
Abstract
Since 1972, Drug Metabolism Reviews has been recognized as one of the principal resources for researchers in pharmacological, pharmaceutical and toxicological fields to keep abreast of advances in drug metabolism science in academia and the pharmaceutical industry. With a distinguished list of authors and editors, the journal covers topics ranging from relatively mature fields, such as cytochrome P450 enzymes, to a variety of emerging fields. We hope to continue this tradition with the current compendium of mini-reviews that highlight novel biotransformation processes that were published during the past year. Each review begins with a summary of the article followed by our comments on novel aspects of the research and their biological implications. This collection of highlights is not intended to be exhaustive, but rather to be illustrative of recent research that provides new insights or approaches that advance the field of drug metabolism.
| Abbreviations | ||
| NAPQI | = | N-acetyl-p-benzoquinoneimine |
| ALDH | = | aldehyde dehydrogenase |
| AO | = | aldehyde oxidase |
| AKR | = | aldo-keto reductase |
| CES | = | carboxylesterase |
| CSB | = | cystathionine β-synthase |
| CSE | = | cystathionine γ-lyase |
| P450 | = | cytochrome P450 |
| DHPO | = | 2,3-dihydropyridin-4-one |
| ESI | = | electrospray |
| FMO | = | flavin monooxygenase |
| GSH | = | glutathione |
| GSSG | = | glutathione disulfide |
| ICPMS | = | inductively coupled plasma mass spectrometry |
| i.p. | = | intraperitoneal |
| MDR | = | multidrug-resistant |
| NNAL | = | 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol |
| NNK | = | 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone |
| oaTOF | = | orthogonal acceleration time-of-flight |
| PBK | = | physiologically based kinetic |
| PCP | = | pentachlorophenol |
| SDR | = | short-chain dehydrogenase/reductase |
| SULT | = | sulfotransferase |
| TB | = | tuberculosis |
Current articles covered in this review.
Abstract
Source: Drug Metabolism Disposition 43:1303–1306, 2015
Synopsis: Benzbromarone (1) is a potent uricosuric agent that has been used in treating hyperuricemia and chronic gout. Although 1 is highly effective and considered to be superior to allopurinol, it is associated with serious adverse reactions including fulminant or fatal liver injury. As a result, it was not approved in the US and has been withdrawn from European markets. This article describes the P450-mediated bioactivation of 1 in liver microsomal systems. In this study Kitagawara et al. (Citation2015) have identified mono-debrominated catechol (2-ethyl-3-(3-bromo-4, 5-dihydroxybenzoyl) benzofuran (M1) and a novel truncated metabolite, 2,6-dibromohydroquinone (M2), following incubation of 1 with rat and human liver microsomes and have shown that M1, M2 and 2,6-dibromobenzoquinone M3 () are cytotoxic in HepG2 cells. Previous metabolism studies have indicated that 1 is bioactivated to an orthoquinone M6 by sequential oxidation of 6-hydroxybenzbromarone (M4, ).
Metabolism studies were performed by incubation of 1 with phenobarbital induced rat liver microsomes and human liver microsomes. The metabolites were analyzed by LC-MS and GC-MS (for M2 after trimethylsilylation). The metabolites were confirmed by matching them with their respective synthetic standards. Phenotyping studies with recombinant enzymes and incubation with P450 inhibitors suggested that both, M1 and M2 were formed by CYP2C9.
Commentary: The novelty of the paper is the formation of M1 and especially M2, which involves a carbon-carbon bond cleavage. Kitagawara et al. propose that both these metabolites are formed via the ipso-substitution reaction. While no evidence for this mechanism has been provided, the hypothesis is based on the previous work that describes the formation of substituted phenols by P450-mediated ipso-substitution (Ohe et al., Citation1997).
Oxidative dehalogenation of halogenated para- or ortho-substituted phenols is a known P450-mediated metabolic process (den Besten et al., Citation1993; Li et al., Citation1985; Park & Kitteringham, Citation1994; Rietjens & Vervoort, Citation1992). Reaction mechanisms for the formation of M1 may involve formation of a quinol intermediate 2 bearing both the bromo-substitutent and a hydroxyl originating from the iron-oxenoid (FeIV+•=O) (Ohe et al., Citation1997) or the hydroperoxo-iron complex (FeIII-OOH) (Vatsis & Coon, Citation2002), intermediate (). Subsequent elimination of the bromo group can afford the corresponding catechol metabolite M1 either via the ortho-benzoquinone intermediate (3) resulting from the loss of Br as an anion and subsequent reduction (, pathway A) or formation of M1 directly via elimination of Br as a cation ( pathway B). The low yield of M1 is consistent with the metabolism studies with model halogenated phenols and anilines and that the ipso substitution and decrease in hydroquinone formation follows the order p-fluorophenol > p-chlorophenol > p-bromophenol (Ohe et al., Citation1997). This also argues against a mechanism proceeding via a loss of a halogen cation. An alternative explanation is the potential covalent binding of quinone intermediates to microsomal proteins that can result in low recovery/yield of the product, as described in the paper.
The elimination of benzofuranyl group from 1 to yield M2 via ipso-substitution as proposed by the authors is quite unique given that the substituent is attached by a carbon-carbon bond. These substituents are generally stable and resistant to cleavage. A reaction mechanism involving formation of quinol intermediate similar to 4 has been proposed by Ohe and co-workers (Ohe et al., Citation1997). Similar to the oxidative dehalogenation described earlier, its formation involves the reactive oxygen species, FeIII-OOH or FeIV+•=O, as the electrophilic oxidant. Hydrolytic cleavage of 4 by the attack of water on the carbonyl carbon can yield the benzofuran carboxylic acid derivative (M8) and the corresponding hydroquinone M2 ().
An alternative mechanism can be readily envisioned for C–C bond cleavage and formation of M2. The scission can take place via nucleophilic addition of the peroxy anion (FeIII-O-O-), on the carbonyl carbon (). The corresponding tetrahedral intermediate formed can either rearrange to an ester (8) either via the Baeyer–Villiger pathway (pathway A) or via an epoxide intermediate 7 (Pathway B). Subsequent hydrolytic cleavage of the ester under the incubation conditions can yield M2. P450 enzymes can catalyze the oxidation of many substrates via the peroxide shunt pathway as postulated by Akhtar et al. (Citation2011) and Varfaj et al. (Citation2014) for nabumetone. Such reactions can be easily supported using cumene peroxide as a reagent to carry out the reaction.
As noted by the authors, metabolites M1, M2 and M3 are all cytotoxic in HepG2 cells when compared to 1, suggesting that bioactivation of benzbromarone may be a prerequisite for the potential cytotoxic effect. While M2 may be relevant to benzbromarone induced toxicity, as the authors suggest, the potential of M1 should not be underestimated given that it is possibly formed via the orthoquinone 3 (). Experiments to trap these reactive metabolites as glutathione conjugates have not been conducted in this study. Overall, the paper suggests novel metabolites of benzbromarone that can potentially result in toxic events elicited by the drug.
Figure 1. Metabolic Scheme of benzbromarone in rat and human liver microsomal systems (Kitagawara et al., Citation2015). Metabolites M4, M5 and M6 were previously reported (McDonald & Rettie, Citation2007; Kobayashi et al., Citation2012).
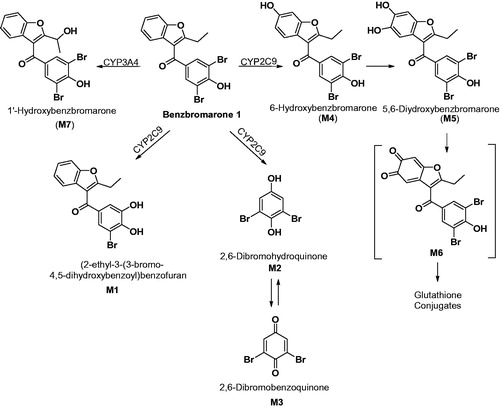
Figure 2. Proposed mechanisms for formation of metabolite M1 (A) and M2 (B).
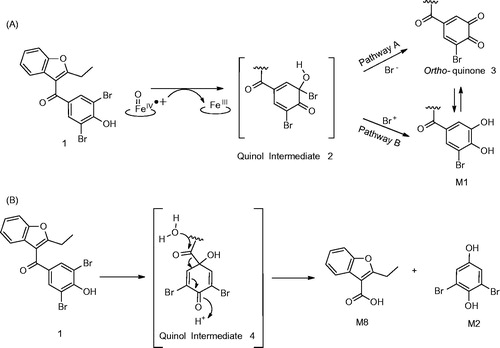
Figure 3. Alternate mechanism for formation of M2.
References
- Akhtar M, Wright JN, Lee-Robichaud P. (2011). A review of mechanistic studies on aromatase (CYP19) and 17alpha-hydroxylase-17,20-lyase (CYP17). J Steroid Biochem Mol Biol 125:2–12.
- den Besten C, van Bladeren PJ, Duizer E, et al. (1993). Cytochrome P450-mediated oxidation of pentafluorophenol to tetrafluorobenzoquinone as the primary reaction product. Chem Res Toxicol 6:674–680.
- Kitagawara Y, Ohe T, Tachibana K, et al. (2015). Novel bioactivation pathway of benzbromarone mediated by cytochrome P450. Drug Metab Dispos 43:1303–1306.
- Kobayashi K, Kajiwara E, Ishikawa M, et al. (2012). Identification ofisozymes involved in benzbromarone metabolism in human liver microsomes. Biopharm Drug Dispos 33:466–473.
- Li JJ, Purdy RH, Appelman EH, et al. (1985). Catechol formation of fluoro- and bromo-substituted estradiols by hamster liver microsomes. Evidence for dehalogenation. Mol Pharmacol 27:559–565.
- McDonald MG, Rettie AE. (2007). Sequential metabolism and bioactivation of the hepatotoxin benzbromarone: formation of glutathione adducts from a catechol intermediate. Chem Res Toxicol 20:1833–1842.
- Ohe T, Mashino T, Hirobe M. (1997). Substituent elimination from p-substituted phenols by cytochrome P450. ipso-substitution by the oxygen atom of the active species. Drug Metab Dispos 25:116–122.
- Park BK, Kitteringham NR. (1994). Effects of fluorine substitution on drug metabolism: pharmacological and toxicological implications. Drug Metab Rev 26:605–643.
- Rietjens CM, Vervoort J. (1992.) A new hypothesis for the mechanism for cytochrome P-450 dependent aerobic conversion of hexahalogenated benzenes to pentahalogenated phenols. Chem Res Toxicol 5:10–19. [https://doi.org/10.1021/tx00025a004]
- Varfaj F, Zulkifli SN, Park HG, et al. (2014). Carbon–carbon bond cleavage in activation of the prodrug nabumetone. Drug Metab Dispos 42:828–838.
- Vatsis KP, Coon MJ. (2002). Ipso-substitution by cytochrome P450 with conversion of p-hydroxybenzene derivatives to hydroquinone: Evidence for hydroperoxo-iron as the active oxygen species. Arch Biochem Biophys 397:119–129.
Abstract
Source: Drug Metabolism Disposition 43:1441–1449, 2015
Synopsis: AZD9819 (1) is an inhibitor of human neutrophil elastase and is being developed as an inhaled drug for potential treatment of chronic obstructive pulmonary disease (Gu et al., Citation2015). This article describes the rearrangement of 1 to a lipid peroxide–mediated rearrangement product, a five-membered oxazole derivative (2), which was formed in plasma of preclinical species and humans, ex-vivo (). The study focuses on the cause/factors that catalyze the formation of 2 and the mechanism of this unusual rearrangement. While this is not a typical “biotransformation/bioactivation” study, it describes the role of human low-density lipoprotein (LDL) in the radical mediated oxidation of compounds and also provides information regarding the means of prevention, which could be valuable for biotransformation and bioanalysis scientists.
Commentary: The two main reasons that makes this report interesting could be summarized by; first, the cause/factors of oxidation of the pyrazinone moiety in 1 and second, the conversion of 1 to an oxazole derivative 2 (Gu et al., Citation2015). The distinct feature in this paper is the finding that formation of 2 is mediated by lipid peroxides in the plasma and not P450. This is intriguing since P450-catalyzed epoxidation of heteroaromatic rings yielding the corresponding hydroxylated metabolites is well precedented in the literature (Guengerich, Citation2001a, Citation2001b, Citation2007). Additionally, P450-mediated rearrangements of pyrazinone epoxide to products that have five membered rings as a core have also been published (Singh et al., Citation2003; Subramanian et al., Citation2003), as noted by the authors. The lack of involvement of P450 in the oxidation of 1 was shown by its absence in incubation of 1 with hepatocytes from rat, dog and human. That the product is only formed ex vivo or in vitro in the plasma was further recognized by the authors since it was absent in the biological matrices (plasma, urine and feces), collected during the ADME studies in rat and dog using radiolabelled 1 and its lack in the human plasma and urine that was collected during clinical studies.
The authors discerned the role of lipid peroxides in this oxidation by the method of elimination. They analyzed the factors and the cause of oxidation co-incubation of 1 in plasma along with different reagents as well as applying various experimental conditions to these incubations. In their experiments, pretreatment of plasma (rat plasma was used) with ascorbic acid and glutathione resulted in attenuation of formation of 2. Further, this inhibition was concentration-dependent and greater inhibition was observed with increase in concentration of both these reagents. As noted by the authors, both these reagents are essentially anti-oxidants. Ascorbic acid prevents peroxidation of lipids by scavenging oxygen free radicals while GSH is a co-factor for GSH-dependent seleno-peroxidases, which catalyzes a two-electron reduction of lipid peroxides (LOOH) to the corresponding alcohols and is itself converted to glutathione disulfide (GSSG). To further prove the role of lipid peroxides in this oxidation, the authors also incubated 1 with low density lipoproteins (LDL) and detected 2 in this incubation mixture.
As noted above, the other unique aspect of this paper is the formation of a rearranged oxidation product. While the overall MS results reveal a simple oxidation of 1 with an addition of 16 Da, in-depth structural elucidation and analysis suggested an unusual rearrangement of the pyrazinone motif. Based on the results of ascorbic acid and GSH experiments and the fact that LDL catalyzes the formation of 2, mostly under basic conditions, Gu et al. (Citation2015) propose that the first step is a nucleophilic attack of the lipid peroxide on the electron-deficient double bond of the pyrazinone moiety to yield the epoxide 3 as depicted in (Step 1). The corresponding imine, formed as an intermediate in the process, is then hydrated thereby allowing intramolecular nucleophilic attack of the carboxamide oxygen on the epoxide. This leads to the rearranged oxazole core with subsequent loss of a water molecule (, Step 2). The authors envision the ultimate step of the reaction to be an intramolecular transfer of an acetyl group. The possible driver for this transfer is conversion of the high energy anion to a reasonably low energy anion and therefore the formation of the more stable tertiary amide moiety to afford the corresponding oxazole product 2. The authors also conducted 18O2 experiments helped in the structural elucidation. These experiments indicated incorporation of 18O in the acetyl group as described by the authors. This confirmed that the primary driver of this complex rearrangement was a simple mono-oxygenation of the compound to an epoxide.
Another facet of the paper that is worth noting was the structural characterization of 2. While the conversion of 1 to 2 is interesting, the identification and structural elucidation of 2 can be pretty challenging. This is because there is a lack of similarity and fragmentation between the two compounds. Given this lack of common fragmentation pattern between 1 and 2 in the MS/MS spectra and the difficulty to identify the oxazole core by NMR (given the lack of protons on the core), the key part of structural elucidation was comparison of 2 with the synthetic standard, the 13C-chemical shifts in the carbon NMR with those predicted by the ACD Labs carbon NMR predictor and the crystal structure of the product. Additional information was obtained from the HMBC NMR correlation data in which the proton of the methylene group (H33) correlated with the carbonyl carbon C34 of the acetyl group.
Overall, in this paper, the authors have reported a unique lipid peroxide catalyzed epoxidation of a pharmacological agent in plasma. The authors also describe different methods of preventing this plasma instability, which may prove useful to biotransformation and bioanalytical scientists.
Figure 4. Chemical structures of AZD9819 (1) and its rearranged product 2 (Gu et al., Citation2015).
Figure 5. Step 1: Mechanism of oxidation of 1 to the epoxide (3). Step 2: Rearrangement of epoxide (3) to the corresponding oxazole 2 (Gu et al., Citation2015).
References
- Gu C, Lewis RJ, Wells AS, et al. (2015). Lipid peroxide-mediated oxidative rearrangement of the pyrazinone carboxamide core of neutrophil elastase inhibitor AZD9819 in blood plasma samples. Drug Metab Dispos 43:1441–1449.
- Guengerich FP. (2001a). Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol 14:611–650.
- Guengerich FP. (2001b). Uncommon P450-catalyzed reactions. Cur Drug Metab 2:93–115.
- Guengerich FP. (2007). Mechanisms of cytochrome P450 substrate oxidation: MiniReview. J Biochem Mol Toxicol 21:163–168.
- Singh R, Silva Elipe MV, Pearson PG, et al. (2003). Metabolic activation of a pyrazinone-containing thrombin inhibitor. Evidence for novel biotransformation involving pyrazinone ring oxidation, rearrangement, and covalent binding to proteins. Chem Res Toxicol 16:198–207.
- Subramanian R, Lin CC, Ho JZ, et al. (2003) Bioactivation of the 3-amino-6-chloropyrazinone ring in a thrombin inhibitor leads to novel dihydro-imidazole and imidazolidine derivatives: structures and mechanism using 13C-labels, mass spectrometry, and NMR. Drug Metab Dispos 31:1437–1447.
Abstract
Source: Drug Metabolism Disposition 43: 646–659, 2015
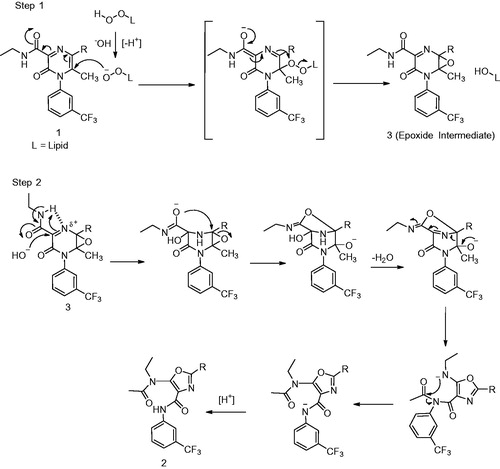
Synopsis: MRX-I (1), an analog of linezolid (2) (), is a novel oxazolidinone antibiotic for treatment of Gram-positive infections, currently in phase II clinical trials in China (Meng et al., Citation2015). In preclinical studies, 1 is superior to 2 and this activity is attributed to its more planar 2,3-dihydropyridin-4-one (DHPO) ring in the molecule. Additionally, 1 has been to shown to have a better safety profile than 2 in preclinical toxicology studies and Phase I clinical trials.
This article describes the metabolism of 1 in humans and depicts the mechanisms underlying the novel oxidative ring opening of DHPO, which was detected to be a principal metabolic pathway leading to the formation of novel metabolites 3 and 4 (). The authors have conducted systematic experiments using incubations with microsomes, cytosol and S9 fractions as well as chemical inhibitors to characterize the pathway(s) leading to the formation of these metabolites. These metabolism/phenotyping studies have demonstrated that formation of metabolites 3 and 4 are catalyzed by flavin monooxygenase (FMO), short-chain dehydrogenase/reductase (SDR), aldo–keto reductase (AKR) and aldehyde dehydrogenase (ALDH). The authors have also conducted studies using 18O-labeled water to decipher the mechanism of formation of these metabolites. Additionally, the authors have uncovered metabolite 5 () in human plasma, urine and feces. Metabolite 5 has the same mass as 3 but its formation is catalyzed by aldehyde oxidase (another cytosolic non-P450 enzyme) rather than P450.
Commentary: The novelty of the paper is the carbon-carbon bond cleavage of DHPO ring in the formation of major metabolites 3 and 4. During the course of identification of metabolites of 1 in humans the authors detected two predominant DHPO-ring opened metabolites (3 and 4) in the plasma and urine. In the process of understanding the enzymes and the mechanism of formation of these metabolites, Meng et al. (Citation2015) have conducted experiments that help in deciphering the routes for formation of these metabolites. Based on their experiments in human liver microsomes, liver S9 fraction and human liver cytosol-fortified with FMO, the authors conclude that multiple enzymes present in the microsomal as well as the cytosolic fraction catalyze the formation of 3 and 4.
Meng et al. (Citation2015) have proposed that the first step in the formation of both these metabolites involves the FMO5-catalyzed Baeyer–Villiger oxidation (). Involvement of FMO5 in the oxidative cleavage was further confirmed when incubation of 1 with cytosol-fortified with FMO5 produced 3 and 4. These metabolites were not observed in cytosolic mixtures that were fortified with FMO1 and FMO3. The mechanism involves a nucleophilic attack of the peroxyflavin (FADOO−) intermediate on the carbonyl group of the ketone. The resulting “classic tetrahedral Criegee intermediate” thus formed rearranges to the enol lactone with concomitant formation and release of a flavin-hydroxide which then regenerates FAD with elimination of water. The resulting enol-lactone is then hydrolyzed to produce the aldehyde intermediate (12) that is then converted to the alcohol 3 and the acid 4.
Human FMO5 has been identified as a selective Baeyer–Villiger monooxygenase in humans. That human FMO5 is the primary enzyme that catalyzes Baeyer–Villiger rearrangements of drugs has been also been reported by Lai et al. (Citation2011). Fiorentini et al. (Citation2016) have recently demonstrated in vitro activity of human FMO5 in catalyzing the oxygenation of both aliphatic and cyclic ketones to the corresponding esters and lactones, respectively. As noted by the authors, it is not surprising given that the Baeyer–Villiger oxidations of α,β-unsaturated ketones favor vinyl migrations over the alkyl migration. Fiorentini et al. (Citation2016) have also demonstrated selective conversion of 3-methyl-pent-3-en-2-one and β-ionone to the corresponding enol acetate in their report.
The authors confirmed the existence of an aldehyde intermediate by incubating 1 with liver cytosol-fortified FMO5 in the presence of H218O. The results of these experiments revealed the presence of two 18O atoms in 3. One was incorporated in the hydroxyethyl group while the other was incorporated in the carboxyethyl group of 3. Similarly, three 18O atoms were incorporated in 4. Mass spectrometric analysis revealed that while one of the 18O atoms was incorporated in the carboxyethyl side chain like in 3, the other two 18O atoms were incorporated in the carboxymethyl group of 4. The authors have rationalized the incorporation of two or three 18O atoms in 3 and 4. The mechanism of incorporation is consistent with the opening of the enol lactone followed by conversion by oxidases such as ALDH. Interestingly, the authors did not attempt to trap the intermediate aldehyde 12 with known aldehyde trapping agents such as methoxylamine or semicarbazide (Rousu et al., Citation2009; Xu et al., Citation2005; Zhang et al., Citation1996). Alternatively, quenching the reaction with sodium borohydride or sodium borodeuteride would theoretically yield the corresponding alcohol 3. The involvement of cytosolic enzymes, SDR, AKR or ALDH in the catalysis of conversion of 12 to 3 and 4 was proven by incubating S9 fraction with 1 in the presence of menadione, flufenamic acid or disulfiram. These three compounds are inhibitors of SDR, AKR and ALDH, respectively. Inhibition of formation of 4 by disulfiram suggested that ALDH is mainly responsible for the conversion of the FMO-mediated intermediate and also indicated that aldehyde is an intermediate following oxidative cleavage of the ring. The formation of 3 was partially reduced by menadione and flufenamic acid which led the authors to suggest that multiple enzymes were involved in the formation of 3.
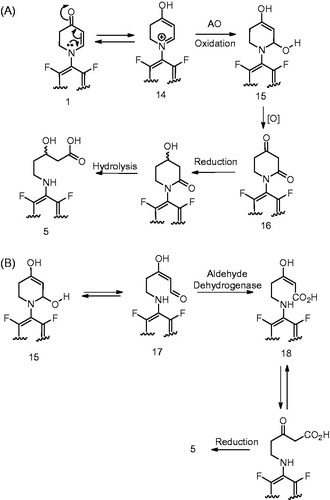
Another interesting metabolite detected by the authors during the metabolism investigation of 1 in humans was the 5-amino-3-hydroxypentanoic acid, 5 (). This metabolite was observed in the plasma, urine and the feces and appeared to be a DHPO ring opened product formed via oxidative carbon-nitrogen bond scission. Like in the case of 3 and 4, metabolite 5 was not observed in NADPH-fortified human liver microsomes but was detected when 1 was incubated with NADPH-supplemented human liver S9 fraction. The fact that 5 was not inhibited by ABT and was also observed when 1 was incubated with cytosol that was fortified with NADPH suggested that this metabolite was not formed by P450. Addition of aldehyde oxidase (AO) inhibitors, menadione and raloxifene to these incubations led to a significant decrease in the formation of 5. This led the authors to conclude that 5 was formed via AO-mediated oxidation and not by oxidative enzymes in the liver microsomes. Additionally, incubation in the presence of H218O, indicated incorporation of two 18O atom into the metabolite and suggested that the two atoms were present in the carboxyl group. This further discerned the metabolic pathway leading to the formation of 5. Based on this analysis, the authors propose a mechanism for the formation of 5 as depicted in . According to this mechanism, 1 is oxidized by 2,4-piperidinedione 16 which probably undergoes sequential reduction by NADPH-dependent carbonyl reductases and hydrolysis to yield the corresponding metabolite 5. For the formation of 16 from 1, one can possibly envisage tautomerism of DHPO to 4-hydroxydihydropyridinium intermediate 14. The intermediate 14 can then undergo AO-catalyzed oxidation (which entails attack of the hydroxyl group from the Moco moiety of AO) to yield a carbinol amine intermediate 15 () (Brandange & Lindblom, Citation1979; Wu et al., Citation1988). An alternative mechanism can also be envisioned in the formation of 5 and is described in . According to this mechanism, the carbinolamine 15 can exist in equilibrium with the ring-opened enolamino aldehyde 17 (Murphy, Citation1973). Oxidation of 17 by ALDH to 18 and subsequent reduction of the enol by cytosolic NADPH-dependent carbonyl reductase could yield the corresponding metabolite 5.
Overall, the authors have presented novel metabolites that involve a carbon-carbon oxidative cleavage of the dihydropiperidinone ring system and the involvement of non-P450 enzymes in their formation. Given the involvement of non-P450 enzymes in addition to P450, in the metabolism of MRX-I (1), the risk of pharmacokinetic interactions with P450 inhibitors and inducers should be possibly minimal.
Figure 6. Proposed metabolic scheme of 1 in humans (Meng et al., Citation2015) and chemical structure of linezolid (2).
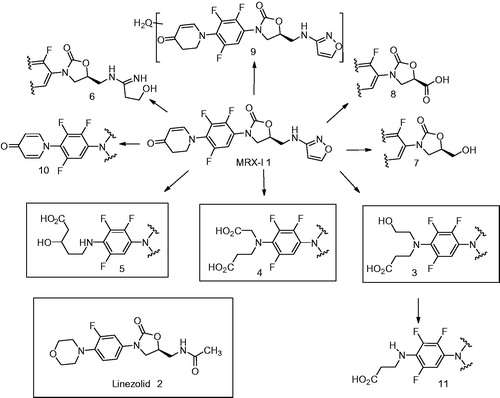
Figure 7. Mechanism of formation of metabolites 3 and 4 by FMO-catalyzed metabolism of 1 (Meng et al., Citation2015).
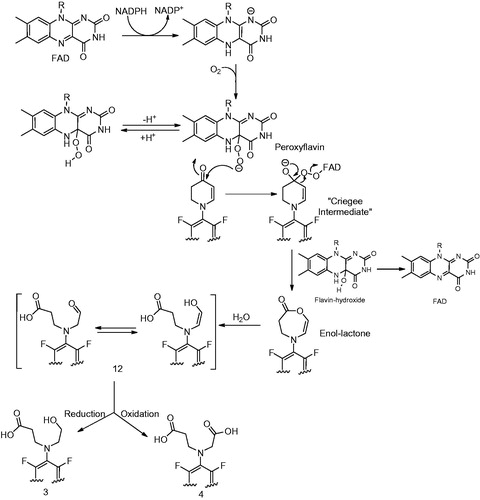
Figure 8. (A) Formation of metabolite 5 (Meng et al., Citation2015). (B) Alternative mechanism for the formation of 5 from intermediate 15.
References
- Brandange S, Lindblom L. (1979). The enzyme “aldehyde oxidase” is an iminium oxidase. Reaction with nicotine delta 1'(5') iminium ion. Biochem Biophys Res Commun 91:991–996.
- Fiorentini F, Geier M, Binda C, et al. (2016). Biocatalytic characterization of human FMO5: unearthing Baeyer–Villiger reactions in humans. ACS Chem Biol. 11:1039–48.
- Lai WG, Farah N, Moniz GA, Wong YN. (2011). A Baeyer–Villiger oxidation specifically catalyzed by human flavin-containing monooxygenase 5. Drug Metab Dispos 39:61–70.
- Meng J, Zhong D, Li L, et al. (2015). Metabolism of MRX-I, a novel antibacterial oxazolidinone, in humans: The oxidative ring opening of 2,3-dihydropyridin-4-one catalyzed by non-P450 enzymes. Drug Metab Dispos 43:646–659.
- Murphy PJ. (1973). Enzymatic oxidation of nicotine to nicotine 1'(5') iminium ion. A newly discovered intermediate in the metabolism of nicotine. J Biol Chem 248:2796–2800.
- Rousu T, Pelkonen O, Tolonen A. (2009) Rapid detection and characterization of reactive drug metabolites in vitro using several isotope-labeled trapping agents and ultra-performance liquid chromatography/time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 23:843–855.
- Wu E, Shinka T, Caldera-Munoz P, et al. (1988). Metabolic studies on the nigrostriatal toxin MPTP and its MAO B generated dihydropyridinium metabolite MPDP+. Chem Res Toxicol 1:186–194.
- Xu S, Zhu B, Teffera Y, et al. (2005). Metabolic activation of fluoropyrrolidine dipeptidyl peptidase-IV inhibitors by rat liver microsomes. Drug Metab Dispos 33:121–130.
- Zhang KE, Naue JA, Arison B, Vyas KP. (1996). Microsomal metabolism of the 5-lipoxygenase inhibitor L-739,010: Evidence for furan bioactivation. Chem Res Toxicol 9:547–554.
Abstract
Source: Chemical Research in Toxicology 28:1796–1802, 2015
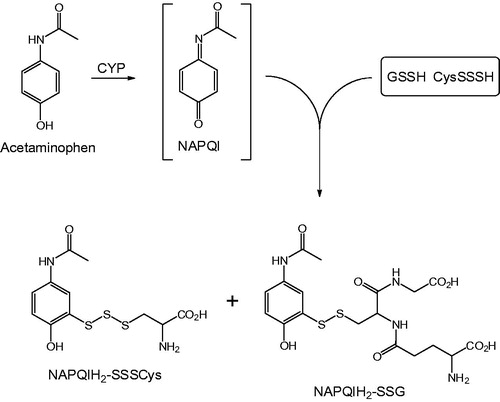
Synopsis: The electrophilic metabolite of acetaminophen, N-acetyl-p-benzoquinoneimine (NAPQI), which is believed to be the primary mediator of acetaminophen-induced liver injury, has long been known to be detoxified in vivo through reaction with glutathione (GSH) to form thioether-linked conjugates. In this publication, evidence is presented for the formation of a group of novel S-linked conjugates in mice injected i.p. with acetaminophen that reflect the interaction of NAPQI with nucleophilic per- and polysulfides produced by the enzymes cystathionine γ-lyase (CSE) and cystathionine β-synthase (CSB). These conjugates, denoted as NAPQIH2-SSSCys (detected in serum, liver and urine) and NAPQIH2-SSG (detected in urine only), are attributed to the reaction between NAPQI and CysSSSH and GSSH, respectively. These and other persulfide adducts were identified in vitro when NAPQI was allowed to react with synthetic persulfides such as CysSSH, GSSH, and Na2S4. Collectively, the data suggest that enzymes such as CSE may play an important protective role in acetaminophen-induced hepatotoxicity through their ability to generate nucleophilic per- and polysulfides that scavenge the reactive metabolite, NAPQI.
Commentary: Previous studies by the authors and their co-workers (Ida et al., Citation2014) demonstrated that small molecule persulfides and polysulfides are formed endogeneously during the metabolism of sulfur-containing amino acids through the action of the transsulfuration enzymes CSE and CSB, and that high levels (>100 μM) of persulfides such as GSSH are maintained in mammalian tissues. Moreover, it was shown that these per- and polysulfides are both highly nucleophilic and reducing, and it was hypothesized that they may play a previously unrecognized role in regulatory functions and redox cell signaling, as well as a key role in the detoxification of electrophilic drug metabolites. Further studies by these investigators (Hagiya et al., Citation2015) revealed that the susceptibility of mice towards acetaminophen-induced liver injury was enhanced by hemizygosity in the CSE and CBS genes; thus, deletion or polymorphism in these genes led to a greater sensitivity to the hepatotoxic properties of the drug, and this effect was ascribed to a diminished availability of per- and/or polysulfides to scavenge NAPQI. Based on the above findings, Abiko et al. sought to investigate whether persulfide conjugates of NAPQI could be identified in tissues and biological fluids from mice given a single dose of acetaminophen (150 mg/kg i.p.). With the aid of LC-MS/MS techniques and reference samples of persulfide conjugates prepared by synthesis, the authors successfully detected adducts that derived from reaction of the quinoneimine with CysSSSH and GSSH (). Parallel in vitro experiments with synthetic NAPQI led to the identification of additional reaction products, such as disulfide-linked dimers of NAPQI. It was concluded, therefore, that low molecular weight persulfides can react efficiently with NAPQI in vivo; this reactivity is ascribed to the lower pKa values of persulfides as compared to those of the sulfhydryl groups of GSH and cysteine, which results in persulfides existing predominantly in their deprotonated, more nucleophilic forms at physiological pH.
Although not the first report of a disulfide-linked metabolite of acetaminophen (Sun et al., Citation2008), this publication is the most detailed account to date of the potential role of per- and polysulfides as protective agents against acetaminophen hepatotoxicity, and it will be important to establish the relevance of these animal and in vitro findings to the human situation. These results raise the possibility that other therapeutic agents also undergo metabolic activation to form conjugates with per- and polysulfides in vivo and, if this proves to be the case, it will be of interest to determine whether such detoxification pathways are quantitatively important relative to conjugation with GSH. Finally, in light of these results, the toxicological implications of genetic variations in the CSE and CSB genes becomes an important issue, since it has been reported that the prevalence of hemizygosity in the human population is in the order of 1:200–300 (Hagiya et al., Citation2015). Clearly, much remains to be understood about the biological roles of this interesting new class of endogenous nucleophiles and reducing agents.
Figure 9. Conjugation of NAPQI with GSSH and CysSSSH.
References
- Hagiya Y, Kamata S, Mitsuoka S, et al. (2015). Hemizygosity of transsulfuration genes confers increased vulnerability against acetaminophen-induced hepatotoxicity in mice. Toxicol Appl Pharmacol 282:195–206.
- Ida T, Sawa T, Ihara H, et al. (2014). Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc Natl Acad Sci USA 111:7606–7611.
- Sun J, Schnackenberg LK, Holland RD, et al. (2008). Metabonomics evaluation of urine from rats given acute and chronic doses of acetaminophen using NMR and UPLC/MS. J. Chrom B: Anal Technol Biomed Life Sci 871:328–340.
Abstract
Source: Clinical Cancer Research 21:2297–2304, 2015
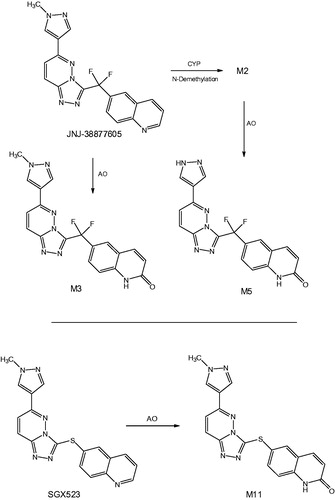
Synopsis: This publication reports on the findings of a first-in-human study with an inhibitor of c-Met tyrosine kinase, JNJ-38877605, that caused renal toxicity in patients, even at sub-therapeutic doses, leading to discontinuation of its development. This toxicity was not predicted by preclinical safety evaluation in rats and dogs, in which the compound was well tolerated, but subsequent studies in rabbits recapitulated the human findings and demonstrated the formation of renal crystals with degenerative and inflammatory changes. Comparative metabolic studies with JNJ-38877605 showed that humans and rabbits exhibited high systemic exposure to two products (M3 and M5) arising from oxidation of the drug by aldehyde oxidase (AO), whereas these metabolites were minor in rats and dogs. The relatively poor aqueous solubility of the AO metabolites, together with the fact that their major pathway of elimination in humans and rabbits was via renal excretion, led the authors to conclude that the toxicity of JNJ-38877605 in human subjects most likely was due to crystallization of M3 and M5 in the kidney.
Commentary: It is becoming increasingly apparent that metabolism of drug candidates by AO can be problematic in the context of clinical development. Thus, a number of compounds that proved to be substrates for this enzyme have been discontinued due to low oral bioavailability and high clearance in humans that was not predicted from animal PK data (notably from studies in rats and dogs) (Sanoh et al., Citation2015). It has also been recognized that the products of AO-mediated oxidation of heterocyclic amines can exhibit lower aqueous solubility than their respective parent compounds, and in cases where urinary excretion is an important route of elimination, this has the potential to result in crystal formation and ensuing renal damage. The subject of the present publication, JNJ-38877605 (), represents the second reported example of a drug candidate whose development had to be halted in early Phase 1 studies as a result of this phenomenon, the first case being the closely related c-Met inhibitor, SGX523, which undergoes metabolism by AO to the sparingly soluble lactam M11 (Diamond et al., Citation2010; Infante et al., Citation2013). In both programs, preclinical toxicology studies employed rats and dogs, which are now known to express only low levels of AO activity compared to humans (Smith & Obach, Citation2009). As a result, systemic exposure to poorly soluble AO metabolites in these animal species probably was significantly less than in humans, leading to the unexpected toxicology findings in man.
Interestingly, Lolkema et al. state that, “On the basis of metabolite patterns in hepatocytes, all human metabolites were present in rat and dog with the exception of an N-glucuronide, which is considered to be nontoxic. Therefore, rat and dog were identified as suitable species to test toxicity.” While the reason(s) for this apparent in vitro/in vivo discrepancy is not clear, and simply may reflect quantitative, as opposed to qualitative, differences in AO metabolite generation across species, the authors speculate that AO activity in the human kidney (Moriwaki et al., Citation2001) may have contributed to the adverse effects of JNJ-38877605. Regardless, the rabbit proved to be a good model for man in this instance, leading the authors to recommend the use of the rabbit as an alternative toxicology species in situations where AO is known, or suspected, to play an important role in the disposition of the compound-of-interest.
Figure 10. Metabolism of quinoline-containing c-Met inhibitors by AO to lactam derivatives.
References
- Diamond S, Boer J, Maduskie TP, et al. (2010). Species-specific metabolism of SGX523 by aldehyde oxidase and the toxicological implications. Drug Metab Dispos 38:1277–1285.
- Infante JR, Rugg T, Gordon M, et al. (2013). Unexpected renal toxicity associated with SGX523, a small molecule inhibitor of MET. Invest New Drugs 31:363–369.
- Moriwaki Y, Yamamoto T, Takahashi S, et al. (2001). Widespread cellular distribution of aldehyde oxidase in human tissues found by immunohistochemistry staining. Histol Histopathol 16:745–753.
- Sanoh S, Tayama Y, Sugihara K, et al. (2015). Significance of aldehyde oxidase during drug development: Effects on drug metabolism, pharmacokinetics, toxicity, and efficacy. Drug Metab Pharmacokinet 30:52–63.
- Smith DA, Obach RS. (2009). Metabolites in safety testing (MIST): Considerations of mechanisms of toxicity with dose, abundance, and duration of treatment. Chem Res Toxicol 22:267–279.
Abstract
Source: Xenobiotica 45: 672–680, 2015
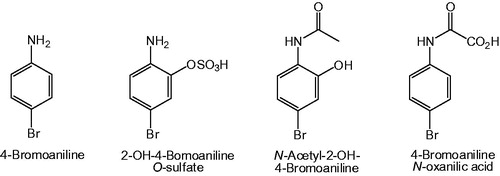
Synopsis: In this proof-of-principle study, inductively coupled plasma mass spectrometry (ICPMS) was employed to obtain both qualitative and quantitative data on the metabolic fate of 4-bromoaniline in the bile duct cannulated rat without the aid of a radiotracer. Following an i.p. dose of 4-bromoaniline (50 mg/kg), bile and urine were collected over a period of 48 hours and analyzed directly by ICPMS set to monitor 79Br and 81Br. By reference to a calibration curve, the excretion of brominated metabolites over the collection period was found to be 68.9 ± 3.6% of the administered dose in urine, and 21.4 ± 1.4% in bile, for a total recovery of some 90%. Subsequent sample analysis by on-line HPLC-ICPMS, again with selective detection of Br, provided metabolic profiles of bromine-containing components in urine and bile, and revealed the presence of 21 brominated species in the 0–12 hours urine and 19 brominated compounds in the 6–12 hours bile. Finally, samples were analyzed by HPLC-MS using an orthogonal acceleration time-of-flight (oaTOF) instrument, equipped with an electrospray (ESI) ion source and operated in the negative ion mode under conditions of high mass resolution. In total, no less than 60 brominated metabolites were detected in urine and 21 in bile, the most abundant being 2-OH-4-bromoaniline O-sulfate (urine) and N-acetyl-2-OH-4-bromoaniline (bile). Elaboration of the N-acetyl side chain of acetamide metabolites was evident in the formation of oxidation products such as the N-oxanilic acid shown in .
Commentary: Although ICPMS is not a new technique, and has been employed previously in studies of drug metabolism, the present investigation represents a particularly impressive example of the power of this technology when employed in conjunction with contemporary HPLC-ESI-MS methodology. Thus, using 4-bromoaniline as a model bromine-containing compound, it was possible to obtain mass balance data, as well as qualitative and quantitative metabolite profiles in rat bile and urine, without recourse to a radiolabeled analog as tracer. An interesting comparison was made of the profiles of brominated metabolites determined by HPLC-ICPMS using Br-selective detection and corresponding “bromine-isotopograms” extracted from the HPLC-oaTOFMS dataset on the basis of the presence of characteristic 79/81Br isotopic doublets; while excellent agreement was obtained between the two independent methods of analysis from a qualitative standpoint, only the ICPMS method provided reliable quantitative information since the ESI response factors of individual metabolites were not known. On the other hand, HPLC-oaTOFMS was necessary to identify metabolites of 4-bromoaniline, illustrating the complementarity of the two mass spectrometric techniques. ICPMS is a highly discriminating technique for the detection of bromine-containing compounds in biological fluids, since background levels of 79/81Br normally are very low, and the large negative mass defect of Br serves to minimize interference when bromine-containing ions are subjected to analysis by ESI-MS using high resolution mass analyzers. Other elements that may be detected by ICPMS include chlorine, iodine, and sulfur, rendering the technique broadly applicable to xenobiotic biotransformation studies, although an obvious requirement for detection of metabolites is that the tracer element in question is retained during the course of metabolism (e.g. oxidative debromination of a brominated drug should not be a major pathway of metabolism).
In view of the growing need to rapidly obtain quantitative metabolic profile information on new chemical entities, it may be anticipated that the combined use of HPLC-ICPMS and contemporary HPLC-ESIMS techniques will find increased application in drug metabolism and related fields since, for appropriate molecules, this approach is quantitative in nature and obviates the need for preparation of a radiolabeled tracer.
Figure 11. Structures of 4-bromoaniline and its major metabolites in rats.
Abstract
Source: Chemical Research in Toxicology 28:2112–2119, 2015
Synopsis: The study investigates the stereospecific clearance of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) using human liver microsomes and HEK293 cells overexpressing wild-type UGT1A9, UGT2B7, UGT2B10, and UGT2B17. NNAL is the metabolite of the most potent and one of the most abundant tobacco specific carcinogenic nitrosamines, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). NNAL exists in two enantiomeric forms which are both tumorigenic while their glucuronidated forms are not (Upadhyaya et al., Citation1999), with the S enantiomer being more carcinogenic in rats and mice than the R-enantiomer (Lao et al., Citation2007; Upadhyaya et al., Citation1999; Zhang et al., Citation2009). Due to this differential carcinogenic potential of the NNAL enantiomers, it is of interest to know which UGT enzymes target the specific NNAL enantiomers for glucuronidation. Kozlovich et al. developed a chiral separation method to isolate the NNAL enantiomers enabling the quantification of the enantiomeric preference for NNAL glucuronidation by different UDPG-glucuronosyl transferase (UGT) enzymes. The results revealed that variations in the expression or activity of specific UGTs may affect the clearance of the NNAL enantiomers known to induce tobacco related cancers.
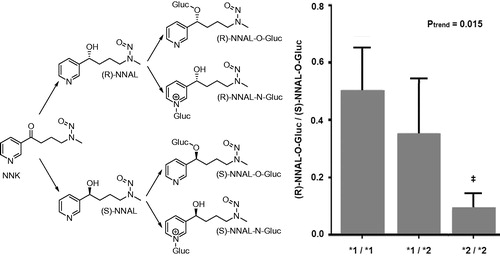
Commentary: Various factors may add to interindividual and interspecies differences in sensitivity towards carcinogens. The study reported by Kozlovich et al. focuses on NNK, the most potent and one of the most abundant nitrosamines in tobacco smoke and smokeless tobacco products. By measuring urinary NNAL in smokers it was estimated before that 39–100% of NNK was converted to NNAL (Carmella et al., Citation1993). It was also shown that NNAL comprised 82–89% of total NNK metabolites in human lung tissue (Richter, Citation2009). Since NNAL is considered to be equally carcinogenic as the parent compound NNK, glucuronidation of NNAL is considered an important detoxification route. NNAL can be conjugated with glucuronide at the oxygen atom of its carbinol group or at the nitrogen atom of its pyridine ring ().
The ratios of urinary levels of glucuronidated compared to unconjugated NNAL (Carmella et al., Citation1995; Hecht et al., Citation1993) and of O- compared to N-glucuronidated NNAL (Carmella et al., Citation2002) were reported to vary substantial, indicating that individual smokers vary greatly in their ability to detoxify NNAL. The study by Kozlovich et al. revealed that there were also substantial differences in the clearance of the S and R enantiomer of NNAL which may contribute further to the interindividual differences in sensitivity to this important tobacco specific carcinogen NNK. Especially UGTs 2B7 and 2B17 appeared to be involved in the glucuronidation of individual NNAL enantiomers, with both enzymes exhibiting high stereospecificity.
To further define the impact of changes in these enzyme activities between individuals for the potential cancer susceptibility more comprehensive studies are required on how changes in the expression and activity of these UGTs affect the production of NNAL glucuronide diastereoisomers and affect the level of free non conjugated NNAL in the target tissues with time and dose. It can be envisaged that the kinetic constants defined by Kozlovich et al. could be used to define the so-called physiologically based kinetic (PBK) models (Rietjens et al., Citation2011). Such PBK models would facilitate modeling of the time-, dose- and enzyme pattern-dependent changes in detoxification of the two NNAL enantiomers in vivo in different species including human. Such PBK models would also allow estimation of the interindividual differences in metabolism, predicting levels of unconjugated NNAL isomers expected in the target tissue of interest for the whole population using Monte Carlo modeling. Validation of the PBK model-based predictions using experimental data on NNAL metabolite patterns in experimental animals and human may validate the models and predictions made.
Figure 12. Regio- and stereoslective glucuronidation of NNAL and (R)-NNAL-O-Gluc/(S)-NNAL-O-Gluc ratio in incubations with human liver microsomes stratified by UGT2B17 genotype. *1 refers to the wild-type UGT2B17 allele; *2 refers to the UGT2B17 gene deletion allele. ‡, p = 0.012.
References
- Carmella SG, Akerkar S, Hecht, SS. (1993). Metabolites of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in smokers’ urine. Cancer Res 53:721–724.
- Carmella SG, Akerkar SA, Richie Jr. JP, Hecht SS. (1995). Intraindividual and interindividual differences in metabolites of the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in smokers’ urine. Cancer Epidemiol Biomarkers Prev 4:635–642.
- Carmella SG, Le K-A, Upadhyaya P, Hecht SS. (2002). Analysis of N- and O-glucuronides of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) in human urine. Chem Res Toxicol 15:545–550.
- Hecht SS, Trushin N, Reid-Quinn CA, et al. (1993). Metabolism of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in the patas monkey: Pharmacokinetics and characterization of glucuronide metabolites. Carcinogenesis 14:229–236.
- Lao Y, Yu N, Kassie F, Villalta PW, Hecht SS. (2007). Formation and accumulation of pyridyloxobutyl DNA adducts in F344 rats chronically treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem Res Toxicol 20:235–245.
- Richter E, Engl J, Friesenegger S, Tricker AR. (2009). Biotransformation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in lung tissue from mouse, rat, hamster, and man. Chem Res Toxicol 22:1008–1017.
- Rietjens IMCM, Louisse J, Punt A. (2011). Tutorial on physiologically based kinetic (PBK) modelling in molecular nutrition and food research. Mol Nutr Food Res 55:941–956.
- Upadhyaya P, Kenney PM, Hochalter JB, et al. (1999). Tumorigenicity and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol enantiomers and metabolites in the A/J mouse. Carcinogenesis 20:1577–1582.
- Zhang S, Wang M, Villalta PW, et al. (2009). Analysis of pyridyloxobutyl and pyridylhydroxybutyl DNA adducts in extrahepatic tissues of F344 rats treated chronically with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem Res Toxicol 22:926–936.
Abstract
Source: Chemical Research in Toxicology 28:831–837, 2015
Synopsis: Halogenated quinones are carcinogenic intermediates and disinfection byproducts in the production of drinking water (Qin et al., Citation2010; Zhao et al., Citation2010). In a previous study, the authors of this study demonstrated that halogenated quinones could react with H2O2 producing hydroxyl radicals in a metal-independent manner. The study reported by Huang et al. demonstrated that a similar mechanism can generate alkoxyl radicals (RO•) from a reaction between the halogenated quinone and an organic hydroperoxide (ROOH) without the need for a metal catalyst (). The study also provided a pathway for metal independent degradation of ROOH to genotoxic aldehydes like the conversion of the ROOH 13-hydroperoxy-9,11-octadecadienoic acid to 4-hydroxy-2-nonenal (HNE).
Commentary: Generation of hydroxyl radicals (•OH) and alkoxyl radicals (RO•) from H2O2 and organic hydroperoxides (ROOH), respectively, in metal-catalyzed reactions is well known. In the present study, the authors studied the reaction between 2,5-dichloro-1,4-benzoquinone (DCBQ) and tert-butylhydroperoxide (t-BuOOH) as the model reagents to sow that DCBQ could markedly enhance t-BuOOH decomposition and the accompanying generation of t-BuO•. The observation that metal chelators did not affect the reaction indicated that the reaction proceeds in a metal-independent fashion. These findings represent an unusual pathway for alkoxyl radical formation and the study also demonstrated that the reaction is also relevant for conversion of ROOH to genotoxic aldehydes like HNE. The authors indicate that this novel pathway may even in part explain the potential carcinogenicity of pentachlorophenol and other halogenated aromatic carcinogens when they are metabolized in vivo to halogenated quinone metabolites.
It should be noted, however, that the studies reported are limited to in vitro incubations in cell-free models. It therefore remains to be established, as also outlined by the study authors themselves, to what extent the novel pathway will occur and can be detected in biological model systems such as cells in vitro or animals in vivo. Furthermore, it remains to be established in kinetic experiments whether and to what extent the reaction of the halogenated quinones with ROOH can actually compete with their reaction with glutathione, which is likely to be present in much higher concentrations in cellular models than the ROOH. It also remains to be established whether the newly identified carbon-centered quinone ketoxy radical intermediates, or any other reaction products formed in the novel reaction pathway actually react with cellular macromolecules such as DNA, protein or lipids and cause damage.
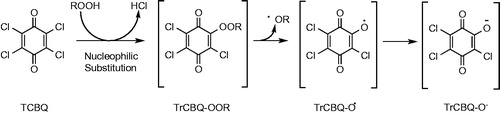
Figure 13. Proposed mechanism for •OH production by tetrachloro-1,4-benzoquinone (TCBQ) and organic hydroperoxides (ROOH), forming a trichlorohydroperoxyl-1,4-benzoquinone (TrCBQ-OOH) intermediate, which can decompose homolytically to produce the •OH and the trichloro-hydroxy-1,4-benzoquinone radical (TrCBQ-O•).
References
- Huang CH, Ren FR, Shan GQ, et al. (2015). Molecular mechanism of metal-independent decomposition of organic hydroperoxides by halogenated quinoid carcinogens and the potential biological implications. Chem Res Toxicol 28:831–837.
- Qin F, Zhao YY, Zhao YL, et al. (2010) A toxic disinfection by-product, 2,6-dichloro-1,4-benzoquinone, identified in drinking water. Angew Chem Int Ed 49:790–792.
- Zhao YL, Qin F, Boyd JM, et al. (2010) Characterization and determination of chloro- and bromo-benzoquinones as new chlorination disinfection byproducts in drinking water. Anal Chem 82:4599–4605.
Abstract
Source: Chemical Research in Toxicology 28:1760–1773, 2015
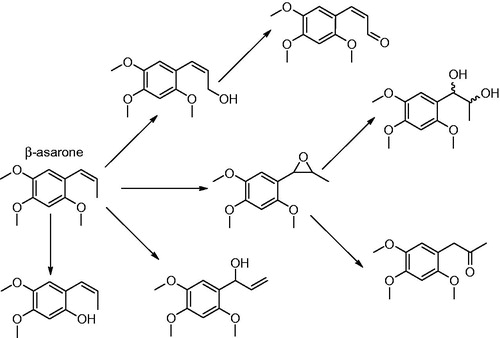
Synopsis: The manuscript reports the hepatic metabolism of the carcinogenic alkenylbenzene β-asarone. The authors performed incubations with liver microsomes of various species including rats, bovines, porcines and humans identifying the metabolites formed by 1H NMR, HPLC-DAD and LC-ESI-MS/MS. This is of interest because the metabolic pathways for β-asarone, including the pathway for its bioactivation to the ultimate carcinogenic metabolite, have so far not been elucidated (SCF, Citation2002; CEO 2005). The results presented suggest that the pathway for bioactivation of β-asarone is different from that of the structurally related genotoxic carcinogenic alkenylbenzenes safrole, methyleugenol and estragole for which bioactivation proceeds by cytochrome P450 mediated 1′-hydroxylation followed by sulfotransferase (SULT)-mediated formation of a reactive 1‘-sulfoxy metabolite which is unstable and able to form DNA adducts (Borchert et al., Citation1973; Drinkwater et al., Citation1976; Miller et al., 1976; Wislocki et al., Citation1976, Citation1977; Wiseman et al., Citation1985). In the present study, it was demonstrated that in incubations with human liver microsomes 71–75% of β-asarone is metabolized via epoxidation, 21–15% via hydroxylation with further oxidation and 8–10% via demethylation. Based on these results, the authors hypothesize that the genotoxic epoxides are the ultimate carcinogens formed from β-asarone.
Commentary: Previous studies have shown that the pretreatment of male mice with the SULT inhibitor pentachlorophenol (PCP) had no effect on the incidence of hepatocellular adenomas and carcinomas induced by β-asarone (Wiseman et al., Citation1987). This is in contrast to results obtained by the same authors for the related alkenylbenzene safrole, for which PCP significantly reduced induction of liver tumors (Boberg et al., Citation1983; Wiseman et al., Citation1987). This already indicated that a mechanism proceeding by side-chain 1‘-hydroxylation followed by sulfonation and formation of a DNA reactive 1‘-sulfoxy metabolite, seems not to be responsible for the carcinogenicity of β-asarone. The difference may be due to a difference in the isomerisation of the double bond in the allyl side chain resulting in safrole, methyleugenol and estragole being allyl alkoxybenzenes whereas β-asarone is a propenyl alkoxybenzene (Rietjens et al., Citation2014). In the microsomal incubations reported in the study by Cartus et al. formation of the epoxide and the diol and ketone metabolites derived from the epoxide was readily observed (). In addition formation of different side chain hydroxyl metabolites was reported including formation of the 1‘-hydroxymetabolite in which hydroxylation was accompanied by isomerisation of the double bond by an as yet unidentified mechanism (). In theory sulfation of this 1‘-hydroxymetabolite could result in a pathway for bioactivation similar to that for other alkenylbenzenes. In addition, metabolites resulting from O-demethylation were detected (). The kinetic data determined for the various metabolic conversions revealed epoxidation of the side chain to be by far the major metabolic route for cytochrome P450 catalyzed metabolism of β-asarone. The authors now conclude that the mutagenicity and carcinogenicity of β-asarone may be the result of the side chain epoxide rather than of a 1‘-sulfooxy metabolite. This conclusion and mechanism could be compared to the pathway for bioactivation of the propenyl alkoxybenzene anethole. Anethole also induces liver carcinogenicity in laboratory animals at high dose levels in the diet (Truhaut et al., Citation1989). These neoplastic effects of anethole have been related to continuous elevated tissue concentrations and resulting hepatotoxicity of its metabolite, anethole epoxide, that in turn leads to regenerative hyperplasia (Rietjens et al., Citation2014 and references therein).
This implies that given the results of the study reported by Cartus et al. it remains to be established whether the liver carcinogenicity of β-asarone is really due to the in vivo mutagenicity of the epoxide or rather to a non-genotoxic mode of action based on the hepatotoxicity of the epoxide metabolite. Thus, further studies, identifying the possible formation and nature of DNA adducts formed upon exposure of animals in vivo to β-asarone are essential to further elucidate the actual mode of action. The authors indicate that unpublished LC-MS/MS data provide spectroscopic evidence for the formation of a β-asarone-epoxide derived DNA adduct with deoxyguanosine formed in rat hepatocytes exposed in vitro to β-asarone, thus providing further support for this unique novel bioactivation pathway for β-asarone as compared to the other alkenylbenzenes. To actually solve the issue probably in vivo studies providing the conditions for continuous elevated tissue concentrations and resulting hepatotoxicity and regenerative hyperplasia may be required to establish whether DNA adduct formation or rather hepatotoxicity of the epoxide causes the liver tumors.
Figure 14. Hepatic metabolism of β-asarone. Formation of the epoxide is suggested to represent the bioactivation pathway to a (geno)toxic metabolite.
References
- Boberg EW, Miller EC, Miller JA, et al. (1983). Strong evidence from studies with Brachymorphic mice and pentachlorophenol that 1'-sulfooxysafrole is the major ultimate electrophilic and carcinogenic metabolite of 1'-hydroxysafrole in mouse liver. Cancer Res 43:5163–5173.
- Borchert P, Miller JA, Miller EC, Shires TK. (1973). 1'-Hydroxysafrole a proximate carcinogenic metabolite of safrole in the rat and mouse. Cancer Res 33:590–600.
- Cartus AT, Stegmüller S, Simson N, et al. (2015). Hepatic metabolism of carcinogenic β-asarone. Chem Res Toxicol 28:1760–1773.
- COE (Committee of Experts on Flavouring Substances of the Council of Europe). (2005). The 1st report on active principles (Constituents of Toxicological Concern) contained in natural sources of flavourings. Available from: http://www.coe.int/t/e/social_cohesion/soc%2Dsp/public_health/flavouring_substances/Active%20principles.pdf.
- Drinkwater NR, Miller EC, Miller JA, Pitot HC. (1976). Hepatocarcinogenicity of estragole (1-allyl-4-methoxybenzene) and 1’-hydroxyestragole in the mouse and mutagenicity of 1'-acetoxyestragole in bacteria. J Natl Cancer Inst 57:1323–1331.
- Rietjens IMCM, Adams TB, Bastaki M, et al. (2014). The impact of structural and metabolic variation on the toxicity and carcinogenicity of hydroxy- and alkoxy-substituted allyl- and propenylbenzenes. Chem Res Toxicol 27:1092–1103.
- SCF (Scientific Committee on Food). (2002). Opinion of the Scientific Committee on food on the presence of β-asarone in flavourings and other food ingredients with flavouring properties, SCF/CS/FLAV/FLAVOUR/9 ADD1 Final. Available from: http://ec.europa.eu/food/fs/sc/scf/out111_en.pdf.
- Truhaut R, Le Bourhis B, Attia M, et al. (1989) Chronic toxicity/carcinogenicity study of trans-anethole in rats. Fd Chem Toxico 27:11704–11720.
- Wiseman RW, Fennell TR, Miller JA, Miller EC. (1985). Further characterization of the DNA adducts formed by electrophilic esters of the hepatocarcinogens 1'-hydroxysafrole and 1'-hydroxyestragole in vitro and in mouse liver in vivo, including new adducts at C-8 and N-7 of guanine residues. Cancer Res 45:3096–3105.
- Wiseman RW, Miller EC, Miller JA, Liem A. (1987). Structure-activity studies of the hepatocarcinogenicities of alkenylbenzene derivatives related to estragole and safrole on administration to preweanling male C57BL/6J × C3H/HeJ F1 mice. Cancer Res 47:2275–2278.
- Wislocki PG, Borchert P, Miller JA, Miller EC. (1976). The metabolic activation of the carcinogen 1'-hydroxysafrole in vivo and in vitro and the electrophilic reactivities of possible ultimate carcinogens. Cancer Res 36:1686–1695.
- Wislocki PG, Miller EC, Miller JA, et al. (1977) Carcinogenic and mutagenic activities of safrole, 1'-hydroxysafrole, and some known or possible metabolites. Cancer Res 37:1883–1891.
Abstract
Source: Drug Metabolism Disposition 43:1267–1276, 2015
Abstract
Source: Drug Metabolism Disposition 43: 1277–1283, 2015
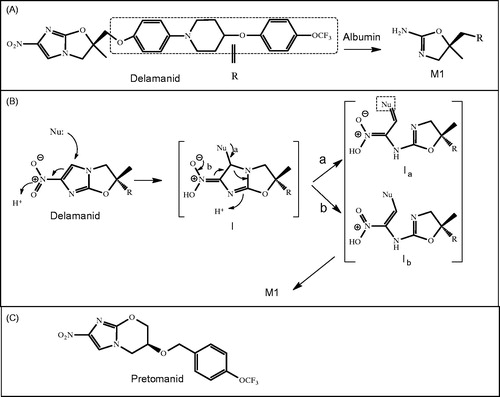
Synopsis: Delamanid is a new anti-tuberculosis (TB) drug for the treatment of multidrug-resistant (MDR)-TB and was approved in 2014 in the European Union, Japan and Korea. It is from a class of nitro-dihydro-imidazooxazole, and it works by inhibiting mycolic acid synthesis in the bacterium M. turberculosis (Matsumoto et al., Citation2006). In these articles, the authors have performed a number of mechanistic studies to better characterize the metabolism and pharmacokinetics of delamanid.
In human plasma, delamanid is rapidly metabolized to M1 by albumin with limited or no contribution from liver microsomes for catalyzing this reaction. The plasma albumin from various species was capable of cleaving the imidazole ring and forming 4,5-dihydrooxazol-2-amine M1. In human plasma, the apparent Km was 51.5 μM. The rate of this reaction is species dependent with a higher rate in human, followed by dog and much less in rodents. It is also the major metabolite found in human circulation. M1 in human is further metabolized to several oxidative metabolites, mainly by CYP3A4, to form several metabolites with the site of oxidation on the dihydrooxazole ring.
A couple of aspects of this reaction remain unclear. One unknown is the specific binding site on albumin that is responsible for M1 formation (). Additionally, the possible intermediates of this reaction have not yet been determined. M1 formation is speculated to proceed via a hard nucleophile such as a lysine or arginine on the albumin, followed by initial nucleophilic attack to the C-5 of the imidazole ring of delamanid (). This position in the imidazole is electron deficient due to its proximity to C-4, which is attached to a nitro group. The reaction then proceeds through a hydrolytic step to finally release M1.
Commentary: The investigators were able to unravel an interesting reaction involving metabolism of delamanid through albumin to form M1. Several reports on albumin-catalyzed reactions exist, but they mostly involve simple hydrolytic reactions such as ester hydrolysis, as is the case with N-trans-cinnamoylimidazoles (Ohta et al., Citation1983). The mechanism of formation of M1 is not straightforward, and multiple steps, which are not yet fully characterized, are assumed to be involved. The investigators speculate that the initial step is nucleophilic attack on the C-5 of the imidazole ring. No intermediates were characterized to confirm this; however, in tests utilizing chemical analogs of delamanid, the absence of a nitro group prevented formation of M1 (data not shown). Based on this information, it is reasonable that C-5 is the site of nucleophilic attack. In order for the initial step of the reaction to occur at C-5 (), the aromaticity of the imidazole must be interrupted, meaning that relatively high activation energy is required. However, the newly formed intermediate has several double bond conjugates, which perhaps lowers the activation energy and allows the reaction to proceed (intermediate I). This reaction proceeds further to cleave the C-N bond of the imidazole (intermediates Ia and Ib). The investigators propose that either Ia accepts a lone pair of electrons from the amine on lysine (pathway a) or the nitro group donates electrons to cleave the C-N bond as shown in pathway b. Finally attack of these intermediates by one or more water molecules would release M1. The fragment that is released from albumin remains unknown, but this could reveal where the attack of the water molecule takes place.
The identity of the amino acid on albumin that is involved in M1 formation has not been determined; however, the authors speculate that either lysine or arginine may be involved. This is indirectly supported by two observations: (1) M1 is formed in the presence of 25% ammonia or alkylamines and (2) the rate of formation of M1 in human albumin increased at higher pH. In the latter experiment, delamanid was stable in the presence of albumin at pH 6, yet a much higher conversion to its metabolite was seen at pH 8. These results are consistent with other published studies. Though lysine and arginine are plausible candidates, one cannot ignore the possible involvement of cysteine-34 (Cys-34) on albumin. Cys-34 is reported to have a wide range of pKa values, between 5 and 8, depending on the analytical tools used (Noble & Williams, Citation2001). Cys-34 could possibly be involved in this reaction considering that a soft electrophile can also be incorporated in the initial Michael’s acceptor. There are several ways of testing this hypothesis to better understand the amino acid involved; one is to selectively derivatize the exposed amino acids on albumin prior to incubation with delamanid. If no structural changes on albumin took place, then changes to the rate of M1 formation due to derivatization could indicate the amino acid involved in the metabolic reaction.
The investigators demonstrated that the rate of hydrolysis in vitro in plasma or albumin is much higher than that reported based on in vivo plasma half-life. The metabolic half-life of delamanid in vitro is less than 1 hour in diluted human plasma (reported as 0.64 hour at 37oC). However the plasma half-life seems to be much longer in vivo and is in the range of many hours. In preclinical species, the in vitro half-life is longer, but not by much. In dog, this value is 0.84 hours and in mouse it is 1.9 hours. The terminal half-life in dog is 18.4 hours and in mouse it is 7.2 hours. These values are puzzling considering the in vitro data. The lack of stability in vitro is the reason that the plasma protein binding studies were performed at a decreased temperature in order to minimize conversion. If the plasma could be so active in catalyzing this reaction, how could the drug last in circulation so long? It is possible that the in vivo concentrations are much lower and in vitro, and this may explain the discrepancies.
Another consideration about catalytic reactions by albumin is that the intermediate formed may be slowly and thus not completely released. This means that it is possible that after M1 is released that there are still conjugates covalently bound to the amino acid on albumin (be it lysine or cysteine). If the release is slow, it is possible that the same amino acid may not be able to perform this hydrolysis. Depending on the rates, if it is very slow (hours to days), this may result in changes in rates of M1 following consecutive exposure to delamanid. Considering that there are high concentrations of albumin in plasma (in millimolar ranges) and also possibly several amino acids on each albumin molecule responsible for this reaction, it is also possible that a slow release may not result in slowing down the rate of delamanid degradation in vivo, while this could be observed in vitro.
As described, human liver microsomes have limited or no capacity for the formation of M1 and most likely plasma is the main component for the formation of this metabolite. This means that there should be little concern with co-medication with other drugs that may inhibit P450 or in the case of rifampicin induction of CYP3A4. In a clinical drug–drug interactions study, this result was confirmed with no change to delamanid exposure upon treatment with a weak P450 inducer such as efavirenz (600 mg/day) for 10 days in combination with delamanid 100 mg twice daily. However, in the case of a strong P450 inducer RIF (300 mg), delamanid exposure dropped by 45% (EMA summary; Lewis and Sloan 2015). The RIF results may suggest that there is a significant P450 component for the clearance of delamanid unless RIF could interact directly at the plasma level as well. At this time based on the information given it is not possible to know the reasons behind the drop in delamanid exposure in the presence of RIF. What will help with this assessment is knowing the circulating levels of M1 and perhaps other oxidative metabolites.
5-Nitroimidazole is considered by some medicinal chemists especially for antiprotozoal and antibacterial properties. For example, a close analog of delamanid containing nitro-imidazo-dihydro-oxazole is now in clinical trial called pretomanid (PA-824; ) (Olaru et al., Citation2014). It is thought to mainly be eliminated by P450. For pretomanid, it is important to consider the P450-mediated DDI. There are clinical studies underway to specifically address this.
Figure 15. (A) Catalytic conversion of delamanid to M1 by albumin, (B) proposed mechanism of this reaction proceeding via a nucleophile (Nu) from albumin, and (C) the chemical structure of pretomanid.
References
- Matsumoto M, Hashizume H, Tomishige T, et al. (2006). OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med 3:e466.
- Ohta N, Kurono Y, Ikeda K. (1983). Esterase-like activity of human serum albumin II: reaction with N-trans-cinnamoylimidazoles. J Pharm Sci 72:385–8.
- Olaru ID, von Groote-Bidlingmaier F, Heyckendorf J, et al. (2014). Novel drugs against tuberculosis: a clinician’s perspective. Eur Respir J 45:1119–1131.
- EMA summary. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002552/WC500166232.pdf)
- Noble DR, Williams DLH. (2001). Formation and reactions of S-nitroso proteins. J Chem Soc Perkin Trans 2:13–17.
Abstract
Source: Drug Metabolism Disposition 43:1929–1933, 2015
Synopsis: The investigators previously reported the formation of two unusual metabolites, M13 and M14, in multiple species from the MAPK/ERK inhibitor GDC-0623, which is mainly cleared by conjugation via glucuronides. M14 is formed at a higher level than M13, and in rat and dog excreta, M14 is mainly found in its glucuronide form (M2) at 7% and 1% of the administered dose, respectively. Though these levels were modest in preclinical species, the circulating levels of M2 and M14 were estimated (not based on authentic standards) to be 10% of the parent in circulation in Phase 1 clinical studies, and therefore further investigations were warranted.
Based on interpretation of mass spectrometry fragments, it was clear that both M13 and M14 had lost the alkyl side chain in addition to a loss of 2 Da as a result of ring formation. For M14, NMR was useful to confirm the presence of a newly formed pyrazol-3-ol ring. The structure of M14 was also conducive toward performing a deuterium exchange experiment, which led to an increase of 2 Da from one exchangeable hydrogen plus one proton from the mass spectrometric ionization. M13 was less conducive toward use of these techniques since the proposed structure included an imidazole-2-one, which requires the breaking and forming of several bonds. For this reason, an authentic standard was synthesized to confirm the structure of M13.
Both M13 and M14 were reported to be formed by P450 enzymes in human liver microsomes. M14 formed directly from GDC-0623, but M13 formed from M15, an amide metabolite. In both cases, CYP2C19 was the major enzyme involved in the formation of these metabolites in human, followed by CYP2C9. In the case of M14 formation from GDC-0623, the authors speculated that the aniline amine is the nucleophile that attacks the nitrogen atom of hydroxamate to form the pyrazol-3-ol. In order for this to happen, the alkoxyl of the hydroxamate needs to be a good leaving group, and, for this reason, the investigators proposed that cytochrome P450 oxidation occurs at the C-1 position of the side chain.
In the case of M13 formation from the amide metabolite M15, the synthesis of imidazole-2-one requires breaking one C-C bond and forming two new C-N bonds. For this reason, the investigators hypothesized that the reaction mechanism involves a Hofmann-type degradation. The aniline is proposed to undergo oxidization to form the intermediate, IB1 (). This is followed by abstraction of a hydride from the amide nitrogen, which is the driving force for breaking the C-C bond and forming the C-N bonds. The newly formed formamide intermediate, IB2, is then attacked by aniline nitrogen to form M13.
Commentary: MEK inhibitors are a new class of kinase inhibitors in oncology that inhibit the MAPK/ERK pathway and are represented by the approved drug entities trametinib (GSK) and cobimetinib (Genentech/Roche). GDC-0623 is an experimental MEK inhibitor that made it to the clinical stage. Like many MEK inhibitors, such as cobimetinib, GDC-0623 has a dihalogenated aniline attached to an aromatic core plus an amide or hydroxamate side chain.
In the case of GDC-0623, the investigators reported on two novel metabolites, M13 and M14. Though these are minor contributors to the total clearance of this compound in rat and dog, in human, M14 and M2 (its glucuronide conjugate) contribute up to about 10% of parent in circulation. These metabolites were not expected to retain any pharmacological activities against MEK, however, the presence of these metabolites in the circulation triggered further studies to elucidate their structures and understand the mechanisms of their formation.
M13 and M14 are isomeric metabolites that have formed a new fused ring. Based on molecular weights, the ethane-1,2-diol side chain was lost and the new fused ring was formed from the amide moiety. Though the numbers of available isomeric structures are limited, the metabolites had to be confirmed by NMR and synthetic standards. The studies indicated the newly formed fused rings are a pyrazol-3-ol in the case of M14 and an imidazole-2-one in the case of M13.
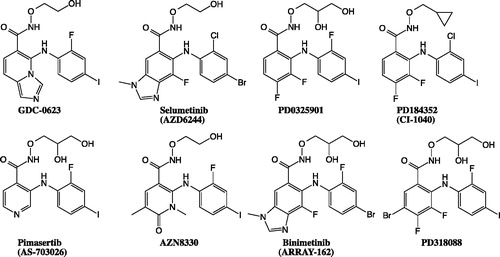
The reaction mechanisms proposed by the investigators are reasonable, and each of the metabolites are formed from different starting material. M14 is generated from GDC-0623 and M13 is generated from the amide metabolite, M15. Interestingly, both of these metabolites are formed by CYP2C enzymes. A more detailed study of the docking poses of these probes at the active sites of CYP2C19 and CYP2C9 could inform our understanding of the reaction mechanisms. Several MEK inhibitors have hydroxamate side chains, similarly to GDC-0623. Six of them are shown in . Based on the proposed mechanisms, some of these inhibitors could also possibly form fused ring metabolites. CI-1040 is the only compound on this list that has undergone metabolism studies; however, in this case, no such fused ring metabolites were reported. The dihalogenated combination on aniline may affect metabolism, and further study is necessary to better understand the mechanism of fused ring formation.
Figure 16. Proposed reaction mechanisms for the formation of M14 and M13 from GDC-0623.
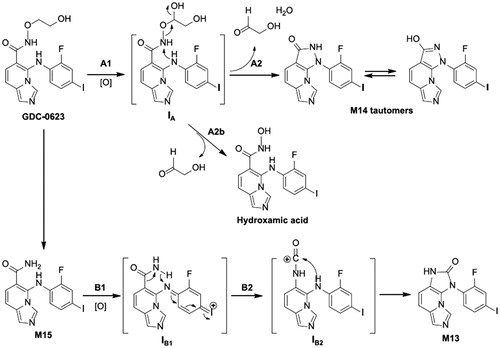
Figure 17. The structure of MEK inhibitors that include hydroxamate side chains, similarly to GDC-0623.
Reference
- Wabnitz PA, Mitchell D, Wabnitz DA. (2004). In Vitro and in vivo metabolism of the anti-cancer agent CI-1040, a MEK inhibitor, in rat, monkey, and human. Pharm Res 21:1670–1679.
Abstract
Source: Drug Metabolism and Disposition 43:908–915, 2015
Synopsis: In the clinic, GDC-0834 (a BTK inhibitor), had poor oral systemic exposure, most likely due to high metabolic clearance via amide hydrolysis (). Interestingly, aldehyde oxidase (AO) was determined to be the enzyme responsible for this metabolic reaction: hydrolysis by AO is a novel reaction that has not been previously reported. A series of in vitro studies using AO inhibitors and substrates confirmed the contribution of AO. In human liver cytosol, the Michaelis-Menten kinetic values of the hydrolytic reaction were measured, yielding a Km of 0.8 μM and a Vmax of 409 pmol/min/mg protein. Several AO inhibitors inhibited this reaction with single digit micromolar IC50 values. On the other hand, the IC50 values of GDC-0834 in human liver cytosol against multiple AO substrates was in the range of 0.33 μM for raloxifene to 2.14 μM for DCPIP.
The specific contribution of AO versus carboxylesterase (CES) could not be clearly dissected. Interestingly, dog liver cytosol was also capable of hydrolyzing GDC-0834, yet it is known that dog does not have active AO. After further investigations, the main enzyme responsible for this dog liver catalysis was determined to be CES. This result was confirmed using CES and AO inhibitors. AO inhibitors diminished the hydrolysis of GDC-0834 in human liver cytosol; however, AO inhibitors did not modulate the metabolism of GDC-0834 in dog liver cytosol. Another interesting finding by the investigators was that in human plasma the overall hydrolytic conversion capability for GDC-0834 was limited. Human plasma is known to have some CES activity but no AO activity, and, consistent with this, AO inhibitors did not change the rate of the reaction in human plasma. However, CES inhibitors significantly decreased this reaction in plasma.
Though the investigators demonstrated the involvement of AO in this hydrolytic reaction, the exact mechanism is not yet elucidated. The oxidative reaction by this enzyme is typically initiated by a hydroxymolybdenum species attacking an electrophilic sp2 carbon center (Barr et al., Citation2014). The investigators speculated that the same nucleophilic center might be involved in the hydrolysis; however, since an amide bond is less prone to nucleophilic attack, the investigators propose that the molybdenum center is in a reduced form (MoIV) as opposed to the oxidated form (MoVI) that is present when AO is involved in oxidation ().
Commentary: The discovery of a new activity for an enzyme that has been known for decades is not common. The earliest studies on characterizing AO date back to 1940 with the isolation of AO from pig liver (Gordon et al., Citation1940). AO is involved in aldehyde oxidation; however, whether this is the most important enzyme for this type of reaction is unclear (Sonah et al., Citation2015). The enzyme is also involved in other oxidation and reduction reactions including the reductase activity of sulfoxide, N-oxides and oxazole plus the oxidation of iminium ions. Now hydrolysis can be added to the list of activities for this enzyme. The oxidation of azaheterocyclic compounds has recently been the focus of research activities in the pharmaceutical industry (Garattini and Terao, Citation2013). This oxidation is mediated by AO at the carbon atom in the adjacent or para position to the nitrogen.
As for the mechanism of this reaction, the investigators speculate that the hydrolysis takes place at the active site using a reduced form of hydroxymobdenum, namely MoIV. Unlike the oxidation process mediated by AO, the hydrolysis should not require an electron transfer through iron-sulfur clusters to a flavin center. This should be a testable hypothesis. It is possible that perturbation to the electron transfer may not impact hydrolysis but would block oxidation and reduction reactions mediated by this enzyme.
Finally, the investigators discovered an enzymatic role that has gone undetected before. In the case of GDC-0834 hydrolysis to M1, it is clear that AO is playing a significant role. There seems to be some synergy between AO and CES for this reaction that makes it difficult to dissect the relative contribution of each enzyme. The number of different drugs that are hydrolysed by AO is unknown, but AO might play a role in the clearance of many drugs other than the oxidation and reduction reactions that have been documented previously.
Figure 18. Aldehyde oxidase hydrolysis of GDC-0834 to M1 and M2.
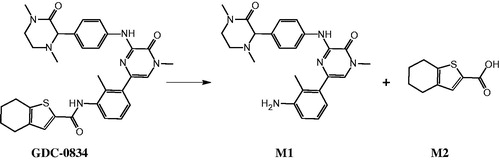
Figure 19. Proposed reaction mechanism for hydrolysis of GDC-0834 by aldehyde oxidase (AO).
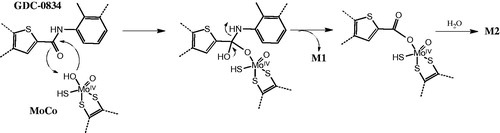
Disclosure statement
The authors report no conflicts of interest.
References
- Barr JT, Choughule K, Jones JP. (2014). Enzyme kinetics, inhibition, and regioselectivity of aldehyde oxidase. Methods Mol Biol 1113:167–186.
- Garattini E, Terao M. (2013). Aldehyde oxidase and its importance in novel drug discovery: present and future challenges. Expert Opin Drug Discov 8:641–654.
- Gordon AH, Green DE, Subrahmanyan V. (1940). Liver aldehyde oxidase. Biochem J 34:764–74.
- Sanoh S, Tayama Y, Sugihara K, et al. (2015). Significance of aldehyde oxidase during drug development: Effects on drug metabolism, pharmacokinetics, toxicity, and efficacy. Drug Metab Pharmacokinet 30:52–63.