic rats
Dragomir I. Draganov1, Teresa L. Wright2, William Avery2, Terry L. Johnson1, Daniel W. Sved1, and Ian Fier2
1Metabolism, WIL Research Laboratories, LLC, Ashland, OH, USA, 44805
2Momenta Pharmaceuticals, Inc., Cambridge, MA, 02142
M118 is a next-generation low molecular weight heparin product that was rationally engineered to provide flexible supportive anticoagulant therapy to patients with acute coronary syndromes. This study was conducted to evaluate the pharmacokinetics of M118 as determined by the plasma Anti-factor Xa (Anti-Xa) and Anti-factor IIa (Anti-IIa) activities in normal and uremic (5/6 nephrectomized) male rats after intravenous or subcutaneous administration in comparison with unfractionated heparin (UFH, for the intravenous route) and enoxaparin sodium (EPS, for the subcutaneous route). The results for UFH and EPS concur with published data on pharmacokinetics/pharmacodynamics of these anticoagulants in experimental or clinical renal insufficiency. The pharmacokinetic parameters measured by plasma Anti-Xa and Anti-IIa activities for UFH administered intravenously were comparable between normal and 5/6 nephrectomized rats, whereas systemic exposure to EPS was about 2-fold higher in the 5/6 nephrectomized rats than in the normal rats. After IV administration, exposure to M118 in the uremic rats was about 50% higher than in normal rats (AUClast 5.48 and 8.20 Anti-Xa IU×h/mL for normal and uremic rats, respectively). However, after subcutaneous administration and in contrast to EPS results, the PK parameters for M118 were comparable between normal and 5/6 nephrectomized rats (AUClast 3.62 vs. 4.69 Anti-Xa IU×h/mL, Cmax 1.59 vs. 1.27 Anti-Xa IU/mL and half-life 0.96 vs. 0.87 h for normal vs uremic rats, respectively). In conclusion, renal insufficiency may not require adjustment in M118 dosage when the drug is administered subcutaneously.
134. Classification of drugs according to presence or absence of CNS activity based on mechanistic QSAR models of the rate and extent of brain delivery
Kiril Lanevskij, Pranas Japertas, and Remigijus Didziapetris
ACD/Labs, Inc., Vilnius, Lithuania, LT-08117
This study presents a simple classification scheme for in silico evaluation whether brain penetration of novel compounds is sufficient to exhibit central action. Classification is performed taking into account both kinetic and thermodynamic characteristics of drug transport across blood-brain barrier. The calculation procedure for the brain penetration rate expressed by permeability-surface area product (log PS) was described previously (Lanevskij K, Japertas P, Didziapetris R, Petrauskas A. Ionization-specific prediction of blood-brain permeability. J Pharm Sci. 2009 Jan;98(1):122-34). The extent of brain penetration was represented by experimentally determined steady-state brain/blood distribution ratios (log BB) for about 500 compounds collected from literature. These data were split into two terms corresponding to drug binding to plasma proteins and brain constituents – the two major processes that influence partitioning between brain and plasma under the assumption of passive diffusion-driven transport. Brain tissue binding affinity of drugs was then described by a nonlinear model in terms of key physicochemical determinants – octanol/water log P and pKa. Prediction of CNS activity was performed on the basis of calculated log BB and supplementary parameter brain/plasma equilibration rate defined as log PS corrected for unbound fraction in brain. It was shown that a simple combination of the respective models allows correctly classifying more than 90% of drugs in the literature data set comprised of about 1600 diverse molecules with experimentally assigned CNS activity categories (CNS+/CNS-). Moreover, as demonstrated by several examples, the proposed classification scheme provides an insight on the onset and duration of action of central drugs.
135. Carbamazepine (CBZ) distribution in the central nervous system of freely moving rats: Correlation between brain Extracellular Fluid (ECF) and Cerebrospinal Fluid (CSF) exposure
Huadong Sun, Victor Saldivia, Henrianna Pang, Yingo Yang, and Ines A.M. de Lannoy
NoAb BioDiscoveries Inc., Mississauga, ON, Canada, L5N 8G4
To investigate whether exposures of the anticonvulsant CBZ in the brain ECF and CSF are comparable, brain ECF concentrations of CBZ were determined by quantitative intracerebral microdialysis and compared to results from CSF collection studies.1 A brain probe was surgically implanted into the striatum and the femoral vein and artery were catheterized for drug administration/blood replacement and blood sampling, respectively. The probe was perfused with artificial CSF (0.5 μL/min) and dialysate samples collected every 20 min. Plasma was sampled at the midpoint of the dialysate collection interval. CBZ and its active metabolite, 10,11-epoxide CBZ (ECBZ), in plasma and dialysate samples were quantified by LC-MS/MS analysis. Calibration of the probes was first evaluated in vitro: the relative loss (RL) of CBZ, ECBZ and deuterated CBZ (d10-CBZ) from the probes equalled the respective relative recoveries (RR), and RL of d10-CBZ equalled the RR of CBZ and ECBZ. The unbound plasma and brain ECF concentrations of CBZ and ECBZ, were determined both after a single i.v. bolus dose of 4 mg/kg CBZ and at steady-state during constant rate i.v. infusion of 1 mg/h/kg CBZ. The RL of d10-CBZ was used as a calibrator throughout the study. The RL of CBZ, ECBZ and d10-CBZ in vivo were also determined following a 24 hour wash-out period. In vivo, the RL of d10-CBZ equalled the RL of CBZ, however, the RL of ECBZ was only ~half of that of d10-CBZ and CBZ. Following a bolus dose of CBZ, both CBZ and ECBZ exhibited good brain distribution. The ratios of the area under the unbound concentration versus time curves (AUC) for brain ECF and plasma were 0.76 ± 0.13 and 0.84 ± 0.11 for CBZ and ECBZ, respectively (n=7). These values are consistent with the ratios of the unbound brain ECF to unbound plasma concentrations determined at steady-state (0.60 ± 0.15 for CBZ and 0.64 ± 0.12 for ECBZ, n=7). The CSF to unbound plasma AUC ratio for CBZ determined previously (0.71 ± 0.08 (n=7)), compares favourably to these values, indicating that CSF exposure is an appropriate surrogate for brain ECF exposure of CBZ.
References
- Saldivia, V. et al. Pharmacokinetics of Carbamazepine in Serial Samples of Plasma and CSF Following Intravenous Administration to Conscious Rats. Drug Metab. Rev. 40 (Suppl. 3): 108–109 (2008).
136. Mass balance, metabolism and excretion of 14C-Thalidomide in healthy human subjects
Sekhar Surapaneni1, Maxine Stoltz2, Martijn Hilhorst3, Gondi Kumar1, and Oscar Laskin2
1Drug Metabolism & Pharmacokinetics, Celgene Corporation, Summit, NJ, USA, 07901
2Early Drug Development, Celgene Corporation, Summit, NJ, USA, 07901
3PRA international, Assen, Netherlands
Thalidomide has immunomodulatory and antiangiogenic effects and clinical benefits have been shown in a number of clinical conditions. The purpose of this study was to evaluate the metabolism and excretion of thalidomide in healthy human subjects (n = 8) following a single oral dose of 400 mg (~3.7 MBq) 14C-Thalidomide. Following administration, plasma, urine and fecal samples were collected for 7 days. Mass balance and metabolic profiling were conducted using liquid scintillation counting and radio-HPLC-MS/MS, respectively. Radioactivity was mainly excreted in urine. (>90% of the dose), with only a small amount excreted in feces (<1.3% of the dose). The majority of the urinary radioactivity consisted of two hydrolysed compounds N-(o-carboxybenzoyl) glutarimide and phthaloyl isoglutamine, accounting for approximately 36% and 33% of the dose, respectively. A third hydrolytic metabolite, pthaloyl glutamine, was also present in urine (3.7% of the dose). In addition to these singly hydrolyzed metabolites, hydrolysis products of both glutarimide and pthalimide rings, N(o-carboxybenzoyl) glutamine and N(o-carboxybenzoyl) isoglutamine, were also detected in urine to a minor extent. In plasma, unchanged thalidomide was the major component (>80% of the plasma radioactivity). In addition to thalidomide, hydrolytic products N-(o-carboxybenzoyl) glutarimide and phthaloyl isoglutamine were also present in plasma (<15% of plasma radioactivity). Although hydroxylated forms of thalidomide or their hydrolysis products were observed, they accounted for a negligible fraction of both circulating and excreted radioactivity and the levels were below the detection limit (8.9 ng Eq/mL). Overall, hydrolysis of pthalimide and glutarimide rings is predominant clearance mechanism of thalidomide in humans, with minimal contribution of oxidative metabolism.
137. Characterization of the hepatobiliary disposition of the anti-parasitic prodrug, pafuramidine, in isolated perfused rat livers provides a mechanistic explanation for the prolonged hepatic accumulation of the active metabolite furamidine
Zhixia (Grace) Yan1, Mary F. Paine1, Richard R. Tidwell2, James E. Hall2, and Kim L.R. Brouwer1
1Division of Pharmacotherapy and Experimental Therapeutics, UNC Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC, USA, 27599
2Department of Pathology and Laboratory Medicine, School of Medicine, University of North Carolina, Chapel Hill, NC, USA, 27599
Purpose: Pafuramidine, a prodrug of furamidine, is the only orally-active agent that has shown promise in clinical trials for the treatment of early-stage African trypanosomiasis. Clinical development of pafuramidine recently was suspended due, in part, to delayed hepatotoxicity. An improved understanding of the hepatic disposition of this prodrug is, therefore, imperative. As such, the hepatobiliary disposition of pafuramidine and derived metabolites was characterized in isolated perfused rat livers (IPLs).
Methods: IPLs were prepared from male Wistar rats using standard techniques; perfusion was conducted ex situ over designated times (up to 2h) in a temperature-controlled chamber with recirculating Krebs-Henseleit buffer containing 20% (v/v) rat blood. Pafuramidine (10 μM) was added as a bolus dose to the perfusate reservoir. Bile and perfusate were collected during perfusion; livers were harvested and homogenized at the end of perfusion. All samples were analyzed by LC-MS/MS. Stepwise nonlinear least-squares regression analysis was used to fit a compartmental model to pafuramidine and derived metabolites mass vs. time data from perfusate, liver, and bile. The unbound fraction of furamidine in liver homogenate was determined by ultrafiltration.
Results: Hepatic clearance of pafuramidine (18 ml/min) was comparable to perfusate flow rate (20 ml/min); the extraction ratio of pafuramidine was 0.88. The extent of conversion of pafuramidine to furamidine increased with perfusion time (41% at 2h). The rate constant for biliary excretion of furamidine was 4-fold greater than that for basolateral (liver to perfusate) efflux (0.12 vs. 0.03 h−1). Hepatic accumulation of furamidine over 2h was extensive (99% of total formed). The hepatic unbound fraction of furamidine was <1%.
Conclusions: Pafuramidine was taken up and biotransformed rapidly to the active metabolite, furamidine, in rat IPLs. Basolateral efflux was rate limiting in furamidine hepatic excretion. The significant and prolonged hepatic accumulation of furamidine is consistent with previous in vivo data demonstrating that furamidine binds to intracellular compartment(s) with high affinity. Hepatocellular sequestration may limit the systemic exposure of furamidine and predispose the liver to toxicity.
Supported by the Bill and Melinda Gates Foundation and NIH GM41935
138. A Cross-species comparison of plasma stability using amide, ester and phosphoester substrates
Ainslie R. Ainslie, Erika Manyak, Michael Rooney, and Simon Roberts
Drug Metabolism & Pharmacokinetics, AstraZeneca, Waltham, MA, USA, 02451
The stability of a new chemical entity (NCE) in plasma is highly relevant in drug discovery as poor stability may cause poor in vivo pharmacokinetics (PK), or in the case of prodrugs, provide an additional means of biotransformation to the efficacious drug form. It is important to understand the stability of a NCE in human plasma as well as preclinical species to accurately interpret PK results. We evaluated the ability of plasma from mouse, rat, dog and human to metabolize several substrates of esterase, amidase and alkaline phosphatase (ALP) enzymes. Additionally we evaluated assay conditions that impact the measurement of plasma stability in vitro. Incubations with substrates for esterase, amidase, and ALP enzymes were incubated at 1μM at 37°C for 4 h in the presence of plasma from each of the four species. At designated time-points aliquots were removed from the incubations and reaction quenched by precipitating proteins with cold acetonitrile. Quantitation was done using LC/MS/MS analysis monitoring for the disappearance of substrate. We did not observe a clear trend between species in the case of amidase and esterase substrates. However, the hydrolysis rates of ALP substrates were consistent between all preclinical species and were 4-fold faster than the rates from human plasma. Using either plasma thawed after storage at -20°C for 72 h or freshly collected plasma for incubations showed no observable difference in amidase and esterase probe stability. In contrast, ALP was nearly completely inactivated under these storage conditions. Furthermore, we observed that plasma prepared via filtration does not retain enzyme activity as well as that by which was prepared through centrifugation. We conclude that there is an observed difference in amidase, esterase, and ALP activity dependent on plasma preparation and storage.
139. Reactive metabolite screening by UPLC/MS using multiple acquisition functions in a serial and parallel mode
John Shockcor, Jose Castro-Perez, Kate Yu, and Henry Shion
Waters Corporation, Milford, MA, USA, 01757
The study of reactive metabolites has been gaining more and more attention in modern drug development due to its difficulty (not stable) and high potential for toxicology. A common way to study the reactive metabolites is by tapping them with a nucleophilic reagent such as Glutathione (GSH). By detecting the GSH conjugates, potential reactive metabolites can be identified. One of the common practices of GSH trapping is to perform in vitro microsome incubation with GSH reagent added into the reaction mix. The GSH conjugates can be quickly screened using an LC/MS/MS approach, thus provide a quick assessment of the potential toxicology effect for the parent drug. A few MS experiments are commonly used to help screening the GSH conjugates. In positive ESI, a loss of m/z 129 (pyroglutamic acid), 307 (aliphatic and benzylic thioethers), or 147 (thioesters) can be monitored. In negative ESI mode, monitor the precursor ion at m/z 272 (gama glutamyldehydroanalyl-glycine). In this work, we will present an approach where multiple MSMS data can be obtained during a single UPLC injection, thus allowing effective screening for reactive metabolites at very high throughput. An antidepressant drug nefazodone was used as the example drug. The drug was incubated using human liver microsome with GSH reagent for 60 minutes. The sample was analyzed by the Waters ACQUITY UPLC/Xevo TQ MS system. The new feature from the TQ system, ScanWave, allows ions to be trapped in the collision cell and released according to their m/z ratios. The release of these ions is synchronized with the scanning of MS2, which greatly improves the duty cycle and enhances the sensitivity in scanning acquisition mode. Compared with the standard mode of operation, significant sensitivity enhancement was observed during experiments when ScanWave was enabled, this was specifically helpful for data dependent analysis such as precursor scan triggered product ion scan, neutral loss triggered product ion scan etc. Multiple examples will be shown during the presentation. This significantly enhanced speed and sensitivity obtained from ScanWave also allowed us to perform MS full scan, MRM and multiple product ion scans within the same LC injection. As a result, using ACQUITY UPLC/Xevo TQ MS system with ScanWave enabled, the screening of GSH conjugates of Dlicofenac can be accomplished at very high throughput as multiple experiments can be performed with a single LC injection.
140. Challenges in metabolite identification in-vivo: the use of high resolution TOF MS with sub 1 ppm mass accuracies & extended dynamic range combined with intelligent software tools
Jose Castro-Perez1, John Shockcor1, Kate Yu1, Henry Shion1, and Yasuhiro Yamada2
1Waters Corporation, Milford, MA, USA, 01757
2Showa University, Tokyo, Japan
With the current publication from the FDA on the guidance of metabolites in safety testing (MIST), there is added emphasis on the need to have adequate LC/MS platforms to be able to accommodate these new challenges. In order to address these challenges, we will present an LC/MS workflow that consisted of a hybrid quadrupole oa TOF with enhanced dynamic range and improved spectral resolution. The improved dynamic range is achieved via a novel ADC based detection system which allows us to obtain a dynamic range of over 104. The enhanced spectral resolution of over 40,000 FWHM is achieved using a folded oa-ToF geometry with high field extraction and multiple dual stage and single stage reflectrons. Rat plasma, bile and urine samples taken at different time points were used for these analyses. The animals were administered a 10 mg/kg dose of Ritonavir. Chromatographic separation was accomplished by using UPLC at a flow rate of 0.5 ml/min with ammonium bicarbonate buffer at pH 5.0 in Water (A) and in Acetonitrile (B). An Acquity HSS T3 column (1.8 mm, 2.1 × 100 mm) was employed. The gradient time was 20 min. An MSE strategy using two MS acquisition functions was used during a single LC injection, function one acquired the low collision energy data whilst function two acquired the high collision energy data.
For these experiments, a model compound Ritonavir a petidometic inhibitor of the HIV-1 protease was analyzed in plasma, urine and bile. For in-vivo metabolite identification, the key method development criterion is good chromatographic resolution, sensitivity and accuracy of the results. By this approach, the results revealed all metabolites of Ritonavir in the three different biological matrices. It metabolized extensively to a number of Oxidative metabolites at m/z 737. In the bile sample the predominant metabolic route manifested as the glucuronidated conjugation pathway. The MSE strategy allowed for an easy –to-review data analysis of fragment ions and corresponding alignment with the low energy data so that structural information could be derived from a single injection. Metabolite identification was performed automatically by the use of the application software provided by the instrument vendor (MetaboLynx XS). During data processing, the software first identifies the major dealkylated fragments that can be generated by the parent drug and uses this information to generate and targeted mass defect filters to eliminate false positives. It then uses the parent drug as well as the major dealkylated fragments to compare the dosed sample with the control sample and extract expected and unexpected metabolites.
141. A Generic ultra fast and sensitive UPLC/TOF MSE strategy to determine in-vitro rates and routes on metabolism
Stephen McDonald1, Jose Castro-Perez2, John Shockcor2, Kate Yu2, and Henry Shion2
1Waters, Beverly, MA, USA, 94568
2Waters Corporation, Milford, MA, USA, 01757
Metabolite identification (Met ID) is a crucial component of the modern drug development process. Resolution plays a key role for positive identification of metabolites from complex matrices. In addition, speed and sensitivity are two critical factors to consider for successful met ID studies. In this work, we will present a simple and generic strategy for met ID studies using high resolution TOF MS, high speed and high sensitivity. A serotonin 5-HT4 receptor agonist Cisapride was used as the example parent drug to demonstrate this methodology. The sample used was an in-vitro incubation of cisapride in rat liver microsomes (RLM) at 0.1 uM incubation level. A Waters ACQUITY UPLC/Xevo Qtof system was used for the sample analysis. The data acquisition strategy was conducted utilizing the TOF MSE functionality of this particular MS system, in which the mass spectrometer acquires data into two separate discrete functions. One acquisition function is set at a low collision energy and the other acquisition function is set at high collision energy. This resulted in the generation of parent drug and metabolites information (both intact and fragment ions) within a single LC injection. The UPLC offers fast separations with superior chromatographic resolution, and the XevoQtof MS offers analyte detection with exact mass measurement at very high data acquisition rate (up to 20 spectra/second) perfectly accommodating narrow UPLC peaks with sufficient data points across the chromatogram/s. Major metabolites were detected which corresponded to single hydroxylations and metabolic cleavage of the 4-amino-5-chloro-2-methoxy-N-(3- methoxypiperidin-4-yl) benzamide moiety losing the 1-fluoro-4-propoxybenzene motif. These metabolites were detected by the use of a software algorithm MetaboLynx XS which allows for the generation of metabolic cleavages and mass defect filters in an automated fashion with the aim of removing false positives to leave only drug-related material.
142. Full scan data acquisition for rapid quantitative and qualitative analysis using the exactive LC-MS high resolution mass spectrometer
Lester Taylor, Yingying Huang, Rose Herbold, and Seema Sharma
Thermo Fisher Scientific, San Jose, CA, USA, 95134
Introduction: Current approaches to discovery stage drug metabolism studies (pharmacokinetics, hepatocyte stability, etc) have focused on the use of targeted analysis (MRM) based approaches for quantitative analysis. This necessitates the optimization of parameters such as Q1 and Q3 m/z values, collision energy and interface voltages. These studies only detect the specified compound and information about other components, such as metabolites, is lost. The ability to do full scan acquisition for quantitation eliminates the need for compound optimization while enabling the detection of metabolites and other non-drug related endogenous components.
Method: Samples from a range of studies (hepatocyte stability, pharmacokinetics) were analyzed using the Exactive non-hybrid Orbitrap LC-MS. Sample introduction used UHPLC and fast generic gradients to enable rapid cycle times and high throughput analysis. The mass spectrometer consisted of a compact single stage Orbitrap mass analyzer capable of operating at scan speeds compatible with UHPLC peak widths while maintaining characteristic Orbitrap mass accuracy and resolution.
Preliminary data: Initially, microsomal stability samples were analyzed. Preliminary results indicate that the instrument provides sufficient quantitative capability together with the ability to identify and confirm metabolites. In addition, structure elucidation of metabolites was facilitated by the collection of high resolution accurate mass fragmentation data. The use of extracted ion chromatograms using narrow m/z windows provides selectivity comparable to MRM analysis. Sample sets were further processed to show that metabolites were present in the plasma samples. Further results will be presented that demonstrate that the quantitative-qualitative workflow is possible with this new bench-top single stage mass spectrometer.
Novel Aspect: A bench-top single stage UHPLC-Orbitrap system is used for combined high throughput qualitative and quantitative analysis.
143. A minimalist approach to attenuating bioactivation and improving in vivo tolerability of drug candidates in rodents
Collette D. Linder1, F. Barclay Shilliday1, Shridhar Hegde2, Mark G. Obukowicz2, David Thompson2, and J. Scott Daniels2
1Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development. Pfizer, Inc., Chesterfield, MO, USA, 63017
2Pharmacokinetics Dynamics and Metabolism, Pfizer Global Research and Development. Pfizer, Inc., Chesterfield, MO, USA, 63017
Following oral administration (50- 300 mg/kg) of the aryl amide 1 ([M+H]+ 603), Sprague-Dawley rats experienced an acute liver toxicity that was characterized by increases in serum biomarkers commonly associated with hepatotoxicity; particularly noteworthy was an elevation in total bilirubin (5 fold) and serum transaminases (e.g., ALT, 10-100 fold). Moreover, histopathology indicated mid-zonal lesions and apoptosis consistent with a metabolism-mediated hepatotoxicity. Accompanying this observation was an ADME appraisal which indicated extensive oxidative metabolism of the scaffold and subsequent bioactivation in rat, dog and human liver S9. Results from liquid chromatography, tandem mass spectrometric analysis (LC/MS/MS) demonstrated the formation of several glutathione-derived metabolites ([M+H]+ 926 and 908), producing loss of glutamic acid as the major fragment ion (-129 Da). The type and quantity of sulfhydryl conjugates detected for 1 (net + 323 Da) were indicative of an epoxidation of the aryl amide moiety; these findings were also consistent with the metabolic stability data generated for 1 and ensuing analogs. We surmised that simple disruption of the aromaticity of the aryl moiety - while conserving the integral features critical to pharmacology – would successfully mitigate the bioactivation, and thus result in compounds with improved in vivo tolerability. Indeed, following a 300 mg/kg oral administration of compounds 2 and 3 to Sprague-Dawley rats, the findings from serum analysis were markedly improved. A closer examination of the histopathology would also indicate the absence of hepatotoxicity for 2 and 3. In addition to providing mechanistic insight into the hepatotoxicity of 1, ensuing structural analogs proved successful in mitigating bioactivation of the core scaffold while maintaining exceptional in vitro and in vivo pharmacology.
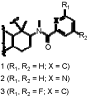
144. Development of a rapid small scale NMR approach that enables metabolite structure determination in sync with the cycle time of drug discovery
Kim Anne Johnson, Xiaohong Liu, Stella Huang, W. Griffith Humphreys, and Yue-Zhong Shu
Pharmaceutical Candidate Optimization, Biotransformation, Bristol-Myers Squibb, Wallingford, CT, USA, 06492
The fast changing drug discovery process requires biotransformation scientists to identify metabolite efficiently and address metabolism-related issues such as metabolic softspots, and reactive and potentially active metabolites in sync with the cycle time of lead-seeking and optimizing activities. Although LC/MS and high resolution LC/MS are widely used as front-line tools for metabolite identification, the CID fragmentation often does not produce spectra informative enough for localizing the site of metabolism. In addition, LC/MS has inherent limitations in elucidating regio- and stereo-chemistry. NMR can be a highly valuable and complimentary tool, particularly due to improved probe technology in recent years. However, NMR is still much less sensitive than LC/MS, and typically requires time-consuming metabolite scale-up, sample clean-up and purification that ultimately restrict its practical use in a drug discovery setting. The present study reports an improved sample preparation workflow in conjunction with the use of state-of-the-art NMR instrumentation. A single 2-5 mL in vitro incubation of drug candidate (20 uM) in microsomes, hepatocytes, or recombinant CYPs was used for metabolite generation. Following precipitation of proteins with acetonitrile and solvent removal, metabolite mixtures were chromatographed with repeated injections using the same LC/MS conditions, and fractions of metabolite collected into a 96-well plate, dried, and reconstituted in NMR solvents. 1H NMR of isolated metabolites was performed on a Varian 600 MHz spectrometer equipped with a 5 mm cryo probe. This methodology has been successfully employed as an extension of LC/MS based metabolite ID and applied frequently to metabolites with quantities of 0.5-10 ug. Most structure determinations on the small scale metabolites were rapidly achieved by 1D 1H-NMR with satisfactory signal/noise ratio, whereas some required 2D 1H-NMR. Numerous examples from our laboratories have demonstrated real impact to medicinal chemistry in a timely manner by avoiding large scale metabolite generation and purification. The presentation will highlight method development and technical details of the approach using a model compound.
145. Effect of co-administration of efavirenz on the pharmacokinetics of proguanil in healthy volunteers
Julius O. Soyinka, and Cyprian O. Onyeji
Pharmaceutical Chemistry, Obafemi Awolowo University, Ile-Ife, Nigeria
Aims: Efavirenz, a HIV non-nucleoside reverse transcriptase inhibitor, and proguanil, an antimalarial agent effective for prophylactic treatment of malaria, are likely to be administered concurrently for treatment of patients with HIV and malaria. The metabolism of proguanil is principally mediated by CYP 2C19 and this isozyme is known to be inhibited by efavirenz. This study, therefore, was aimed at evaluating the pharmacokinetic interaction between efavirenz and proguanil.
Methodology: This was an open-label, randomized, crossover study. Proguanil alone (300 mg single dose), efavirenz alone (400 mg daily for 9 days), and proguanil plus efavirenz were administered to fifteen healthy volunteers. Proguanil was coadministered with the 9th dose (day 9) of efavirenz. Blood samples were collected at pre-determined time intervals over 48 hours following the single doses of proguanil alone and the concurrent drug administration, for determination of proguanil and cycloguanil in the absence and presence of efavirenz, using validated HPLC methods.
Results: The AUCT, Tmax and Cmax of proguanil increased by 112%, 71% and 47% respectively. (AUC: 45.58 ± 12.75 vs 97.00 ± 23.33 mg/L.hr; Tmax: 2.80 ± 0.99 vs 4.80 ± 0.99 h; Cmax: 2.55 ± 0.24 vs 3.75 ± 0.48 mg/L) following concurrent efavirenz administration. Efavirenz administration also caused a significant increase (41%) in the elimination half-life of proguanil (16.50 ± 4.55 vs 23.24 ± 4.08 hr). The metabolism of proguanil to its major metabolite, cycloguanil, was markedly decreased by efavirenz as indicated by a pronounced decrease in the AUC(metabolite)/AUC(unchanged drug) ratio of proguanil from 0.38 ± 0.15 (proguanil alone) to 0.12 ± 0.05 (proguanil plus efavirenz) along with a significant decrease in Cmax of the metabolite (0.61 ± 0.13 vs 0.42 ± 0.09 mg/L) (p < 0.05) Conclusions: There is a significant pharmacokinetic interaction between proguanil and efavirenz and downward dosage adjustment of proguanil may be necessary when the drug is co-administered with efavirenz. Keywords: cytochrome P450 2C19, proguanil, pharmacokinetic interaction, efavirenz.
146. Analysis of the contribution of circulating metabolites to inhibitory drug-drug interactions: Literature evaluation based on the metabolism and transport drug interaction database
Yasushi Fujioka, Catherine K. Yeung, Houda Hachad, Rene H. Levy, and Nina Isoherranen
Department of Pharmaceutics, University of Washington, Seattle, WA, USA, 98195
The aim of this study was to assess the contribution of circulating metabolites to inhibitory drug-drug interactions. The Metabolism and Transport Drug Interaction Database was queried to retrieve inhibitors reported with 1) in vivo interactions (130 of 1323 drugs on the U.S. market), 2) circulating metabolites (106 of 130), and 3) in vitro reversible inhibition for both parent and metabolites with specific P450 isoform (19 of 106). For extracted 19 drugs, [I]/Ki of parent (Pi) and metabolites (Mi) were calculated using plasma total Css (or Cmax) for [I] and in vitro Ki. The Mi/Pi ratio was used to predict the contribution of metabolites to observed in vivo CYP inhibition. The Mi/Pi values ran a quite wide range from 0.0002 (quinidine) through 25 (amiodarone and bupropion). Using a cutoff value Mi/Pi of > 0.2 as possible metabolite effect, metabolites were predicted to contribute to interactions caused by 15 of the 19 inhibitors (79%). Detailed quantitative analyses using [I]/Ki of parent and metabolites were conducted for 12 drugs (59 interaction studies) that were selected because AUC changes of appropriate marker substrates were available. Based on the FDA guidance and the calculated [I]/Ki ratios, 3 of 12 drugs (bupropion, sertraline, and sulfinpyrazone) were categorized into “remote” zone ([I]/Ki < 0.1) when considered parent [I]/Ki only, although in vivo interactions were observed (AUC ratios of 1.4-5.2). On the other hand, incorporation of metabolite [I]/Ki for these 3 drugs improved the prediction, resulting in “possible” zone (0.1 < [I]/Ki < 1). The quantitative analysis considering [I]/Ki and fmCYP of victim drug also demonstrated that the prediction was improved for some drugs by incorporation of metabolites. In conclusion, although the available literature data does not allow for comprehensive evaluation, this study suggests that circulating metabolites contribute to interactions with some drugs and further study will be required for in-depth understanding of their overall clinical significance.
147. Predicting the metabolic interaction potential of zolpidem with CYP3A drugs
Thomas M. Polasek, Janani S. Sadagopal, David J. Elliot, and John O. Miners
Dept of Clinical Pharmacol, Flinders Univ, Adelaide, Australia, AU-5042
Zolpidem is a widely used hypnotic that causes time-dependent inhibition of recombinant CYP3A4 (rCYP3A4) in vitro. The metabolic interaction profile of zolpidem in vivo is largely unknown. The objectives of this study were to evaluate zolpidem as a mechanism-based inactivator of human CYP3A, and to predict its interaction potential with CYP3A drugs (in vitro-in vivo extrapolation; IV-IVE). A co- versus pre-incubation strategy was used to quantify time-dependent inhibition of human liver microsomal (HLM) and rCYP3A4 by zolpidem. Experiments involving a 10-fold dilution step were employed to determine the kinetic constants of inactivation (KI and kinact) and to assess the in vitro mechanism-based inactivation (MBI) criteria. Inactivation data were entered into the Simcyp® population-based ADME simulator (V8.1) and virtual clinical trials in healthy volunteers were conducted to predict the increase in the area under the plasma concentration-time curve (AUC) for orally administered midazolam. Consistent with MBI, the inhibitory potency of zolpidem toward CYP3A was greater following 30 min pre-incubation, with IC50 values decreasing from 43 to 21 μM and from 48 to 13 μM, using HLMs and rCYP3A4 as the respective enzyme sources. In HLMs, the maximal rate of inactivation (kinact) was 0.094 min−1 and the concentration required for half maximal inactivation (KI) was 122 μM. In comparison, kinact and KI values with rCYP3A4 were 0.229 min−1 and 50 μM, respectively. Zolpidem fulfilled all other in vitro MBI criteria, including irreversible inhibition. The mean oral AUC for midazolam in healthy volunteers was predicted to increase 1.1- to 1.7-fold due to the inhibition of metabolic clearance by zolpidem (range = 1.0- to 1.4-fold for HLM IV-IVE and 1.2- to 4.0-fold for rCYP3A4 IV-IVE). These findings demonstrate that zolpidem is a relatively weak mechanism-based inactivator of human CYP3A in vitro. Virtual clinical trails exclude significant interactions between zolpidem and CYP3A drugs in most individuals.
148. Quantification of the pharmacokinetic inhibition of a CYP3A4 substrate with low hepatic first pass effect and long terminal half life; importance of study design and inhibition mechanism: a simulation study
Nassim Djebli, Clémence Rauch, David Fabre, and Xavier Boulenc
Global Metabolism and PharmacoKinetics, Sanofi-Aventis R&D, Montpellier, France, 34000
Introduction: In clinical drug development, CYP3A4 involvement in total clearance is generally investigated after co-administration of strong (e.g. ketoconazole, reversible inhibitor) or moderate (e.g. erythromycin, mechanism-based inhibition) CYP3A4 inhibitors. Recent results in the literature have demonstrated the importance of considering the pharmacokinetic features of both substrate and inhibitor when designing a clinical interaction study (Zaho et al, 2009). In particular, these authors have shown that for compounds with a long terminal half-life, it is essential to maintain ketoconazole plasma concentration over the pharmacokinetic profile of the substrate.
Methods and results: A PBPK model in Simcyp (Massoud et al, 2009) software was set up and validated for a CYP3A4 and CYP2D6 substrate (SARXX) that exhibited a low clearance and a long terminal half-life in humans. Simulations in CYP2D6 PM and EM subjects assessed the impact of CYP3A4 inhibition through mechanism-based inhibition (e.g. erythromycin) and through the effect of a strong reversible inhibitor dosing regimen (200 mg bid versus 400 mg o.d. ketoconazole, 1 week) on the pharmacokinetics of SARXX. With a mechanism-based CYP3A4 inhibitor, even if the pharmacokinetic profile of this substrate is not covered by the inhibitor concentration, the inhibition is maintained suggesting that for such inhibition mechanism, the administration frequency is less critical. For a such combined CYP2D6 and CYP3A4 substrate (with low hepatic first pass-effect and long terminal half-life), eryhthomycin, considered as moderate CYP3A4 inhibitor provided higher inhibition compared to ketoconazole, considered as strong inhibitor, whatever the frequency of dosing (see table 1).
Conclusion: The in vivo CYP3A4 inhibitors classification, proposed several years ago, with the corresponding classical study designs (e.g. 400 mg od ketoconazole as strong inhibitor; 500 mg tid erythromycin, as moderate inhibitor) is based on the effect towards the CYP3A4 probe substrate midazolam (high first pass effect and short terminal half-life). This classification cannot be applied totally for CYP3A4 substrates with a low first pass effect and long terminal half-life. Mechanism types of the inhibitor, in addition to the pharmacokinetic features of both substrate and inhibitor have to be considered when designing a clinical interaction study to investigate the effect of CYP3A4 inhibitors and when classifying CYP3A4 inhibitors.
Table 1. Simulated steady-state AUC0-24 interactions ratios in CYP2D6 EM and PM after repeated co-administration of SARXX with ketoconazole (400 mg o.d. or 200 mg b.i.d.) or erythromycin 500 mg t.i.d.
References:
- Zaho et al, J. Clin. Pharmacol, 49, 351–359, 2009.
- Massoud et al, Expert Opinion, Drug Metabol, Tox, 5(2), 211–223, 2009
149. Midazolam interactions with ketoconazole in dogs can be attributed to CYP2B11, not CYP3A12
Chuck Locuson1, Matthew Zaya1, Kenneth Feenstra1, Rodney Walters1, Beth Miskimins-Mills2, and Julie White1
1Metabolism & Safety, Pfizer Animal Health, Kalamazoo, MI, USA, 49001
2Division of Medicinal and Natural Products, University of Iowa, Iowa City, IA, USA, 52240
Current recombinant cytochrome P450 (P450) phenotyping methods were applied to midazolam-ketoconazole drug interaction studies in dogs. Published midazolam-ketoconazole studies in dogs have led to appreciable increases in plasma midazolam AUC values and the results have been attributed to an interaction with the CYP3A12/26 pathway. However, enzyme phenotyping studies demonstrated higher intrinsic clearances (CLint,app) of midazolam with canine CYP2B11 and CYP2C21. Application of microsome activity correction factors and isoform hepatic abundance further implicated CYP2B11 (fm=0.88 - 0.99) as the primary dog enzyme responsible for midazolam-ketoconazole interactions in vivo. It was also shown that mean AUCi/AUC ratios from intravenous and oral interaction studies were predicted well with unbound Ki, and estimates of unbound Chep,inlet and Fg,i/Fg.
150. The inhibition of noscapine to CYP3A4 and CYP2C9: explanation for clinical DDI between noscapine and warfarin
Zhong-Ze Fang1, and Ling Yang2
1Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China
2Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China, 116023
Noscapine, a naturally occurring phthalideisoquinoline alkaloid isolated from opium, has been used as an antitussive drug for more than 30 years. Recently, noscapine is drawing more and more attention as a promising chemotherapeutic agent. However, drug-drug interactions between noscapine and warfarin reported to Swedish adverse drug interaction register (SWEDIS) make researchers worry about its future application to treat cancer. Therefore, clarification of mechanism of drug-drug interaction between noscapine and warfarin is important and necessary. In this study, the inhibition of noscapine to seven CYP isoforms was investigated and the results demonstrated that noscapine strongly inhibited the activity of CYP3A4 and CYP2C9 with both reversible inhibition and time-dependent inhibition type. The kinetic parameters of reversible inhibition (Ki) are 5.2 μM and 8.82 μM for CYP3A4 and CYP2C9, respectively. The results also displayed that the inactivation kinetic parameters (KI and kinact) are 9.3 μM and 0.06 min−1 for CYP3A4, 8.9 μM and 0.014 min−1for CYP2C9. According to the reversible inhibition prediction equation, the value of AUCi/AUC is 1.05 and1.33 for oral administration and intravenous influsion, repectively; While Employing the TDI prediction equation, the value of AUCi/AUC is 2.95 and 7.51 for oral administration and intravenous influsion, respectively. In conclusion, significant increase in AUC of warfarin when coadministrated with noscapine clearly explained the clinical DDI between noscapine and warfarin. Our results also reminded cancer researchers of potential weakness of noscapine which benefits its future application as a chemotherapy agent.
151. Inhibition of curcumenol to human liver cytochrome P450 enzymes
Dong-Xue Song1, Zhong-Ze Fang2, Yan-Yan Zhang1, and Ling Yang1
1Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China, 116023
2Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China
Abstract: Curcumenol, one of the major components of Zedoary Turmeric Oil, has been widely used to treat cancer and inflammation. In Chinese Pharmacopoeia, curcumenol is also recognized as a phytochemical marker for the quality control of Zedoary Turmeric Oil. As antibiotics or anticancer drug, curcumenol is highly likely to be used in combination with various synthetic drugs in most cases, thus it is necessary to evaluate potential pharmacokinetic drug-drug interactions induced by curcumenol. In this article the inhibition of curcumenol to seven CYP isoforms was investigated and the results demonstrated that only CYP3A4 was strongly inhibited (IC50 = 15.6 ± 0.1 μM). Kinetic analyses showed the inhibition type was competitive with Ki value of 11.6 μM. Time- and NADPH-dependent inhibitions were also investigated to show curcumenol is not a mechanism-based inhibitor. Employing these in vitro data and dog’s in vivo pharmacokinetic data, [I]/Ki was predicted to be 0.00156 which suggested that curcumenol as antibiotics and antitumor agent may be safely used without inducing drug-drug interaction. Nevertheless, due to the limited pharmacokinetic data of Curcumenol in humans, it is nigh impossible to evaculate its potential effects to human from in vitro data. Further work will be done in the future.
152. Biotransformation of gomisin a by cytochrome P450 3A4 leads to metabolite–P450 complex formation and irreversible inhibition
Yan-Yan Zhang, and Ling Yang
Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China, 116023
Gomisin A is one of the major dibenzocyclooctadiene lignans isolated from the traditional Chinese medicine Fructus Schisandrae. Pharmacological studies have revealed that gomisin A shows protective effects on chemicals-induced hepatotoxicity and glutamate induced oxidative neuronal damage. In the present study, to gain insight into the potential of schisandra-prescription drug interactions, a detailed study on the metabolic pathway of gomisin A was conducted. The influence of gomisin A as a CYP3A4 reversible inhibitor or irreversible inactivator was also evaluated. In human liver microsomes, gomisin A was oxidized to demethylenation gomisin A and one monohydroxylated metabolites. A combination of correlation analysis, chemical inhibition studies, assays with recombinant CYPs and enzyme kinetics indicated that both metabolites were generated predominantly by CYP3A4. IC50 shift studies demonstrated that gomisin A was a time dependent inhibitor of CYP3A4. An apparent Ki value of 1.9 μM without preincubation and an NADPH-dependent inhibition with a Kinact of 0.032 ± 0.001 min−1 and a KI of 0.65 ± 0.06 μM were determined. Spectral scanning of recombinant CYP3A4 with gomisin A suggested that the formation of a metabolite intermediate complex was responsible for CYP3A4 inactivation. These results collectively demonstrated that gomisin A might exhibit significant modulatory effects on CYP3A4 activities. The presence of these methylenedioxyphenyl lignans in Fructus Schisandrae may substantially undermine the clinical value of this herb medicine.
153. Effect of subunit composition and agonist of GABAA receptor on the noncompetitive antagonist binding
Ligong Chen, and John E. Casida
Department of Environmental Science, Policy, and Management, University of California at Berkeley, California, CA, USA, 94720
The γ-aminobutyric acid type A (GABAA) receptor subunit composition determines the sensitivity and selectivity of noncompetitive antagonists (NCA) including some major insecticides. Recent studies have shown β3 subunit is highly sensitive to NCA binding and play an important role in receptor assembly. β1 homomer retains only about 10% binding activity of β3 homomer although the putative binding sites from 2’to 9’ are the same except for 15’ in the channel-lining segment M2 i.e. S 15’ for β1 and N 15’ for β3. This study test the potency difference of β1 and β3 homomer using mutagenesis, chimeragenesis, heteromer combined with α1 subunit and agonist GABA as modulator. Chimer 1(CH1) containing extracellular half of the β3 and β1 transmembrane domains retained about 70% binding level of β3 homomer. Chimer 2 (CH2) with extracellular half of the β1 and β3 transmembrane domains had similar binding activity to β1 homomer. Mutations β1S15’N and β3N15’S aimed to exchange the only different homologous residue at 15’ for β1 and β3 subunit did not change the binding activity. GABA stimulated heteromer α1β1 and α1β3 binding level to 2.0-2.5 folds at 3 mM. Addition of α1 subunit rescued the inactive CH2 close to wildtype α1β1 and 3 mM GABA also increased α1CH2 to high binding activity. However, the binding activity of α1β1S15’N showed insensitivity to GABA. Inhibitory effect of general anesthetics etomidate on NCA binding was reduced more than 5-fold by mutation β3N15’S. Therefore, NCA binding site is tightly regulated by the open-state conformation occupied by receptor that largely decides the sensitivity of GABAA receptor.
154. Impact of intestinal concentrations simulated by an ACAT model on the prediction of P-glycoprotein drug interactions
Tsuyoshi Mikkaichi, Tomoki Imaoka, Ken-ichi Itokawa, Noriko Okudaira, and Osamu Okazaki
Drug Metabolism and Pharmacokinetics Research Laboratories, Daiichi Sankyo Co., Ltd., Tokyo, Japan, 140-8710
Clinically relevant drug-drug interactions (DDI) mediated by transporters, especially P-glycoprotein (P-gp), are of increasing concern in drug development. As a possible in vivo interaction criterion via inhibition of P-gp, [I2]/IC50 ([I2]: dose divided by a volume of 250 mL) has been proposed. Here, we evaluated advantage of using dynamic intestinal concentration simulated by an Advanced Compartmental Absorption and Transit (ACAT) model, requiring only in vitro solubility and membrane permeability data (Papp), for DDI risk assessment via P-gp inhibition. The in vitro IC50 values of marketed drugs for P-gp mediated vectorial transport of digoxin in Caco-2 cells were collected. Papp in Caco-2 cells and the solubility at pH6.8 were evaluated and used in an ACAT model. A new index, “average P-gp inhibition”, was introduced, which was calculated using the simulated intestinal concentration profile (Isim) and in vitro IC50 (Isim/(IC50+ Isim)). In addition, to clarify the advantage of this method, four virtual model compounds exhibiting high/low permeability and high/low solubility were evaluated. They were assumed to possess the same inhibitory potential. Using the four model compounds, we revealed that although they have the same [I2]/IC50 value, the calculated “average P-gp inhibition” varied among the model compounds: the compound with low membrane permeability and high solubility exhibited the highest inhibitory potential on intestinal P-gp due to longer retention in the intestine with high concentration. The calculated “average P-gp inhibition” of marketed drugs moderately correlated with the logarithmic value of [I2]/IC50 and the compounds with low permeability tended to show higher “average P-gp inhibition”. The index was also compared with the AUC change of digoxin in clinical trials. We conclude that DDI risk assessment using Isim, considering not only the dose but also its solubility and permeability, could give us additional information for better insight into P-gp mediated DDI.
155. Inhibition of codeine glucuronidation in vitro by fluconazole, ketamine, ketoconazole and valproic acid: Prediction of in vivo drug-drug interaction potential
John O. Miners1, Pritsana Raungrut2, Verawan Uchaipichat3, David J. Elliot4, and Benjamas Janchawee5
1Dept of Clin Pharmacol, Flinders Univ Sch of Med, Adelaide, Australia
2Biomedical Sciences, Prince of Songkla University, Hat Yai, Thailand,
3Clinical Pharmacy, Khon Kaen University, Khon Kaen, Thailand
4Dept of Clinical Pharmacol, Flinders Univ, Adelaide, Australia, AU-5042
5Pharmacology, Prince of Songkla University, Hat Yai, Thailand
Codeine is widely used as an analgesic, separately or in combination with other drugs (e.g. acetaminophen). Glucuronidation is the principal elimination pathway of codeine in humans, accounting for approximately 70-80% of the dose. In contrast, metabolic activation to morphine via O-demethylation accounts for 5-10% of codeine biotransformation. Codeine glucuronidation is catalyzed by UGT2B4 and UGT2B7. Inhibition of UGT2B7 has been implicated as the mechanism of several drug-drug interactions. Thus, this work sought to identify potential drug-drug interactions in vivo arising from inhibition of codeine glucuronidation. Initial studies characterized the kinetics of codeine glucuronidation by human liver microsomes (HLM), in the absence and presence of bovine serum albumin (BSA). Addition of BSA to incubations sequesters inhibitory long chain unsaturated fatty acids, thereby providing ‘true’ estimates of Km and Ki values. Kinetic parameters generated here were corrected for the binding of codeine and potential drug inhibitors for binding to HLM and BSA. Mean Km values for codeine glucuronidation determined in the absence and presence of BSA were 2320μM and 290μM, respectively. Inhibition screening studies conducted at codeine concentrations corresponding to these Km values identified fluconazole, ketamine, ketoconazole and valproic acid as inhibitors of human liver microsomal codeine glucuronidation. Kinetic studies were subsequently performed to generate Ki values for each drug. Ki values obtained for fluconazole, ketamine and ketoconazole in the presence of BSA were 202μM, 3.5μM and 0.7μM, respectively. Comparison of these data to plasma concentrations observed in vivo suggest that ketoconazole is likely to impair codeine clearance via glucuronidation, increasing the proportion of the dose converted to morphine. Valproic acid was a weak inhibitor. Interestingly, however, addition of valproic acid to incubations converted the kinetics of human liver microsomal codeine glucuronidation from hyperbolic to sigmoidal, suggesting that this compound my activate UGT2B7 via an allosteric site.
156. Effect of Yin Qiao San/Sang Ju Yin and their components on the metabolism and absorption of oseltamivir
Xiaoan Wang1, Siukwan Wo1, Qi Chang2, Moses S.S. Chow3, and Zhong Zuo1
1School of Pharmacy, Faculty of Medicine, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong, N/A
2Institute of Medicinal Plant Development, Chinese Academy of Medical Science and Peking Union Medical College, Beijing, China, 100094
3Center for Advancement of Drug Research and Evaluation, College of Pharmacy, Western University of Health Science, Pomona, CA, USA, 91766-1854
Oseltamivir (O) is the pro-drug of a neuraminidase inhibitor Oseltamivir Carboxylate (OC). O is currently recommended for the prophylaxis and treatment of avian influenza together with four Chinese Medicine Formulae (CMF) in Hong Kong. Pharmacokinetic and pharmacodynamic herb-drug interactions between O and Yin Qiao San/Sang Ju Yin (CMF1) have been observed in our previous animal studies. The current study aims to provide the mechanistic explanations for such interactions from the perspective of absorption and metabolism.
Effects of CMF1/CMF1 components on the metabolism of O in GI mucosa and plasma were investigated by in vitro incubation systems. Dose response curves were obtained and compared among different components. In vitro Caco-2 monolayer model was employed to study the absorption transport of O as well as the effect of CMF1/CMF1 components on its absorption. Samples were analyzed by a developed LC/MS/MS for the concentration of O and OC.
Chlorogenic Acid, Glycyrrhizic Acid, Forsythin, Forsythoside A, Liquiritin, and Caffeic Acid were identified and quantified from CMF1 by HPLC, and tested for their effects on the metabolism of O. Metabolism of O to OC in plasma was much more prominent than that in GI mucosa. Such metabolism was found to be inhibited by CMF1/CMF1 components in a dose-dependent manner. Among the studied components, Forsythoside A and Forsythin have demonstrated relatively strong inhibition on the metabolism of O to OC in plasma with estimated IC50 of 230 μM and 400 μM, respectively. Results from Caco-2 monolayer model showed that O is a substrate of P-gp in GI tract with an efflux ratio of 32 at30 μg/ml and 15 at 150 μg/ml of O. OC was found in both apical and basalateral chambers with certain intracellular accumulation after loading O at the apical side of the Caco-2 model. In presence of 5 mg/ml CMF1, absorption transport of O was significantly increased for ten times (from a Papp of 1.13E-7 ± 1.16E- 8 cm/sec to 1.31E-6 ± 1.71E- 7 cm/sec at 30 μg/ml of O, and from 8.86E-8 ± 7.3E- 9 cm/sec to 6.17E-7 ± 2.27E- 7 cm/sec at 150 μg/ml of O) and the corresponding formation of OC was decreased.
In conclusion, our results demonstrated that metabolism of O could be inhibited and the absorption of O could be increased in the presence of CMF1/CMF1 components. (Acknowledgement: CUHK direct grant 2041449)
157. Assessing and minimizing time-dependent inhibition of CYP3A in drug discovery
Wei Tang
Drug Metabolism & Pharmacokinetics, Merck @ Co., Rahway, NJ, USA, 09065
Assessing and Minimizing Time-Dependent Inhibition of CYP3A in Drug Discovery W Tang, RA Stearns, RW Wang, RR Miller and Q Chen Department of Drug Metabolism and Pharmacokinetics, Merck & Co., Rahway, NJ 07065 A number of compounds with amino functional groups exhibited time-dependent inhibition (TDI) of CYP3A during drug discovery screening. Subsequent study revealed that incubations of these compounds with recombinant CYP3A4 exhibited the maximum UV/Vis absorbance at ~450 nm. The absorbance increased with increasing incubation time and decreased following adding K3FeCN to the incubation mixture, suggesting formation of MI complex. Supporting evidence of this conclusion was the detection by LC-MS/MS of nitroso intermediates trapped by cyanide in human liver microsomal incubations. These data enabled project team to circumvent TDI via introducing alkyl group(s) that is alpha to the amino group, and the resulting compounds indeed did not show TDI of CYP3A. With a representative compound, it was demonstrated that the TDI observed in vitro could be quantitatively correlated to drug-drug interaction determined in vivo in a rat model, and the exercise increased confidence in projecting clinical risk based on preclinical data. This case study highlights the importance of mechanistic studies in support of drug discovery and decision-making processes.
158. Total bilirubin concentrations in plasma of renal transplant patients may reflect differences in MRP2 inhibition by cyclosporine and tacrolimus
Benedetta C. Sallustio, and R. G. Morris
Dept of Clinical Pharmacology, Queen Elizabeth Hospital, Woodville, Australia, AU-5011
Maintenance immunosuppression following renal transplantation typically consists of prednisolone, mycophenolic acid (MPA) and a calcineurin inhibitor, cyclosporine (CsA) or tacrolimus (Tac). Both CsA and Tac are substrates for CYP3A enzymes and P-glycoprotein, but only CsA inhibits the canalicular efflux transporter MRP2, resulting in an apparent increase in MPA clearance, due to inhibition of the biliary secretion of MPA’s glucuronide metabolites and subsequent decrease in its enterohepatic recirculation. The bilirubin glucuronides are also MRP2 substrates. This study investigated whether the different effects of CsA and Tac on MRP2 are reflected in total bilirubin (bilirubin + bilirubin glucuronide) concentrations in plasma. Fiftyseven renal transplant recipients (38 CsA, 19 Tac) were followed for 6 months post-transplantation. Demographic data such as age, sex, ethnicity, donor type, graft ischaemic time, panel reactive antigens, HLA matching and delayed graft function were recorded for each subject. Liver function test results were assessed pre- and 1, 3 and 6 months post-transplant. There were no differences in any pre-transplant liver function tests or demographics, except a slightly higher ratio of Caucasian versus Aboriginal/Asian subjects in the CsA group. CsA subjects had significantly higher (median, interquartile range) total bilirubin concentrations (μmol/L) at 1(16.5, 11.5-20.0), 3 (13.0, 9.5-20.0) and 6 (13.0, 9.0-21.0) months, compared to Tac subjects at 1 (9.0, 7.0-11.0, p< 0.001), 3 (7.5, 6.0-8.5, p<0.001) and 6 (8.5, 6.0-12.0, p<0.05) months, although, only 7-8% of CsA subjects had total bilirubin concentrations above the normal range. At 1 month, CsA subjects also had higher aspartate aminotransferase and alanine aminotransferase, and lower albumin levels compared to those on Tac, but there were no differences by 6 months. The persistently higher total bilirubin concentrations in CsA patients are consistent with its inhibition of canalicular MRP2, and suggest a modest effect as <8% of patients attained concentrations above normal. The modest but significant differences in other markers of liver function suggest that, in this study population, CsA was associated with a slightly increased risk of early hepatotoxicity.
159. In vitro drug-drug interaction of domperidone and thiazolidinediones (Pioglitazone or Rosiglitazone) in human liver microsomes
Din Ung1, Henry P. Parkman2, and Swati Nagar1
1Pharmaceutical Sciences, Temple University School of Pharmacy, Philadelphia, PA, USA, 19140
2Gastroenterology Section, Department of Medicine, Temple University School of Medicine, Philadelphia, PA, USA, 19140
The prokinetic agent domperidone is co-prescribed with thiazolidinedione antidiabetic drugs pioglitazone or rosiglitazone to treat diabetic gastroparesis patients. These drugs are metabolized by cytochrome P450 (CYP) isozymes. Domperidone is metabolized by CYP3A4, which is known to be inhibited by pioglitazone. This study examines potential drug-drug interaction of domperidone and either glitazone in pooled human liver microsomes (HLM). Time- and concentration-dependent enzyme assays were performed in HLM to evaluate drug-drug interaction. CYP activity towards domperidone was characterized in HLM pre-incubated at varying times (0-40 min) and concentrations of pioglitazone (0-75 μM) or rosiglitazone (0-100 μM). After pre-incubation at 37°C, 100μM or 400μM domperidone was added and further incubated for 30min. Reactions were terminated with acetonitrile containing internal standard encainide. Samples were analyzed for the major 5-hydroxydomperidone metabolite via HPLC assay. The kinetic constants of inactivation were calculated graphically and by nonlinear regression. Apparent rates of inactivation (kobs) were estimated from slopes of a plot of the log of enzyme activity remaining after pre-incubation with respect to pre-incubation time. The KI and kinact parameters were estimated from the equation: kobs = kobs[I]=0 + ((kinact•[I])/(KI + [I])). At 400 μM domperidone, the remaining enzyme activity after pre-incubation with 50 or 75 μM pioglitazone for 20, 30, and 40min were significantly different than the control (n = 6, p <0.05 with ANOVA and posthoc Tukey-Kramer test). At 400 μM domperidone, treatment with 100 μM rosiglitazone was found to have a slope or kobs significantly different from the control. At 100 μM domperidone, treatment with 25 or 100 μM rosiglitazone had slopes significantly different from the control. These in vitro results demonstrate an interaction between domperidone and pioglitazone or rosiglitazone. The type of interaction –reversible (competitive, noncompetitive, uncompetitive, mixed) or mechanism-based inactivation will be determined in future studies. Interactions could result in altered therapeutic efficacy or toxicity in patients. Acknowledgment: This work was partially funded by a seed grant to support Temple University faculty research collaborations.
160. Investigation of metabolic DDI between ranolazine and amiodarone in vitro
Nancy I. Chu, Jia Hao, and Kwan H. Leung
Pre-Clinical Development, Gilead Sciences, Inc., Palo Alto, CA, USA, 94304
Amiodarone (AMI), an antiarrhythmic agent and ranolazine (RAN), an antianginal agent, may be co-administered in the clinic. The potential of metabolic DDI between these two drugs and their major metabolites was investigated in vitro using human liver microsomes. Km values for the metabolism of AMI to its major CYP3A4-mediated metabolite mono-deethylated amiodarone (DEA) and for the metabolism of RAN to its major CYP3A4-mediated metabolite, CVT-2738, and major CYP2D6-mediated metabolite, CVT-2514, were determined. At the apparent Km (25 μM) for AMI, IC50 values to inhibit the formation of DEA by RAN and its four major metabolites CVT-2514, CVT-2738, CVT-2512 and CVT-4786, were determined. Similarly, at the apparent Km (50 μM) for RAN, IC50 values to inhibit the formation of CVT-2738 and CVT-2514 by AMI and DEA were determined. Levels of DEA, CVT-2514 and CVT-2738 in microsomal incubates were determined by LC/MS/MS assays. Under the experimental conditions, IC50 values for RAN and CVT-2514 to inhibit the metabolism of AMI to DEA were 149 and 217 μM, respectively. The other three major metabolites CVT-2512, CVT-2738, and CVT-4786, did not show significant inhibition at concentrations up to 500 μM. The IC50 for AMI and DEA to inhibit RAN metabolism to the CYP3A4 metabolite, CVT-2738, were 917 and 107 μM, respectively. The IC50 for the inhibition by AMI and DEA of RAN conversion to the CYP2D6 metabolite, CVT-2514, were 51 and 88 μM, respectively. In summary, the metabolism of AMI was weakly inhibited by RAN and the CYP2D6-mediated metabolite, CVT-2514. Reciprocally, the metabolism of RAN to CVT-2514 was weakly inhibited by AMI and DEA. DEA also weakly inhibited metabolism of RAN to CVT-2738, a CYP3A4-mediated reaction. The IC50 values for both drugs and their metabolites were well above therapeutic concentrations and hence it can be concluded that pharmacokinetic DDI will be weak when both drugs are co-administered to patients.
161. Dynamic in vitro approach to prediction of mixture interactions
Jonathan Boyd1, Holly Williams1, Nathan Boggs2, Julia Patrone2, Mellisa Theodore2, and Huong Le2
1Chemistry, West Virginia University, Morgantown, WV, USA, 26506
2Applied Physics Lab, Johns Hopkins Univ., Laurel, MD, USA, 20723
While exposure to chemical mixtures is an everyday reality, an understanding of their combined effects, and any potential prediction thereof, is extremely limited. Realistic exposures potentially consist of hundreds to thousands of chemicals per day, but even relatively simple binary mixture interactions can be inherently difficult to predict based upon the lack of temporal and spatial mechanisms for the individual constituents. To this end my laboratory has been developing in vitro assays to enable a high-throughput means of defining general toxicodynamic response pathways in vitro, with a full dosing regimen, which has the potential to identify and predict possible interactions. A central aspect of our assays elucidates altered activity for cell signaling cascades following multiple time-course exposures to toxicants which allows for the identification of both their common and disparate response pathways. Rather than assess all possible interactions between the compound and multiple cellular targets, our approach is to determine the cell signaling response to multiple classes of toxicants and thereby map interactions in time and space for prediction of effects. Preliminary results will be presented with data gathered from hepatocytes exposed to deguelin (0.001-100 uM), potassium cyanide (0.001-100 uM), staurosporine (10 uM) and SB202190 (350 nM), alone and in combination, for 24 hours. Oxygen consumption kinetic profiles suggested key changes in ATP production at 400 minutes post-dose which initiated an investigation into the activity of the signaling cascades via a bead-based multiplexed (8-plex) immunoassay at that time point. Dose-dependent cascade initiation indicates a clear identification of threshold response to low-level exposures, and crosstalk amongst selected proteins correctly predicts interactions leading to “other-than-additive” mixtures interactions. In this study, we demonstrate the potential of a new in vitro approach for the prediction of toxic mixtures interactions that is fundamentally driven by the interdependence of energy metabolism, signal transduction, and cell survival.
162. In vitro and in situ interactions of CVT-3619 with Pgp, BCRP, and nucleoside transporters
Eve-Irene Lepist1, Jing Zhang2, Tracey Tackaberry3, Vijaya Damaraju2, Wendy P. Gati4, Sylvia Yao4, James D. Young4, Jennifer Tang1, Nevena Mollova1, Nancy I. Chu1, Carol E. Cass2, and Kwan H. Leung1
1Pre-Clinical Development, Gilead Sciences, Inc., Palo Alto, CA, USA, 94304
2Cross Cancer Institute and University of Alberta, Edmonton, AB, T6G 2E1
3Cross Cancer Institute, Edmonton, AB, T6G 2E1
4University of Alberta, Edmonton, AB, T6G 2E1
CVT-3619 [(2-{6-[((1R, 2R)-2-hydroxycyclopentyl)amino]purin}-yl} (4S, 5S, 2R, 3R) 5-[2 fluorophenylthio methyl) oxolane-3,4-diol)] is a partial A1 adenosine receptor agonist being developed for the treatment of hypertriglyceridemia.
We have investigated the role of P-glycoprotein (Pgp), breast cancer resistance protein (BCRP), and nucleoside transporters (NTs) in the transport of CVT-3619. MDR1-MDCK, wild type Caco-2, and Caco-2 BCRP knockdown, Pgp knockdown, and vector control cells were used to assess the susceptibility of CVT-3619 to Pgp and BCRP. Relative affinities of nucleoside transporters (NTs) for CVT-3619 were determined using competitive inhibition of [3H]uridine transport in yeast expressing recombinant human hENT1, hENT2, hCNT1, hCNT2, or hCNT3. Transportability of CVT-3619 was tested in voltage clamp experiments using Xenopus oocytes expressing recombinant hCNT1/2/or 3 and an in situ rat brain perfusion model by using a known ENT1 inhibitor nitrobenzylmercaptopurine ribonucleoside (NBMPR). The efflux ratios of CVT-3619 in bidirectional transport studies were 76.7, 2.6, 3.5, 1.2, and 3.8 in MDR1-MDCK, Caco-2, BCRP knockdown, Pgp knockdown and vector control cells, respectively. The Pgp transport of CVT-3619 in Caco-2 cells exhibited Km and Vmax values of 214 ± 41.2 mM and 246 ± 26.8 pmol/min/cm2, respectively. CVT-3619 did not inhibit Pgp-mediated digoxin efflux in MDR1-MDCK cells. CVT-3619 bound to all NTs with Ki values for hCNT1 (42.4 ± 5 mM) < hENT1 (57.4 ± 6.9 mM) < hCNT2 (78.3 ± 8.8 mM) < hCNT3 (122 ± 14 mM) < hENT2 (659 ± 70.1 mM). In uptake studies, no transport of CVT-3619 via hCNT1/2/or 3 was seen in Xenopus oocytes. In situ, rat brain uptake of CVT-3619 was not affected by NBMPR.
In summary, CVT-3619 is a substrate, but not a potent inhibitor, of Pgp. It is not a BCRP substrate. CVT-3619 exhibited low to moderate affinities for binding to the five recombinant hNT proteins but showed no transportability.
163. Dose-dependent effects of itraconazole and its metabolites on oral midazolam exposure
Ian E. Templeton1, Chi-Chi Peng1, Kenneth Thummel1, Connie L. Davis2, and Nina Isoherranen1
1Department of Pharmaceutics, University of Washington, Seattle, WA, USA, 98195
2Department of Medicine, Division of Nephrology, University of Washington, Seattle, WA, USA, 98195
Itraconazole (ITZ) is a potent inhibitor of fungal P450s. It is also a potent inhibitor of human CYP3A4 and is metabolized by CYP3A4 in vitro and in vivo. The magnitude of CYP3A4 inhibition caused by ITZ has been investigated by numerous groups, but never has ITZ and its three sequentially CYP3A4-mediated metabolites OH-ITZ, keto-ITZ and ND-ITZ been measured concurrent with determination of CYP3A4 inhibition. This in vivo study was designed to assess whether the historic under-prediction of CYP3A4 inhibition by ITZ in vivo could be explained by the inhibition of CYP3A4 by ITZ metabolites. The aim of this study was to determine the magnitude of CYP3A4 inhibition following escalating doses of ITZ. We hypothesized that due to the saturation of ITZ metabolism by CYP3A4 at higher doses, the contribution of the metabolites to CYP3A4 inhibition would decrease when ITZ dose was increased. Six healthy volunteers, one woman and five men, participated in the study. All subjects were CYP3A5*3/*3 carriers, indicative of lack of functional CYP3A5. Midazolam clearance was measured in four sessions after a 2 mg dose of midazolam oral solution. ITZ was administered in three of these sessions in single escalating doses of 50 mg, 200 mg and 400 mg po. The AUC of midazolam was increased 2.2-, 5.1- and 5.9-fold after 50 mg, 200 mg and 400 mg doses of ITZ, respectively. This observed dose dependent increase was in good agreement with predicted change in midazolam AUC. When the measured concentrations of ITZ, OH-ITZ, keto-ITZ and ND-ITZ in the subjects were used together with a midazolam fm,CYP3A4 of 0.9 and Fg of 0.55 to predict the in vivo interaction, a 2.2-, 4.5- and 6.1-fold increase in midazolam AUC was predicted. In contrast, if only ITZ was used to predict in vivo interaction, the magnitude of MDZ AUC change was underpredicted. These results suggest that metabolites may contribute significantly to in vivo drug-drug interactions.
164. Nonclinical and clinical assessment of pharmacokinetic drug-drug interactions with toremifene
Juhyun Kim1, Concepción Peraire2, Juergen Froelich2, Josep Sola2, Gary K. Barnette1, James T. Dalton1, and Karen A. Veverka1
1GTx Inc, Memphis, TN, USA, 38163
2Ipsen Pharma, S.A., San Feliu de Llobregat, Spain, 08980
Toremifene is a SERM being evaluated for the prevention of bone fractures in men with prostate cancer taking androgen deprivation therapy (ADT). The impact of toremifene citrate on the catalytic activity of CYP 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4 (midazolam) and 3A4 (testosterone) isoforms was determined in human liver microsomes in the presence and absence of standard inhibitors or toremifene citrate (0.03 to 30 μM). Induction of CYP1A2 and 3A4 by toremifene was also investigated in human hepatocytes. Toremifene (up to 30 μM) did not significantly inhibit CYP1A2 or 2D6; however, CYP2A6, 2B6, 2C8, 2C9, 2C19, 2E1, and 3A4 (testosterone and midazolam) isoforms were inhibited (Ki values ranging from 6.3 to 26 μM). Toremifene did not induce CYP1A2, but increased CYP3A4 enzyme activity and expression in vitro. These data indicate that toremifene is unlikely to play a role in clinical drug-drug interactions related to CYP1A2 and 2D6 metabolism. A clinical study was performed to assess the magnitude of potential pharmacokinetic drug:drug interactions when toremifene was co-administered with drugs that are metabolized by CYP3A4 and 2C9. A single-center, single-sequence, open-label Phase I pharmacokinetic study was conducted in 20 healthy male volunteers. Midazolam (CYP3A4 substrate) and tolbutamide (CYP2C9 substrate) plasma concentrations were determined before toremifene was administered to the subjects, and after steady state toremifene was achieved. Tolbutamide exposures (peak and total) were slightly higher following co-administration with toremifene multiple doses suggesting net inhibition of CYP2C9 by toremifene, while the peak and total exposures of midazolam and toremifene were decreased after multiple doses of toremifene, suggesting a net induction of CYP3A4. However, due to the interindividual variability of CYP3A4, the magnitude of change is not clinically relevant according to current industry and regulatory standards. Importantly, toremifene should not produce clinically significant pharmacokinetic drug-drug interactions related to inhibition or induction of CYP3A4, CYP2C9 or any of the other isoforms characterized during in vitro or in vivo studies.
165. Simultaneous determination of CYP450 induction and metabolism in metabolically competent human hepatocyte cell line hepaRG
Robert R. Annand1, David Steen2, Mary Jacewicz3, Katherine Tsaioun1, and Christophe Chesne2
1Apredica, Watertwon, MA, USA, 02472
2R & D, Biopredic international, Rennes, France, 35000
3Cell Biology, Apredica, Watertown, MA, USA, 02472
Understanding metabolism and characterizing Cytochrome P450 (CYP) induction by xenobiotics are important parts of the drug discovery process and of characterizing the potential for drug-drug interactions. Traditional studies of metabolism and induction using primary hepatocytes are hampered by the scarcity of the cells and their short lifetime in culture. HepaRG human hepatic cells are highly differentiated and display many hepatocyte-like functions, including CYP expression and induction. In an effort to speed the characterization of CYP metabolism and induction, we investigated the use of HepaRG cells to study metabolism and CYP induction in the same well. Nifedpine, a dihydropyridine calcium channel blocker, is known to be both a potent inducer of, as well as a substrate for, CYP3A4. To measure metabolism, nifedipine was incubated with HepaRG cells, and aliquots at various time points were analyzed by LC/MS/MS to determine the amount remaining parent. Subsequent to the metabolism determination, the cells were washed and incubated for two days with nifedipine. CYP activity was determined by measuring testosterone 6b-hydroxylase activity of induced cells compared to a vehicle control using an LC/MS/MS-based assay. This experiment demonstrated both the rapid metabolism (half-life about 1 hr) of nifedipine, as well as the induction of CYP3A4 activity (greater than five-fold induction of activity). Studies with other compounds known to be both inducers of and substrates for CYPs confirmed the utility of this assay. By using the combined metabolism-induction assay, we are able to speed drug discovery by rapidly identifying compounds that have potential for metabolism or CYP induction in a single, rapid assay using cells that are readily available with reproducible characteristics. Routine use of this assay will allow the identification of surprises early in the drug discovery process, thereby reducing the amount of time and money invested in compounds with liabilities.
Reference:
- Kanebratt K.P. and T.B. Andersson (2008). “Evaluation of HepaRG Cells as an In Vitro Model for Human Drug Metabolism Studies”. Drug Metabolism and Disposition 36(7) 1444–52.
166. Drug interaction between ranolazine and metoprolol: In vitro and in vivo correlation
Eve-Irene Lepist, Daniel Soohoo, Nancy I. Chu, Xuegong Wang, Justus Bingham, William Chen, Nevena Mollova, Peter Staehr, and Kwan H. Leung
Pre-Clinical Development, Gilead Sciences, Inc., Palo Alto, CA, USA, 94304
Ranolazine (Ran), an inhibitor of the cardiac late sodium current (INa), has been approved in the US and Europe for the treatment of patients with chronic angina. Ran is a weak inhibitor of and in part metabolized by CYP2D6. Metoprolol (Met), a CYP2D6 substrate and widely-prescribed cardioselective β-blocker, may be used concomitantly with Ran. The potential of drug-drug interaction between these two drugs was investigated in vitro using human liver microsomes, and confirmed in vivo in healthy volunteers. The Km value for the metabolism of Met to its major CYP2D6-mediated metabolite, alpha-hydroxymetoprolol (a-OH-Met), was determined. At Km of Met (rounded to 100 μM), IC50 values to inhibit the formation of a-OH-Met by Ran and its four major metabolites CVT-2514, CVT-2738, CVT-2512 and CVT-4768 were determined. In a multiple dose, open-label study, 25 healthy volunteers received an oral dose of Ran 750 mg bid from Day 3 through Day 8. All volunteers additionally received a 100 mg oral dose of Met on Days 1 and 7. Levels of Met, Ran, and their primary hepatic metabolites were determined by LC/MS/MS. IC50 values to inhibit the metabolism of Met to a-OH-Met by RAN and CVT-2514 were 608 and 830 μM, respectively. The other three major metabolites, CVT-2512, CVT-2738, and CVT-4786, did not show significant inhibition at concentrations up to 1000 μM. The IC50 value for Ran was well above its therapeutic concentration range of 1-10 μM. Based on the in vitro results, the predicted AUCMet with Ran /AUCMet alone ranged between 1.2-1.5. In comparison, in vivo AUC0-inf and Cmax of Met increased 1.82-fold (95% CI) and 1.48-fold (95% CI), respectively, in the presence of Ran. In summary, in vitro estimations predicted the weak pharmacokinetic drug interaction between Ran and Met observed in vivo in healthy volunteers reasonably well.
167. Comparison of clinical drug-drug interaction study results for sunitinib and axitinib with an in silico tool
Sadayappan V. Rahavendran, Yazdi K. Pithavala, Carlo Bello, and Karen J. Klamerus
Pharmacokinetics, Dynamics and Metabolism, Pfizer, Inc, San Diego, CA, USA, 92121
Computer based modeling (in silico) has the potential to supplement clinical trials by quickly and cost effectively providing answers to drug-drug interaction (DDI) questions. SimCYP® software contains databases of ADME properties of many drugs including specific inhibitors/inducers of CYP3A enzyme such as ketoconazole and rifampin. In this study, we illustrate the use of SimCYP® (version 6.2) to determine the DDI risk associated with co-administering ketoconazole or rifampin separately with sunitinib (Sutent) and axitinib. Both of these oncology compounds are receptor tyrosine kinase inhibitors that target VEGFR. Sunitinib is primarily (>90%) metabolized by CYP3A and axitinib is metabolized by CYP3A with minor contributions from CYP1A2, CYP2C19 and UGT1A1. Two PK parameters, AUC and Cmax were compared with and without the inducer/inhibitor. SimCYP® DDI predicted in the presence/absence of ketoconazole an AUCi/AUC and Cmaxi/ Cmax increase of 75% and 30% compared to clinical results of 85% fold and 60% for sunitinib. SimCYP® DDI predicted in the presence/absence of ketoconazole an AUCi/AUC and Cmaxi/ Cmax increase of 100% and 41% compared to clinical results of 100% and 50% for axitinib. SimCYP® DDI predictions of sunitinib AUC and Cmax in the presence of rifampin indicated a decrease of 70% and 46% respectively for sunitinib compared to clinical results demonstrating 47% and 22% decreases. SimCYP® DDI predictions of axitinib AUC and Cmax in the presence of rifampin indicated 52% and 66% decreases respectively compared to clinical results demonstrating 79% and 71% decreases. The concordance between modeled DDI and observed clinical results for axitinib and sunitinib indicates value in the use of in silico simulations in predicting clinically relevant DDI.
168. Physiologically-based model for ketoconazole disposition and prediction of its drug-drug interactions
Grazyna Fraczkiewicz1, Neil Parrott2, Viera Lukacova3, Michael B. Bolger3, John R. Crison3, Walter S. Woltosz3, and Thierry Lavé4
1Simulations Plus, Inc., Lancaster, CA, USA, 93534-7059
2Non-clinical development - Drug safety, F.Hoffmann-La Roche Ltd., Basel, Switzerland, 4070
3Simulations Plus, Inc., Lancaster, CA, USA, 93534
4Dept of Non-Clin Drug Safety 69/154, Hoffmann-La Roche Ltd, Basel, Switzerland, CH-4070
Purpose. Ketoconazole is a potent inhibitor of the major drug-metabolizing enzyme, CYP3A4, and as the result of that is involved in many drug-drug interactions (DDIs). Pharmacokinetic information available for ketoconazole is limited since no intravenous human study has been reported to date, which is probably the main reason behind the lack of a published comprehensive PK model for this drug. The aim of our study was to simulate the human pharmacokinetics of ketoconazole using physiologically based pharmacokinetics (PBPK) and to predict the magnitude of its DDIs.
Methods. GastroPlus™ (Simulations Plus, Inc.) was used to build a PBPK model of ketoconazole’s distribution and clearance in humans using oral Cp-time profiles for 200, 400, 600 and 800 mg doses obtained from the literature. A modified Rodgers and Rowland predictive method based upon drug properties and tissue composition was applied to calculate tissue:plasma distribution coefficients (Kps). Clearance was fitted to oral doses with the PKPlus™ module within GastroPlus using the nonlinear kinetics solution. ADMET Predictor™ (Simulations Plus, Inc.) was used to predict human intestinal permeability for ketoconazole. DDIs were predicted using the DDI Module in GastroPlus.
Results. PBPK models for 200, 400, 600, and 800 mg ketoconazole oral doses using Kps obtained with the modified Rodgers and Rowland method provided a very close fit to the experimental plasma concentration-time profiles. Volume of distribution, half-life, and fraction bioavailable were reproduced with high accuracy for all doses. DDI predictions (as AUC ratios) obtained for 7 substrates (alprazolam, loratidine, midazolam, nisoldipine, sirolimus, tacrolimus, and triazolam) were within 10% of the observed in vivo values for 4 drugs and within 30% for the remainder.
Conclusions. Accurate predictions of the unbound ketoconazole liver concentrations using PBPK models were of particular importance in predicting the magnitude of its drug-drug interactions. Application of the steady-state inhibitor concentration [I] in the liver together with unbound in vivo or in vitro Ki values provided exceptional predictions of AUC ratios.
169. Evaluation of bioavailability enhancement by kajjali, an ayurvedic proprietary herbomineral product
Devang P. Shah1, Vijay Zala2, Anagha Damre3, and Sadhana S Sathaye1
1Department of Pharmaceutical Sciences and Technology, Institute of Chemical Technology (formerly UDCT), Matunga, Mumbai, India, 400 019
2Veeda clinical research, Plymouth, United Kingdom, PL6 5HH
3Drug Metabolism and Pharmacokinetics, Piramal Life sciences, Goregaon, Mumbai, India, 400 063
Evaluation Of Bioavailability Enhancement By Kajjali, An Ayurvedic Proprietary Herbomineral Product Shah Devanga, Zala Vijayb, Damre Anaghac, Sathaye Sadhanaa a: Dept. of Pharmaceutical Sciences, Institute of Chemical Technology, Matunga, Mumbai-400 019, India b: Veeda Clinical Research, Plymouth, PL6 5HH, UK c: Drug Metabolism and Pharmacokinetics, Piramal Life Sciences Limited, Goregaon (E), Mumbai-400 063, India Kajjali, an ayurvedic proprietory herbo-mineral product, is reported in ancient Ayurvedic literature to have yogavahi property, i.e. it enhances the activity of drugs co-administered with it. This study examined the effect of Kajjali on the single dose oral pharmacokinetics of Rifampicin in healthy Wistar rats. Two groups (n=6) were employed one receiving single dose of Rifampicin alone and other received Kajjali (15 mg/kg, p.o.) with Rifampicin (10 mg/kg, p.o.). Blood samples were collected up to 10 h to estimate the plasma levels of Rifampicin. Further effect of Kajjali on testosterone metabolism was investigated in vitro in rat liver microsomes. The formation of 6?-hydroxy testosterone from testosterone was used as an index of CYP3A activity in rat liver microsomes1 under control conditions and in the presence of varying concentrations of Kajjali. Kajjali was found to be a weak inhibitor of CYP3A with a 50% decrease in enzyme activity occurring at a concentration of ~50 μg/ml (IC50) in rat liver microsomes. Ketaconazole was used as a standard inhibitor of CYP3A. In rats, co-administration with Kajjali increased the Cmax, AUC and t1/2 of Rifampicin by ~1.75, 1.5 and 1.35-fold, respectively. Thus, bioavailabilty enhancement is the possible mechanism of increased activity of drugs co-administered with Kajjali, which could be due to inhibition of first-pass intestinal metabolism as in the case with Rifampicin. Reference: 1. Vilasinee H, Benjabhorn S, Hitoshi S, Inhibitory Effects of Saturated and Polyunsaturated Fatty Acids on the Cytochrome P450 3A Activity in Rat Liver Microsomes. Biological & pharmaceutical bulletin 30(8) pp.1586-1588 (2007)
170. Effects of MAOI and CYP2D6 status on 5-Methoxy-N,N-dimethyltryptamine metabolism and pharmacokinetics
Hongwu Shen1, Chao Wu2, Xiling Jiang2, and Ai-Ming Yu3
1Department of Pharmaceutical Sciences, School of Pharmacy and Pharmaceutical Sciences, The State University of New York at Buffalo, Buffalo, NY, USA, 14260
2Department of Pharmaceutical Sciences, School of Pharmacy and Pharmaceutical Sciences, University at Buffalo, The State University of New York, Amherst, NY, USA, 14260
3Department of Pharmaceutical Sciences, University at Buffalo, SUNY, Buffalo, NY, USA, 14260-1200
5-methoxy-N,N-dimethyltryptamine (5-MeO-DMT) is a psychoactive indolealkylamine substance of abuse(1, 2). While 5-MeO-DMT mainly undergoes monoamine oxidase (MAO)-catalyzed inactivation process, a small fraction is O-demethylated to active bufotenine by polymorphic cytochrome P450 2D6 (CYP2D6). Bufotenine shows 10-fold higher affinity to 5-HT2A receptor than 5-MeO-DMT itself. Furthermore, concurrent use of 5-MeO-DMT and monoamine oxidase inhibitor (MAOI) harmaline causes severe or even fatal hyperserotonergic effects(3). Therefore, current study aimed to delineate the impact of CYP2D6 phenotype/genotype and MAOI on 5-MeO-DMT metabolism and pharmacokinetics. First, CYP2D6 allelic isozymes showed significantly different 5-MeO-DMT O-demethylase activities. Estimated intrinsic clearance values for CYP2D6.2 and CYP2D6.10 were about 40% and 3% of that for CYP2D6.1, respectively. Second, bufotenine formation was not found in incubation with human CYP2D6 poor metabolizer (PM) hepatocytes but only in incubation with extensive metabolizer (EM) hepatocytes. Co-incubation with harmaline sharply reduced 5-MeO-DMT depletion and elevated bufotenine production. Third, a 1.67-fold higher exposure (AUC) to bufotenine was found in CYP2D6-humanized (Tg-CYP2D6) mice(4) than wild-type (WT) mice after i.p. administration of 20 mg/kg of 5-MeO-DMT. Co-administration of harmaline (5 mg/kg) with 5-MeO-DMT (2 mg/kg) led to 3.8- and 4.0-fold increase in exposure to 5-MeO-DMT in Tg-CYP2D6 and WT mice, respectively. The higher and long exposure to 5-MeO-DMT was translated into a more severe hyperthermic effect in both Tg-CYP2D6 and WT mice. These findings may provide helpful insight into risks of indolealkylamine intoxication.
Acknowledgements
This project is supported by the award (R01DA 021172) from National Institute on Drug Abuse, National Institutes of Health.
References:
- A. M. Yu, AAPS J 10, 242 (Jun, 2008).
- R. A. Glennon, Pharmacol Biochem Behav 64, 251 (Oct, 1999).
- D. E. Brush, S. B. Bird, E. W. Boyer, J Toxicol Clin Toxicol 42, 191 (2004).
- A. M. Yu, J. R. Idle, F. J. Gonzalez, Drug Metab Rev 36, 243 (May, 2004).
171. Effect of interferon-alpha2b on various drug-metabolizing enzymes and transporters in regular cultures and co-cultures of fresh human primary hepatocytes
Cliff Chen, Yong-Hae Han, Zheng Yang, and A. David Rodrigues
Bristol-Myers Squibb Co., Princeton, NJ, USA, 08536
Interferon-alpha2b (IFN-alpha2b) is known to alter gene expression in different cell types. Therefore, its effect on the expression (mRNA) of various human drug-metabolizing enzymes and transporters was studied using regular- and co-cultures of fresh human primary hepatocytes. When added at therapeutically relevant concentrations (1.0 to 1000 IU/mL), IFN-alpha2b up-regulated its target gene STAT1 (signal transducer and activator of transcription factor 1) in both culture systems. At the same time, its effect on various drug-metabolizing enzymes and transporters was broad but relatively selective. For example, a number of cytochrome P450s (CYP1A2, CYP2B6, CYP2E1), a UDP-glucuronosyltransferase (UGT2B7), a sulfotransferase (SULT1A1), a carboxylesterase (CES1), organic anion transporter (OAT2), and a multidrug resistance associated protein (MRP4) were significantly down-regulated by IFN-alpha2b (up to ~60%; p < 0.05), whereas two peroxisome proliferator activator receptor alpha (PPARalpha)-controlled genes (pyruvate dehydrogenase kinase 4, PDK4; and adipose differentiation-related protein, ADFP), CYP3A4, and MRP2 were significantly up-regulated (p < 0.05). Other genes, such as SULT2A1, CES2, organic anion transporting peptides (OATP-C, OATP8, OATP-B), organic cation transporter 1 (OCT1), P-glycoprotein (P-gp, ABCB1) and breast cancer resistance protein (BCRP) were not significantly affected by the IFN-alpha2b treatment. In addition, a number of genes (e.g., ADFP, PDK4, CYP3A4, CYP1A2, CYP2B6, SULT1A1, CES1, MRP2, and MRP4) responded differently to IFN-alpha2b in the regular versus co-cultures. The present results demonstrated complex interactions between IFN-alpha2b and liver cells and the observed down-regulation of CYP1A2, OAT2 and UGT2B7 is consistent with reports of drug interactions between IFN-alpha2b and drugs such as theophylline, clozapine and gemfibrozil.
172. Induction effects of chemical inducers on CYP3A enzyme activity in fresh dog and rabbit hepatocytes
Zhiming Wen, Nicole Harker, Carol Kissa, Joseph Rager, Jibin Li, Sid Bhoopathy, Albert Owen, and Ismael Hidalgo
Absorption Systems LP, Exton, PA 19341, USA
Induction effects of chemical inducers on CYP3A enzyme activity in fresh dog and rabbit hepatocytes were estimated using LC-MS/MS methods. Fresh dog (beagle, male) and rabbit (New Zealand White, male) hepatocytes were seeded at 150,000 cells/cm2 in 48-well plates and incubated with cell culture media in a 95% air/5% CO2 incubator at 37°C for 24 hours prior to the induction experiments. Subsequently, the cells were treated with known chemical inducers (rifampicin and phenobarbital) at various concentrations and the treatment was lasted for three days with daily replacement of cell culture media containing the chemical inducers. CYP3A activities (expressed as metabolite formation normalized by the cell viability) were determined by measuring the formation of 6beta-hydroxytestosterone (probe substrate, 250 μM testosterone), 1’-hydroxymidazolam (probe substrate, 20 μM midazolam), or N-demethyl erythromycin (probe substrate, 250 μM erythromycin). The results indicated that the inductive effects of rifampicin and phenobarbital on CYP3A enzyme activity in dog and rabbit hepatocytes were substrate and inducer dependent. For dog hepatocytes, phenobarbital significantly increased the formation of 6beta-hydroxytestosterone and exhibited up to 8-fold induction compared with the vehicle-treated control. Rifampicin also demonstrated inductive influence on the formation of 6beta-hydroxytestosterone and 1’-hydroxymidazolam. However, the fold-inductions by rifampicin were less than 2-fold in comparison with the vehicle-treated controls. Rifampicin and phenobarbital did not show significant induction effects on the formation of N-demethyl erythromycin. For rabbit hepatocytes, rifampicin significantly increased the formation of 1’-hydroxymidazolam with up to 3-fold induction and the formation of 6beta-hydroxytestosterone with less than 2-fold induction compared with the vehicle-treated controls. Rifampicin and phenobarbital did not show significant induction effects on the formation of N-demethyl erythromycin. In addition, phenobarbital demonstrated some inhibitory effect on the formation of 6beta-hydroxytestosterone. Conclusively, phenobabital at 2000 μM can be used as a positive inducer for CYP3A in dog hepatocytes and rifampicin at 50 μM can be used as a positive inducer for CYP3A in rabbit hepatocytes.
173. Can dietary phytochemical combinations induce glucuronidation in a human intestinal cell model (CACO-2)?
Otito F. Iwuchukwu, Din Ung, and Swati Nagar
Pharmaceutical Sciences, Temple University, Philadelphia, PA, USA, 19140
Glucuronidation by UDP-glucuronosyl transferases (UGTs) is a main inactivation pathway for carcinogens. We hypothesized that cis or trans-resveratrol alone and in combination with other polyphenols can synergistically induce UGT (1A1 and 1A6) expression levels in the intestine - the first exposure site for oral xenobiotics. Cell proliferation assays were conducted using the AlamarBlue™ assay in 96 well plates (1000 cells/well) for 72h with the polyphenols trans and cis-resveratrol, curcumin, quercetin and chrysin (0-100 uM) in 0.1% DMSO (n = 4). Caco-2 cells were grown in Eagle’s minimal essential medium (EMEM) containing 20% fetal bovine serum and 1% penicillin/streptomycin (3.5 unit/3.5 μg/ml) in 10cm plates. For induction experiments, 25uM of trans-resveratrol (t-RES) alone, 20uM curcumin alone and 20uM t-RES in combination with 1, 5, and 10 uM curcumin were used. Preconfluent (24h post seeding) and confluent (9 days post seeding) cells were treated with phytochemicals for 72 h (n = 3). Cells were collected and lysed by sonication. UGT activity in treated and control cell lysates was determined using a highly sensitive fluorescent assay with 4-methyl umbelliferone (4-MU) as substrate. Quenching of fluorescence signal upon conversion of 4-MU to its glucuronide was monitored over time (2h). Quercetin was seen to induce cell proliferation at all concentration and time points studied, and chrysin also induced cell proliferation at lower concentrations (0-10uM) over the 72h period. Curcumin (>50 uM) was cytotoxic while cis and trans-resveratrol (≤ 100 uM) were non-toxic to cells (48 and 72h). T-RES alone (p<0.05) and curcumin alone (p<0.01) significantly increased glucuronidation activity compared against untreated controls. T-RES (20uM) + curcumin (10uM) significantly increased glucuronidation activity compared with t-RES alone (p<0.01). The increase in UGT activity observed with t-RES, curcumin (and their combination) will be further probed at both the protein and mRNA levels by immunoblotting and quantitative real time PCR techniques. Parallel studies using cis-resveratrol and the same combination of phytochemicals will also be conducted and any differences in UGT inducibility will be compared.
174. Induction of hepatic drug-metabolizing enzymes in mice with a disrupted glutathione S-Transferase Mu 1 gene
Shingo Arakawa1, Yukari Shibaya1, Yumi Nishiya2, Naoki Kiyosawa1, Takanori Maejima1, Takashi Yamaguchi1, Jun Hirao1, Kazunori Fujimoto1, Wataru Takasaki1, and Ryo Atsumi1
1Medicinal Safety Research Laboratories, Daiichi Sankyo Co., Ltd., Fukuroi, Shizuoka, Japan, 437-0065
2Drug Metabolism and Pharmacokinetics Research Laboratories, Daiichi Sankyo Co., Ltd., Tokyo, Japan, 140-8710
We investigated the toxicological responses to 1,2-dichloro-4-nitrobenzene (DCNB), a specific substrate to Mu class glutathione S-transferase (GST), in mice with a disrupted GST Mu 1 gene (Gstm1-null mice) focusing on methemoglobinemia. As for other changes noted in this study, we observed the unexpected induction of hepatic drug-metabolizing enzymes in a repeated dose study for 14 days and consider the implications here. First, we confirmed the higher exposure to DCNB in Gstm1-null mice than in the wild-type control. Western blot analysis and measurement of isoform-specific activities showed the induction of Cyp1a2 and Cyp2c. While induction of Cyp1a2 was observed in both wild-type and Gstm1-null mice, induction of Cyp2c was specifically observed in Gstm1-null mice. These results suggested that the induction of Cyp2c was dependent on higher exposure to DCNB in Gstm1-null mice, but the induction of Cyp1a2 was not. Additionally, it was suggested that the induction mechanism by DCNB was different between Cyp1a2 and Cyp2c. To confirm the induction by typical inducers, we performed oral administration of 3-methylcholanthrene (3-MC) and phenobarbital (PB), which are not specific substrates of GSTM1, for 4 days. According to the results, induction of Cyp1a2 by 3-MC and induction of Cyp2b by PB were similar in wild-type and Gstm1-null mice. In conclusion, it is suggested that compounds which are both substrates of GSTM1 and inducers of drug-metabolizing enzymes, such as DCNB, might cause marked induction in Gstm1-null mice.
175. Metabolism mediated CYP 2B induction by LX6171 (3′-chlorobiphenyl-4-yl)-1-(pyrimidine-2-yl) piperidin-4-yl) methanone) in the rat
Ling Li1, Jeffrey A. Kramer1, William E. Heydorn2, Amr Nouraldeen1, James Schmidt1, Jianghong Jiang1, Liam Moran1, and Alan GE Wilson1
1Drug Metabolism, Pharmacokinetics, Toxicology and Pathology, Lexicon Pharmaceuticals, The Woodlands, TX, USA, 77401
2PreClinical Development, Lexicon Pharmaceuticals, Princeton, NJ, USA
LX6171 (3′-chlorobiphenyl-4-yl-1-(pyrimidine-2-yl) piperidin-4-yl) methanone is a drug candidate for cognitive disorder. Carbonyl reductase was shown to be the major enzyme involved in the metabolism of LX6171 to its major chiral metabolite (metabolite A) in preclinical species and human. In rat liver S9 fraction, LX6171 was exclusively converted to the S-form of metabolite A, namely S-(+)-(3’-chloro-biphenyl-4-yl)-(1-pyrimidine-2-yl-piperidin-4-yl)-methanol)), with no evidence of the R-form. The in vitro finding agrees with the in vivo observation that only the S-enantiomer of metabolite A was formed in rats administered LX6171. In human liver S9 fraction, the S-form of metabolite A was also the predominant enantiomer. Upon chronic treatment (28 days) of rats with LX6171 at 450 mg per kg, a statistically significant increase in liver weight and an approximately 6-fold increase in cytochrome P450 2B activity (CYP 2B) was observed. The CYP 2B enzyme induction was further characterized using rat primary hepatocytes. An approximate 2-fold induction of CYP 2B activity was observed in rat primary hepatocytes treated with LX6171 for 48 hours. Under these conditions LX6171 was almost completely converted to the S-form of metabolite A (> 90%). A similar level of CYP 2B induction was observed by incubation of rat hepatocytes with the S-form of metabolite A. In contrast, under similar conditions, incubation of LX6171 or the S-form of metabolite A, failed to induce CYP 2B activity in human primary hepatocytes. Our results indicate that while the overall metabolism of LX6171 is similar between rat and man, the induction observed in rats does not translate to humans. These studies further indicate the caution that should be applied in extrapolating findings of rodent CYP induction to man.
176. Fast LC-MS/MS method for determination of CYP1A, CYP2B and CYP3A induction in primary human hepatocytes
Mary A. Palacios, Karl B. Frank, and David J. Moore
Non-Clinical Safety, Hoffmann-La Roche Inc., Nutley, NJ, USA, 07110
The potential of new molecular entities (NMEs) to induce Cytochrome P450s in human liver is evaluated early in the development process in order to predict drug-drug interaction liabilities. Primary human hepatocyte cultures are incubated with NMEs for a period of time to permit enzyme induction, then probe substrates specific for CYP1A and CYP3A are added. This is consistent with current FDA guidance, which emphasizes the induction of CYP1A and CYP3A. CYP1A and CYP3A are controlled primarily by the transcription factors AhR and SXR, respectively. Activation of the major transcription factor CAR is detected indirectly because of cross-talk between CAR and SXR. Bupropion is a selective probe substrate for the CAR-activated CYP2B6 and is used in order to distinguish compounds which activate CAR preferentially from compounds which activate SXR. The combination of tacrine (CYP1A2), bupropion (CYP2B6), and midazolam (CYP3A4) permits detection of AhR, CAR and SXR activation. Analysis of metabolites from probe substrate cocktails can require lengthy LC-MS/MS runs, so we investigated methods to reduce the analytical time. Short HPLC columns containing small, fused-core particles, along with faster flow rates dramatically reduce the amount of time per sample. The fast analytical method permits quantitation of three metabolites plus internal standard using run times of 0.75 minutes per sample, a four-fold increase in throughput over previous methods.
177. Development of in vitro higher throughput CYP1A, CYP2B6 and CYP3A induction assays using probe cocktail approach
Jingjing Yu, and Jun Tang
Cerep, Redmond, WA, USA, 98052
CYP enzymes, such as CYP1A2, CYP2B6, and CYP3A4 are susceptible to induction. In vitro CYP induction assays using plateable hepatocytes are widely used to identify compounds that may induce CYP activities. To increase the throughput in CYP induction study, we evaluated a probe cocktail approach for CYP1A, CYP2B6, and CYP3A. Cryopreserved human hepatocytes were treated with known CYP inducer omeprazole (50 μM, for CYP1A), phenobarbital (1000 μM, for CYP2B6), or rifampin (10 μM, for CYP3A) for 48 hours. CYP activities were analyzed using the following four probe substrates as either single form or a cocktail: 100 μM phenacetin (P) or 2 μM ethoxyresorufin (E) for CYP1A, 200 μM bupropion (B) for CYP2B6, and 10 μM midazolam (M) for CYP3A. For CYP1A induction assay, there were substantial increases in ethoxyresorufin or phenacetin metabolism when analyzed using either probe cocktails (P/B, P/M, P/B/M, E/B, E/M, and E/B/M) or single substrate (ethoxyresorufin or phenacetin only). The increase in ethoxyresorufin metabolism (21-28 fold induction) was greater than in phenacetin metabolism (6-15 fold induction). For CYP2B6 induction assay, there were considerable increases in bupropion metabolism when analyzed using either probe cocktails (P/B, P/B/M, E/B, and E/B/M) or single substrate (bupropion only) (8-21 fold induction). The increase in midazolam metabolism (in cocktails P/B/M and E/B/M) (17-20 fold induction) was even greater than that in bupropion metabolism in CYP2B6 induction assay, verifying that phenobarbital also induces CYP3A activity. For CYP3A induction assay, the increase in midazolam metabolism using probe cocktails (P/M, P/B/M, E/M, and E/B/M) was similar to that using single substrate (midazolam only) (14-18 fold induction). The increase in bupropion metabolism was also observed in CYP3A induction assay when analyzed using cocktail P/B/M or E/B/M (4-7 fold induction), less than that of midazolam metabolism. The results indicated that probe cocktail approach can be used to screen compounds for their CYP induction potential although it may not be able to differentiate CYP2B6 and CYP3A inductions.
178. Modeling and simulation of concentration-response relationships of CYP induction in human primary hepatocytes
Mike Hayashi1, Yuping Chen1, Gary L. Skiles2, and Magang Shou1
1Department of Pharmacokinetics and Drug Metabolism, Amgen Inc., Thousand Oaks, CA, USA, 91320-1799
2Department of Pharmacokinetics and Drug Metabolism, Amgen Inc., Thousand Oaks, CA, USA, 91320
Enzyme induction can lead to undesirable drug interactions that alter the efficacy of co-administered drugs. During drug development, drug candidates are often tested for their propensity to cause enzyme induction in primary hepatocyte cultures; however, in these studies there is typically a limited understanding of the dynamic relationship between changing concentrations of the inducer and levels of induction. In the present study the induction of CYP1A2 and CYP3A4 was studied in cultures of human hepatocytes from two donors using the prototypical inducers omeprazole and rifampin. The time course of inducer concentrations, mRNA transcript levels, and catalytic activity was monitored. An indirect model was used to link drug clearance to induction response. Clearances of omeprazole (50 μM) and rifampin (10 μM) in human hepatocytes (106 cells/mL, volume = 0.5 mL) from two donors were determined to be 50-60 and 14-22 μL/hr, respectively. Half lives (0.693/kout) for degradation of transcript and catalytic activity of the two enzymes were resolved. The simulated EC50 and Emax in cells from Donor1/Donor 1 were determined to be 5/16 μM and 25/45 fold increase for CYP1A2 and 0.1/0.8 μM and 16/23 fold increase for CYP3A4. This indirect model makes it possible to account for turnover of the inducers as well as changes in mRNA and enzyme levels for a more complete kinetic assessment of induction. Accounting for these dynamic changes and improving the accuracy of the induction kinetics should aid in the prediction of clinical drug-drug interactions.
179. A Comprehensive evaluation of the in vitro tools used for determining hPXR activation, CYP3A4 induction, time dependent inhibition (TDI) and the effects of protein binding on several classes of antibacterials
Suzzette Haney, Karen Stams, Garrett Ainslie, Selvi Pradeepan, Patrick Brassil, and C. Edwin Garner
Discovery DMPK, AstraZeneca R&D Boston, Waltham, MA, USA, 02451
Abstract: Hypothesis: The aim of the present studies was to investigate the utility of several in vitro tools to predict the potential for drug-drug interactions of several classes of early and late generation antibacterials in a drug discovery setting.
Methods: Profiles of hPXR activation were assessed using cell lines transfected with hPXR and a luciferase reporter gene. Sandwich cultured human hepatocytes were treated with both prototypical CYP3A4 inducers and inhibitors and selected antibacterials at concentrations ranging from 0.1uM to 100uM. CYP3A4 enzyme activity was determined in situ using testosterone as a probe substrate. RNA was isolated from hepatocyte cultures and CYP3A4 expression was measured by TaqMan analysis. Plasma protein binding was performed using human plasma to estimate the free fraction values using rapid equilibrium dialysis method.
Results: CYP3A4 activity was observed to significantly increase with rifampicin, dicloxacillin and nafcillin treatment. Piperacillin, ceftriaxone and vancomycin were observed as moderate inducers and the fluoroquinolones, macrolides and aztreonam were observed as weak inducers. CYP3A4 induction EC50 was calculated for using both enzyme activity analysis and mRNA TaqMan analysis. With some macrolides, TDI was found to drive a lack of correlation that was observed between CYP3A4 mRNA levels and CYP3A4 enzyme activity. The hPXR activation seen in some antibacterials correlated roughly with the induction measured in human hepatocytes. Conclusions: The present study evaluated a successful strategy for predicting induction of hPXR-mediated drug metabolism employing a combination of integrated in vitro approaches. These data suggest that several early and late generation antibacterials commonly in use have moderate to potent CYP3A4 induction potential. These results suggest that a combination of in vitro tools should be used to identify antibiotics that are most likely to cause the potential for drug-drug interactions mediated via CYP3A4 induction. Early use of hPXR activation may be used to rank order and identify CYP450 inducers in the drug discovery process.
180. Relative potential of anti-HIV protease inhibitors to induce cytochrome P450 enzymes and transporters in human hepatocytes
Li Liu1, Ganesh M. Mugundu2, Divya Samineni2, Brian J. Kirby1, Pankaj Desai2, and Jashvant Unadkat1
1Department of Pharmaceutics, University of Washington, Seattle, WA, USA, 98195
2College of Pharmacy, University of Cincinnati, Cincinnati, OH, USA, 45267
Anti-HIV protease inhibitors (PRIs) produce profound and unpredictable drug drug interactions (DDIs) that cannot be fully explained by their inhibition/inactivation of CYP3A enzymes. Many of these DDIs appear to be due to their concurrent, yet less well-characterized induction of CYP450 enzymes and transporters. Identification of the proteins inducible by PRIs, and the magnitudes of their induction are crucial in developing an integrative mechanistic understanding and prediction of PRI DDIs. Primary human hepatocyte induction studies were conducted at 37°C for 72hrs, with media containing 10μM PRIs (Ritonavir/RTV, Nelfinavir/NFV, Saquinavir/SQV, Amprenavir/APV, Indinavir/IDV, Lopinavir/LPV, Tipranavir/TPV and Atazanavir/AZV), and Rifampin/RIF (positive control) changed every 24hrs. Changes in the expressions of hepatic CYP450s (3A4/5, 2B6, 2C8/9/19, 1A1/2, 2E1, 2A6, and 2D6; activities and transcripts), and transporters (OATP1B1/1B3/1A2, P-gp, MRP2/4; transcripts) were evaluated using relative RT-PCR, and two validated LCMS/MS cocktail assays developed for microsomal CYP450 activity determinations. All results were expressed relative to the induction produced by RIF, with >40% increase in activity designated as significant induction. CYP2B6 activity was induced by all PRIs, ranging 40% - 95%. Except for RTV and SQV, CYP3A activity was significantly induced 40% - 85% by all PRIs. CYP2C8 activity was induced 40% - 63% by APV, LPV, TPV, and IDV, with other PRIs eliciting 35% - 40% induction. mRNA induction for CYP3A4/5, 2B6, and 2C8 varied between 25% - 355%. No significant induction was observed for other CYP450s. Only OATP1B1 and P-gp showed significant mRNA induction, ranging 64% - 442% and 36% - 141%, respectively. Amprenavir consistently ranked highest in PRI induction potentials. Our results provided the much needed induction profiling of hepatic CYP450s and transporters by the PRIs. The significant induction of CYP2B6, 3A, 2C8, OATP1B1, and P-gp by the PRIs likely explains many of the inductive DDIs such as those with methadone (CYP2B6, P-gp), loperamide (P-gp, CYP3A4, 2C8), and pravastatin (OATP1B1, P-gp).
181. In situ evaluation of CYP1A2, CYP2B6 and CYP3A4/5 induction in human hepatocytes with a cocktail of probe substrates
Immaculate Amunom, Sarah Brawner, Jennifer Simpson, Janet Sawi, Rebecca Campbell, Jason Neat, and Andrew Parkinson
XenoTech, LLC, Lenexa, KS, USA, 66219
In situ (cell-based) incubations of cultured human hepatocytes with specific probe substrates are an industry-accepted approach to measuring enzyme induction (Chu et al., 2009). The objective of this study was to develop an in situ incubation method to screen for the induction of CYP1A2, CYP2B6 and CYP3A4/5 with a cocktail of marker substrates: phenacetin (100 μM), bupropion (500 μM) and midazolam (30 μM), respectively. Freshly isolated human hepatocytes were cultured in a sandwich configuration and treated with the vehicle control, dimethyl sulfoxide (DMSO; 0.1%, v/v) or prototypical inducers, namely omeprazole (CYP1A2), phenobarbital (CYP2B6) or rifampin (CYP3A4/5). Specific probe substrates were incubated either individually or as a cocktail mixture to investigate the induction of CYP enzyme activity following treatment of the hepatocytes with prototypical inducers. The metabolites, namely acetaminophen, hydroxybupropion and 1′-hydroxymidazolam, and the corresponding deuterated internal standards were quantified by HPLC-MS/MS. The data show that the CYP activities determined with the individual substrates in control and inducer-treated hepatocytes were only slightly different from those determined with the cocktail mixture of substrates. In addition, in reaction phenotyping experiments with 18 individual samples of human liver microsomes, CYP2B6 and CYP3A4/5 activity determined with the substrate cocktail was the same as that determined with bupropion and midazolam alone, whereas CYP1A2 activity determined with the cocktail was slightly less (~27%) than that determined with phenacetin alone. The sample-to-sample variation in CYP activity determined with the cocktail of probe substrates was remarkably similar to that determined with the individual substrates. These results suggest that in human hepatocytes and human liver microsomes there are negligible interactions between 100 μM phenacetin, 500 μM bupropion and 30 μM midazolam when co-incubated together, which supports the use of a cocktail of these three substrates to screen simultaneously for induction of CYP1A2, CYP2B6 and CYP3A4 in human hepatocytes.
References:
- Chu V, Einolf HJ, Evers R, Kumar G, Moore D, Ripp S, Silva J, Sinha V, Sinz M and Skerjanec A. In vitro and in vivo induction of cytochrome P450: A survey of the current practices and recommendations: A Pharmaceutical Research & Manufacturers of America (PhRMA) perspective. Drug Metab. Dispos. April 2009. Doi:10.1124/dmd.109.027029
182. CAR (constitutive androstane/active receptor)-mediated Up-regulation of Cytochrome P450 (CYP) 2B6 Expression by 17β-estradiol (E2)
Kyunghee Yang1, Kwihye Koh1, Suyoung Choi2, and Hyunyoung Jeong1
1Pharmacy Practice, University of Illinois at Chicago, Chicago, IL, USA, 60612
2Center for Pharmaceutical Biotechnology, University of Illinois at Chicago, Chicago, IL, USA, 60612
Oral contraceptives are known to alter the expression and the activity of CYP isozymes. Estrogen is a major component of oral contraceptives, but its effects on CYP expression have not been fully understood. CYP2B6 is one of the major drug metabolizing enzymes responsible for metabolism of clinically important drugs. Recently, estrogen has been reported to induce the expression of Cyp2b10, a mouse homologue of human CYP2B6. Therefore, we hypothesized that estrogen affects CYP2B6 expression in humans. In this study, we investigated the effects of E2 on the CYP2B6 expression and the underlying regulatory mechanism. To this end, we performed quantitative real-time PCR and functionality assays to determine the mRNA and the activity levels of CYP2B6, respectively. To identify the regulatory mechanism involving PXR and CAR, luciferase promoter reporter assays and nuclear translocation assays were performed. Treatment of primary human hepatocytes with E2 (100 nM) increased mRNA expression of CYP2B6 by 12.3-fold; the induction of CYP2B6 expression was E2-concentration dependent (EC50 = 56.6 nM). Also, the activity of CYP2B6 (S-mephenytoin N-demethylation) was increased by 3.4-fold upon E2 treatment. In HepG2 cells expressing hPXR, the luciferase promoter activity of CYP2B6 was not influenced by treatment with E2. In addition, the expression level of CYP3A4, a known PXR target gene, was not influenced by E2 treatment in primary human hepatocytes. In primary rat hepatocytes, E2 triggered nuclear translocation of green fluorescent protein-tagged human CAR. Taken together, our data indicate that E2 up-regulates CYP2B6 at the transcriptional level, and this regulatory mechanism is mediated by CAR, not by PXR. These findings suggest potential increase in CYP2B6 expression level in users of estrogen-based oral contraceptives.
183. The effects of 17 β-estradiol on activities of cytochrome P450s in human hepatocytes
Su-Young Choi1, and Hyunyoung Jeong2
1Center for Pharmaceutical Biotechnology, University of Illinois at Chicago, Chicago, IL, USA, 60612
2Department of Pharmacy Practice, College of Pharmacy, University of Illinois at Chicago, Chicago, IL, USA, 60612
Results from clinical studies suggest that estrogen may have regulatory effects on cytochrome P450 (CYP). 17β-Estradiol (E2), the predominant endogenous estrogen, is potentially responsible, but the effects of E2 on expression and activities of major CYP enzymes in human tissues remain unknown. The objective of this study is to investigate the effects of E2 on individual CYP activity. We treated primary human hepatocytes with vehicle (ethanol), estradiol (1 μM), or positive controls (rifampin 10 μM, CITCO 100 nM, phenobarbital 200 μM or ethanol 5 mM). After 72 hour incubation, media were replaced to contain a pathway-specific probe drug: phenacetin (CYP1A2), S-mephenytoin (CYP2B6 and -2C19), diclofenac (CYP2C9), bufuralol (CYP2D6), midazolam (CYP3A4), or p-nitrophenol (CYP2E1). Media were sampled every 30 min for 6 hours. The metabolite concentrations were determined by using LC/MS/MS, and CYP enzyme activities were estimated by calculating the rates of metabolite formation. E2 increased the activities of CYP2B6 by 3.4 fold (p=0.0008), -2C9 by 1.7 fold (p=0.0009), -3A4 by 1.9 fold (p=0.04), and -2E1 by 1.8 fold (p=0.006). The E2-mediated induction of CYP activities was intermediate to strong as compared to the induction by positive controls; 43% of CITCO-mediated CYP2B6 induction, 113% of phenobarbital-mediated CYP2C9 induction, 41% of rifampin-mediated CYP3A4 induction, and 112% of ethanol-mediated CYP2E1 induction. E2 did not affect the activities of CYP1A2, -2C19, and -2D6. Taken together, our results suggest that E2 influences CYP activities in a pathway-dependent manner in human hepatocytes.
184. Mechanism-based inhibition of CYP2C8: Differentiation by IC50 shift approach and enzyme inactivation kinetics
Zhiming Wen, Carol Kissa, Nicole Harker, Joseph Rager, Jibin Li, Sid Bhoopathy, Chris Bode, Albert Owen, and Ismael Hidalgo
ABSORPTION SYSTEMS LP, Exton, PA, USA, 19341
Mechanism-based inhibition of cytochrome P450 (CYPs)-mediated reactions leads to irreversible or quasi-irreversible inactivation and has greater clinical impact than reversible inhibition. In this study, the inhibitory effects of known reversible (quercetin and montelukast) and mechanism-based (phenelzine, amiodarone, and gemfibrozil glucuronide) inhibitors of human CYP2C8 were investigated with pooled human liver microsomes by LC-MS/MS. Two IC50 shift approaches were compared: (i) a 30-minute pre-incubation with inhibitor and microsomes in the presence or absence of NADPH followed by an enzyme activity incubation with the probe substrate amodiaquine (pre-incubation approach), and (ii) a 30-minute co-incubation with inhibitor, amodiaquine, microsomes, and NADPH (co-incubation approach). Mechanism-based inhibitors demonstrated significant leftward IC50 shift after pre-incubation with NADPH, in comparison with the IC50 after pre-incubation in the absence of NADPH. In contrast, reversible inhibitors exhibited a rightward IC50 shift or no changes in IC50 after pre-incubation with NADPH. Similar IC50 alterations were observed by the co-incubation approach coupled with the 30-minute pre-incubation in the presence of NADPH, suggesting that the IC50 shift estimated by either the pre-incubation or co-incubation approaches can be used to differentiate reversible and mechanism-based inhibitors of CYPs in vitro. The enzyme inactivation kinetics of mechanism-based inhibitors of CYP2C8 were assessed using multiple pre-incubation times (0, 5, 10, 15, 20, and 30 min) and multiple inhibitor concentrations with human liver microsomes (1 mg/ml) followed by an enzyme activity assay using amodiaquine (at a concentration approximately 5-fold above the Km) with a 20-fold dilution from the pre-incubated solutions. The estimated KI (inhibitor concentration at half-maximal inactivation) and kinact (maximum inactivation rate constant) values were 36 μM and 0.085 min−1 for phenelzine, 286 μM and 0.051 min−1 for amiodarone, and 52 μM and 0.23 min−1 for gemfibrozil glucuronide.
185. In vitro-to-in vivo extrapolation (IVIVE) of cytochrome P450 (CYP) inhibition results: Test-system-dependent outcomes with gemfibrozil and ezetimibe
Brian W. Ogilvie, Phyllis Yerino, Faraz Kazmi, David B. Buckley, and Andrew Parkinson
XenoTech, LLC, Lenexa, KS, USA, 66219
The potential of new drug candidates to inhibit CYP enzymes is frequently assessed in vitro with human liver microsomes (HLM) or recombinant CYP enzymes, per FDA recommendations. HLM lack many of the drug-metabolizing enzymes and transporters present in human hepatocytes (either because the protein is not present in microsomes or because the appropriate cofactor is not added). Accordingly, CYP inhibition studies in hepatocytes would be expected to yield more clinically relevant data than studies in HLM. However, there are surprisingly few compelling examples to support this viewpoint. In the present study, we provide two examples where in vitro studies in hepatocytes, but not in HLM, predicted the outcome of clinical drug-drug interaction studies with gemfibrozil and ezetimibe. In the case of gemfibrozil, studies with NADPH-fortified HLM under-predicted the clinically relevant inhibition of CYP2C8, whereas similar studies with ezetemibe over-predicted the degree of inhibition of CYP3A4 observed clinically. In the case of gemfibrozil, the inhibition of CYP2C8 involves the metabolism-dependent, irreversible inhibition of CYP2C8 by gemfibrozil (an example of glucuronidation-dependent activation). In the case of ezetemibe, the rapid and extensive metabolism-dependent, irreversible inhibition of CYP3A4 was not observed in hepatocytes due to its rapid glucuronidation (an example of glucuronidation-dependent protection). These CYP inhibition studies with gemfibrozil and ezetimibe provide examples of system-dependent outcomes and illustrate exceptions to the “rank-order” approach to interpreting in vitro CYP inhibition results from HLM.
186. The super-proportional impact of inhibition of multiple pathways of diclofenac metabolism in cryopreserved human hepatocytes
Kah Hiing John Ling1, David B. Buckley2, Phyllis Yerino2, Brian W. Ogilvie2, Dale Yu1, Diane Tang-Liu1, and Andrew Parkinson2
1Clinical Pharmacology & Quantitative Sciences, Allergan, Irvine, CA, USA, 92612
2XenoTech, LLC, Lenexa, KS, USA, 66219
The impact of multiple perpetrators on the disposition of a victim drug is often far greater than the sum or product of the individual perpetrator due to the inhibition of two or more metabolic pathways, or a metabolic and transport-mediated pathway, and are difficult to examine in vitro. Diclofenac 4′-hydroxylation is an in vitro marker of CYP2C9 activity, and the potent CYP2C9 inhibitor, sulfaphenazole, inhibits diclofenac 4′-hydroxylation in human liver microsomes. Diclofenac is also directly glucuronidated (UGT2B7), with subsequent oxidation by CYP2C8 to form 4′hydroxydiclofenac glucuronide in vivo. We hypothesize that diclofenac can serve as a model substrate to examine potentially disproportionate effects of UGT and CYP inhibitors on its metabolism in human hepatocytes. Among the several CYP and UGT inhibitors examined, sulfaphenazole (3 μM) and androsterone (100 μM) were found to be adequately selective for CYP2C9 and UGT2B7, respectively. These inhibitors (singly, and in combination) were used for time-course (120 min) experiments with cryopreserved human hepatocytes (0.7 million cells/mL) with diclofenac (8 μM) and the study results are summarized in the following table.
These results show that glucuronidation is the more important pathway in diclofenac metabolism since the reduction in clearance with sulfaphenazole is approximately half of that effected by androsterone. There was an approximately three-fold increase in the production of diclofenac acyl glucuronide in the presence of sulfaphenazole, indicative of shunting to the UGT pathway. Both CYP2C9 and UGT pathways were completely inhibited in the presence of both sulfaphenazole and androsterone leading to a 96.7% decrease in diclofenac metabolism supporting the super-proportional effect of two perpetrators on the disposition of a victim drug. This approach may have value in attempts to predict the clinical effects of multiple inhibitors on the pharmacokinetics of a victim drug.
187. Interaction of human Bax inhibitor-1 with anthracycline-ring compound
Eun Yi Cho1, Ho-Zoon Chae1, Hyung-Ryong Kim2, Han-Jung Chae3, Taeho Ahn4, and Chul-Ho Yun5
1School of Biological Sciences and Technology and Hormone Research Center, Chonnam National University, Gwangju, South Korea
2Department of Dental Pharmacology, School of Dentistry, Wonkwang University, Iksan, South Korea
3Department of Pharmacology and Institute of Cardiovascular Research, School of Medicine, Chonbuk National University, Jeonju, South Korea
4Department of Biochemistry, College of Veterinary Medicine, Chonnam National University, Gwangju, South Korea
5School of Biological Sciences and Technology, Chonnam National University, Gwangju, South Korea
Bax inhibitor-1 (BI-1) is an evolutionarily conserved cell death suppresser in both animals and plants. We examined the effect of clinically important anthracycline-ring compounds such as doxorubicin (DXR) and daurorubicin (DNR) on the functional regulation of recombinant human Bax inhibitor-1 (BI-1) reconstituted into model membranes. DXR and DNR specifically interacted with the reconstituted BI-1 and the dissociation constants were calculated as 1.42 ? 0.28 uM and 1.67 ? 0.32 uM, respectively, using the radioactively labeled-compounds. When DXR or DNR was added to the proteoliposomes containing encapsulated Ca2+ and the external acidity was decreased from pH 7.4 to 6.0, they also inhibited the proton-induced Ca2+ release from the membranes in a dose-dependent manner. The half maximal inhibitory concentration (IC50) values were calculated as approximately 0.76 uM for DXR and 0.93 uM for DNR, respectively. In vivo effects of the anthracycline drugs in relation with BI-1 functions were also discussed. Keywords: Bax inhibitor-1 (BI-1), anthracycline-ring, doxorubicin (DXR), daurorubicin (DNR)
188. CTP-347: A deuterated analog of paroxetine with greatly reduced CYP2D6 mechanism-based Inactivation
Richard Gallegos
Concert Pharmaceuticals, Inc., Lexington, MA, USA, 02421
Paroxetine, a frequently-prescribed serotonin reuptake inhibitor, is a potent CYP2D6 mechanism-based inactivator (1). As such, paroxetine is associated with significant drug-drug interactions due to the role of CYP2D6 in drug metabolism. CTP-347 is a new chemical entity that differs from paroxetine by specific deuterium incorporation at key positions of the molecule. CTP-347 possesses an in vitro pharmacology profile similar to paroxetine, but shows an increased rate of clearance in vitro and in vivo. This difference in clearance is due to a significant decrease in the mechanism-based inactivation of CYP2D6 relative to paroxetine. To our knowledge, CTP-347 is the first compound to have deuterium isotope effects upon mechanism-based inhibition. These results suggest that the potential for drug-drug interactions for CTP-347 may be substantially reduced compared to paroxetine.
189. Pitfalls in the design of CYP inhibition studies incorporating a dilution step to examine time-dependent inhibition (TDI) or metabolism-dependent inhibition (MDI)
Brandy L. Paris, Faraz Kazmi, David B. Buckley, Brian W. Ogilvie, Amy E. Gipson, and Andrew Parkinson
XenoTech, LLC, Lenexa, KS, USA, 66219
A recent PhRMA publication (Grimm et al., 2009), advocates the use of a dilution step in experiments to determine the kinetics of TDI/MDI of CYP enzymes of drug candidates in human liver microsomes (HLM). Conducting such studies to determine KI and kinact is useful because it reduces the direct inhibitory effects of the drug candidate. However, the results of the present study argue strongly against the use of a dilution step in IC50-shift experiments to assess the ability of test drugs to cause TDI/MDI of CYP enzymes. Dilution experiments (10-, 20-, 30- and 40-fold) with the direct-acting CYP3A4 inhibitor fluconazole established that when IC50 values are based on the pre-dilution concentration, they increase in direct proportion to the fold dilution of HLM. Consequently, the IC50 value for direct inhibition of CYP3A4 with fluconazole should be based on the “final” nominal concentration of inhibitor in the post-dilution incubation with marker substrate. In contrast, dilution experiments with several MDIs of CYP3A4 established that, following a 30-min pre-incubation with NADPH-fortified HLM, IC50 values for MDI decrease in direct proportion to the fold dilution of HLM, whereas IC50 values for direct inhibition increase in direct proportion to fold dilution. Consequently, IC50 values for MDI should be based on the pre-dilution concentration of inhibitor (concentration of inhibitor during the preincubation with HLM prior to dilution). Therefore, unless the IC50 value obtained without pre-incubation is based on the post-dilution inhibitor concentration, and unless the IC50 value obtained with a 30-min pre-incubation is based on the pre-dilution concentration, then the shift in IC50 will be exaggerated by a factor approximately equal to the fold dilution. Furthermore, when the concentration of HLM is increased 10-fold, certain potent MDIs appear to lose their ability to inactivate CYP enzymes because there is insufficient inhibitor to inactivate the 10-fold higher concentration of CYP enzyme and/or because there is inhibitor depletion during the preincubation period. In summary, a dilution step has a certain value in studying the kinetics of CYP inactivation, but it should not be used in the initial assessment (e.g., IC50 shift experiments) of whether a drug candidate is a time- or metabolism-dependent inhibitor, and it may have limitations when determining KI and kinact for potent metabolism-dependent inhibitors.
References
- Grimm SW, Einolf HJ, Hall SD, He K, Lim HK, Ling KH, Lu C, Nomeir AA, Seibert E, Skordos KW, Tonn GR, Van Horn R, Wang RW, Wong YN, Yang TJ and Obach RS. The conduct of in vitro studies to address time-dependent inhibition of drug metabolizing enzymes: a perspective of the Pharmaceutical Research and Manufacturers of America (PhRMA). Drug Metab Dispos. April 9, 2009 doi:10.1124/dmd.109.026716.
190. Natural products as Inhibitors of CYP1 enzymes
Simone Badal1, Sheri-Ann Williams2, Helen Jacobs2, and Rupika Delgoda1
1Natural Products Inst, Univ of the West Indies, Mona, Jamaica, JM-J
2Dept of Chemistry, Univ of the West Indies, Mona, Jamaica, JM-J
The involvement of CYP1 family of enzymes in the bioactivation of procarcinogens such as polycyclic aromatic hydrocarbons (PAHs), heterocyclic amines, aromatic amines and nitro polycyclic hydrocarbons, in addition to the biotransformation of drugs including anti-cancer drugs, have made them targets for drug discovery. Inhibitors of CYP1 enzymes are thought to be potential anti-carcinogens if they could inhibit the ability of these enzymes to metabolize PAHs to toxic intermediates and/or decrease their ability to detoxify cancer drugs. Such thinking has led to the search for potent inhibitors of CYP1 family. In this study, we screened purified and related compounds from the plant Amyris plumieri for the inhibition of CYP1 family. Six chromene amide compounds were isolated and purified from this tropical Rutaceae genus and monitored for their inhibition of the activities of heterologously expressed CYP microsomes (CYPs 1A1, 1A2, 1B1) using a fluorescent assay. Moderate inhibition was found against CYP1A1 with IC50 values for chromene amide 1 (see figure 1) being the most potent at 1.537 ± 0.995 μM. Chain elongation in these amides significantly reduced their potency with both CYP1A2 and CYP1B1. The addition of a propyl residue at R1 increased the IC50 of chromene amide 1 six fold against CYP1A2 and tenfold against CYP1B1. Such structure-activity relationship studies provide a useful basis for the search for more potent inhibitors of CYP1 enzymes which may have implications in cancer chemoprevention. (Supported by the International Foundation for Science, Postgraduate Research Fund and Study and Travel grant, UWI)
Figure 1. Strucuture of chromene amide 1 where R1=CH3
191. Optimization of in vitro UGT inhibition studies
Lois J. Haupt, Phyllis Yerino, Brian W. Ogilvie, Brandy L. Paris, David B. Buckley, Amy E. Gipson, and Andrew Parkinson
XenoTech, LLC, Lenexa, KS, USA, 66219
The design of in vitro UDP-glucuronosyltransferase (UGT) inhibition experiments can be problematic because the degree of inhibition is influenced by the incubation conditions, and it can be system-dependent (i.e., it can differ between human liver microsomes (HLM) and recombinant UGT enzymes). For example, 1-naphthol inhibits UGT1A4 (measured by trifluoperazine glucuronidation) in native HLM with an IC50 value of 135 μM; however, the IC50 value increases by more than an order of magnitude when recombinant UGT1A4 is used. The difference reflects the rapid glucuronidation of 1-naphthol in HLM (by UGT1A6), which leads to a decrease in the cofactor UGPGA and an increase in its competitive inhibitor UDP; hence, there is inhibitor (UDP) formation with HLM but not with rUGT1A4. Conversely, 1-naphthol is a potent inhibitor of recombinant UGT1A9 (measured by propofol glucuronidation) but not of UGT1A9 in HLM because in the latter system, 1-naphthol is rapidly glucuronidated by microsomal UGT1A6; thus, there is inhibitor (1-naphthol) depletion with HLM but not with rUGT1A9. The concentration of UDPGA can influence the results of UGT inhibition experiments. Higher concentrations of UDPGA lessen the inhibitory effects of UDP and, therefore, decrease the inhibitory effects of high turnover compounds like 1-napthol. For example, the IC50 value for inhibition of UGT1A4 by 1naphthol increases approximately four-fold after increasing the concentration of UDPGA from 0.2 to 20 mM. Similarly, the IC50 value for inhibition of UGT1A9 by 1naphthol increases approximately three-fold after increasing the concentration of UDPGA from 0.2 to 20 mM. While the addition of activators such as alamethicin or CHAPS may be appropriate for use in studies evaluating potential drugs as substrates for UGT enzymes, the use of activators in UGT inhibition experiments offers no advantage, particularly because adequate analytical response for measurement of inhibition can be readily achieved without their use. Finally, it is important to consider experimental conditions for UGT inhibition experiments that minimize inhibitor depletion, especially if glucuronidation of the inhibitor is possible.
192. Effects of plasma on cytochrome P450 (CYP) inhibition studies in human liver microsomes (HLM): Consequence on in vitro-in vivo extrapolations (IVIVE)
Faraz Kazmi, David B. Buckley, Phyllis Yerino, Brian W. Ogilvie, and Andrew Parkinson
XenoTech, LLC, Lenexa, KS, USA, 66219
For direct, reversible inhibitors of CYP enzymes, the potential for clinical drug-drug interactions is based on [I]/Ki, where Ki is the inhibition constant determined with HLM and, based on FDA recommendations, [I] is the total concentration of drug in plasma at Cmaxss, not the pharmacologically relevant free (unbound) drug concentration. Theoretically, if in vitro CYP inhibition experiments are performed in the presence of plasma (Lu et al., 2007), such that the total and free drug concentration in vitro are the same as those in vivo, then IVIVE can be correctly based on total drug concentration (Cmaxss), as required by the FDA, without even knowing the free concentration of drug. We conducted studies with NADPH-fortified HLM in the presence of ~95% human plasma to evaluate the IVIVE of three CYP inhibitors, namely, ketoconazole, montelukast, and terbinafine. In general, the apparent CYP activity decreased with increased binding of the probe substrate to plasma proteins, resulting in large shifts in apparent Km (e.g. diclofenac Km shifts from 9 μM (– plasma) to ~700 μM (+ plasma). As expected, addition of plasma to incubations decreased the inhibitory potential of CYP inhibitors that bind extensively (> 95%) to plasma proteins (ketoconazole, ritonavir, terbinafine). However, there was little change in the inhibitory potential of drugs not extensively bound to plasma proteins (< 90%), such as erythromycin and quinidine. Results of in vitro CYP inhibition studies performed in HLM (plasma versus buffer) were compared with clinical outcomes for known CYP inhibitors ketoconazole (CYP3A4), montelukast (CYP2C8), and terbinafine (CYP2D6). For ketoconazole, the correct clinical outcome was predicted by HLM in both buffer and plasma. In the case of montelukast, HLM in buffer over-predicted a potential interaction, whereas HLM in plasma correctly predicted the lack of clinical interaction with CYP2C8 substrates. In the case of terbinafine, HLM in plasma under-predicted the clinical interaction, whereas HLM in buffer correctly predicted a clinical interaction with CYP2D6 substrates. In conclusion, CYP inhibition studies with HLM in plasma can improve IVIVE in some cases but does not uniformly improve IVIVE.
Reference:
- Lu C, Miwa GT, Prakash SR, Gan LS, Balani SK. (2007) A novel model for the prediction of drug-drug interactions in humans based on in vitro cytochrome P450 phenotypic data. Drug Metab Dispos 35:79–85.
193. Inhibition of cytochrome P450 (CYP) enzymes, CYP1A2 and CYP2C8, by oligonucleotides in human liver microsomes (HLM): A system-dependent outcome
David B. Buckley, Faraz Kazmi, Phyllis Yerino, Brian W. Ogilvie, and Andrew Parkinson
XenoTech, LLC, Lenexa, KS, USA, 66219
Oligonucleotide-based molecules represent an evolving class of new drug candidates with diverse structures, functions and therapeutic applications. Oligonucleotides are large polyanionic molecules not metabolized by CYP enzymes; accordingly, oligonucleotides would not be expected to inhibit CYP enzymes. Currently, there are no reported cases of clinical interactions of oligonucleotide-based drugs with co-medications cleared primarily by CYP-dependent metabolism. Unexpectedly, in HLM, we observed inhibition of CYP1A2 and CYP2C8 by oligonucleotides. In the present study, four oligonucleotides, two with phosphodiester backbones (oligos #1 and #2) and two with phosphorothioate backbones (oligos #3 and #4) were evaluated (10 μM) as direct-acting, time-dependent (TDI) and metabolism-dependent inhibitors of seven human CYP enzymes, namely CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4. Little or no CYP inhibition was observed with oligonucleotides containing a phosphodiester backbone (oligos #1 and #2). However, in HLM, both phosphorothioate oligonucleotides (oligos #3 and #4) were relatively potent direct inhibitors of CYP1A2 (IC50 4.3 and 1.0 μM, respectively) and CYP2C8 (IC50 12 and 1.2 μM, respectively) and were mixed-type inhibitors of CYP1A2 (Ki values 3.7 and 0.8 μM, respectively). In the case of oligo #4, there was some evidence of TDI (not NADPH-dependent) of CYP2C8 (IC50 shift from 1.0 to 0.5 μM), which was reversible after re-isolation of microsomes by ultracentrifugation. Both oligonucleotides were filtered (MW < 3000); the low molecular weight filtrate caused no CYP inhibition, suggesting that CYP inhibition is attributable to the parent molecule. In contrast to HLM, neither phosphorothioate-containing oligonucleotide (oligos #3 and #4) inhibited CYP1A2 in human hepatocytes (suspensions), although they did cause weak inhibition of CYP2C8 (IC50 74 and 37 μM, respectively). The weak inhibition of CYP2C8 by oligos #3 and #4 in hepatocytes was further reduced in the presence of human plasma. Although it remains to be determined whether oligonucleotide-based drugs can cause clinically significant inhibition of CYP1A2 and/or CYP2C8, the large difference in inhibitory potency between HLM and human hepatocytes with oligonucleotides provides another example of system-dependent CYP inhibition.
194. Does U73122 Inactivate PLCβ3 In MDCK-II Cells while activating it in a cell free system?
Chester L. Costales1, Ryan R. Klein2, and Dhiren R. Thakker1
1Eshelman School of Pharmacy-Division of Molecular Pharmaceutics, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA, 27599
2Drug Metabolism and Pharmacokinetics, GlaxoSmithKline Inc., Research Triangle Park, NC, USA, 27709
Phospholipase C (PLC) is an intracellular signaling molecule that has been implicated in the regulation of tight junctions, which can limit the transport of hydrophilic molecules via the paracellular pathway. 1-(6-((17b-3-methoxyestr-1,3,5(10)-trien-17-yl)amino)hexyl)-1H-pyrrole-2,5-dione (U73122) is the prototypical PLC inhibitor commonly employed to demonstrate involvement of PLC in cellular processes. We have recently shown that U73122 activates recombinant PLCβ3 in a cell free system. However, at similar concentrations, formation of PLC dependent products in both MDCK and Caco-2 cells is inhibited by U73122 while tight junction permeability is increased. The purpose of this study is to elucidate the direct molecular interactions between PLCβ3 and U73122 in MDCK-II cells. Irreversible binding of U73122 and recombinant PLCβ3 was assessed by ultrafiltration (Ultracel YM-30 membrane – 30,000 NMWL, 4°C, 14,000 rpm, 24 min). MDCK-II cells were transfected with 6XHistidine-tagged PLCβ3 (his-PLCβ3) and were treated with U73122 or vehicle control. Batch protein purification of his-PLCβ3 was performed from cell lysates by nickel affinity gel. Activity of purified enzyme was determined by measuring [3H]inositol phosphate formation after incubation with [3H]phosphatidylinositol-4,5-bisphosphate in a dodecylmaltoside micellar system. Possible alkylation of his-PLCβ3 in MDCK-II was assessed by liquid chromatography coupled to high resolution mass spectrometry (LCMS-TOF). Treatment of recombinant PLCβ3 with U73122 resulted in 2.5-fold activation compared to controls, and activation was irreversible following ultrafiltration. his-PLCβ3 was expressed in MDCK-II cells as evidenced by Western blot analysis. Treatment of MDCK-II cells containing his-PLCβ3 with U73122, followed by isolation of the enzyme and determination of its activity will indicate if U73122 inhibits intracellular PLC activity by direct modification of intracellular PLC or through other indirect mechanisms. This work was designed to elucidate the interaction between U73122 and PLCβ3. No change in the activity of U73122-treated PLCβ3 upon ultrafiltration indicated U73122 activates the enzyme in a cell free system by covalent modification. Effect of U73122 on the activity of PLCβ3 expressed in MDCK-II cells would clarify whether U73122 inhibits cellular PLC by direct covalent modification or another indirect mechanism.
195. Metabolite inhibitory complex formation by alkylamines may involve formation of a methyleneamine, not a nitroso, metabolite
Joanna E. Barbara, David B. Buckley, Faraz Kazmi, Phyllis Yerino, Brian W. Ogilvie, Mark J. Horrigan, Paul C. Toren, and Andrew Parkinson
XenoTech, LLC, Lenexa, KS, USA, 66219
Secondary and tertiary amines containing at least one N-methyl group, such as troleandomycin, erythromycin and diltiazem, are known metabolism-dependent inhibitors of cytochrome P450 (CYP) enzymes, particularly CYP3A4, through formation of a metabolite inhibitory complex (MIC) involving the ferrous heme iron. It is generally accepted that the inhibitory metabolite that binds tightly, but noncovalently, to the ferrous heme iron is a nitroso metabolite formed from a (mono)- or (di)-methylamine by the following steps:
R-N(CH3)2 → R-NHCH3 → R-NH2 → R-NHOH → R-N=O
In an attempt to identify the inhibitory metabolite, diltiazem was incubated with NADPH-fortified HLM under conditions that lead to MIC formation, after which the microsomes were isolated by ultracentrifugation to remove diltiazem and its soluble metabolites. Samples were analyzed by UPLC and accurate mass spectrometry before and after treatment of the isolated microsomes with potassium ferricyanide to dissociate the MIC. The microsomes isolated by ultracentrifugation contained detectable levels of diltiazem and several of its metabolites (formed by N-demethylation, O-demethylation, ester hydrolysis or a combination thereof), but no hydroxylamine or nitroso metabolites were detected. Only one metabolite increased appreciably following potassium ferricyanide treatment, and was consistent with a methyleneamine metabolite. Accordingly, we propose that in the case of diltiazem MIC formation involves the following steps:
R-N(CH3)2 → R-NHCH3 → R-N=CH2
This pathway to MIC formation requires at least one N-methyl group, which could account for the observation that MI complexes can form with diltiazem and nordiltiazem but not with di-nordiltiazem (Zhao et al., 2007).
Reference:
- Zhao, P., Lee C.A., Kunze, K.L. (2007). Drug Metab. Disp. 35(5):704–712.
196. Impact of model selection on drug interaction predictions: Competitive inhibition versus partial inhibition
Rucha S. Sane, Eleanore Seibert, and Mitchell E. Taub
Drug Metabolism & Pharmacokinetics, Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT, USA, 06877-0368
Competitive inhibitors of CYP3A4 can cause complete inhibition of enzyme activity at an infinitely high inhibitor concentration. The equation: AUCi/AUC = 1 + I/Ki, is widely used to predict the potential for clinical drug-drug interactions (DDI) in such cases. However, if CYP3A4 is only partially inhibited, some of its catalytic activity is retained, even at infinitely high concentration of inhibitor. In studies conducted with BI-C, selection of the most appropriate inhibition kinetic model and the impact of this selection on the prediction of DDI potential are described. Inhibition of CYP3A4 by BI-C was assessed in human liver microsomes using midazolam as a probe substrate. Ki values were generated via nonlinear regression of the data using a competitive inhibition and a partial inhibition model. Based on visual examination of the goodness of fit, the AIC values, and the run’s test, the data fit better to the partial inhibition model compared to the competitive inhibition model. The Ki values for competitive inhibition and partial inhibition were 7 and 0.2 μM, respectively and the ‘α’ value for partial inhibition of CYP3A4 by BI-C was ~3. The change in AUC of a concomitantly administered CYP3A4 substrate due to partial inhibition by BI-C was predicted using the following equation: AUCi/AUC = (1+I/Ki)/(1+I/α*Ki). Based on the two described methods, at α > 1, for any value of I/Ki, competitive inhibition predicts a greater change in AUC of a CYP3A4 substrate compared to partial inhibition. The maximal predicted change in substrate AUC based on partial inhibition of CYP3A4 did not exceed ‘α’, i.e. 3, for BI-C. On the other hand, the predicted change in the substrate AUC based on competitive inhibition increases with increasing concentrations of BI-C. Accurate prediction of CYP450-based potential DDIs is highly dependent upon the inhibition model used, and as such critical evaluation of the data and application of the appropriate inhibition model is necessary.
197. Evaluation of fipexide for time-dependent inhibition (TDI) of 5 major human CYP450 enzymes
Carlo Sensenhauser, Jose Silva, and Heng Keang Lim
Dmpk, Johnson & Johnson Pharmaceutical Research and Development, Raritan, NJ, USA, 08869
Fipexide is a nootropic drug that was withdrawn from the market as a result of idiosyncratic hepatotoxicity observed in man. Previous metabolism work identified acyl glucuronide and acyl CoA thioester conjugates of the 4-chlorophenoxyacetic acid metabolite resulting from amide hydrolysis of fipexide, and glutathione conjugates derived from a quinone reactive intermediate following demethylenation and further oxidation of fipexide, as potential causes for hepatotoxicity. In this study, we evaluated fipexide for its potential to act as a time-dependent inhibitor (TDI) of the 5 major human CYP450 isoforms since it has the 3,4-methylenediophenyl structural alert for CYP450 TDI. Time-dependent inhibition of a particular CYP450 isoform was determined by incubating fipexide in pooled human liver microsomes, with or without an NADPH regenerating system followed by a 20-fold dilution in the aliquot transfer to a secondary incubation mixture containing NADPH regenerating system and an excess of a known marker substrate to assess residual CYP-specific activity. The following marker substrates were used: phenacetin for CYP1A2, tolbutamide for CYP2C9, S-mephenytoin for CYP2C19, dextromethorphan for CYP2D6 and testosterone and midazolam for CYP3A4. In a single time point experiment (45 minutes), residual activity was plotted as a function of inhibitor concentration for the determination of the apparent partition ratio (APR, i.e. the number of inactivator molecules turned over by each enzyme molecule prior to being inactivated). Fipexide was found to inactivate to varying degrees, all the 5 major CYP isoforms screened. CYP3A4 and CYP2D6 were the isoforms inactivated more significantly with over 75% loss of activity after 45 minutes in the presence of 30 μM fipexide. APR values were 130 and 1000, for the inactivation of CYP3A4 and CYP2D6, respectively, compared to values of 5 and 150 for the known inactivators, mifepristone (CYP3A4) and paroxetine (CYP2D6), respectively. CYP1A2, CYP2C9 and CYP2C19 were also inactivated by fipexide, although to a lesser extent. Kinetic constants determinations yielded KI values of 5 and 3 μM and inactivation rates kinact of 0.018 and 0.040 min-1, for CYP3A4 and CYP2D6, respectively, which gave an inactivation clearance of 3.6 and 13.3 min-1 mM-1. Incubations of fipexide with recombinant enzymes of each of the five major CYP isozymes resulted in formation of two aryl thioether conjugates of a quinone reactive intermediate. CYP2C19 seems to play a major role in the formation of these reactive species and further work will focus on determination of inactivation kinetic constants of remaining CYPs and the reactive species binding to the microsomal protein following time-dependent inhibition.
198. Perpetrator-perpetrator-victim interactions: The super-proportional effect of two perpetrators on the disposition of a victim drug
Brian W. Ogilvie, David B. Buckley, Brandy L. Paris, and Andrew Parkinson
XenoTech, LLC, Lenexa, KS, USA, 66219
This presentation draws attention to the impact of two independent inhibitory perpetrators (such as two drugs that inhibit different routes of elimination of a victim drug, or a drug and a genetic polymorphism that affect different routes of elimination of a victim drug), which are far greater than the sum or even the product of the individual perpetrator effects (based on observed fold increases in exposure to the victim drug). The focus of research on parallel pathways of metabolism has typically been on the over-prediction that can occur when a parallel metabolic pathway is not inhibited, or the effects of two perpetrators (for example, a drug and its metabolite, such as itraconazole and hydroxyitraconazole) acting on the same pathway of elimination of the victim drug. If combined inhibitory effects on different elimination pathways are ignored, a gross under-prediction of the effects of two perpetrators on the disposition of a victim drug can result, as illustrated for voriconazole (victim) and ritonavir (perpetrator 1) with and without the CYP2C19 poor metabolizer phenotype (perpetrator 2) (from Mikus et al., 2006):
This type of scenario has been termed “maximum exposure”, which occurs in poor metabolizers when there is concurrent inhibition of one or more nonpolymorphic pathways (Collins et al., 2006). This dramatic effect of two perpetrators is nevertheless predictable from the sum of the fractional metabolism (fm) values, as outlined by Collins and highlighted here. Several examples of this type of perpetrator-perpetrator-victim interaction will be highlighted.
References:
- Collins C, Levy R, Ragueneau-Majlessi I and Hachad H (2006) Prediction of maximum exposure in poor metabolizers following inhibition of nonpolymorphic pathways. Current Drug Metabolism 7:295–299.
- Mikus G, Schowel V, Drzewinska M, Rengelshausen J, Ding R, Riedel KD, Burhenne J, Weiss J, Thomsen T and Haefeli WE (2006) Potent cytochrome P450 2C19 genotype-related interaction between voriconazole and the cytochrome P450 3A4 inhibitor ritonavir. Clinical Pharmacology and Therapeutics 80:126–135.
199. Investigating the reactive metabolite(s) responsible for the mechanism-based inactivation of CYP3A4 by the anti-human immunodeficiency virus protease inhibitor, ritonavir
Brooke M. VandenBrink, Brian J. Kirby, Jashvant D. Unadkat, and Kent L. Kunze
Medicinal Chemistry, University of Washington, Seattle, WA, USA, 98195
Administration of the anti-HIV protease inhibitor ritonavir (RTV) causes inhibitory drug-drug interactions with multiple CYP3A4 substrates in vivo. CYP3A4 inhibition by RTV was previously proposed to be due to mechanism-based enzyme inactivation via the formation of a metabolic-intermediate (MI) complex to the heme iron.3 We investigated the metabolism of RTV in 3A4 supersomes™, 3A4 purified enzyme, and HLMs in an effort to identify the reactive intermediate(s) and/or metabolite(s) that are responsible for enzyme inactivation. The kinetic parameters for inactivation (KI= 0.4 uM, kinact= 0.6 min−1) we observe are similar to literature reports. This inactivation is not attenuated by co-incubation with the trapping agent, glutathione. We have been unable to obtain spectrophotometric confirmation of the formation of an MI complex; however, we observe significant covalent binding of radioactive 3H-RTV to CYP3A4 protein. This suggests that an electrophilic reactive intermediate is responsible for mechanism-based inactivation of CYP3A4. An extensive search for glutathione adducts of the reactive metabolite(s) resulted in no conclusive hits using a variety of LC/MS/MS techniques. This finding, along with a low partition ratio for RTV, leads to the conclusion that negligible concentrations of the putative reactive intermediate(s) are released into solution. We hypothesize that oxidative bioactivation of one of the two thioazolyl rings (located on opposite ends of RTV) leads to covalent binding to the enzyme. To this end, we have obtained an analog of RTV, desthiazolylmethyloxycarbonyl ritonavir, which lacks one of the thioazolyl rings. Together, the results of ongoing studies with this analog, and whole protein mass spectrometry of the RTV-CYP3A4 adduct will aid in elucidating the mechanism of CYP3A4 inactivation. Knowledge of the mechanistic features of the inactivation will provide additional insights into CYP3A4 biochemistry and its vulnerability to the effects of reactive intermediates, as well as ultimately improving our understanding of CYP3A4 drug-drug interactions.
Reference:
- Ernest, C. S., II; Hall, S. D.; Jones, D. R., Mechanism-Based Inactivation of CYP3A by HIV Protease Inhibitors. J Pharmacol Exp Ther 2005, 312, (2), 583–591.
200. Glucuronidation of dihydrotestosterone and trans-androsterone by recombinant UGT1A4: evidence for multiple UGT1A4 aglycone binding Sites
Jin Zhou1, Timothy S. Tracy2, and Rory P. Remmel1
1Department of Medicinal Chemistry, University of Minnesota, Minneapolis, MN, USA, 55455
2Department of Experimental and Clinical Pharmacology, University of Minnesota, Minneapolis, MN, USA, 55455
Approximately 20% of the top 200 prescribed drugs that are cleared to some degree through glucuronidation are metabolized by UGT1A4-catalyzed glucuronidation. Though atypical kinetic profiles (non-hyperbolic, non-Michaelis-Menten) of UGT1A4-catalyzed glucuronidation have been occasionally reported, systematic kinetic studies to explore the existence of multiple aglycone binding sites on UGT1A4 have not been conducted. To this end, the positional isomers dihydrotestosterone (DHT) and trans-androsterone (t-AND) were used as probe substrates and the kinetics of UGT1A4 glucuronidation were evaluated both alone and in the presence of the high affinity UGT1A4 substrate tamoxifen (TAM). Additionally, the effects of DHT and t-AND on TAM glucuronidation were also evaluated. DHT glucuronidation exhibited a hyperbolic kinetic profile whereas t-AND glucuronidation exhibited an atypical kinetic profile indicative of substrate inhibition kinetics. TAM also displayed substrate inhibition kinetics, suggesting that both t-AND and TAM may exhibit dual binding orientations within the UGT1A4 active site. When co-incubated, TAM activated DHT glucuronidation with mixed effects on t-AND glucuronidation (concentration-dependent activation or inhibition). Conversely, DHT and t-AND inhibited TAM glucuronidation. To determine if this heteroactivation by TAM was substrate- dependent, the effects of TAM on lamotrigine (LTG) glucuronidation by UGT1A4 (hyperbolic kinetics) were investigated. TAM competitively inhibited LTG glucuronidation, in contrast to the activation observed with DHT glucuronidation and the mixed effects observed on t-AND glucuronidation. Our results suggest that multiple aglycone binding sites exist within UGT1A4, which can result in atypical kinetic profiles (both homotropic and heterotropic) in a substrate-dependent fashion.(Supported by Bristol-Myers Squibb and NIH: # GM063215).
201. Metabolic switching of BILR 355 in the presence of ritonavir II: elucidation of metabolic pathway for the formation of BILR 516
Yongmei Li1, George Lai2, Jun Xu1, Andrea Applegreen1, and Donald Tweedie1
1Drug Metabolism & Pharmacokinetics, Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT, USA, 06877-0368
2Drug Disposition, Eisai Research Institute, Andover, MA, USA, 01810-2441
BILR 355 BS is a dipyridodiazepinone non-nucleoside reverse transcriptase inhibitor (NNRTI) of the human immunodeficiency virus HIV-1. BILR 355 had a very short half life (2 to 5 h) and low exposure after oral administration to humans. Ritonavir is a potent CYP3A4 inhibitor and was used as a boosting agent to increase the exposure of BILR 355. However, while it was anticipated that the levels of metabolites would decrease upon addition of ritonavir, a metabolite, BILR 516, which had previously been a minor component, was now a disproportionate human metabolite with levels exceeding the parent at steady-state. The metabolic pathway for the formation of BILR 516 from BILR 355 is described. BILR 516 can not be generated from BILR 355 directly. It was shown that BILR 355 was reduced to an intermediate, BILR 402, most probably by intestinal flora, which was further oxidized to BILR 516 by aldehyde oxidase. The involvement of aldehyde oxidase was confirmed based on selective chemical inhibitors, 18O incorporation study using H218O, and incubation of BILR 402 with recombinant human aldehyde oxidase. Both BILR 355 and BILR 402 were extensively metabolized by CYP3A4. With co-administration of ritonavir, the CYP3A4 metabolism of BILR 355 and BILR 402 was significantly inhibited and the alternative metabolic pathway, i.e. formation of BILR 516, became predominant. In addition, the implications of the involvement of aldehyde oxidase are discussed. Aldehyde oxidase activity is generally high in humans, low in rats, and deficient in dogs. This would explain the low exposure of BILR 516 in Toxicology species administered BILR 355/ritonavir. As a result of these species differences, a metabolite which is formed through aldehyde oxidase may have greater potential to become a disproportionate human metabolite.
202. Human skin metabolism of topical retinoid tazarotene
Hansen L. Wong, Leandro Santos, and Xue Ge
Preclinical Research and Development, Stiefel Laboratories, Inc., Palo Alto, CA, USA, 94304
Topical drugs and environmental toxicants can penetrate or deposit in the skin. However, there are few literature reports for xenobiotic metabolism in human skin. In the present study, human skin metabolism was investigated for tazarotene (ethyl 6-[2-(4,4-dimethyl-3,4-dihydro-2H-1-benzothiopyran-6-yl)ethynyl]pyridine-3-carboxylate), a well-known topical retinoid marketed as Tazorac and Avage for the treatment of psoriasis, acne, and sun-damaged skin. Madhu and co-workers reported that tazarotene was quickly converted to an active metabolite, tazarotenic acid, by esterases in human liver microsomes and blood (1). Tazarotenic acid was further oxidized by liver cytochrome P450s and flavin monooxygenases to the corresponding sulfoxide and sulfone (2). Therefore tazarotene is a good probe compound for the study of skin metabolism because several different types of metabolizing enzymes are involved in its hepatic metabolism. There are no previous reports of tazarotene metabolism in the skin, the target tissue and site of absorption. In our study, tazarotene metabolites were identified in human skin microsomes and S9 fractions using liquid chromatography with tandem mass spectrometry. Metabolite profile of tazarotene in skin and that in the liver were compared. We showed that tazarotene was hydrolyzed to tazarotenic acid by skin microsomes and S9 fractions; similar to that observed in liver microsomes. Esterase-catalyzed hydrolysis to tazarotenic acid was confirmed as it was inhibited by paraoxon, an esterase inhibitor. Acyl glucuronidation of tazarotenic acid was observed in both human liver and skin microsomes. The rates of ester hydrolysis and glucuronidation were substantially lower in skin relative to liver. Studies are ongoing to verify metabolism of tazarotenic acid to the tazarotenic acid sulfoxide and sulfone.
References:
- Madhu, C.; Duff, S; Baumgarten, V.; Rix, P.; Small, D.; Tang-Liu, D. Metabolic deesterification of tazarotene in human blood and rat and human liver microsomes. J. Pharm. Sci., 1997, 86, 972–974.
- Attar, M.; Dong, D.; Ling, K. J.; Tang-Liu, D. Cytochrome P450 2C8 and flavin-containing monooxygenases are involved in the metabolism of tazarotenic acid in humans. Drug. Metab. Disp., 2003, 31, 476–481.
203. Functional consequences of the unique structure of human cytosolic sulfotransferase (SULT) 2B1b
Emily D. Salman, and Charles N. Falany
Pharmacology and Toxicology, University of Alabama at Birmingham, Birmingham, AL, USA, 35294
Hydroxysteroid sulfotransferase (SULT) 2B1b is a cytosolic SULT that catalyzes the transfer of the sulfonate moiety from 3’-phosphoadenosine 5’-phosphosulfate (PAPS) to an acceptor substrate. SULT2B1b is unique compared to other SULT isoforms since it has a 16 aa N-terminal extension and a 56 aa C-terminal extension not present in other SULT isoforms. Further, SULT2B1b is catalytically unstable as it readily loses sulfation activity. It is also the only SULT isoform that translocates to the nucleus. Structural analysis has revealed clues as to the functional significance of the unique N- and C-terminal extensions of SULT2B1b. We hypothesize that the distinctive structure of SULT2B1b contributes to its catalytic instability, aids in nuclear translocation, and affect subunit interactions. Gel filtration chromatography was used to determine the molecular weight of purified recombinant human 6His-SULT2B1b. The calculated monomeric MW for SULT2B1b is 42 kD, however, the enzyme was catalytically active at 78 kD, demonstrating that the enzyme is active as dimer and that the N- and C-terminal extensions do not interfere with dimerization. Transfection of mammalian cells with SULT2B1b with and without the 56 aa C-terminal extension have shown that this peptide is required for sulfation activity and nuclear translocation. Radioimmunoprecipitation assays with active or inactive recombinant 6His-SULT2B1b were used to evaluate the ability of the enzyme to bind substrate and PAPS. While PAPS binding is unaffected by loss of activity, substrate binding is diminished when the enzyme is no longer active. This finding shows that loss of activity is associated with structural changes that prevent access of substrates into the active site binding pocket. Molecular modeling was used to build an energy minimized structure of SULT2B1b with both the N- and C-terminal extensions, as these structures are absent in resolved crystal structures. The models suggest that the N-terminal extension of SULT2B1b is directly adjacent to the substrate-binding pocket. Taken together, these results show that the unique structural properties of SULT2B1b contribute enzyme stability, nuclear localization and subunit interactions.
204. Effect of benzo(a)pyrene and 3-methylcholanthrene on the target genes of hormonal carcinogenesis
Lyudmila F. Gulyaeva, Pystylnyak Vladimir, and Chanyshev Michail
Molecular carcinogenesis, Institute of MolBiol & Bioph SB RAMS, Novosibirsk, Russia, 630117
In most countries the incidence rates of female cancers have trend to increase every year. Environmental factors especially xenoestrogens may have an important role in increasing the risk of these hormone-related cancers. These chemicals have estrogenic effect that may be one of the causes of cancer development. Polycyclic aromatic hydrocarbons (PAHs) are candidates possessing estrogen mimicry effect in target tissues. To confirm this statement we have studied the gene expression of CYP1A1, CYP1B1, CYP19, ERα, cyclin D1 in liver, ovary and uterus of female rats treated with benzo(a)pyrene (BP) and 3-methylcholanthrene (MC) using RT-real time PCR. The obtained results are presented in the table.
Gene expression in various organs of female rats treated with MC and BP (fold of induction, *- significant meaning)
Both studied PAHs are well known to be powerful inducers of CYP1A1 and 1B1 in rat liver as also shown by us. Our results demonstrate very prominent induction of CYP1A1 in ovary and uterus and to smaller extent of CYP1B1. Treatment of rats with MC and BP doesn’t have any remarkable effect on aromatase expression in liver and ovary; however, MC significantly suppresses its expression in uterus. BP, but not MC, has enhanced gene expression of ERα in rat liver and stronger in ovary. In addition, BP decreases gene expression of cyclin D1 in liver and ovary whereas MC decreases cyclin D1 gene expression in liver only. Thus, studied PAHs affect gene expression of both estrogen-metabolizing enzymes (CYP19, CYP1A1, and CYP1B) and cell cycle regulating genes (cyclin D, ERα) that may be a very important initial step in hormonal carcinogenesis.
205. Role of microRNAs in drug metabolism and disposition
Ai-Ming Yu
Department of Pharmaceutical Sciences, University at Buffalo, SUNY, Buffalo, NY, USA, 14260-1200
MicroRNAs (miRNAs) represent a large group of newly-recognized, short, noncoding RNAs that control the expression of target genes. Currently, more than 600 miRNAs have been identified in humans, which are thought to regulate at least 30% of coding genes in human genome. MicroRNA target genes investigated are involved in almost all biological processes, including the gene-xenobiotic interactions. However, in contrast to active investigation of nuclear receptor (NR)-mediated transcriptional gene regulation of drug-metabolizing enzymes (DMEs) and drug transporters (DTs) is the lack of understanding miRNA-governed posttranscriptional gene regulation in drug metabolism and disposition. The latter is critical since advanced understanding of DME and DT gene regulatory pathways shall provide increased understanding and better prediction of potential drug-drug interactions in clinical pharmacotherapy and in early stage of drug discovery and development. Recent studies have revealed that the expression and function of certain miRNAs are largely altered by xenobiotic drugs, and some DMEs, DTs and NRs may be directly or indirectly regulated by miRNAs. Herein we present our findings that a number of miRNAs regulate human breast cancer resistance protein (BCRP/ABCG2) and cytochrome P450 3A4 (CYP3A4), and consequently, affect the capacity of drug metabolism and disposition, and the sensitivity of cells to xenobiotic agents.
206. Comparative expression of genes encoding drug metabolizing enzymes, drug transporters, and transcription factors in the Caco-2 and LS513 human colorectal cancer cell lines
Emile G. Plise1, Jinfeng Liu2, and Laurent Salphati1
1Drug Metabolism and Pharmacokinetics, Genentech, Inc, South San Francisco, CA, USA, 94080
2Bioinformatics, Genentech, Inc, South San Francisco, CA, USA, 94080
Caco-2 cells are well characterized and widely used to examine passive permeability, efflux, uptake, and metabolism of xenobiotics. Recently it was shown that LS513 cells could be grown on permeable membranes for transport studies. They were also found to express functional levels of MDR1, BCRP, MRP2, and Cytochrome P450 (CYP) 3A4/5 that could be induced with PXR/CAR/VDR agonists1, 2. To further examine LS513 cells as a potential in vitro model system, the mRNA expression profiles of various ABC and SLC transporters, CYPs, and transcription factors were compared to those in Caco-2 cells. The cell lines were seeded in 6 well plates (1.0 • 106 cells/well), grown to confluence, and harvested on day 4 for mRNA analysis on an Affymetrix HGU133P GeneChip (n=3). The expression level of 80 genes was compared by Student’s t-tests. Significantly different expression for 21 genes (39 probesets) was observed in the two cell lines using a threshold of p-value < 0.05 and false discovery rate < 0.1. Caco-2 cells had markedly higher expression of MRP2 (63-fold), MRP4 (7-135 fold), and MRP6 (3-5 fold). They also showed considerably higher levels of the OATPs SLCO2B1 (12-19 fold) and SLCO4C1 (31-fold), whereas expression of SLCO1B3 was ~4-fold lower than in the LS513 cells. CAR (NR1I3) expression was approximately 2-fold higher in the Caco-2, while VDR expression was 5-fold higher in LS513 cells. Expressions of ABCB1 (MDR1), BCRP (ABCG2), MRP3 (ABCC3), and MRP5 (ABCC5) were 2- to 3- fold higher in the LS513 cells. LS513 displayed also notably higher expression of CYP2B6 (2-19 fold), CYP3A4 (~3-fold), CYP3A5 (2-10 fold), CYP3A7 (14-fold). The two cell lines did not have significantly different levels of NR1I2 (PXR), AhR, and many of the other OCTs, OATPs, and CYPs examined. The expression profiles suggested LS513 cells may be a suitable alternative model to Caco-2 for studying the interplay between transporters and drug metabolizing enzymes as well as VDR-dependent induction.
References:
- Salphati, L., Plise, E. G, Li, G. (2009). Expression and activity of the efflux transporters ABCB1, ABCC2 and ABCG2 in the human colorectal carcinoma cell line LS513. Eur. J. Pharm. Sci. 37:463–468.
- Plise E. G., Boggs J., Wong S., Salphati L. (2008). Drug Met. Rev. 40:177.
207. Nicotine induces a single CpG methylation within the StAR promoter region leading to reduced gene expression and cortisol/dehydroepiandrosterone production
Wang Ting-ting1, Man Chen1, Huai-yan Cheng2, Hui Wang1, and Ying-hong Feng2
1Department of Pharmacology, Basic Medical School of Wuhan University, Wuhan, China
2Department of Pharmacology, Uniformed Services University of the Health Sciences, Bethesda, Maryland, USA, Maryland, MD, USA, 20743
Steroidogenic acute regulatory protein (StAR) is a rate-limiting enzyme for the production of glucocorticoid, essential to fetal development. We have reported that the StAR expression in fetal adrenal is inhibited in a rat model of nicotine-induced intrauterine growth retardation (IUGR). Here using primary human fetal adrenal cells and a human fetal adrenal cell line NCI-H295A, we recapitulate the nicotine-induced inhibition of StAR expression and uncover a single site methylation within the StAR promoter region responsible for the reduced expression. Nicotine inhibited StAR expression in both the primary cells and NCI-295A cells in a dose and time dependent manner with comparison to control treatments. Analysis of CpG methylation within the StAR promoter region uncovered no alterations in global methylation status, but a single CpG methylation within the binding motif (CGCCTGA) of the transcription factor Pax6. Consistently, the frequency of this point methylation also increased in a time dependent manner. Interestingly, this single CpG methylation remained unchanged 15 days after withdrawal of nicotine treatment and the expression of StAR remained suppressed with comparison to the control treatment. Knocking down the expression of Pax6 reduces the StAR expression in the absence of nicotine treatment, suggesting the single CpG methylation may prevent the Pax6 binding to the StAR promoter. These data identify for the first time that a single CpG methylation within the StAR promoter region reduces the gene expression and suggest an inheritable epigenetic mechanism that may contribute to nicotine-induced fetal toxicity and IUGR-related late onset of diseases or disorders such as metabolic syndrome. Supported by Chinese Scholarship Council (20073020); Chinese Nature Science Foundation (30800931, 30672566 and 30830112); NIH R01 Grant (HL065492). *Corresponding authors: [email protected] or [email protected].
208. Dysplasia of hypothalamic-pituitary-adrenal axis in caffeine-induced rat intrauterine growth retardation: Based on an up-regulated expression of glucocorticoid receptor in hippocampus
Xu Dan, Wen-wen HE, Gai Liang, Ben-jian Zhang, Jie Ping, Fang Liu, and Hui Wang
Department of Pharmacology, Basic Medical School of Wuhan University, Wuhan 430071, China, Wuhan, China
Adverse intrauterine environment (such as ischemia, stress) would cause fetal hypothalamus-pituitary-adrenal (HPA) axis dysplasia and intrauterine growth retardation (IUGR). It is known that the occurrence of fetal hippocampus glucocorticoid receptors (GR) is closely related to the development of HPA axis. This study was designed to investigate the alteration of HPA axis in caffeine-induced IUGR rats and its possible neuroendocrine mechanism. Caffeine (0, 20, 60, 180 mg/kg d) was administered orally to pregnant rats from gestational day (GD) 12 to GD 20. Maternal rats were sacrificed on GD 20 and fetal rats were excised. Fetal developmental parameters were recorded. Adrenocorticotrophin (ACTH) and corticosterone levels in fetal peripheral blood were determined by radioimmunoassay and ELISA method. The expressions of hippocampus GR, hypothalamus corticotropin releasing hormone (CRH), adrenal steroidogenic acute regulatory protein (StAR), cytochrome P450 cholesterol side chain cleavage (P450scc) and placental 11b-hydroxysteroid dehydrogenase type 2 (11b-HSD-2) were determined using real-time quantitative RT-PCR and Western blot. And the expressions of GR and DNA methyltransferases (DNMTs) were also determined in hippocampal neurons culture system in vitro, after caffeine treatment. The results showed that prenatal chronic caffeine exposure causes IUGR in rats (P<0.01); expressions of GR increased in fetal hippocampus (P<0.05), while expressions of CRH, StAR and P450scc decreased in fetal adrenals (P<0.01 or P<0.05). Furthermore, expressions of placental 11b-HSD-2 decreased (P<0.05) but levels of ACTH and corticosterone increased in fetal peripheral blood (P<0.01). Further study in vitro found that caffeine can cause a direct increase in hippocampus GR expression (P<0.01), while Dnmt3a and Dnmt3b expressions were decreased (P<0.05). These results suggest that caffeine-induced IUGR is associated with the disturbance of glucocorticoid homeostasis in fetal rats. A possible underlying mechanism is that long term caffeine administration leads to fetal overexposure to maternal glucocorticoid by the impaired placental barrier to it, which eventually leads to the fetal HPA axis dysfunction and IUGR. And the increased fetal hippocampus GR expression may involved in the caffeine-induced HPA axis dysplasia, which might be due to the altered epigenetic modification in hippocampus GR by caffeine. 1Supported by the grants from the China Nature Science fund (30830112£»30672566£»30800931). 2Correspondence author: [email protected].
209. Effects of in utero and/or postnatal exposure to mixtures of blood contaminants on the adulthood glucocorticoid stress response in male rat
Gong-hua Xiao, Cathy Cummings-Lorbetski, and Daniel Desaulniers
Health Canada, Ottawa, ON, Canada, K1A 0K9
Rodent studies suggest that perinatal events could reprogram expression of glucocorticoid receptor for an entire lifespan, creating abnormal hormone levels and predisposition to diseases. As part of a larger study, we tested a hypothesis of a link between perinatal exposure to environmental contaminants and abnormal adulthood corticosterone (CS) stress response (CSR) in rats. The experiment included 9 treatment groups. From gestation-day 0 (day of vaginal plug) and until postnatal day (PND) 20, dams were fed daily cookies laced with corn oil (control) or a chemical mixture (M: polychlorinated biphenyls, organochlorine pesticides, and methylmercury) at 0.5 or 1.0 mg/kg/day (0.5M, and 1M). At birth, some control (C) and 1M litters were crossfostered to create 4 groups of pups with the following in utero/postnatal exposure: C/C, C/1M, 1M/C, 1M/1M. Other dams received cookies with a dose of 1.7 ng/kg/day of a mixture of aryl hydrocarbon receptor (AhR) agonists (AhR: non-ortho PCBs, dibenzodioxins and furans) without or with 0.5M. A CSR was induced in male offspring at PND85 (10 min exposure to a heating lamp to induce vasodilation and 5 min bag-restraint to facilitate the collection of 500 μl of blood from the tail vein). Then, the CS decay was assessed from the trunk blood collected at decapitation 30 min later (T30). The concentrations of CS returned to normal at T30 in the group C, 0.5M, 1M, C/C and 1M/C, but it remained elevated in the group AhR, AhR+0.5M, C/1M, and 1M/1M. Interestingly, 1M had no effect on its own but it prevented the CS drop in adulthood in the 1M/1M group, in which the perinatal exposure is associated with the postnatal stress created by the crossfostering procedure, suggesting that rats can tolerate exposure to 1M with no consequences unless they are subjected to early postnatal stress. Liver is a metabolic target organ for corticosterone. Abundances of hepatic glucocorticoid receptor (GR) mRNA and protein were significantly reduced by the C/M and MM treatments. In contrast, abundances of GR mRNA and protein were significantly elevated by the AhR treatment compared with 1M. Globally, the crossfostering procedure permitted us to demonstrate that both CSR, and the abundances of hepatic GR mRNA and protein are modified by the postnatal period of exposure to 1M and not by in utero exposure, and that postnatal stress might modify the effects of exposure to chemicals. These results are important in our understanding of perinatal influence of contaminant exposure on stress-induced diseases.
210. Investigation of novel gene regulation caused by low dose exposure of benzo[a]pyrene in Jurkat cells using suppressive subtractive hybridization
Minh Phuong Nguyen1, and William K. Chan2
1Department of Labour Physiology, Vietnam Military Medical University, Hadong, Hanoi, Vietnam
2Department of Pharmaceutics & Medicinal Chemistry, Thomas J. Long School of Pharmacy and Health Sciences, University of the Pacific, Stockton, CA, 95211
We are interested in investigating the differential gene expression caused by a carcinogen. Our hypothesis is that by employing suppressive subtractive hybridization PCR (SSH PCR), we would identify novel rare up- and down-regulated transcripts that might be missed in conventional microarray and proteome analyses. We exposed a non-toxic dose of benzo[a]pyrene (BaP) to AhR-deficient Jurkat cells and investigated the cellular response in genome-wide gene expression. After treating the cells with 2.5 μM BaP for 48 hours, we isolated mRNA for SSH PCR analysis. We performed blue/white and DNA dot blot hybridization screenings of approximately 15,000 clones to obtain a final of 41 positive clones (20 up- and 21 down-regulated clones). The validity of this approach to identify the regulated transcripts was confirmed by real-time qPCR using the Jurkat cDNA library with or without BaP treatment as the template. The identity of these regulated transcripts was determined by BLAST search as follows: the up-regulated transcripts are SIGMA1, NEU3, ELOVL5, RPL7, PTP4A2, GTF2H2, RNASEH1, ZNF107, SNRPD1, C1orf151, RPS11, DKC1, HSPA8, PSME1, PPA2, FDPS, BRD3, UTP18, G3BP2, and TUBB2C; the down-regulated transcripts are: C19orf48, RNF146, HIRA, ZAP70, EEF1D, ERGIC3, TCF7, DOCK2, LOC100129034, SART1, BCL2, CAT, SUSD4, C10orf4, XAB2, WDR6, BBS9, SFRS4, RNF130, SNRK and KEAP1. Among these 41 transcripts, 27 of them are related to cell growth, DNA repair, and gene regulation; 3 of them (TCF7, DOCK2, and ZAP70) are related to immune response. Two of our down-regulated transcripts BCL2 and TCF7 have been reported to be also down-regulated by cigarette smoke and the rest are novel BaP-regulated transcripts. Our data suggested that Jurkat cells are sensitive to DNA damage at a non-toxic dose of BaP because Jurkat cells down-regulate genes (SART1, SNRK, HIRA, and BCL2) that promote cell growth and up-regulate DNA repair related genes (GTF2H2 and RNASEH1). In addition, immune response is suppressed even though no apparent change in morphology has been observed when comparing the BaP-treated cells to the untreated cells.
211. Caffeine induces a single CpG demethylation within the StAR promoter region leading to increased gene expression
Jie Ping1, Ting-ting Wang1, Ying-hong Feng2, and Hui Wang1
1Department of Pharmacology, Basic Medical School of Wuhan University, Wuhan 430071, China, Wuhan, China
2Department of Pharmacology, Uniformed Services University of the Health Sciences, Bethesda, Maryland, USA, Maryland, MD, USA, 20743
Our previous studies proved that prenatal caffeine exposure can result in fetal developmental toxicity. As the rate-limiting enzyme for adrenal steroidogenesis, steroidogenic acute regulatory protein (StAR) is essential to fetal development. This study was designed to investigate the molecular mechanisms of caffeine-induced alteration of StAR expression and the hereditability of its developmental toxicity. The human fetal adrenal cell line NCI-H295A was cultured with 4 ¦ìM, 40 ¦ìM and 400 ¦ìM caffeine for 48 h or 10 days. The mRNA and protein expressions of StAR were measured using real-time quantitative RT-PCR and Western blot, respectively. The methylation state of CpG sites within the StAR promoter region were detected by bisulfite-sequencing PCR (BSP). Then caffeine was removed from cells and the expression of StAR was measured in 3, 5, 7 passage cells. Methylation-specific PCR (MSP) was used to determine the methylation state of -368 site on the StAR promoter region. The results showed that both acute and chronic treating cells with caffeine can dose-dependently increase the mRNA and protein expressions of StAR. Meanwhile, caffeine can induce demethylation on -368 site within the StAR promoter region. This single CpG demethylation remained unchanged 7 passages (21 days) after withdrawal of caffeine treatment and the expression of StAR remained increased with comparison to the control. These results indicated that a single CpG demethylation within the StAR promoter region increases its gene expression and suggest an inheritable epigenetic mechanism that may contribute to caffeine-induced fetal developmental toxicity and related late onset of diseases or disorders such as metabolic syndrome. Supported by the National Natural Science Foundation of China, No. 30800931, 30830112 and 30672566; *Correspondence author: [email protected] and [email protected].
212. Effects of commonly used excipient compounds on the expression of CYP3A4 in colon and liver
Leslie M. Tompkins, Caitlin Lynch, James Polli, and Hongbing Wang
Department of Pharmaceutical Sciences, University of Maryland School of Pharmacy, Baltimore, MD, USA, 21201
Pharmaceutical excipient compounds are presumed to be inert constituents of solid drug formulations chosen to aid in the manufacturing, delivery, or absorption of the active ingredient. A myriad of excipients have been used for decades in drug formulations without significant concerns, but the current debate over the extension of biowaivers for in vivo bioavailability and bioequivalence studies for immediate-release (IR) solid dosage forms has raised questions about excipient contributions to the in vivo behavior of drug formulations. Our investigation asked whether a cross-sectional panel of 19 excipients, including fillers, lubricants, plasticizers and surfactants were able to regulate drug metabolizing enzymes, i.e. CYP3A4. Excipients were tested in multiple cell systems for their ability to activate the CYP3A4 promoter, regulate CYP3A4 gene expression and alter CYP3A4 protein expression. In a cell-based reporter assay, all excipients failed to activate hPXR and increase promoter activity. Upon treatment of colon adenocarcinoma-derived LS174T cells, most excipient treatments showed little regulatory effect on CYP3A4 expression. However, 4 of the 19 excipients caused a significant decrease in CYP3A4 mRNA, with maximal repression greater than 60% of control. Although cell culture systems can’t mimic in vivo mechanisms, human primary hepatocytes (HPH) provide an in vitro system which closely resembles in vivo hepatic conditions. In HPH cultures, 4 out of 15 tested excipients demonstrated significant repression of CYP3A4 mRNA expression. Furthermore, western blot analysis of these treatments showed only one excipient able to decrease CYP3A4 protein expression, while the others showed no effect. Polysorbate 80 was the most potent repressor of CYP3A4 mRNA and the only excipient to decrease both mRNA and protein expression of CYP3A4. Collectively, these results suggest that excipient compounds may repress the expression of hepatic drug metabolizing enzymes such as CYP3A4, and affect the metabolism and toxicity of drug products. The realities of polypharmacy in treatment and the occurrence of deleterious drug-drug interactions are well-known, but the impact of excipient regulation on this complex system has been largely overlooked.
213. Role of c-Jun/AP-1 in oxidative stress-mediated induction of murine aldehyde dehydrogenase 1a1 (Aldh1a1)
Ngome L. Makia1, Immaculate Amunom1, Daniel J. Conklin2, Aruni Bhatnagar2, and Russell A. Prough1
1Biochemistry & Molecular Biology, U. Louisville School of Medicine, Louisville, KY, USA, 40292
2Medicine/Cardiology, U. Louisville School of Medicine, Louisville, KY, USA, 40292
The lipid aldehydes, 4-hydroxy-2-nonenal (4-HNE) and propene-2-al (acrolein) are reactive α,β-unsaturated aldehydes generated during the peroxidation of lipids and are implicated in pathologic conditions, including myocardial infarction and inflammation. We established that purified recombinant mouse liver Aldh1a1 efficiently metabolizes HNE (Km 95 μM) and acrolein (Km 727 μM), indicating that Aldh1a1 may protect hepatocytes and cardiomyocytes against their cytotoxic effects. The purpose of the study was to investigate whether oxidative preconditioning regulates expression of Aldh1a1 and to elucidate the signaling pathway involved. We performed expression microarray analyses to establish the effect of oxidants on expression of hepatic Aldh1a1. Mice were administered AIN76A (control) diet, diet containing 0.45% butylated hydroxyanisole (BHA) or 5 mg/kg acrolein per os. Significant induction of hepatic Aldh1a1 and other xenobiotic metabolizing enzymes were observed by treatment with acrolein and BHA and was confirmed by quantitative real time PCR. To decipher the signaling pathways involved in Aldh1a1 induction, we analyzed the mRNA levels of hepatic Aldh1a1 in WT and Nrf2−/- mice exposed to BHA, a synthetic phenolic antioxidant and inducer of Nrf2 and AP-1 transcription factors. The exposure of mice to BHA resulted in ≈2-fold increase in mRNA levels of hepatic Aldh1a1 in WT and Nrf2−/- mice compared to control. However, the basal expression of Aldh1a1 was significantly reduced in Nrf2−/- mice compared to WT mice. We hypothesized that BHA- and acrolein-induced expression of hepatic Aldh1a1 is mediated by AP-1 transcription factor. Transient transfection experiments were conducted in HepG2 cells with Aldh1a1 5’-flanking luciferase reporter vectors. While co-transfection with Nrf2 expression plasmid alone or in the presence of t-butylhydroquinone had no effect, over-expression of c-Jun resulted in ≈4-fold induction in Aldh1a1 transcriptional activity. We further showed that c-Jun mediates Aldh1a1 promoter activity as a homodimer, but not as c-Jun/c-Fos heterodimer. We also established by deletion analysis that two AP-1 sites at position -758 and -1069 relative to Aldh1a1 transcription start site are responsible for induction by c-Jun. Supported in part by USPHS ES11860.
214. MicroRNA-328 controls posttranscriptional regulation of breast cancer resistance protein (BCRP/ABCG2)
Yuzhuo PAN1, Marilyn E. Morris2, and Ai-Ming Yu3
1Pharmaceutic science, University at Buffalo,Suny, Amherst, NY, USA, 14260
2Sch of Pharmacy and Pharmaceutical Sciences, Univ at Buffalo, State University of New York, Amherst, NY, USA, 14260-1200
3Department of Pharmaceutical Sciences, University at Buffalo, SUNY, Buffalo, NY, USA, 14260-1200
Breast cancer resistance protein (BCRP/ABCG2) is an ATP-binding cassette membrane transporter expressed ubiquitously in humans, controlling the absorption, distribution and clearance of numerous xenobiotics including pharmaceutical agents, dietary carcinogens and conjugated metabolites. In addition, overexpression of ABCG2 and other drug transporters in tumorigenic stem cells represents an important mechanism for multidrug resistance (MDR). Two most recent studies suggest that miR-519c and 520h may regulate ABCG2, in which miR-520h was nicely assessed by luciferase reporter assay, and miR-519c inhibitor or mimic were clearly shown to alter ABCG2 protein expression in A549 cells. In the present study, however, we show that human miR-328 would readily target ABCG2, concerning the target-site accessibility, and given the finding that expression of ABCG2 protein and mature miR-328 in drug-resistant MCF-7/MX100 cells and parental drug sensitive MCF-7 cells are inversely related. Furthermore, our data show that miR-328 negatively regulates ABCG2 protein expression by acting on the 3’-UTR segment, and repression of ABCG2 expression via miR-328-mediated pathway is translated into significantly increased drug accumulation in MCF-7/MX100 cells, and the sensitivity of cells to anticancer drug. Together, our results suggest that miR-328 targets the 3’-UTR of ABCG2 and negatively regulates ABCG2 protein expression, and suppressed miR-328 expression may be another underlying mechanism for ABCG2 overexpression in drug-resistant breast cancer cells (MCF-7/MX100). The findings also indicate that ABCG2-mediated MDR may be modified via interference of miRNA pathway in cancer cells.
215. Regulation of CYP2A13 expression in CYP2A13-transgenic Mice
Hong Wu, Guoyu Ling, Yuan Wei, Jaime D’Agostino, Xiuling Zhang, and Xinxin Ding
Wadsworth Center, NYSDOH, Albany, NY, USA, 12201-0509
Human CYP2A13, expressed selectively in the respiratory tract, is highly efficient in the metabolic activation of NNK (4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone), a lung carcinogen. CYP2A13 is also active toward many other xenobiotic compounds. Interindividual differences in CYP2A13 expression are believed to contribute to the differing susceptibility to lung cancer among smokers. The aim of this study is to investigate whether CYP2A13 expression can be induced by exposures to xenobiotic compounds, or suppressed by lung inflammation. The effects of DMBA (7,12-dimethylbenzene[a]anthracene), phenobarbital, pyrazole, 3-MC (3-methylcholanthrene), or lipopolysaccharide(LPS) treatment on CYP2A13 expression were examined in a CYP2A13-transgenic mouse, or a CYP2A13-humanized (CYP2A13-tansgenic and Cyp2a5-null) mouse, in which human CYP2A13 is selectively expressed in the respiratory tract. We found that, while lung CYP2A13 expression was not changed by treatment with any of the known P450 inducers studied, pulmonary expression of CYP2A13 protein and CYP2A13 mRNA, as well as lung microsomal metabolic activity toward NNK, was substantially decreased by LPS treatment. The LPS-induced repression of CYP2A13 transcription was further confirmed in vitro, in cultured human lung tumor H441 cells. Reporter gene assays identified two critical CYP2A13 promoter regions for the response to LPS. The involvement of NF-κB in LPS-mediated CYP2A13 down-regulation was established by the addition of an NF-κB inhibitor. Further gel-shift assays revealed that both CAAT/enhancer-binding protein (C/EBP) and NF-κB could bind to the CYP2A13 promoter at the sites implicated in mediating LPS-response. We conclude that CYP2A13 is not inducible by DMBA, phenobarbital, pyrazole, or 3-MC, under the experimental conditions used, in the CYP2A13-transgenic mouse model, and that CYP2A13 expression is suppressed by LPS-mediated lung inflammation, through mechanisms involving NF-κB and C/EBP. These data suggest that the levels of CYP2A13 proteins present in normal human lung will likely be much higher than the levels detected in tissue samples from patients with lung inflammation, a notion that is very important for assessing the role of CYP2A13 in chemical-induced lung toxicity. (Supported in part by NIH grant CA-092596.)
216. Nrf2 is required for TCDD induction of AhR battery genes Nqo1, Ugt1a6, and Gsta1 in livers of mice
Ronnie L. Yeager, Scott A. Reisman, Lauren M. Aleksunes, and Curtis D. Klaassen
Pharmacology, Toxicology and Therapeutics, The University of Kansas Medical Center, Kansas City, KS, USA, 66160
2, 3, 7, 8-Tetrachlorodibenzo-p-dioxin (TCDD) induces genes via the transcription factor aryl hydrocarbon receptor (AhR), including Cyp1a1, NAD(P)H: quinone oxidoreductase 1 (Nqo1), UDP-glucuronosyltransferase 1a6 (Ugt1a6), and glutathione S-transferase a1 (Gsta1). These genes are included in the classical ‘AhR gene battery’. Interestingly, the AhR gene battery overlaps with the gene battery of another transcription factor, nuclear factor erythroid 2-related factor 2 (Nrf2). There has been speculation that Nrf2 may be involved in TCDD induction of drug processing genes; however, the data are not definitive. Therefore, to address whether Nrf2, in addition to AhR, is necessary for TCDD induction of Nqo1 and most Ugts and Gsts in livers of mice, we conducted the definitive experiment by administering TCDD (50 μg/kg, i.p.) to Nrf2-null and WT mice and collecting livers 24 h later to quantify the mRNA expression of coinciding AhR and Nrf2 target genes. TCDD induced mRNA expression of the prototypical AhR target gene Cyp1a1 in livers of both WT and Nrf2-null mice. Thus, TCDD induction of Cyp1a1 is not dependent on Nrf2. In contrast, TCDD induced Nqo1, Ugt1a6, and Gsta1, as well as many other Ugt and Gst isoforms in livers of WT mice, but not in Nrf2-null mice. TCDD induction of drug processing genes via both AhR and Nrf2 is among the first examples of two transcription factors being necessary, yet not sufficient by themselves, for induction of drug processing genes in vivo. Collectively, the results from the present study demonstrate that AhR and Nrf2 are both required for TCDD induction of the classical AhR battery genes Nqo1, Ugt1a6, and Gsta1, as well as most Ugt and Gst isoforms in livers of mice. (Supported by NIH Grants ES007079, ES009649, ES009716, ES013714, DK081461, and RR021940).
217. Transcriptional regulation of the human karyopherin α2 (KPNA2) promoter by cyproterone acetate: role of the glucocorticoid receptor
Manraj S. Cheema, G. Gordon Gibson, Nick J. Plant, and Kathryn E. Plant
Centre for Toxicology, Faculty of Health and Medical Sciences, University of Surrey, Guildford, United Kingdom, GU2 7XH
Xenobiotics exposure can induce a number of genomic responses, which are typically mediated by the nuclear receptor family of ligand-activated transcription factors. Nuclear translocation of these key factors, whether before or after ligand binding is imperative to their activity and is under the control of a group of transport factors known as the karyopherin (importin) family. The karyopherin α family of adapter proteins forms a molecular bridge between nuclear cargoes and the nuclear import machinery, thus playing a role in mediating translocation of at least some of the nuclear receptors. Studies on the rat karyopherin α have shown that these genes are responsive to several xenobiotics which in turn are ligands for several nuclear receptors (Plant et al., 2006), although the mechanism of regulation in human is poorly understood. Our initial analysis has shown that the human KPNA2 promoter is repressed by cyproterone acetate (CPA) in the human hepatoma (HuH7) cell line with an EC50 of 46μM±1.15 and that there are putative pregnane X receptor (PXR) and glucocorticoid receptor (GR) binding sites within the promoter which might account for this response. We hypothesized that one or both of these nuclear receptors may be responsible for mediating the response of the KPNA2 promoter to CPA; the objective of this study was to investigate the functionality and molecular mechanism(s) of these sites in rendering the response elicited by this promoter. A reporter gene assay system was used to study the KPNA2 promoter and was further characterized by deletion and mutagenesis analysis. Results showed that following deletion or mutation of the putative PXR site, CPA still represses the KPNA2 promoter whereas, this repression is lost when the GR site is deleted or mutated. To further consolidate this finding, electrophoretic mobility shift assay showed that there was no binding to the putative PXR binding site within the KPNA2 promoter, however the GR binding site was confirmed as being viable. In conclusion, we have shown that the regulation of the human KPNA2 promoter by CPA is mediated via the GR and not the PXR. By identifying the presence of a functional GR binding site within the human KPNA2 promoter, we provide a rational explanation in the mechanistic basis for human KPNA2 gene repression by CPA.
Reference
- Plant, K.E., Everett, D.M., Gibson, G.G., Lyon, J., and Plant, N.J. (2006) Transcriptomic and Phylogenetic Analysis of Kpna Genes: A Family of Nuclear Import Factors Modulated in Xenobiotic-Mediated Liver Growth. Pharmacogenetics and Genomics. 16; 647–658.
218. Transcription factor-dependent regulation of hepatic drug metabolizing and transport genes in response to chemical activation
Lauren M. Aleksunes, and Curtis D. Klaassen
Pharmacology, Toxicology and Therapeutics, The University of Kansas Medical Center, Kansas City, KS, USA, 66160
Chemical activators of the transcription factors aryl hydrocarbon receptor (AhR), constitutive androstane receptor (CAR), pregnane X receptor (PXR), peroxisome proliferator-activated receptor α (PPARα), or nuclear factor erythroid 2-related factor 2 (Nrf2) regulate drug metabolizing and transporter encoding genes in livers of mice. However, the specificity of this transcriptional regulation has not been determined in a systematic manner. The purpose of this study was to identify drug metabolizing and transport genes induced by chemical activators in a transcription factor-dependent manner using wild-type and null mice. Chemical activators (TCDD, AhR; TCPOBOP, CAR; PCN, PXR; CFB, PPARα; and OPZ, Nrf2) were administered ip to wild-type and transcription factor-null mice for 4 days. Livers were collected 24h after the final dose, and total RNA was isolated for multiplex suspension analysis. Expression profiles for each chemical inducer-transcription factor pair were generated for 14 phase-I, 24 phase-II, and 7 transporter genes. As expected, chemical activators increased hepatic mRNA expression of prototypical target genes (AhR, Cyp1a2; CAR, Cyp2b10; PXR, Cyp3a11; PPARα, Cyp4a14; and Nrf2, Nqo1) in wild-type, but not the respective null mice. In addition, these target genes were induced by multiple chemical activators. For example, Nqo1 was up-regulated not only by the Nrf2 activator, but also by ligands of AhR, CAR, and PPARα. Select isoforms of aldehyde dehydrogenases, glutathione-S-transferases, sulfotransferases, UDP-glucuronosyltransferases, as well as transporters including organic anion transporting polypeptides and multidrug resistance-associated proteins were increased by chemical activators in wild-type, but not in null mice. Activation of CAR and PPARα induced the highest number of genes (31) compared to AhR (24), PXR (22), and Nrf2 (26). Collectively, these data indicate transcription factor specificity and overlap in the regulation of mouse hepatic drug metabolizing and transporter genes by chemical inducers. Coordinated regulation of phase-I, -II, and transport genes by activators of transcription factors has implications in the development of novel pharmaceuticals as well as risk assessment of environmental contaminants. (Supported by NIH grants DK080774, DK081461, ES009649, ES009716, ES007079, ES013714, RR021940.)
219. Novel technology used for effective tissue preparation
John Tobin, Hamid Khoja, Jennifer Wu, and James Laugharn
Covaris, Inc., Woburn, MA, USA, 01801
Novel Technology Used for Effective Tissue Preparation John Tobin, Hamid Khoja, Jennifer Wu, James Laugharn Covaris Inc., 14 Gill Street, Unit H, Woburn, MA 01801 One of the first key steps for tissue preparation is to deconstruct the initial tissue sample to very fine pieces, sometimes nanometer scale particles, in order to release target molecules. These target molecules can be nucleic acids, peptides or metabolites. Some target molecules are labile and require processing without metabolic or enzymatic degradation. Primary tissue sample preparation has not been updated with high-tech tools, as well as an industrial style approach, to enable highly sensitive analytical instruments to perform at optimal levels. Current mechanical-based sample preparation processes cannot effectively break tissue pieces to particles small enough. Thus, most target molecules are still entrapped within the tissue mass. Typically, the sample preparation process is a combination of mechanical disruption to partially increase the surface area and followed by chemical stabilization. The chemical stabilization step has limited effectiveness since it is diffusion-dependent. Comparing to commonly used sample preparation methods, such as pressure-based, chemical-based, shear/stress-based or non-focused, low frequency “sonicators”, the Covaris process delivers unique benefits, including non-contact processing in closed vessel to avoid potential cross contamination and isothermal processing with no heat damage to samples. This poster shows how to apply the Covaris proprietary AFA (Adaptive Focused Acoustics) technology to various sample preparation applications. We will show the material, methods, and data on how to effectively disrupt different types of tissues and release target biomolecules. Using Covaris CP02 and S2 instruments, tissue preparation gives substantial higher recovery, better reproducibility, speed and ease of use. For high throughput processing needs, the Covaris method is automation friendly and can be integrated with commonly used automation platforms, as well as other reagent kits. Covaris the sample prep advantage www.covarisinc.com
220. Not all C57BL/6 mice are created equal
Mohammed Bourdi, John S. Davies, Khalid Sendide, and Lance R. Pohl
Molecular and Cellular Toxicology, Laboratory of Molecular Immunology, National Heart,Lung and Blood Institute, National Institutes of Health, DHHS, Bethesda, MD, USA, 20892
C57BL/6 inbred mouse strain is one of the most widely used animals for research models. However, their popularity has led to the creation of several C57BL/6 mice substrains maintained within and among different vendors. In this regard, major discrepancies between C57BL/6 mice substrains have been shown in several areas of research including behavioral studies, diabetes, cancer and oxidative stress, among others. We present evidence here describing similar problems in the field of toxicology. When a hepatotoxic dose of acetaminophen (APAP) was administered to substrains of C57BL/6 mice from 4 differents vendors (Taconic Farms, Charles River, Harlan and The Jackson Laboratories), significant differences were found in their susceptibility to liver injury and survival. Comparing APAP bioactivation of C57BL/6J (The Jackson Laboratory; the least susceptible substrain) with C57BL/6Tac (Taconic Farms; one of the most susceptible substrains), we found by immunoblot analysis a reduced level of mitochondrial APAP protein-adducts in C57BL/6J mice compared to C57BL/6Tac that was correlated with mitochondrial levels of CYP2E1. Moreover, APAP treatment caused less mitochondrial glutathione (GSH) depletion in C57BL/6J mice compared to C57BL/6Tac. Interestingly, the levels of APAP protein-adducts and GSH in whole liver homogenates did not differ segnificantly between the two substrains. Overall, these findings suggest, for the first time, that susceptibility differences exist between different C57BL/6 mice substrains in APAP-induced liver injury model and possibly other forms of injury. It also stresses that researchers should carefully consider the appropriate C57BL/6 mice control when using genetically engineered mice on a C57BL/6 background, not only for toxicological research, but also for other biomedical studies.
221. A genomic analysis of small intestinal NADPH-cytochrome P450 reductase function in a conditional knockout mouse model
Jaime D’Agostino, Fang Cheng, Xinxin Ding, and Qing-Yu Zhang
Wadsworth Center, NYSDOH, Albany, NY, USA, 12201-0509
The NADPH-cytochrome P450 reductase (CPR) is an essential component for the functioning of a number of enzymes, including microsomal cytochrome P450 (P450) monooxygenases and heme oxygenases. An intestinal epithelium (IE)-specific Cpr-knockout mouse model (IE-Cpr-null) has been described recently (Zhang et al., 2009, Drug Metab. Dispos. 37:651-7), as a useful model for elucidation of the roles of the small intestine in the first-pass clearance of orally administered drugs and dietary compounds. The aim of the current study is to identify, through microarray analysis, intestinal gene-expression changes in response to the IE-specific Cpr gene knockout, in efforts to discover the biological functions of CPR in this tissue. Significant differences in expression were found between IE-Cpr-null mice and wild-type (WT) littermates for gene ontology terms related to P450s, transporters, cholesterol biosynthesis, iron metabolism, and antigen presentation/processing. Selected genes with expression changes were confirmed either at the mRNA level (by real-time RT-PCR) or at the protein level (by immunoblot analysis). In addition to increases in gene expression for numerous microsomal P450 enzymes, many genes in the cholesterol biosynthesis pathway were up-regulated in IE cells, presumably due to the deficiency in cholesterol biosynthesis in Cpr-null enterocytes. However, no difference in cholesterol levels was observed, either in the plasma or in IE cells, between the IE-Cpr-null and WT mice. These findings suggest that IE cholesterol biosynthesis does not make a significant contribution to circulating or tissue cholesterol levels. Nevertheless, the disruption to de novo cholesterol synthesis seems to be mechanistically linked to expression changes in genes important for immunity in IE cells, an idea that we are actively pursuing. The expression of several genes related to iron homeostasis was also changed, presumably because of the inactivation of heme oxygenases, and consequent attempts by the cells to decrease iron export and increase heme export. Studies to measure plasma and tissue levels of iron and other endogenous compounds, including P450 metabolites, are underway. (Supported in part by NIH grant GM082978)
222. Comparison of uptake clearance between suspended and sandwich-cultured human hepatocytes
Yi-an Bi1, Jennifer Reeve2, Emi Kimoto3, and David B. Duignan1
1Pharmacokinetics, Dynamics & Metabolism, Pfizer, Inc., Groton, CT, USA, 06379
2Brown University, Providence, RI, USA, 02912
3Pharmacokinetics, Dynamics & Metabolism, Pfizer, Inc., Groton, CT, USA, 06340
Sandwich-cultured hepatocytes are widely recognized as a useful model to measure in vitro biliary clearance. We have previously reported the ability to use cryopreserved human hepatocytes in sandwich-culture and have shown that cryopreserved hepatocytes are able to form bile canaliculi and that uptake and efflux transporter activities are similar to those in fresh hepatocytes. To date, much emphasis has been placed on using sandwich-cultured hepatocytes to measure efflux, but their utility in measuring hepatic uptake has not received as much attention. The purpose of these studies is to compare the uptake clearance between suspended hepatocytes and sandwich-cultured (SC) hepatocytes for a set of well-characterized compounds. Cryopreserved hepatocytes were purchased from commercial vendors. The uptake clearance of four compounds was determined in both suspended hepatocytes and SCH: [3H]digoxin (OATP1B3), fexofenadine (OATP1B3), rosuvastatin (OATPs/NTCP), and [3H]taurocholate (NTCP). Compounds were incubated at 2 μM for 0.5, 2 and 5 minutes in suspension and in SC. Suspension experiments were terminated by quick centrifugation though oil, while SC studies were terminated by quickly washing the cells with cold buffer. Quantification was determined by scintillation spectroscopy or by LC/MS. With the exception of TC, the apparent accumulation of drug was 2-3-fold higher in suspended hepatocytes compared to SC hepatocytes. However, the apparent uptake clearance for each compound was similar between suspended hepatocytes and SC hepatocytes. For [3H]digoxin, fexofenadine, rosuvastatin and [3H]taurocholate, the mean apparent uptake clearances (μl/min/mg) in suspended hepatocytes and SC hepatocytes, respectively, were 1.4 and 3.0, 2.7 and 1.9, 7.5 and 4.6, and 11 and 14. Further studies to assess uptake kinetics are underway. The results suggest that cryopreserved sandwich-cultured human hepatocytes can be used to measure hepatocyte uptake clearance and that uptake clearances in SC hepatocytes are similar to those obtained in suspended hepatocytes. These data further show the utility of SC hepatocytes, since one can measure both uptake clearance and efflux clearance in the same model. This should allow for better predictions of hepatic transporter-mediated clearances in vivo.
223. Comparison of metabolic capacities of fresh and cryopreserved human hepatocytes isolated from the same donor: Metabolic stability, plated metabolism, metabolite ID applications
Cornelia Smith1, Christina Nolan1, Manda Edwards1, Todd Stewart1, Jean Hatfield1, Shiloh Barfield1, Jeanette Hill2, James Hill2, Jasminder Sahi1, and Edward LeCluyse1
1Life Technologies, Durham, NC, USA, 27703
2Life Technologies, Austin, TX, USA, 27703
Fresh and cryopreserved hepatocytes are accepted by the FDA for in vitro ADME studies. Convenience and commercial availability make the use of cryopreserved hepatocytes ideal for metabolic stability, metabolic profiling, metabolite ID and drug-drug interaction studies. To test the hypothesis that the metabolic capacity of cryopreserved hepatocytes is similar to that of fresh hepatocytes prepared from the same individual human donor tissues, metabolic activities were determined using probe substrates phenacetin (CYP1A2), bupropion (CYP2B6), paclitaxel (CYP2C8), diclofenac (CYP2C9), S-mephenytoin (CYP2C19), dextromethorphan (CYP2D6), testosterone (CYP3A4/5), midazolam (CYP3A4/5) and benzydamine (FMO). 7-Hydroxycoumarin was used to probe for UGT and SULT activities. Assays for the intrinsic clearance of midazolam, phenacetin, dextromethorphan, benzydamine and the low-turnover substrates tolbutamide and diazepam were performed in both suspensions and cultures of human hepatocytes. Prototypical CYP substrates and the glutathione-S-transferase (GST) substrate, 3-methylindole were incubated for two hours to compare metabolites generated in fresh and cryopreserved human hepatocytes. Metabolites and parent compounds were detected by LC-MS/MS or HPLC analyses. No statistically significant differences between fresh and cryopreserved metabolic activities were observed for all CYP isoforms [e.g. mean CYP3A4/5 activity 576 (fresh) vs. 650 (cryo) pmol/min/106 cells]. Predicted clearances from hepatocyte suspensions of moderate- to high-turnover compounds were similar in fresh and cryopreserved hepatocytes isolated from four donors (e.g. mean midazolam clearance 12.3 vs 14.4 mL/min/kg, respectively). Predicted clearances from hepatocyte cultures were also similar [e.g. mean midazolam clearance 10.8 (fresh) vs 6.09 (cryo) mL/min/kg]. Reliable intrinsic clearance data for low-turnover compounds was not attainable in short-term suspension assays. However, mean predicted diazepam clearances were 3.50 and 1.73 mL/min/kg in fresh and cryopreserved hepatocyte cultures, respectively, isolated from four donors. Major metabolites from prototypical CYP substrates were observed in fresh and cryopreserved hepatocyte metabolite ID incubations. Our results using direct comparisons show that cryopreserved human hepatocytes possess metabolic capacities comparable to freshly isolated hepatocytes. Therefore, cryopreserved hepatocytes are a valuable in vitro model useful in the research and development of new drug candidates.
224. Decreases in CYP450 observed during in vitro induction and inactivation studies: Predicting clinical outcomes
Diane (Ely) Ramsden, Eleanore Seibert, and Jinling Xie
Drug Metabolism and Pharmacokinetics, Boehringer Ingelheim, Ridgefield, CT, USA, 06877
In drug discovery and development, compounds are routinely screened for drug-drug interaction (DDI) potential utilizing in vitro methodologies in order to predict the propensity towards clinical DDIs. As such, BI_F was evaluated as a CYP450 inducer in 3 lots of freshly isolated human hepatocytes following a standard induction protocol. Surprisingly, BI_F exhibited concentration dependent decreases of both CYP3A4 mRNA levels and enzyme activities. Additionally, pre and co-incubation of BI_F with Rifampicin (25 μM) a potent prototypical inducer of CYP3A4 was able to abolish the inductive effect of Rifampicin. Decrease in CYP3A4 was not associated with a drug induced cytotoxicity event as demonstrated by 6 of the most commonly utilized toxicity assays. Further in vitro data demonstrated BI_F to be a CYP3A4 inactivator thereby complicating conclusions from a clinical DDI risk assessment. Utilizing the CYP3A4 enzyme activity analyses from the human hepatocyte studies, a prediction of the expected clinical AUC change, resulting from the dual effects of inactivation and decreases in CYP levels was made using a novel enzyme kinetic mathematical model.
225. A distribution analysis of cytochrome P450 Enzyme activities in cryopreserved human hepatocytes from over 400 individuals
Diane (Ely) Ramsden1, Gillian Wallace2, Cornelia Smith2, and Tom S. Chan1
1Drug Metabolism and Pharmacokinetics, Boehringer Ingelheim, Ridgefield, CT, USA, 06877
2Hepatic Biology Division, Life Technologies Corporation CellzDirect, Durham, NC, USA, 27703
Human cryopreserved hepatocytes are increasingly being used for drug development research to compensate for the inconsistent availability of freshly isolated hepatocytes. Despite the stresses of cryopreservation and reconstitution, human cryopreserved hepatocytes maintain the original levels of drug metabolizing enzyme activities1,2. This bestows an added advantage to the use of cryopreserved hepatocytes in that enzyme activity levels can be pre-screened to identify lots that are either most representative of the general population or a specific group of individuals. In the current study, we retrospectively analyzed the distribution of vendor-provided cytochrome P450 (CYP450) activity data from over 400 donors. Enzyme ranges encompassing the 25% and 75% quartiles around the median were chosen to depict the most common range of enzyme activities in the sample population for a given substrate. Log transformation of CYP450 activity resulted in a more normalized distribution that allowed the visualization of polymorphic subgroups. We have also presented evidence that pooled hepatocytes from different donors can increase the probability of achieving CYP450 activities that fall +/- 25% of the median values. In summary, these data will help to determine the best range of enzyme activities most representative of the general population.
References:
- Brown HS, Griffin M, Houston JB. (2007). Evaluation of cryopreserved human hepatocytes as an alternative in vitro system to microsomes for prediction of metabolic clearance. Drug Metab. Disp. 35(2); 293–301.
- Gomez-Lechon MJ, Donato MT, Castell JV, Jover R. (2004). Human hepatocytes in primary culture: the choice to investigate drug metabolism in man. Curr. Drug.Metab. 5;443–462.
226. Characterization of drug uptake into cryopreserved human hepatocytes
Greg Loewen, Tiffini Hensley, Maciej Czerwinski, Brian Ogilvie, and Andrew Parkinson
XenoTech, LLC, Lenexa, KS, USA, 66219
Cryopreserved hepatocytes retain a high level of drug-metabolizing enzyme activity and serve as an in vitro system to support drug metabolism studies. However, the clearance of some drugs is determined largely by their rate of hepatic uptake. Compounds can enter hepatocytes by passive diffusion and, in some cases, by active (energy-dependent) processes either involving transporters or endocytosis, which results in a liver-to-blood ratio greater than one. A high liver-to-blood ratio is also seen with various lipophilic amines that enter hepatocytes largely by passive diffusion and then become ionized and trapped in lysosomes which maintain a low pH (approximately 5) in an ATP-dependent process. The organic anion transporting polypeptide 1B1 (OATP1B1), OATP1B3, OATP2B1, sodium taurocholate co-transporting polypeptide (NTCP), organic cation transporter (OCT1) and organic anion transporter (OAT2) are uptake transporters expressed on the sinusoidal membrane of hepatocytes. In the present study, we used cryopreserved human hepatocytes to characterize the uptake of the OATP1B1 substrate 3H-estrone-3-sulfate (E3S), the NTCP substrate 3H-taurocholic acid (TCA), the OCT1 substrate 3H-1-methyl-4-phenylpyridinium iodide (MPP+) and the lipophilic amine 3H-propranolol. Hepatocytes suspended in Krebs-Henseleit buffer were incubated with radiolabeled substrates from 15 sec to 10 min, after which the cells were isolated by centrifugation through a layer of silicone oil (Parker and Houston. Drug Metab. Dispos. 36: 1375-1384, 2008) and analyzed by liquid scintillation counting. The hepatocytes were incubated with each substrate at 4 and 37°C, and the difference in uptake was taken to reflect active uptake or an energy-dependent process like lysosomal trapping. The uptake of all four substrates was rapid (and proportional to time for about one minute) and was concentration dependent. The Eadie-Hofstee plots for all four substrates revealed biphasic kinetics which probably represents a high affinity/low capacity active transporter-mediated component and a passive perfusion or non-specific binding component. The high affinity Km values of all four substrates were below 10 μM. The Vmax values for MPP+, TCA, E3S and propranolol were ~100, ~100, ~300 and ~2000 pmol/million cells/min. The effect of the inhibitors ketoconazole, rifampin and verapamil on uptake was evaluated. The uptake of prototypical substrates into several lots of cryopreserved hepatocytes was determined. Cryopreserved hepatocytes retain uptake transporter activity and can be used to determine the transporter-mediated uptake and lysosomal trapping of drugs.
227. Assessing compounds for liver distribution properties in a drug discovery setting
Leena Laitinen, Shaolan Li, Pamela McCarthy, Craig Zetterberg, Lei Win Tung, Arthur Rugg, Hong Tsao, Wojciech Dworakowski, Christopher Brummel, David Howe, and Francoise Berlioz-Seux
Drug Innovation Pharmacokinetics, Vertex Pharmaceuticals, Inc., Cambridge, MA, USA, 02139
For therapeutic indications where the target tissue for pharmacological response is the liver, it is important to understand the interplay of ADME processes that influence liver drug levels. In particular, metabolism, active uptake, efflux, and free fraction are important ADME properties that affect systemic pharmacokinetics of a drug. As part of discovery efforts at Vertex Pharmaceuticals, the liver levels for various compounds were determined in Sprague-Dawley rats. The ADME properties of various compounds with a wide-range of liver concentrations were investigated. The metabolic rate was determined in rat liver S9 fraction. The free fraction was determined in rat plasma and rat liver homogenates by equilibrium dialysis. No correlation between fraction unbound in liver homogenate or metabolic stability and in vivo liver to plasma ratio was observed. The hepatic uptake of these compounds was evaluated in rat hepatocyte suspensions. The partition ratio, Kp, of the test set of compound at an initial concentration of 1 mM in rat hepatocytes at 37°C ranged from 4 to 14. No correlation was observed between rat hepatocyte uptake values at 37°C and liver concentrations. However, the liver levels were found to correlate to the ratio of the hepatocyte partition ratios obtained at 37°C and 4°C. These results suggest that it may be possible to rank order compounds with respect to liver levels in a discovery setting.
228. Development of pooled human cryopreserved hepatocytes using a predictive algorithm to target specific metabolic activities
Caitlin Ailes, Ji Young Lee, Ana Miller, and Timothy Moeller Celsis
In Vitro Technologies, Halethorpe, MD, USA, 21227
Traditionally, metabolic properties of new chemical entities (NCEs) have been determined with liver microsomes in the discovery and development of drugs. However, the data are limited to selected metabolic enzymes. To provide more relevant data, hepatocytes can be used as a more physiologically appropriate system. However, hepatocytes from individual donors retain the inherent variability present in a population. To overcome the diverse metabolic capacities, researchers have independently pooled several hepatocyte donors to mimic average activities, such as obtained from pooled microsomes. Due to time, cost and effort, these pools were usually small. To address the need for average activities in hepatocytes, we developed LiverPool™, 5-, 10- and 20-donor pooled human cryopreserved hepatocytes, to gain the benefits of an average human response as in multi-donor microsomes, yet with the full complement of phase I and phase II metabolic enzymes, transporters and other activities that better represent in vivo responses. In an advance beyond this, we developed Purpose-Pooling™, which creates pools of cryopreserved human hepatocytes with specific enzymatic criteria. Herein, we show the testing of individuals and LiverPool lots for CYP1A2, 2A6, 2C9, 2C19, 2D6, 2E1 and 3A4, along with glucuronidation and sulfation activities. The coefficient of variation for individual donors (n>100) ranged from 0.60 – 3.20 across the metabolic activities. LiverPool lots that were created from a random selection of individual donors reduced the coefficient of variation range to 0.35 – 0.80. LiverPool lots created from selected individuals further reduced the range of the coefficient of variation to 0.11 – 0.35, thereby providing confidence in the prediction of measured activities. Further, we created LiverPool lots with measured activities that closely matched targeted values. In all, we have shown that through the use of a proprietary algorithm, pooled human hepatocytes may now be blended to obtain specific activities for the purpose of creating consistently performing lots or to meet an individual researcher’s criteria.
229. Characterization of digoxin uptake in sandwich-cultured human hepatocytes
Emi Kimoto1, Yi-an Bi2, Jonathan Chupka1, Yongling Xiao1, and David B. Duignan2
1Pharmacokinetics, Dynamics & Metabolism, Pfizer, Inc., Groton, CT, USA, 06340
2Pharmacokinetics, Dynamics & Metabolism, Pfizer, Inc., Groton, CT, USA, 06379
Purpose. Digoxin is a cardiac glycoside which is commonly used in clinical practice to treat congestive heart failure and atrial fibrillation. Digoxin is primarily cleared by the kidney; however, a significant component of clearance is due to hepatic transport into bile. Digoxin has a narrow therapeutic index; consequently, patients can be particularly susceptible to DDI-mediated cardiotoxicity. It has been reported that digoxin uptake into human hepatocyte suspensions involves active transport, and that digoxin is a substrate for the organic anion transporting polypeptide 1B3 (OATP1B3). However, further characterization of uptake is needed. The purpose of the present study is to investigate the uptake mechanisms of digoxin into sandwich-cultured human hepatocytes (SCHH) using cryopreserved hepatocytes.
Methods. We previously reported the ability to use cryopreserved human hepatocytes in sandwich-culture and have shown that uptake and efflux transporter activities are similar to those in fresh hepatocytes. Therefore, for this study the uptake of digoxin in the presence or absence of inhibitors was measured using SCHH.
Results. The uptake of 1 μM digoxin in SCHH was 9.5 ± 2.2 pmol/mg protein/5 min (N=3). Digoxin (0.05 – 1 μM) uptake involved active processes, since uptake at 4°C was not observed. The uptake of 1 μM digoxin was not inhibited by 10 μM cyclosporin A or 100 μM rifampicin – two compounds known to broadly inhibit human OATPs. In addition, 50 μM quinidine (an inhibitor of OCT1) had no significant effect on digoxin uptake into hepatocytes. These results are not consistent with OATP1B3-mediated active uptake of digoxin. Studies with SCHH and with OATP-transfected cells are ongoing in order to understand this discrepancy and to further elucidate the mechanism of digoxin active uptake.
230. The hepaRG human hepatocyte cell line is a reliable control cell line for ADME-Related gene expression induction studies
David Steen1, Christophe Chesne1, Rob Shipman2, Jodi Morrison2, and David K.H. Lee2
1R & D, Biopredic international, Rennes, France, 35000
2Genomics, NoAb BioDiscoveries, Mississauga, ON, Canada, L5N 8G4
Inter-individual variability can confound in-vitro analyses using human hepatocytes, so human DTEx™ microarray analysis of gene expression induction was initiated to assess the uniformity of response to prototypical inducers by HepaRG human hepatic cells and primary human hepatocytes. Three lots of HepaRG cells were obtained from BioPredic and six lots of primary human hepatocytes were obtained from CellzDirect, and all were treated with prototypical inducers for 24 hours according to each supplier’s protocol. CYP gene expression was assessed using the human DTEx™ microarray [145 ADME-related genes]. Total RNA was isolated from each sample using the RNeasy kit [Qiagen] and amplified using the MessageAmpII aRNA amplification kit [ABI-Ambion]. Labelled cDNA was generated from the amplified RNA using SuperScript II reverse transcriptase [Invitrogen] and CY5-labelled random pentadecamers [Eurofins MWG Operon]. Microarrays were hybridised for 18 hours at 60°C. Microarray images were acquired with ScanArray [Perkin Elmer] and image analysis was performed with QuantArray [Perkin Elmer]. Data analysis, cluster dendrograms and matrix plots were generated in GeneLinker Gold [Improved Outcomes Software] and Excel [Microsoft Inc.]. Human DTEx™ microarray analysis indicated that induction of ADME-related gene expression was more uniform in HepaRG cells than in primary human hepatocytes. Induction of CYP3A4 gene expression in Rifampicin-treated samples ranged from 0.7 to 2.1 fold in human hepatocytes tested and 2 -2.3 fold in HepaRG cells. Gene expression levels for most of the ADME-related genes on the human DTEx™ microarray were inducible in drug-treated HepaRG cells and primary human hepatocytes. Inter-individual variability in CYP gene expression induction, due to liver donor demographic and genetic factors, was more pronounced in primary human hepatocytes. HepaRG human hepatic cells are a reliable control cell line and suitable substitute for primary human hepatocytes in ADME-related gene expression induction studies.
Reference:
- Kanebratt, K.P. and T.B. Andersson (2008). “HepaRG Cells as an in Vitro Model for Evaluation of Cytochrome P450 Induction in Humans.” Drug Metabolism and Disposition 36(1): 137–45
231. Time-course disposition of ritonavir and prototypical hepatic inducers in cultures of primary human hepatocytes: context for induction & inhibition concentration responses
Kimberly Freeman, Jonathan Jackson, and Stephen S. Ferguson
CellzDirect/Invitrogen Corporation (a part of life technologies), Durham, NC, USA, 27703
Various experimental methods use cell free and/or cell-based assays to study molecular interactions. In these studies, a chemical is added to the assay system (typically in some form of diluted solvent) at a targeted concentration, and all data processing thereafter reference the targeted exposure concentrations. However, certain cell types such as cultures of primary human hepatocytes maintain and/or establish active uptake and efflux pathways as well as metabolic capacity more akin to the hepatocytes’ in vivo environment where transporter handling and metabolism can significantly affect the exposure concentrations within the culture monolayer. A notable drug that we have studied in our research projects is ritonavir. Ritonavir is a clinically-prescribed HIV therapeutic that is known to both inhibit and induce CYP3A. This multidimensional effectiveness in humans has made it and related compounds useful to de-convolute simultaneous process (e.g. induction, inhibition, and metabolism) in whole-cell model systems. For ritonavir, studies using cell-free and cell line systems have shown that the binding affinity and EC50 for these systems is approximately 2 μM with respect to the published findings with the nuclear receptor PXR. However, in cultures of primary human hepatocytes (that more closely resemble the adult liver with regard to metabolism and transport), we find that ritonavir is actually concentrated within the hepatocyte monolayers (e.g. 0.1 μM addition of ritonavir to cultures after 24 hours results in ~15 μM monolayer concentrations, and 1 μM addition results in ~102 μM monolayer concentrations) relative to the concentrations added to these cell cultures. These responses also account for the metabolic turnover of ritonavir due to the inherent metabolic capacity of the hepatocyte cultures to eliminate ritonavir, and when correlated with observed CYP3A4 mRNA expression responses demonstrate the utility of these data in cell-based vs. cell-free assays. These results and preliminary studies with a few notable prototypical inducers will be described.
232. Throughput advantages of multiple parallel LC systems utilizing a single data file
Matthew J. Berube
Thermo Fisher Scientific, Franklin, MA, USA, 02038
Introduction: Sample analysis time is a major limiting factor in high-throughput environments. This study increased sample throughput by using multiple parallel chromatography systems and a single data file for each sample batch. Data processing was performed using a novel software program that extracts individual sample data from the single data file quickly and accurately.
Methods: Atenolol and warfarin were spiked into neat samples along with cabemazepine as the internal standard to make a standard curve. Staggered injections from multiple parallel two-dimensional chromatography systems were monitored using a single mass spectrometer and data file. The data was then processed using QuickCalc chromatographic viewer software which is able to match sample and analyte information to individual peaks within a single data file. Standard curves were quantitated and the coefficient of determination was evaluated.
Preliminary Results: Using multiple parallel chromatography systems and a single data file, the sample analysis time was cut in half compared to the same sample batches run with separate data files for each sample. The sample analysis took approximately forty seconds using a data file for each sample injection, compared to twenty seconds using a single data file for all of the sample injections. The data processing software accurately chose the retention times and peaks and produced reports quickly and accurately.
Novel Aspect: Combining chromatograms from multiple parallel chromatography systems into a single MS data file increases throughput without compromising data quality.
233. Development and validation of a higher throughput in vitro CYP inhibition assay to simultaneously assess the reversible and time dependent inhibitory potencies of new chemical entities
Maria D. Bacolod, Kimloan Nguyen, and Gamini Chandrasena
Drug Metabolism and Pharmacokinetics, Lundbeck Research USA, Inc., Paramus, NJ, USA, 07652
Inhibition of cytochrome P450 (CYP) enzymes has the potential to cause serious drug-drug interactions (DDI) in clinic due to large number of drugs being metabolized by these enzymes. Hence, early inhibition screening of new drug candidates is needed for mitigating DDI risk. In this study, an in vitro CYP inhibition assay was developed and validated to simultaneously assess the reversible and time dependent inhibitory potencies of test compounds towards 7 major human CYP isozymes, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. To determine the reversible inhibitory effects, various concentrations of test compounds were co-incubated with 0.16 mg/mL pooled human liver microsomes (HLM), NADPH, and a cocktail of the selective CYP probe substrates (phenacetin-CYP1A2, bupropion-CYP2B6, amodiaquine-CYP2C8, diclofenac-CYP2C9, omeprazole-CYP2C19, dextromethorphan-CYP2D6, and midazolam-CYP3A4) in a 96-well plate format using TECAN liquid handling system for 10 min at 37°C. To evaluate the time dependent inhibitory effects, in parallel, the test compounds were pre-incubated in HLM and NADPH at 37°C for 25 minutes. After 25 min of pre-incubation, a cocktail of the selective CYP probe substrates was added and allowed to incubate for 10 min. The enzyme activities (metabolite formation) were measured using LC-MS/MS. and the corresponding IC50 values were estimated for each isozyme. The IC50 values obtained from well-characterized reversible and mechanism-based CYP inhibitors were found to be in good agreement with the literature data. Similarly, good correlations in the data between individual and cocktail incubations were observed. In general, > 2 fold downward shift in IC50 from co-incubation to pre-incubation assays suggest time dependent inhibitory effects by the test compounds. Overall, this assay offers a fast and a reliable means of assessing the DDI liabilities of discovery compounds in a higher throughput mode.
234. Comparison of rapidfire® ultra high throughput MS with traditional LCMS/MS for cytochrome P450 inhibition testing
Elke S Perloff1, Shangara S. Dehal1, Andrew K Mason1, Andrew P Blanchard1, Charles L. Crespi1, Can C. Ozbal2, William A Lamarr2, Vaughn P Miller2, and David M. Stresser1
1BD Gentest Contract Research Services, BD Biosciences Discovery Labware, Woburn, MA, USA, 01801
2BioTrove, Inc., Woburn, MA, USA, 01801
Objective: Assessment of cytochrome P450 inhibition by NCEs has moved into earlier phases of drug discovery/development, and recent guidance from the FDA recommends routine in vitro IC50 determinations for at least six P450 isoforms. The resulting increase in the number of samples generated in P450 inhibition screens has led to a demand for higher throughput analysis options. The objective of this study was to apply RapidFire LC/MS/MS technology (BioTrove, Woburn MA) to in vitro cytochrome P450 inhibition testing and compare the results to traditional LC/MS/MS methods validated in house.
Methods: Seven-point IC50 values for a range of inhibitors were determined in individual incubations in human liver microsomes using the following FDA recommended drug probe substrates with previously validated assay methods [Perloff ES et al (2009) Xenobiotica 39:99-112]: CYP1A2/ phenacetin (tacrine for RapidFire); CYP2B6/ bupropion; CYP2C8/ amodiaquine; CYP2C9/ diclofenac; CYP2C19/ S-mephenytoin; CYP2D6/ dextromethorphan; CYP3A4/ testosterone, midazolam. Samples were analyzed individually by RapidFire technology as well as by traditional LC/MS/MS methods developed and validated in house. Stable-labeled isotope internal standards were used for all probe substrate metabolites except for 1-hydroxytacrine, where bucetin was employed. Percent remaining activity was calculated for each inhibitor concentration, and IC50 values were calculated by linear interpolation.
Results: IC50 values obtained using RapidFire ultra high throughput LC/MS/MS analysis were consistent with the data obtained using traditional LC/MS/MS methods. Greater than 90% of corresponding IC50 values were within 2.0-fold of each other. A run time of approximately 7 seconds per injection with RapidFire technology compared to 2 to 4 minutes for traditional LC/MS/MS methods provided a significant decrease in analysis time. Conclusions: P450 inhibition IC50 results obtained using RapidFire were comparable to those obtained using traditional LCMS methods validated according to FDA guidance. The increased analysis speed represents a >20-fold improvement in cycle time, thereby permitting rapid data delivery to project teams and clients. In addition, compared to typical drug discovery inhibition assays, ultra-rapid analysis allows acquisition of more data points per unit time providing for the option of conducting robust, multipoint assays typical of drug development.
235. Ultra-performance liquid chromatographic-electrospray mass spectrometric determination (UPLC-ESI-MS) of protocatechuic aldehyde (3,4-dihydroxybenzaldehyde, PAL) glucuronidation in vitro
Hui-Xin Liu, Ying Hu, and Ling Yang
Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, The Chinese Academy of Sciences, Dalian, China, 116023
PAL has been considered as one of the major active constituents of Salvia miltiorrhiza, a commonly used traditional Chinese medicine for treating coronary heart disease, cerebrovascular disease and chronic renal failure. A number of pharmacological studies showed that PAL possessed arrays of biological activities as anti-cardium colic, reducing atherosclerosis and inhibiting the aggregation of platelets. However, detailed information on the metabolism of PAL and their pharmacokinetic fate in mammals are still scant. Drug metabolic profile plays an important role in discovering and developing the novel drug from the metabolites possessed the pharmacological activities. An understanding of the enzymology of the metabolic clearance of a drug, whether by phase I or phase II mechanisms, is pivotal to new drug development. Early knowledge of the potential biotransformations of drug in the target species is of great interest. In mammals, glucuronidation is a major conjugation reaction providing for metabolic elimination of exogenous compounds and endogenous compounds. This reaction is catalyzed by a family of enzymes known as UDP-glucuronosyltransferases (UGTs, EC 2.4.1.17). We have previously identified UGT1A6 was the major isoform responsible for PAL glucuronidation. Planar phenol derivatives like PAL, such as 4-methylumbelliferone and 4-nitrophenol, have been reported to be mainly conjugated by UGT1A6 isoform in mammals. However the contribution ratio differed extensively among the animal species. Therefore, an accurate and specific method for UGT activities toward PAL in mammalian tissues will be an important initial step in understanding the role of PAL glucuronidation pathway. Recently, UPLC was introduced as commercially available instrument, which has been applied for the pharmaceutical, toxicological and biochemical analysis. It has the advantages of the fast analysis, high peak capacity, good sensitivity and low consumption of samples. Furthermore, MS techniques play an important role in the metabolism study, because the high sensitivity of MS as an LC detector facilitates the detection of metabolites which are difficult to obtain by conventional means. In this study, a rapid and specific UPLC-MS method was developed for the determination of PAL glucuronidation activities in liver microsomes from different species. Liver microsomes incubation system with PAL resulted in the formation of two product peaks. Following the purification, the peaks were respectively confirmed as PAL meta-glucuronide (M1) and PAL para-glucuronide (M2) by MS and NMR. The method was validated for the determination of M1 and M2 with respect to specificity, linearity, detection limit, recovery, stability, precision and accuracy. The chromatographic separation was achieved on a UPLC BEH C18 column (50 mm × 2.1 mm i.d., 1.7 μm), with phase of acetonitrile-water (ratio 10:90). Selective ion reaction (SIR) monitor was specific for PAL, M1, M2 and I.S.. The method was linear over the concentration range 0.5-100 μM for M1 and M2 in spiked HLMs, respectively. Acceptable precision and accuracy were obtained for concentrations over the standard curve range. The method was successfully used to determine the kinetics of glucuronidation activities toward PAL in liver microsomes from different speices. The developed method was appropriated for rapid screening PAL glucuronidation activities in liver microsomes from different species.
236. Evaluation of accurate mass TOF-MS for use in high throughput metabolic stability screening
Michelle V. Romm, Nikunj Parikh, Vaughn P. Miller, William A. LaMarr, and Can C. Ozbal
BioTrove, Woburn, MA, USA, 01801
The metabolic half-life or stability of a drug candidate has important pharmacokinetic and clinical significance, because it influences both oral bioavailability and plasma concentration of a compound, ultimately affecting efficacy. Large compound libraries and advancements in liquid handling have placed demands on the throughput, reliability and reproducibility of high throughput screening of in vitro metabolic stability assays. While it is possible to prepare compound incubations in a high throughput manner using robotic liquid handlers, analysis of the resulting samples, typically accomplished by LC/MS/MS techniques, is a bottleneck in the process. Though advances in auto-injectors and other technologies have increased the sample load capacities of common MS workflows; the development MRM methods, a pre-requisite of LC/MS/MS analysis, requires a significant portion of human resources and time. We propose that the use of accurate mass analysis, offered by time of flight (TOF) mass spectrometers, may act as an alternative protocol for compound selectivity, eliminating the need for MRM method development. A set of diverse compounds was incubated with human liver microsomes over a time course of 0-60 min. Specific MRM methods for each of thirty-seven compounds we optimized using Agilent’s Optimizer software. All samples were analyzed using specific MRM methods on an Agilent 6410 QqQ-MS, or utilizing generic source parameters and exact mass windows on an Agilent 6220 ESI-TOF. All samples were processed and injected onto the MS instruments using the RapidFire system with cycle times of 6-8 secs per sample. The metabolic half-life values predicted by the two different MS platforms are equivalent, supporting the use of exact mass MS as part of a high-throughput workflow for metabolic stability testing. The results presented here may have implications for significantly increasing the throughput of a variety of in vitro ADME applications.
237. Integrated high throughput MS-based CYP time-dependent inhibition assay (5 Major CYPs and 6 Probes) using robotics and analytical time saving techniques
Sophie Mukadam, Erlie Delarosa, Daniel Tran, Cornelis E. Hop, S. Cyrus Khojasteh, Jane Kenny, and Jason S. Halladay
Dmpk, Genentech, Inc., South San Francisco, CA, USA, 94080
Integrated High Throughput MS-Based CYP Time-Dependent Inhibition Assay (5 Major CYPs and 6 Probes) Using Robotics and Analytical Time Saving Techniques Sophie Mukadam, Erlie Marie Delarosa, Daniel Tran, Cornelis E. Hop, S Cyrus Khojasteh, Jane Kenny, and Jason S Halladay* Department of Drug Metabolism and Pharmacokinetics, Genentech, Inc., 1 DNA Way (MS 412a), South San Francisco, CA 94080 USA In vitro assays to assess time-dependent inhibition (TDI) of cytochrome P450 (CYP) enzymes are used to assess the risk for potential drug-drug interactions at the lead ADME optimization stage. In our laboratory, we have developed a 384-well high-throughput IC50 shift assay using well-known mass spectrometry-based (MS) probes to provide relevant TDI data at the early drug discovery stage1. In our method, we have utilized the time saving techniques of cassette analysis and column switching to yield a robust process that reduces data turnaround time. This method has been optimized for the following six probes to determine IC50 shift values for inhibition of the five major CYPs: 1A2 (phenacetin), 2C9 (S-warfarin), 2C19 (S-mephenytoin), 2D6 (dextromethorphan), 3A4/5 (testosterone), and 3A4/5 (midazolam). The pre-incubation plate contains human liver microsomes (HLM), NADPH or buffer, and test compound at four concentrations, 1, 10, 50, and 100 uM, plus a vehicle control (no test compound; 100% activity). After 30 min, the incubation is then diluted 10X with NADPH and probe substrates and incubated for an additional 10, 30, or 40 min depending on the probe substrate. Samples are analyzed by mass spectrometry and TDI IC50 (termed TDI IC50 to differentiate from IC50 generated in reversible CYP inhibition assays) and percent change in AUC between data with and without NADPH in the pre-incubation are reported. Here we describe our findings following incubation of positive controls in HLM in a 384-well plate format. A significant analysis time savings was achieved by pooling six samples (eventually eight) with different probes to minimize the number of injections and using a column switching system to analyze the samples. Both percent change in AUC and TDI IC50 following pre-incubation correlate with kinact/KI ratio generated in traditional TDI kinetic evaluations allowing preliminary prediction of the magnitude of DDI for discovery compounds. Future experiments will include 2B6 (bupropion) and 2C8 (paclitaxel/amodiaquine/in-house compound).
References
- Obach RS. et al, (2007) Mechanism-Based Inactivation of Human Cytochrome P450 Enzymes and the Prediction of Drug-Drug Interactions. Drug Metab Dispos 35: 246-255.
238. Time-dependent inhibition (TDI) of human cytochromes P450: Evaluation of screening methods utilizing rapidfire® MS technology
Vaughn P. Miller1, Elke S. Perloff2, Andrew K. Mason2, Shangara S. Dehal2, Andrew P. Blanchard2, David M. Stresser2, Charles L. Crespi2, Can C. Ozbal1, and William A. LaMarr1
1BioTrove, Woburn, MA, USA, 01801
2BD GentestSM Contract Research Services, Woburn, MA, USA, 01801
Recently there has been a greater recognition of the importance of screening for time-dependent inhibition (TDI) of human cytochrome P450s earlier in drug discovery. This has lead to the demand for higher throughput analysis options for this assay. The objective of this study was to evaluate single vs multi-concentration methods for in vitro TDI screening and compare the results from RapidFire® LC/MS/MS technology to traditional LC/MS/MS analysis. IC50 curve shift assays were performed on a range of inhibitors in individual incubations of human liver microsomes using FDA recommended drug probe substrates and previously validated methods [Perloff et al. (2009), Xenobiotica, 39:99]. IC50 values for CYP3A4, CYP1A2, CYP2D6, and CYP2C9 were determined. Samples were analyzed individually by RapidFire MS technology as well as by traditional LC/MS/MS methods. The RapidFire technology provided a >20-fold decrease in analysis time compared to traditional LC/MS/MS methods. The utility of using a single inhibitor concentration (e.g. 10 uM or 50 uM) with a 30 min preincubation time was analyzed within the IC50 curve data. Significant changes in inhibition potential as a result of the preincubation were detectable regardless of experimental design. IC50 values from the RapidFire LC/MS/MS technology were comparable to traditional LC/MS/MS methods. The data in this study suggests that a single inhibitor concentration provides relevant outcomes for initial screening, however for some inhibitors it would be difficult to distinguish between direct inhibition and TDI. As expected, full IC50 shift determinations improve assay robustness, but increase sample load. However, the >20-fold decrease in assay analysis times using the RapidFire system may offset concerns with the turnaround time required for full inhibition curves for this important ADMET screen.
239. Rapid screening of LC-MS/MS data to identify drug metabolites using meteor metabolite prediction software
John W. Firth1, Lee Boyling1, Gareth L. Shackleton1, and Sian Ives2
1Gmpk, Sanofi-Aventis, Alnwick, United Kingdom, NE66 1UA
2Knowledge Base, Lhasa Limited, Leeds, United Kingdom, LS2 9HD
In February 2008, the US Food and Drug Administration issued a guidance document on the safety testing of drug metabolites. According to this guidance, human metabolites representing more than 10% plasma exposure of the parent compound at steady state are considered relevant, and of safety concern if they are exposed more highly in humans than in animals used in toxicity assessment. A metabolite meeting these criteria should undergo additional safety testing prior to the conduct of large scale clinical trials, potentially delaying drug development. The early identification of metabolites to avoid such delays has therefore become a priority. Many vendor supplied LC/MS data analysis tools, contain list of phase I and II metabolites, but they are not predictive and manual selection of metabolites is required by the user. In-silico metabolism expert prediction system (e.g. METEOR) offers advantages of predicting more likely metabolites and are compatible with these analysis tools. Examples will be presented how a variety of approaches can be applied sequentially to filter large LC-MS data sets to facilitate metabolite identification. Such approaches include a simple manual selection of metabolite transformations (MH+16, MH-14, etc.); specific mass spectrometry metabolism packages (e.g. MASSLYNX, METABOLYNX) which include a “pick-list” of metabolites and expert metabolite prediction software (e.g. METEOR).
240. In Silico prediction of biotransformation pathways: Amines and related compounds that generate isocyanates and nitrile oxides
Ernest Murray, Mohammed A Ali, and Anthony Long
Knowledge Base, Lhasa Limited, Leeds, United Kingdom, LS2 9HD
There is increasing interest in metabolic bioactivation pathways that lead to the formation of intermediates that are associated with the expression of toxicological endpoints or the observation of idiosyncratic adverse drug reactions. To be of value as a tool which informs the chemical basis for the prediction of such intermediates, an expert system for the in silico prediction of mammalian xenobiotic pathways should, at least, contain knowledge about all such bioactivation processes as are known in the literature. This paper highlights the formalisation and implementation of biotransformations regarding amines and related compounds and also demonstrates the added value that knowledge of electrophilic intermediates brings to the prediction of metabolism. Candidate biotransformations were identified by reviewing the literature. Benzylamines, N-nitrosoureas and N-amido sulphonamides all form glutathione adducts and, for all three compound classes, evidence exists for the involvement of electrophilic isocyanate or nitrile oxide intermediates. Three structure-metabolism relationships were developed and implemented as biotransformations in an expert system. A comparison between the new and a previous version of our biotransformation dictionary was carried out using the following test compounds: benzylamine, DPC-423, fotemustine and tolbutamide. Three new entries were registered in the biotransformation dictionary: conjugation of benzylamines with glutathione, glutathione addition to N-amido sulphonamides and glutathione conjugation of N-nitrosoureas. In all three cases, the test compounds, above, activated one or more new biotransformations in the updated dictionary and highlighted the potential adduct-forming intermediates in the predicted pathways. Structures of predicted intermediates and metabolites were consistent with those reported in the literature. Metabolic intermediates, significant in mechanisms of toxicity, are often present transiently, at low concentrations and can be difficult to detect. The results of this work suggest that sufficient evidence exists in the literature to support the inclusion of intermediates in a predictive knowledge base, at least for some compound classes. This can be of use in understanding phenomena such as dose-related toxicity, as in the case of tolbutamide.
241. In silico prediction of biotransformation pathways: Furans and thiophenes that generate epoxides, aldehydes and S-Oxides
Ernest Murray, Mohammed A Ali, and Anthony Long
Knowledge Base, Lhasa Limited, Leeds, United Kingdom, LS2 9HD
There is increasing interest in metabolic bioactivation pathways that lead to the formation of intermediates that are associated with the expression of toxicological endpoints or the observation of idiosyncratic adverse drug reactions. To be of value as a tool which informs the chemical basis for the prediction of such intermediates, an expert system for the in silico prediction of mammalian xenobiotic pathways should, at least, contain knowledge about all such bioactivation processes as are known in the literature. This paper will illustrate the development of four biotransformations that express bioactivation pathways of thiophenes and furans. Biotransformation mechanisms were identified and confirmed by literature review. Both thiophenes and furans undergo ring opening and conjugation with glutathione. For both compound classes evidence exists for the involvement of aldehydes, epoxides and, for thiophenes, thiophene-S-oxides. Three structure-metabolism relationships (SMR) were reviewed and their corresponding implementations in an expert system were updated. One new SMR was also developed and implemented. Three test compounds: 2-phenylthiophene, (R)-(+)-menthofuran and tienilic acid were evaluated against the updated biotransformation dictionary. In all three cases, the test compounds activated one or more of the new or updated biotransformations (glutathione conjugation of furans, S-oxidation and glutathione conjugation of thiophenes, ring opening of furans and thiophenes, and glutathione conjugation of thiophenes) and in all cases the intermediates and metabolites in the predicted pathways were consistent with those reported in the literature. Heteroaromatic structures are often used as bioisosteres in drug candidates and are therefore of great interest to medicinal chemists. Intermediates present during the metabolism of such compounds are often present transiently, at low concentrations and are, therefore, difficult to detect. Early evaluation of such metabolic liabilities in the drug development process remains a priority as a low rate of attrition at later stages is desirable. The results of this work suggest that sufficient evidence may exist in the literature to support the inclusion of intermediates in a predictive knowledge base for many classes of heteroaromatic compounds.
242. Comparison of metasite and stardrop prediction of CYP3A4, CYP2C9 and CYP2D6
V. Sashi Gopaul, Young Shin, Hoa Le, Matthew Baumgardner, Cornelis Hop, and Cyrus Khojasteh
Drug Metabolism & Pharmacokinetics, Genentec, Inc, South San Francisco, CA, USA, 94080
Metabolite identification studies play an important role in determining the sites of metabolic liability of new chemical entities (NCEs) in drug discovery. However, generating these complex and detailed studies in a highthroughput environment is often a challenge. Therefore, the use of in silico tools that can predict the sites of metabolism of an NCE could enhance the drug design process. In this study we compare the utility of MetaSite and Stardrop, two predictive softwares available for this purpose. MetaSite is a predictive software for the identification of regioselectivity of metabolism by major P450 isoforms. StarDrop is a data mining software that includes an in silico modeling feature to predict the regioselectivity and site of metabolism by CYP3A4, CYP2D6 and CYP2C9 only. Neither software can predict non-P450 catalyzed metabolism nor the rates of metabolism. Our objective was to evaluate the accuracy of MetaSite and StarDrop to predict the site of oxidation by CYP3A4, CYP2D6 and CYP2C9. Altogether, 12 substrates of CYP3A4, 9 substrates of CYP2C9 and CYP2D6 were each analyzed by each software and the results were comparedFootnote1. To measure the degree of prediction by each software, we assigned 3 points if the first major metabolite reported is predicted correctly, 2 points for the second choice and one point for the 3rd choice. No points were given for the 4th choice and beyond. The total points assigned for each enzyme experimentally were compared as a percentage of the total points assigned theoretically for a first choice prediction for all substrates for each enzyme. Our results show that MetaSite and StarDrop are similar in predicting the correct site of metabolism for CYP3A4 (86% vs 83%). StarDrop appears to do better in predicting the correct site of metabolism by CYP2C9 and CYP2D6 metabolism (89% and 93%, respectively) compared to MetaSite (59% and 70%, respectively). We are currently assessing the accuracy of MetaSite and StarDrop predictions of NCEs with in-house experimental observations.
243. GALAS modeling methodology applications in the prediction of the drug safety related properties
Andrius Sazonovas, Remigijus Didziapetris, Justas Dapkunas, Liutauras Juska, and Pranas Japertas
ACD/Labs, Inc., Vilnius, Lithuania, LT-08117
Early computational evaluation of drug candidate properties related to its pharmaceutical safety is becoming increasingly important in the drug discovery process. Yet the effective use of any available third-party predictive algorithms for these properties in the pharmaceutical industry is severely hindered by a number of problems. E.g. the training set rarely covers the specific part of the chemical space occupied by the compounds that a certain company is working with or a specific experimental protocol is used to measure the corresponding properties or activities ‘in house’. Therefore the need arises for a method that would allow any company to tailor a third-party predictive algorithm to its specific needs using proprietary ‘in house’ data. Here we present a novel GALAS (Global, Adjusted Locally According to Similarity) modeling methodology that provides a possibility for a researcher to expand the Applicability Domain of the resulting models with the help of a custom database of experimental values for the property of interest. A Reliability Index (RI) is also calculated as a measure of the quality of the particular prediction. The use of the method is illustrated with examples of its application in predicting CYP3A4 and hERG inhibition which figure among the major factors attributing to the rising attrition rate, being responsible for the various unwanted drug-drug interactions and cardiotoxicity respectively. It is shown that a relatively small amount (5 to 10) of similar compounds has to be added to substantially improve the prediction for a group of problematic compounds that is not represented in the original training set. Similarly the models are shown to be able to utilize ‘in house’ data obtained using different protocol compared the experimental training set data. Most importantly all of the above benefits are obtained without time consuming statistical retraining of the initial GALAS models.
244. Guiding the decision-making process to identify high quality compounds
Matthew D. Segall, Edmund Champness, Olga Obrezanova, and Christopher Leeding
Optibrium Ltd., Cambridge, United Kingdom, CB25 9TL
Key decisions in drug discovery involve the design and selection of compounds with an appropriate balance of many properties and hence a high chance of achieving the therapeutic goals of a project. In early drug discovery, data on a large number of compounds and properties from a wide variety of sources, in silico and in vitro, must be integrated and assessed against a project’s requirements. We have previously described an approach for scoring compounds, based on their likelihood of success against a profile defining the property requirements and their relative importance to a project’s objectives [1,2]. Within this assessment, uncertainty in the data is explicitly considered; most sources of drug discovery data have significant statistical or experimental uncertainties and we should consider the impact on our ability to confidently choose between different compounds. Defining a property profile often leads to lengthy (and interdisciplinary) discussions about the criteria and their relevance. For example, is it worth sacrificing some potency to gain additional metabolic stability or solubility? However, a question that is rarely asked is, “What impact does making that sacrifice have on the final outcome?”, particularly given the underlying uncertainty. A rigorous scoring approach allows this question to be addressed directly. By assessing the sensitivity of the ultimate compound selection to the property profile, criteria that have a significant effect on the ultimate decision can be identified. This, in turn, can focus attention on critical experiments, e.g. in vivo studies, that will help to identify the most appropriate profile to select high quality compounds with greater accuracy.
References:
- M. D. Segall, A. P. Beresford, J. M. R. Gola, D. Hawksley, M. H. Tarbit, “Focus on Success: Using in silico optimisation to achieve an optimal balance of properties” Expert Opin. Drug Metab. Toxicol. 2006, 2, p. 325
- M. D. Segall, E. Champness, O. Obrezanova, C. Leeding, “Beyond Profiling: Using ADMET models to guide decisions” Chemistry and Biodiversity 2009 (in press)
245. Structure - brain exposure relationships in the rat using a novel dataset of the unbound brain-to-plasma ratio, Kp,uu,brain
Markus Fridén1, Susanne Winiwarter2, Ola Bengtsson3, Gunilla Jerndal2, Hong Wan4, Ulf Bredberg2, Margareta Hammarlund-Udenaes1, and Madeleine Antonsson2
1Department of Pharmaceutical Biosciences, Division of Pharmacokinetics and Drug Therapy, Uppsala University, Uppsala, Sweden
2Discovery DMPK, AstraZeneca R&D Mölndal, Mölndal, Sweden, 431 83
3AstraZeneca R&D Lund, Lund, Sweden, 431 83
4Lead Generation, AstraZeneca R&D Mölndal, Mäölndal, Sweden, 431 83
Purpose: Current understanding of structure-brain exposure relationships is largely based on measurements of the total brain-to-plasma ratio known as logBB or Kp,brain. This study evaluates the relationship between compound structure and the unbound brain-to-plasma ratio, Kp,uu,brain, being the pharmacologically relevant measure of brain exposure.
Methods: Forty-three drugs were selected to represent the chemical drug space. Kp,uu,brain was measured in the rat by combining total brain, plasma and cerebrospinal fluid (CSF) concentrations with the unbound fraction in plasma and brain slice estimates of brain tissue binding. Due to its long history of use Kp,brain (logBB) was calculated for comparison. Structure-brain exposure relationships were investigated with 16 common molecular descriptors and multivariate data-analysis using Projection to Latent Structures (PLS) with SIMCA-P+.
Results: There was no relationship between Kp,uu,brain and lipophilicity or ion class (acid/base). Kp,uu,brain was best predicted by descriptors related to hydrogen bonding. The predictivity was however modest: only 23 out of 41 compounds were predicted within a 3-fold range of experimental data. Descriptors of hydrogen bonding was also important for prediction of LogBB, but unlike Kp,uu,brain, logBB was also correlated with lipophilicity and dependent on the ion class. This was explained by lipophilicity and basicity driving brain tissue binding and acidity driving binding to plasma albumin.
Conclusion: The use of prediction models based on data of logBB in lead optimization is strongly discouraged since this is likely to result in the design of unnecessarily lipophilic and basic compounds. As would be expected from active transport determining brain exposure, it is difficult to predict Kp,uu,brain. Nevertheless, hydrogen bonding potential influences Kp,uu,brain by promoting the interactions with efflux transporters and reducing passive permeability. As a rule of thumb, addition or removal of 2 hydrogen bond acceptors results in a 2-fold reduction and increase, respectively, in Kp,uu,brain. Proportional (2-fold) changes in dose requirements of centrally acring drugs are expected, as are the changes in therapeutic window of peripherally acting drugs having critical CNS side effects.
246. Optimization of the pore-forming peptide alamethicin in UGT incubations at low human microsomal protein concentrations
Robert L. Walsky
Pharmacokinetics Dynamics & Metabolism, Pfizer Global R & D, Groton, CT, USA, 06340
The UDP-glucuronosyltransferases (UGTs) represent an important superfamily of membrane-bound enzymes responsible for the phase II glucuronidation of many drugs. In an effort to increase enzyme activity during the in vitro assessment of UGT-mediated glucuronidation in human liver microsomes, the pore forming peptide, alamethicin is frequently added at a specific ratio of 50 μg/mg microsomal protein. However, sensitive analytical tools, such as LC/MS/MS, now allow very low microsomal protein concentrations to be utilized, particularly for inhibition assessments, and we have found limited increases in activity following alamethicin preincubation. By preincubating human liver microsomes (0.01 – 0.5 mg/mL) at multiple alamethicin concentrations and evaluating estradiol-glucuronosyltransferase (UGT1A1), trifluoperazine-glucuronosyltransferase (UGT1A4), and AZT-glucuronosyltransferase (UGT2B7) activities, we found that a solution concentration of 10 μg/mL was optimal for achieving maximal enzyme activity regardless of microsomal protein concentration. Furthermore, at low microsomal protein concentrations (<0.1 mg/mL), adding alamethicin at a ratio of 50 μg/mg provided little or no benefit.
247. A solution to predict drug candidate entrance to the brain based on a physiologic primary Blood-Brain Barrier in vitro model
Nicolas Perrière
VigiCell, Villejuif, France, 94800
Context: The Blood-Brain Barrier (BBB) is a complex endothelium which protects the brain from toxic compounds. It also restricts CNS drug candidates entrance to the brain. A relevant in vitro BBB model is required for early screening of drug candidates in development. Such a model should present identical properties to the in vivo brain endothelium which is characterized by: Endothelial cell monolayer (thickness of the cells, membrane lipid composition and density) with specific passive diffusion properties; The expression of tight junction complexes which limit the paracellular pathway; The expression of functional polarized specific influx and efflux transporters, particularly P-gp and Bcrp, described as prominent hindrance to brain entrance.
Aims: The aims were to develop and characterize a RAT PRIMARY model constituted of a syngenic co-culture of selected brain endothelial cells and astrocytes maintaining the specifications of the BBB.
Results: First, we characterized this model in terms of biological properties, focusing on the preservation of brain endothelial specificities:
We validated a standardized method of positive selection in order to obtain pure capillary rat brain endothelial cells (RBECs) with the BBB phenotype.
RBECs showed expression of functional tight junction complexes similar to those described in vivo, conferring low permeability coefficients to small hydrophilic compounds (i.e. 0.12 10− 3 cm/min for fluorescein).
RBECs displayed expression of functional efflux transporters (P-gp, Bcrp), correlated with a preferential apical localization, as observed for example with daunorubicine, showing an efflux ratio (B to A vs A to B) value of 11, partially reversed by PSC833 treatment.
We obtained high correlations between in vitro permeabilities and in vivo data collected using the in situ brain perfusion technique, on a panel of compounds known to enter the brain with different mechanisms (pure passive diffusion, paracellular pathway, via Influx or Efflux transporters, and mixed mechanisms).
Second, we validated this model in term of reproducibility and reactivity for industrial service purposes:
We developed standardized protocols for the endothelial cells isolation and co-culture, in order to minimize cell culture intra- and inter-variability.
We validate the monolayer integrity of each insert with an internal control, as well as each culture with a separate P-gp functionality test.
We obtained very low standard deviations over several months (i.e. PAB[fluorescein] = 0.12 ± 0.02 10− 3 cm/min, n=40).
Lastly, service-wise, we commit to deliver permeability experiments within 8 days after reception of the drug candidate.
Conclusion: This in vitro BBB model presents all the expected in vivo characteristics. As such, it allows an early permeability prediction of drug candidate through the BBB combining passive diffusion and active or facilitated transport. Data obtained from this model constitute a strong base to estimate the global bioavailability of a drug candidate into the brain parenchyma without the constraints of in vivo experimentations. Implementation of this in vitro model is also compatible with early stages of drug development (screening).
248. An In-vitro equilibrium dialysis technique for simultaneous protein binding assessment of multiple tissues vs. plasma
Cindy Xia1, Mark G. Qian1, Susan Chen1, Tai-Nang Huang2, and Frank W. Lee1
1Drug Metabolism and Pharmacokinetics, Millennium Pharmaceuticals, The Takeda Oncology Company, Cambridge, MA, USA, 02139
2Dept of DMPK, Linden Bioscience, Cary, NC, USA, 27513
In drug discovery, it is desirable to have a balanced tissue distribution profile for lead compounds, higher uptake into the targeted tissue (e.g., solid tumor) and low accumulation in sensitive organs (e.g., liver). Regular in-vivo pharmacokinetic studies can only provide a convoluted assessment of the property based on the parameter of the volume of distribution. To evaluate the distribution in specific tissues, individual tissue samples must be collected, homogenized, and analyzed. The processes involved are tedious, costly, and at best only suited for few final lead compounds. It is a widely accepted concept that the drug distributes into various tissues through plasma circulation. At steady state, drug in plasma reaches partition equilibrium with tissues. The free concentration in the tissue interstitial space equals to the free concentration in plasma. The total tissue concentration, however, could be different depending on the binding property of the compound with tissue proteins, membrane efflux, and uptake transporters. Based on this theory, a multiplex equilibrium dialysis device was constructed for simultaneous assessment of the drug distribution at steady state between multiple tissue homogenates and the corresponding plasma spiked with the compound of interest. This presentation will provide results that demonstrate the capability of the simple and cost-effective technique for differentiating the drug binding characteristics of different tissue homogenates against plasma proteins. Various tissues from multiple species were tested. The results generated using the novel technique are comparable to those from the in-vivo assay.
249. Highly bound drugs may need longer dialysis times to achieve equilibrium in plasma protein binding assays
Adrian Sheldon, Angela Shen, Tuyen Nguyen, and Xin Zhang
Agilux Laboratories, Worcester, MA, USA, 01605
Binding of drugs to plasma proteins is often measured using either equilibrium dialysis or ultracentrifugation approaches. Using the equilibrium dialysis method, accurate measurement of % protein bound depends on equilibrium being achieved across the dialysis membrane. If equilibrium is not reached in the assay system, the drug concentration in the donor (buffer) compartment is under-estimated and the calculated % protein bound value is thus overestimated. Using multiple drug compounds, we investigated whether equilibrium was achieved following dialysis for 6 and 24 hours. Human K3EDTA plasma was spiked with 2 μM drug and dialyzed against blank plasma for 6 or 24 hours at 37°C with shaking using Thermo R.E.D. inserts. The drug concentrations in the donor and receiver compartments were then measured using LC/MS/MS. If equilibrium is achieved, the ratio of these concentrations should be 1. Compounds with moderate binding, such as propranolol (approx. 80% bound), demonstrated equilibrium ratio (ER) values of 1.1, confirming that equilibrium was reached. However, we observed that the ERs for several drugs were >1, and that the ER values increased as the % binding increased. For example, warfarin was 99% bound with an ER value of 10.3 at 24 hours, and naproxen showed 100% binding with an ER of 61.2 at 24 hours. The ER values at 24 hours were consistently lower than those at 6 hours, demonstrating that equilibrium was promoted with longer dialysis times. We conclude that longer dialysis times (e.g., 24 hours or more) may be needed for highly bound compounds, and suggest that controls should be included in plasma protein binding assays to confirm acceptable equilibrium and plasma stability under the assay conditions.
250. Quantity production of metabolites using hepaRG human hepatic cells
Christophe Chesne1, David Steen1, Katherine Tsaioun2, Robert Annand3, Mary Jacewicz3, and Fabrice Guillet4
1R & D, Biopredic international, Rennes, France, 35000
2Apredica, Watertwon, MA, USA, 02472
3Cell Biology, Apredica, Watertown, MA, USA, 02472
4Metabolism, Xenoblis, Saint-Gregoire, France
Limited availability of a metabolite to perform structural, toxicological, or pharmacological evaluation can be a drug development bottleneck. When chemical synthesis is impractical, several biological systems can be considered for metabolite production, including recombined or native systems (microsomes), perfused liver, recombined cells including yeasts, E.coli and mammalian cells, and hepatocytes. Many factors affect the decision including production capacity, ease-of-implementation, concentration of metabolite in supernatant, and the nature of the supernatant in facilitating isolation of the metabolite. We hypothesized that HepaRG human hepatic cells could be useful for such production; differentiated HepaRG cells are equipped with functional CYPS and other metabolic enzymes at levels that generally resemble, and occasionally exceed, those in primary human hepatocytes. Reports have explored their use as a multi-purpose cell to study metabolism, induction, inhibition, toxicity, and transport function, but thus far no published study explored an expansion of their use into metabolite production. We studied this, using as one probe substrate 200um of nifedipine, a high clearance, highly-soluble, CYP3A4 substrate with minimal toxicity, which is transformed into oxidized nifedipine. We additonally used 20uM of paclitaxel, a low clearance compound transformed into 6-alpha-OH paclitaxel; production of the paclitaxel metabolite presents a challenge since it relies on CYP2C8 for its biotransformation. Differentiated HepaRG cells were maintained in 75cm2 flasks with culture medium renewed every two days; Rifampicin was added to one to increase the 2C8 activity and resulting production of oxidized paclitaxel. The resulting concentration of those nifedpine and paclitaxel metabolites exceeded 50uM and 1uM respectively, and after two weeks of incubation, more than 1mg of oxidized nifedpine and 100ug of OH paclitaxel were produced. We conclude that HepaRG cells can be a practical and efficient system for production of human metabolites.
Reference:
- Kanebratt, K.P. and T.B. Andersson (2008). “Evaluation of HepaRG Cells as an In Vitro Model for Human Drug Metabolism Studies.” Drug Metabolism and Disposition 36(7): 1444–52.
251. The application of 3D alginate scaffolds for liver tissue modeling
Alexandria V. Sams, Zhensheng Li, and Mark J. Powers
Primary & Stem Cell Systems, Invitrogen Corporation, Frederick, MD, USA, 21704
We have developed a well-defined porous alginate matrix (AlgiMatrix™) to establish a 3D cell culture environment that facilitates the formation of more in vivo-like tissue structures. The addition of divalent cationic salts (e.g. calcium, barium) to a sodium alginate solution produces a highly porous (>90%) and highly interconnected biocompatible scaffold. Using multiple cell types including C3A human hepatocyte cell line, primary hepatocytes, C6 glioma cell line, and mesenchymal stem cells we have shown that the matrix pores (50 - 150μm diameter) act as localized compartments in which cells form reproducible spheroidal aggregates of similar diameter. Non-enzymatic spheroid recovery is simplified by the addition of a Dissolution Buffer that dissolves the alginate matrix within a few minutes while the cell structures remain intact. Hepatocellular spheroids are of particular interest due to the observation that such structures provide for more physiologically relevant performance. Phase contrast microscopy revealed that hepatocyte spheroid formation is faster and more efficient in AlgiMatrix™ cultures compared to other currently available platforms (e.g. low bind polystyrene). Live/dead staining and intracellular ATP levels confirmed that the AlgiMatrix™ cultures retain a high viability through at least 14 days in culture. Cytochrome P450 enzyme (e.g. CYP1A) activity was assessed using ethoxyresorufin substrate and commercially-available luminescent kits. AlgiMatrix™ culture of hepatocytes (e.g. primary rat, C3A) demonstrated significantly increased enzyme activity at both early and late time points compared to 2D culture. Additional data demonstrating the recapitulation of numerous morphological and/or functional features indicates that AlgiMatrix™ is a platform for creating more physiologically relevant models. We conclude that the 6-well, 24-well, and 96-well AlgiMatrix™ format represents a powerful new predictive tool in xenobiotic metabolism research.
252. The mining of ADME data from a toxicity perspective
Jianhua Liu1, Christopher L. Shaffer2, and Dennis O. Scott1
1PDM, ADME Science and Technology, Pfizer Inc, Groton, CT, USA, 06340
2PDM/Neuroscience, Pfizer Inc, Groton, CT, USA, 06340
Toxicology and DMPK have great overlaps in the pharmaceutical research and development field. Drug metabolism concepts and techniques are often used to investigate the bioactivation of compounds and the mechanism(s) of toxicities. Toxicokinetics is the application of pharmacokinetics to determine in experimental animals the relationship between the systemic exposure of a compound and toxicity. In this presentation, the in vitro IC50 of transformed human liver epithelial cell viability (THLE cell ATP depletion) assay was chosen as a toxicology indicator to investigate the possible relationship between compounds’ ADME properties and their in vitro cell toxicities. All data utilized in the presentation were retrieved from our proprietary HTS database. The results indicate that compounds with high hepatic clearance and/or low free fraction of human liver microsomal protein binding are more likely to have lower cell viability IC50s. Although compounds that tested “positive” in GSH reactive metabolite screening resulted in more cell damage than those deemed “negative” compounds, certain reactive metabolite bioactivation pathways did decrease cell viability IC50s more than the others. These results demonstrated that compounds with better ADME properties (as defined by CLint, reactive metabolite liability, permeability and P-gp liability) reduce the likelihood of cell damage in cell viability studies.
253. Halothane-induced liver injury is mediated by interleukin-17 in Mice
Tsuyoshi Yokoi, Eisuke Kobayashi, Tatsuki Fukami, and Miki Nakajima
Graduate School of Medical Science, Kanazawa University, Kanazawa, Japan, 920-1192
Drug-induced liver injury is a major problem in drug development and clinical drug therapy. In most cases the mechanisms are still unknown, thus it is difficult to predict or prevent these reactions. It has been known that halothane, a widely used inhalation anesthetics, induced mild and severe liver injury. To investigate the mechanisms of halothane-induced liver injury, we used recently established liver injury mice model. We observed the increase of plasma interleukin-17 (IL-17) level and hepatic macrophage inflammatory protein (MIP-2) expression in halothane-administrated BALB/c mice. Neutralization of IL-17 prevented hepatotoxic effect of halothane. Administration of recombinant IL-17 (1 μg/mouse, single ip) to the halothane-treated mice resulted the remarkable increase of alanine aminotransferase and aspartate aminotransferase. In conclusion, marked increase of hepatic MIP-2 expression and the following increase of IL-17 would be responsible for halothane-induced liver injury. This is the first study to demonstrate that IL-17 involved in drug-induced liver injury.
254. A zone classification system for risk assessment of idiosyncratic drug toxicity using daily dose and covalent binding
Shintaro Nakayama1, Ryo Atsumi2, Hideo Takakusa1, Yoshimasa Kobayashi1, Atsushi Kurihara1, Yoko Nagai1, Daisuke Nakai1, and Osamu Okazaki1
1Drug Metabolism and Pharmacokinetics Research Laboratories, Daiichi Sankyo Co., Ltd, Tokyo, Japan
2Medicinal Safety Research Laboratories, Daiichi Sankyo Co., Ltd, Shizuoka, Japan
The risk of idiosyncratic drug toxicity (IDT) is of great concern to the pharmaceutical industry. Current hypotheses based on retrospective studies suggest that the occurrence of IDT is related to covalent binding and daily dose. We determined the covalent binding of 42 radiolabeled drugs in three test systems-human liver microsomes and hepatocytes in vitro and rat liver in vivo-to assess the risk of IDT. On the basis of safety profiles given in official documentation, tested drugs were classified into the safety categories of safe, warning, black box warning, and withdrawn. The covalent binding in each of the three test systems did not distinguish the safety categories clearly. However, when the log-normalized covalent binding was plotted against the log-normalized daily dose, the distribution of the plot in the safety categories became clear. An ordinal logistic regression analysis indicated that both covalent binding and daily dose were significantly correlated with safety category, and that covalent binding in hepatocytes was the best predictor among the three systems. When two separation lines were drawn on the correlation graph between covalent binding in human hepatocytes and daily dose by a regression analysis in order to create three zones, 30 of 37 tested drugs were located in zones corresponding to their respective classified safety categories. In conclusion, we established a zone classification system using covalent binding in human hepatocytes and daily dose for the risk assessment of IDT.
255. Novel fluorescent assays to identify adverse responses to sub-cellular compartmentalization of pre-clinical drug candidates using microplate readers
Yue Jun Xiang1, Jack Coleman1, Zaiguo Li1, Nyaya Kelkar1, Praveen Pande1, Dee Shen1, JoAnne Schultz1, Paul Held2, Peter Banks2, and Wayne F. Patton1
1R&D, Enzo Life Sciences, Farmingdale, NY, USA, 11735
2R&D, BioTek Instruments, Winooski, VT, USA, 05404
Cationic drugs often exhibit large apparent volumes of distribution, which is consistent with various forms of sequestration within cells and tissues. This form of drug uptake can occur in intact cell types that are not necessarily specialized for the handling of xenobiotics. The drugs are sequestered into cells by a variety of metabolically-driven, but receptor-independent means including mitochondrial membrane potential-driven concentration, nuclear concentration via DNA affinity and vacuolar-ATPase-driven trapping into lysosomes that subsequently swell by an osmotic mechanism. Drug concentration within cells may arise from the innate membrane permeability of the uncharged forms of these drugs, as well as by means of specific transporters (organic cation transporters, choline transporters, etcetera). Such drug sequestration could contribute to overall toxicity, as well as prolong the duration of drug action. A panel of fluorescence-based assays was devised to assess the impact of xenobiotics on overall cell function, with particular emphasis on the lysosomal, mitochondrial and nuclear compartments. The semi-automated 96-well cell-based assay workflow presented provides a rapid and quantitative high-throughput approach for determining drug- or toxic agent-induced live cell response, offering throughput advantages relative to methods based upon electron microscopy, fluorescence microscopy or flow cytometry. Early secondary screening of candidate drugs for potentially adverse cell activity in the drug discovery phase could predict later risks in drug development arising from drug safety issues. Such a screening approach could aid in selecting the best candidate compounds for further drug development efforts, as well as provide preliminary benchmarking of dosing limits in preclinical toxicity studies.
256. Risk assessment of drug-induced liver injury based on in vitro covalent binding to human liver proteins
Toru Usui1, Masashi Mise1, Takanori Hashizume1, Masashi Yabuki1, and Setsuko Komuro2
1Pharmacokinetics Research Laboratories, Dainippon Sumitomo Pharma Co.,Ltd., Osaka, Japan, 554-0022
2Pharmacokinetics Research Laboratories, Dainippon Sumitomo Pharma Co.,Ltd., Osaka, Japan
At present, prediction of idiosyncratic drug-induced liver injury (DILI) is difficult and the underlying mechanisms are not fully understood. However, the trigger of DILI is considered to be formation of reactive metabolites and sequential covalent binding (CB) to cellular macromolecules in the liver. The objective of this study was to clarify whether the risk of idiosyncratic DILI can be estimated by comparing CB levels between 12 positive and 12 negative compounds. The radio-labeled compounds were incubated with human liver microsomes and hepatocytes. To determine CB, the radioactivity covalently bound to proteins was measured by a method in which a glass filter was used to collect and wash the proteins. After incubation with microsomes in the presence of NADPH, there was large overlap in the distribution of CB amounts between the positive and negative groups. On addition of UDP glucuronic acid (UDPGA) as a cofactor for glucuronidation, the CB levels of bromfenac and diclofenac were increased. With addition of nucleophilic GSH, values for most compounds were decreased. However, separation of the two groups on the basis of CB could not be improved by addition of either UDPGA or GSH. CB with hepatocytes also failed to discriminate positive from negative compounds. Therefore, the CB level alone is not sufficient for risk assessment of DILI. Next, whether pharmacokinetic parameters (Cmax and daily dose) are useful for prediction of DILI was investigated. The CB amount multiplied by Cmax, which may reflect systemic exposure, did not improve the discrimination of the two groups. On the other hand, when the CB amount was multiplied by the maximum daily dose, which may reflect maximum hepatic exposure, the two groups did become discriminated. Taken together, these findings suggest that the combination of CB amount and daily dose can estimate the risk of idiosyncratic DILI and is very useful for risk assessment.
257. Measurement of reactive acyl glucuronide metabolites in in vitro and in vivo drug discovery systems
Anila Desai, Yueh-Tyng Chien, Wilmin Bartolini, and Robert Busby
Analytical Pharmacology / DMPK, Ironwood Pharmaceuticals, Cambridge, MA, USA, 02141
Small molecules with carboxylic acid moieties can readily form acyl glucuronides through phase II biotransformation. These acyl glucuronides can be unstable and re-arrange which can lead to covalent modification of proteins and possible regeneration of the parent compound in a futile cycle. The reactive acyl glucuronides can cause in vivo toxicity leading to a failure in the clinic or drug withdrawal from the market. For a series of carboxylic acid-containing compounds, acyl glucuronide formation was monitored and a qualitative measure of reactivity was assessed based on the amount of glucuronide formed and the observed migration of the acyl group. An in vitro system to generate phase I and phase II biotransformations was prepared using rat, dog and human liver microsomes. The presence of a glucuronide metabolite was monitored by LC/MS/MS analysis as well as its stability in the presence of plasma and microsomal proteins. Compounds with low glucuronide stability exhibited a signature pattern in the LC/MS/MS chromatogram. Lead compounds with low in vitro reactivity were dosed in rats for pharmacokinetic analysis. Follow up analysis for potential reactive metabolites were assessed by rate of clearance of the parent compound, the presence of an acyl glucuronide and the qualitative attributes of the glucuronide signal in the LC/MS/MS chromatogram. Compounds showing less glucuronide formation and less migration of the acyl group exhibited higher maximum tolerated doses in both the rat and the dog.
258. In Vitro biotransformation studies of nomifensine mono-hydroxylation
Jian Yu, Xin (Cindy) Shen, and Doug Burdette
Discovery DMPK, Astrazeneca Pharmaceuticals LP, Wilmington, DE, USA, 19850
Nomifensine, an effective antidepressant, was withdrawn from the market due to the increased incidence of hemolytic anemia, as well as kidney and liver toxicity. Bioactivation has been postulated as a potential mechanism for its toxicity. Our recent studies showed that multiple GSH adducts, with the incorporation of one oxygen atom and a GS moiety to the nomifensine molecule, were formed in the incubations of nomifensine with NADPH and GSH-supplemented human and animal liver microsomes. While modifications to both the aniline and the arene groups were identified, our previous studies could not provide additional insight into the regio chemistry of the arene C-linked GSH conjugates due to their mass fragmentation patterns. Reports of metabolite identification in humans in vivo revealed hydroxylation modifications only at the phenyl ring (Fig. 1, C ring). Also, the enzymes that catalyze the formation of the corresponding reactive intermediates remain unknown. Because one oxygen atom was introduced into the GSH conjugates, the structures of the mono-hydroxylated metabolites may provide critical clues to the reactive intermediates that were trapped by GSH. The present studies were conducted to elucidate the sites of arene mono-hydroxylation in human and rat liver microsomes supplemented with NADPH and to identify CYP isoenzymes potentially responsible for the formation of these metabolites using recombinant human cytochrome P450s. The structures of the mono-hydroxylated metabolites of nomifensine in liver microsomal incubations were characterized with respect to mass, elemental composition, and fragmentation pattern on LC/MS/MS. Our results indicated that multiple mono-hydroxylated metabolites were formed in human and rat liver microsomes. In addition to the reported modification of the C ring in nomifensine, we also detected A ring mono-hydroxylations, suggesting the formation of benzoquinone imines as a potential route of bioactivation. The CYP450 isozymes involved in the formation of each of these mono-hydroxylated metabolites were also identified. These findings provide new insights into the possible mechanisms of nomifensine bioactivation.
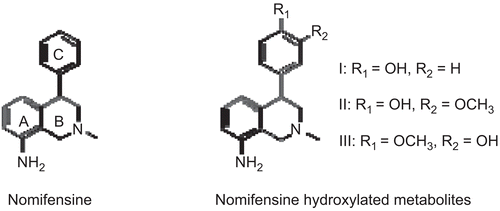
Reference:
- Yu et al., 2008, 15th North American regional ISSX meeting
259. Metabolism of glucokinase activators: Formation of a reactive metabolite responsible for hepatic lipidosis
Jagdish Racha1, Wanping Geng1, Michael Pignatello1, Zhenmin Liang1, Ramakanth Sarabu2, Joseph Grimsby3, and David Moore1
1Non Clinical Safety, Hoffmann-La Roche Inc., Nutley, NJ, USA, 07110
2Discovery Chemistry, Hoffmann-La Roche Inc., Nutley, NJ, USA, 07110
3Metabolic & Vascular Diseases, Hoffmann-La Roche Inc., Nutley, NJ, USA, 07110
Glucokinase (GK) activators represent a novel class of antidiabetic agents currently in development for the treatment of type 2 diabetes. GK activators improve dysglycemia by augmenting glucose stimulated insulin release and by suppressing hepatic glucose production. GK activators, Compound 1 and 2, are 1st generation candidates for development. Based on the in vitro, in vivo, and DMPK profiles, the team advanced these two compounds towards dose-range finding (DRF) studies in rats and dogs. Both compounds had low solubility, but high permeability, and good PK profiles in both rat and dog. Compound 1 in rat DRF study was found to cause necrosis of the liver, and was quickly abandoned. Compound 2 induced hepatic lipidosis in both rat and dog DRF studies which was reversible upon cessation of the treatment. In order to find out whether lipidosis was due to the structure-based toxicity or due to GK activation, the team investigated if the formation of a reactive metabolite from either the parent molecule or from its metabolite could be the causative agent. The metabolite profile of Compound 2 indicated formation of the corresponding thiourea via the oxidative ring opening of thiazole and formation of hydroxyl or keto metabolites related to the cyclopentyl ring. To eliminate for GK mechanism-based effects, the “S” stereoisomer of Compound 2, which has no ability to activate GK, was scaled up for use in toxicology studies. A 5-day rat toxicology study was conducted with the racemic thiourea metabolite and “S” isomer of Compound 2. Both compounds caused hepatic lipidosis even in the absence of GK activation. These findings strongly suggest that hepatic lipidosis is due to the formation of a thiourea metabolite of Compound 2.
260. Simultaneous detection and quantitation of iminium ions from bioactivation of compounds by radiolabeled chemical tagging prior to LC/dynamicflow ARC/MS analysis
Rongfang Fran Xu, Jose Silva, and Heng-Keang Lim
Biotransformation, DMPK, GPCD, Johnson & Johnson PRD, Raritan, NJ, USA, 08869
Toxicity is the leading cause of drug attrition in preclinical and clinical development. There has been considerable progress made in detection and structural elucidation of reactive metabolites trapped as glutathione and cyano conjugates by LC/MS analysis. However, there will be instances where both the toxicophore and pharmacophore are localized to the same region of the molecule and hence, making it difficult to design out the problematic functionality. Under such circumstance, the quantitative difference in the formation of the reactive metabolite can be used to differentiate between the drug candidates. We now report the simultaneous detection and quantitation of iminium ions from bioactivation of compounds using a mixture of native and 14C-labeled cyano conjugates by hyphenated LC with radioactivity detector and mass spectrometry (LC/DynamicFlow ARC/MS).
Typical incubation, 0.5-mL final total volume, consisted of 1 mg/mL human liver microsomes, 1 mM EDTA, 0.8 mM MgCl2, 10 mM drug (verapamil, proclorperazine, nefazodone, trozodone and aripiprazole), 0.35 NADP+, 0.8 mM glucose-6-phosphate, 2.5 U/mL glucose-6-phosphate dehydrogenase and KCN:K14CN in 0.1M phosphate buffer (pH 7.4). The mixture was incubated at 37 °C for 60 minutes and reaction terminated by centrifugation of CaCl2 precipitated microsomal proteins. The supernatant was loaded onto SPE cartridge and the excess KCN:K14CN was removed by repeated elution until radioactivity in eluant reached background level. The cyano adducts were eluted with 1 mL acetonitrile and evaporated to dryness prior to analysis. Cyano conjugates were formed with all compounds investigated. Two previously reported cyano conjugates of verapamil with m/z 480 and 466 were detected at 9.9 and 0.7 ng equivalent/mg protein. High-resolution accurate mass measurements provided confirmation provided evidence for trapping of the reactive N-methylene iminium ions from bioactivation of verapamil. Also, a previously reported cyano conjugate of procloperazine with m/z 405 was formed at 1.9 ng equivalent/mg protein. Interestingly, there was differentiation in the quantitative formation of iminium ions from bioactivation of nefazodone, trozodone and aripiprazole in the following order: nefazodone > trazodone > aripiprazole. A method for simultaneous elucidation of structure and quantitation of cyano conjugates by radiolabeled chemical tagging and LC/DynamicFlow have been developed.
261. Comparison of sprague dawley and dark agouti rats as animal models of perhexiline-induced hepatotoxicity
John Licari1, A A Somogyi1, John Pierides2, and Benedetta C. Sallustio3
1Discipline of Pharmacology, University of Adelaide, Adelaide, Australia
2Institute of Medical and Veterinary Science, Adelaide, Australia
3Dept of Clin Pharmacol, Queen Elizabeth Hosp, Adelaide, Australia
Perhexiline, 2-(2,2-dicyclohexylethyl)piperidine, a potent antianginal agent, improves myocardial function via inhibition of carnitine palmitoyltranferase-1, switching myocardial energy metabolism from fatty acid to carbohydrate utilization. At high concentrations, perhexiline causes hepatotoxicity, characterised by steatosis, phospholipidosis, and glycogen accumulation. Patients’ dosage is titrated to maintain plasma concentrations between 0.15-0.60 mg/L. Perhexiline is metabolised by CYP2D6 and poor metabolisers require significantly lower doses to minimise the risk of clinical toxicity. Sprague Dawley (SD) and Dark Agouti (DA) rats are animal models of CYP2D poor and extensive metabolism. This study investigated the use of SD and DA rats as models of perhexiline-induced hepatotoxicity. Animals (n=4 per group) were administered vehicle or 200 mg/kg perhexiline daily for 8 weeks, subsequently sacrificed and blood and livers collected. Plasma and liver concentrations of perhexiline and its metabolites were quantified by HPLC. Blood samples were also used for biochemical liver function assessments, and liver tissue morphology was analysed by electron microscopy. Within-strain comparisons were performed by 2-tailed, unpaired t-test. All SD rats attained plasma perhexiline concentrations <0.15 mg/L, whilst concentrations in DA rats ranged from 0.16-1.13 mg/L, consistent with lower CYP2D-catalysed metabolism. Similarly, hepatic perhexiline concentrations in SD were lower (0.9-8.2 ug/g) than in DA (9.4-54.7 ug/g). SD but not DA rats treated with perhexiline had higher mean±sem alkaline phosphatase (U/L) (146.8 ± 21.2) compared to controls (91.0 ± 4.9, p=0.04). DA rats treated with perhexiline had higher lactate dehydrogenase (U/L) (268.3 ± 29.2) compared to controls (137.0 ± 16.4, p=0.02). No effects of perhexiline were observed for other liver function tests. No marked changes in liver histology were observed, however, in DA rats there was evidence for increased glycogen storage with increasing plasma perhexiline concentrations (p=0.06). In this study, both strains appeared relatively resistant to perhexiline’s hepatotoxic effects, showing only mild changes in histology or liver function tests. However, the trend for increased glycogen storage in DA rats was consistent with clinical histology reports of perhexiline toxicity. Thus the DA rat may be a more suitable model for perhexiline-induced hepatotoxicity.
262. Genetic polymorphism of xenobiotic metabolizing enzymes and pesticide induced DNA damage
Satyender Singh1, Vivek Kumar2, Sachin Thakur1, B. D. Banerjee2, S.S Grover1, D. S. Rawat1, S. T Pasha3, S. K. Jain4, and Arvind Rai1
1Department of Biochemistry & Biotechnology, National Institute of Communicable Diseases, Delhi, India, 110054
2Department of Biochemistry, UCMS & GTB Hospital, Delhi, India, 110095
3Ministry of Health & Family Welfare, National Programme for Prevention and Control of Fluorosis, DGHS, New Delhi, India, 110011
4Department of Epedemiology, National Institute of Communicable Diseases, Delhi, India, 110054
Pesticides pose a clearly identifiable risk to those who are occupationally exposed to this carcinogen causing acute to chronic health impacts including various neoplastic diseases and congenital malformations. Studies have indicated that pesticides are metabolized by various phase I and phase II metabolizing enzymes which provide critical defence against carcinogen. Their polymorphism has been found to be associated with various forms of cancer. Therefore, it is important to identify the potential genetic susceptibility factors affecting individual responses to carcinogen exposure. Hence, the present study was designed to evaluate genetic polymorphism in CYP1A1 (m1, m2 and m4), CYP3A5 (A44G), CYP2C9 (Arg144Cys and Ile359Leu), CYP2D6 (*3 and *4) and PON1 (Gln192Arg and Leu55Met), GSTM1, GSTT1, GSTP1 and NAT2 and its correlation with DNA damage in pesticides exposed workers. Using the comet assay and PCR-RFLP, the extent of DNA damage and genotype frequencies were evaluated in the PBMC of 80 pesticides exposed workers and equal number of age, ethnicity and gender matched control subjects. Exposed workers showed significantly higher levels of DNA damage compared to controls as measured through various comet parameters. The prevalence of m1, m2, m4 CYP1A1, A44G CYP3A5, CYP2C9, CYP2D6, PON1, GSTM1, GSTT1, GSTP1 and NAT2 genotype frequencies did not differ significantly in pesticide exposed subjects and controls. Cys144Cys CYP2C9, CYP2D6*4, PON1 (Arg-Gln, Gln-Gln, Leu-Leu), Ile-Ile GSTP1 individuals had higher comet assay parameters as compared to other forms of these enzymes in pesticide exposed subjects. The prevalence of slow acetylation was found significantly higher in smokers as compared to non-smokers in pesticide exposed subjects which was not seen in control subjects who were smokers. In conclusion, the results show that unfavourable alleles of these enzymes are more susceptible to genotoxic effects than those with favourable alleles which could be helpful to identify individual markers of susceptibility and toxicity toward pesticides. Further studies are required in this context in order to form the larger database in different population.
263. Mechanisms of olfactory toxicity of the herbicide Dichlobenil: essential roles of CYP2A5 and nasal inflammation in dichlobenil-induced neurodegeneration
Fang Xie1, Xin Zhou2, Melissa Behr3, James Schwob4, Kurunthachalam Kannan5, and Xinxin Ding6
1Laboratory of Molecular Toxicology, Wadsworth Center, NYSDOH, Albany, NY, USA, 12201-0509
2Laboratory of Molecular Toxicology, Wadsworth Center, Albany, NY, USA, 12201-0509
3Wisconsin Veterinary Diagnostic Lab, Madison, WI, USA, 53706
4Department of Anatomy & Cellular Biology, Tufts University School of Medicine, Boston, MA, USA, 02111
5Wadsworth Center, NYSDOH, Albany, NY, USA, 12201
6Dept HTME, Wadsworth Ctr, Albany, NY, USA, 12201-0509
Dichlobenil (2,6-dichlorobenzonitrile, or DCBN), causes permanent loss of olfactory receptor neurons (ORNs) in the dorsal olfactory epithelium (OE) in mice. The aims of this study were to identify the P450 enzymes responsible for DCBN metabolic activation in the OE, and to explore the mechanisms by which DCBN causes permanent ORN degeneration and respiratory metaplasia. To study DCBN metabolic activation in vivo, we utilized two novel knockout mouse models: one (named liver-Cpr-null) with liver-specific suppression of all microsomal P450 activities; the other (named Cyp2a5-null) with germline deletion of Cyp2a5, a gene expressed most abundantly in the olfactory mucosa. We observed that loss of hepatic P450 activity (in the liver-Cpr-null mouse) did not protect mice against DCBN olfactory toxicity, despite a substantial decrease in hepatic DCBN metabolism. In contrast, a global loss of CYP2A5 (in the Cyp2a5-null mouse) blocked DCBN toxicity completely, with no effect on systemic clearance of the toxicant. Therefore, we conclude that CYP2A5, most likely the set expressed in the olfactory mucosa, is essential for DCBN-induced olfactory toxicity. For studies on the mechanism of DCBN-induced ORN degeneration, we observed that DCBN treatment (at 50 mg/kg, single i.p. dose) led to detachment of the layer of horizontal basal cells (HBC; known as neural stem cells in the OE), as well as nasal inflammation, at 24 hr after the treatment. Further studies revealed that DCBN-induced nasal inflammation, but not HBC detachment, can be prevented by treatment of mice with an anti-inflammatory agent, a result indicating that inflammation was not responsible for the HBC detachment. Moreover, at 8 weeks after DCBN exposure, the dorsal OE was replaced by a non-neuronal epithelium, despite reappearance of keratin-5-positive and Pax6-positive (stem-like) cells in the basal cell layer. These results suggest that nasal inflammation induced by DCBN can interfere with the differentiation of nascent neural stem cell in the OE, leading to replacement of the OE with respiratory epithelium. (Supported in part by NIH grant ES07462)
264. Gamma-glutamyldehydroalanylglycine is an electrophilic metabolite of glutathione
Cody J. Peer1, Islam R. Younis2, Valerie C. Bukowski3, Stephen S. Leonard3, William P. Petros1, and Patrick S. Callery1
1Pharmaceut & Pharmacol Sci, School of Pharmacy, West Virginia University, Morgantown, WV, USA, 26506-9530
2Office of Clinical Pharmacology, CDER, US Food and Drug Administration,, Silver Spring, MD, USA, 20993
3Pathology and Physiology Research Branch, Health Effects Laboratory Division, National Institute for Occupational Safety and Health, Morgantown, WV, USA, 26505
Gamma-Glutamyldehydroalanylglycine (EdAG) is a dethiolated, electrophilic metabolite of glutathione (GSH) derived from the Phase II conjugation of GSH with busulfan catalyzed by glutathione S-transferase (GST), specifically isoform A1-1. Electrophilic substrates of GST, such as ethacrynic acid and 1-chloro-2,4-dinitrobenzene, have been shown to be irreversible inhibitors of glutathione transferase activity at high concentrations in vitro. EdAG at high concentration (10 mM) was found to be an irreversible inhibitor of human GSTA1-1, and at lower concentrations showed noncompetitive inhibition (Ki = 11 μM). Conversion of GSH to EdAG represents a loss of thiol-related redox properties and the gain of a dehydroalanine group with the potential to scavenge ROS. EdAG competed with DMPO for hydroxyl radical generated in the Fenton reaction in a concentration-dependent manner. The results suggest a stabilized carbon-based captodative radical intermediate in the reaction of EdAG with hydroxyl radical. In support of a captodative mechanism was the identification of a dimerized gamma-glutamylserylglycine as a product in the reaction of hydroxyl radical with EdAG. In summary, it was determined here that EdAG can inhibit GSTA1-1 and also scavenge hydroxyl radical in vitro through a proposed captodative mechanism. The results indicate that the chemical instability of a busulfan metabolite results in the conversion of GSH into EdAG, a reactive compound that is a Michael acceptor and free radical scavenger. The biological implications of EdAG reactivity may have an impact on GSH biochemistry, cellular free radical reactivity, and busulfan toxicology.
265. Alternative non-targeted methods for metabolite screening and identification on a rapid scanning linear ion Trap/QqQ mass spectrometer
Jeffrey D. Miller1, James A Ferguson2, Sai Y. Chang3, and Nanqun Zhu4
1Applied Biosystems, Framingham, MA, USA, 01701
2Psm, Applied Biosystems, Framingham, MA, USA, 01701
3MSMS Science, LLC, Sedona, AZ, 86336
4Genzyme, Framingham, MA, USA, 01701
Conventional non-targeted approaches commonly use high-resolution MS4 detection followed by data mining software processing, usually comparing one sample vs. control at a time. This process is time consuming and can lack the ability to directly compare multiple data sets.
Alternative non-targeted metabolomics approaches presented here are applicable for finding metabolites in complex biological matrices (in vivo rat plasma and urine) and in vitro samples. The data are processed by batch, group or subject by statistical analysis (PCA or t-tests). This approach does not predict or introduce any user-bias into the selection process, and is simple and easy to implement. Linking the urine, plasma, bile study information as well as comparing cross-species studies, dosage levels and semi-quantification of metabolites could be very useful to investigators, when data are processed together in one file format.
Method: We present two different non-targeted approaches; the first method uses a MIM1 experiment as the survey covering a broad mass range in either positive or negative ion mode. The second method uses full-scan positive/negative switching surveys. Both methods then trigger ion-trap dependent scans of ER (enhanced resolution for isotope definition) and EPI (enhanced product ion scan) for product ion spectra. The increased scanning speed of a hybrid linear ion trap mass spectrometer allows for full-scan survey MSMS experiments in one second cycle times.
Dosage and sample extraction: Male Spraque-Dawley rats were dosed orally with carbamazepine at a level of 3 mg/kg. Blood samples were collected via the jugular vein catheter and urine samples were also taken at t = 0 and t = 8 hrs post-dose. Preliminary Data: Preliminary data shows good coverage of xenobiotic metabolites found compared to literature2, 3 (or targeted approaches), using both the MIM and Full Scan as surveys.
Triplicate injections were performed on each set of plasma samples (control and post-dosed time course samples). Statistical analysis (PCA) software was utilized to mine the large amount of data generated for metabolites. DMPK profiles of the parent carbamazepine as well as the major metabolites, including an iminoquinone (MW 207) and sulfonation-oxidation (MW 332) metabolites were profiled over the time course study. Metabolite identification software was used to provide fast confirmation for the presence of the xenobiotics as well as to give potential structural formula for those metabolites based on the MS/MS spectra generated. The metabolite list comparing the MIM approach and the pos/neg approach showed similar number of major metabolites (> 1% total peak area) and also similar sensitivity.
Novel Aspect: Non-targeted metabolomics approaches to metabolite analysis using either a broad range MIM survey or a positive/negative full-scan surveys in a fast-scanning mass spectrometer coupled with high-resolution LC.
References:
- Yao, M, Duchoslav, E, Zhu, M, Rapid Commun. Mass Spectrom. 2009; 23; 1683–1693.
- Uetrecht JP, Ju, C, J. Pharm. Exp. Therap. 1999, 288, 51–56.
- Madden, S., Maggs, J.L., Park, B.K. Drug Metab. & Disposition, 1996, 24,469–479.
- Zhang H and Yang Y. J. Mass Spectrom. 2008, 43: 1181–1190
266. Metabolism and pharmacokinetics of GS-8374: A novel phosphonate-containing HIV protease inhibitor with high barrier to resistance
Eugene J. Eisenberg, Bernard P. Murray, Melody S. Lee, Lani M. Wieman, Kelly Wang, Yujin Wang, Jianhong Wang, Leah Tong, Xubin Zheng, and Gerry Rhodes
Drug Metabolism, Gilead Sciences, Inc., Foster City, CA, USA, 94404
GS-8374 is a novel diethyl-phosphonate containing nonpeptidic human immunodeficiency virus (HIV) protease inhibitor (PI) intended to be used in treatment of HIV-1 infection. Compared with early-generation PI’s, GS-8374 has a high genetic barrier to resistance and is active against highly-PI-resistant HIV isolates. Here we describe the metabolism and preclinical pharmacokinetics of GS-8374. In vitro metabolism experiments were performed with hepatic microsomal fractions and primary hepatocytes. In vivo studies were carried out in rats and dogs. Identification and characterization of GS-8374 was via HPLC with detection by tandem and ion-trap mass spectrometry (LC/MS/MS). GS-8374 was significantly more stable in human microsomes and hepatocytes, than in those of other species. Its metabolic stability was greater than that of all currently marketed HIV PI’s. The major putative metabolites of GS-8374 (shown below) resulted from carbamate amide bond cleavage, hydroxylation at the benzylic position and O-demethylation at the methoxyphenyl moiety (M5, M4 and M9, respectively). There was no detectable loss of the phosphonate moiety. An additional major metabolite in dog microsomes resulted from hydroxylation on the isobutyl moiety (M6). Several minor metabolites in all species resulted from glucuronidation of the hydroxy group (hepatocytes), hydroxylations on the bis-tetrahydrofuran, isobutyl moieties and from combinations of the above metabolic reactions. All metabolites observed in human in vitro experiments were also observed in other species at higher levels. In vivo, GS-8374 has a moderate steady state volume of distribution. The clearance values were relatively high and were well predicted from in vitro metabolic stability. Since the metabolic stability of GS-8374 in human microsomes and hepatocytes was relatively high the clearance in humans is expected to be significantly lower than in preclinical species. Data from these studies suggest that GS-8374 warrants further investigation as a promising new agent for treatment of HIV-1 infection.
267. Metabolic profiles of CVT-3619 in human plasma and urine
Nevena Mollova, Jennifer Tang, Claire Bramwell-German, Eve-Irene Lepist, Nancy Chu, William Chen, Belinda Wong, Peter Staehr, and Kwan H. Leung
Pre-Clinical Development, Gilead Sciences, Inc., Palo Alto, CA, USA, 94304
CVT-3619 [(2-{6-[((1R, 2R)-2-hydroxycyclopentyl)amino]purin}-yl} (4S, 5S, 2R, 3R) 5-[2-fluorophenylthio)methyl]oxolane-3,4-diol)] is a partial A1 adenosine receptor agonist being developed for the treatment of hypertriglyceridemia. The purpose of this study was to characterize in vivo metabolic profiles and to quantitate the major metabolites in human plasma and urine. Plasma and urine were collected from 77 healthy and obese patients during CVT-3619 clinical study CVT 7011. Plasma samples were prepared using protein precipitation. Urine samples were analyzed by an LC-MS/MS on-line extraction procedure. Mass spectrometric detection and quantitation were carried out on an API-3000 or API-5000 LC-MS/MS system with positive electroSpray ionization using multiple reaction monitoring (MRM). The combination of UPLC-QTRAP5500 was applied for separation and characterization of the most polar isomeric glucuronides of CVT-3619. Seven mono-hydroxylated metabolites were identified in human plasma and urine. Their structures were previously characterized in vitro and in vivo. These include products resulting from oxidation of sulfur to sulfoxide (M1, M3) which were further oxidized to sulfone (M2), and products following hydroxylation of the 2-hydroxycyclopentyl ring (M4, M5 and M11) and the fluorophenyl group (M6). In this study, we report the quantitation of the major phase I metabolites M1, M3, M4 and M5 in human plasma and urine and an estimate of CVT-3619 urinary excretion. In addition, we provide structural identification of CVT-3619 glucuronides characteristic of human urine and plasma. Glucuronidation of the parent ribose forming major metabolite G1 and minor G2 previously observed in vitro and in animal species were also present in human urine and plasma. Another two major glucuronides were G4-1 and G10-1 which were tentatively assigned as M4- and M2-glucuronides in the ribose moiety, respectively. Several isomers of G3, G4, G7, G8 and G9 glucuronides were also found in human plasma and urine. In summary, major metabolites of CVT-3619 were characterized and quantitated in human plasma and urine. Results will allow continued evaluation of the role of metabolism on the efficacy and safety of CVT-3619 in future clinical studies.
268. Identification and characterization of GSH conjugates of mifepristone in human liver microsomes
Hoa Le, V. Sashi Gopaul, Cornelis Hop, and Cyrus Khojasteh
Drug Metabolism & Pharmacokinetics, Genentec, Inc, South San Francisco, CA, USA, 94080
Mifepristone is an antiprogesterone and antiglucocorticosteroid agent with potential anti-cancer activity in humans. A recent report1 demonstrated that mifepristone, whose structure contains an acetylene group, is not only a mechanism-based inactivator (MBI) of CYP3A4 but of CYP2B6 as well. The MBI is proposed to occur via heme destruction and covalent binding of a reactive intermediate to the P450 protein1. Structure of the reactive intermediate is proposed to be an oxidation of the acetylene moiety (possibly an oxirene) that can also be trapped by GSH possibly via a ketene intermediate. Also identified were GSH adducts proposed to form via the epoxidation of the double bond at a different position in the molecule.
The objective of this study was to characterize the GSH conjugates of mifepristone in human liver microsomes (HLM). Mifepristone was incubated at 10 and 20, and 50 μM with HLM for 0 and 1 hour. Incubations were at 37°C in the presence of a NADPH fortified with alamethicin, GSH, and UDPGA. Samples were processed and analyzed by LC-MSn analysis using a ThermoFinnigan LTQ XL mass spectrometer. Similar to reported observations, three GSH conjugates of the oxidized mifepristone, each characterized by an [MH]+ at m/z 753, m/z 769, and m/z 751 were identified. In addition, we report here the identification of three new GSH conjugates of mifepristone with [MH]+ corresponding to m/z 737, m/z 723, and m/z 709, respectively. The structures of these newly discovered metabolites were consistent with those of the GSH adducts of mifepristone itself and those of the N-dealkylated and the N,N-didealkylated products of mifepristone, respectively. The mechanism by which these three metabolites are formed warrants further investigation.
269. Metabolic switching of BILR 355 in the presence of ritonavir I: Identification of a disproportionate human metabolite BILR 516
Yongmei Li
Drug Metabolism & Pharmacokinetics, Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT, USA, 06877-0368
BILR 355 BS is a dipyridodiazepinone non-nucleoside reverse transcriptase inhibitor (NNRTI) of the human immunodeficiency virus HIV-1. BILR 355 had a very short half life (2 to 5 h) and low exposure after oral administration to humans. BILR 355 is primarily metabolized by CYP3A4. Ritonavir is a potent CYP3A4 inhibitor which has been used as a boosting agent to increase the exposure of other HIV drugs. Coadministration of ritonavir with BILR 355 increased systemic exposure and half-life of BILR 355. A total of eighteen metabolites were identified in human plasma samples obtained from Phase Ib clinical studies, after co-administration of ritonavir with BILR 355. Their structures were proposed based on LC/MS/MS technologies and seven metabolites were confirmed after comparison with authentic standards. The metabolic pathways of BILR 355 have been proposed based on the identified metabolites.
The metabolites for which authentic standards were available, were quantified using LC/MS/MS. A major human metabolite was identified, BILR 516, which had a long half-life and higher exposure than the parent compound at steady state. Since there was inadequate coverage for BILR 516 in Toxicology species (rat and dog) with administration of BILR 355/ritonavir, BILR 516 was identified as a disproportionate human metabolite.
270. Metabolism and excretion of BG12 in rats and humans following oral administration of a single oral dose of [14C]BG12
Lin Xu1, Kate Dawson2, James Woodworth1, Liyu Yang1, Lewis J Klunk1, Scott Stecher3, and Chandra Prakash1
1Dmpk, BiogenIdec, Cambridge, MA, USA, 02142
2Neurology, BiogenIdec, Cambridge, MA, USA, 02142
3Clinic Operation, BiogenIdec, Cambridge, MA, USA, 02142
BG12 [dimethyl fumarate (DMF)] displays immunomodulatory effects, including inhibition of the expression of pro-inflammatory cytokines and adhesion molecules involved in the inflammatory cascade. It is in phase III trials and being development for the oral treatment of multiple sclerosis (MS). The objectives of this study were to determine the disposition and metabolic profiles of BG12 in Long-Evans rats and humans after administration of a single oral dose of [14C]BG12. Total recoveries of radioactivity were 89% in rats and 75% in humans. The majority of dose was recovered in the expired air for both rats (~63%) and humans (~59%). The absorption of the BG12 radioactivity was rapid, as the plasma concentrations for BG12 radioactivity peaked within 1 h after oral administration. Based on AUC(0-24) values, the majority of the circulating radioactivity was comprised of metabolites. BG12 is extensively metabolized in rats and humans since no unchanged drug was detected in urine; Recovery in expired air indicates that the BG12 is primarily converted to CO2 as an end metabolite via the tricarboxylic acid (TCA) cycle. Glucose (M8) was the predominant circulating metabolite, accounting for 50% and 60% of the total extractable radioactivity in rat and human plasma, respectively. Other most abundant metabolites, fumaric acid and citric acid combined accounted for 33% and 27% in rat and human plasma. The hydrolytic metabolite, monomethyl fumarate, accounts for less than 0.2 % in rat and 4.9% in human plasma. Cysteine and/or N-acetylcysteine conjugates of mono- and di-methyl succinate were found as major urinary metabolites, representing a total 10% and 7% of the administered dose in rat and human, respectively. In conclusion, human and rat showed similar metabolic profile. Identified radiolabelled endogenous glucose, citric acid, fumaric acid, and CO2 in air suggested that the major route of metabolism of BG12 was by the endogenous TCA cycle pathway.
271. Intelligent workflows (MSM) for metabolite screening and characterization on a hybrid dual-cell linear trap and orbitrap mass spectrometer
Yingying Huang1, Ji Ma2, Nicolaie Eugen Damoc3, Thomas Moehring3, Robert Cho4, Jae Schwartz1, Yan Chen1, and Tim Carlson2
1Thermo Fisher Scientific, San Jose, CA, USA, 95134
2Dept of PKDM, Amgen, South San Francisco, CA, USA, 94080
3Thermo Fisher Scientific, Bremen, Germany
4Dept of PKDM, Amgen, South San Francisco, CA, 94080
The characterization of drug metabolites is an integral part of drug discovery and development since it identifies compounds which may have pharmacological activity or specific toxicity. LC-MS is typically carried out for both discovery phase metabolite screening and for definitive biotransformation characterization. While the focus and the data requirements for each may differ, the general challenges remain the detection and identification of metabolites in the presence of highly complex biological matrices. There are business demands for increased automation, throughput, sensitivity and accuracy. We describe the use of MSM, which utilizes multiple collision cells, dissociation methods, scan modes, mass analyzers and detectors to perform intelligent metabolite identification experiments.
Method: A new hybrid mass spectrometer with a duel-cell linear ion trap (LIT), an orbitrap mass analyzer, and an HCD (Higher-energy Collisional Dissociation) cell was used. The isolation mass window up to 600amu for HCD scans was enabled. The experiment was designed such that a high resolution full scan was acquired, followed by a high resolution HCD MS/MS of all incoming ions within a specified 600amu window. In parallel, the LIT acquired data dependent MSn spectra. UHPLC and 1.9mm C18 column were used. Verapamil and haloperidol (10mM and 1mM) were incubated with rat and human hepatocytes and the resulting samples analyzed.
Preliminary Data: The HCD cell can excite a large number of precursors simultaneously. Fragmentation information for all ions within the 600amu window was acquired. By comparing the HCD MS/MS with the full MS, it is possible to mimic conventional neutral loss scanning, precursor ion scanning, and multiple reaction monitoring experiments. This was achieved by data mining from datasets with high resolution and accurate mass. Once potential metabolites were identified using such comparisons, the parallel data dependent CID MSn data allowed unambiguous structure interpretation. In each scan cycle, 10 scan events was completed in 1.8 seconds with two high resolution scans from the Orbitrap (full scan and the HCD MS/MS) and eight CID MSn (n=2-3) scans from the dual-cell LIT. Using m/z 165.0910, a diagnostic fragment ion of verapamil, accurate mass precursor ion analysis of high resolution full scan and HCD MS/MS of 10mM rat hepatocytes sample led to identification of more than 16 putative metabolites. The results are similar to those from precursor ion scanning experiments using a triple quadrupole mass spectrometer. Similar analysis for the fragment ion at m/z 151.0753 resulted in the discovery of 5 additional metabolites. Definitive structural elucidation of these putative metabolites was achieved by analysis of the data dependent LIT CID MS2 and MS3 data collected in parallel. Analysis of 1mM rat hepatocytes sample using the same approach also led to the identification of all Phase I and II metabolites.
Novel Aspect: Utilizes multiple collision cells, dissociation methods, scan modes, mass analyzers and detectors together for intelligent metabolite identification and structural elucidation.
272. In vitro metabolism of CP-690,550 in liver microsomes of mouse, rat, and human: Formation of a novel metabolite via the loss of a nitrile group
V. Sashi Gopaul, Hoa Le, Matthew Baumgardner, O. Helen Chan, Cornelis Hop, and Cyrus Khojasteh
Drug Metabolism & Pharmacokinetics, Genentec, Inc, South San Francisco, CA, USA, 94080
CP-690,550 (A) is reported to be a Janus kinase 3 (Jak3) inhibitorFootnote1 and currently undergoing clinical trial.Footnote2 The objective of this study is to compare the metabolism of A in liver microsomes of mouse (MLM), rat (RLM), and human (HLM).LM (0.5 mg/mL) were incubated with 10 μM of A and diclofenac at 37 °C for 0 and 1 hr in the presence of NADPH (1 mM) and fortified with alamethicin (100 μg/mL), UDPGA (5 mM), and GSH (5 mM). Samples were processed and analyzed by LC-MSn analysis (ES+) using ThermoFinnigan LTQ XL and LTQ-Orbitrap mass spectrometers. The extent of metabolism of A varied according to species and in the following order: mouse > rat > human. No human specific metabolite was observed. Metabolites M1 and M2 were formed following N-demethylation and oxidation of A, respectively, in all species. M3 was characterized with an [MH]+ at m/z 304, nine mass units lower than that of the parent (m/z 313), and consistent with an elemental composition of C15H22O2N5. The product ion spectrum of m/z 304 produced informative fragment ions at m/z 274 consistent with the loss of H2CO, the portion of the molecule undergone metabolism and at m/z 246 following the additional loss of the adjacent CO group. Further fragmentation of m/z 246 (MS3) produced the same fragment ions as observed for the parent indicating the portion of the molecule unaffected by this rarely reported biotransformation reaction. Assessed altogether, M3 appeared to be formed following the displacement of the terminal cyano moiety by a hydroxyl group. This unique metabolite was found at 6.4% of sample in the mouse and at 1% in the rat and human, respectively. Studies are currently underway to investigate the mechanism of formation of this metabolite. The glucuronide of M3 was also identified in the mouse and rat only.
273. A new strategy based on UHPLC-LTQ/Orbitrap for simultaneous metabolite identification and quantitation of in vivo PK samples
Yanou Yang, Chiuwa Emily Luk, Mary Grubb, Haiying Zhang, William G. Humphreys, and Jonathan Josephs
Dept of Biotransformation/HW21-1-15, Bristol-Myers Squibb Co, Pennington, NJ, USA, 08543
The integrated approach for the semi-quantitation of both a drug candidate and its metabolites without the requirement for authentic standards has been proven to be a powerful tool in drug discovery. This approach uses diluted “metabolite standards” generated from high concentration in vitro incubations as single point calibration standards for the quantitation of these metabolites in both in vitro and in vivo samples based on a UV/MS response factor. The current practice of this approach involves separate runs using two different instrument methods for metabolite identification and metabolite quantitation respectively. The present study is to establish a new strategy for simultaneous identification and quantitation of all metabolites in the same run as the quantitation of the parent drug candidate of the in vivo PK samples. This is achieved by full scan accurate mass data acquired with a high resolution LTQ/Orbitrap coupled to an Ultra High Pressure Liquid Chromatograph (U-HPLC). The “metabolite standards” generated from a 30 mM incubation in liver microsomes are diluted in the corresponding in vivo matrix and run together with the in vivo PK samples to calculate UV/MS response factors. The conversion factor for each metabolite is calculated based on the UV peak area of the metabolite in the 30 mM incubation sample and the MS peak area of the exact mass extracted ion chromatogram of the same metabolite under the same LC conditions. The concentrations of the drug candidate and its metabolites are calculated from their UV/MS conversion factors. Comparison is made with the pharmacokinetic profiles of the drug candidate and its metabolites obtained from this approach to those obtained from the same samples but quantitated with authentic standards and a full calibration curve. The validity of this approach is demonstrated for the model compound buspirone for which we have multiple authentic standards and extensive biotransformation data.
274. Increase in sensitivity of on-line radioactivity detection with microbore LC-MS coupled with dynamicflow ARC system
Wing W. Lam1, Heng Keang Lim1, Jose Silva1, Aaron Young2, and Dian Y. Lee2
1Dmpk, Johnson & Johnson PRD, Raritan, NJ, USA, 08869
2AIM Research Company, Hockessin, DE, USA, 19707
The detection, characterization and quantitation of low levels of systemic circulating radiolabeled metabolites present an analytical challenge in the pharmaceutical industry. The need for more sensitive on-line detection of small amount of radioactivity in plasma is even more critical for the definitive human absorption, elimination and metabolism study because of the low dose of radiolabeled drug. In addition, the need to quantify circulatory metabolites, in the absence of reference standard, driven by requirements from the regulatory agencies (e.g. MIST document) will result in more studies with radiolabeled compounds. This study is to investigate the application of microbore LC/RAD/MS system to profile samples from drug metabolism study. The microbore LC/DynamicFlow ARC/MS system utilizes a microbore column (0.5 or 1 mm ID), a smaller radioactivity detection flow-cell (< 100 uL) and an LTQ/Orbitrap. With the changes to a smaller flow cell and a smaller column ID, there was a substantial increase in sensitivity. Our preliminary results indicates that a limit of detection of <20 DPM (14C) has been achieved using this method. Other advantages in using microbore LC include 1) smaller sample size is needed for injection, which is particularly useful for plasma analysis because of limited samples availability, 2) less endogenous compounds from matrices are introduced to MS and thus, less ion suppression is expected, and 3) cost savings from low consumption of mobile phase and scintillation cocktail. Consequently, metabolite identification can be greatly facilitated with the benefits of an increase in sensitivity in both radiochemical detection and mass spectrometry, and with less ion suppression.
275. Screening for reactive drug metabolites in establishing structure metabolite relationship during drug development
Gopinadh Bhyrapuneni, Ranjithkumar Ponnamaneni, Arunkumar Manoharan, Shantaveer Irupannavar, Raghavachaudary Palacharla, Koteshwara Mudigonda, Vishwottam Kandikere, and Ramakrishna Nirogi
Discovery Research, Suven Life Sciences Ltd, Hyderabad, India
Characterization of reactive metabolites is critical step in developing new drug candidates with an improved safety profile. Most reactive metabolites are electrophiles which can react with nucleophilic sites of proteins and subsequently cause various levels of toxicities (Wen and Fitch, 2009). HPLC coupled with MS is the first tier technique to screen for reactive metabolites. Over the past few years various scanning approaches were evolved to avoid false positives and to increase the selectivity. In-vitro assays were employed to validate these techniques with carbamazepine and imipramine which forms arene oxide reactive intermediate. In our internal CNS discovery programme, compounds were screened for reactive metabolite assay. Few of those would able to form reactive intermediate metabolite like arene oxide and dihydrodiol etc. In in-vitro the conjugates of glutathione and its ethyl ester derivatives were identified with API-4000 Q-trap LC-MS/MS using IDA method employing negative precursor ion scan and subsequent scanning with enhanced product ion scan. With this in-vitro data we could able identify those functional groups which form reactive metabolites. From these studies we established structure-metabolite relation ship which helped to drive the SAR with appropriate functional modification. The evaluation and structure-metabolite relationship will be discussed.
Reference
- Wen B, Fitch WL. Screening and characterization of reactive metabolites using glutathione ethyl ester in combination with Q-trap mass spectrometry. J. Mass. Spectrom. 44, 90–100:2009.
276. Semi-quantification of metabolites using a rapid scanning hybrid QqQ/linear ion trap mass spectrometer while doing non-targeted metabolite profiling
James A. Ferguson1, Sai Y. Chang2, Jenny E. Moshin3, Jeffrey D. Miller3, and Sandeepraj Pusulkar4
1Application Laboratory, Applied Biosystems, Inc., Framingham, MA, USA, 01701
2MSMS Science, LLC, Sedona, AZ, 86336
3Applied Biosystems, Framingham, MA, USA, 01701, 4Millennium Pharmaceuticals, Cambridge, MA, USA, 02139
Novel Aspect—Non-targeted metabolite ID and semi-quantification in one experiment. Quantification of the metabolites has been difficult due to differences in the ionization efficiencies of compounds and their metabolites. Often quantification requires synthesis of the metabolites and sometimes labeled or analog internal standards. Good qualitative and quantitative (or at least semi-quantitative) data from one experiment would be highly desirable. We present an MIM[1]–triggered information-dependent acquisition experiment using a wide mass range to cover both Phase I and Phase II metabolites for both metabolite discovery and confirmation (based on the dependent LIT product ion scan) and quantification using the area under the MIM curve for the respective metabolites. We will present several compounds for which the parent drug will be quantified by both traditional MRM with internal standard and by MIM without. The MIM method provides a less noisy chromatogram for integration than an extracted ion chromatogram from a full scan experiment. The assumption would be that if the parent can be quantified using MIM, a good relative quantification could also be obtained for the metabolites. Male Spraque-Dawley rats were dosed orally with verapamil and haloperidol at several levels from 0.1 to 10 mg/kg. Blood samples were collected via the jugular vein catheter and urine samples were also taken at several time points post-dose. Preliminary data shows good discovery and confirmation of xenobiotic metabolites, using the MIM-triggered IDA experiments. Standard curves for the compounds were prepared in the appropriate matrices and the incubated samples included in the curves. Quantification was by optimized MRM methods and by the MIM-triggered IDA experiments used for metabolite ID. In addition to the quantification results, the incubated samples were processed using standard metabolite ID software to search for metabolites, which were confirmed by comparison of their product ion spectra to those of the parent compound.
References:
- Yao, M.; Duchoslav, E.; Zhu, M., Rapid Commun. Mass Spectrom. 2009, 23, 1683–1693.
277. Structure elucidation of thioether conjugates of ethacrynic acid in NADPH- and glutathione-fortified human liver S9 by microbore LC-LTQ/Orbitrap
Heng Keang Lim, and Jose Silva
Dmpk, Johnson & Johnson PRD, Raritan, NJ, USA, 08869
The Michael addition of glutathione to the a,b-unsaturated keto functionality of the diuretic drug, ethacrynic acid, has been reported and this Michael addition reaction is reversible via retro Michael cleavage of (glutathion-S-yl)ethacrynic acid. Ethacrynic acid and its glutathione conjugate are potent inhibitor of all classes of glutathione S-transferase enzymes via Micahel addition to cysteine residue 47. The inhibition of glutathione S-transferase enzymes by ethacrynic acid is again reversible via retro Michael cleavage. In the investigation of ethacrynic acid as inhibitor of glutathione S-transferase in human liver S9, several thioether conjugates of ethacrynic acid were detected. This report described the structural elucidation of these thioether conjugates of ethacrynic acid, following incubation in NADPH-and glutathione-fortified human liver S9, cytosol and microsomes, by microbore LC-LTQ/Orbitrap analysis. Typical incubation, 0.5 mL final volume, consisted of 2 mg/mL human liver S9, cytosol or microsomes, 1 mM EDTA, 5 mM MgCl2, 50 mM ethacrynic acid and 5 mM glutathione in 0.1M phosphate buffer (pH 7.4). The mixture was incubated at 37 °C for 60 minutes and reaction terminated by centrifugation of CaCl2 and ZnSO4precipitated proteins. The thioether conjugates of ethacrynic acid were chromatographically separated using HyPurity Aquarstar column (150x 1 mm ID, 3m) at 50°C using a nonlinear gradient of 0.2% formic acid and acetonitrile at flow rate of 200 mL/min. The structure of each thioether conjugate was elucidated using chemical formulae of protonated molecule and its product ions from chlorine isotope triggered data-dependent scan. The conjugate from direct Michael addition of glutathione to ethacrynic acid at m/z 610.1027 (C23H30O10N3Cl2S, 9.5 RDB, 0.58 ppm) further metabolized in vitro to form (g-glutamyl-glutathion-S-yl)ethacrynic acid (m/z 739.1452: C28H37O13N4Cl2S, 11.5 RDB, 0.35 ppm) presumably catalyzed by g-glutamyltranspeptidase. In addition, metabolites from mercapturic acid pathway were also detected. Another (glutathion-S-yl)ethacrynic acid with m/z 608 (C23H28O10N3Cl2S, 10.5 RDB, 0.50 ppm) was also detected and postulated to derive from addition of glutathione to ethacrynic acid epoxide followed by dehydration. Also, thioether conjugates from direct Michael addition of glutathione to hydroxylated ethacrynic acid were also detected.
278. Desipramine metabolism is comparable in CYP2D6-humanized mouse and human liver microsomes
Hongwu Shen1, and Ai-Ming Yu2
1Department of Pharmaceutical Sciences, School of Pharmacy and Pharmaceutical Sciences, The State University of New York at Buffalo, Buffalo, NY, USA, 14260
2Department of Pharmaceutical Sciences, University at Buffalo, SUNY, Buffalo, NY, USA, 14260-1200
Genetically modified mice expressing human cytochrome P450 enzymes are possible models to overcome species difference in drug metabolism between human and animals(1). The CYP2D6-humanized (Tg-CYP2D6) mice have been shown to express human CYP2D6 in the liver, kidney, and intestine, as a similar distribution pattern as that in human. Studies in Tg-CYP2D6 versus wild-type mouse models also successfully indicate the impact of CYP2D6 status on pharmacokinetics, dynamics and metabolism of CYP2D6-metabolized drugs(2-4). In the present study, we investigated the metabolite production from desipramine (DMI) in Tg-CYP2D6 mouse and human liver microsomes. After incubation with Tg-CYP2D6 mouse liver microsomes, three metabolites were found by LC-MS analyses. One major metabolite was 2-hydroxyldesipramine (2-OH-DMI), and two minor metabolites were 10-hydroxyldesipramine (10-OH-DMI) and desmethyldesipramine. The same three metabolites were identified in incubations with pooled human liver microsomes. The relative abundance of the metabolites, according to peak areas in LC-MS analyses, was estimated as 9.9:1.0:2.4 in transgenic mouse liver microsomes and 12:1.0:1.9 in human liver microsomes, respectively. These results suggest that DMI metabolic profiles are comparable in Tg-CYP2D6 mouse and human liver microsomes.
References:
- F. J. Gonzalez, A. M. Yu, Annu Rev Pharmacol Toxicol 46, 41 (2006).
- J. Corchero et al., Mol Pharmacol 60, 1260 (Dec, 2001).
- A. M. Yu et al., Pharmacogenetics 13, 173 (Mar, 2003).
- C. Wu, X. L. Jiang, H. W. Shen, A. M. Yu, Biochem Pharmacol (May 13, 2009).
279. In vitro metabolic activation of nevirapine: Dehydrogenation and Inactivation of cytochrome P450 3A4
Bo Wen, Yuan Chen, and William Fitch
Drug Metabolism and Pharmacokinetics, Roche Palo Alto, Palo Alto, CA, USA, 94304
Nevirapine, a non-nucleoside HIV-1 reverse transcriptase inhibitor, has been associated with incidences of skin rash and hepatotoxicity in patients. While the mechanism of idiosyncratic hepatotoxicity remains unknown, it is proposed that metabolic activation of nevirapine and subsequent covalently binding of reactive metabolites to cellular proteins play a causative role. Studies were initiated to determine whether nevirapine undergoes cytochrome P450 (P450)-mediated bioactivation in human liver microsomes to electrophilic intermediates. LC-MS/MS analysis of incubations containing nevirapine and NADPH-supplemented microsomes in the presence of glutathione (GSH) revealed the formation of a GSH conjugate derived from the addition of the sulfydryl nucleophile to nevirapine. No other GSH conjugates were detected including conjugates of oxidized metabolites of nevirapine. These findings are consistent with a bioactivation sequence involving initial P450-catalyzed dehydrogenation of the aromatic nucleus with 4-methyl group in nevirapine to an electrophilic quinone methide intermediate, which is subsequently attacked by glutathione yielding the sulfydryl conjugate. Formation of the nevirapine GSH conjugate was primarily catalyzed by heterologously expressed recombinant CYP3A4, and to a less extent, CYP2D6, CYP2C19 and CYP2A6. In addition, the quinone methide reactive metabolite was a mechanism-based inactivator of CYP3A4, with inactivation parameters KI = 31 μM and kinact = 0.029 min−1 respectively. It is proposed that formation of the quinone methide intermediate may represent a rate-limiting step in the initiation of nevirapine-mediated hepatotoxicity.
280. Identification of human enzymes involved in the metabolism of BMS-690514, an ErbB/VEGF receptor inhibitor
Haizheng Hong1, Hong Su1, Alban Allentoff2, Haojun Sun2, Ramaswamy A. Iyer1, W. Griffith Humphreys1, and Lisa J. Christopher1
1Department of Biotransformation-PCO, Bristol-Myers Squibb, Princeton, NJ, USA, 08543
2Chemical Synthesis, Bristol-Myers Squibb, New Brunswick, NJ, USA, 08903
BMS-690514 ((3R,4R)-4-amino-1-((4-((3-methoxyphenyl)amino)pyrrolo[2,1-f][1,2,4]triazin-5-yl)methyl)-3-piperidinol) is a potent inhibitor of ErbB and VEGF tyrosine kinases that is currently in clinical development for treatment of solid tumor cancers. From previous metabolism studies in human, BMS-690514 was primarily cleared by metabolism with the primary metabolic pathways being direct glucuronidation (M6), hydroxylation (M1, M2), and O-demethylation (M3). Incubations of [14C]BMS-690514 with a panel of cDNA-expressed human CYP enzymes showed that M1 and M2 were mainly formed by CYP3A4 and CYP2D6, whereas CYP2C9, 2C19 and 3A4 were capable of forming M3. The formation of M1 and M2 in human liver microsomes (HLM) was significantly inhibited by ketoconazole (a CYP3A4/5 inhibitor) and quinidine (a CYP2D6 inhibitor), whereas the formation of M3 was extensively inhibited by sulfaphenazole (a CYP2C9 inhibitor) and to a lesser extent ketoconazole, but was not appreciably inhibited by benzylnirvanol (a CYP2C19 inhibitor). Consistently, the intrinsic clearance (Vmax/km) in HLM, CYP3A4, CYP2D6 and CYP2C9 further confirmed that CYP3A4 and CYP2D6 were the two key CYP enzymes responsible for the formation of M1 and M2, while CYP2C9 was the major CYP enzyme for the formation of M3. In incubations with a panel of cDNA-expressed UGT enzymes, M6 was only formed by UGT2B4 and UGT2B7. The formation of M6 in HLM, UGT2B4 and UGT2B7 incubation mixtures was significantly inhibited by hyodeoxycholic acid (HDCA), fluconazole and Maxipost, competitive inhibitors of UGT2B7 or UGT2B4. These results confirmed that UGT2B4 and UGT2B7 were two major UGT enzymes responsible for the formation of M6. The formation of M6 was similar in pooled HLM and HLM obtained from UGT1A1*28*28 (UGT1A1 poor metabolizer), confirming that UGT1A1 was not involved in the formation of M6. In summary, reaction phenotyping experiments suggested that multiple enzymes (CYP3A4, CYP2D6, CYP2C9, UGT2B4 and UGT2B7) were involved in the biotransformation of BMS-690514. In vivo results demonstrated that the multiple enzymes determined as important in vitro did mediate important metabolic clearance reactions. This conclusion was supported by profiling of an individual bile sample from a CYP2D6 poor metabolizer, which suggested that when the metabolism through the CYP2D6 pathway was reduced, other metabolic pathways were able to participate in the metabolic clearance of BMS-690514. The overall conclusion of these experiments is that inhibition of one of these metabolic pathways either via co-administered drugs or gene polymorphisms is unlikely to result in significant (>2-fold) increases in BMS-690514 exposure.
281. Metabolism of BMS-561388 in rats, dogs, monkeys and humans
Ramaswamy A. Iyer, Peggy Liu-Kreyche, and Griff Humphreys
Department of Biotransformation-PCO, Bristol-Myers Squibb, Princeton, NJ, USA, 08543
Metabolism of BMS-561388 in rats, dogs, monkeys and humans Authors: R. Iyer, P. Liu-Kreyche, L. Zhou, J. Pursley, D. Parker, R. Dockens, R. Burrell, J. Easter, and W. G. Humphreys Bristol-Myers Squibb Company, Research & Development, Princeton, New Jersey Abstract BMS-561388 (N,N-bis(2-methoxyethyl)-8-(4-methoxy-2-methylphenyl)-2,7-dimethylpyrazolo[1,5-a][1,3,5]trazin-4-amine) is a potent CRF1 receptor antagonist. In previous studies, biotransformation profiling of BMS-561388 was carried out in liver microsomal and hepatocyte preparations from mouse, rat, dog, monkey and human, and in bile-duct cannulated rats. The major routes of metabolism were identified as O-demethylation, oxidation, N-dealkylation and glucuronidation. This study investigated the biotransformation of [14C]BMS-561388 following single oral dose to rats (60 mg/kg, 60 μCi/kg), dogs (50 mg/kg, 10 μCi/kg), monkeys (60 mg/kg, 40 μCi/kg) and humans (200 mg, 100 μCi). Urine, feces and plasma samples were collected and metabolites of [14C]BMS-561388 were profiled using HPLC-MS and radioactivity detection. Following a single oral dose to rats, dogs, monkeys and humans, compound-related radioactivity was eliminated both in urine and feces. Percent recoveries of radioactive dose in urine ranged from 14.5% in rats, 5.9% in dogs, 48.6% in monkeys and 50.4% in humans. Fecal recoveries ranged from 67.8% in rats, 82.6% in dogs, 30.1% in monkeys and 28.3% in humans. In Human, the maximum mean concentration of [14C]BMS-561388-derived radioactivity in plasma was observed at 1.5 h posedose. Plasma levels declined slowly after reaching maximum concentration. The Cmax for BMS-561388 was 1.7 μM, approximately 40% lower than that of total radioactivity (TRA). The AUC (INF) of BMS-561388 was only 5.7% that of TRA. These data indicated that metabolites contributed significantly to the circulating drug-related components. The plasma profiles were qualitatively similar across species. BMS-561388, M19 and M23 were the prominent circulating metabolites in plasma. In all species, except for dog, no parent compound was detected in urine and feces, indicating that BMS-561388 was extensively metabolized prior to elimination. Oxidative metabolism played a major role in the elimination of BMS-561388, suggesting that CYP enzymes may contribute significantly to the metabolism of BMS-561388.
282. Metabolism of BMS-562086 in rats, dogs and humans
Peggy Liu-Kreyche, and Ramaswamy A. Iyer
Department of Biotransformation-PCO, Bristol-Myers Squibb, Princeton, NJ, USA, 08543
Metabolism of BMS-562086 in rats, dogs and humans Authors: P. Liu-Kreyche, L. Zhou, D. Parker, J. Zeng, J. Pursley, R. Dockens, J. Sun, S. Traeger, D. Wu, R. Burrell, J. Easter, B. Brock, W. G. Humphreys and R. Iyer Bristol-Myers Squibb Company, Research & Development, Princeton, New Jersey Abstract BMS-562086 [Pexacerfont, 8-(6- methoxy-2-methl-2-methyl-3-pyridinyl)-2,7-dimethyl-N-((1R)-1-methylpropyl)pyrazolo[1,5-a][1,3,5]triazine-4-amine) is a potent CRF1 receptor antagonist. In previous studies, biotransformation profiling of BMS-562086 was carried out in liver microsomal and hepatocyte preparations from mouse, rat, dog, monkey and human, and in bile-duct cannulated rats. The major routes of metabolism were identified as O-demethylation, oxidation, N-dealkylation and glucuronidation. This study investigated the biotransformation of [14C]BMS-562086 following single oral dose to rats (33.3 mg/kg, 100 μCi/kg), dogs (30 mg/kg, 20 μCi/kg) and humans (100 mg, 100 μCi). Urine, feces and plasma samples were collected from each species at various time intervals. Radioactivity in urine, feces and plasma was quantified by liquid scintillation counting and metabolites of [14C]BMS-562086 were profiled using HPLC-MS and radioactivity detection. Percent radioactive dose recovered in urine ranged from 29.1% in rats, 17.8% in dogs, and 47.3% in humans. Percent radioactive dose recovered in feces ranged from 55.5% in rats, 65.7% in dogs and 40.8% in humans. The maximum concentration of [14C]BMS-562086-derived radioactivity in plasma was observed between 1 to 2 h postdose across species. Plasma concentrations declined slowly after reaching maximum concentrations. The overall exposure due to BMS-562086 was much lower than the total radioactivity (TRA), indicating that metabolites contributed significantly to TRA. Biotransformation profiles of plasma samples were qualitatively similar across species. The most prominent components in the plasma included unchanged parent drug and metabolites M12 and M16b. In urine samples, the prominent components detected included M3, M22, and M32, M1, M24 and M25. The fecal metabolite profiles varied across species. In human, unchanged parent and M35 were the prominent components. In rat and dog, M35 and M25 were the prominent components, respectively. In all species, oxidative metabolism played a major role in the elimination of BMS-562086, suggesting that CYP enzymes may contribute significantly to the metabolism of BMS-562086.
283. Bupropion metabolism by human and baboon hepatic and placental microsome
Xiaoming Wang1, Olga L. Zharikova1, Doaa R. Abdelrahman1, Svetlana Patrikeeva1, Gary D. V. Hankins1, Mahmoud S. Ahmed1, Tatiana N. Nanovskaya1, and Charles Timchalk2
1Department of Obstetrics & Gynecology, University of Texas Medical Branch at Galveston, Galveston, TX, USA, 77555-0587
2Msin:P7-59, Battelle-Pacific Northwest Lab, Richland, WA, 99352
Bupropion is an antidepressant that is also used for smoking cessation in men and non-pregnant women. Limited data on its pharmacokinetics during pregnancy and possible adverse effects on fetal growth and development prevents its use in treatment of the pregnant patient seeking smoking cessation. Therefore, information on its effectiveness and safety during pregnancy is needed. The aim of this investigation is to provide information on the metabolism of bupropion determined by human and baboon hepatic and placental microsomes. The baboon (Papio cynocephalus) was chosen to validate its use as a non-human primate animal model of human pregnancy well suited for investigations of bio-disposition of bupropion during pregnancy.
Methods: Term human placental and baboon placental and hepatic microsomes were prepared from villus and hepatic tissue homogenates by differential centrifugation. Human hepatic microsomes are commercially available and were purchased.
The metabolites of bupropion formed by the microsomes in reaction solutions were identified and quantified utilizing HPLC-UV.
Results: The data obtained in this investigation revealed that bupropion was biotransformed by human and baboon hepatic and placental microsomes to three major metabolites namely, hydroxybupropion, erythro- and threohydrobupropion. The metabolism of bupropion by human and baboon hepatic microsomes revealed saturation kinetics with apparent Km values of 78 ± 8 μM and 100 ± 12 μM, respectively. The major enzyme responsible for the hydroxylation of bupropion by human and baboon hepatic microsomes was CYP2B6. On the other hand, the major metabolite of bupropion formed by human and baboon placentas as well as baboon fetal microsomes was threohydrobupropion. These data suggest that the enzymes and metabolic pathway of bupropion metabolism by placenta and fetal liver were different from that by adult liver of both humans and baboons. Moreover, the formation of threohydrobupropion in placental tissue of smokers was significantly higher than in nonsmoking (p<0.05).
Conclusions: The data obtained suggest that hepatic and placental enzymes metabolizing bupropion are different and that the baboon could be used to investigate its bio-disposition during pregnancy.
284. Metabolism and disposition of [14C]brivanib alaninate after oral administration to rats, monkeys and humans
Jiachang Gong1, Jinping Gan1, S. Nilgun Comezoglu1, W. Griff Humphreys1, Janice Pursley1, Janet CaceresCortes1, Eric Masson2, Chi-Yuan Wu2, Alban Allentoff3, Michael Lago3, Scott Tran3, and Ramaswamy Iyer1
1Pco, Bristol-Myers Squibb, Princeton, NJ, USA, 08543
2Dmcp, Bristol-Myers Squibb, Princeton, NJ, USA, 08543
3Chemistry, Bristol-Myers Squibb, Princeton, NJ, USA, 08543
Brivanib is a novel dual inhibitor for both VEGF and FGF signaling pathways that regulate angiogenesis and tumor cell proliferation. Brivanib alaninate, the oral alanine prodrug of brivanib, is currently in phase III clinical trials in hepatocellular carcinoma and colorectal cancer. Preclinical and clinical studies have demonstrated that brivanib alaninate was highly effective and was well tolerated in humans when administrated on a once-a-day schedule. Following a single oral dose of [14C]brivanib alaninate to rats, monkeys and humans, the majority of the radioactivity was excreted through feces. Brivanib underwent extensive metabolism prior to excretion. The prominent biotransformatiom pathways include methyl oxidation, sulfation and O-dealkylation. Using recombinant enzymes and selective enzyme inhibitors, it was demonstrated that the oxidation of brivanib in humans was mainly catalyzed by CYP1A2 and 3A4, and multiple sulfotransferases involved in the sulfation of brivanib. Overall, brivanib was extensively metabolized in all species, and the metabolism was catalyzed by multiple enzymes.
285. A novel biotransformation of alkyl amino pyrrolidine to amino piperidine
Yuping Chen, Magang Shou, Dean Hickman, Gary Skiles, and Feng-Yin Hsieh
Pharmacokinetics and Drug Metabolism, Amgen Inc, Thousand Oaks, CA, USA, 91320
Here we describe a novel biotransformation of aminopyrrolidine to an aminopiperidine. AMG657417, 5-(4-chlorophenyl)-3-methyl-2-((2R)-2-(((1-methylethyl)amino)methyl)-1-pyrrolidinyl)-6-(4-pyridinyl)-4(3H)-pyrimidinone was incubated in human S9 fraction with NADPH. The major metabolite (M18) had a protonated molecular ion (m/z 438) identical to that of AMG657417, but it eluted earlier than the parent drug on a reverse phase HPLC. M18 had product ions at m/z 126 and 84, corresponding to isopropyl-amino-piperidinyl and amino-piperidinyl moieties, respectively, which were absent in the product ion mass spectrum of AMG657417. The structure of M18 was confirmed with a synthesized standard 5-(4-chlorophenyl)-3-methyl-2-(((3S)-1-(1-methylethyl)-3-piperidinyl)amino)-6-(4-pyridinyl)-4(3H)-pyrimidinone. The following reaction mechanism is proposed. An aminoaldehyde intermediate was formed from the pyrrolidinyl moiety of the parent molecule via hydroxylation and ring-opening. A piperidinyl iminium ion (6-membered ring) is subsequently formed via an intramolecular Schiff reaction between the aldehyde and the exocyclic isopropylamine nitrogen. This product is further reduced to the piperidine product (M18). The aldehyde intermediate was confirmed by the formation of an adduct (m/z 511) with semicarbazide in human liver microsomes (HLM), and S9 incubations of AMG657417. The Schiff base was confirmed when incubations of AMG657417 were supplemented with potassium cyanide (KCN) in HLM and S9. Cyanide conjugates (m/z 463) at both carbons alpha to the piperidine nitrogen were found indicating tautomeric iminium intermediates.
286. Characterization of human metabolic pathway of daphnetin: UDP-glucuronosyltransferase involved in the methylated daphnetin
Si-Cheng Liang1, Guang-Bo Ge1, Hui Xin Liu1, Jiang Wei Zhang1, Zhong-Ze Fang2, and Ling Yang3
1Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China, 116023
2Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China
3Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, AK, China
Daphnetin (7, 8-dihydroxycoumarin) was used for treatment against coagulation disorders since 1980s. Like other coumarin analogs, daphnetin has been found to exhibit broad biological effects including anti-inflammatory, antimicrobial, anti-malarial, anti-cancer activities. Previous studies indicated that daphnetin was metabolized rapidly in vivo. However its metabolic pathway in human is not fully characterized. According to the structure of daphnetin, there are two phenolic hydroxyl groups in the coumarin skeleton, indicating the potential phase II enzymes mediated clearance. Our previous study revealed that the UGT1A9 and UGT1A6 involved in the glucuronidation of daphnetin. In this study, we investigated other phase II metabolic pathway of daphnetin. One metabolite (M-1) was detected when daphnetin was incubated with human liver S9 in the presence of 3’-phosphoadenosine 5’-phosphosulfate, indicating the existence of methylation pathway of daphnetin by Catechol-O-methyltransferase (COMT). M-1 was indentified as the C-7 methylated daphnetin by liquid chromatography/mass spectrometry (LC/MS) and nuclear magnetic resonance (NMR). Notably, M-1 was found can be glucuronidated in human liver microsomes (HLMs) in the presence of UDPGA. Further study showed that UGT1A1, UGT1A3, UGT1A7, UGT1A8 and UGT1A9 involved in M-1 glucuronidation by using 12 commercially available recombinant human UGTs. These results indicated that daphnetin can be metabolized by COMT and the methylated metabolite (M-1) can be further metabolized by several UGTs, which revealed that the phase II metabolic pathway plays a crucial role in metabolic clearance of daphnetin.
287. Characterization of human UDP-Glucuronosyltransferase isoforms responsible for the in vitro glucuronidation of esculetin
Guang-Bo Ge, Si-Cheng Liang, Hui-Xin Liu, Yan-Yan Zhang, and Ling Yang
Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China, 116023
Esculetin (6,7-dihydroxycoumarin), is one of coumarin analogs isolated from various plants including Cichorium intybus, Artemisia scoparia and Fraxinus japonica Blume, has been found to exhibit broaden biological effects, such as analgesic, anti-inflammatory, anti-tumor, anti-arrhythmic, and scavenging activity against reactive oxygen species. Esculetin has been widely used in many Asia countries, and a previous study showed that it can be metabolized by recombinant human UDP-glucuronosyltransferase (UGT) 2B15. However, the metabolic pathway of esculetin in human has not been well-characterized. In the present study, the glucuronidation pathway of esculetin was investigated by using human liver microsomes (HLMs) and 12 commercially available recombinant human UGTs. The results showed that only one metabolite (M-1) was rapidly formed in the presence of UDPGA with HLMs, which was identified as a C-6 mono glucuronidation metabolite by liquid chromatography/mass spectrometry (LC/MS) and nuclear magnetic resonance (NMR), indicating that C-6 phenolic group of esculetin was a preferred glucuronidation site. Recombinant human UGTs study revealed that many UGT isoforms (including UGT1A6, UGT1A1, UGT1A7, UGT1A8 and UGT2B15) involved in C-6 glucuronidation of esculetin, and further study revealed that Km value of M-1 in human liver microsomes was 208 ± 31 μM. These results indicated that many UGT isoforms have contributed glucuronidation clearance of esculetin via the selective glucuronidation of C-6 phenolic group.
288. Contributions of enzyme(s) other than CYP2E1 to chlorzoxazone 6-hydroxylation in human liver microsomes
Drané O’Brien
Drug Metabolism and Pharmacokinetics, Boehringer-Ingelheim Pharmaceuticals Inc., Ridgefield, CT, USA, 06877
Historically, chlorzoxazone 6-hydroxylation has been used as a selective marker of CYP2E1 activity in in vitro assays, and clinically for the assessment of drug-drug interactions. However, there have been several reports in the literature suggesting that CYP2E1 may not be solely responsible for this reaction. Thus, we have performed a comprehensive characterization of chlorzoxazone 6-hydroxylation in human liver microsomes. In pooled human liver microsomes, 6-hydroxychlorzoxazone formation rates could not be adequately described by Michaelis Menten kinetics. A visual inspection of the Eadie-Hofstee plot and a comparison of the regression analyses using statistical methods suggested that at least two enzymes were involved. When studies were carried out in recombinant human CYP2E1, Michaelis Menten kinetics were observed. In order to determine which other enzyme(s) are involved in chlorzoxazone 6-hydroxylation in human liver microsomes, the following studies were performed:
Evaluation of non-P450 mediated chlorzoxazone 6-hydroxylation in heat treated (50°C) and untreated microsomes.
Reaction phenotyping using a variety of recombinant human cytochromes P450 (rCYPs) and a preliminary assessment of the relative contribution of each enzyme to the formation of 6-hydroxychlorzoxazone.
Determination of Km and Vmax values in incubations with rCYPs for each of the additional enzymes that contribute to chlorzoxazone 6-hydroxylation.
The Km and Vmax values were used together with reported hepatic abundances of each isoform to predict the in vivo contribution of each isoform to chlorzoxazone 6-hydroxylation. Based on these results, the suitability of chlorzoxazone as a selective probe substrate of CYP2E1 is questioned.
References:
- Ono S et al. Chlorzoxazone is metabolized by human CYP1A2 as well as by human CYP2E1. Pharmacogenetics. 1995; 5 143–150.
- Gorski CJ et al. Contribution of human CYP3A subfamily members to the 6-hydroxylation of chlorzoxazone. Xenobiotica. 1997; 3: 243–256.
289. Identification of the enzymes involved in the oxidative metabolism of saxagliptin and kinetics of formation of its major hydroxylated metabolite
Lisa J. Christopher1, Hong Su1, Anthony Barros Jr.1, Lifei Wang1, Kai Cao2, Samuel J. Bonacorsi Jr.2, Ramaswamy A. Iyer1, and William G. Humphreys1
1Department of Biotransformation, Bristol-Myers Squibb, Princeton, NJ, USA, 08543
2Department of Radiochemistry, Bristol-Myers Squibb, Princeton, NJ, USA, 08543
Saxagliptin ((S)-3-hydroxyadamantylglycine-L-cis-4,5-methanoprolinenitrile) is a potent, selective dipeptidyl peptidase-4 (DPP-4) inhibitor, specifically designed for extended inhibition of the DPP-4 enzyme. It is currently in Phase III clinical trials for the treatment of type-2 diabetes. Saxaglitpin is primarily cleared by oxidative metabolism and excretion of unchanged parent drug in the urine. BMS-510849 (M2), an active, hydroxylated metabolite is the major metabolite formed in vivo and in in vitro incubations of saxagliptin with human liver microsomes (HLM). The current study was conducted to determine the enzymes responsible for the oxidative metabolism of saxagliptin and use the information in conjunction with excretion data from the human ADME study to estimate the potential for drug interactions. In incubations with a panel of individually expressed CYP enzymes, saxagliptin was only metabolized by CYP3A4/5 to M2 and other minor monohydroxylated metabolites. Consistently, the formation of M2 in human liver microsomes was inhibited by ketoconazole (1 mM), troleandomycin (20 mM), 1-aminobenzotriazole (1-ABT, 1 mM), and an anti-CYP3A4 antibody. In incubations with HLM from 16 different donors, the rate of M2 formation closely correlated (r ≥0.949) with the reported CYP3A activity. Kinetic experiments indicated that the formation of M2 in HLM, and expressed CYP3A4 and CYP3A5 followed Michaelis-Menten kinetics. The catalytic efficiency (Vmax/Km) for CYP3A was about 4-fold higher than for CYP3A5. Therefore, it is likely that even in subjects with high expression levels of CYP3A5, the formation of M2 will be predominantly mediated by CYP3A4. The predicted AUCi/AUC ratios obtained with reaction phenotyping results in conjunction with data from a human ADME study with [14C]saxagliptin correlated well the actual findings from a clinical study with ketoconazole, in which the exposure of saxagliptin increased about 2.5-fold when co-administered with ketoconazole. Additional clinical drug-drug interaction studies showed a 2.1-fold increase in the AUC of saxagliptin when co-administered with diltiazem and a 76% reduction of saxagliptin AUC when co-administered with rifampin, confirming the major role of CYP3A in the clearance of saxagliptin.
290. Cis-resveratrol glucuronidation kinetics in human and recombinant UGT1A sources
Otito F. Iwuchukwu, and Swati Nagar
Pharmaceutical Sciences, Temple University, Philadelphia, PA, USA, 19140
Cis-resveratrol is the less studied geometric isomer of Resveratrol (3,4′,5-trihydroxy stilbene). We hypothesized that the contribution of its glucuronidation to the disposition of total resveratrol may be greater than previously assumed. To this end, we have fully characterized its glucuronidation kinetics in human liver and intestinal microsomes (HLM and HIM) as well as in recombinant human UGT1A1, 1A6, 1A9 and 1A10 Supersomes. Pooled human liver and intestinal microsomes and relevant human recombinant Supersomes were characterized for their catalytic activity towards cis-resveratrol with a validated RP- HPLC assay. Conditions for protein and time linearity were optimized. Substrate (0-2000uM), purified protein, MgCl2, alamethicin and UDPGA were incubated at 37°C for 60 minutes. Reactions were quenched with a methanolic solution of the internal standard acetaminophen. Data were analyzed by non-linear regression using GraphPad Prism 4. Relevant kinetic parameters were estimated for the major cis-resveratrol-3-O-glucuronide formed via atypical kinetic pathways in protein sources catalyzing its formation. Partial substrate inhibition was observed with apparent Vmax, Km and Ki of 6.1 ± 0.3/27.2 ± 1.2 nmol/min/mg, 415 ± 48.1/989.9 ± 92.8 uM and 789.6 ± 76.3/1012 ± 55.9 uM in HIM and UGT1A6 respectively (Data expressed as estimate ± standard error). Biphasic kinetics was observed in HLM. Sigmoidal kinetics were seen in UGT1A9, with estimates Vmax = 11.92 ± 0.2 nmol/min/mg; Km = 360uM; n = 1.27 ± 0.07. The 4′-O-glucuronide also exhibited atypical enzyme kinetics in HLM, HIM, UGT1A1 and 1A10 as determined by Eadie-Hofstee plots but kinetic parameters could not be estimated due to low rates of formation. UGT1A9 catalyzed 4′-O-glucuronide formation at higher substrate concentrations (> 200uM) via Michaelis-Menten kinetics (Vmax = 0.33 ± 0.015 nmol/min/mg; Km = 537.8 ± 67.8 uM). The rate of glucuronidation of cis-resveratrol-was seen to be higher than that of the trans isomer in HLM and UGT1A9Footnote1. In vivo formation clearances (and contribution to total resveratrol clearance) will be estimated in future studies.
291. Application of chimeric mice with humanized liver for study of human-specific drug metabolism
Thomas J. Bateman1, Vijay G.B. Reddy1, Yoshio Morikawa2, Masakazu Kakuni2, and Sanjeev Kumar1
1Drug Metabolism and Pharmacokinetics, Merck Research Laboratories, Rahway, NJ, USA, 07065
2PhoenixBio Co., Ltd., Higashihiroshima, Japan, 739-0046
Human-specific or disproportionately larger human metabolites of drug candidates that are not formed to a significant extent by the nonclinical species used for their toxicological evaluation (and, hence, are not adequately qualified in the preclinical safety assessment program) pose an important drug development challenge. This risk can be effectively mitigated if an accurate assessment of significant human metabolites of the drug candidate can be made early in the development program. However, the currently available in vitro models (e.g., liver microsomes, hepatocytes) do not always provide an adequate picture of the potential in vivo metabolic profile either due to the low metabolic turnover observed or lack of a good in vitro-in vivo correlation. Furthermore, the conduct of actual human ADME studies is an expensive and time-consuming endeavor that is more suited for later in development when the risk of failure has been reduced. We evaluated a recently developed chimeric mouse model with humanized liver for its ability to predict human disposition of four model drugs that are known to exhibit human-specific metabolism routes (Table).

The results from these studies demonstrate that chimeric mice were able to able to reproduce the human-specific metabolite profile for lamotrigine, MRK-A and diclofenac. In the case of propafenone, however, the human-specific C-5 hydroxylation was not detected as a predominant pathway and the metabolite profile in non-humanized vs humanized mice was similar; this could either be due to expression of suboptimal CYP2D6 activity or presence of residual propafenone-metabolizing mouse enzymes in chimeric mice. Overall, the data indicate that the chimeric mice with humanized liver have the potential to be a useful tool for the prediction of human-specific metabolism of xenobiotics and warrant further investigation.
292. Strategies to eliminate CYP3A4 inhibition and reactive metabolite formation by phenol-based pyrido[4,3-d]pyrimidin-4(3H)-one derivatives and orally active calcium sensing receptor antagonists
Jonathan N. Bauman, Amit S. Kalgutkar, Hao Sun, Miao Zhuang, Mary T. Didiuk, Hang T. Nguyen, and Kosea S. Frederick
Pharmacokinetics, Dynamics, and Metabolism, Pfizer Global Reseach and Development, Groton, CT, USA, 06340
Osteoporosis is a life-debilitating and prevalent disease associated with significant mortality and morbidity, particularly after hip fracture. Teriparatide (Forteo®), a synthetic 1-34 amino acid peptide fragment of human parathyroid hormone (PTH) is the only FDA-approved anabolic agent that, upon daily subcutaneous administration, has been shown to increase bone mineral density and reduce fracture rates in humans. As a viable alternative to subcutaneous PTH therapy, one can envision stimulation of the secretion of endogenous PTH from the parathyroid glands, which is regulated by the calcium sensing receptor (CaSR). Proof-of-concept in vivo studies in osteopenic, ovariectomized rats as well as humans with CaSR antagonists have demonstrated that blockade of CaSR results in transient PTH release and stimulation of new bone formation. Pyrido[4,3-d]pyrimidin-4(3H)-one derivative 1 is a de novo orally active CaSR antagonist currently in preclinical development for first-in-human studies. Two ADME/Toxicological liabilities with 1 include potent reversible CYP3A4 inhibition and propensity to form glutathione conjugates (consistent with reactive metabolite formation) in the presence of CYP3A4. While the low projected human dose mitigates CYP3A4 drug-drug interaction and immune-mediated ADR risks, we felt it was necessary to eliminate/reduce the liability in back-up compounds. Mechanistic studies on 1 indicated that bioactivation occurred on the pendant phenol ring and that the reactive species was an ortho-quinone derivative. Based on the information, back-up compounds were designed to eliminate/reduce reactive metabolite liability, while retaining primary pharmacology and other ADME attributes. Information from CYP3A4 molecular docking and Ab Initio calculations proved invaluable in the design strategy, which yielded several analogs with reduced propensity towards bioactivation and CYP inhibition. The collective results are summarized herein.
293. In Vitro investigation of the bioactivation potential of various substituted benzothiophenes
Mukesh K. Mahajan1, Rebecca L. Clark2, and Christopher A. Evans3
1Drug Metabolism and Pharmacokinetics, Immuno-Inflammation Centre of Excellence for Drug Discovery, GlaxoSmithKline, King of Prussia, PA, USA, 19426
2Waters, Beverly, MA, USA, 01915
3Drug Metabolism and Pharmacokinetics, Preclinical Developement, GlaxoSmithKline, King of Prussia, PA, USA, 19426
The link between metabolic activation leading to the formation of reactive electrophilic intermediates and occurrence of idiosyncratic adverse drug reactions has forced the withdrawal of several marketed drugs. Therefore, it has become common practice to screen new-chemical entities for the formation of reactive electrophilic intermediates early in drug discovery. This laboratory previously reported the formation of reactive arene oxide intermediates on a series of benzothiophene-containing discovery compounds. In an effort to further substantiate the involvement of substituted benzothiophene moieties in the formation of reactive arene oxide intermediates, nearly 30 commercially available benzothiophene containing drug-like compounds (average MW – 362.1 and average cLogP – 3.6) were selected for investigation. These compounds were incubated with fresh rat hepatocytes and subjected to various LCMS techniques to screen for evidence of reactive intermediate formation derived from the various substituted benzothiophenes. The culmination of the data generated suggest the formation of reactive arene oxide pathways for a multitude of the compounds; as indicated by the formation of dihydrodiol and glutathione conjugation byproducts. When detected, these byproducts were then subjected to accurate mass LC-MS/MS structure elucidation experiments in an attempt to further correlate the arene oxide intermediate with the benzothiophene moiety; in most instances formation correlated with the benzothiophene. It is postulated that the electrophilic intermediate is formed through the initial P450-catalyzed epoxidation of the benzothiophene moiety, followed by nucleophilic attack of water or glutathione. Extending our previous findings, this new dataset broadly indicate the increased propensity of substituted benzothiophenes to undergo bioactivation through a reactive arene oxide intermediate.
294. Determination of the stereochemistry of two CYP2C19-mediated metabolites M4 and M5 of CVT-3619
Nancy I. Chu1, Jia Hao1, Tetsuya Kobayashi2, Elfatih Elzein2, Jeff Zablocki2, and Kwan H. Leung1
1Pre-Clinical Development, Gilead Sciences, Inc., Palo Alto, CA, USA, 94304
2Medicinal Chemistry, Gilead Sciences, Inc., Palo Alto, CA, USA, 94304
CVT-3619 [2-{(6-[((1R, 2R)-2-hydroxycyclopentyl)amino]purin)-yl}-(4S, 5S, 2R, 3R)-5-[(2-fluorophenylthio)methyl]oxolane-3,4-diol)] is a partial adenosine A1 receptor agonist being developed for the potential treatment of hypertriglyceridemia. Four major Phase I metabolites have been identified following in vitro and in vivo studies in rats, dogs and humans. M1 and M3 are CVT-3619 enantiomeric sulfoxides mediated by CYP3A4 whereas M4 and M5 are monohydroxylated products at the 2-hydroxycyclopentyl ring of CVT-3619 mediated by CYP2C19. ref. 1,2 There are 3 possible positions (*) on the 2-hydroxycyclopentyl ring that could be hydroxylated and the hydroxyl group could exist either as R or S; hence there are 6 possible structures in all.
To determine the stereochemistry of M4 and M5, all six possible stereoisomers were synthesized, mostly as mixtures of diastereoisomers. M4 and M5 were also generated using recombinant human CYP2C19 expressed in E.coli. The stereochemistry of M4 and M5 were determined using (a), NMR spectroscopy; (b), HPLC retention time following chiral separation using supercritical fluid chromatography. Results indicate that M5 is ((3R,4S,5S)-2-{6-[(1R,2R,5S)-2,5-dihydroxycyclo pentylamino]-9H-purin-9-yl}-5-[(2-fluorophenylthio) methyl]tetrahydrofuran-3,4-diol) and M4 may be ((3R,4S,5S)-2-{6-[(1R,2R,4R)-2,4-dihydroxycyclopentylamino]-9H-purin-9-yl}-5- [(2-fluorophenylthio) methyl] tetra hydro furan-3,4-diol). M4’s definitive structure will be confirmed by X-ray crystallography. Structures:
References
- In Vitro metabolism of CVT-3619. 15th Northern America Regional Meeting 2008 ISSX Poster. N. Chu, J. Hao, D. Soohoo, D. Lustig, L. Bajpai, N. Mollova and K. H. Leung.
- Biotransformation of CVT-3619. 15th Northern America Regional Meeting 2008 ISSX Poster. L. Bajpai, N. Mollova, D. Lustig, D.Soohoo, J. Hao, E. Elzein, N. Chu and K. H. Leung
295. Hepatic metabolism of cediranib, a potent VEGF inhibitor: Interspecies comparison and human enzymology
Michael Spear, Chris RJ. Pollard, Christine Pattison, Sarah Kelly, Helen Rollison, Michelle Ward, Mike P. Harrison, and Timothy Schulz-Utermoehl
Clinical Pharmacology and DMPK, AstraZeneca, Macclesfield, United Kingdom
The metabolism of cediranib (4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-[3-(1-pyrrolidinyl)propoxy]quinazoline; RECENTIN(tm)), a vascular endothelial growth factor (VEGF) tyrosine kinase inhibitor (TKI) of all three VEGF receptors in phase III development for the first line treatment of colorectal cancer and the treatment of recurrent glioblastoma was investigated in human in vitro systems using radiolabeled material and compared with that in a range of pre-clinical species. The drug-metabolising enzymes involved in the metabolism of cediranib were also identified. Cediranib primarily underwent oxidation (on the propyl pyrrolidine side chain and the indole ring) and N-glucuronidation in human hepatocytes to form five metabolites, with the N-glucuronide metabolite being the major metabolite. All metabolites with the exception of the N-glucuronide metabolite were observed in hepatocytes from rat and cynomolgus monkey pre-clinical species. Further metabolism studies in liver microsomes from these or other preclinical species (CD-1 mouse, Han Wistar rat, Dunkin Hartley guinea-pig, Göttingen mini-pig, New Zealand White rabbit, Beagle dog, cynomolgus and rhesus monkey) indicated that the N-glucuronide metabolite was also not formed in these species. Based on results of cytochrome P450 (CYP450) non-selective chemical inhibition using 1-aminobenzotriazole and metabolism by recombinant flavin-containing monooxygenase (FMO) and uridine glucuronosyltransferase (UGT) enzymes studies, FMO1 and FMO3 contributed to cediranib N-oxidation, whilst UGT1A4 had a major role in cediranib N-glucuronidation. In conclusion, with the exception of the N-glucuronide metabolite, all human metabolites of cediranib were observed following incubation with rat and cynomolgus monkey hepatocytes. Non-CYP450 enzymes are predominantly involved in the metabolism of cediranib, suggesting that clinical drug interactions involving this system are unlikely.
296. Characterisation and identification of the N-glucuronide metabolite of cediranib
Eva Lenz1, Chris Drake2, Michael Spear1, Sarah Kelly1, Mike P. Harrison1, and Timothy Schulz-Utermoehl1
1Clinical Pharmacology and DMPK, AstraZeneca, Macclesfield, United Kingdom
2Novacta Biosystems Ltd, Welwyn Garden City, United Kingdom
Cediranib (4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-[3-(1-pyrrolidinyl)propoxy]quinazoline; RECENTIN(tm)), a vascular endothelial growth factor (VEGF) tyrosine kinase inhibitor (TKI) of all three VEGF receptors, is currently in phase III clinical trials for the first-line treatment of colorectal cancer and the treatment of recurrent glioblastoma. It has been determined during its clinical development that an N-glucuronide metabolite was a major circulating metabolite. Given the possibility of four sites for the conjugation of the glucuronic acid moiety, determination of the location of the conjugation site on cediranib was warranted. A small quantity of the N-glucuronide metabolite of cediranib was generated using recombinant human uridine glucuronosyltransferase 1A4 (UGT1A4) enzymes. The metabolite generated was characterised and identified by HPLC with ultraviolet (HPLC-UV) and mass spectrometric (HPLC-MSn) detection and by nuclear magnetic resonance (NMR) spectroscopy. The N-glucuronide metabolite generated in the UGT1A4 scale-up experiment was confirmed by HPLC-MSn to have the same retention time, molecular mass and mass fragmentation data as a reference sample of the metabolite generated in a human liver microsomal incubate sample. Using 1H-NMR analysis, a characteristic anomeric doublet at approximately 4.7 ppm was observed. In a series of selective Rotating frame Overhauser Effect Spectroscopy (ROESY) experiments, irradiation of the anomeric proton at 4.7 ppm showed no enhancement in the aromatic region of the spectrum, but enhancement in the aliphatic region. A 2D 1H-1H Correlation Spectroscopy (COSY) experiment confirmed the identity of these enhanced signals. In conclusion, the site of glucuronidation on cediranib was on the pyrrolidine nitrogen and not on the aromatic nitrogens, suggesting that cediranib N+-glucuronide formation is only likely in human and higher primates (great apes). Significant species differences exist in the N+-glucuronidation of cyclic tertiary amines.
297. Monitoring metabolic adaptation of HepG2 cells to achieve new steady state of oxygen consumption
Nathan Boggs1, Huong Le1, Julia Patrone1, Mellisa Theodore1, Holly Williams2, Stanley Strawbridge2, and Jonathan Boyd2
1Applied Physics Lab, Johns Hopkins Univ., Laurel, MD, USA, 20723
2Bennett Department of Chemistry, West Virginia University, Morgantown, WV, USA, 26506
Energy metabolism is a crucial element of the toxicodynamic response of cells to chemical insults, and is related to pathways of both proliferation and apoptosis. Understanding metabolic response to insults, including adaptation to achieve a new ‘steady state’ of energy production following a chemical challenge, is vital to understanding the potential toxicodynamic interactions which may exist between low doses of compound mixtures. To this end, we defined metabolic steady state as a function of respiration and monitored the 24 hour oxygen consumption patterns of whole HepG2 cells post exposure to low doses (< 10 nM) of mitochondrial inhibitors and uncouplers alone (rotenone, deguelin, pyridaben, potassium cyanide, oligomycin, antimycin a, chloroform, TTFA and FCCP), and in combination with a glycolysis inhibitor (2-deoxy-D-glucose). Our hypothesis is that by monitoring the oxygen consumption of cells, we may be able to define critical time points in the toxicodynamic response necessary for determination of potential interaction pathways (between mixtures) that may be further explored by alternative proteomic techniques. This study utilizes an extracellular fluorescent oxygen probe (A65N) that offers near real-time elucidation of metabolic response as a function of dose and time, resulting in a unique means to define key time points and metabolic trajectories associated with acute cellular response. In this study we demonstrate the ability to extend the measurement timeframe to 24 hours, and clearly identify significant metabolic responses at all non-lethal doses. Additionally, co-exposure with 2-deoxy-D-glucose results in unique respiratory patterns (compared to inhibitor or uncoupler alone) which could indicate alternate metabolic and ultimately toxicodynamic response to mixtures. In conclusion, we provide data that suggest a commercially available oxygen consumption assay can be used as a first step in mechanistic studies to outline the time course of acute toxicodynamic response to metabolic insults.
298. Probabilistic model of regioselectivity of metabolism in human liver microsomes
Justas Dapkunas, Andrius Sazonovas, and Pranas Japertas
ACD/Labs, Inc., Vilnius, Lithuania, LT-08117
Here we present a model for in silico prediction of the most probable sites of human liver microsomal metabolism in a molecule. The developed models calculate the probabilities of being a target of human cytochrome P450 enzymes (CYP3A4, CYP2D6, CYP2C9, CYP2C19, CYP1A2) for any atom in a molecule and allow forecasting of the most probable phase I metabolites. The novel GALAS (Global, Adjusted Locally According to Similarity) modeling methodology was used for development of probabilistic models. It provides a possibility to estimate the reliability of prediction. Moreover, the Applicability Domain of the models can be easily expanded to cover compound classes of user interest by incorporating ‘in house’ databases containing experimental metabolism data. Experimental data for >600 compounds with >6000 different carbon atoms were used for modeling. Four baseline models were developed for four types of atoms (aromatic carbon, aliphatic carbon, carbon near nitrogen, carbon near oxygen). GALAS modeling methodology finds most similar metabolism sites in the training set, according to which corrections to the baseline predictions are made and final prediction quality is estimated in the form of Reliability Index (RI). The numbers of mispredictions and inconclusive results reduce significantly when only results of high quality (RI>0.5) are taken into account, demonstrating that RI reflects accuracy of prediction. The regioselectivity models are shown to be trainable using experimental data for compounds not present in the training set.
299. Ring-Opening of carbamates in rat liver microsomes: A mechanisitic study
Silvi Ann Chacko1, Mary Grubb2, and Jonathan Josephs3
1Biotransformation, Bristol-Myers Squibb, Pennington, NJ, USA, 08534
2Biotransformation, Bristol-Myers Squibb Co, Princeton, NJ, USA, 08543
3Biotransformation, Bristol-Myers Squibb Research&Development, Princeton, NJ, USA, 8540
Carbamate esters have been widely used as insecticides and to a lesser extent as pharmaceutical agents. Metabolism of carbamate esters usually involve a combination of hydrolysis, conjugation and oxidation reactions. In our study, an integrated approach was used to carry out in vitro metabolite characterization and quantitation of compounds containing cyclic carbamate esters. Incubations were conducted in an automated fashion using a Tecan platform. Compounds were incubated at 0.5 and 30 μM with liver microsomes (0.5 mg/mL) and 2 mM NADPH. Aliquots were removed at timepoints (0 to 60 for 0.5 μM, 0 and 45 min for 30 μM). The reaction was terminated by the addition of an equal volume of acetonitrile. Following centrifugation of quenched samples, metabolites were analysed by LC-MS using an electrospray source operated in the positive ion mode. Quantitative analysis was carried out for parent drug and metabolites in the 0 to 60 min samples of the 0.5 μM incubates using extracted ion chromatogram from full scan MS data. Qualitative analysis of the metabolites was done with the 45 min samples of the 30 μM incubates. High resolution MS data for identification of metabolites was achieved using an LTQ Orbitrap (Thermo). Among the series of the carbamates studied, ring opening to form the alcohol and the aldehyde via oxidation was unique to rodent liver microsomes and was not observed in dog or human liver microsomes. Two possible pathways for ring opening of the carbamate esters are either hydrolysis or oxidation alpha to the carbonyl resulting in an unstable intermediate. However, an analog containing two deuterium atoms at the alpha position resulted in loss of only one, favoring oxidation. Additionally, the ring-opened metabolite does not occur in incubates not containing NADPH. A final piece of evidence is that in metabolite concentration-time profiles, the aldehyde appears prior to the hydroxylated ring-opened metabolite, suggesting subsequent reduction of the aldehyde that is initially formed.
300. Assessment of pharmacokinetic linearity of metabolites from a microdose to a normal dose
Carmai Seto1, Takeo Sakuma1, Jinsong Ni2, Fred Ouyang3, Louis Lo3, Devin Welty3, Gabriella Szekely-Klepser4, and Andrew Acheampong5
1MDS Analytical Technologies, Concord, ON, Canada, L4K 4V8
2Dept of PKDM/RD-2B, Allergan, Irvine, CA, 92612
3Dept of PKDM/RD-2B, Allergan, Irvine, CA, USA, 92612
4Allergan, Irvine, CA, USA, 92612
5Bioanalytical Mass Spectrometry, Allergan, Irvine, CA, USA, 92612
Microdosing studies may not predict the pharmacokinetic behavior of a drug at clinical doses due to nonlinear pharmacokinetics between a microdose and a clinically-relevant dose. Non-linear exposure may result from dose-dependent effects on absorption, metabolism, or saturation and /or inhibition of other elimination pathways. Identifying unique and major human metabolites early in the drug development process allows for timely assessment of potential safety issues and therapeutically active metabolites. The work presented here investigates the use of a new fast scanning hybrid mass spectrometer with enhanced LIT (define acronym) sensitivity for the quantification of metabolites in microdosing studies in rats.
Methods: PK Studies: Atorvastatin, methimazole, ofloxacin, omeprazole, and tamoxifen were administered to jugular vein-cannulated male Sprague-Dawley rats via oral gavage. All compounds except atorvastatin were administered at two different doses, 1.67 μg/kg (microdose) and 5000 μg/kg (normal dose). Atorvastatin was administered at four different doses, 1.67, 25, 350 and 5000 μg/kg. Plasma samples were taken at five different time points and were prepared using protein precipitation, dried and reconstituted for analysis. Urine samples were collected for 24 hours and were prepared by centrifugation and dilution. LC/MS/MS Analysis: Multiple Reaction Monitoring-Information Dependant Acquisition (MRM-IDA) was used to quantify metabolites in plasma and urine. A method was created for each compound using the automated method creation tool in LightSight® software. The analyses were conducted using a QTRAP® 5500 LC/MS/MS system.
Preliminary data: Following metabolite detection in either the microdose and/or normal dose, pharmacokinetic curves were generated for that metabolite at both doses. Pharmacokinetic curves were generated using the MRM survey scan of the method. Semi-quantitation was used to estimate the levels of the metabolites which do not have synthetic reference standard for quantitation. The MS/MS confirmation of the MRM-IDA was necessary to confirm the identity of the metabolite without synthetic standard. The pharmacokinetic curves were then used to determine if there is linear exposure for the metabolites between the microdose and normal dose. Using the pharmacokinetic data we were able to calculate some pharmacokinetics parameters such as half life, AUC, Cmax, etc.
For all compounds, metabolites were detected in plasma and urine at microdosing levels. For example - atorvastatin, several metabolites were detected in the microdose. Two of these metabolites (p- and o-hydroxyatorvstatin) appear to have linear exposure between the microdose and normal dose, while another metabolite (atorvastatin lactone) did not. Conclusions: Quantification of metabolites in plasma and urine at microdosing levels can be achieved using the AB SCIEX QTRAP® 5500 system. Using this system, it was observed that the metabolic profile between the normal and microdose is compound dependant. As well, the linearity of pharmacokinetics of metabolites between a normal and microdose is individual metabolite dependant.
301. The absorption, distribution, metabolism and excretion of HPN-100 in the cynomolgus monkey; A novel pharmaceutical under development for the potential treatment of hyperammonemia
Webber Colin1, Shaun Johnson1, Brian A. John1, and Michael J. Lees2
1Drug Metabolism, Huntingdon Life Sciences, Huntingdon, United Kingdom
2Bioanalysis, Huntingdon Life Sciences, Huntingdon, United Kingdom
HPN-100 (glyceryl tri-(4-phenylbutrate)) is a triglyceride oil containing three molecules of phenyl butyric acid (PBA) linked to a glycerol backbone. HPN-100 is considered a pro-drug of PBA and a pre-pro-drug of phenyl acetic acid (PAA) which scavenges nitrogenous substances in the form of glutamine (phenylacetyl glutamine, PAGN) and provides an alternate pathway to the urea cycle for the elimination of waste nitrogen. Like humans, the cynomolgus monkey also metabolises PAA to PAGN and so provides a suitable model to assess the disposition and safety profile of HPN-100. This poster compares aspects of the absorption, distribution, metabolism and excretion of [14C]HPN-100 with that of [14C]PBA. Studies were conducted after a single oral (gavage) dose of neat oil [14C]HPN100 (600 mg PBA equivalents/kg) to three male animals. Data were compared with that following both oral (600 mg/kg) and intravenous (150 mg/kg) administration of an aqueous (or saline) solution of [14C]PBA to the same three animals. Concentrations of parent molecules (HPN-100 and PBA) and known metabolites (PAA, PAGN, PAG, PBG and PBGN) in plasma, were measured using a validated LC-MS/MS assay following intravenous administration of PBA and oral administration of HPN100. Metabolites in plasma, urine, faeces, bile, liver and kidney were assessed using radiochromatographic techniques and tissue concentrations of radioactivity were measured following [14C]HPN100 dosing. HPN100 related material (ie 14C-radioactivity) was absorbed from the gut into the systemic circulation with a mean Tmax of 8 hours, but known metabolites were measurable in the plasma by 1.5 hours post-dose indicating rapid metabolism once absorbed. HPN100 related material was widely distributed with tissue concentrations generally highest at 8 hours post-dose. Drug-related material was predominately cleared renally with PAGN representing ca 88% of the urinary radioactivity (ca 39% dose). The data to be presented demonstrated that HPN100 acts as a slow release source of PBA and appears to give increased exposure to PBA and its known metabolites (PAA and PAGN) compared to administration of PBA.
302. In Vitro Hepatic Metabolism of CMX001, a Lipid Conjugate of Cidofovir
Timothy K. Tippin, Merrick R. Almond, George R. Painter, and Bernhard M. Lampert
Chimerix, Inc., Durham, NC, USA, 27713
The lipid conjugate of the acyclic nucleoside phosphonate cidofovir, phosphonic acid, [[(S)-2-(4-amino-2-oxo-1(2H)-pyrimidinyl)-1-(hydroxymethyl)ethoxy]methyl]mono[3-(hexadecyloxy) propyl]ester (synonym: hexadecyloxypropyl-cidofovir, CMX001), is efficacious in cell-based and animal models of double-stranded DNA viral infection. The purpose of the present study was to identify the metabolites produced by hepatocytes, and to determine which liver enzymes are involved in the degradation of CMX001. After incubation of 14C- CMX001 with human hepatocytes and analysis by LC-MS-MS/radiochemical detection, the most abundant metabolite measured was cidofovir comprising 33% of the total radioactivity administered to the cells. An additional three metabolites were identified, including a hexadecyl alkyl chain-shortened carboxylic acid (CMX064, 11%), a metabolite monohydroxylated on the hexadecyl alkyl chain (CMX108, 3%), and the glucuronide conjugate of CMX108 (6%). These four metabolites were also produced in mouse, rat, rabbit and monkey hepatocytes. Using the intrinsic clearance value derived from monkey hepatocytes, an accurate prediction of in vivo monkey plasma clearance was obtained (19 versus 22 mL/min/kg). Multiple cytochrome P450 (CYP) enzymes were involved in the metabolism of CMX001, including CYP3A4, CYP2C19, and CYP2C8, based on a significant decrease in metabolism of CMX001 by human liver microsomes in the presence of selective inhibitors of these isozymes. These data suggest that liver metabolism is likely to play a major role in CMX001 elimination, and that CYP enzymes are involved in the metabolism of CMX001 in humans and other animal species.
303. A comparative metabolic study of the phase I and II metabolism of haloperidol and its silicon analogue sila-haloperidol
Tove Johansson1, Lars Weidolf2, Friedrich Popp3, Reinhold Tacke3, and Ulrik Jurva4
1Discovery DMPK, AstraZeneca R&D Molndal/University of Gothenburg, Molndal, Sweden, 431 83
2Clinical Pharmacology & DMPK, AstraZeneca R&D Molndal, Molndal, Sweden, SE-431 83
3University of Würzburg, Würzburg, Germany
4Lead Generation, AstraZeneca R&D Molndal, Molndal, Sweden
The pyridinium metabolite has been proposed to be the cause of the severe side effects of haloperidol, including parkinsonism and tardive dyskinesia. A silicon analogue of haloperidol (sila-haloperidol) has been synthesized where the carbon at the 4-position of the piperidine ring of haloperidol was replaced by a silicon atom. The aim of the study was to investigate how this carbon/silicon exchange would influence the metabolic fate of the compound, e.g. the pathway to form the pyridinium ion. The metabolism was studied in rat and human liver microsomes as well as rat, dog and human hepatocytes. For the metabolite identification, an ultra-performance liquid chromatography system was used in combination with high resolution mass spectrometry. The pyridinium metabolite was a major metabolite of haloperidol. For sila-haloperidol, the corresponding (sila)pyridinium metabolite was not formed. Instead, three metabolites originating from opening of the piperidine ring were formed, a mechanism that has not been observed for haloperidol. One of the significant phase II metabolites of haloperidol was the glucuronide of the hydroxy group bound to the piperidine ring. For sila-haloperidol the corresponding glucuronidation of the silanol function (SiOH) was not observed in the hepatocyte incubations. To further support the observed differences in metabolic pathways between haloperidol and sila-haloperidol, the phase I and phase II metabolism of another pair of structurally related carbon/silicon analogues were investigated. In these studies with trifluperidol and sila-trifluperidol the same differences in metabolic pathways were observed as between haloperidol and sila-haloperidol. In conclusion, by replacing one single carbon atom in haloperidol by a silicon atom the metabolic pathways, including the pyridinium metabolite formation but also the glucuronide formation, changed dramatically. In addition to these significant changes in metabolic pathways, this study represents a metabolic investigation of silicon drug analogues in different in vitro systems.
304. Hydroxylation of liquiritigenin in human liver microsomes is specifically catalyzed by cytochrome P450 1A2
Hui-Xin Liu, Ying Hu, and Ling Yang
Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China, 116023
Flavonoids are part of a family of naturally occurring polyphenolic compounds and represent one of the most prevalent classes of compounds in vegetables, nuts, fruits and beverages such as coffee, tea, and red wine as well as medical herbs. More than 8000 compounds with a flavonoid structure have been identified and categorized into flavonols, flavones, flavanones, isoflavones, catechins, anthocyanidins, and chalcones. Liquiritigenin (7,4’-dihydroxyflavanone) is found most notably in Glycyrrhizae radix (G. radix), which is one of the oldest and most frequently used botanicals in the polyherbal preparations. G. radix exerts anticancer, antitussive, expectorant and antipyrotic actions and is often used to treat cough, pharyngitis, bronchitis, bronchial asthma. Liquiritigenin have been shown to have a inhibition of xanthine oxidase activity in vitro, dose related anti-allergic activities, and growth-inhibitory effect on cancer cell. It also has the anti-angiogenic effect, and antioxidant, anti-inflammatory activities. Six oxidative metabolites of liquiritigenin (7,3’,4’-trihydroxyflavone, a hydroxyl quinine metabolite, two A-ring dihydroxymetabolites, 7,4’-dihydroxyflavone, and 7-hydroxychromone) have been detected in rat liver microsomes (HLMs), and one CYP3A4-catalyzed metabolite (7,4’-dihydroxyflavone) has been identified in human liver microsomes (HLMs) recently. In the present study, a novel mono-hydroxylated metabolite was detected in reaction catalyzed by HLMs, and was identified as naringenin (4’,5,7-Trihydroxyflavanone) by comparing the tandem mass spectra and the chromatographic retention time with that of the standard compound. To identify and characterize the CYP isozymes involved in liquiritigenin hydroxylation, we examined liquiritigenin hydroxylation activities in HLM and rCYP incubation systems and compared the enzyme kinetic parameters between them. In addition, CYP reaction phenotyping of liquiritigenin was performed using a combination of three basic approaches as described by Bjornsson et al.: to determine whether heterologously expressed rCYPs were capable of metabolizing liquiritigenin; to examine the metabolic reaction in the absence and presence of CYP-selective chemical inhibitors; and to correlate the rate of the reaction with a CYP-selective marker activity across a panel of well characterized liver microsomal samples from individual donors. The kinetic profile followed Michaelis-Menten kinetics in HLM incubation system. The Eadie-Hofstee plot for liquiritigenin hydroxylation by HLMs was monophasic, suggesting that a single CYP isozyme is responsible for the hydroxylation. Furthermore, the Km value of liquiritigenin hydroxylation in HLMs was very close to that in rCYP2A6 (24.3 versus 25.6 μM). Among recombinant CYP isozymes examined in the present study, only the CYP1A2 isozyme exhibited liquiritigenin hydroxylation activity. In addition, the activities of liquiritigenin hydroxylation in 11 individual human liver microsomes were significantly correlated with the activities toward phenacetin, a proposed CYP1A2-selective probe substrate. Moreover, it is noteworthy that in HLMs, only furafylline (CYP1A2-selective inhibitor), but not other CYP-selective chemical inhibitors, inhibited the production of metabolite almost completely. These results suggest that CYP1A2 would specifically catalyze the liquiritigenin hydroxylation in human liver microsomes. Idenficiation of CYP1A2 as being responsible for liquiritigenin hydroxylation will greatly improve future investigations of CYP1A2 interindividual differences associated with liquiritigenin clinical trials and the magnitude of drug-drug interactions.
305. Effects of phenobarbital on thyroid hormone catabolism in rat hepatocytes
Vicki M. Richardson, and Michael J. DeVito
ORD/Nheerl/Istd/PB, USEPA, Research Triangle Park, NC, USA, 27711
Hepatic enzyme inducers such as phenobarbital (PB) decrease circulating thyroid hormone (TH) concentrations in rodents. The induction of hepatic xenobiotic metabolizing enzymes by PB increases the catabolism and biliary elimination of thyroid hormones. It is this study’s objective to examine the catabolism and clearance of THs by using an in vitro method. Sandwich-cultured hepatocytes from male Sprague-Dawley rats were treated for 72 hours in serum-free media with or without 1mM PB. After removal of media, 0.05 uM [I125]-T4, which approximates rat serum thyroxine concentrations, was added to rat hepatocytes and incubated for up to 24 hours at 37°C. After 24 hours, media was removed and cell lysates were collected for analysis. 4.2 and 5.3% of total I125 was shown to accumulate within hepatocytes pretreated with 0mM or 1mM PB, respectively. To examine T4 metabolites in media of PB-treated hepatocytes, the media was analyzed using Ultra Performance Liquid Chromatography (UPLC). Eluent from the UPLC was allowed to flow through a fraction collector and the fractions were then counted on a gamma counter. T4-glucuronide (T4G) was the predominant metabolite found in the media of control and PB-treated hepatocytes. In comparison to control, T4G in media increased 90% following PB treatment. The results indicate that pretreatment with 1mM PB increased the accumulation of I125 into rat hepatocytes and increased the formation of a major thyroid hormone metabolite, T4G. These observations also demonstrate that rat hepatocytes may be useful for screening thyroid hormone disruptors. (This is an abstract of a proposed presentation and does not necessarily reflect USEPA policy.)
306. Investigation of the esterases involved in the in vitro metabolism of Compound X by rat, minipig and human
Danny S. Paul, Stephane Barassin, Anthony K. Bartlett, and Michael Hall
Dept of In Vitro Metabolism, Huntingdon Life Sciences Ltd, Cambridgeshire, United Kingdom, PE28 4HS
Compound X is a short-acting intravenous anaesthetic currently under clinical development. The purpose of this study was to identify the esterases that make the most significant contribution to the metabolism of [14C]Compound X to its de-esterified product, Compound Y in vitro. This was attempted by incubating [14C]Compound X with liver microsomes and whole-blood from Han Wistar rat, Göttingen minipig and human, and plasma from human only, in the presence of selective esterase inhibitors. On the basis of the results of a series of preliminary incubations, 20 μM (minipig and human) and 200 μM (rat) [14C]Compound X was incubated in the presence of huperzine A (0.05, 0.5 and 5 μM; acetylcholinesterase inhibitor), tetra(monoisopropyl)pyrophosphortetramide (iso-OMPA; 1, 10 and 100 μM; butyrylcholinesterase inhibitor), EDTA (1, 10 and 25 mM; aryldialkylphosphatase (paraoxonase) inhibitor), bomin-1 (0.5, 5 and 50 μM; carboxylesterase inhibitor) and diazepam (10, 25 and 50 μM; an example ligand of albumin). In order to generate measurable production of Compound Y, rat liver microsomes were incubated for 1 minute, rat whole-blood for 5 minutes, minipig and human liver microsomes for 10 minutes and minipig and human whole-blood and plasma for 60 minutes. At the end of the incubations, samples were extracted and analysed by radio-HPLC. >70% inhibition of formation of Compound Y was elicited by incubation of 100 μM iso-OMPA with human whole-blood and plasma and 25 mM EDTA with rat liver microsomes. >25 - ≤70 % inhibition of formation of Compound Y was elicited by incubation of 0.05 μM and 0.5 μM huperzine A with rat whole-blood and 5 μM huperzine A with rat liver microsomes and minipig whole-blood; 1 μM iso-OMPA with rat liver microsomes, 10 μM and 100 μM iso-OMPA with minipig whole-blood and 100 μM iso-OMPA with human liver microsomes; 10 mM and 25 mM EDTA with human liver microsomes and 25 mM EDTA with rat whole-blood. All other co-incubations, including those of bomin-1 and diazepam with all matrices elicited ≤25% inhibition of formation of Compound Y. These data suggest that several esterases may be involved in the metabolism of [14C]Compound X in vitro. Aryldialkylphosphatase (paraoxonase) and butyrylcholinesterase may be important particularly in rat liver and human blood, respectively. The relative abundance of the esterases in different tissues also needs to be considered when extrapolating these results to in vivo.
307. A novel isoxazole ring opening pathway mediated by non-CYP enzymes in liver microsomes
Wenying Li, Weiping Zhao, Mingshe Zhu, Jonathan Josephs and W. Griff Humphreys
Bristol-Myers Squibb Research&Development, Princeton, NJ, USA, 8540
Reductive ring opening of 1,2-isoxazole, such as zonisamide, was shown to involve various enzymes, including CYP3A4 and aldehyde oxidase. Other isoxazole ring opening reactions, such as the opening of 4,5-isoxazole in leflunomide, are believed to be mediated by CYPs. We encountered a 5-isoxazole compound (P) that underwent ring opening to a-cyanoenol (M1) in liver microsomes. The bioconversion proceeded much faster in the presences of NADH than NADPH, and did not require molecular oxygen, suggesting the involvement of non-CYP enzymes. Findings in the mechanistic studies of the reaction will be discussed.
![]()
308. Aldehyde oxidase catalyzed oxidation of a nitrogen containing heterocyclic compound
Ping Li, Liyu Yang, and Liang-Shang Gan
Drug Metabolism & Pharmacokinetics, Preclinical and Clinical Development Sciences, Biogen Idec, Cambridge, MA, USA, 02142
Metabolism of compound A, a nitrogen containing heterocyclic molecule, was investigated. Studies using human hepatocytes suggested that oxidative metabolisms are the primary metabolic pathways. CYP mapping studies indicated that CYP3A4, 2C8, 2C9, and 2C19 are responsible for the formation of observed major oxidative metabolites with exception of the metabolite M2. Studies on the formation of M2 were further carried out using human liver microsomes and S9 fractions. M2 formation was found independent of NADPH, and the level was 9-fold higher in S9 incubations, as compared to that of microsomal incubations. It appears that a NADPH independent cytosolic enzyme is responsible for the formation M2. The O18 isotope was found incorporated to the metabolite M2 when compound A was incubated with human S9 fractions in the presence of the O18 labeled water (e.g. H2O18). Furthermore, the M2 formation in human S9 incubations was inhibited by menadione, a selective aldehyde oxidase inhibitor, but was not affected by methotrexate, a xanthine oxidase/dehydrogenase inhibitor. In summary, results from the present study strongly suggest that aldehyde oxidase is responsible for the formation of M2.
309. Human flavin monoxygenase 1 (FMO1) as a catalyst of nicotine N-Oxidation
Sharon E. Murphy1, Linda B. von Weymarn1, and Anthony L. Hinrichs2
1Masonic Cancer Center, University of Minnesota, Minneapolis, MN, USA, 55455
2Department of Psychiatry, Washington University, Saint Louis, MO, 98195
(S)-Nicotine [(S)-3-(1-methylpyrrolidin-2-yl)pyridine] is the major active agent in tobacco. The major pathway by which it is metabolized in smokers is P450 2A6-catalyzed oxidation to the 5’-iminium ion, followed by further metabolism to cotinine. Nicotine is also metabolized by either N-oxidation of the pyrrolidine ring or N-glucuronidation of the pyridine ring. FMO3 is known to catalyze the stereo specific N-oxidation of nicotine. However, some recent data from a genome-wide association and candidate gene study on nicotine dependence led us to investigate whether human FMO1 is a catalyst of nicotine N-oxidation. In that study, a polymorphism in a splice site of FMO1, rs10912675, showed significant association with nicotine dependence (p=0.0065, OR=1.25). Follow-up genotyping in an independent dataset replicated the association (p=0.0067, OR=1.18). To begin to assess a possible role for FMO1 in nicotine dependence in smokers we determined the catalytic efficiency and product specificity of FMO1-catalyzed nicotine metabolism. The metabolism of [5’-3H]-(S)- nicotine by FMO1 and FMO3 Supersomes™(BD Bioscience Woburn MA) was compared. As expected, the product of FMO3 catalyzed (S)-nicotine metabolism was > 95% trans nicotine-N-oxide. The product of FMO1 metabolism was an approximately equal mixture of cis and trans nicotine N-oxide. FMO1-catalyzed N-oxidation followed classic Michaelis-Menten kinetics with a Km of 1.2 mM and a Vmax of 35 nmol/min/mg protein (1 nmol FAD/mg protein). However, at a concentration of 1.2 mM nicotine the rate of FMO3-catalyzed N-oxidation was 9.2 nmol/min/mg and was still increasing. Therefore, FMO1 is a more efficient catalyst of nicotine N-oxidation than is FMO3. FMO3 is the predominant FMO enzyme in the liver, while FMO1 may be more abundant in extrahepatic tissues. We hypothesize that FMO1 may play a significant role in nicotine metabolism in extrahepatic tissues where P450 levels are relatively low.
310. Oligomeric and kinetic characterization of purified human FMO3 And FMO5 expressed as maltose binding protein fusions
Robert R. Reddy1, Erik Ralph1, Meike Motika2, Jun Zhang1, and John R. Cashman1
1Human BioMolecular Research Institute, San Diego, CA, USA, 92121
2Pharmaceut Inst, Universitat Kiel, Kiel, Germany, DE-24118
The flavin-containing monooxygenase (FMO) family of enzymes oxygenates nucleophilic xenobiotics and endogenous substances. Because FMOs are membrane associated, FMO functional activity is commonly assessed in vitro during ADMET studies using either microsome preparations derived from hepatocytes, or Supersomes derived from insect cells transduced with baculovirus encoding recombinant FMO. Of the five human forms, FMO5 and FMO3 are most prominent in adult human liver. To improve solubility and facilitate characterization of FMO3 and FMO5, N-terminus MBP fusions of both enzymes were prepared to >90% purity in the presence of detergent, and then compared with commercially available FMO Supersomes. Based on size-exclusion chromatography, MBP-FMO3 was found to exist as a monomer in the presence of 0.5% Triton-X-100 and as a hexamer in the presence of 0.5% CHAPS. The different oligomeric states of MBP-FMO3 correlated to different steady-state kinetic parameters (i.e., kcat and kcat/Km). Addition of 0.5% Triton-X-100 to MBP-FMO3 incubation mixtures resulted in improved kcat/Km values for methimazole and L-methionine (i.e., 2.5- and 2.2-fold, respectively). Similarly, addition of 0.5% Triton-X-100 to FMO3 Supersomes resulted in a 2.4-fold improvement in kcat/Km for methimazole. The apparent Km values were the same for FMO Supersomes and corresponding MBP-fused FMO enzymes but MBP-FMO enzymes afforded a lower kcat compared with Supersome FMO3 (i.e., kcat of 11.5 min−1 vs. 22.0 min−1 using methimazole as a substrate) and Supersome FMO5 (i.e., kcat of 0.64 min−1 vs. 1.01 min−1 using 10-(N,N-dimethylaminooctyl)-2-(trifluoromethyl) phenothiazine as a substrate). A comparison of kinetic parameters and sensitivity to detergents suggest the overall protein folding, coenzyme association, and oxygenation properties of Supersome FMOs are retained in the MBP-FMO fusion enzymes. Because altered xenobiotic clearance is often manifested differently in populations due to polyomorphisms in drug metabolizing enzymes such as FMO, the ease of constructing and producing variant forms of MBP-FMO in high purity should be of great utility for studying both drug efficacy and drug-metabolism induced idiosyncratic disease.
311. Disposition and metabolism of an MAO-A substrate CP-409092 in rat and human
Aarti Sawant1, Rebecca Lebowitz1, Emily Hudson1, Klaas Schildknegt1, and Deepak Dalvie2
1Pharmacokinetics, Dynamics and Metabolism, Pfizer, Groton, CT, USA, 06340
2Pharmacokinetics, Dynamics and Metabolism, Pfizer, La Jolla, CA, USA, 92121
CP-409092 (1) was developed for General Anxiety Disorder (GAD) and presents an interesting phenomenon of human pharmacokinetic predictions. In vitro, (1) was extensively metabolized in rat and human systems predominantly to acid metabolite formed by oxidative deamination of the N-methyl group of (1) mediated by MAO-A (as deduced from inhibition experiments). Male SD rats dosed with 100 mg/kg of 14C-(1) afforded a moderate to high plasma clearance and a moderate volume of distribution with a terminal elimination half-life of 3h. Majority of the radioactivity was excreted in feces as unchanged drug and was attributed to poor absorption or high biliary clearance. This data combined with the moderate half life of ~ 3-4 h demonstrated that MAO was not the major route of clearance for (1) in rat. To determine the extent of MAO mediated clearance we determined the intrinsic clearance of (1) @ 0.1- 50 uM in RLMs and HLMs (± NADPH) using the well-stirred model (Obach 2001; Obach et al. 1997). At concentration of 0.1-1 uM (well below it’s Km of 70-90 uM) the intrinsic clearance of (1) was predominantly mediated by CYP450 as demonstrated by no turnover in the absence of NADPH and was predicted to be low to moderate; at substrate concentrations of ≥10 uM, the intrinsic clearance with and without NADPH were identical. Thus at low substrate concentrations [S<(1) for MAO, this pathway played a dominant role. Scaling the human liver microsomal intrinsic clearance with appropriate scaling factors yielded a plasma half life of ~ 6h in humans. In humans following a p.o. dose of (1), the apparent oral plasma clearance (Cl/F) was 273 mL/min/kg and a moderate plasma half life of ~7-8h. A pooled analysis of human plasma demonstrated that the acid metabolite was the major circulating metabolite indicating that MAO-A may have played a role in vivo. But, the high Cl/F may also be attributed partly to low oral bioavailability, observed pre-clinically and in in vitro permeability assays. But in absence of human radiolabel ADME data, this cannot be concluded concretely. Based on the data presented here we conclude that a high Km of (1) for MAO observed in vitro may have factored in MAO playing a less significant role in the clinic resulting into a moderate elimination t1/2 in human (and rat).
312. Effect of CYP3A5 genotypes on 6β-OH-Cortisol/Cortisol ratio in north indians
Naushad Rais1, and Krishn K. Kohli2
1Biotechnology, Manipal University, Dubai, United Arab Emirates
2Biochenistry, PGIMER, Chandigarh, India
The aim of present work was to study correlation between CYP3A phenotypes and genotypes in North Indians. CYP3A isoforms CYP3A4 and CYP3A5 are capable of 6β-hydroxylation of cortisol (CS). Both CS and 6β-OH-CS are excreted in the urine. Hence, urinary 6β-OH-CS to free CS ratio has been proposed as a measure of CYP3A activity in liver. Three hundred North Indians were phenotyped for CYP3A activity by measuring CS and 6β-OH-CS in the morning spot urine. One hundred and fifty North Indians were genotyped for CYP3A5*3 and *6 by PCR-RFLP. Mean 6β-OH-CS/CS ratio in North Indians was 61. North Indians demonstrated unimodal distribution of 6β-OH-CS/CS ratio as analyzed by a histogram. The allele frequency of CYP3A5*3 was 0.71 in North Indians. CYP3A5*6 was absent in this population. Out of 75 subjects genotyped for CYP3A5*3 in low activity group, 4 (5 %) were normal homozygotes, 28 (37 %) were heterozygotes and 43 (57 %) were mutant homozygotes. Thus frequencies of CYP3A5*1 and CYP3A5*3 were 0.24 and 0.76, respectively in low activity group. Out of 75 subjects genotyped for CYP3A5* 3 in high activity group 8 (11 %) were normal homozygotes, 34 (45 %) were heterozygotes and 33 (44 %) were mutant homozygotes. Thus frequencies of CYP3A5*1 and CYP3A5*3 were 0.33 and 0.67 in high activity group. Out of total 150 subjects genotyped for CYP3A5*3, 12 (8 %) were normal homozygotes (CYP3A5*1/*1), 62 (41 %) were heterozygotes (CYP3A5*1/*3) and 76 (51 %) were mutant homozygotes (CYP3A5*3/*3). Thus frequencies of CYP3A5*1 and CYP3A5*3 were 0.29 (95 % CI, 0.22-0.36) and 0.71 (95 % CI, 0.64-0.78) in 150 North Indians genotyped. Genotypes and allele frequencies in high activity group were not statistically different from genotypes and allele frequencies in the low activity group. This is the first report establishing the presence of CYP3A5*3 in North Indians.
313. Genetic polymorphisms of cytochrome P450 oxidoreductase gene (POR) in a japanese population
Yoshiro Saito1, Hiromi Fukushima-Uesaka1, Noriko Katori1, Kouichi Kurose1, Keiko Maekawa1, Nahoko Kaniwa1, Ryuichi Hasegawa2, Jun-ichi Sawada1, Noboru Yamamoto3, Hideo Kunitoh3, Hiroshi Nokihara3, Ikuo Sekine3, Yuichiro Ohe3, Teruhiko Yoshida4, Tomohide Tamura3, Nagahiro Saijo5, Yasuhiro Matsumura5, and Haruhiro Okuda1
1National Institute of Health Sciences, Tokyo, Japan, 158-8501
2National Institute of Health Sciences, Tokyo, Japan
3National Cancer Center Hospital, Tokyo, Japan
4National Cancer Center Research Institute, Tokyo, Japan
5National Cancer Center Hospital East, Kashiwa, Japan
Backgrounds and purpose: P450 oxidoreductase (POR) is a protein containing both flavine adenine dinucleotide and flavin mononucleotide that transfers electrons from NADPH to microsomal cytochrome P450 (CYP) enzymes. POR activity is prerequisite for CYP activities to catalyze drug oxidations. Genetic variations that affect the POR activity might influence broad ranges of CYPs by changing their activities. Several reports, many of which were focusing on non-synonymous variations, have been published recently. However, there is no report that comprehensively screened the genetic variations of the POR gene in a Japanese population.
Methods: The 5’- and 3’-flanking regions, all 16 exons, and their surrounding introns of POR gene were directly sequenced for 235 Japanese subjects.
Results and Discussion: We found 75 variations including 37 novel ones: 7 in the 5’-flanking region, 2 in the 5’-untranslated region (non-coding exon 1), 16 in the coding exons (10 non-synonymous and 6 synonymous), 45 in the introns, 4 in the 3’-untranslated region, and 1 in the 3’-flanking region. All the variations were in Hardy-Weinberg equilibrium. Among the 10 non-synonymous variations, novel T29M, R550W, R570C, E580Q, and A659T were found as heterozygotes in single subjects (minor allele frequency = 0.002 for each variation). As for previously reported variations, P228L, G413S, A485T, A503V, and G504R were detected at 0.002, 0.009, 0.002, 0.434, and 0.002 frequencies, respectively. The Pro to Leu substitution at codon 228 was known to reduce POR activity, resulting in decreased activities of several CYPs. The other four variations were reported to be associated with reduced POR activities but not with the alterations of CYP activities significantly. Using the detected variations, the analyzed regions were divided into two linkage disequilibrium blocks, and their haplotypes were estimated.
Conclusion: In the POR gene, we detected 75 variations including 5 novel and 5 known non-synonymous ones from 235 Japanese subjects. This information on POR variations is useful for genotyping and association studies for drug responsiveness where CYP activity is involved in the drug metabolism.
314. CYP3A5 genotypes affect pharmacokinetics of immediate-released nifedipine, but not for extended-released dosage form
Jin-Ding Huang1, Yi-Hua Chen1, and Ming-Liang Lai2
1Dept of Pharmacol, Natl Cheng Kung Univ Med Col, Tainan, Taiwan, TW-70101
2Department of Neurology, National Cheng Kung University Medical Center, Tainan 701, Taiwan, Tainan City 701, Taiwan
CYP3A5 is polymorphically expressed in human liver and small intestine. The nonfunctional CYP3A5*3 is primarily due to a SNP, g.6986A>G. People with at least one CYP3A5*1 allele express a higher amount of CYP3A5. Subjects without significant CYP3A5 expression was supposed to have higher plasma drug concentration and a lower clearance. However, literature data are controversial when effects of CYP3A5 genotype on drug disposition were examined. Our previous report showed no difference in midazolam pharmacokinetics between CYP3A5-negative and CYP3A5-positive subjects, whereas a significantly higher alprazolam, sirolimus, and tacrolimus plasma concentration in CYP3A5*3/*3 subjects was found in other studies. We postulated that there is a CYP3A4 up-regulation in the small intestine for some CYP3A5 substrates. To investigate the possible CYP3A4 up-regulation in different parts of the small intestine, we investigated the effect of CYP3A5 genotype on nifedipine pharmacokinetics with different dosage forms in healthy male Chinese subjects. One group is given a single-dose of Adalatò(immediate-release form of nifedipine), and the other group is given a single-dose of Coractenò(extended-release form of nifedipine). Each group contains 12 CYP3A5*1/*3 subjects and 12 CYP3A5*3/*3 subjects. Blood samples were collected to analyze the pharmacokinetics of nifedipine. We found that CYP3A5 genotype has no effects on nifedipine pharmacokinetics in the Coractenò study. However, in the Adalatò study, CYP3A5*3/*3 subjects exhibited lower peak concentrations (71.5°”54.6 ng/mL) than CYP3A5*1/*3 subjects (128.5°”73.8 ng/mL; P=0.04). The immediate-released nifedipine is rapidly absorbed in the upper part of gastrointestinal tract. We therefore concluded a substantial up-regulation of CYP3A4 in the region, the up-regulation is less in the lower gastrointestinal tract where the most of the extended-released nifedipine is absorbed.
315. Association of carboxylesterase 1A genotypes with irinotecan pharmacokinetics in japanese cancer patients
Kimie Sai1, Yoshiro Saito1, Naoko Tatewaki1, Masakiyo Hosokawa2, Nahoko Kaniwa1, Tomoko Nishimaki-Mogami1, Mikihiko Naito1, Jun-ichi Sawada1, Kuniaki Shirao3, Tetsuya Hamaguchi3, Noboru Yamamoto3, Hideo Kunitoh3, Yuichiro Ohe3, Tomohide Tamura3, Yasuhide Yamada3, Teruhiko Yoshida4, Hironobu Minami5, Atsushi Ohtsu5, Yasuhiro Matsumura5, Nagahiro Saijo5, and Haruhiro Okuda1
1National Institute of Health Sciences, Tokyo, Japan, 158-8501
2Chiba Institute of Science, Choshi, Japan, 288-0025
3National Cancer Center Hospital, Tokyo, Japan
4National Cancer Center Research Institute, Tokyo, Japan
5National Cancer Center Hospital East, Kashiwa, Japan
Backgrounds and purpose: Human carboxylesterase 1 (CES1) is highly expressed in the liver and capable of hydrolyzing irinotecan, an anticancer prodrug, to produce an active metabolite SN-38. Recent research on CES1 gene structure has revealed two functional CES1 genes, CES1A1 and 1A2, were inversely located on chromosome 16q13-q22.1 in a tail-to-tail orientation (CES1A2 - 1A1) (Hosokawa et al. Drug Metab Pharmacokinet. 2008; 23: 73-84). The CES1A1 and CES1A2 genes have a 98% homology with 5 nucleotide (4 amino acid) changes in exon 1 which encodes a signal peptide. In addition, recombinations of CES1A1 with CES1A2 (var1A1) and of CES1A2 with CES1A3 (pseudogene) have been also identified (Tanimoto et al., Pharmacogenet Genomics. 2007; 17:1-10, Fukami et al. Pharmacogenet Genomics. 2008; 18: 911-20). In this study, to clarify the role of the CES1 genotypes in irinotecan therapy, we analyzed effects of diplotypes of CES1A genes on irinotecan pharmacokinetics in Japanese cancer patients.
Methods: Effects of CES1A diplotypes [combinations of the haplotypes A (1A3-1A1), B (1A2-1A1), C (1A3-var1A1) and D (1A2-var1A1)] on the in vivo CES activity, AUC ratio of (SN-38 + SN-38G) to irinotecan, were investigated for 58 Japanese cancer patients treated by irinotecan monotherapy (100 mg/m2 or 150 mg/m2).
Results: Frequencies of CES1A1 (A and B) and var1A1 (C and D) were 69.0% and 31.0%, and those of CES1A2 (B and D) and 1A3 (A and C) were 33.6% and 66.4%, respectively. Regarding the total number of active CES1 genes (1A1, 1A2 and var1A1), frequencies of 2 (A/A, A/C and C/C), 3 (A/B, A/D or B/C, and C/D) and 4 active genes (B/B and B/D) were 39.7%, 53.4% and 6.9%, respectively. The AUC ratio [(SN-38+SN-38G)/CPT-11] in the patients with 3 or 4 active genes (N=35) were 1.24-fold higher than those with 2 genes (N=23)(P=0.0134). No significant difference was found between CES1A1 and var1A1 (1A1/1A1, 1A1/var1A1 and var1A1/var1A1).
Conclusion: This study suggests that the total number of functional CES1A genes might affect formation of the active metabolite of irinotecan in Japanese cancer patients.
316. Genetic polymorphisms of ORM1 and ORM2 genes encoding α1-acid glycoprotein in a Japanese population
Noriko Katori1, Yoshiro Saito1, Kimie Sai1, Hiromi Fukushima-Uesaka1, Kouichi Kurose1, Chikako Yomota1, Toru Kawanishi1, Jun-ichi Sawada1, Noboru Yamamoto2, Hideo Kunitoh2, Hiroshi Nokihara2, Ikuo Sekine2, Yuichiro Ohe2, Teruhiko Yoshida3, Tomohide Tamura2, Nagahiro Saijo4, Yasuhiro Matsumura4, and Haruhiro Okuda1
1National Institute of Health Science, Tokyo, Japan, 158-8501
2National Cancer Center Hospital, Tokyo, Japan
3National Cancer Center Research Institute, Tokyo, Japan
4National Cancer Center Hospital East, Kashiwa, Japan
Backgrounds and purpose: Alpha-1-acid glycoprotein (AGP), an acute-phase response protein, functions as a drug-binding protein, especially for basic drugs. AGP is encoded by two genes, ORM1 and ORM2, located in tandem on chromosome 9. In order to identify genetic polymorphisms affecting the expression levels and drug-binding affinity, we analyzed ORM1 and ORM2 genes for a Japanese population.
Methods: First, duplications of the ORM1 and ORM2 genes were detected for 165 Japanese subjects by PCR amplification using primers specific for ORM1 and ORM2. Then, using the samples without duplications, the 5’- and 3’-flanking regions, all 6 exons, and their surrounding introns of both ORM1 and ORM2 genes were directly sequenced.
Results: We found the duplication of ORM1 in 51 subjects (frequency: 0.309) and that of ORM2 in 1 subject (0.006). Then, genetic polymorphisms of ORM1 and ORM2 were surveyed for the 113 subjects without duplications. Regarding the ORM1 gene, we detected 43 variations including 17 novel ones: 13 in the 5’-flanking region, 4 in the exons (3 non-synonymous and 1 synonymous ones), 22 in the introns, 1 in the 3’-untranslated region, and 3 in the 3’-flanking region. The 3 non-synonymous variations Q38R, V144F and K170R were found at frequencies of 0.173, 0.004, and 0.009, respectively. As for ORM2, 40 genetic variations were found including 15 novel ones: 14 in the 5’-flanking region, 4 in the exons (2 non-synonymous and 2 synonymous ones), 19 in the introns, and 3 in the 3’-flanking region. Among two non-synonymous variations, a novel substitution E110K was found at a frequency of 0.013, and the known polymorphism G141R at 0.044. Using the detected variations, haplotypes covering the two genes were estimated because strong linkage disequilibrium between the variations was observed throughout the analyzed regions.
Conclusion: Current findings on the ORM1 and ORM2 duplications and variations could be useful for genotyping of these genes and also for association studies on responsiveness to drugs that bind to AGP.
317. A new mechanism of altered phenytoin clearance in CYP2C19*2 subjects
Amarjit S. Chaudhry1, Jatinder K. Lamba2, Stephen C. Strom3, J. H. You4, Angela Birnbaum5, Rory P. Remmel6, and Erin G. Schuetz1
1Department of Pharmaceutical Sciences, St. Jude Children’s Research Hospital, Memphis, TN, USA, 38105
2Department of Experimental and Clinical Pharmacology, University of Minnesota, Minneapolis, MN, USA
3Department of Pathology, University of Pittsburgh, Pittsburgh, PA, 15213
4School of Pharmacy, Faculty of Medicine, The Chinese University of Hong Kong, China
5Department of Experimental and Clinical Pharmacology, University of Minnesota, Minneapolis, MN, 55455
6Department of Medicinal Chemistry, University of Minnesota, Minneapolis, MN, 55455
We previously found that subjects on long term phenytoin (PHT) maintenance therapy who had a combined CYP2C9*1/*1 & CYP2C19*1/*2 genotype had a urinary PHT metabolite (S)/(R) ratio that was paradoxically low. We hypothesized that CYP2C9 regulatory polymorphisms (rSNPs), G-3089A and -2663TG del, in linkage disequilibrium (LD) with CYP2C19*2 were responsible for the low activity CYP2C9 phenotype. We tested for the association of the CYP2C9 rSNPs with dosing requirements for the CYP2C9 substrate warfarin in vivo and with CYP2C9 mRNA expression and activity in liver microsomes. However, the CYP2C9 rSNPs did not correlate with warfarin dose requirements in patients on this anticoagulation therapy. Moreover, CYP2C9 rSNPs did not correlate with human liver CYP2C9 mRNA levels and the PHT metabolite (S)/(R) ratio was actually slightly higher in liver microsomes from donors with the CYP2C19*2 genotype. To explain this additional paradox we further hypothesized that the CYP2C9 promoter rSNPs differentially affected induction of CYP2C9 transcription by PHT because PHT is a known activator of PXR, CAR and NRF2 and the subjects were on maintenance PHT (induction) therapy. In silico analysis predicted the G-3089A created a YY1 site and the -2663TG del destroyed an Nrf2 site. Transfection of HepG2 cells with wild-type (WT) and variant (VAR) CYP2C9 promoter-luciferase reporters revealed similar basal activity for the two haplotypes but a significantly greater magnitude of CYP2C9 induction of the WT versus the VAR haplotype (p=0.02) by PHT activated PXR. Site directed mutagenesis of each site indicated that both rSNPs contributed to the phenotype: The extent of inhibition by YY1 was greater for the VAR-HAP (p=0.02) compared to WT-HAP and PXR and Nrf2 mediated PHT induction was significantly greater (p=0.008) for the WT-HAP compared to VAR-HAP. PHT treated human hepatocytes corroborated these findings with hepatocytes from donors with CYP2C9 WT-HAP demonstrating a greater induction compared to those with the CYP2C9 VAR-HAP. We conclude that regulatory polymorphisms in the CYP2C9 promoter affect PHT induction of CYP2C9 and subsequent metabolism of PHT.
318. Genotyped, cryopreserved suspensions of human hepatocytes as a tool for investigating drug metabolism in polymorphic individuals
Cornelia Smith1, Jonathan Jackson2, Marie Gonzalez2, Manda Edwards1, and Stephen Ferguson3
1Life Technologies, Durham, NC, USA, 27703
2Life Technologies, Austin, TX, USA, 78701, 3Cellzdirect/Invitrogen, Durham, NC, USA, 27703
Cryopreserved suspensions of human hepatocytes are widely accepted as an effective model system to assess hepatic metabolic stability in vitro. An advantage of cryopreserved human hepatocytes is that they are often pre-characterized for enzymatic activity, allowing researchers to select lots that conform to certain specifications. Invitrogen (a part of Life Technologies) is a supplier of commercially available cryopreserved human hepatocytes for in vitro use. To test the hypothesis that the metabolic profiles of cryopreserved human lots supplied by Invitrogen (a part of Life Technologies) mimic those of the general population, cryopreserved lots isolated from forty-seven donor tissues were phenotyped and genotyped. Metabolic activities CYP2C9, CYP2C19, CYP2D6 and CYP3A were determined using probe substrates diclofenac, S-mephenytoin, dextromethorphan and testosterone, respectively. Genotypes for thirteen SNPs within the CYP2C9, CYP2C19, CYP2D6 and CYP3A5 genes were determined by using TaqMan® Primer/Probe sets using qRT-PCR. Amongst the forty-seven lots, the median activities of CYP2C9, CYP2C19, CYP2D6 and CYP3A were 112, 17.3, 20.6 and 439 pmol/min/106 cells, respectively. Allele frequencies within the set were 3.2% (CYP2C9*2), 1.1% (CYP2C9*3), 17% (CYP2C19*2), 1.1% (CYP2D6*3), 18% (CYP2D6*4), 3.2% (CYP2D6*6), 2.1% (CYP2D6*9), 90%(CYP3A5*3) and 1.1% (CYP3A5*6). A distinct pattern was observed from the CYP2D6 phenotype-genotype correlation where donor preparations homozygous for CYP2D6*4 or CYP2D6*6 exhibited the lowest enzymatic activities (<2.50 pmol/min/106 cells). Hepatocytes lots lacking the CYP3A5*3 SNP were only observed in individuals of African-American ethnicity, consistent with reported population frequencies for this allele. Overall, the observed allele frequencies discovered in this study agree with other reports published on various populations. These results support the utility of genotyped cryopreserved human hepatocytes to assess drug disposition in polymorphic donors, especially in cases where candidate drugs are CYP2D6 substrates.
319. The role of dna repair gene XRCC1 Arg194Trp and Arg399Gln polymorphisms as risk factor for the development of childhood acute lymphoblastic leukemia: A study on turkish population including the case only analysis of interactive effect of cigarette smoke
Emel Arinç, Tugba Boyunegmez Tumer, Duygu Yilmaz, Cihan Tanrikut, and Gulen Ulusoy
Biochemistry Graduate Programme and Department of Biological Sciences, Middle East Technical University, Ankara, Turkey
Maintenance of genomic stability in mammalian cells is achieved by the efficient work of DNA repair mechanisms. Polymorphisms of DNA repair genes impair the DNA repair capacity and may lead to genetic instability and carcinogenesis. X-ray repair cross-complimenting group 1 (XRCC1) is one of the most important genes functioning in base excision repair pathway and a number of studies suggest that polymorphic forms of XRCC1 has a role in susceptibility to various kinds of cancer including breast, prostate, lung and bladder cancers. However, there are limited and contradictory data on the association between the XRCC1 polymorphisms and childhood acute lymphoblastic leukemia (ALL). In this study, we investigated the possible association of XRCC1 Arg399Gln and Arg194Gln variants with the risk of incidence of childhood ALL in Turkish population. The genotypes were determined with the use of PCR/RFLP techniques on 190 healthy controls and 167 ALL patients. For Arg399Gln polymorphism, the heterozygous (Arg/Gln) and homozygous mutant (Gln/Gln) genotypes were significantly more common in the ALL patients than the controls (OR: 1.6; 95% CI 1.0-2.4, p=0.04), resulting in a defective allele (399Gln) frequency of 39% in patients and 31% in controls (p=0.02). Particularly, the homozygote mutant genotype for codon 399 polymorphism significantly increased the risk of disease up to 2.0 fold (OR: 2.0; 95% CI 1.0-3.8, p=0.04). However, no significant associations have been found between codon 194 polymorphism and risk of childhood ALL. In addition, in this study, interactions of Arg399Gln and Arg194Trp variants with parental, prenatal and postnatal smoking exposure of children have been investigated, but no significant associations have been found. To conclude, in the current work, we provide evidence that XRCC1 Arg399Gln polymorphisms, on its own, significantly contribute to the risk of development of childhood ALL.
320. Exploratory study of genetic biomarkers associated with drug-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in japanese patients
Masahiro Tohkin1, Nahoko Kaniwa1, Kouichi Kurose1, Yoshiro Saito1, Michiko Aihara2, Kayoko Matsunaga3, Yukitoshi Takahashi4, Hirokazu Furuya5, Masaaki Muramatsu6, Shigeru Kinoshita7, Zenro Ikezawa2, and Ryuichi Hasegawa1
1National Institute of Health Sciences, Tokyo, Japan, 158-8501
2Yokohama City University Graduate School of Medicine, Yokohama, Japan
3Fujita Health University School of Medicine, Toyoake, Japan
4National Epilepsy Center, Shizuoka, Japan
5National Omuta Hospital, Omuta, Japan
6Tokyo Medical and Dental University, Tokyo, Japan
7Kyoto Prefectural University of Medicine, Kyoto, Japan
Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are rare severe blistering skin diseases, that are mainly caused by drugs. Human leukocyte antigen (HLA) alleles, located on chromosome 6, have been shown to predispose to SJS associated with carbamazepine (CBZ) and serious cutaneous adverse reactions associated with allopurinol (ALP). However, CBZ-induced SJS, associated with HLA-B*1502 in Han Chinese patients, does not appear to be associated with this HLA allele in populations of European origin. Therefore, an HLA allele that may be a highly specific marker of susceptibility in certain population may not be such a good marker in other populations. In order to explore the genetic biomarkers associated with SJS/TEN in Japanese patients, we previously determined the HLA class I alleles with SJS/TEN in 58 Japanese patients. In this study, we analyzed the HLA alleles in 90 Japanese patients with SJS/TEN. Furthermore, we carried out a genomewide association study using approximately 250,000 single nucleotide polymorphisms (SNPs) in 82 definitively diagnosed SJS/TEN patients and 183 controls. There were no HLA-B*1502 carriers among the Japanese patients with CBZ-induced SJS/TEN. HLA-B* 5801, which has been reported to be associated with serious ALP-induced cutaneous adverse reactions, was found in 5 among 15 Japanese patients with ALP-induced SJS/TEN (odds ratio = 32.7). The genomewide scanning yielded a significant association between loxoprofen-induced SJS/TEN and the SNP located on chromosome 3 (Pc = 2.53 × 10 −7 ) among the Japanese subjects, from the association test with the genotype (Fisher’s exact test). Four other SNPs and 3 other SNPs, respectively, were found to be significantly associated with CBZ-induced SJS/TEN (Pc = 2.26 × 10 −7 ) and loxoprofen-induced SJS/TEN (Pc = 1.18 × 10 −7 ), among Japanese subjects, from the trend test of association with the genotype (Cochrane-Armitage test). From these results, we consider that HLA-B* 5801 and the SNPs found from the genomewide association studies might be potential genetic biomarkers of drug-induced SJS/TEN in the Japanese population. To confirm these associations with potential SNPs, second stage of association analyses must be conducted in new patient series and control groups.
321. Gemcitabine pharmacogenetics: Characterization of genetic variants of cytidine deaminase
Hwei Li1, Jessica A. Roseberry Baker1, Olukayode A. Oluyedun1, Yue-Wei Qian2, Bomie Han2, Barbara J. Ring1, Enaksha Wickremsinhe1, Anne H. Dantzig3, Stephen D. Hall1, Steven A. Wrighton1, and Yingying Guo1
1Drug Disposition, Eli Lilly and Company, Indianapolis, IN, USA, 46285
2Integrative Biology, Eli Lilly and Company, Indianapolis, IN, USA, 46285
3Cancer Research, Eli Lilly and Company, Indianapolis, IN, USA, 46285
Gemcitabine (dFdC), a nucleoside anticancer drug, is rapidly metabolized to an inactive metabolite, 2’,2’-difluorodeoxyuridine (dFdU), by cytidine deaminase (CDA). Two nonsynonymous single nucleotide polymorphisms (SNPs) have been identified in CDA, 79A>C (Lys27Gln) and 208G>A (Ala70Thr), and haplotypes harboring them were designated CDA*2 and CDA*3, respectively. Previous studies have shown that CDA*3 was associated with lowered CDA activity in plasma and decreased clearance of gemcitabine in vivo1. In the present study, the functionality of these two SNPs was further investigated. Recombinant enzymes of wild type CDA and its two variants were expressed and purified. The purity and identities of the preparations were confirmed by SDS PAGE and mass spectrometry, respectively. Further, all three enzymes were shown to have comparable levels of tetramers and incorporated zinc. In addition, compared with the wild type protein, CDA*2 was found to have similar Km and Vmax values for ara-C metabolism, and CDA*3 had significantly decreased Km, Vmax and intrinsic clearance. This is consistent with the literature, in which CDA*3 was also observed to have much lower activity towards cytidine and cytosine metabolism2. However, surprisingly, all three enzymes yielded comparable kinetic parameters for the conversion of gemcitabine to dFdU. This is the first time that recombinant enzymes have been utilized to examine the impact of CDA*2 and CDA*3 on gemcitabine metabolism. These results suggest that there exists substrate-dependent activity for CDA*3, and factors other than A70T may have contributed to the low in vivo CDA activity towards gemcitabine that has been associated with CDA*3 haplotype1. For example, the expression levels of CDA may be lowered in patients carrying CDA*3 haplotype due to additional genetic changes in the regulatory region or unknown epigenetic regulation of CDA. References 1. Sugiyama et al. Pharmacokinetics of gemcitabine in Japanese cancer patients: the impact of a cytidine deaminase polymorphism. J of Clinical Oncology 25:32-42, 2007 2. Yue et al. A functional single-nucleotide polymorphism in the human cytidine deaminase gene contributing to ara-C sensitivity. Pharmacogenetics 13:29-38, 2003
322. Cytochrome P450 CYP2C19 genotypes in sickle cell disease patients and normal controls in an african population
Chinedum Peace Babalola1, Olufunmilayo Adejumo2, Din Ung3, Zhiheng Xu4, Abayomi Odetunde1, Taiwo Kotila1, Adeyinka G. Falusi1, and Swati Nagar5
1Department of Pharmaceutical Chemistry, University of Ibadan, Ibadan, Nigeria
2Pharmaceutical Chemistry, Faculty of Pharmacy, Olabisi Onabanjo University, Ago-Iwoye, Nigeria
3Pharmaceutical Sciences, Temple University, Philadelphia, PA, USA, 19140
4Biostatistics and Bioinformatics, Winship Cancer Institute, Emory University, Atlanta, USA, GA 30322
5Pharmaceutical Sciences, Temple University School of Pharmacy, Philadelphia, PA, USA, 19140
Genetic variability in CYP2C19 is an important cause for inter-individual variability in the pharmacokinetics of numerous clinically important drugs such as proguanil, omeprazole, diazepam and voriconazole. CYP2C19 genotypes might have implications in proguanil pharmacokinetics, an important pro-drug for malaria prophylaxis in sickle cell patients (SCP). PG undergoes oxidative metabolism to cycloguanil (CG), the active metabolite which inhibits the enzyme dihydrofolate reductase. An estimated 12 - 15 million people with sickle cell disease (SCD) reside in Sub-Saharan Africa, with approximately 6 million cases in Nigeria alone, where malaria is a life-threatening disease. The present study evaluated CYP2C19 *1, *2, and *3 allele and genotype frequency in Nigerian normal controls (n = 43) and sickle cell disease (SCD) patients (n = 115) for the first time. Genomic DNA samples were genotyped for CYP2C19 *1 (‘wild-type’), *2 (681 G>A), and *3 (636 G>A) variants with previously published PCR and restriction fragment length polymorphism (PCR-RFLP) methods of analysis.
CYP2C19 *1 (84.3 vs. 84.9%) or *2 allele frequency (15.7 vs. 15.1%) was not significantly different between SCD vs. normal groups. No *3 allele was detected in the cohort. The SCD group exhibited a statistically significantly lower frequency of *1/*1 genotype (69.6%) compared to normal controls (74.4%). Frequency of *2/*2 was significantly lower in SCD (0.9%) compared to normal controls (4.7%). Frequencies of *1/*2 (29.6% versus 20.9%) were comparable in SCD and normal controls. We have previously reported the existence of PG phenotypic PMs to be 4.8% in a Nigerian population. The present study, conducted in an independent Nigerian population, exhibited a 4.7% CYP2C19 *2/*2 genotype frequency. This genotype might completely represent the CYP2C19 PM phenotype in Nigerians. This hypothesis will be tested in future CYP2C19 genotype studies in conjunction with PG pharmacokinetic evaluation.
In summary, CYP2C19 *1 and *2 allele and genotype frequencies are reported for the first time in normal Nigerian controls as well as the SCD population. Significant differences were observed in genotype frequencies between normal controls and SCD patients. Our data compared well with reports in other African populations, with no significant differences among the various populations. Future work will focus on PG pharmacokinetics and anti-malarial activity in conjunction with CYP2C19 pharmacogenetics.
323. Cytochrome P450 oxidoreductase genotypes associated with human liver CYP enzyme activities
Ranjit K. Thirumaran1, Emma Jarvis1, Xia Yang2, Bin Zhang2, Cliona Molony2, Eugene Chudin2, Ke Hao2, Jun Zhu2, Christine Suver2, F. Peter Guengrich3, Stephen C. Strom4, Thomas Rushmore5, Roger Ulrich2, J. Greg Slatter2, Eric Schadt2, Andrew Kasarskis2, Pek Y. Lum2, and Erin Schuetz1
1Department of Pharmaceutical Sciences, St. Jude Children’s Research Hospital, Memphis, TN, USA, 38105
2LLC, a wholly owned subsidiary of Merck & Co., Inc.,, Rosetta Inpharmatics, Seattle, WA, USA
3Department of Biochemistry and Center in Molecular Toxicology, Vanderbilt University School of Medicine, Nashville, TN, USA
4Department of Pathology, University of Pittsburgh, Pittsburgh, PA, USA, 15213
5Drug Metabolism, Merck Research Laboratories, West Point, PA, USA, 19486
The microsomal flavoprotein Cytochrome P450 oxidoreductase (POR) is the required electron donor to all microsomal CYP enzymes, which catalyze the biosynthesis of steroids, fatty acids, and bile acids, as well as metabolism of more than 80% of prescription drugs. We genotyped 46 common sequence variations across the POR gene and determined their effect on eight human drug metabolizing CYP activities in 164 human liver microsomes. The human liver samples were phenotyped for nine drug metabolizing P450 enzyme activities using model substrates: CYP1A2 (phenacetin), CYP2A6 (coumarin), CYP2B6 (bupropion), CYP2C8 (paclitaxel), CYP2C9 (tolbutamine), CYP2C19 ((S)-mephenytoin), CYP2E1 (chloroxazone) and CYP3A4 (testosterone and midazolam). Eight POR polymorphisms, including many intronic variants, were significantly associated with one or more drug oxidation activities. Four POR intronic variants significantly affected 1’-OH midazolam hydroxylase and testosterone 6beta-hydroxylase activities, two markers of CYP3A4 activity. Investigations into possible mechanistic explanations demonstrating how POR variants affect POR mRNA processing (e.g., splicing) are in progress. Our results identified human POR genetic polymorphisms significantly associated with CYP-catalyzed drug metabolism. Whether POR can prove to be a predictive pharmacogenetic factor will depend on in vivo validation studies. Our data provide a basis for further studies towards inclusion of POR polymorphisms in pharmacogenomic strategies.
324. Comparative pharmacokinetics of Ritonavir and BILR 516 in animals
Dale E. Sharp1, Susan Hattox1, Dongmei Qiang2, and Haizhou Zhang3
1Drug Metabolism and Pharmacokinetics, Boehringer Ingelheim Pharmaceuticals Inc., Ridgefield, CT, USA, 06877
2Pharmaceutics, Boehringer Ingelheim Pharmaceuticals Inc., Ridgefield, CT, USA, 06877, 3Shanghai ChemPartner Co. Ltd., Shanghai, China
During the development of the non-nucleoside reverse transcriptase inhibitor BILR 355, a metabolite (BILR 516) was discovered in plasma from human subjects in a Phase Ib study which employed Ritonavir (RTV) as a boosting agent. BILR 516 was not detected when RTV was not administered and was a minor metabolite in rat and dog, even with RTV coadministration. Due to the long half-life of BILR516 in humans (~40 h), its AUC was approximately twice that of BILR 355 at steady state. In order to establish the safety of BILR 516, it was necessary to administer BILR 516 in a toxicity study. RTV was coadministered in order to decrease the metabolism of BILR 516. A number of species were screened in order to find one in which the BILR 516 Cmax and AUC approximated that observed in humans. All species received 10 mg/kg of both RTV and BILR 516. BILR 516 was extremely insoluble, and crystalline BILR 516 had very low bioavailability. Thus, an amorphous solid dispersion formulation was developed to increase exposure. A commercial liquid formulation of RTV was diluted in water for administration. There was a >21,000 fold difference in RTV AUC0-t among the species examined. The rank order was ferret>>beagle≈Sprague Dawley rat> male Göttingen minpig> cynomolgus monkey> female Göttingen minpig>guinea pig ≈ New Zealand White rabbit. The range of half-lives of RTV was much narrower (1.9-4 h), suggesting that differences in bioavailability or distribution rather than differences in systemic metabolism accounted for the range of RTV AUCs. The rank order of BILR 516 AUC0-t was ferret ≈ guinea pig>cynomolgus monkey> beagle ≈ male Göttingen minpig> New Zealand White rabbit>Sprague Dawley rat> female Göttingen minpig. The values for BILR 516 AUC0-t varied by as much as 50-fold among species. As with RTV, the terminal half-lives varied over a much narrower range (2.7-6.3 h). In general, the species with higher RTV exposure had higher BILR 516 exposure. The exceptions were guinea pig, which had high BILR 516 exposure despite very low plasma RTV concentrations, and rat, which had relatively low BILR 516 exposure despite relatively high RTV concentrations. Additional studies in rats at higher RTV dose levels showed that the plasma concentrations achieved at the 10 mg/kg RTV dose were insufficient to inhibit BILR 516 metabolism completely. The results of the study identified ferrets and guinea pigs as the species most likely to achieve BILR 516 exposure similar to that of humans.
325. Application of TMDD model to a single dose pharmacokinetic study of a monoclonal antibody in monkeys via a population pharmacokinetic approach
Jian-ping Tang1, and Christophe Meille2
1Non-Clinical Safety, Hoffmann-La Roche, Nutley, NJ, USA, 07110
2Non-Clinical Safety, Hoffmann-La Roche, Basel, Switzerland
Objective: To develop a population pharmacokinetic model to best describe the disposition of R1507, a monoclonal antibody for the treatment of patients with advanced solid tumors, in monkeys; to simulate human systemic exposures in attempting to aid selection of dosing regimen.
Method: The data were collected from 24 animals (6 male monkeys/group) that received a single intravenous injection of the test compound at doses of 3, 15, 50 or 150 mg/kg. The concentrations of R1507 were continuously monitored at various time points over a 7-week period. Data analyses were performed by non-linear mixed-effect modeling with the MONOLIX software. Two structural pharmacokinetic models, target mediated drug disposition (TMDD) and Michaelis-Menten model, were evaluated. Objective function and goodness of fit were used for model selection.
Results: The TMDD model best described the disposition of R1507 in monkeys with a -2x log likelihood value of 8457 and Akaike information criteria (AIC) of 8479 as compared to 9167 and 9183, respectively, in the Michaelis-Menten model. In addition, the TMDD model is consistent with the mode of pharmacological action of R1507.
Conclusion: Although additional pharmacokinetic parameters (7 vs. 5) were required in the TMDD model, it best described the disposition of the test compound evidenced by the minimum -2x log likelihood and AIC values. Human systemic exposure was simulated with this model, which attempted to assist selection of dosing regimen. The TMDD model will also be used to facilitate dose optimization for a upcoming 13-week toxicity/toxicokinetic study.
326. Metabolism, excretion and pharmacokinetics following an oral dose of 14C-INCB018424 to healthy human subjects
Adam D. Shilling1, Frank Nedza1, Thomas Emm1, Edward McKeever2, Naresh Punwani2, William Williams2, and Swamy Yeleswaram1
1Drug Metabolism and Biopharmaceutics, Incyte Corporation, Wilmington, DE, USA, 19880
2Clinical Development, Incyte Corporation, Wilmington, DE, USA, 19880
INCB018424 is an inhibitor of the Janus kinase family of protein tyrosine kinases (JAKs). Six healthy human subjects received a single dose of 25 mg of INCB018424 containing 100 μCi 14C-INCB018424 as an oral solution. INCB018424 was absorbed rapidly, attaining peak plasma concentrations within 1 hour after dosing. Plasma concentrations subsequently declined in a monophasic or biphasic fashion with a mean terminal-phase disposition t1/2 of 2.32 hours. Recovery of the administered radioactivity was 96%, with 74% and 22% in urine and feces, respectively, indicating urine was the major route of excretion. Excretion was fairly rapid with > 70% of the dosed radioactivity excreted within 24 hours post-dose. Parent compound was the predominant entity in circulation, representing 58% to 74% of the total radioactivity between 1 and 6 hours postdose, indicating that the overall circulating metabolite burden was low (< 50% of parent). Two hydroxylated metabolites in plasma (INCB027598 and INCB025264) were identified as the only major metabolites (30% and 14% of parent based on AUC0-24). Other INCB018424-related peaks in human plasma were at levels less than 10% of INCB018424 and consisted of mono- and di-hydroxylated metabolites. In urine and feces, the metabolite profiles also consisted of various hydroxylated and ketone metabolites and subsequent glucuronide conjugates. Parent drug accounted for < 1% of the administered dose in urine and feces. Data from previous clinical studies in healthy subjects indicated that there was no gender difference in INCB018424 levels when normalized for body weight and there were minimal differences in metabolite levels between single and multiple dosing. Importantly, no metabolites unique to humans have been identified, while the exposure (AUC) of the two major metabolites in human plasma after oral dosing of INCB018424 is lower than observed in plasma from oral studies in species used in toxicology studies at doses equal to or below the no-adverse-effect levels.
327. Preclinical disposition and pharmacokinetics-pharmacodynamic modeling of biomarker response and tumor growth inhibition in xenograft mouse models with G-573, a MEK inhibitor
Edna F. Choo1, Marcia Belvin2, Klaus Hoeflich3, Christine Orr3, Kirk Robarge4, and Jason W. Boggs1
1Dept of Drug Metabolism and Pharmacokinetics, Genentech Inc, South San Francisco, CA, USA, 94080
2Cell Signaling Pathways, Genentech Inc., USA
3Translational Oncology, Genentech Inc.
4Chemistry, Genentech Inc.
The mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase kinase (MEK) pathway is a key signaling pathway that regulates cell proliferation, cell cycle regulation, cell survival, angiogenesis, and cell migration. G-573 is an oral small molecule allosteric inhibitor of MEK that has been found to be potent and selective in various in vitro and cellbased assays. The objectives of these studies were to characterize the disposition of G-573 in preclinical species and to determine the relationship of G-573 plasma concentrations to pERK inhibition and tumor growth inhibition in K-ras mutant xenograft tumors, HCT-116 (colon) and H2122 (lung; NSCLC). The clearance of G-573 was low in mouse (7.7 mL/min/kg), rat (2.24 mL/min/kg), dog (10 mL/min/kg) and cynomolgus monkey (0.754 mL/min/kg) with low to moderate volumes of distribution (0.114-1.77 L/kg), resulting in moderate half-lives across species (~2 to 9 h). Bioavailabilities ranged from 50 to 100%. Indirect response models were used to characterize the relationship between plasma concentration of G-573 and both pERK inhibition and tumor growth inhibition. The IC50 value for pERK inhibition in HCT116 tumors by G-573 was estimated to be 0.406 mM and the IC50 values for tumor growth inhibition in HCT116 and H2122 were estimated to be 3.43 and 2.56 mM, respectively. ED50 estimates in HCT116 and H2122 models were estimated to be ~4.6 and 1.9 mg/kg/day, respectively. It appears that the in vivo potency of G-573 was similar in both HCT116 and H2122 xenograft models. These results are useful in estimating the target concentrations/doses required in the clinic and for determining appropriate clinical dosing regimens.
328. Hepatobiliary disposition of deoxyschizandrin in perfused rat livers in situ
Jingjing Wu, Yanyan Zhang, Guangbo Ge, and Ling Yang
Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China
Deoxyschizandrin (Schizandrin A, Sin A), is one of the major active lignans isolated from Schisandra fruits. In vitro studies have indicated that Sin A was selectively metabolized by CYP3A4 in human liver microsome. However, except for metabolism, blood flow, protein binding and transporter activity have also been identified as the determinants of organ clearance. Aim to determine the respective contributions of enzyme and transporter to hepatic clearance, and further develop Sin A as a new in vivo CYP3A probe, we investigated the disposition of deoxyschizandrin in recirculated perfused rat livers, descibed as a simple physiologically based pharmacokinetic (PBPK) liver model. Sin A was introduced as a bolus into the perfusate reservoir at a concentration of 25 μM. The perfusate and bile samples were collected over 60 min, and the concentrations of Sin A and it’s metabolite in perfusate and bile were measured using ultra fast liquid chromatograph assay-diode array detection (UFLC-DAD). Both Sin A and its C-7 hydroxylation metabolite, schizandrol A (Sol A), were identified in perfusate and bile. The levels of formation of other metabolites except for Sol A were negligible during our experimental procedure. Sin A was eliminated from the perfusate in a biexponential manner. The hepatic clearance was characterized with a high hepatic extraction ratio of approximately 63% for the parent drug. However, Sin A exhibited minimal biliary excretion, representing around 0.003% of the dose administered. Over 32% of the starting Sin A could be converted to Sol A at steady state. In conclusion, Sin A is unitarily metabolized in rat liver and hepatic metabolism is the major elimination pathway for Sin A, which improve the probability that Sin A might be served as a in vivo probe to evalute the elimination capability of liver via CYP3A.
329. Translational pharmacokinetic-pharmacodynamic modeling of a potent and selective MEK inhibitor
Harvey Wong1, Laurent Vernillet2, Amy Peterson2, Joseph A. Ware2, Lillian Lee3, Jean-Francois Martini3, Peiwen Yu3, Connie Li4, Geoffrey Del Rosario4, and John Prescott2
1Dept of Drug Metab & Pharmacokinetics, Genentech Inc, South San Francisco, CA, USA, 94080
2Genentech Inc, South San Francisco, CA, USA, 94080
3Exelixis, South San Francisco, CA, USA
4Exelixis (Current address Genentech Inc.), South San Francisco, CA, USA
The Raf/MEK/ERK signaling pathway is involved in cellular responses relevant to tumorigenesis. MEKi is a potent and selective MEK inhibitor that accumulates in xenograft tumor tissue. In preclinical studies with MEKi, pharmacodynamic response (i.e. tumor pERK inhibition) correlated well with tumor rather than plasma concentrations. The objective of this study is to characterize the accumulation kinetics of MEKi in tumor tissue and to estimate a plasma concentration needed for 85% tumor pERK inhibition. Briefly, MEKi was dosed orally to WM-266-4 xenografts (a partial-responder) at 1, 3, and 10 mg/kg for 1 or 14 days. Samples (plasma and tumor) were collected up to 168 hours post-dose on Days 1 and 14, and concentrations assessed by LC/MS/MS. Tumor pERK inhibition was determined by Western blot. Plasma and tumor concentration, and tumor pERK inhibition were fitted to a PK-PD model where pERK inhibition was linked to a tumor rather than a plasma compartment. The rate constants characterizing the movement of MEKi from plasma to tumor (kpt) and tumor to plasma (ktp) were estimated as 0.0383 hr−1 and 0.0324 hr−1, respectively. Pharmacodynamic parameters in WM-266-4 on Day 1 were estimated as follows: Emax (Maximum level of pERK inhibition) – 104%, EC50 (Tumor concentration associated with pERK inhibition equal to ½ Emax) – 1.03 μM. Similar pharmacodynamic parameters were estimated for responder tumor types (A375, A2058, Colo205, and MDA MB-231) following the oral administration of single doses of MEKi in xenografts (Dose range: 0.3 to 100 mg/kg). Pharmacodynamic parameters from responder xenografts and anticipated human pharmacokinetic parameters were incorporated into the PK-PD model and simulations were performed. Based upon these simulations, a minimum trough plasma concentration of 0.11 μM plasma concentration was determined to be needed for a sustained pERK inhibition of 85%. This study demonstrates the utility of preclinical PK-PD modeling in aiding the selection of a target clinical plasma concentration for compounds where the pharmacodynamic response is closely associated with concentrations at the site of action.
330. Pharmacodynamics of a potent and selective MEK inhibitor: An integrated model linking systemic and tumor concentrations, pERK inhibition and tumor growth inhibition
Harvey Wong1, Marcia Belvin2, Edna F. Choo3, Klaus Hoeflich4, John Prescott5, and Joseph A. Ware5
1Dept of Drug Metab & Pharmacokinetics, Genentech Inc, South San Francisco, CA, USA, 94080
2Cell Signaling Pathways, Genentech Inc., USA
3Dept of Drug Metabolism and Pharmacokinetics, Genentech Inc, South San Francisco, CA, USA, 94080
4Translational Oncology, Genentech Inc.
5Genentech Inc, South San Francisco, CA, USA, 94080
The Raf/MEK/ERK signaling pathway is involved in cellular responses relevant to tumorigenesis, including cell proliferation, invasion, survival and angiogenesis. MEKi is a potent and selective MEK inhibitor. The objective of the current study is to better understand the relationship between pERK inhibition and tumor growth inhibition in Colo205 xenografts following oral administration of MEKi. This was accomplished through PK-PD modeling of MEKi using an integrated PK-PD model linking MEKi tumor concentrations to pERK inhibition, and pERK inhibition to tumor growth inhibition. The disposition of MEKi in plasma and tumor was characterized in PK studies using WM-266-4 xenografts. The relationship between pERK and MEKi tumor concentrations was characterized in single dose studies (0.3 to 30 mg/kg) in Colo205 xenografts. An integrated PK-PD model combining these two elements with efficacy data from a Colo205 xenograft experiment dosed daily at 1, 3, and 10 mg/kg was used to understand the relationship between pERK inhibition and tumor growth inhibition. The following parameters were estimated from the integrated PK-PD model: kng (Net growth rate constant) - 0.0037 hr−1, Kmax (Maximum value of rate constant describing tumor growth inhibition [K]) - 0.998 hr−1, K(I%)50 (Percent pMEK1 inhibition where K is ½ Kmax) – 65%, and n (Hill coefficient) – 4.7. A simulated plot of K versus pERK inhibition generated using these parameter estimates, showed a steep pERK inhibition-response curve consistent with the estimated Hill coefficient of ~ 5. The observed relationship between pathway modulation and efficacy is consistent with the reported “ultrasensitive” behavior of the Raf-MEK-ERK pathway.
331. Pharmacokinetic-pharmacodynamic modeling of tumor growth inhibition and biomarker modulation by the novel PI3K inhibitor GDC-0941
Laurent Salphati1, Marcia Belvin2, Kyle Edgar2, Wei Wei Prior3, Deepak Sampath3, Jeffrey Wallin2, and Harvey Wong1
1Drug Metabolism and Pharmacokinetics, Genentech, Inc, South San Francisco, CA, USA, 94080
2Cancer Signaling, Genentech, Inc, South San Francisco, CA, USA, 94080
3Translational Oncology, Genentech, Inc, South San Francisco, CA, USA, 94080
The phosphatidylinositol 3-kinase (PI3K) pathway is a major determinant of cell cycling and proliferation. Its deregulation, by activation or transforming mutations of the p110α subunit, is associated with the development of many cancers. In recent years, this pathway has emerged as a major target for the investigation of anticancer drugs. GDC-0941 is a novel small molecule inhibitor of PI3K currently being evaluated in the clinic as an anticancer agent. The objectives of these studies were to characterize the relationships between GDC-0941 plasma concentrations and tumor growth inhibition in PC3-NCI prostate and MDA-MB-361.1 breast cancer xenografts, and to evaluate the relationship between the pharmacodynamic biomarker (pAKT) response in tumor and the antitumor efficacy. Female athymic nu/nu (nude) mice implanted subcutaneously with PC3-NCI or MDA-MB-361.1 cells were treated for up to 3 weeks with GDC-0941 at various doses (12.5 to 200 mg/kg) and dosing schedules (daily to weekly). GDC-0941 plasma concentrations were described by a one compartment model. Indirect response models were fitted to tumor growth and inhibition of AKT phosphorylation data. Estimates of GDC-0941 concentrations required for tumor stasis were approximately 4 and 0.3 μM in the PC3-NCI and MDA-MB-361.1 cancer xenografts, respectively. In the MDA-MB-361.1 model, the IC50 (plasma concentration achieving 50% of the maximum inhibition of AKT phosphorylation) was estimated to be approximately 0.4 μM. These results in the breast cancer xenograft model suggested that plasma concentrations associated with a 50% suppression of pAKT would achieve tumor stasis. Simulations of tumor growth and AKT phosphorylation using projected human PK parameters provided guidance to predict the doses and exposures likely to achieve suppression of AKT phosphorylation and efficacy.
332. Preclinical evaluation of a novel PI3K/mTor inhibitor and prediction of its human pharmacokinetics
Jodie Pang1, Matthew Baumgardner1, Xiao Ding1, Kyle Edgar2, Emile G. Plise1, Jeffrey Wallin2, Susan Wong1, Xiaolin Zhang1, and Laurent Salphati1
1Drug Metabolism and Pharmacokinetics, Genentech, Inc, South San Francisco, CA, USA, 94080
2Cancer Signaling, Genentech, Inc, South San Francisco, CA, USA, 94080
The phosphatidylinositol 3-kinase/mammalian target of rapamycin (PI3K/mTor) pathway is a major determinant of cell survival and proliferation. Its deregulation, by activating mutation of the p110alpha subunit, is associated with many cancers. In recent years, this pathway has emerged as a major target for the investigation of anticancer drugs. Compound I is a novel small molecule dual inhibitor of PI3K and mTor that displays anti-tumor activity in preclinical cancer models. It has been shown to be potent in PC3-NCI prostate cancer and MDA-MB-361.1 breast cancer cells with IC50 values of 0.31 and 0.47 μM, respectively. The pharmacokinetic properties of Compound I were assessed in pre-clinical species following intravenous and oral administrations. The permeability, plasma protein binding and metabolism were also examined in vitro. Compound I had low plasma clearance (Clp) in mice, rats, and dogs at 9, 15, and 6 mL/min/kg, respectively. In the monkey, Clp was moderate (19 mL/min/kg). The terminal half-life (t1/2) ranged from 0.6 hr in monkey to 6.3 hr in dogs. The steady-state volume of distribution (Vss) varied from approximately total body water volume in the monkey, to 6-fold total body water volume in the rat. Bioavailability ranged from 23% to 100%. In vitro, Compound I displayed low binding (47%-72%) to plasma proteins and the apparent permeability determined in MDCK cells was high.
Human PK parameters were predicted using different approaches, such as allometric scaling and PBPK modeling. Predicted human Clp and Vss were approximately 4 mL/min/kg and 1.7 L/kg, respectively.
333. The PK/PD relationship of an S1P1 agonist in rats as a tool to guide FIH dose selections
Ellen Rohde, Cheryl Black, Ping He, Ivan Nestorov, Qin Wang, and Liang-Shang Gan
Drug Metabolism and Pharmacokinetics, BiogenIdec, Cambridge, MA, USA, 02142
Agonists of the S1P1 receptor decrease circulating lymphocytes by blocking their egress from peripheral lymphoid tissues, thereby limiting their access to sites of inflammation in the body. Hence, S1P1 agonists have been used in preclinical models of autoimmune diseases. One molecule, FTY720, has shown efficacy in patients with multiple sclerosis. The lowered number of lymphocytes in circulation is used as a pharmacodynamic (PD) marker for receptor interaction but poses also a potential risk for opportunistic infections due to immunosupression. Mathematical modeling of the exposure-response relationship may be able to correlate dose levels that show efficacy with lymphocyte levels that provide optimal safety in chronic use against autoimmune diseases. Compound A (CpdA) is phosphorylated in vivo to yield metabolite 1 (M1) that acts as a potent S1P1 agonist (in vitro EC50 of 8nM). We describe an iterative process of pharmacokinetic (PK) and pharmacodynamic (PD) response modeling in preclinical species with the goal to guide decisions in clinical development. In a SAD study male Sprague-Dawley rats received a single oral bolus dose at 7 dose levels from 0.01mg/kg to 10mg/kg. Whole blood samples were collected up to 336h and absolute lymphocyte counts were measured in an aliquot of each. The rest of the samples were used to analyze for CpdA and M1 by LC-MS. The exposure to M1 was modeled with a 2-compartment model with first order absorption and elimination. Since M1 is thought to cause the inhibition of lymphocyte re-entry into the circulation the correlation between M1 concentration and absolute lymphocyte counts was modeled with an indirect response model. Assuming Imax=1 the response curves were fitted for all dose levels and an IC50 of 3ng/ml (6nM) was determined. Using the model we simulated differential lymphocyte levels and corresponding dose levels for a MAD study in rats. Results from this study will be compared with the prediction to create a data set that supports a comparable iterative modeling approach to studies in higher preclinical species and man.
334. Pharmacokinetics of Sarpogrelate following single intravenous or oral administration in rats
Ramakrishna Nirogi, Gopinadh Bhyrapuneni, Nageswararao Muddana, Ganesh Budige, Devender Ajjala, Satyanaryana Chintanippula, Koteshwara Mudigonda, and Vishwottam Kandikere
Discovery Research, Suven Life Sciences Ltd, Hyderabad, India
Sarpogrelate is a novel 5-HT2A and 5-HT2B receptors antagonist, which blocks serotonin-induced platelet aggregation (Shinohara et al., 2008; Uchiyama et al, 2007). The objective of the present investigation is to study the pharmacokinetics of Sarpogrelate in rats after a single intravenous or oral administration. Sarpogrelate was administered intravenously or orally at 5 mg/kg to male Wistar rats as a solution. The jugular vein of male wistar rats (n=6) were cannulated and a catheter was implanted for blood collection. The blood samples were collected from the jugular vein at predose, 0.08, 0.25, 0.5, 1, 2, 4, 6, 8 & 24 hr post dosing using isoflurane as an anesthetic agent. Blood samples were immediately centrifuged after withdrawal and plasma was frozen on dry ice. Sarpogrelate concentrations in plasma were analyzed by validated LC-MS/MS method. Data were analyzed using noncompartmental analysis model. Sarpogrelate was well tolerated during the course of study. Sarpogrelate after oral administration reached peak plasma concentration (Cmax) of 11514 ng/mL at 0.25 h. The clearance was 218 mL/h/kg and volume of distribution was 392 mL/kg after intravenous administration. After intravenous administration, the plasma concentration of sarpogrelate declined monoexponentially with a terminal half-life of 1.2 h. Sarpogrelate was absorbed rapidly after oral dosing with 47% bioavailability.
References:
- Shinohara Y, Nishimaru K, Sawada T, Terashi A, Handa S, Hirai S, Hayashi K, Tohgi H, Fukuuchi Y, Uchiyama S, Yamaguchi T, Kobayashi S, Kondo K, Otomo E, Gotoh F; S-ACCESS Study Group. Sarpogrelate-Aspirin Comparative Clinical Study for Efficacy and Safety in Secondary Prevention of Cerebral Infarction (S-ACCESS): A randomized, double-blind, aspirin-controlled trial. Stroke 39(6):1827–33, 2008.
- Uchiyama S, Ozaki Y, Satoh K, Kondo K, Nishimaru K. Effect of sarpogrelate, a 5-HT(2A) antagonist, on platelet aggregation in patients with ischemic stroke: clinical-pharmacological dose-response study. Cerebrovasc Dis. 24(2–3):264–70, 2007.
335. Evaluation of blood draw free Solid Phase Microextraction (SPME) sampling to determine the pharmacokinetics of R,R-Fenoterol (Fen) and R,R-Methoxyfenoterol (Mfen) in conscious rats
Ines A. M. de Lannoy1, Joanne Chung Yan Yeung2, Brad Gien1, Dajana Vuckovic2, Henrianna Pang1, Yingbo Yang1, and Janusz Pawliszyn2
1NoAb BioDiscoveries Inc., Mississauga, ON, Canada, L5N 8G4
2Department of Chemistry, University of Waterloo, Waterloo, ON, Canada, N2L 3G1
A blood draw free SPME in vivo sampling method was developed and compared to conventional plasma sampling for the determination of the pharmacokinetics of the β2-adrenergic receptor agonist, Fen, and a pharmacologically active analog, MFen. Five mg/kg Fen and Mfen were administered intravenously to groups of rats. For conventional sampling, plasma was isolated following collection of serial arterial blood samples. Plasma concentrations of Fen and MFen were determined by protein precipitation and LC-MS/MS analysis. Plasma clearances (CLp) for Fen and Mfen were similar (2969 ± 310 and 2726 ± 242 mL/h/kg (n=3), respectively), whereas their steady-state volumes of distribution (Vss) differed considerably (1928 ± 503 and 6374 ± 720 mL/kg, respectively). The terminal half-lives of Fen and Mfen were also similar (2.54 ± 0.21 and 2.63 ± 0.74 h, respectively). The fraction of Mfen metabolized to Fen averaged 0.96 ± 0.40%. For SPME sampling, prototype SPME probes (Supelco) with biocompatible extractive coating were used. A sampling interface was designed to facilitate insertion of the SPME probes for repeated sampling from arterial blood. A diffusion-based, pre-equilibrium, internal standard-free calibration method for quantitative analysis of the 2 analytes by SPME was employed. A constant sampling rate (1.4 mL/min) was accomplished by an AccuSampler®. Total blood concentrations of Fen and Mfen were determined by SPME sampling and LC-MS/MS analysis. Total blood concentrations were consistently greater than corresponding plasma concentrations determined by conventional sampling. Estimated blood clearances (CLb) for Fen and Mfen were lower than CLp (1205 ± 129 and 812 ± 97.7 mL/h/kg (n=5), respectively), as were Vss values (560 ± 177 and 1592 ± 491 mL/kg, respectively). This is primarily a result of red blood cell partitioning/binding since the blood to plasma concentration ratios (B/P) of Fen and Mfen were 1.71 ± 0.10 and 2.03 ± 0.15, respectively. CLp’s calculated from the corresponding B/P and CLb values were 2061 ± 221 and 1649 ± 198 mL/h/kg, respectively, indicating that SPME sampling holds promise as a valuable method for blood draw free sampling from rodents.
336. Poor correlation between phenytoin urinary metabolites and oral bioavailability in epilepsy patients
Theresa Aliwarga1, Varun Goel2, Richard Brundage2, Ilo E. Leppik2, John O. Rarick2, James C. Cloyd2, and Rory P. Remmel1
1Medicinal Chemistry, University of Minnesota, Minneapolis, MN, USA, 55455
2Experimental and Clinical Pharmacology, University of Minnesota, Minneapolis, MN, USA, 55455
Phenytoin (PHT) has been shown to be one of the most effective anticonvulsants for treating generalized tonic-clonic seizuresFootnote[1] and status epilepticusFootnote[2]. PHT is known to be extensively bound to plasma proteins (~90%), to be almost entirely excreted from the body as metabolites in the urine, and to exhibit a non-linear pharmacokinetics profile. In this study, urine samples from epilepsy patients who were on maintenance therapy of PHT were collected from 0-12 hours and 12-24 hours after a single daily dose. Stable-labeled PHT was administered intravenously (IV) or intramuscularly in order to directly determine the pharmacokinetic parameters of volume and half-life. Pharmacokinetic parameters (IV and oral) were used to determine the absolute oral bioavailability by NONMEM software. There were two principal PHT urinary metabolites, 5-(4-hydroxyphenyl)-5-phenylhydantoin (p-HPPH) and 5-(3,4-dihydroxy-1,5-cyclohexadien-1-yl)-5-phenylhydantoin (DHD), which reportedly account for 65% to 90% of administered PHTFootnote[3] [4]. An isocratic HPLC-NI-APCI-MS method was used to quantify both of the principal urinary metabolites and to obtain the 24-hour dose recovery. With the Spearman Rank Correlation test, a positive correlation was observed between the total urinary metabolites recovered and the oral bioavailability. Although the correlation coefficient was unexpectedly low (0.422), the p-value was significant (0.00697). The percent of dose recovered ranged from 13.5% to 62.2% in young patients (age 21-49 years old) and 11.8% to 85.3% in elderly patients (age 65-93 years old) indicating highly variable absorption.
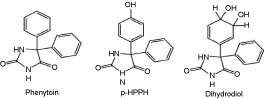
337. A pharmacogenetics-based pharmacokinetic model explains the difference in harmaline drug effects between wild-type and CYP2D6-humanized mouse models
Xiling Jiang1, Chao Wu1, Hongwu Shen2, and Ai-Ming Yu3
1Department of Pharmaceutical Sciences, School of Pharmacy and Pharmaceutical Sciences, University at Buffalo, The State University of New York, Amherst, NY, USA, 14260
2Department of Pharmaceutical Sciences, School of Pharmacy and Pharmaceutical Sciences, The State University of New York at Buffalo, Buffalo, NY, USA, 14260
3Department of Pharmaceutical Sciences, University at Buffalo, SUNY, Buffalo, NY, USA, 14260-1200
Harmaline, 4-dihydro-7-methoxy-1-methyl-β-carboline, is a β-carboline alkaloid that selectively inhibits the activity of monoamine oxidase-A, and displays neuroprotective and neurotoxic properties [1-4]. Our recent studies have revealed an important contribution of polymorphic cytochrome P450 2D6 (CYP2D6) to harmaline O-demethylation metabolism [5, 6]. Current study, therefore, aimed to develop a pharmacogenetics-based pharmacokinetic (PK) model to delineate the impact of CYP2D6 phenotype/genotype on harmaline PK and pharmacodynamics (PD). Comparative studies in CYP2D6-humanized (Tg-CYP2D6) versus wild-type control mice, which differ in CYP2D6 drug-metabolizing activity [7, 8], showed that Tg-CYP2D6 mice had lower exposure to harmaline (5 and 15 mg/kg; i.v. and i.p). Consistently, wild-type mice showed more severe harmaline-induced hypothermia. Then a pharmacogenetics-based PK model involving the clearance of drug by CYP2D6 (CLCYP2D6) and other mechanisms (CLother) was linked to an indirect response PD model, which described well the effect of CYP2D6 status on the PK/PD of harmaline in the two genotyped mice. These results indicate that distinct CYP2D6 status may result in significant variations in harmaline PK and PD. In addition, the pharmacogenetics-based PK model may be applied to define the PK variations caused by other polymorphic drug-metabolizing enzymes in different populations.
Acknowledgments
This project is supported by the award (R01DA 021172) from National Institute on Drug Abuse.
References:
- Abdel-Fattah, A.F., et al., Central serotonin level-dependent changes in body temperature following administration of tryptophan to pargyline- and harmaline-pretreated rats. Gen Pharmacol, 1997. 28(3): p. 405–9.
- Hilber, P. and P. Chapillon, Effects of harmaline on anxiety-related behavior in mice. Physiol Behav, 2005. 86(1–2): p. 164–7.
- Yu, A.M., Indolealkylamines: biotransformations and potential drug-drug interactions. AAPS J, 2008. 10(2): p. 242–53.
- Moura, D.J., et al., Antioxidant properties of beta-carboline alkaloids are related to their antimutagenic and antigenotoxic activities. Mutagenesis, 2007. 22(4): p. 293–302.
- Yu, A.M., et al., Contribution of individual cytochrome P450 isozymes to the O-demethylation of the psychotropic beta-carboline alkaloids harmaline and harmine. J Pharmacol Exp Ther, 2003. 305(1): p. 315–22.
- Zanger, U.M., S. Raimundo, and M. Eichelbaum, Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch Pharmacol, 2004. 369(1): p. 23–37.
- Corchero, J., et al., The CYP2D6 humanized mouse: effect of the human CYP2D6 transgene and HNF4alpha on the disposition of debrisoquine in the mouse. Mol Pharmacol, 2001. 60(6): p. 1260–7.
- Gonzalez, F.J. and A.M. Yu, Cytochrome P450 and xenobiotic receptor humanized mice. Annu Rev Pharmacol Toxicol, 2006. 46: p. 41–64.
338. Toxicokinetics of tirapazamine analogs in rats and its use for prediction of therapeutic activity against hypoxic tumor cells by spatially resolved PKPD modeling
Jagdish K. Jaiswal, Kevin O. Hicks, Frederik B. Pruijn, William A. Denny, Michael P. Hay, and William R. Wilson
Auckland Cancer Society Research Centre, The University of Auckland, Auckland, New Zealand, 1142
Tirapazamine (TPZ) is a benzotriazine-di-N-oxide with selective toxicity to hypoxic cells, which is known to limit the efficacy of radiotherapy in head and neck squamous cell carcinoma (HNSCC). TPZ recently failed to achieve its primary endpoint in a pivotal randomized phase III trial with cisplatin and radiotherapy for advanced HNSCC. Preclinical evidence suggests one of the limitations of TPZ is its poor micropharmacokinetics due to limited penetration into hypoxic tissue. We have developed and validated a spatially resolved (SR) PKPD model that describes PK and PD (cell killing) at each point in a tumor microvascular network, and have used this to guide a lead optimization program for hypoxic cell killing in human xenografts in CD-1 nude mice. The two most active compounds from this program were the morpholides SN 29751 and SN 30000, which have improved ability to penetrate hypoxic tissue. The SR-PKPD model can in principle be used to evaluate therapeutic potential of these analogs in other species for which appropriate tumor models are less readily available. To this end, we determined the plasma PK of TPZ and the two analogs at their respective maximum tolerated doses (MTD) in Sprague Dawley rats following single intraperitoneal doses. The MTD values were 178/178 (male/female) μmol/kg for TPZ, 316/316 μmol/kg for SN 29751, and 316/316 μmol/kg for SN 30000. Non-compartmental plasma PK values, were: Cmax 80 ± 1/95 ± 2 μM for TPZ, 202 ± 16/203 ± 8 μM for SN 29751 and 112 ± 5/183 ± 8 μM for SN 30000, and AUC0-4h72 ± 6/122 ± 4 μM.h for TPZ, 387 ± 6/391 ± 21 μM.h for SN 29751 and 118 ± 2/281 ± 3.2 μM.h for SN 30000. Using the plasma PK as input to the tumor compartment, the SR-PKPD model predicted log10 cell kill in hypoxic regions of a (virtual) HT29 tumor in rats treated at MTD as 0.5/1.0 for TPZ, 2.8/2.8 for SN 29751 and 2.4/4.6 for SN 30000. Thus, the achievable plasma PK in rats leads to SR-PKPD predictions of activity that indicate clear superiority of the novel analogs over TPZ, as was seen in mice.
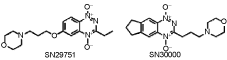
339. PXR and CAR regulate expression of carboxylesterase 6 in mouse liver and small intestine
Chenshu Xu, Xinkun Wang, and Jeff L. Staudinger
Department of Pharmacology and Toxicology, School of Pharmacy, University of Kansas, Lawrence, KS, USA, 66045
Pregnane X receptor (PXR) and constitutive androstane receptor (CAR), two members of the nuclear receptor superfamily of ligand-activated transcription factors, mediate gene activation in response to xenobiotic stress. Together, these receptors comprise a protective response in mammals that coordinately regulate hepatic transport, metabolism, and elimination of numerous xenobiotic compounds. The liver- and intestine-enriched carboxylesterase 2 (CES2) enzyme catalyzes the hydrolysis of several clinically important anticancer agents administered as prodrugs. In the present study, microarray analysis was used to identify PXR target genes in duodenum in mice. Here, we show that a gene encoding a member of the CES2 subtype of liver- and intestine-enriched CES enzymes, called Ces6, is induced after treatment with pregnenolone 16a-carbonitrile (PCN) in a PXR-dependent manner in duodenum and liver in mice. Treatment of mice with the CAR activator 1,4-bis[2-(3,5-dichloropyridyloxy)] benzene (TCPOBOP) also induced expression of Ces6 in duodenum and liver in a CAR-dependent manner. Taking together, these data identify a key role for PXR and CAR in regulating the drug-inducible expression and activity of an important CES enzyme in vivo.
340. The molecular mechanism by which ERK1/2 regulates CAR activation
Makoto Osabe
Lrdt, NIH/NIEHS, Durham, NC, USA, 27709
The nuclear xenobiotic receptor CAR is the key transcription factor that regulates drug metabolism and excretion. CAR is sequestered in the cytoplasm and translocates into the nucleus following drug activation. We previously characterized the activated extracellular signal-regulated kinase1/2 (ERK1/2) as the endogenous signal that regulates cytoplasmic retention of CAR in mouse primary hepatocytes (1). To decipher the molecular mechanism of this ERK1/2-mediated regulation, we have now investigated the interaction of CAR with ERK1/2 by co-immunoprecipitation: ectopically expressing human CAR (hCAR) with endogenous ERK1/2 in Huh7 cells. Based on our recent finding that threonine 38 within the hCAR DBD can be phosphorylated and that this phosphorylation sequesters hCAR in the cytoplasm, we constructed Flag-tagged T38D and T38A mutants. These mutants and wild type hCAR were expressed in Huh7 cells, from which hCAR proteins were immunoprecipitated by anti-phospho-ERK1/2 (P-ERK1/2) antibody and subjected to Western blot analysis for co-immuoprecipitation, of which, only the T38D mutant, but not the wild type nor the T38A mutant, co-immunoprecipitated with P-ERK1/2. Various deletions were constructed within the context of Flag-tagged T38D mutant and expressed in Huh7 cells for co-immunoprecipitation with P-ERK1/2, by which the P-ERK1/2 binding is delineated to the XRS region (xenobiotic response signal; residues from 309 to 328) within the hCAR LBD. A specific co-immunoprecipitation of ectopic GFP-tagged XRS with P-ERK1/2 has further confirmed the interaction of P-ERK1/2 with XRS: XRS was previously defined as the peptide required for CAR nuclear translocation in mouse liver (2). These results are consistent with the hypothesis that P-ERK1/2 specifically interacts with the phosphorylated form of hCAR and prevents it from dephosphorylation, thus retaining hCAR in the cytoplasm.
References:
- Koike et al., Extracellular Signal-Regulated Kinase Is an Endogenous Signal Retaining the Nuclear Constitutive Active/Androstane Receptor (CAR) in the Cytoplasm of Mouse Primary Hepatocytes. Mol. Pharmacol. 71:1217–1221 2007
- Zelko et al., The Peptide Near the C Terminus Regulates Receptor CAR Nuclear Translocation Induced by Xenochemicals in Mouse Liver. Mol. Cell. Biol. 21:2838–2846 2001
341. Molecular mechanism of cytochrome P450 induction by atorvastatin and atorvastatin metabolites
Ella Hoffart1, Andreas K. Nuessler2, Wolfgang E. Thasler3, Thomas Weiss4, Matthias Schwab1, and Oliver Burk1
1Dr. Margarete Fischer-Bosch - Institute of Clinical Pharmacology, Stuttgart, Germany, 70376
2Traumatology, TU Munich, Munich, Germany
3Surgery, LM University Munich, Hospital Grosshardern, Munich, Germany, 81377
4Surgery, University of Regensburg, Regensburg, Germany, 93040
The HMG-CoA reductase inhibitor atorvastatin (ATV), one of the most frequently prescribed drugs worldwide, was shown to induce the expression of drug metabolizing enzymes. As the complex interplay between hydroxylation and lactonization, involving CYP3A and UGT enzymes, determines the ratio of pharmacologically active and inactive metabolites, autoinduction of ATV metabolism may be a critical factor for the variable disposition of active metabolites. We thus aimed to elucidate the molecular mechanism of ATV-dependent enzyme induction, which we hypothesized to involve PXR and/or CAR. The treatment of primary human hepatocytes with ATV and ATV metabolites demonstrated differential induction of CYP3A4 and CYP2B6, with p-OH-ATV being the weakest inducer. Furthermore, induction was significantly reduced as compared to prototypical PXR and CAR activators. Mammalian two-hybrid nuclear receptor-coactivator interaction and nuclear receptor assembly assays demonstrated that ATV and all of its metabolites function as ligands of PXR, however with significant lower affinity than the prototypical ligand rifampin. Among the metabolites, ATV lactone showed the highest affinity to this receptor. In CYP3A4 promoter/reporter gene analysis, ATV and its metabolites further proved to activate PXR. In contrast, we could not prove ligand binding of ATV metabolites to CAR. ATV treatment of primary human hepatocytes further resulted in a significant increase of SREBP2 expression, which we could show to negatively interfere with PXR-mediated induction by co-transfection of SREBP2 expression plasmids with CYP3A4 promoter/reporter genes into HepG2 cells stably expressing PXR. The respective molecular mechanism may involve protein-protein interaction between SREBP2 and PXR, as both proteins showed to interact directly in a GST pull-down assay in vitro. In conclusion, these data show that ATV and its metabolites induce hepatic CYP3A4 and CYP2B6 expression by binding as ligands to PXR. The significantly reduced induction, as compared to prototypical ligands, is most likely explained by a combined effect of the reduced affinity of ATV binding to PXR and the ATV-dependent increase in SREBP2, which negatively interferes with PXR-mediated induction.
342. 3-Indoxyl sulfate is a high-affinity agonist of the human aryl hydrocarbon receptor
Jennifer C. Schroeder, Iain A. Murray, Colin A. Flaveny, Brett C. DiNatale, and Gary H. Perdew
Veterinary and Biomedical Sciences, Penn State University, University Park, PA, USA, 16802
The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that has historically been studied in terms of its ability to induce a number of drug metabolizing enzymes (i.e. CYP1A1/2, UGT1A1, SULT1A1). Recent studies by our laboratory and others have demonstrated that the AHR is involved in the regulation of a number of genes involved in the immune system, including interleukin-6 (IL6) (Hollingshead BD, Beischlag TV, Dinatale BC, Ramadoss P, Perdew GH. Inflammatory signaling and aryl hydrocarbon receptor mediate synergistic induction of interleukin 6 in MCF-7 cells. Cancer Res. 2008 May 15;68(10):3609-17). Characterization of AHR-null mouse models has further shown physiological relevance for the AHR. While not embryonic lethal, AHR-knock-out mice exhibit a decreased immune response, as well as compromised vascular formation in the liver and reproduction deficiencies. Thus far, the search for an endogenous AHR ligand has yielded few compounds that elicit an adequate response at physiologically-relevant levels. In the data presented here, we show that 3-indoxyl sulfate (I3S), an indole metabolite formed in the liver after absorption from the gut (Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A. 2009 Mar 10;106(10):3698-703), binds to the AHR and acts as a partial agonist for the human AHR at low concentrations (EC50 = 12 nM). Activation of the mouse AHR was also observed, but at a much higher dose of the compound (EC50 = 5.8 μM) in a stable reporter cell line. This response was specific to I3S, as other indole-derived metabolites (i.e. tryptophan, serotonin, and indole-3-propionic acid) were tested and did not activate the AHR in the reporter assay. Enhanced expression of AHR-mediated genes was observed in both Huh7 cells and primary human hepatocytes, as determined by real-time RT-PCR. Functional assays further demonstrated an increase in CYP1A activity following treatment. Additionally, I3S mediated the repression of the SAA1 gene through the AHR (Patel RD, Murray IA, Flaveny CA, Kusnadi A, Perdew GH. Ah receptor represses acute-phase response gene expression without binding to its cognate response element. Lab Invest. 2009 Jun;89(6):695-707). I3S and IL1B synergistically induce IL6 expression in MCF-7 tumor cell line, which may results in increased cell survival and invasive potential. As patients undergoing dialysis can develop elevated levels (upwards of 800 μM) of this compound (Sun H, Huang Y, Frassetto L, Benet LZ. Effects of uremic toxins on hepatic uptake and metabolism of erythromycin. Drug Metab Dispos. 2004 Nov;32(11):1239-46), we suggest that certain adverse side effects associated with this treatment may actually result from the continuous activation of the AHR.
343. The roles of nuclear xenobiotic receptor CAR in DDC-induced Oval cell proliferation in mouse liver
Yuichi Yamazaki, Rick Moore, and Masahiko Negishi
Pharmacogenetics Section, LRDT, NIEHS, NIH, Research Triangle Park, NC, USA, 27709
Liver is endowed with ability to regenerate hepatocytes in response to injury. When this regeneration ability is impaired during liver injury, oval cells, which are considered to be postnatal hepatic progenitors, proliferate and differentiate into hepatocytes. Here we have investigated the role of nuclear xenobiotic receptor CAR in the regulation of oval cell proliferation and differentiation using the 3, 5-diethoxycarbonyl-1, 4-dihydrocollidine (DDC)-induced mouse model (1). To this end, Car+/+, Car-/-, Pxr+/+, and Pxr-/- mice were fed with control and DDC-containing diets for 2 weeks, of which only the Car-/- mice did not develop hepatomegaly, cholestasis (an increase of serum ALT and bilirubin levels) and the atypical ductular reaction (an indication of oval cell proliferation). This CAR-dependent oval cell proliferation was further substantiated by immunohistochemical staining of ductular regions using an anti-A6 antibody as well as by the increase of TROP2 mRNA, the gene specifically expressed in oval cells. In addition, real time PCR analysis revealed that the Cyp2b10, Cyp2c55, Tnfa, Il6 and Gadd45b genes are all up-regulated in a CAR-dependent manner, whereas the Oatp1, Oatp2, Oat4, Ntcp, Bsep,Ugt1a1, Fxr and Hnf1 genes are all down-regulated in a CAR-dependent manner. These genes are the subjects for further investigation for their involvement in CAR-mediated liver injury, cholestasis, regeneration and oval cell proliferation in DDC-fed mice.
Reference:
- Okabe et al. Potential hepatic stem cells reside in EpCAM+ cells of normal and injured mouse liver. Development 136, 1951–1960, 2009
344. Nuclear receptor PXR Elicits p38 MAPK signal via activation of the GADD45β gene
Susumu Kodama, and Masahiko Negishi
Pharmacogenetics Section, LRDT, National Institute of Environmental Health Sciences, NIH, Research Triangle Park, NC, USA, 27709
The nuclear pregnane X receptor (PXR) was originally characterized as a key transcription factor of hepatic endo-/xeno-biotic metabolism and excretion (1). Here we have demonstrated a novel role of PXR in the regulation of the immediate-early response GADD45β gene, eliciting a p38-mediated signal and cell migration. Rifampicin treatment causes morphological changes and increases cell migration in ShP51 cells (HepG2 stably expressing human hPXR). Western blot analysis has revealed that treatments with rifampicin and SR12813 results in a sharp increase of p38 MAPK phosphorylation. The phosphorylayion of the up- and down stream factors of p38 (MKK3/6, MK2 and ATF2) are also up-regulated. Treatment with the specific p38 inhibitor SB239063 represses this p38 phosphorylation by hPXR and abolishes both the hPXR-dependent morphological changes and the migration of ShP51 cells. Drug activation of hPXR was also found to increase the expression of GADD45β gene. Ectopically expressing GADD45β increases p38 phosphorylation in parental HepG2 cells, whereas trasnfection of GADD45β siRNA decreases hPXR-dependent p38 phosphorylation in ShP51 cells. Therefore, these results suggest that hPXR induces the GADD45β gene to activate the p38 signal pathway, causing morphological changes and cell migration.
Reference:
- Kliewer et al., An orphan nuclear receptor activated by pregnane defines a novel steroid signaling pathway. Cell 92, p73–82, 1998.
345. The molecular mechanism by which ERK 1/2 regulates CAR activation
Makoto Osabe, Shingo Mutoh, Rick Moore, Tatsuya Sueyoshi, and Masahiko Negishi
Pharmacogenetics Section, LRDT, NIEHS, NIH, Research Triangle Park, NC, USA, 27709
The nuclear xenobiotic receptor CAR is the key transcription factor that regulates drug metabolism and excretion. CAR is sequestered in the cytoplasm and translocates into the nucleus following drug activation. We previously characterized the activated extracellular signal-regulated kinase 1/2 (ERK1/2) as the endogenous signal that regulates cytoplasmic retention of CAR in mouse primary hepatocytes (1). To decipher the molecular mechanism of this ERK1/2-mediated regulation, we have now investigated the interaction of CAR with ERK1/2 by co-immunoprecipitation: ectopically expressing human CAR (hCAR) with endogenous ERK1/2 in Huh7 cells. Based on our recent finding that threonine 38 within the hCAR DBD can be phosphorylated and that this phosphorylation sequesters hCAR in the cytoplasm, we constructed Flag-tagged T38D and T38A mutants. These mutants and wild type hCAR were expressed in Huh7 cells, from which hCAR proteins were immunoprecipitated by anti phospho-ERK1/2 (P-ERK1/2) antibody and subjected to Western blot analysis for co-immunoprecipitation, of which, only the T38D mutant, but not the wild type nor the T38A mutant, co-immunoprecipitated with P-ERK1/2. Various deletions were constructed within the context of Flag-tagged T38D mutant and expressed in Huh7 cells for co-immunoprecipitation with P-ERK1/2, by which the P-ERK1/2 binding is delineated to the XRS region (xenobiotic response signal; residues from 309 to 328) within the hCAR LBD. A specific co-immunoprecipitation of ectopic GFP-tagged XRS with P-ERK1/2 has further confirmed the interaction of P-ERK1/2 with XRS: XRS was previously defined as the peptide required for CAR nuclear translocation in mouse liver (2). These results are consistent with the hypothesis that P-ERK1/2 specifically interacts with the phosphorylated form of hCAR and prevents threonine 38 from dephosphorylation, thus retaining hCAR in the cytoplasm.
References:
- Koike et al., Extracellular Signal-Regulated Kinase Is an Endogenous Signal Retaining the Nuclear Constitutive Active/Androstane Receptor (CAR) in the Cytoplasm of Mouse Primary Hepatocytes. Mol. Pharmacol. 71:1217–1221 2007
- Zelko et al., The Peptide Near the C Terminus Regulates Receptor CAR Nuclear Translocation Induced by Xenochemicals in Mouse Liver. Mol. Cell. Biol. 21:2838–2846 2001
346. Nuclear receptor signaling in HepaRG cells
Dan E. Brobst1, and Jeffery L. Staudinger2
1Dept of Pharmacology & Toxicology, University of Kansas School of Pharmacy, Lawrence, KS, USA, 66045
2Dept of Pharmacology & Toxicology, University of Kansas School of Pharmacy, Lawrence, KS, 66045
HepaRG cells (Biopredic, France) represent a novel cell line derived from a human hepatocellular carcinoma. These cells exhibit two unique features; (a) when seeded at low density they acquire an elongated undifferentiated morphology, (b) they actively divide and after reaching confluency form typical ‘hepatocyte-like’ colonies that are surrounded by biliary ‘epithelial-like’ cells (1). HepaRG cells express high levels of various liver-enriched cytochrome P450 enzymes including CYP1A2, CYP2B6, CYP2C9, CYP2E1, and CYP3A4. HepaRG cells also express high levels of the nuclear receptor proteins constitutive androstane receptor (CAR, NR1I3) and pregnane X receptor (PXR, NR1I2). (2). The farnesoid x receptor (FXR, NR1H4) is another liver-enriched member of the nuclear receptor superfamily of ligand activated transcription factors that is involved in the regulation of bile acid, lipid, and glucose metabolism. The FXR protein is increasingly thought of as a potential pharmacological target for treatment of metabolic syndrome. Data will be presented regarding the expression levels of FXR, small heterodimer partner (SHP), CYP7A1, bile salt export pump (BSEP), apolipoprotein E (APOE), phosphoenolpyruvate carboxykinase (PEPCK) and Na+-taurocholate cotransporting polypeptide (NTCP) in both HepaRG cells and primary human hepatocytes following treatment with the highly selective synthetic FXR agonist, GW4064. These results reveal that the HepaRG cell line represents a novel tool that is useful in the examination of FXR’s role in bile acid, lipid and glucose metabolism, as well as in drug metabolism. Taken together, these results show that the HepaRG cells more thoroughly recapitulate the hepatocyte phenotype when compared with other cell lines such as HepG2. (1) The human hepatoma HepaRG cells: a highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Guillouzo A, Corlu A, Aninat C, Glaise D, Morel F, Guguen-Guillouzo C. Chemico-Biological Interactions. 2007 May 20;168(1):66-73. Review. PMID: 17241619 (2) Functional expression, inhibition and induction of CYP enzymes in HepaRG cells. Turpeinen M, Tolonen A, Chesne C, Guillouzo A, Uusitalo J, Pelkonen O. Toxicology In Vitro. 2009 Jun;23(4):748-53. Epub 2009 Mar 26. PMID: 19328226.
347. Evaluation of in vivo P-glycoprotein activity by in situ brain perfusion technique
Takashi Ueno1, Yoshitane Nozaki1, Osamu Takenaka1, and Tsutomu Yoshimura2
1Drug Metabolism and Pharmacokinetics Research, Eisai Co., Ltd., Tsukuba, Japan, 300-2635
2Drug Metabolism and Pharmacokinetics Research, Eisai Co., Ltd., Tsukuba, Japan
P-glycoprotein (P-gp) is responsible for limiting the brain penetration of its substrates. P-gp deficient mouse has been a useful in vivo tool for evaluating the impact of P-gp on the drug distribution across the blood-brain barrier (BBB). However, restricted availability of P-gp deficient mouse sometimes makes the assessment of P-gp effect less efficient especially at the early stage of drug development. Thus, we investigated the effect of P-gp inhibitors on the brain uptake of P-gp substrates by in situ brain perfusion technique using normal mice. To optimize perfusion conditions, the brain distribution of the typical P-gp substrate, [3H]quinidine, was evaluated. [3H]Quinidine was taken up by the brain in a perfusion time-dependent manner for up to 120 sec, and influence of flow rate on its uptake was not observed at more than 1.0 mL/min. The distribution volume of [3H]quinidine increased 9.5 to 16.1-fold in the presence of P-gp inhibitors. The brain penetration of [3H]quinidine in P-gp deficient mouse was similar to that in normal mouse with P-gp inhibitors, suggesting that P-gp inhibitors could inhibit completely P-gp activity on the BBB. Based on these results, inhibition assay conditions, which maximally inhibited P-gp, were determined: perfusion time = 60 sec; flow rate = 1.0 mL/min; and inhibitor = verapamil (100 μM). Brain penetration of 9 compounds including typical P-gp substrates was evaluated by measuring brain concentrations by LC-MS/MS except for [3H]quinidine, and brain uptake of P-gp substrates was increased in the presence of verapamil. There was a good correlation between the ratio of initial brain uptake clearance in situ with and without verapamil and the ratio of Kp,brain in P-gp deficient and wild-type mouse for tested compounds. The present study suggested that the in situ brain perfusion method with and without verapamil is useful to assess the influence of P-gp in vivo on the brain penetration of compounds, and moreover effective technique for the evaluation of in vivo P-gp effect in an early stage of drug development.
348. Fluorescence-based microplate assay for assessing the interaction of drugs with human organic anion transporters 1 and 3
Arpine Vapurcuyan, Imad Hanna, and Ryan M. Pelis
Drug Metabolism and Pharmacokinetics, Novartis Institutes for BioMedical Research, East Hanover, NJ, USA, 07936
The organic anion transporters 1 (OAT1) and 3 (OAT3) contribute to the renal clearance of numerous drugs and drug-related metabolites. Thus, inhibition of the OATs could influence the body distribution and elimination of the drugs they handle. Here, an assay was established to examine the potential of drugs to inhibit the OATs. 6carboxyfluorescein (6CF) was chosen as a substrate since it is transported by both proteins (Cihlar and Ho, 2000)(Zhang et al., 2004). The human orthologs of OAT1 and OAT3 were expressed in human embryonic kidney (HEK) cells. Uptake of 6CF by HEK/OAT1 and HEK/OAT3 cells was done in 96-well plates, and the cellular content was determined with a fluorescence microplate reader. Uptake of 1.5 μM 6CF by HEK/OAT1 and HEK/OAT3 cells was linear for 25 min, but undetectable in HEK parental cells. The Jmax of 6CF transport by OAT1 and OAT3 were 27.6 ± 0.39 and 15.4 ± 0.38 pmol mg protein−1min−1, respectively. The Km values for 6CF transport were 18.1 ± 1.0 (OAT1) and 17.7 ± 1.7 μM (OAT3). The potential for cimetidine, para-aminohippurate (PAH) and estrone-3-sulfate (ES) to inhibit OAT1- and OAT3-mediated 6CF transport was tested, as these are known substrates/inhibitors. The rank ordering for inhibition of OAT1 was PAH>ES>cimetidine, and for OAT3 was ES>cimetidine>PAH. We have established a cell based assay for examining the potential for drugs to inhibit OAT1 and OAT3. This assay will be useful for further determining if identified drugs are substrates of OAT1 and OAT3, and the possible impact this may have on their renal clearance in humans.
References
- Cihlar T and Ho ES, Fluorescence-based assay for the interaction of small molecules with the human renal organic anion transporter 1. Anal Biochem 283: 49-55, 2000.
- Zhang X, Groves CE, Bahn A, Barendt WM, Prado MD, Rödiger M, Chatsudthipong V, Burckhardt G, Wright SH. Relative contribution of OAT and OCT transporters to organic electrolyte transport in rabbit proximal tubule. Am J Physiol 287: F999-1010, 2004.
349. Validation of OATP1B1 transporter assays for regulatory submissions
Pradeep Sharma, Victoria E. Holmes, Robert Elsby, Craig Lambert, and Dominic Surry
Clinical Pharmacology and DMPK, AstraZeneca R&D Charnwood, Loughborough, United Kingdom
Studying OATP1B1 (SLCO1B1)-mediated transport of drugs and the influence of pharmacogenetic factors as well as the potential for drug-drug interaction is of great importance in the development of new medicines. Substrates of OATP1B1 may demonstrate variable exposures in the clinic due to polymorphisms resulting in a “poor transporter” or CC phenotype, whereas inhibition of the uptake of such drug substrates may result in clinically significant drug interactions as exemplified with the statin class of lipid lowering agents (1). This inhibition of statin uptake may have two clinical outcomes – a decrease in efficacy due to reduced access to the target organ (the liver) and an increase in toxicity (myopathy) due to increased systemic availability (2). Regulatory guidance for the characterisation and validation of transporter assays and their outcomes is currently limited and primarily focussed on P-glycoprotein (P-gp) (3). We have previously published an approach using the Caco-2 cell line for studying compounds as substrates and inhibitors of P-gp (4). Here we describe a validation of a cell-based system to assess new drug candidates as substrates and inhibitors of OATP1B1. The study included passage-to-passage variability of the kinetics for the prototypical substrate estradiol 17ß-glucuronide and assessments of the known OATP1B1 substrates estrone 3-sulfate and pravastatin (2). Inhibition of OATP1B1mediated transport was studied over a range of passages using rifamycin sv and on single occasions with gemfibrozil and bezafibrate (2). The data generated has demonstrated the procedures to be robust and reproducible. Subsequently we have developed criteria to monitor future performance in a GLP environment whilst allowing for elimination of the assay repetition currently proposed by the FDA.
References
- Neuvonen PJ, Niemi M, Backman JT. Drug interactions with lipid-lowering drugs: mechanisms and clinical relevance. Clin.Pharmacol.Ther. 2006 Dec;80(6):565–581.
- Niemi M. Role of OATP transporters in the disposition of drugs. Pharmacogenomics 2007 Jul;8(7):787–802.
- Huang SM. Draft Guidance for Industry: Drug Interaction Studies — Study Design, Data Analysis, and Implications for Dosing and Labeling. 2006.
- Elsby R, Surry DD, Smith VN, Gray AJ. Validation and application of Caco-2 assays for the in vitro evaluation of development candidate drugs as substrates or inhibitors of P-glycoprotein to support regulatory submissions. Xenobiotica 2008 Jul;38(7–8):1140–1164.
350. Clearance of perfluorooctanoate in isolated perfused kidneys from male and female rats - Effects of protein binding and renal organic anion transport inhibitors
Diane L. Nabb, Morgan B. Golt, Robert T. Mingoia, and Xing Han
DuPont Haskell Global Centers for Health and Environmental Sciences, Newark, DE, USA, 19714
Rat renal clearance of perfluorooctanoate (PFO) is sex-dependent, being more than 20 times faster in females than in males. In rats, PFO is metabolically inert, primarily binds to serum albumin in the blood, and is actively secreted and reabsorbed in the kidney via organic anion transport proteins. In order to better understand the renal clearance mechanism of PFO in rats, we studied PFO clearance kinetics in isolated perfused kidneys (IPK) from male and female rats under a range of bovine serum albumin (BSA, 0.25 to 6%) concentrations. At 6% BSA, PFO renal clearance (CLR) values were averaged to be 11.47 ± 1.72 and 0.85 ± 0.37 ml/h/kg for female and male rat kidneys, respectively, which were relatively comparable to in vivo CLR values. Unbound PFO fractions (fu) were determined to be 3.8, 5.9, 9.8, and 26.8% in perfusates at 6, 4, 2, and 0.25% of BSA, respectively. In general, proportionally higher CLR values for male rat kidneys were observed at lower BSA concentrations. However, for female rat kidneys, higher PFO unbound fractions at lower BSA concentrations did not result in expected higher CLR. We are currently working on IPK experiments in the presence of inhibitors specific to organic anion transporters (Oats) and organic anion transporting polypeptides (Oatp1a1). Our results will shed light on the role of protein binding, tubular basolateral secretion and apical reabsorption of PFO renal clearance in rats.
351. Contribution of different microRNAs to the regulation of breast cancer resistance protein (BCRP/ABCG2)
Xin Li, Yuzhuo Pan, and Aiming Yu
Department of Pharmaceutical Sciences, University of Buffalo, SUNY, Buffalo, NY, USA, 14260
MicroRNAs (miRNAs) represent a large group of small noncoding RNAs that control the expression of target genes involved in many critical biological processes including drug disposition mediated by ATP-binding cassette (ABC) efflux transporters. Using luciferase reporter assay, Liao et al. have shown that miR-520h may act on the 3’-untranslated region (3’UTR) of ABCG2 mRNA (1). To et al. have demonstrated that miR-519c inhibitor and mimic causes up- and down-regulation of ABCG2 in A549 cells, respectively (2). Meanwhile, our gene overexpression and knockdown experiments have revealed that ABCG2 may be readily regulated by miR-328 (3). An immediate question is whether any of these miRNAs dominate posttranscriptional regulation of ABCG2. To answer this question, we investigated the potential role of five miRNAs, miR-328, -519c, 520d, -520g, and -520h, in regulation of ABCG2. Immunoblot analyses indicated that ABCG2 was not down-regulated in MCF-7/MX100 cells transfected with plasmids expressing miR-520d, -520g or 520h, but in cells transfected with plasmids expressing miR-328 or -519. Consistently, ABCG2 protein was sharply up-regulated in MCF-7 cells transfected with selective miR-328 or -519c antagomir. ABCG2 mRNA degraded at a faster rate in cells transfected with plasmid expressing miR-328 or -519c, as compared to cells transfected with control plasmid. In addition, lower ABCG2 protein expression in MCF-7/MX100 cells caused by interference of miR-328 and -519c pathways was translated into significantly increased cellular accumulation of mitoxantrone and doxorubicin. The data suggest that miR-328 and -519c have important role in regulation of ABCG2 in human breast cancer cells. (*Corresponding author)
References;
- R. Liao et al., J Cell Biochem 104, 805 (Jun 1, 2008).
- K. K. To, Z. Zhan, T. Litman, S. E. Bates, Mol Cell Biol 28, 5147 (Sep, 2008).
- Y. Z. Pan, M. E. Morris, A. M. Yu, Mol Pharmacol 75, 1374 (Jun, 2009).
352. Deconvoluting the contribution of multiple efflux transporters in monolayer permeability studies: Consideration of multiple in vitro methods in tandem
Kirsten Mease, Rucha Sane, and Mitchell E. Taub
Drug Metabolism & Pharmacokinetics, Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT, USA, 06877-0368
MDCK-MDR1 and Caco-2 cell monolayers are routinely used to evaluate the interaction of drugs and drug candidates with efflux transporters such as P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and the multidrug resistance-associated protein (MRP2). However, due to the expression of multiple transporters in these cell lines and the lack of commercially available selective substrates and inhibitors for studying the activity of individual transporters, data interpretation can often be challenging. In this study, alternative in vitro methodologies were used in conjunction with routine methods to determine the interaction of BI-G with P-gp, BCRP, and MRP2. MDCK-MDR1 and Caco-2 monolayer studies yielded a BL-AP/AP-BL permeability directional ratio (PDR) for BI-G of >2. In MDCK-MDR1 and Caco-2 cells, active efflux of BI-G was eliminated in the presence of LY335979 (5 μM), demonstrating that BI-G is a P-gp substrate. In Caco-2 cells, FTC (10 μM) and MK571 (50 μM) were used as selective inhibitors to determine the relative contribution of BCRP and MRP2, respectively, to the efflux transport of BI-G. In the presence of FTC or MK571, the PDR of BI-G decreased but was not normalized to 1. These results suggested that BI-G interacted with P-gp, BCRP, and MRP2; however, careful analysis of the data led us to determine that additional mechanistic studies were required. Using BCRP- and MRP2-expressing Sf9 membrane vesicles, it was demonstrated that ATP-mediated uptake of BI-G did not occur. Additionally, BI-G did not inhibit the uptake of prototypical probe substrates into BCRP- and MRP2-expressing vesicles, providing further evidence that BI-G does not interact with these transporters. Thus, using a variety of in vitro tools, it was concluded that BI-G is a P-gp substrate, but does not interact with BCRP and MRP2. Studies using cell lines that express multiple transporters, such as Caco-2, can be used to generate valuable data. However, interpretation of the data improves when supported by experiments conducted in tandem with cell lines and/or inverted membrane vesicles expressing a single efflux transporter.
353. Suppression of OATP1B1, OATP1B3, and OATP 2B1 transporters in primary cryopreserved human hepatocytes following lipid delivery of stealth siRNA
Susan Grepper1, Laura Vozza-Brown2, and Jasminder Sahi1
1CellzDirect/Life Technologies, Durham, NC, USA, 27703
2Invitrogen/Life Technologies, Carlsbad, CA, USA, 92008
The Organic Anion Transporting Polypeptide (OATP) transporters play an important role in the basolateral hepatic uptake of a wide range of drugs and endogenous compounds. Primary hepatocytes (fresh isolated or cryopreserved) cultured appropriately in the sandwich configuration retain the major hepatic basolateral and apical membrane transporters and drug metabolizing enzymes. The absence of transporter - specific substrates and inhibitors makes it difficult to characterize the contribution of specific transport proteins. We have developed a model using cryopreserved human hepatocytes and Stealth Select siRNA™ to specifically knock down OATP1B1, OATP2B1 and OATP1B3 expression while not affecting the other two transporters. The methods used were 1) determining the best lipid delivery system (Lipofectamine2000™ versus RNAiMAX™), 2) exploring the effects of cell confluency and antibiotics in culture medium on transfection efficiency and potential hepatotoxicity and 3) determining the best Stealth Select oligos for knockdown of the three OATP transporters. Three separate hepatocyte preparations were used and mRNA expression and transporter activity using tritiated estradiol 17-β-glucuronide (all three transporters) and digoxin (more specific to OATP1B3) were assessed. Our results indicate that RNAiMAX™ was more effective than Lipofectamine-2000™ for primary hepatocyte siRNA transfection, as we achieved >70% mRNA knockdown of each OATP tested. Antibiotics did not interfere with transfection efficiency, while cell confluency was an important consideration, with lower transfection efficiency being achieved at >80% confluence. When all three OATP transporters were knocked down simultaneously, the effect was partially additive, as we could not knock down complete expression or activity. Our in vitro siRNA model can serve as an extremely valuable research tool for investigating the substrate and inhibition potential of specific hepatic transporters.
354. IC50 determinations in an in vitro P-Glycoprotein inhibition assay: Papp values vs. efflux ratios
Jianrong Lin, and Scott Grimm
Clinical Pharmacology and DMPK, Astrazeneca Pharmaceuticals LP, Wilmington, DE, USA, 19850-5437
Hypothesis: In vitro Pgp inhibition assay has been used to guide the potential clinical drug interaction studies. Since Pgp has broad substrate recognition, in this study, we examined IC50s of known Pgp inhibitor, ketoconazole, using 3Hdigoxin and 14Ccompound 1 as probe substrates. We also compared the use of both Papp values and efflux ratios for calculation of IC50 values. Method: MDCK-II and human MDR1-trasfected MDCK-II cell lines were grown in DMEM supplemented with 10% fetal bovine serum. Cells were seeded onto polycarbonate Transwellò filter membranes at a density of 150,000 cells/cm2. Pgp substrate digoxin and compound 1 were tested at 2 and 0.1 μM, respectively. The test concentrations for ketoconazole were from 0.001 to 50 μM. The bi-directional transport of 3Hdigoxin and 14C-compound 1 was measured in cells.
Data and Results: IC50 value for ketoconazole was 1.89 μM using digoxin as the substrate. The IC50 value for ketoconazole was 2.42 μM when compound 1 was used as the probe substrate. These IC50s were obtained based on the percent of inhibition of the active Pgp mediated transport of the probe substrate using efflux ratios that generated from the experiments. A notable difference was observed when Papp values were used to calculate IC50 values ketoconazole with digoxin and compound 1 as substrates. The IC50 (Papp) for ketoconazole was 2.65 μM using digoxin as a substrate. For compound 1, the IC50 (Papp) for ketoconazole was greater than 50 μM.
Conclusion: This study shows that ketoconazole IC50 values for Pgp inhibitor when digoxin and compound 1 were probe substrates were not different when efflux ratios were used for the calculations. Whereas there were large differences when Papp was used to calculate the IC50 values. These observations raise questions about the various strategies used to evaluate the potential of compounds to inhibit Pgp.
355. Comparison of two cell-based assays for examining P-glycoprotein inhibition
Natalya Alexander, Adrienne Natrillo, Heidi Einolf, Ryan M. Pelis, and Imad Hanna
Drug Metabolism and Pharmacokinetics, Novartis Institutes for BioMedical Research, East Hanover, NJ, USA, 07936
The inhibition of P-glycoprotein (P-gp) represents a drug-drug interaction mechanism that should be addressed during the course of preclinical drug development. Multiple In vitro assay systems aimed at examining P-gp inhibition are currently used by the pharmaceutical industry. Cell-based assays that examine the effect of drugs on either the flux of model P-gp substrates across cell monolayers, or their intracellular accumulation, are widely used. Both assay formats can assess the potential of developmental compounds to inhibit P-gp by providing an estimate of the concentration necessary to block 50 % of efflux activity (IC50). However, it is not clear if the IC50 values obtained from either assay format are comparable in value, or whether they even maintain the same rank-order of inhibition potency. In these studies, the IC50 values associated with the inhibition of [3H]digoxin efflux across Caco-2 cells grown on transwell inserts, and the inhibition of rhodamine123 extrusion from P-gp expressing MDA435 cells using flow cytometry, was determined for a number of drugs for which clinical data are available regarding their potential for eliciting drug-drug interactions. Although the absolute IC50 values did vary, the rank order of inhibition potency with the two different assays was similar, e.g., carvedilol (IC50s for Caco-2 vs. MDA435 cells; 25 vs. 7 μM) > verapamil (26 vs 27 μM) > quinidine (37 vs. 29 μM) > nitrendipine (47 vs. 164 μM). These data should be useful for modeling efforts aimed at assessing the potential of new drug candidates to elicit drug-drug interactions via P-gp inhibition.
356. Application of fluorescent labeled bile salts to evaluate alterations in bile acid hepatobiliary disposition
Christopher J. Parsons, William R. Smith, Cassandra H. Perry, Robert L. St. Claire III, and Kenneth R. Brouwer
Research and Development, Qualyst, Inc., Durham, NC, USA, 27713
Drug-induced liver toxicity is the single most common reason for withdrawal of FDA approved drugs from the market. Cholestasis is a common observation in drug-induced liver toxicity. Transporter based drug – drug interactions can result in cholestasis, and depending on the site of interaction (basolateral uptake and/or canalicular efflux), accumulation of bile salts within the hepatocyte may occur, leading to toxicity. Previously we have demonstrated the ability of B-CLEAR® technology to assess the cholestatic potential of drugs by inhibition of hepatic bile acid transport (uptake and efflux) using radiolabeled and stably labeled taurocholate. The use of fluorescent probes for evaluation of alterations in the hepatobiliary disposition of bile salts would increase the throughput and utility of the assay. We evaluated two fluorescent probes, cholyl-lysyl-fluorescein (CLF) and cholyl-glycylamido-fluorescein (CGamF) for their potential to predict the hepatobiliary disposition of bile salts. Sandwich-cultured rat hepatocytes (Day 4 of culture; 24-well format) were incubated with 2.5 μM CLF or CGamF in the presence or absence of calcium (Ca++) for 15 minutes at 37°C. Studies using 3H-taurocholate were used as a control. Cyclosporine (50 μM) was used as a probe inhibitor to evaluate the effect on the hepatobiliary disposition of CLF. Following incubation, cells were lysed and the resulting lysates were analyzed for CLF and CGamF using a fluorescent plate reader. Relative fluorescence units were converted to accumulation values (pmol/mg protein); CLF and CGamF biliary excretion indices (BEIs) and in vitro biliary clearance (Clbiliary) were calculated. All studies with the exception of CGamF were performed in hepatocytes obtained from three different livers (n=3). At a dose concentration of 2.5 μM, the BEI and in vitro biliary clearance for CLF were 27.2 ± 8.88 % and 5.18 ± 3.44 mL/min/kg, respectively. The total accumulation (cell + bile) and intracellular mass (accumulation – cell) were 83.4 ± 15.8 and 59.1 ± 6.89 pmol/mg protein, respectively. CGamF accumulated to a much greater extent with total accumulation (cell + bile) and intracellular mass (accumulation – cell) of 376 ± 20.6 and 2541 ± 14.3 pmol/mg protein, respectively. The BEI and in vitro biliary clearance for CGamF were greater than CLF at 32.5 % and 26.1 mL/min/kg, respectively. For taurocholate, the BEI and in vitro biliary clearance were 77.5 ± 1.98 % and 24.7 ± 9.56 mL/min/kg, respectively. The accumulation (cell + bile) and (cell) were 39.8 ± 15.0 and 8.9 ± 3.2 pmol/mg protein. Co-incubation with cyclosporine decreased the accumulation (cell + bile) and (cell) of CLF to a similar extent (43 % of control). The BEI and in vitro biliary clearance for CLF were decreased to 11.0 and 4.23 % of control, respectively. These changes for CLF in the presence of cyclosporine were similar to those observed for taurocholate when co-incubated with cyclosporine. CLF and CGamF may have utility in the prediction of alterations in the hepatobiliary disposition of bile acids.
357. In vitro and in vivo evaluation of impact of metabolism and drug transport on clearance of a novel aurora kinase inhibitor
Bianca M. Liederer, Jason S. Halladay, Laurent Salphati, Savita Ubhayakar, and Susan Wong
Department of Drug Metabolism and Pharmacokinetics, Genentech, Inc., South San Francisco, CA, USA, 94080
The objective of this study was to use in vitro and in vivo techniques to investigate the contribution of transporters and drug metabolizing enzymes to the clearance of compound X (4-(2-(4-(2-(4-acetylpiperazin-1-yl)-2-oxoethyl)phenylamino)-5-fluoropyrimidin-4-ylamino)-N-(2-chlorophenyl)benzamide), a novel small molecule Aurora kinase inhibitor. In rat liver microsomes and hepatocytes compound X showed poor metabolic stability with CLint = 290 and 149 mL/min/kg, respectively. In rat hepatocytes, the cytochrome P450 (P450) inactivator 1-aminobenzotriazole (ABT) inhibited its metabolism by 47%, suggesting that compound X was a substrate for P450. The uptake of compound X in rat hepatocytes was temperature dependent (i.e., 4°C vs. 37°C) and inhibited by cyclosporine A (CsA), a general transporter inhibitor, indicating that active transport mechanism was involved in hepatic uptake. In contrast to MDR1-transfected MDCK cells, bcrp-transfected MDCK cells showed a marked efflux ratio of 35. Bcrp inhibitor Fumitremorgin C inhibited this polarized efflux to 4. These data suggest that compound X is a substrate for bcrp but not for MDR1. Consistent with in vitro results, CsA and/or ABT or GF120198 (P-gp and bcrp inhibitor) pretreatment in rats resulted in an increase in compound X (administered IV) exposure. When CsA or/and ABT, individually or in combination were administered, the clearance of compound X decreased ~2, 1.5 and 5-fold respectively, compared to the control value (21.3 ± 3.65 mL/min/kg). In addition, the half-life increased ~2, 3 and 5-fold, respectively compared to the control (0.262 ± 0.01 hr). GF120198 co-administration resulted in a ~2-fold decrease in clearance and a ~2-fold increase of the half-life compared to the control. In conclusion, these results indicate that P450 and bcrp but not MDR1 play an important role in the in vivo clearance of compound X. Pretreatment combination of ABT and CsA was most effective in maximizing and prolonging systemic exposure of compound X. These initial characterizations of the clearance mechanisms and how to circumvent those using inhibitors provided options to enhance exposure for proof-of-concept safety/efficacy studies and were helpful in compound optimization.
358. The application of An OATP1B1 inhibition assay within drug discovery: Use of in vitro and in silico approaches to aid drug design
Matt G. Soars1, Patrick Barton2, Ken Grime3, and Robert J. Riley3
1Discovery DMPK, AstraZeneca, Loughborough, United Kingdom, LE11 5RH
2Medicinal Chemistry, AstraZeneca, Leics, United Kingdom, LE11 5RH
3Discovery DMPK, AstraZeneca, Loughborough, United Kingdom
The Application Of An OATP1B1 Inhibition Assay Within Drug Discovery: Use Of In vitro and In silico Approaches To Aid Drug Design. The organic anion transporting polypeptides (OATPs), and more specifically OATP1B1 have been shown to transport a large range of anionic drugs, most notably statins. It is therefore imperative that any potential drug-drug interactions mediated via OATP1B1 are assessed. Consequently an OATP1B1 inhibition assay using pitavastatin as a non-radiolabelled substrate has been developed. The selection of an appropriate substrate has been shown to be critical in producing accurate assessment of inhibition potential. For example the IC50 determined for simvastatin using either pitavastatin or estradiol-17-glucuronide as a substrate was 6 μM whereas a value of 40 μM was obtained using estrone-3-sulfate. The screening of over 200 inhibitors from both the literature and in house chemistry has allowed the development of an in silico model which has subsequently been used to predict accurately the inhibition potential of a range of statins and angiotensin II antagonists. Furthermore, OATP1B1 inhibition data has been shown to relate to hepatic uptake rates determined from studies with both human and rat hepatocytes. This assay may therefore highlight when hepatic uptake may contribute significantly to the clearance of new chemical entities. The integration of OATP1B1 inhibition assay data into current decision trees for the prediction of both clearance and drug-drug interaction potential for new chemical entities will also be discussed.
359. Assessment of P-gp, BCRP and MRP activity in Caco-2 and MDR1-LLC-PK1 cell monolayers
Sudarshan Kapadnis, Elke S. Perloff, Wendy Khun, Lisa Fox, Charles L. Crespi, and David M. Stresser
BD Gentest Contract Research Services, BD Biosciences Discovery Labware, Woburn, MA, USA, 01801
Drug intestinal permeability and drug transporter interactions are frequently studied in-vitro. One of the most well accepted and FDA recommended models is the assessment of bidirectional transport across polarized cell monolayers such as Caco-2 cells or transfected cell lines like MDR1-LLC-PK1. In this study, the activity of P-gp, BCRP, and MRP transporters was assessed in Caco-2 and MDR1-LLC-PK1 cell monolayers by measuring bidirectional permeability of digoxin, estrone-3-sulfate (E3S), and leukotrien C4 (LTC4) as probe substrates for P-gp, BCRP, and MRP transport, respectively. Efflux of the probe substrates was assessed in the presence or absence of various prototypical efflux transporter inhibitors (ketoconazole, elacridar, verapamil, quinidine, fumitremorgin C, novobiocin, benzbromarone, MK571).
In Caco-2 cells we observed active efflux of digoxin, which significantly decreased upon addition of P-gp inhibitors, ketoconazole, elacridar, verapamil, and quinidine. Similarly, efflux of E3S decreased considerably upon addition of BCRP inhibitors, novobiocin and FTC. Limited efflux (efflux ratio of 1.2) was observed for the MRP probe substrate LTC4. In MDR1-LLC-PK1 cells we observed active efflux of digoxin, which significantly decreased upon addition of the P-gp inhibitors. Limited efflux (efflux ratio <1.5) was observed for the BCRP probe substrate E3S and for the MRP probe substrate LTC4. Consistent with literature data, there was considerable overlap in inhibitor selectivity.
In conclusion, Caco-2 cells showed strong P-gp and BCRP efflux activity and negligible MRP efflux. In comparison, MDR1-LLC-PK1 cells showed strong P-gp efflux activity and negligible BCRP and MRP efflux. The functional activity of various transporters should be considered when assessing drug candidate inhibition response in cell monolayer models when testing for drug-drug interaction potential.
360. An approach for deconstructing dual substrate mechanisms involved in efflux transport of BI-H in MDCK-MDR1 and Caco-2 Cell monolayers
Lalitha Podila, and Mitchell E. Taub
Drug Metabolism & Pharmacokinetics, Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT, USA, 06877-0368
The breast cancer resistance protein (BCRP), P-glycoprotein (P-gp), and the multidrug resistance protein-2 (MRP2) are ATP binding cassette (ABC) efflux transporters that have been shown to interact with numerous drugs. MDCK-MDR1 cells are of canine origin, and mature monolayers overexpress human P-gp. Caco-2 cells are of human origin, and mature monolayers endogenously express P-gp, BCRP, and MRP2. Both cell lines were utilized in bidirectional transport assays to deconstruct the mechanisms responsible for efflux of BI-H in vitro. BI-H was soluble in the assay buffer at concentrations exceeding 2 mM. Data from transport studies indicated that BI-H is a substrate of P-gp, as evidenced by a high secretory/absorptive permeability directional ratio (PDR >20). The secretory apparent permeability (Papp) of BI-H remained constant at incubation concentrations up to 2 mM, indicating that P-gp was not saturated. LY 335979 (5 μM), a P-gp selective inhibitor, completely inhibited the efflux transport of BI-H across MDCK-MDR1 cell monolayers. However, in Caco-2 cell monolayers, only partial inhibition of BI-H efflux transport occurred. Thus, these data suggested that BI-H is a substrate for another efflux transporter expressed in Caco-2 cells, such as BCRP or MRP2. BCRP-mediated efflux transport of BI-H in Caco-2 cells was confirmed by evaluating its transport in the presence of prazosin, a known BCRP inhibitor. Efflux of BI-H was inhibited by 34% in the presence of LY 335979 (5 μM) and by 50% in the presence of prazosin (80 μM). Incubation of BI-H in the presence of both LY 335979 (5 μM) and prazosin (80 μM) generated an additive effect, inhibiting efflux of BI-H by 83%. Additional experiments conducted in MDCK-MDR1 cells demonstrated that prazosin did not inhibit P-gp mediated efflux of BI-H. Thus, these studies demonstrated that BI-H is a substrate of both P-gp and BCRP.
361. Evaluation of LS-180 human intestinal cells as an in vitro model of Pgp induction
W. Zhang, Y. Wang, E.A. Weiskircher, R.A. George, J. Li, A. Owen, C. Bode, and I.J. Hidalgo
Absorption Systems LP, Exton, PA, USA, 19341
Purpose: To develop a rapid, stable, reliable and highly sensitive in vitro model to identify compounds which are likely to induce Pgp expression.
Methods: Wild type LS-180 cells (from ATCC) were treated with the Pgp inducers rifampicin (10 μM), ritonavir (10 μM) and vinblastine (11 and 55 μM) for 3 days. Following this treatment, RNA and protein were extracted. The expression of Pgp, CYP3A4 and CYP1A1 mRNA was determined by quantitative PCR (qPCR) and the expression of Pgp protein determined by western blot (WB) analysis. The functional activity of Pgp was determined by measuring the uptake of rhodamine 123 (Rh123) fluorescence in the cells.
Results: In the qPCR assay, LS-180 cells treated with 0.1% DMSO (vehicle) were used as the control group and the expression level (2−ΔΔCT values) relative to untreated cells was close to one, indicating that there was no change in the expression level of the Pgp, CYP3A4, and CYP1A1 genes. Rifampicin, ritonavir and vinblastine increased expression of Pgp mRNA 5.02-, 5.93-, and 4.42-fold, respectively, compared to DMSO-treated cells. In parallel experiments, rifampicin caused higher (14-fold) induction of CYP3A4 mRNA expression, compared to ritonavir (~4-fold) and vinblastine (~4-fold), whereas vinblastine induced higher expression of CYP1A1 mRNA (86-fold) than rifampicin (~15-fold) and ritonavir (~5-fold). WB analysis results showed an increase in the expression of Pgp in the treated cells. The order of Pgp induction based on the level of expression of the corresponding mRNA and protein was: ritonavir (10 mM) > rifampicin (10 mM) > vinblastine (55 mM). The uptake of R123 in the cells treated with the three Pgp inducers was lower than in the control cells, indicating that the functional activity of Pgp increased after 3 days of treatment with the Pgp inducers. This increase in the functional activity of Pgp was associated with increased expression of Pgp mRNA and protein.
Conclusion: Pgp was significantly induced in terms of increased expression of mRNA, protein and functional activity after LS-180 cells were treated with rifampicin, ritonavir or vinblastine for 3 days. Our results support the notion that these three drugs share a common mechanism, most likely PXR, in the induction of Pgp and CYP3A4. The greater induction of CYP 1A1 may involve aryl hydrocarbon receptors (AhR), especially in cells treated with vinblastine. The LS-180 cell line can be useful as a stable and highly sensitive in vitro model to identify compounds that induce intestinal Pgp.
362. OATP expression in human intestinal epithelial cells and its influence on the transport of statin drugs across Caco-2 cell monolayers
Jibin Li, Wei Zhang, Samantha M. Allen, Yuehua Huang, Chris Bode, Albert Owen, and Ismael J. Hidalgo
ABSORPTION SYSTEMS LP, Exton, PA, USA, 19341
Organic anion transporting polypeptides (OATPs) constitute an uptake transporter family that affects the systemic pharmacokinetics and subsequent pharmacological and toxicological effects of many drugs. OATP1B1 (also known as OATP2, OATP-C, or LST-1) and OATP1B3 (OATP8, or LST-2) are selectively expressed at the basolateral (sinusoidal) surface of hepatocytes and facilitate hepatic uptake of substrates from the blood. In this study, expression of OATP1B1 and OATP1B3, thought to be hepatocyte-specific transporters, was detected in several human intestinal cell lines (Caco-2, HT29, T84, and LS180) by RT-PCR. Functional activity of OATP1B1/3 in Caco-2 cells was assessed using a fluorescent substrate, chenodeoxycholyl-(Nϵ-NBD)-lysine (CDCA-NBD). CDCA-NBD was preferentially transported in the basolateral-to-apical (B-A) direction in Caco-2 cells, whereas polarized efflux was not observed in MDR1-MDCK cells, indicating the expression of unique transporter system(s) in Caco-2 cells. CDCA-NBD transport in Caco-2 cells was inhibited by rifampicin (an OATP1B1/3 inhibitor) and several known OATP1B1/3 substrates. CDCA-NBD efflux was also inhibited by cyclosporine A, an inhibitor of several apical efflux transporters, in Caco-2 cells and partially diminished in BCRP knockdown Caco-2 cells. The results suggest that CDCA-NBD was taken up by basolateral transporter(s) and subsequently secreted by apical efflux transporter(s). In addition, several statin drugs (atorvastatin, fluvastatin, and pitavastatin) were found to exhibit vectorial transport in Caco-2 cells with B-A transport rates 3- to 12-fold higher than A-B rates. Rifampicin on the basolateral side of the monolayers produced greater inhibition of statin transport than on the apical side, which further suggests basolateral localization of uptake transporter(s). In summary, the current study discovered unexpected expression of OATP1B1 and OATP1B3 in human intestinal cell lines. Results from transport and inhibition experiments in Caco-2 cells indicate the existence of uptake transporter(s), presumably OATP1B1/3, facilitating drug uptake at the basolateral membrane. The study demonstrated potential applications of the Caco-2 cell model for studying the interplay of basolateral uptake and apical efflux transporters, and the utility of efflux transporter knockdown cells in identifying the role of efflux transporters in drug disposition.
363. Amino acids in the transmembrane domain 1 (TM1) of the sodium-dependent bile acid transporter ASBT (SLC10A2) are critical for function
Tatiana Claro da Silva, Naissan Hussainzada, Chandra M. Khantwal, and Peter W. Swaan
Pharmaceutical Sciences, University of Maryland, Baltimore, Baltimore, MD, USA, 21201
The apical sodium-dependent bile acid transporter (ASBT, SLC10A2) is a relatively small integral membrane glycoprotein (38 – 41 KDa, 348 amino-acid residues), which plays a critical role in the enterohepatic circulation of bile acids (BA). In the gut, BAs facilitate digestion and absorption of dietary lipids and fat-soluble vitamins, thus establishing the importance of proteins involved in bile acid transport. The present study investigates functional contributions of the relatively amphipatic transmembrane domain 1 (TM1) of the human ASBT (hASBT). Presently, successive cysteine substitutions were incorporated along TM1 (Ile27-Gly50) via site-directed mutagenesis, against a C270A template. Mutant function was assessed by transient expression in COS-1 cells followed by [3H]-taurocholate uptake at physiological (137 mM) and equilibrating (12 mM) extracellular Na+ concentrations. Cysteine substitution affected approximately 40% of TM1. As expected, N27, which is located at the membrane aqueous interface of TM1 and the N-terminus, was significantly affected by mutation, due to its critical position in the protein. The same occurs with G50C, which delineates TM1 and the intracellular loop 1 (IL1). This observation tempts speculation that mutation on these residues leads to structural changes that would interfere with substrate permeation. For each mutant, solvent accessibility was probed by thiol modification using methanethiosulfonate (MTS) reagents. Specifically, the positively charged MTSET reagent affects the majority of residues lining the first half of TM1, which indicates accessibility to the aqueous environment, and a possible interaction with the substrate. Ongoing sodium and bile acid competition experiments will help us understand which of these substrates would be involved in this interaction. Overall, TM1 amino acids appear critical for hASBT transport function. Supported by NIH RO1 DK61425.
364. Induction of transporter expression and modulation of hepatobiliary disposition using sandwich cultures of human hepatocytes
Jonathan P. Jackson1, Rob Taylor1, Ganesh Rajaraman1, LaHoma Easterwood1, Jeanette Hill1, Christie Baucom2, and Stephen Ferguson2
1Cellzdirect/Invitrogen, Austin, TX, USA, 78653
2Cellzdirect/Invitrogen, Durham, NC, USA, 27703
Transporter-related drug interactions are emerging as an important consideration in the clearance of drugs. In particular, hepatic clearance is important due to the liver’s role in xenobiotic defense. However, few in vitro assays are available to quantify hepatobiliary transport, and even fewer studies have described the effect of altered transporter expression on hepatic transport. Historically, most studies investigating transporter modulation have focused on mRNA content providing little data on transporter activity following drug treatment. The current study examines the use of a sandwich cultured hepatocyte model (B-CLEAR®, Qualyst, NC) to quantify the effects of various transporter expression modulators on hepatobiliary transport. Multiple models have been used to assess hepatic transporter activity (e.g. transiently and stably transfected cell lines); however, cell lines traditionally have been shown to lack many of the hepatic regulatory pathways (e.g. nuclear receptors) necessary to model hepatic induction. The sandwich-culture hepatocyte model maintains many of these critical cell-signaling pathways and also forms functional bile canalicular networks with liver-like polarization of hepatic uptake and efflux transporters. This provides an integrated and physiologically relevant model to quantify changes in transporter activity as a result of alterations in expression. Primary human hepatocytes in sandwich-culture were treated for 3 days with DMSO (0.1%), PB (1 mM), RIF (10 μM), CDCA (100 μM), Colchicine (0.5 μM), Mancozeb (40 μM), or Bensulfuron-methyl (5 μM). Messenger RNA content of efflux (ABCB1, ABCG2, ABCB11, ABCC1, ABCC2, and ABCC3) and uptake transporters (SLCO1B1) was measured by TaqMan® Assays. Hepatobiliary transport activity was quantified by using selective transporter probe substrates (e.g. digoxin, taurocholate, and estradiol). Following treatment for 3 days with xenobiotics, preliminary data showed a 2.2-fold increase in the biliary excretion index of digoxin (2.2-fold) in RIF treated hepatocytes. Similarly, changes in transporter mRNA expression were detected. This approach has the potential to become an effective tool in the prediction of transporter-related drug-drug interactions resulting from altered hepatobiliary transport.
365. Factors influencing placental transfer of lopinavir: Binding, uptake and efflux
Abhishek Gulati1, and Phillip M. Gerk2
1Pharmaceutics, Virginia Commonwealth University, Richmond, VA, USA, 23298-0533
2Dept of Pharmaceutics, Virginia Commonwealth Univ, Richmond, VA, USA, 23298-0533
The aim of the therapy in treating HIV-infected pregnant patients is the treatment of the mother and prevention of viral transmission to the fetus. Among antiretroviral drugs used, HIV protease inhibitors are particularly poorly transferred. The purpose of this study was to characterize the factors limiting placental passage and fetal exposure to lopinavir. The primary objective was to increase the transfer of HIV protease inhibitors to the fetus, in order to lower the vertical HIV transmission in pregnant HIV positive women. We examined lopinavir uptake, binding and efflux using BeWo human trophoblast cell culture model. BeWo cells were treated with 3H-lopinavir in the absence or presence of inhibitors of ABC transporters. The radioactivity was then measured in the buffer and the cells after incubating for various time intervals and at two temperatures. Verapamil (100μM) stimulated apparent efflux of 3H-lopinavir by two fold, possibly due to ABCC2. In addition, this efflux process was 75% inhibited by reduced temperature (4°C). Ritonavir (10μM) also stimulated 3H-lopinavir efflux, whereas GF120918 (1μM) had no effect. To examine potential displacement of lopinavir from its cellular binding sites, we examined the effects of verapamil, ritonavir, and reduced temperature on lopinavir binding to BeWo cell homogenates. Reduced temperature (4°C), verapamil (100μM) or ritonavir (10μM) individually did not significantly affect the binding of 3H-lopinavir to cell homogenates. We also investigated the uptake of lopinavir into BeWo cells. Lopinavir uptake was not sensitive to verapamil, bromosulfophthalein, taurocholate or to reduced temperature, suggesting uptake involves diffusion rather than OATP transporters. The results suggested that interplay between cellular binding and ABC efflux transporters, in addition to simple diffusion, determines the extent of 3H-lopinavir distribution into BeWo cells.
366. Identification of a near infrared fluorescence probe as a potential imaging agent for assessing P-Glcyoprotein function in vitro and in vivo
Ngoc On, Fang Chen, Martha Hinton, and Donald Miller
Pharmacology and Therapeutics, University of Manitoba, Winnipeg, MB, Canada, R3E0T6
Drug efflux transport proteins, such as P-glycoprotein (P-gp) have an important role in drug absorption, distribution and elimination. The development of effective and affordable imaging systems for assessing P-gp activity in the whole animal setting would be of potential importance for understanding physiological and/or pathophysiological alterations in P-gp function. In the present study, we report our initial efforts in utilizing near infrared fluorescence (NIRF) spectroscopy to image P-gp function. Studies were performed to evaluate the suitability of the NIRF probe, rhodamine 800 (R800), to assess P-gp function both in vitro and in the whole animal setting. Cell culture studies consisted of cell accumulation and bi-directional permeability studies using MDCK MDR cells grown on a 6-well insert membrane. Studies were performed under control conditions (with functional P-gp) and conditions in which P-gp was inhibited using GF120918. Under control conditions, R800 was applied to either the apical or the basolateral compartment of the membrane at a concentration of 10 mM. The treated cells were exposed to 1mM of GF120918 prior to the administration of R800. Samples were taken from the corresponding compartments at various times (15-60 minutes) and quantitatively scanned for R800 content using an Odyssey near infrared imager. The distribution of R800 in mouse brain tissue was also examined under control conditions (functional P-gp), and following pre-treatment with GF 120918. At various times (0-20 minutes) whole body scans of the mice were taken. In addition at the conclusion of the experiment, mice were sacrificed, brains removed and the amount of R800 present in brain tissue quantitated using an Odyssey infrared imager. Bi-directional permeability studies in MDCKMDR1 showed that the passage of R800 in the presence of functional P-gp from the basolateral to the apical side of the membrane is significantly higher (3-fold) than the passage of R800 from the apical to basolateral side. Following pretreatment with GF120918 the passage of R800 from either side of the membrane is the same. In vivo data from healthy mice showed that by inhibiting the function of P-gp with GF120918, the distribution of R800 in the brain increased by approximately 4-fold. In conclusion, both in vitro and in vivo experiments established that R800 is substrate for P-gp and that the NIRF properties of R800 may be utilized to provide imaging of P-gp function in the BBB and other tissues. Research Supported by a grant from the National Science and Engineering Council of Canada.
367. Albumin effect on the P-Glycoprotein mediated drug transport
Yasuhisa Adachi, Chikako Honma, Yoshihiro Ohzone and Shin-ichi Ninomiya
ADME & Tox. Research Institute, Sekisui Medical Co., Ltd., Ibaraki, Japan, 319-1182
Various transporters are involved in the occurrence of drug-drug interactions. In particular, P-glycoprotein (P-gp) is attracting much attention because of P-gp related clinically important drug-drug interactions such as loperamide vs quinidine, digoxin vs cyclosporine etc. For the in vitro evaluation of P-gp related drug-drug interactions, concrete methodology and decision tree are shown in recent FDA draft guidance in which bi-directional transport studies are recommended. However, recent new chemical entities (NCE) often show poor solubility and/or severe absorption to labware that make it difficult to perform the bi-directional transport studies. Addition of BSA might improve the apparent solubility and absorption of NCE, however, effects of BSA in the bi-directional transport studies have not yet been well-characterized. We have tested the addition of BSA in the bi-directional transport studies on P-gp using digoxin (as the probe substrate), verapamil and cyclosporine (as inhibitors). IC50 values of verapamil and cyclosporine against digoxin transport were 0.5 and 0.2 microM, respectively, in the absence of BSA. Addition of BSA at physiological blood albumin concentration (4%) gave IC50 value of 1.6 for verapamil and 0.6 microM for cyclosporine. The suppressed inhibition of verapamil and cyclosporine in the presence of BSA suggests the reduced concentrations of free inhibitors by the protein binding. The effect of protein binding of drugs in the bi-directional transport studies will be discussed in order to clarify the efficacy of BSA for the evaluation of NCE with high absorption to labware.
368. Characterization of drug transporters responsible for the disposition of mesna: in vitro to in vivo
Murray J. Cutler1, Bradley L. Urquhart1, Thomas J. Velenosi1, Henriette E. Meyer Zu Schwabedissen2, Rommel G. Tirona1, Richard B. Kim3, and David J. Freeman3
1Physiology & Pharmacology, The University of Western Ontario, London, ON, Canada, N6A5A5
2Pharmacology, Ernst Moritz Arndt University of Greifswald, Greifswald, Germany
3Medicine, The University of Western Ontario, London, ON, Canada, N6A5A5
Background: Mesna (sodium 2-sulfanylethanesulfonate) is widely utilized during oxazaphosphorine-based chemotherapy to prevent urothelial injury such as hemorrhagic cystitis. However, there is little data regarding the mechanisms of mesna disposition. Upon entering the systemic circulation, mesna is rapidly oxidized to its inactive dimer, dimesna. Subsequently, dimesna is selectively reduced to mesna in the kidney, thereby producing its uroprotective effect. We hypothesized that renal drug transporters are responsible for the delivery of dimesna to the kidney. To determine the role of transporters to dimesna disposition, an array of drug uptake and efflux transporters known to be expressed in organs such as the kidney were examined.
Methods: Twenty-seven unique transporters were screened for dimesna uptake or efflux using a recombinant vaccinia-mediated over-expression system in HeLa cells. In addition, the in vivo role of the organic anion transporters (OATs) in the secretion of mesna by the kidney was determined by administration of the OAT inhibitor probenecid to healthy subjects, at steady-state, in a three-armed cross-over trial.
Results: Significant and saturable uptake by renal OAT1 (SLC22A6), OAT 3 (SLC22A8), and OAT4 (SLC22A11), but not the predominantly hepatic transporter OAT2 (SLC22A7) was observed. Efflux transporters P-glycoprotein (MDR1/ABCB1), MATE1 (SLC47A1), MRP1 (ABCC1), MRP4 (ABCC4), and MRP5 (ABCC5) significantly reduced intracellular dimesna accumulation. Probenecid exerted dose-dependent inhibition of dimesna uptake by OATs in vitro. The co-administration of probenecid with oral mesna decreased its urinary excretion and renal clearance, subsequently increasing the area under the concentration-time curves approximately two-fold (90.5 ± 33.6%).
Conclusions: Apical renal uptake transporter OAT4 in concert with apical renal efflux transporters represents a yet unidentified mechanism of intracellular dimesna flux. OAT transporters appear to be involved in the uptake of dimesna whereas MPR transporters and the recently identified MATE1 appeared to mediate its efflux. Our results suggest the urinary disposition of mesna depends significantly on facilitated transport. Furthermore, the strategy outlined here provides an integrated in vitro to in vivo approach for the identification and prediction of transporter-mediated drug interactions.
369. Effect of short and middle chain fatty acids on the cell-surface expression and transport capacity of Bile Salt Export Pump (BSEP/ABCB11)
Takuya Kato, Hisamitsu Hayashi, and Yuichi Sugiyama
Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan
Bile salt export pump (BSEP/ABCB11) is a member of the ATP-binding cassette (ABC) superfamily transporters. BSEP is localized in the canalicular membrane, and plays an essential role in the biliary excretion of monovalent bile acids. The reduced expression of BSEP at the canalicular membrane, caused by a mutation in the BSEP gene or endotoxins or drug treatment, is associated with cholestasis-induced hepatotoxicity due to the accumulation of bile acids in hepatocytes. So, the restoration of BSEP expression at the cell-surface is a pharmaceutically attractive target for the treatment of cholestasis. Previously, we reported that 4-phenylbutyrate (4PBA), approved drug for urea cycle disorders, is a promising agent for intrahepatic cholestasis due to the increase in BSEP expression on the cell-surface accompanied by enhancement of the transport capacity of BSEP (Hayashi et. al. Hepatology. 2007 45(6):1506-16.). However, from the results of in vivo animal studies, it appears that a high dose is needed for an adequate effect due to its poor efficacy. So, in this study, we searched for other compounds with a more potent effect than 4PBA. The change in BSEP expression at the cell surface by the test compounds was initially evaluated in terms of BSEP-mediated transport function by calculating the efflux clearance of [3H]taurocholic acid across the apical membrane of BSEP-expressing MDCKII cells. The BSEP-mediated transport was enhanced by all the tested short and middle chain fatty acids except for formate, acetate and hexanoic acid, compared with 4PBA. In addition, for butyrate and octanoic acid, there was a good correlation between the increase in BSEP expression at the cell surface and the enhancement of BSEP-mediated transport as shown by the cell-surface biotinylation study. These studies suggest that almost all short and middle chain fatty acids enhance the transport capacity of BSEP by increasing BSEP expression at the cell surface. Further studies are underway to clarify the efficiency of these agents in vivo and the additive effect of these agents when combined with 4PBA.
370. In vivo role of human organic anion transporting polypeptide 1B1 (OATP1B1/SLCO1B1) and its murine orthologue Oatp1b2 to atorvastatin disposition
Marianne K. DeGorter1, Ute I. Schwarz2, Rommel G. Tirona2, and Richard B. Kim3
1Physiology and Pharmacology, University of Western Ontario, London, ON, Canada, N6A 5A5
2Physiology and Pharmacology, University of Western Ontario, London, ON, Canada, N6A3A5
3Division of Clinical Pharmacology, Department of Medicine, The University of Western Ontario, London, ON, Canada, N6A 5A5
Elevated systemic exposure to the 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins) may increase the risk of developing a rare but life-threatening form of muscle injury known as rhabdomyolysis. A common loss-of-function polymorphism in hepatic organic anion transporting polypeptide 1B1 (OATP1B1/SLCO1B1), c.521TC, has been shown to be an important risk factor for statin-induced muscle toxicity. The widely prescribed atorvastatin is an established in vitro substrate of OATP1B1 and its murine orthologue Oatp1b2, and SLCO1B1 polymorphisms have been shown to affect its pharmacokinetic profile in healthy volunteers. Here we examined the effect, in our patients at London Health Sciences Centre on long-term atorvastatin therapy, of SLCO1B1 c.521TC and c.388AG genotypes on the steady-state trough plasma concentrations of atorvastatin acid and its lactone as determined using LC-MS/MS. In the initial set (n=15) of patients taking 10 – 80 mg atorvastatin daily, we did not observe a statistically significant effect of SLCO1B1 polymorphisms in relation to atorvastatin trough concentration. A more formal statistical analysis is planned when the sample size reaches 100. Nevertheless, we already note a marked (30-fold) interpatient variability in plasma levels among patients taking the same dose, indicating that regardless of OATP1B1 genotype, some patients may be at greater risk of developing statin-associated side effects. To further examine the in vivo contribution of hepatic uptake to atorvastatin distribution, we administered single dose atorvastatin to Slco1b2−/- mice (1 mg/kg) by tail vein injection and harvested tissue after 30 minutes. The liver-to-plasma ratio of atorvastatin and atorvastatin lactone showed only a modest trend to higher liver to plasma ratios in both male and female wildtype vs. knockout mice (n=3-4/group), suggesting that other hepatic Oatp transporters may be compensating for the absence of Oatp1b2 in the knockout mice. To our knowledge, this is the first characterization of atorvastatin concentrations in a general patient population. A larger sample size will clarify the contribution of OATP1B1 genetic polymorphisms as a determinant of the observed variable exposure.
The authors declare no conflicts of interest. This work is supported by the Canadian Institutes of Health Research (MOP-89753), the Natural Sciences and Engineering Research Council of Canada, and the Canadian Foundation for Innovation.
371. Intestinal segment-specific upregulation of xenobiotic transporter genes by chemical activation of transcription factor pathways in mice
Rachel R. Buckley, and Curtis D. Klaassen
Pharmacology, Toxicology, and Therapeutics, The University of Kansas Medical Center, Kansas City, KS, USA, 66160
The intestine serves as a major site of absorption for ingested xenobiotics. In turn, intestinal transporters function in the luminal absorption and excretion of therapeutic drugs and other xenobiotics. Despite this critical role in pharmacokinetics, little is known about chemical transport into and out of enterocytes in mice, nor whether they are induced by microsomal enzyme inducers. Thus, the present study quantifies the mRNA expression, as well as chemical and transcriptional regulation of 9 uptake and 10 efflux drug transporters in small and large intestines of mice. Adult wild-type male C57BL/6 and null mice lacking the aryl hydrocarbon receptor (AhR), constitutive androstane receptor (CAR), pregnane X receptor (PXR), peroxisome proliferator-activated receptor α (PPARα), or nuclear factor erythroid 2-related factor 2 (Nrf2) received either vehicle (corn oil, ip) or prototypical microsomal enzyme inducers (TCDD: AhR, TCPOBOP: CAR, PCN: PXR, CFB: PPARα, OPZ: Nrf2) for 4 days. Both uptake and efflux transporter genes involved in the systemic absorption and luminal excretion of chemicals were induced throughout the intestine via ligand-activated transcription factors. Specifically, duodenum had the highest number of genes (8) induced in a transcription factor-dependent manner, compared to jejunum (3), ileum (4), and colon (4). PCN treatment induced the highest number of transporter genes (8), including Oatp2b1, Mrp3, Mdr1a, in a PXR-dependent manner. Furthermore, segments of the intestine (duodenum, jejunum, ileum, and colon) demonstrate somewhat independent transcriptional regulation by these transcription activators, as many transporter genes were induced in only a single segment, not in all segments. Thus, each segment of the intestine should be considered as a separate regulatory unit. In conclusion, chemical activation of xenobiotic-responsive transcription factors induces the expression of some drug transporters in some segments of the small intestine and colon of mice. These data provide insight into mechanisms of drug transporter regulation in enterocytes and may aid in predicting drug-drug interactions. (Supported by NIH grants ES009649, ES009716, ES007079, ES013714, RR021940, DK081461.)
372. Sequence networks reveal functional similarities in the Solute Carrier (SLC) Superfamily
Pär Matsson, Avner Schlessinger, Sook Wah Yee, Libusha Kelly, Leonard Apeltsin, Thomas E. Ferrin, Kathleen M. Giacomini, and Andrej Sali
Department of Bioengineering and Therapeutic Sciences, University of California San Francisco, San Francisco, CA, USA, 94143
The Solute Carrier (SLC) gene superfamily encodes more than 350 human transport proteins in 47 subfamilies that maintain the cellular homeostasis of nutrients and metabolites, regulate signaling processes, and are major determinants of the absorption, distribution and elimination of many clinically used drugs (1, 2). SLC transporters have traditionally been assigned to subfamilies based on a combination of overlapping substrate specificity and sequence similarity with other members. However, detailed functional knowledge is only available for a subset of all SLC transporters, and studies of the overall organization of the SLC superfamily in terms of functional and sequence similarity have been lacking. Here, we aimed to delineate functional and evolutionary similarity patterns within the SLC superfamily using a sequence similarity network approach. We used sensitive profile-profile alignments to determine the pairwise sequence similarities of all available human SLC protein sequences. By mapping functional traits onto the resulting sequence similarity network, patterns of similarities in substrate specificity, transport mechanism, tissue distribution and evolutionary conservation emerge. The 407 distinct SLC sequences grouped into 26 major clusters of structurally related transporters, of which 7 contained multiple traditional subfamilies. Several cases of intuitively appealing, but so far largely unexplored connections between different subfamilies were observed, e.g.: organic anion transporters from the SLC16, 17, 37 and SLCO families cluster together, as do amino acid transporters from the SLC6, 36 and 38 families and inorganic ion transporters from the SLC8 and SLC24 families. Since the similarity network is solely based on sequence, such connections give rise to testable hypotheses of sequence-substrate specificity relationships. Also, this birds-eye view of the SLC transporter superfamily provides valuable guidance for future structural genomics and protein structure modeling efforts.
References:
- Hediger MA, Romero MF, Peng JB, Rolfs A, Takanaga H and Bruford EA. The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteins. Pflugers Arch. 447: 465–8, 2004.
- Dresser MJ, Leabman MK and Giacomini KM. Transporters involved in the elimination of drugs in the kidney: organic anion transporters and organic cation transporters. J Pharm Sci. 9: 397–421, 2001.
373. ABCB6 mediates lead resistance through a heme dependent pathway
John A. Lynch, and John D. Schuetz
Pharmaceutical Science, St. Jude Children’s Research Hospital, Memphis, TN, USA, 38105
ABCB6 is a half-transporter localized to the outer mitochondrial membrane (1). Previously we have shown that ABCB6 is an important regulator of heme biosynthesis, controlling the activity of key hemoproteins (2). Consequently, ABCB6 influences essential cellular activities, including respiration, ROS dissipation, and hemoglobin synthesis (2). Heavy metals, such as lead, constitute a major source of toxic environmental exposure. The prominent disruption of hematopoeisis and mitochondrial respiration by these xenobiotic metals suggests that ABCB6 might be important in resisting challenges by these agents. We demonstrate that ABCB6 overexpression provides resistance against lead toxicity. Conversely, absence of ABCB6 sensitizes MEFs to lead cytotoxicity. Overexpression of ABCB6 does not reduce total lead accumulation, which is similar between vector and ABCB6 overexpressing cells. ABCB6 protection is related to heme, as succinylacetone abrogates the survival advantage conferred by ABCB6. Resistance to cadmium was also observed, possibly involving different pathways, and will be discussed elsewhere. We have further explored the mechanisms involved in lead resistance. ABCB6 plays two roles in heme metabolism, transporting porphyrin precursors into the mitochondria to drive heme assembly, and, also, transporting mature heme into the mitochondrial intermembrane space. We propose that both functions are important in lead resistance. Our data shows that lead is less effective in inhibiting heme biosynthesis in ABCB6-overexpressing cells. This indicates that higher levels of ABCB6-mediated coproporphyrinogen transport, a late stage in heme biosynthesis, can compensate for inhibition of ALAD by lead, an early step. We also found evidence for the importance of heme transport, as administration of exogenous heme only enhanced the survival advantage in ABCB6-overexpressing cells. Binding studies have demonstrated that lead dose-dependently disrupts the interaction between ABCB6 and heme, suggesting that lead acts directly upon ABCB6, which may account for its effects upon both heme and porphyrin transport. These studies reveal a new mechanism of lead toxicity that is likely related to inhibition of ABCB6 function.
Reference:
- Identification of a mammalian mitochondrial porphyrin transporter. Krishnamurthy PC, Du G, Fukuda Y, Sun D, Sampath J, Mercer KE, Wang J, Sosa-Pineda B, Murti KG, Schuetz JD. Nature. 2006 Oct 5;443(7111):586–9. Epub 2006 Sep 27.
- Cell survival under stress is enhanced by a mitochondrial ATP-binding cassette transporter that regulates hemoproteins. Lynch J, Fukuda Y, Krishnamurthy P, Guoqing D, and Schuetz JD. Cancer Research. (In press, 2009)
374. Risperidon (risperdal) therapy in schizophrenia: metabolic effects
Tulin Yanik1, Canan Kursungoz1, Levent Sutcigil2, and Fuat Ozgen2
1Biological Sciences, MIDDLE EAST TECHNICAL UNIVERSITY, Ankara, Turkey, 2Psychiatry Department, GULHANE MILITARY MEDICAL SCHOOL, Ankara, Turkey
Schizophrenia is a common psychotic disorder that has effects in all over the world. While currently used atypical antipsychotics drug treatments offer important relief from some symptoms, they also cause serious adverse effects in many individuals. Those effects are resulted in long terms and there is none early indicators of them. Also, when patients face with the adverse effects, no time to change the drug to another and as a result patients leave the treatment which cause both economical and emotional problems. DNA information in the pathways for drug metabolism may be an important predictor of treatment response in schizophrenia. In this study, we investigated one of the atypical antipsychotic drug, Risperidon (Risperdal) adverse effects in psychotic patients. Risperidone, a serotonin (5-HT) receptor antagonist which is used efficiently in the treatment of schizophrenia with side effect of weight gain. Weight gain mechanisms are controlled by the arcuate nucleus of hypothalamus which secrete the neurohormones such as pro-opiomelanocortin (POMC), cocaine-amphetamine regulated transcript (CART) that diminish the apetite, and neuropeptide Y (NPY) and agouti-related peptide (AgRP) which increase the apetite. We hypothesized that risperidone, by inhibiting the 5-HT receptors, might be causing a change in the gene expression of POMC, NPY and CART which could serve as early predictor for weight gain. We used case-control association design to compare the candidate gene expressions and their serum levels in treatment responders (case) versus nonresponders (controls) from Psychiatry Department of Gülhane Millitary Medical School in Ankara, Turkey. The case group has gained avarage of 9 pounds in a month versus controls. Quantitative real-time PCR (qRT-PCR) experiments revelaed that there is a decrease in the expression of POMC and CART and increase in NPY expression in patients. Furthermore, enzyme-linked immunosorbent assay (ELISA) shows that leptin serum levels has increased and α-MSH, derived from POMC and CART levels have been shown to decreased. This finding provides a molecular basis for the obesity of these patients fairly in short term.
References:
- Goldner, E.M., Hsu, L., Waraich, P., Somers, J.M. Prevalence and incidence studies of schizophrenic disorders: a systematic review of the literature. Canadian Journal of Psychiatry 47: 833–43. (2002)
- Konturek, P.C., Konturek, J.W., Czeœnikiewicz-Guzik, M., Brzozowski, T., Sito, E., Konturek, S.J. Neuro-hormonal control of food intake: basic mechanisms and clinical implications. Journal of Physiology and Pharmacology, 56(6):5–25. (2005)
- Heisler, L.K., Cowley, M.A., Tecott, L.H., Fan, W., Low, M.J., Smart, J.L., Rubinstein, M., Tatro, J.B., Marcus, J.N., Holstege, H., Lee, C.E., Cone, R.D., and Elmquist JK. Activation of central melanocortin pathways by fenfluramine. Science 297:609–611. (2002)
- Schwatrz, M.W., Woods, S.C., Porte, Jr D., Seeley, R.J., Baskin, D.G. Central Nervous System Control of Food Intake. Nature 404(6778): 661–671. (2000)
- Charles U. Nnadi, and Anil K. Malhotra Individualizing Antipsychotic Drug Therapy in Schizophrenia: The Promise of Pharmacogenetics. Curr Psychiatry Rep. 9(4): 313–318. (2008)
Notes
1David F.V. Lewis* (2003) Current Medicinal Chemistry, 2003, 10, 1955–1972
1Jiang JK et al. (2008) Journal of Medicinal Chemistry, 51 (24), 8012-8018
2E. van Gurp et al. (2008) American Journal of transplantation, 8, 1711-1718 In Vitro Metabolism of CP-690,550 in Liver Microsomes of Mouse, Rat, and Human: Formation of a Novel Metabolite via the Loss of a Nitrile Group
1Iwuchukwu, OF and Nagar, S - Resveratrol (trans-Resveratrol, 3,5,4-Trihydroxy-trans-stilbene) Glucuronidation Exhibits Atypical Enzyme Kinetics in Various Protein Sources-Drug Metabolism and Disposition 2008 vol 36 (2)322-330.
[1] DeLorenzo, R.J., Dashefsky, L., Anticonvulsants. Hand Neurochem 1985, 9, 363-403.
[2] DeLorenzo R.J., Status epilepticus. Curr Ther Neurol Dis 1990, 3, 47-53.
[3] Browne, T.R., LeDuc, B., Phenytoin and other hydantoins: chemistry and biotransformation. In: Levy RH, Mattson RH, Meldrum BS, Perucca E, eds. Antiepileptic drugs, 5th ed. Philadelphia: Lippincott Williams and Wilkins, 2002, 565-580.
[4] Kadar,D., Fercycz, T.D., Kalow, W., Can J Physiol Pharmacol 1983, 61, 403-407.