
Abstract
Pharmacogenetic research has resulted in the identification of a multitude of genetic variants that impact drug response or toxicity. These polymorphisms are mostly common and have been included as actionable information in the labels of numerous drugs. In addition to common variants, recent advances in Next Generation Sequencing (NGS) technologies have resulted in the identification of a plethora of rare and population-specific pharmacogenetic variations with unclear functional consequences that are not accessible by conventional forward genetics strategies. In this review, we discuss how comprehensive sequencing information can be translated into personalized pharmacogenomic advice in the age of NGS. Specifically, we provide an update of the functional impacts of rare pharmacogenetic variability and how this information can be leveraged to improve pharmacogenetic guidance. Furthermore, we critically discuss the current status of implementation of pharmacogenetic testing across drug development and layers of care. We identify major gaps and provide perspectives on how these can be minimized to optimize the utilization of NGS data for personalized clinical decision-support.
Introduction
Genetic variation in genes encoding drug targets or enzymes and transporters involved in drug disposition have long been considered as promising biomarkers to predict toxicity and identify patients that will benefit most from the therapy in question. Germline variations, i.e. inherited variants that are passed on to offspring, are mainly used to predict drug pharmacokinetics whereas somatic mutations, i.e. variants that change the DNA sequence of a somatic cell but are not inherited and not passed on to offspring, guide therapy selection in oncology. In recent years, a plethora of studies have described pharmacogenetic associations and as of February 2021, at least 82 and 91 drugs carry actionable germline and somatic biomarkers, respectively (Table of Pharmacogenomic Biomarkers in Drug Labeling. FDA Citation2021). The testing of somatic variations has become increasingly common in routine clinical care, often in the form of companion diagnostics; in contrast, the clinical implementation of most germline biomarkers lags behind and <10% of patients who are prescribed a medication that contains germline pharmacogenomic labeling receive preemptive testing (Young et al. Citation2021). So far, only one variant allele requires preemptive testing (HLA-B*57:01 for abacavir), while screening for a few additional variants is mandated only for certain ethnogeographic groups (e.g. HLA-B*15:02 for carbamazepine in patients of South East Asian descent). Furthermore, certain variants with mounting evidence of their clinical utility and cost-effectiveness might soon be incorporated into routine testing prior to initiation of therapy, including reduced function alleles in DPYD and TPMT for fluoropyrimidine and thiopurine toxicity, respectively.
Early successes of pharmacogenomics were made possible using forward genetics, in which studies aimed to identify genetic differences that might explain a given phenotype. However, this approach proves difficult for rare phenotypes and for complex genetic associations that comprise a multitude of variants with individually small effect sizes. Recent advances in sequencing technologies have opened new possibilities for reverse genetics, in which large-scale genetic data forms the basis for functional studies. In this review, we provide an updated overview of current pharmacogenetic biomarkers of clinical relevance, highlight the advantages and limitations of emerging sequencing methods, and discuss how the resulting genomic datasets can facilitate precision medicine in clinical care and drug development.
Key examples of germline pharmacogenomic biomarkers
Germline variations in genes that are involved in drug pharmacokinetics and pharmacodynamics (PK/PD) and drug-induced immunological responses are estimated to explain approximately 20–40% of the interindividual variability in drug response and toxicity (Sim et al. Citation2013; Clarke and Cherrington Citation2012; Davies and O'Mahony Citation2015; Lauschke and Ingelman-Sundberg Citation2016). In past decades, numerous genetic variants have been identified that can serve as germline pharmacogenomic biomarkers () and it has been estimated that these germline variants could have an overall effect on PK or drug response for 18% of all outpatient prescriptions (Relling and Evans Citation2015). Many of these biomarkers localize to genes encoding cytochrome P450 (CYP) drug metabolizing enzymes, which account for 80% of phase I drug metabolism (Zhou et al. Citation2017). Well-established examples include duplications of the functional CYP2D6 gene, which are associated with codeine intoxication (Gasche et al. Citation2004), the decreased function alleles CYP2C9*2 and *3, which are associated with the requirement for warfarin dose adjustments (Aithal et al. Citation1999), and the loss-of-function allele CYP2C19*2, which causes reduced clopidogrel bioactivation and poorer cardiovascular outcomes in patients undergoing percutaneous coronary intervention (Shuldiner, Citation2009). Furthermore, clinically relevant associations have been identified between CYP2C19 genotypes and exposure as well as therapeutic failure rates of the antidepressants sertraline and escitalopram (Jukić et al. Citation2018; Bråten et al. Citation2020).
While most of the currently identified pharmacogenomic germline biomarkers affect CYP genes, variations in other phase I and phase II drug metabolism genes as well as in genes encoding drug transporters can also be useful to guide drug or dose selection. Well-established examples include DYPD reduced function alleles (mainly HapB3, *2 A or D949V) as biomarkers of capecitabine, fluorouracil, and tegafur toxicity in cancer patients (Van Kuilenburg et al. Citation2000), UGT1A1 promoter polymorphisms for irinotecan-induced myelosuppression and neutropenia (Innocenti et al. Citation2004), as well as the reduced function variant rs4149056 (part of SLCO1B1*5 and SLCO1B1*15) that is associated with simvastatin-induced myopathy and rhabdomyolysis (SEARCH Collaborative Group et al. Citation2008). Furthermore, emerging data suggest that genetic variation in SLC22A1 (encoding the OCT1 transporter) constitutes an important contributor to the interindividual variability in efficacy of opioids and other analgesics (Tzvetkov Citation2017).
Compared to variation in genes involved in drug disposition, the importance of pharmacodynamic variability (i.e. the genetic variation in drug target genes) on interindividual drug response is overall less-well understood. Seminal work on G protein-coupled receptors (GPCRs), one of the most common drug target categories, demonstrated that naturally occurring variants in this gene family can have pronounced effects on drug response, causing biased downstream signaling or drug resistance in a ligand-specific way (Hauser et al. Citation2018). In addition, several studies have explored genetic variability in select drug targets and discussed their population differences and predicted functional consequences (Nelson et al. Citation2012; Dewey et al. Citation2016; Schärfe et al. Citation2017; Yan et al. Citation2017). To systematically evaluate effects of drug target variability on drug response to the US Food and Drug Administration (FDA)-approved drugs, variant data from 138,632 individuals was recently mapped onto all available drug target crystal structures (Zhou et al. CitationUnder consideration). This analysis revealed that one in six individuals carried at least one variant that affected an amino acid in the binding pocket in close proximity (<6Å) to the bound drug. Furthermore, the predicted effects on drug function were experimentally confirmed using three targets from cardiology, oncology and neurology, demonstrating the functional importance of rare variants located in drug-target binding sites, further incentivizing the utilization of such information for guiding personalized drug selection and drug development.
In addition to variations that affect drug response by altering drug binding sites, germline variants can alter the pharmacodynamics of drugs developed for specific congenital disease mutations. Arguably, the most prominent example is ivacaftor, which improves functions of the cystic fibrosis transmembrane conductance regulator (CFTR, encoded by ABCC7) in patients with cystic fibrosis. Cystic fibrosis is caused by genetic variants in the ABCC7 gene that cause misfolding or mislocalization of the transporter and reduced CFTR function. Ivacaftor augments transporter function of CFTR in patients carrying the reduced function variant CFTRG551D (Davies and O'Mahony Citation2015; Young et al. Citation2021). In contrast, ivacaftor is not effective in homozygous CFTRF508del carriers, as the F508del mutation abrogates CFTR localization to the plasma membrane, thus rendering cells unresponsive to the effects of CFTR potentiators (Flume et al. Citation2012). Instead, detrimental effects of CFTRF508del can be rescued using the chemical chaperone lumacaftor, which reduces arrest of the variant protein in the endoplasmic reticulum, restoring its expression at the plasma membrane and allowing for the rescued mutant transporters to subsequently respond to ivacaftor (Van Goor et al. Citation2011).
Recently, a two-stage genome-wide association study (GWAS) identified the intronic variant rs2205986 as a strong risk factor of interferon-β (IFN-β)-induced liver injury (OR = 8.3 irrespective of the co-variants adjustment), an adverse drug reaction commonly observed in IFN-β-treated multiple sclerosis patients (Kowalec et al. Citation2018). The authors demonstrated that rs2205986 is a useful germline biomarker that is predictive of liver injury, similar to elevations in levels of circulating aspartate aminotransferase and alanine aminotransferase. Thus, this finding suggests that preemptive genotyping of rs2205986 might constitute a promising method to stratify patients for multiple sclerosis treatment and minimize IFN-β-induced liver injury.
Human leukocyte antigen (HLA) genes are extremely polymorphic and a large number of HLA variants have been identified as germline biomarkers for drug hypersensitivity (Lauschke et al. Citation2019). The most well-established clinical associations include HLA-B*57:01 with abacavir-induced hypersensitivity syndrome (Mallal et al. Citation2008), HLA-B*15:02 and HLA-A*31:01 with carbamazepine-induced Stevens–Johnson syndrome and toxic epidermal necrolysis (Chung et al. Citation2004; McCormack et al. Citation2011) and HLA-B*58:01 with allopurinol-induced severe cutaneous adverse reactions (Hung et al. Citation2005). Notably, HLA biomarkers are being increasingly discovered. One recent GWAS identified the intronic variant rs114892859, a tag SNP of HLA-B*55:01, as a predictor of penicillin allergy in a European population (OR = 1.47, p = 1.29*10−29; Krebs et al. Citation2020). Furthermore, the direct association between HLA-B*55:01 and penicillin allergy was corroborated in another independent cohort containing more than 1 million subjects of European ancestry (OR = 1.3, p = 1*10−47). Taken together, these findings strongly suggest that HLA-B*55:01 can identify individuals with penicillin allergy among Europeans. However, as the odds ratios are relatively low, the value of the clinical implementation of HLA-B*55:01 testing remains to be determined.
Another GWAS associated HLA-DQA1*05 with the formation of antibodies against the tumor necrosis factor (TNF) inhibitors infliximab and adalimumab, which can result in resistance against these treatments in patients with Crohn’s disease (Sazonovs et al. Citation2020). After 2 years of follow-up, the number of patients that developed anti-drug antibodies was significantly higher in the HLA-DQA1*05 carriers compared to non-carriers (p = 5.88*10−13). The authors demonstrated that HLA-DQA1*05 status, together with other factors such as sex and immunomodulator use, could explain 18% of the variability in immunogenicity to anti-TNF treatment.
Key examples of somatic pharmacogenomic biomarkers
Somatic mutations are not passed on to progeny and can occur in any cell. If such mutations occur directly within the gene body or regulatory region of a so-called driver gene, they can contribute to oncogenesis (Martincorena and Campbell Citation2015). Importantly, the variant proteins resulting from somatic mutations constitute common targets in oncological therapy, including but not limited to hematological disorders, non-small cell lung carcinomas (NSCLC), and breast cancer.
Hematological disorders
Overall survival from hematological malignancies has been improving in both pediatric and adult patients (Pui et al. Citation2019; Pulte et al. Citation2020). In part, these developments can be attributed to the evolution of sequencing technologies and the discovery of new somatic mutational targets. Imatinib was the first drug in a long list of small molecule tyrosine kinase inhibitors (TKIs) that target the oncogenic BCR-ABL chimeric tyrosine kinase (Rossari et al. Citation2018). BCR-ABL is the result of the t(9;22) chromosomal translocation known as Philadelphia chromosome. Despite the hopes that imatinib brought to leukemia patients, drug resistance emerged in almost one third of patients with high-risk chronic myeloid leukemia (Hasford et al. Citation2011). Resistance mechanisms are heterogeneous and involve altered cancer cell drug disposition resulting from reduced OCT1-mediated drug uptake or increased drug efflux due to elevated MDR1 activity, BCR-ABL hyperexpression, and the acquisition of escape mutations that render the fusion kinase itself resistant to imatinib (Hochhaus et al. Citation2002; Thomas et al. Citation2004). Resistance mechanisms are intrinsic or they are acquired after treatment. In acquired resistance, BCR-ABL escapes inhibition due to the accumulation of additional point mutations, most commonly T315I, that confer resistance to treatment with conventional TKIs. This resulted in the introduction of second and third generation of TKIs like dasatinib, nilotinib, and ponatinib, which have higher binding affinity to the mutant forms of BCR-ABL. Somatic mutations play increasingly important roles in the myeloid system, as exemplified by mutations in TP53 that can serve as independent predictors of overall survival in myelodysplastic syndrome (Crisà et al. Citation2020). Furthermore, somatic mutations are believed to precede the occurrence of acute myeloid leukemia, thus paving the path for future surveillance tools in hematological malignancies (Desai et al. Citation2018).
Lung cancer
The use of somatic pharmacogenomic biomarkers is arguably most extensively established for epidermal growth factor receptor (EGFR) inhibitors in NSCLC. EGFR is a receptor tyrosine kinase that upon binding of its ligand, dimerizes and autophosphorylates tyrosine residues in its intracellular domain, which enables downstream signaling via the RAS-RAF-MEK-MAPK and PI3K-PTEN-AKT axes (da Cunha Santos et al. Citation2011). Importantly, the kinase activity of EGFR can be affected by oncogenic mutations in the EGFR tyrosine kinase domain, resulting in ligand-independent EGFR activation and subsequent suppression of cancer cell apoptosis (Bethune et al. Citation2010). Overall, such somatic pharmacogenomic markers in EGFR are prevalent in 15–50% of NSCLC patients (Chan et al. Citation2019). Gefitinib and erlotinib were the first orally active EGFR inhibitors approved for treatment of locally advanced or metastatic NSCLC after failure of at least one prior chemotherapeutic regimen. Additional EGFR inhibitors have been approved in recent years, including afatinib (FDA approval: 2013) and dacomitinib (FDA approval: 2018). Importantly, however, cancers frequently acquire resistance to these medications by accumulation of further mutations. To counter these resistance mechanisms, mutation-specific EGFR inhibitors that specifically target emerging resistance mutations have recently been approved. Specifically, osimertinib significantly improved progression free survival in T790M–positive NSCLC patients who had failed first line therapy compared to platinum–pemetrexed therapy (hazard ratio = 0.3; p < 0.001) (Mok et al. Citation2017).
In addition to EGFR mutations, genomic rearrangements resulting in gene fusions of the receptor tyrosine kinase ALK and EML4 are common, in which the amino terminal of the microtubule associated protein EML4 fuses to the intracellular kinase domain of ALK, occurring in 4–6% of NSCLCs, primarily adenocarcinomas (Pikor et al. Citation2013). Constitutive dimerization of EML4-ALK mediated by a dimerization motif of EML4 results in enhanced kinase activity that results in hyperactivation of RAS/MAPK, PI3K/AKT, and JAK/STAT signaling (Sabir et al. Citation2017). Several approved drugs target ALK rearrangement, including crizotinib, ceritinib, alectinib, brigatinib, and loralitnib, which act by binding to the ATP-binding pocket of ALK and subsequently prevent autophosphorylation required for enzyme activation.
Besides EGFR mutations and ALK rearrangements, somatic mutations of the serine-threonine protein kinase BRAF constitute important therapeutic targets for NSCLC in 3% of patients with V600E being the most common mutation (Gautschi et al. Citation2015). The mutated BRAF phosphorylates and activates MEK/MAPK signaling stimulating cell proliferation. In NSCLC, the combination of BRAF inhibitors (dabrafenib) and MEK inhibitors (trametinib) was able to overcome resistance associated with the use of single agent therapy and provided a clinically meaningful response and safety outcomes in patients with BRAFV600E mutation (Planchard et al. Citation2017; Khunger et al. Citation2018).
Breast cancer
Breast cancer is commonly categorized into four intrinsic subtypes: (i) luminal A, (ii) luminal B, (iii) HER2 positive, and (iv) triple negative. Luminal A and B are usually estrogen and/or progesterone positive, whereas HER2 positive tumors are typically hormone-receptor negative and tend to have worse prognoses (Harbeck et al. Citation2019). Triple negative breast cancer is negative for both hormone-receptors and HER2 and is usually associated with BRCA1 mutations (Chen et al. Citation2018).
The somatic mutation profile in breast cancer is heterogenous with mutations most commonly found in PIK3CA and TP53, of which the former is associated with hormone receptor positive cancers, whereas somatic TP53 mutations are mostly found in HER2 positive tumors and tumors with BRCA1 mutations (Pereira et al. Citation2016; Li et al. Citation2018). Furthermore, breast cancers commonly feature somatic hyperactivation of cyclin dependent kinases (CDKs), particularly in hormone receptor positive tumors where estrogen increases the rate of cell cycle progression (Spring et al. Citation2019). This has led to the development of the specific CDK4/6 inhibitors ribocilib (Hortobagyi et al. Citation2016), palbociclib (Finn et al. Citation2015) and abemaciclib (Johnston et al. Citation2020). Lapatinib is another example of a small molecule that specifically targets HER1 and HER2 and is approved by the FDA for use in previously treated metastasized HER2 positive breast cancers in combination with capecitabine (Geyer et al. Citation2006). Luminal HER2 positive breast cancer typically benefits from a combination neoadjuvant therapy of trastuzumab and laptinib (Xu et al. Citation2017).
Triple negative breast cancer constitutes the most aggressive subtype with overall poor prognosis. The mainstay of treatment has been chemotherapy, however, addition of targeted treatment to BRCA1/2 mutations by poly-(ADP-ribose) polymerase (PARP) inhibitors (olaparib and talazoparib) in combination with cisplatin or carboplatin has improved therapeutic outcomes (Nicolas et al. Citation2018). Furthermore, programmed death-ligand 1 positive (PD-L1+) triple negative breast cancers benefit from treatment with the checkpoint inhibitor atezolizumab in combination with paclitaxel (Schmid et al. Citation2018). Safety and efficacy trials of additional immune checkpoint inhibitors are ongoing and further FDA approvals are expected in the near future.
Emerging sequencing technologies
The landscape of genotyping technologies has drastically changed since the completion of the Human Genome Project in 2003 (). This project incentivized the development of novel Next Generation Sequencing (NGS) technologies and contributed indirectly to the shrinking costs of sequencing (Schwarze et al. Citation2018). Within the context of pharmacogenomics, NGS has proven to generate clinically relevant data, whether using targeted sequencing or whole genome/exome sequencing. In addition to its ability to detect single nucleotide polymorphisms (SNPs), copy number variations (CNVs), and complex structural variations, NGS enabled the discovery of a plethora of rare and novel variants with potential clinical relevance.
Table 1. Selection of clinically relevant pharmacogenomic germline biomarkers.
Table 2. Overview of technological sequencing platforms.
Conventional NGS utilizes short-read sequencing (SRS) that typically amplifies DNA stretches of 50–250 base pairs, which are then assembled to larger contigs. Importantly however, SRS faces major limitations for the profiling of complex or repetitive genetic loci, as short reads are often difficult to unambiguously align or assemble, resulting in issues with the detection of large structural variation and variant phasing (Russell and Schwarz Citation2020; Onishi-Seebacher and Korbel Citation2011). These shortcomings are particularly relevant in pharmacogenomics, as many relevant genes, such as CYP2D6, CYP2A6, ABCB1, SLC22A1, and HLA genes, are highly polymorphic with nearby considerable intervals of low complexity regions, as well as segmental duplications or variable number tandem repeats (Lauschke et al. Citation2017).
These drawbacks of SRS were tackled by the introduction of long-read sequencing (LRS), which is capable of covering up to 100 Kb (van Dijk et al. Citation2018). Two major platforms are available for LRS application: PacBio single-molecule real-time sequencing (SMRT Seq) and nanopore sequencing. SMRT Seq is an optical method in which the integration of fluorescently labeled nucleotides into a nascent nucleic acid chain by an immobilized DNA polymerase is monitored in real time (Eid et al. Citation2009). In contrast, nanopore sequencing quantifies the fluctuations in ionic currents that differ between nucleotides upon translocation of a nucleic acid chain through a nanopore (Feng et al. Citation2015). While both methods can overcome the technical limitations of SRS, the substantially higher cost and longer turnaround times still limit their routine use for clinical applications.
LRS has been successfully established as an emerging genetic testing tool for HLA and CYP2D6 (Ammar et al. Citation2015). One recent example of its successful use is the high-resolution confirmation of an association between HLA-C*07:01 and clozapine-induced myocarditis in patients with schizophrenia (Lacaze et al. Citation2020). In contrast to HLA genes, CYP2D6 does not serve essential endogenous functions (Ingelman-Sundberg Citation2005), albeit some effects on brain physiology and function have been discussed (Kirchheiner et al. Citation2011; Viviani et al. Citation2020). However, the encoded drug metabolizing enzyme is responsible for the metabolism of around 20% of all clinically approved medications (Zanger and Schwab Citation2013). While already >130 distinct CYP2D6 haplotypes with further suballeles have been described, nanopore sequencing of a small sample of 64 CYP2D6 alleles revealed 12 novel alleles/suballeles (Liau et al. Citation2019). Similarly, LRS helped to confirm and refine ten novel CYP2D6 haplotypes identified by targeted short-read sequencing in a large cohort of 990 Japanese samples (Fukunaga et al. Citation2021). These results demonstrate that SRS and LRS currently fill separate synergistic roles in which SRS can flag samples of potential interest in a larger cohort, which are then analyzed further at higher resolution using LRS. However, we envision that within the next decade, LRS will become the predominant sequencing platform for pharmacogenomic studies.
Rare genetic variants
In the last decades, many common variants have been identified and characterized for function and expression in vitro and in vivo. The advent of high throughput technologies for genome sequencing has further accelerated pharmacogenomic research and has resulted in the identification of genetic variants at a record pace. Particularly, such sequencing-based approaches have unveiled a plethora of rare genetic variations whose functional impacts remain unknown. Traditional methods to assess activity and expression of novel variants are not equipped for detecting the functional effects of such a vast number of rare variants of unknown clinical significance. However, the emerging strategies that combine concepts from classic gene expression and function systems with newer, high-throughput approaches and in silico predictions are promising to better understand effects of rare pharmacogenetic variants as potential liabilities for adverse drug events or altered drug response.
Prevalence of rare genetic variants in pharmacogenes
Common genetic variants are more likely to be identified and easier to parse by genetic association studies than rare variants, due to the smaller number of individuals that need to be analyzed to find sufficient numbers of carriers. As such, common variations have been most extensively analyzed. However, recent studies have begun to elucidate the prevalence of rare genetic variation in pharmacogenes. Sequencing data analysis of 806 genes in more than 60,706 exomes suggested that approximately 80% of individuals carry at least one genetic variant in a gene involved in drug response or toxicity (Schärfe et al. Citation2017). Similarly, an analysis of the prevalence of single nucleotide variants (SNVs) in 146 pharmacogenes using exome and genome sequencing data for 7,595 individuals from the 1000 Genomes Project and the Exome Sequencing Project revealed 12,152 SNVs, of which 93% were rare with minor allele frequencies <1% (Kozyra et al. Citation2017). Computational predictions of the functional effects of these variants suggested that rare variants are likely responsible for 30–40% of the functional variation of genes involved in drug response, including drug targets, drug metabolizing enzymes, or drug transporters (Kozyra et al. Citation2017). These findings were corroborated by analyses of 1000 Genomes Project data, which indicated that every individual harbored on average three clinically actionable variants (Wright et al. Citation2018).
Rare genetic variability is prominent across important pharmacogene families, including CYP enzymes (Fujikura et al. Citation2015), and transporters of the solute carrier (SLC) (Schaller and Lauschke, Citation2019), solute carrier for organic anions (SLCO) (Zhang and Lauschke Citation2019) and ATP-binding cassette (ABC) families (Xiao et al. Citation2020). With the abundance of NGS techniques used in research, we continue to discover novel rare variants; however, as their functional consequences are unknown, this information is not currently clinically actionable.
Approaches to study genetic variants
Historically, the impact of common pharmacogenetic variants on drug response and adverse events has been studied using candidate gene studies and GWAS. Candidate gene studies are useful to study select genes known or thought to be important in drug metabolism or response pathways. These targeted approaches can be useful and cost-effective to identify important pharmacogenetic variants, however their utility is limited given the bias of only studying select genes (Motsinger-Reif et al. Citation2013). A more comprehensive approach to identifying risk variants for adverse effects or drug response are GWAS, which use genomic information to identify common genetic variants among patients who exhibit subtherapeutic or supratherapeutic drug concentrations upon therapeutic drug monitoring (TDM), or those who have experienced an adverse drug event (Daly Citation2010). Candidate gene studies and unbiased GWAS have contributed to many important pharmacogenetic discoveries and have laid groundwork for exploring effects of pharmacogenetic variants in more depth for a range of common variants that contribute small to large effects on drug response (Giacomini et al. Citation2017). Importantly, however, candidate gene studies and GWAS are unable to uncover associations for rare genetic pharmacogenetic variants, even for those that mediate large effects on drug response (Motsinger-Reif et al. Citation2013). Particularly for rare events, such as adverse drug reactions (ADRs) for which the numbers of patients who have experienced the ADR are limited, association studies are typically underpowered.
Evaluating the impact of rare pharmacogenetic variants of unknown clinical significance is daunting given their abundance and diversity within the human genome. Functional testing of rare variants in vitro using heterologous expression systems can provide valuable information for variants in drug transporters (Schwarz et al. Citation2011; Russell et al. Citation2020), metabolizing enzymes (Hiratsuka Citation2012), and nuclear receptors (Lamba et al. Citation2004). However, this approach remains low throughput and cannot be scaled to assess the hundreds of thousands of pharmacogenetic variants identified to date. In recent years, CRISPR/Cas9 has also been used to genetically engineer cell lines to express pharmacogenetic variants for functional study (Dorr et al. Citation2017; Takechi et al. Citation2018). However, while genome editing constitutes an important tool to study the effects of whole pharmacogene knockout in animals, most commonly mice (van de Steeg et al. Citation2010; Kumar et al. Citation2017; Medwid et al. Citation2019; Russell et al. Citation2021), application of such methods to study point mutations, especially those that are rare, is cost- and time-inefficient.
To overcome these limitations, high throughput assays to study genetic variation have gained traction in recent years. Deep mutational scanning is one such approach, which can be used to test all theoretical genetic variants in a gene of interest (Fowler and Fields Citation2014; Ipe et al. Citation2017; Lauschke and Ingelman-Sundberg Citation2020). In deep mutational scanning, a library of single-mutant plasmids is generated, for example by one-pot saturation mutagenesis (Wrenbeck et al. Citation2016). Expression of this diversity library in vitro results in each cell expressing an individual variant. A variety of cellular phenotypes can be identified using this approach, including protein abundance and binding or metabolism of substrates for variants within the coding region, or fluorescent reporter assays for non-coding variants (Chiasson et al. Citation2019). Such multiplexed approaches present exciting novel opportunities to overcome the challenges of current low throughput methodologies. A recent study utilized deep mutational scanning to study 230 variants in CYP2C9 and CYP2C19 and identified 55 variants altogether that exhibited <25% of wild type protein expression, which could have implications on the metabolism of numerous drugs (Zhang et al. Citation2020). Additionally, deep mutational scanning revealed variants in NUDT15 with reduced activity that were retrospectively associated with thiopurine toxicity from clinical data (Suiter et al. Citation2020).
However, while such highly multiplexed strategies are becoming increasingly adopted, limitations remain, including the necessity to develop and optimize specific selection and screening assays for each gene of interest, and limited expertise in using these approaches (Russell and Schwarz Citation2020; Chiasson et al. Citation2019). Furthermore, these assays cannot assess certain important aspects related to pharmacogenetic variants, including potential implications of post-translational modifications or cellular localization of the protein of interest.
Given the considerable time and monetary investments required to conduct deep mutational scanning, in silico tools that predict functional effects of genetic variants are commonly used to estimate the functional impacts of pharmacogenetic variation and to prioritize variants for functional testing. These tools often use a combination of parameters including evolutionary conservation, sequence context, and physicochemical alterations of the resulting amino acid change to predict whether a variant will alter protein function (Peterson et al. Citation2013). Importantly, however, almost all commonly used algorithms are trained to predict variant pathogenicity, i.e. their propensity to cause disease. Thus, their application to poorly conserved pharmacogenes lacking important endogenous functions has resulted in low predictive performance (Zhou et al. Citation2018). Recent studies have developed quantitative computational tools specifically developed for the functional interpretation of pharmacogenetic variation and showed that these methods substantially outperformed conventional conservation-based approaches (Zhou et al. Citation2019).
Due to their low cost and high throughput, the use of in silico tools is attractive especially for their application to rare genetic variants. However, predictions are not always concordant with pharmacogenetic activity and expression, especially for variants that exhibit substrate specific effects (Russell et al. Citation2020). While discordance between in silico and in vitro tools is generally believed to be due to the inaccuracy of computational predictions, a recent benchmarking study using DPYD and TPMT population-scale genomic data found that computational methods achieved predictive accuracy similar to in vitro experiments (Zhou et al. Citation2020). Interestingly, the variant sets that did not agree with in vivo data differed between in silico and in vitro methods, suggesting that the complementary use of both approaches can increase confidence that the prediction is accurate.
The major drawback to using these prediction models are considerable false positive and false negative rates, both of which reduce the utility of computational pharmacogenomic predictions. While sensitivity and specificity of >90% have by now been achieved using these in silico models, these accuracies still result in unacceptable numbers of incorrect predictions given the thousands of pharmacogenomic variants found in the human genome. Due to the aforementioned limitations, current computational predictions cannot yet be recommended for implementation into clinical practice. However, going forward, in silico predictions will likely become more accurate through increased availability of experimental data for model training coupled with improved artificial intelligence-based algorithms (Lauschke and Ingelman-Sundberg Citation2016; Lauschke and Ingelman-Sundberg Citation2018). To this end, particularly multiplexed high throughput assays promise to contribute substantially. In addition to improved predictive accuracy, stringent prospective trials are required to inform whether NGS-guided treatment can be of clinical utility, thus paving the way for genetically informed medicine that considers a patient’s entire genetic profile, including rare and novel variations.
Pharmacogenomics in drug discovery and development
The discovery and development of transformative medicines for unmet medical needs is challenged by poor clinical translation mainly due to inadequate efficacy and/or safety concerns. One study observed that in a group of 640 investigational new drugs (INDs), 54% failed clinical development partly due to inadequate efficacy (57%) and safety issues (17%) (Hwang et al. Citation2016). Several genetic and non-genetic factors affect the PK/PD profile of drugs with genetic variations accounting for up to 95% of the differences in drug response (Lauschke and Ingelman-Sundberg Citation2019). Hence, the success of clinical drug development programs is partly dependent on the genetic profile of the patient population recruited for clinical trials, as well as the patient population implicated in ADRs during post market evaluation. It is thus beneficial to characterize the core genetic drivers of the PK/PD pathway of INDs early in the drug development program to allow for the determination of potential interindividual variation in drug response in the affected patient population.
Pharmacogenomics can help drug developers understand the impact of genetic variation on drug response by identifying drug responders and non-responders, as well as individuals at risk of ADRs, with the overall goal of expediting drug development and avoiding drug failures. This can be achieved by applying genomic data to different stages of drug discovery and development starting from biological target identification and validation to patient recruitment for clinical trials.
The benefits associated with the incorporation of pharmacogenomic studies in drug discovery and development can be attributed to technological advancements in the field of genomics including NGS (Dugger et al. Citation2018). The three main genomic approaches that have been applied in drug discovery and development include candidate gene studies, GWAS, and whole exome and whole genome sequencing (Burt and Dhillon Citation2013). These genomic studies have led to an increased body of knowledge regarding genetic polymorphisms associated with interindividual variation in drug response, which are commonly used by drug developers and physicians (). Additionally, the FDA provides a list of pharmacogenomic biomarkers in drug labeling (Table of Pharmacogenomic Biomarkers in Drug Labeling. FDA Citation2021), as well as other useful resources related to pharmacogenomics (Other FDA Resources Related to Pharmacogenomics. FDA Citation2021). This information gathered from several pharmacogenomic databases and evidence-based studies can help drug developers make critical decisions regarding (i) inclusion and exclusion criteria for drug targets, (ii) lead compound optimization (iii), recruitment of patient population for clinical trials and (iv) drug labeling.
Table 3. Overview of publically available resources and databases that provide information regarding the occurrence and clinical implication of pharmacogenomic variations.
Pharmacogenomic information has significantly contributed to the success of several drug development programs. A prominent example is the use of companion diagnostic tests to accelerate the approval of drug candidates by directing therapeutic benefits to a specific subgroup of the affected patient population. Acute myeloid leukemia (AML) is the most common acute leukemia in adults ≥65 years and was responsible for the death of about 10,920 patients in 2019 (Shallis et al. Citation2019). The standard of care has been chemotherapy with the possibility of stem cell transplant; however, the clinical outcome of this treatment regimen is very poor in the elderly patient population due to the unique patient and disease attributes resulting in complete remission rates of 40–50% and shorter duration of remission (Buege et al. Citation2018; Almeida and Ramos Citation2016). The development of targeted treatment approaches has mitigated or reduced the shortfalls of the established standard of care. Previous studies revealed that mutated forms of isocitrate dehydrogenase 1 (IDH1), found in 7–14% of AML patients, generated the oncometabolite 2-hydroxyglutarate that disrupts normal cellular development, contributing to oncogenesis (Dang et al. Citation2016; Mondesir et al. Citation2016). These scientific findings paved the way for the development of ivosidenib, an inhibitor of mutated IDH1 that was approved by the FDA in 2018, for adult patients with relapsed or refractory AML with a susceptible IDH1 mutation as detected by an FDA-approved companion diagnostic test (Dhillon Citation2018). The companion diagnostic device, Abbott RealTime IDH1, is a device that is used to identify AML patients with IDH1 mutations for treatment with ivosidenib. The use of companion diagnostic tests to target a specific subgroup of the patient population has accelerated and improved the clinical outcome of drug development in oncology with about 44 FDA-approved companion diagnostic devices for several oncology drugs (List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools Citation2021).
In addition to identifying individuals susceptible to specific treatments, pharmacogenomics can facilitate the identification of genetic polymorphisms that limit the clinical benefits of drugs in specific patient populations. One such case is the implication of genetic variation in ABCG2 (encoding breast cancer resistance protein, BCRP) as a determinant of response to allopurinol (Brackman et al. Citation2019). Gout is the most common inflammatory arthritis with a prevalence of up to 6.8% and an incidence rate of about 2.89% per 1,000 person-years (Dehlin et al. Citation2020). Gout is caused by hyperuricemia – increased serum uric acid levels leading to the formation and deposition of monosodium urate crystals in and around the joints. Allopurinol is the first-line medication for the prevention of recurrent gout; it exerts its pharmacological activity by acting in synergy with its metabolite oxypurinol to inhibit xanthine oxidase, the enzyme that mediates uric acid biosynthesis. Interestingly, a previous 6-month trial that evaluated the efficacy and safety of once daily allopurinol in gout patients with serum uric acid levels ≥8 mg/dL found that only 42% of patients achieved the recommended serum uric acid target of ≤6 mg/dL (Becker et al. Citation2010). Subsequent pharmacogenomic studies identified a reduced function allele rs2231142 (Q141K, G>T) of BCRP with poor response to allopurinol (Wen et al. Citation2015), hyperuricemia (Kannangara et al. Citation2016), and the development of tophaceous disease (Gaedigk et al. Citation2017). Therefore, implementing genetic screening for the aberrant allele can help identify gout patients that will benefit from allopurinol administration and switch variant carriers who fail to meet the rheumatologist recommended serum uric acid target of ≤6mg/dL to pegloticase (FitzGerald et al. Citation2020).
Another interesting example is the use of pharmacogenomic biomarkers for predicting the outcomes of dalcetrapib therapy. Dalcetrapib is a cholesteryl ester transfer protein (CETP) inhibitor that increased HDL cholesterol but failed to reduce the risk of recurrent cardiovascular events in a large phase 3 trial of 15,871 acute coronary syndrome patients (Schwartz et al. Citation2012). However, a subsequent GWAS study in the discovery cohort of the trial with 5,749 participants identified genetic variations in ADCY9 that are significantly associated with dalcetrapib cardiovascular endpoints (p = 2.1*10−8; HR = 0.61 (Tardif et al. Citation2015)). Interestingly, in mice, Adcy9 ablation reduced atherosclerosis upon high cholesterol feeding, but these positive effects were lost upon transgenic introduction of CETP, thus corroborating a direct functional link between ADC9 and CETP function as well as cardiovascular outcomes (Rautureau et al. Citation2018). However, this association was not supported in 19,210 genotyped individuals of European ancestry treated with the CETP inhibitor anacetrapib, suggesting that effects might be drug-specific or dependent on additional factors yet to be identified (Hopewell et al., Citation2019).
Pharmacogenomics can also inform the postmarket evaluation of drugs. An interesting case study in this category is the safety concern associated with the antiretroviral drug efavirenz. Efavirenz is a non-nucleoside reverse transcriptase inhibitor (NNRTI) used in combination antiretroviral therapy (cART), and its efficacy and long half-life allow for once-daily oral formulations for treatment of HIV. Furthermore, it can be coformulated with other antiretroviral drugs into one tablet. Nevertheless, in 2015, the US department of health and human services demoted efavirenz cART treatment from ‘recommended’ to ‘alternative’ due to increased incidence of central nervous system toxicity and drug-drug interactions relative to other HIV-1 integrase strand transfer inhibitor-based treatments (Desta et al. Citation2019). Many of the safety concerns are due in part to the genetic polymorphisms associated with its main metabolic enzyme, CYP2B6. A previous study showed that efavirenz increases hepatic CYP2B6 activity through CAR transactivation (Meyer zu Schwabedissen et al. Citation2012), resulting in autoinduction following prolonged dosing of efavirenz. However, genetic variation in CYP2B6 plays a critical role in determining the extent of efavirenz autoinduction amongst individuals; for example, CYP2B6*1/*1 and *1/*6 is associated with a significant CYP2B6 autoinduction while CYP2B6*6/*6 shows negligible or no CYP2B6 autoinduction (Ngaimisi et al. Citation2010). For example, when Zimbabwe implemented efavirenz as first-line treatment for HIV, unforeseen ADRs were observed due to supratherapeutic drug concentration mediated by CYP2B6*6 allele alone but not CYP2B6 autoinduction. This public health emergency could have been prevented by the use of precision medicine to identify HIV-patients that would benefit from the clinical administration of efavirenz.
Besides flagging at-risk populations, pharmacogenomics can help inform drug repurposing programs with the overall goal of accelerating drug development. One timely example is the use of drug repurposing to accelerate the development of drugs for the coronavirus disease Citation2019 (COVID-19) pandemic. To inform the clinical development of COVID-19 drugs, a recent study investigated the clinical pharmacogenetic implication of drugs that were being repurposed for the treatment of COVID-19 (Takahashi et al. Citation2020). Eight drug candidates were selected based on established treatment guidelines by Infectious Disease Society of America (Bhimraj et al. Citation2020) and the National Institute of Health (Coronavirus Disease Citation2019), as well as a review of on-going clinical trial data. Review of associated pharmacogenomic information led to the identification of genetic variations that may alter the PK of hydroxychloroquine/chloroquine (CYP2C8, CYP2D6, SLCO1A2, and SLCO1B1), azithromycin (ABCB1), ribavirin (SLC29A1, SLC28A2, and SLC28A3) and lopinavir/ritonavir (SLCO1B1, ABCC2, CYP3A) (Takahashi et al. Citation2020). Such information can help accelerate clinical drug development by targeting a specific patient population with the overall goal of developing effective and safe COVID-19 therapies.
Implementation of pharmacogenomics in primary care
Despite advancements in genomic technologies and the well-accepted role genetic variants play in contributing to adverse drug events, adoption of genetic-guided pharmacotherapy into primary care settings has been slow and pharmacogenetic testing is not widely implemented in most primary care settings. Major barriers to integrating pharmacogenetics into routine clinical care include the lack of evidence of clinical benefit (including in large-scale clinical trials), cost effectiveness, and the lack of standardized treatment modification guidelines (Pritchard et al. Citation2017). Furthermore, streamlining clinical testing and reporting for the seamless integration into clinical workflows as well as improving the education and comfort level of healthcare professionals and patients will be required. Pharmacogenetic testing is more common in research programs, which often utilize retrospective pharmacogenetic testing when efficacy is a concern, in situations of unexpected toxicity, or when drug concentrations fall outside the expected range as measured by TDM, which is particularly common for drugs with narrow therapeutic windows. Prospective pharmacogenetic testing is exceedingly rare outside of oncology, and there is no clear consensus on pharmacogenetic markers to implement in a clinical setting. Current evidence is also limited to specific gene-drug pairs.
The advent of NGS in the mid 2000s dramatically reduced costs of sequencing and although genomic data is being increasingly harnessed toward increased understanding of disease and pharmacotherapy, this technological revolution has not yet resulted in an appreciable increase in the adaptation of pharmacogenomic testing in primary care. Reasons lie mostly in the complexity of genomic data, which require substantial time and expertise for meaningful interpretation (Lauschke and Ingelman-Sundberg Citation2016). Furthermore, ethical dilemmas remain regarding which actions to take, if any, when pathological variants are fortuitously discovered.
Many commonly prescribed medications have limited efficacy (Spear et al. Citation2001; Leucht et al. Citation2015) and severe or fatal adverse drug reactions can ensue even when using the empirical dosing guidelines established by clinical trials (Kim Citation2019). The ‘one size fits all’ approach to drug selection and drug dosing often puts certain populations or individuals at risk of undesirable outcomes such as inadequate efficacy or toxicity. Pharmacogenomics-guided therapy is therefore an attractive approach and it has been demonstrated in multiple studies that implementation in primary care settings is associated with improved outcomes. A clinical trial for major depressive disorder noted that in the pharmacogenetic-guided treatment arm, drug response improved, and intensity and frequency of side effects were reduced (AB-GEN Collaborative Group, Citation2017). Similarly, pharmacogenetic testing for polymorphisms in CYP2D6, CYP2C9, CYP2C19, CYP3A and VKORC1 among elderly patients and the accompanying personalized dose adjustments decreased hospitalizations and emergency room visits and providers were highly satisfied with the utility of the employed clinical decision support tool (Brixner et al. Citation2016). Furthermore, pharmacogenetics-guided dosing decreased re-hospitalizations and emergency room visits in polypharmacy patients 50 years of age and older, which amounted to improved outcomes and potential resource savings (Elliott et al. Citation2017).
However, despite these promising data, the implementation of preemptive genotyping to guide drug or dose adjustments in primary care remains the exception. In the following sections we discuss important hurdles that need to be overcome to facilitate pharmacogenomics in primary care.
Challenges associated with pharmacogenomic clinical trials
The implementation of pharmacogenomic testing to guide treatment decisions in primary care is often suggested to require data generated by gold-standard approaches in evidence-based medicine, including randomized-controlled trials (RCTs). However, the use of RCTs in pharmacogenomics is not always feasible or appropriate. For instance, dose adjustments for patient comorbidities that alter drug exposure occur at the discretion of the physician and do not require RCT data, whereas genetically guided dose adjustments do (Pirmohamed and Hughes Citation2013).
Ethical concerns are also important considerations in the design of pharmacogenomics-based RCTs. A patient who harbors a clinically actionable pharmacogenetic variant cannot be ethically randomized to a treatment group that may precipitate an adverse drug event or toxicity (Huddart et al. Citation2019). In addition, the distribution of populations in RCTs is often biased by overrepresentation of subjects with European ancestry, an aspect that will be covered in detail below. Over-representation of one population may result in the minimization of the potential for safety and efficacy concerns presented by population-specific variants that are absent or rare in the studied group.
An additional consideration is whether the benefit of the data obtained from pharmacogenomics-guided RCTs outweighs the financial investment. This is especially true with regard to drugs already on the market, where resource-intensive, large trials are difficult to justify, and pharmaceutical companies are unlikely to be incentivised to fund such trials. Homogenous patient populations, difficulties recruiting participants carrying variant alleles, and the traditional design of RCTs for determining efficacy and not risk of adverse drug events, suggest it may not be helpful to demand RCTs in pharmacogenomics.
Consequently, the FDA uses data collected in non-RCTs to apply pharmacogenomic information to drug labels. These studies, including smaller non-randomized clinical trials, retrospective studies, and meta-analyses, contribute valuable pharmacogenomic information while being less expensive and easier to conduct than RCTs. Some RCTs have even progressed to include pharmacogenomic testing, which will be helpful going forward (Huddart et al. Citation2019). Post hoc genotyping can be performed such that genetic information is obtained after the patient randomization and treatment, and results can subsequently be analyzed by subgroups to determine whether genotype plays a factor in a specific outcome. Genotyping can also be conducted prior to randomization, where excess numbers of reference sequence patients may be excluded to enrich the study population to better compare outcomes for wild type and genetically variant individuals.
Preemptive genotyping can be used to stratify individuals and compare outcomes for genotype-guided dosing to standard of care in an RCT (van der Baan et al. Citation2011). Such approaches have been successfully integrated into RCT designs; one example is the application of genotype-guided dosing to patients on warfarin therapy for atrial fibrillation or venous thromboembolism. This study randomized patients to a pharmacogenomics-guided arm or standard of care arm and found that pharmacogenomics-guided therapy was associated with a higher percentage of time in the therapeutic range (Pirmohamed et al. Citation2013). A recent, prospective, patient- and rater-blinded trial found that pharmacogenetics-guided treatment was superior for treating depression and anxiety when compared to standard of care (Bradley et al. Citation2018). A similar trial design showed that in patients with difficult-to-treat major depressive disorder, pharmacogenetics-guided treatment improved response and remission rates compared to standard of care (Greden et al. Citation2019). Furthermore, a major RCT in Europe (PREPARE study) evaluates the effects of preemptive genotyping on patient outcomes and cost-effectiveness for 43 drugs in 7 countries across ethnicities and health care systems (van der Wouden et al. Citation2017; Wouden et al. Citation2019). Results of this study are currently being analyzed and are expected to be published in the coming months.
Biased population representation
The effective application of pharmacogenomics in the clinic is also hindered by biased data. One pitfall of typical pharmacogenomic studies is that they have been largely conducted in European or Asian adult populations (Need and Goldstein Citation2009). An analysis of RCTs performed in 2014 showed that 86% of participants were of European ancestry, whereas Asian and Black populations only represented 6 and 3% of RCT participants, respectively (Knepper and McLeod Citation2018). The lack of adequate representation of various ethnogeographic populations presents a knowledge gap, especially given the known population-specific differences in allele frequencies of pharmacogenetic variants. One such example is the VKORC1 gene, which encodes the vitamin K epoxide reductase enzyme that plays a critical role in coagulation. The anticoagulant warfarin inhibits the VKORC1 enzyme, resulting in reduced clotting. The 19% allele frequency of VKORC1 variant (rs7200749) in African Americans, which requires an increased dose of warfarin to prevent excessive blood clotting, is a stark difference to the general absence of this variant in European Americans (allele frequency <0.1%). Similarly, certain genetic loss-of-function variants in CYP2C9 (including rs28371686, rs9332131, and rs28371685), encoding the drug metabolizing enzyme responsible for the metabolic inactivation of warfarin, are rare in individuals of European ancestry but common in African Americans (Limdi et al. Citation2007). Therefore, when considering patient-specific outcomes, ethnicity can be an important factor and cause for ordering appropriate pharmacogenetic testing.
Further, lack of pharmacogenomic testing in children has limited harnessing genetic-guided therapy in pediatrics. For example, the genetic variant rs4149056 in SLCO1B1, encoding the hepatic uptake transporter OATP1B1, results in more pronounced differences in systemic simvastatin acid exposure in children when compared to adults. Mean blood concentrations of simvastatin acid increased by ∼6.3-fold in children homozygous for the variant allele whereas a ∼3.2-fold increase was observed in adults of the same genotype (Pasanen et al. Citation2006; Wagner et al. Citation2018). Increased exposure to simvastatin acid has been linked to muscle pain and weakness, in some cases severe and life-threatening rhabdomyolysis (SEARCH Collaborative Group et al. Citation2008). These data support using caution when extrapolating adult data to pediatrics.
Standardization of ‘actionable pharmacogenetic variants’
A further reason for the slow uptake of pharmacogenetic testing into the clinic is the lack of specific guidelines for recommended dose adjustments or alternate choices of medications (Bank et al. Citation2019). Despite the availability of resources to interpret test results, including guidelines from a variety of academic- and industry-lead consortia and government regulatory agencies, there is a lack of consensus, which can hamper decision-making among healthcare providers, can alienate prescribers, and can make it difficult to decide which patients may benefit from pharmacogenetic testing (Hachad et al. Citation2019). This highlights the importance of not only forming but also harmonizing knowledge bases, including the Clinical Pharmacogenetics Implementation Consortium (CPIC), the Dutch Pharmacogenetics Working Group (DPWG), the Canadian Pharmacogenomics Network for Drug Safety (CPNDS), and the Pharmacogenomics Knowledgebase (PharmGKB) for accurate and up to date evidence-driven guidelines for gene-drug pairs.
A recent analysis of pharmacogenomic guidelines issued by pharmacogenetic consortia and government agencies determined that there are large discrepancies between information disseminated by various information sources (Shekhani et al. Citation2020). This study found that only 50% of the 54 actionable drug/gene pairs listed by CPIC and DPWG had the critical accompanying information to instruct healthcare practitioners on safe and effective use of medications for those patients. Furthermore, regulatory bodies such as the FDA and European Medicines Agency (EMA) were only in agreement with the consortia-lead guidelines in 18% of cases, and drug labels had government-issued pharmacogenomic guidance for a total of 126 gene-drug pairs for which no CPIC or DPWG guidelines existed. Of note, consensus between the FDA and EMA was only 54% (Koutsilieri et al. Citation2020; Shekhani et al. Citation2020).
Discrepancies in standards are further exemplified in custom, targeted next-generation sequencing approaches to identify variants in pharmacogenes. Various numbers of genes have been incorporated into such customized panels, where as little as 74 genes and up to 340 genes have been pulled from sources such as CPIC, PharmaADME, and PharmGKB, as well as independent literature searches where genes with preliminary associations to pharmacogenetic response are added (Gordon et al. Citation2016; Han et al. Citation2017; Gulilat et al. Citation2019; Klein et al. Citation2019). Although these approaches may be customized based on research interests, the lack of harmonization facilitates confusion and does not foster efficient uptake of pharmacogenomics into the clinic.
Integration of test results and recommendations into electronic health records
The integration of pharmacogenetic data and guidelines for selection of alternate therapeutics into the workflow of busy physicians, nurses, and pharmacists poses significant challenges, especially when healthcare providers are not pharmacogenomics experts. Freely available resources including textbooks and databases aimed to educate doctors and nurses have become more widespread. Additionally, educational programs that focus on in-person training for pharmacogenetic testing aim to keep healthcare providers up to date with current information (Pritchard et al. Citation2017). If optimized and adopted properly, electronic health records (EHRs) can prove valuable tools for the integration of this information. However, for integration into clinical practice, test results and the ensuing pharmacogenetic information must be communicated in a standardized fashion, including the genotype, predicted phenotype, and how to address potential medication changes (Kalman et al. Citation2016; Collins et al. Citation2019).
Although workflows incorporating pharmacogenetic information into EHRs along with corresponding best practices are gaining traction, these data are not generally accessible in an easy-to-use manner. Recent advances include adding standardized CPIC phenotype terms to Systematized Nomenclature of Medicine – Clinical Terms (SNOMED-CT), which adds the patient metabolizer status into problem lists within EHR workflows (Caudle et al. Citation2017). Further, the Electronic Medical Records and Genomics (eMERGE) Consortium recently highlighted the importance of standardization to harmonize sequencing procedures and data interpretation between multiple centers (eMERGE Consortium Citation2019). The main objectives of the study were to obtain EHR data and return genetic sequencing results to physicians, who further relayed this information to patients. It should be noted that there are significant challenges associated with the use of multiple centers, such as considering differences in collection sites and methodologies to collect, validate, and relay patient information. Additionally, findings suggested that the brevity of clinical visits hampered detailed phenotypes, and that enrollment in specific research studies may not capture information outside the scope of the study in question, thus stressing the importance of standardization of data collection, interpretation, and subsequent return of important findings to the patient, for effective implementation of precision medicine.
Challenges in the interpretation of pharmacogenetic test results
Certain genetic tests are easy to interpret regarding medication selection and dose adjustments. One such example is testing for DPYD, where carriers of established reduced function variants require dose adjustments of fluoropyrimidines to avoid severe possibly life-threatening toxicity. However, most tests are not as straightforward, particularly when additional non-genetic factors, such as co-morbidities, hepatic and renal function, as well as drug-drug interactions, can have pronounced impacts on drug response or toxicity. For example, patients with atrial fibrillation are often prescribed direct-acting anticoagulants that inhibit factor Xa, such as apixaban or rivaroxaban, for stroke prevention. TDM showed that in a routine clinical setting, interpatient variation in plasma concentrations of rivaroxaban and apixaban was 50–60-fold, higher than what was reported in clinical trials. Furthermore, 12–13% of patients on these medications exceeded the 95th percentile for the maximum plasma concentration reported in clinical trials (Gulilat et al. Citation2017). This variability can be, at least in part, explained by the multitude of factors that impact the pharmacokinetics of factor Xa-inhibitors in addition to genetic variants in CYP3A4/5 and ABCG2, including renal dysfunction and interactions with concomitant medications that inhibit CYP3A or MDR1 (Ueshima et al. Citation2017; Gulilat et al. Citation2020), although it has recently been demonstrated that the inhibition of intestinal efflux transporters MDR1 and BCRP do not play a clinically significant role in apixaban disposition (Sodhi et al. Citation2020). Thus, genetic testing should not be understood as the ‘silver bullet’ but rather as one layer of information that has to be integrated with a multitude of other data sources to optimize patient care.
Healthcare practitioner education and comfort
A significant barrier to the implementation of pharmacogenetics into clinical decision-making is related to the comfort of the healthcare provider in interpreting and implementing results. However, few physicians are adequately trained to request, interpret, and recommend treatment modifications related to genetic testing (Kim Citation2019). A survey of physician opinions on pharmacogenomics revealed that over 50% did not expect to or were unsure whether they would integrate any changes to their prescribing practices based on pharmacogenomics alerts, and only 30% altered their prescribing de facto (St Sauver et al. Citation2016). Furthermore, over 50% of participants found the alerts confusing, frustrating, or had difficulty finding further information regarding pharmacogenetic information contained in the alert. Results from this study reflect common themes related to the implementation of pharmacogenomics into routine care, including lack of pharmacogenomic education, which likely decreases the comfort level of physicians to make appropriate recommendations for dose adjustments or alternative medications.
There was also a lack of agreement on which medical professional should act on pharmacogenetic results. This could pose challenges related to efficient roll-out of pharmacogenetic testing into clinical care. If a specialist believes the primary care physician should act on the pharmacogenetic results, and vice versa, appropriate action could become lost in translation, with potentially detrimental outcomes for patients. A recent study reported that healthcare providers report limited knowledge and experience with pharmacogenetic testing and are concerned about the potential misinterpretation of results when integrating pharmacogenetic testing into their workflow (Vest et al. Citation2020). However, they appeared hopeful that pharmacogenetically guided treatment could improve treatment outcomes. Furthermore, this same study identified an additional gap between comfort and education of primary care providers relative to psychiatrists in that the primary care providers were less comfortable with pharmacogenomics-guided treatment for depression relative to psychiatrists. Thus, adequate pharmacogenetic education of doctors, pharmacists, nurses, and other medical personnel across levels of care will be required to bridge these gaps.
Cost-effectiveness of pharmacogenetic testing
Economics play a role in integrating sequencing into the clinic, where cost-effectiveness of pharmacogenetic genotyping or sequencing must be presented to decision-makers (Patrinos and Mitropoulou, Citation2017). The majority of pharmacogenetic-guided precision medicine strategies with available economic evaluations have proven cost-effective or even cost-dominant (Verbelen et al. Citation2017). Currently, the most cost-effective way of testing genetic variation is via genotyping assays that target one or few variants of clinical relevance. However, as costs of sequencing continue to decline, prospective sequencing will likely become increasingly cost-effective, replacing these current methods of variant identification (Hachad et al. Citation2019). Importantly, cost-effectiveness of preemptive testing is highly dependent on a multitude of factors, including the frequency of the variant of interest, as well as cost and safety profiles of treatment alternatives. For instance, a recent meta-analysis with HLA genotype data of up to 6.4 million individuals found major ethnogeographic differences in the number of patients needed to test to prevent one case of severe hypersensitivity reactions to abacavir, carbamazepine, and allopurinol (Zhou et al. Citation2021). HLA-B*15:02 testing was only cost-effective across South East Asia, whereas genotyping of HLA-B*57:01 and HLA-A*31:01 was cost-effective in the majority of countries, again emphasizing the importance of population-specific considerations.
Future perspectives of pharmacogenomic implementation in primary care
Despite the challenges associated with the widespread implementation of pharmacogenomics into routine clinical practice, the potential for significant improvements to patient care continue to excite health care professionals and clinical scientists. With rapidly advancing sequencing technologies, there is momentum toward the possibility of large-scale pharmacogenomic testing in primary care and thus it is thought that the impact of pharmacogenomics on patient outcomes will be greatest when implemented at the population scale. The increase of popularity of commercial direct-to-consumer genetic testing fuels continued optimism toward this eventual possibility. The potential for significant benefit of integrating pharmacogenomics in patient care has been increasingly recognized, as evidenced by recent integration of pharmacogenomics into the curriculum for healthcare professionals, such as doctors (Linderman et al. Citation2018; Kuzelicki et al. Citation2019), nurses (Chair et al. Citation2019), pharmacists (Adams et al. Citation2016; Frick et al. Citation2016; Weitzel et al. Citation2016) and dentists (Zheng et al. Citation2019).
When integrated into EHRs, pharmacogenetics data can be leveraged alongside patient medical history to better predict a patient’s phenotype (). Patient medical record information coupled with informatics is increasingly being utilized to provide a clinical recommendation to health care providers, and only stands to be improved with the incorporation of complementary pharmacogenetic data (Wilke et al. Citation2011; Caraballo et al. Citation2017; Norgeot et al. Citation2019). Recent examples demonstrate the potential for improved patient outcomes as a result of integration of pharmacogenetics and electronic medical records, for example, in tacrolimus dose selection (Birdwell et al. Citation2012), evaluation of the clinical relevance of arrhythmia-related rare genetic variants (Van Driest et al. Citation2016), and in characterizing associations between genotype and adverse drug effects (Tasa et al. Citation2019). Interestingly, a very recent analysis of adverse drug reactions from medical records of a university hospital emergency department found that only two of 125 adverse drug events observed in a 6-month period may have been prevented by pharmacogenetics testing (Kauppila et al. Citation2021), further highlighting the importance of accounting for both genetics and patient history in successfully predicting patient outcomes. Significant advancements have been made in extracting (and de-identifying) relevant data from patient medical records, including the descriptive text contained in clinician notes that allow valuable insights into adverse outcomes, off-label drug use, and nuances of disease that are challenging to directly capture from electronic records (Norgeot et al. Citation2020).
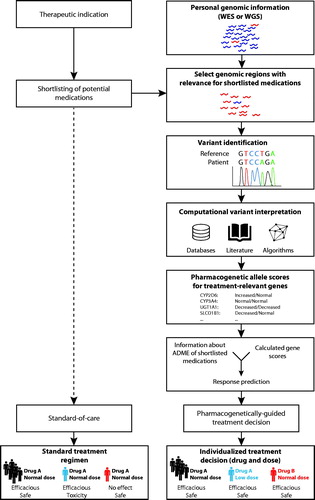
Figure 1. Integration of next generation sequencing data into therapeutic decision-making. Conventional treatment incorporates clinical, demographic and other patient-specific data without however utilizing information about the patient’s genotype (left column). In contrast, pharmacogenetically-guided treatment incorporates genetic information to individualize the drug and dosing regimen (right column). To this end, the patient-specific variant profile is identified from whole exome or whole genome sequencing data (WES and WGS, respectively). Known variants are interpreted based on available guidelines, whereas rare and novel variations with unknown functional consequences are estimated using quantitative computational prediction algorithms specifically trained on pharmacogenomic data. Both known and unknown variants are then integrated into activity scores, which are further translated into individualized predictions of drug efficacy and their propensity to cause adverse drug reactions. Based on this information, individualized predictions of drug efficacy and their propensity to cause ADRs are derived, which is used as guidance for clinical treatment decisions, regarding choice of medication and dose. Figure reprinted with premission from Lauschke and Ingelman-Sundberg 2018.
Beyond an individual’s genetic signature and medical history, interindividual differences in expression levels and activities of drug metabolizing enzymes and transporters constitute important determinants of drug response and toxicity. While these parameters are difficult to measure, recent advancements of minimally invasive liquid biopsies might have the potential to provide quantitative expression information to further stratify patients (Rodrigues and Rowland Citation2019). Some degree of success has been reported when utilizing plasma exosomes to investigate the association between CYP3A4 expression and drug exposure, however reported correlations were only conducted in 6 individuals (pre- and post- CYP3A induction by rifampicin) and in general appear to be primarily driven by a single outlier from the rifampin-induced phase of the trial (Rowland et al. Citation2019). Furthermore, plasma RNA levels of 12 key drug metabolizing enzymes and 4 xenobiotic transporters correlated with the corresponding liver protein levels obtained directly from hepatic biopsies in 29 patients with coefficients of determination (R2) between 0.76 for CYP3A4 to 0.44 for ABCB1 (P-gp, MDR1) (Achour et al. Citation2021). These promising results demonstrate the potential for further patient stratification to complement pharmacogenetic testing toward individualized dosing. Moreover, they show the utility of liquid biopsies to non-invasively characterize hepatic expression of clinically relevant enzymes and transporters.
Conclusions and future perspectives
Currently, the clinical impact of pharmacogenomics remains mostly limited to oncology, where somatic and a few germline biomarkers are routinely used to guide cancer therapy. Advances in genomic technologies allowing for genome-wide studies to be conducted at feasible costs promise to expand the scope of pharmacogenomic studies from the genotyping of candidate variants in small case-control studies to comprehensive discovery studies on a population-scale. This change in perspective has resulted in the identification of a plethora of rare pharmacogenomic variations, many of which constitute unclear functional consequences. Despite these advances, the implementation of pharmacogenomic information into clinical care lags behind. While a multitude of studies showed that the majority of individuals carry actionable pharmacogenetic variants, such information is only rarely generated preemptively and point-of-care testing is not routinely performed in most health care systems. Furthermore, while current careful estimates are that rare variants might contribute to 20–40% of the overall genetically encoded interindividual variability in drug pharmacokinetics, response, and toxicity, functional translation of personalized variant profiles remains difficult, which impedes clinical implementation of NGS-based pharmacogenomic approaches. Several trials that quantify the added value of pharmacogenomically-guided treatment are currently ongoing in Europe, the US, and Asia, which will provide crucial information about the impact on patient outcomes and cost-effectiveness of pharmacogenomics across genes, drugs and healthcare settings.
Disclosure statement
YZ and VML are co-founders and shareholders of PersoMedix AB. In addition, VML is CEO and shareholder of HepaPredict AB and discloses consultancy work for Enginzyme AB. The other authors declare no conflicts of interest. The research conducted and opinions described in this manuscript are their own and are not influenced by the authors’ respective employers. The research work conducted in this publication is not affiliated with Gilead Sciences. No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Achour B, Al‐Majdoub ZM, Grybos‐Gajniak A, Lea K, Kilford P, Zhang M, Knight D, Barber J, Schageman J, Rostami‐Hodjegan A. 2021. Liquid biopsy enables quantification of the abundance and interindividual variability of hepatic enzymes and transporters. Clin Pharmacol Ther. 109(1):222–232.
- Adams SM, Anderson KB, Coons JC, Smith RB, Meyer SM, Parker LS, Empey PE. 2016. Advancing pharmacogenomics education in the core PharmD curriculum through student personal genomic testing. Am J Pharm Educ. 80(1):3.
- Aithal GP, Day CP, Kesteven PJ, Daly AK. 1999. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. The Lancet. 353(9154):717–719.
- Almeida AM, Ramos F. 2016. Acute myeloid leukemia in the older adults. Leuk Res Rep. 6:1–7.
- Ammar R, Paton TA, Torti D, Shlien A, Bader GD. 2015. Long read nanopore sequencing for detection of HLA and CYP2D6 variants and haplotypes. F1000Res. 4:17.
- Bank PCD, Swen JJ, Schaap RD, Klootwijk DB, Baak-Pablo R, Guchelaar H-J. 2019. A pilot study of the implementation of pharmacogenomic pharmacist initiated pre-emptive testing in primary care. Eur J Hum Genet. 27(10):1532–1541.
- Becker MA, Schumacher HR, Espinoza LR, Wells AF, MacDonald P, Lloyd E, Lademacher C. 2010. The urate-lowering efficacy and safety of febuxostat in the treatment of the hyperuricemia of gout: the CONFIRMS trial. Arthritis Res Ther. 12(2):R63.
- Bethune G, Bethune D, Ridgway N, Xu Z. 2010. Epidermal growth factor receptor (EGFR) in lung cancer: an overview and update. J Thorac Dis. 2(1):48–51.
- Bhimraj A, Morgan RL, Shumaker AH, Lavergne V, Baden L, Cheng VC, Edwards KM, Gandhi R, Muller WJ, O’Horo JC, et al. 2020. Infectious Diseases Society of America guidelines on the treatment and management of patients with COVID-19. Clin Infect Dis. ciaa478. DOI:10.1093/cid/ciaa478.
- Birdwell KA, Grady B, Choi L, Xu H, Bian A, Denny JC, Jiang M, Vranic G, Basford M, Cowan JD, et al. 2012. The use of a DNA biobank linked to electronic medical records to characterize pharmacogenomic predictors of tacrolimus dose requirement in kidney transplant recipients. Pharmacogenet Genomics. 22(1):32–42.
- Brackman DJ, Yee SW, Enogieru OJ, Shaffer C, Ranatunga D, Denny JC, Wei W-Q, Kamatani Y, Kubo M, Roden DM, et al. 2019. Genome-wide association and functional studies reveal novel pharmacological mechanisms for allopurinol. Clin Pharmacol Ther. 106(3):623–631.
- Bradley P, Shiekh M, Mehra V, Vrbicky K, Layle S, Olson MC, Maciel A, Cullors A, Garces JA, Lukowiak AA. 2018. Improved efficacy with targeted pharmacogenetic-guided treatment of patients with depression and anxiety: a randomized clinical trial demonstrating clinical utility. J Psychiatr Res. 96:100–107.
- Bråten LS, Haslemo T, Jukic MM, Ingelman-Sundberg M, Molden E, Kringen MK. 2020. Impact of CYP2C19 genotype on sertraline exposure in 1200 Scandinavian patients. Neuropsychopharmacol. 45(3):570–576.
- Brixner D, Biltaji E, Bress A, Unni S, Ye X, Mamiya T, Ashcraft K, Biskupiak J. 2016. The effect of pharmacogenetic profiling with a clinical decision support tool on healthcare resource utilization and estimated costs in the elderly exposed to polypharmacy. J Med Econ. 19(3):213–228.
- Buege MJ, DiPippo AJ, DiNardo CD. 2018. Evolving treatment strategies for elderly leukemia patients with IDH mutations. Cancers. 10(6):187.
- Burt T, Dhillon S. 2013. Pharmacogenomics in early-phase clinical development. Pharmacogenomics. 14(9):1085–1097.
- Caraballo PJ, Bielinski SJ, St Sauver JL, Weinshilboum RM. 2017. Electronic medical record-integrated pharmacogenomics and related clinical decision support concepts. Clin Pharmacol Ther. 102(2):254–264.
- Carr DF, la Porte CJL, Pirmohamed M, Owen A, Cortes CP. 2010. Haplotype structure of CYP2B6 and association with plasma efavirenz concentrations in a Chilean HIV cohort. J Antimicrob Chemother. 65(9):1889–1893.
- Caudle KE, Dunnenberger HM, Freimuth RR, Peterson JF, Burlison JD, Whirl-Carrillo M, Scott SA, Rehm HL, Williams MS, Klein TE, et al. 2017. Standardizing terms for clinical pharmacogenetic test results: consensus terms from the Clinical Pharmacogenetics Implementation Consortium (CPIC). Genet Med. 19(2):215–223.
- Chair SY, Waye MMY, Calzone K, Chan CWH. 2019. Genomics education in nursing in Hong Kong, Taiwan and Mainland China. Int Nurs Rev. 66(4):459–466.
- Chan HT, Chin YM, Low S-K. 2019. The roles of common variation and somatic mutation in cancer pharmacogenomics. Oncol Ther. 7(1):1–32.
- Chen H, Wu J, Zhang Z, Tang Y, Li X, Liu S, Cao S, Li X. 2018. Association between BRCA status and triple-negative breast cancer: a meta-analysis. Front Pharmacol. 9:909.
- Chiasson M, Dunham MJ, Rettie AE, Fowler DM. 2019. Applying multiplex assays to understand variation in pharmacogenes. Clin Pharmacol Ther. 106(2):290–294.
- Chung W-H, Hung S-I, Hong H-S, Hsih M-S, Yang L-C, Ho H-C, Wu J-Y, Chen Y-T. 2004. Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 428(6982):486–486.
- Ciszkowski C, Madadi P, Phillips MS, Lauwers AE, Koren G. 2009. Codeine, ultrarapid-metabolism genotype, and postoperative death. N Engl J Med. 361(8):827–828.
- Clarke JD, Cherrington NJ. 2012. Genetics or environment in drug transport: the case of organic anion transporting polypeptides and adverse drug reactions. Expert Opin Drug Metab Toxicol. 8(3):349–360.
- Collins T, Power K, McCallie D, Owings R. 2019. Finding a place for pharmacogenetics in the electronic health record. Clin Pharmacol Ther. 106(2):295–297.
- Coronavirus Disease. (COVID-19) Treatment guidelines. National Institute of Health (NIH). 2019. [Accessed 2021 Feb 1.]. https://www.covid19treatmentguidelines.nih.gov.
- Crisà E, Kulasekararaj AG, Adema V, Such E, Schanz J, Haase D, Shirneshan K, Best S, Mian SA, Kizilors A, et al. 2020. Impact of somatic mutations in myelodysplastic patients with isolated partial or total loss of chromosome 7. Leukemia. 34(9):2441–2450.
- da Cunha Santos G, Shepherd FA, Tsao M-S. 2011. EGFR mutations and lung cancer. Annu Rev Pathol. 6:49–69.
- Daly AK. 2010. Genome-wide association studies in pharmacogenomics. Nat Rev Genet. 11(4):241–246.
- Dang L, Yen K, Attar EC. 2016. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. 27(4):599–608.
- Davies EA, O'Mahony MS. 2015. Adverse drug reactions in special populations – the elderly. Br J Clin Pharmacol. 80(4):796–807.
- Dehlin M, Jacobsson L, Roddy E. 2020. Global epidemiology of gout: prevalence, incidence, treatment patterns and risk factors. Nat Rev Rheumatol. 16(7):380–390.
- Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee S, Samuel M, Ritchie EK, Guzman ML, Ballman KV, et al. 2018. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med. 24(7):1015–1023.
- Desta Z, Gammal RS, Gong L, Whirl-Carrillo M, Gaur AH, Sukasem C, Hockings J, Myers A, Swart M, Tyndale RF, et al. 2019. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2B6 and efavirenz-containing antiretroviral therapy. Clin Pharmacol Ther. 106(4):726–733.
- Dewey FE, Murray MF, Overton JD, Habegger L, Leader JB, Fetterolf SN, O’Dushlaine C, Van Hout CV, Staples J, Gonzaga-Jauregui C, et al. 2016. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 354(6319):aaf6814.
- Dhillon S. 2018. Ivosidenib: first global approval. Drugs. 78(14):1509–1516.
- Dorr CR, Remmel RP, Muthusamy A, Fisher J, Moriarity BS, Yasuda K, Wu B, Guan W, Schuetz EG, Oetting WS, et al. 2017. CRISPR/Cas9 genetic modification of CYP3A5 *3 in HuH-7 human hepatocyte cell line leads to cell lines with increased midazolam and tacrolimus metabolism. Drug Metab Dispos. 45(8):957–965.
- Dugger SA, Platt A, Goldstein DB. 2018. Drug development in the era of precision medicine. Nat Rev Drug Discov. 17(3):183–196.
- Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. 2009. Real-time DNA sequencing from single polymerase molecules. Science. 323(5910):133–138.
- Elliott LS, Henderson JC, Neradilek MB, Moyer NA, Ashcraft KC, Thirumaran RK. 2017. Clinical impact of pharmacogenetic profiling with a clinical decision support tool in polypharmacy home health patients: a prospective pilot randomized controlled trial. PLOS One. 12(2):e0170905.
- eMERGE Consortium. 2019. Harmonizing clinical sequencing and interpretation for the eMERGE III network. Am J Human Genet. 105:588–605.
- FDA. Other FDA resources related to pharmacogenomics. 2021. [accessed 2021 Feb 20.] https://www.fda.gov/drugs/science-and-research-drugs/other-fda-resources-related-pharmacogenomics.
- FDA. Table of pharmacogenomic biomarkers in drug labeling. 2021. [accessed 2021 Feb 6] https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling.
- Feng Y, Zhang Y, Ying C, Wang D, Du C. 2015. Nanopore-based fourth-generation DNA sequencing technology. Genom Proteom Bioinform. 13(1):4–16.
- Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, et al. 2015. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 16(1):25–35.
- FitzGerald JD, Dalbeth N, Mikuls T, Brignardello‐Petersen R, Guyatt G, Abeles AM, Gelber AC, Harrold LR, Khanna D, King C, et al. 2020. American College of Rheumatology guideline for the management of gout. Arthritis Care Res. 72(6):744–760. 2020).
- Flume PA, Liou TG, Borowitz DS, Li H, Yen K, Ordoñez CL, Geller DE, VX 08-770-104 Study Group. 2012. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest. 142(3):718–724.
- Fowler DM, Fields S. 2014. Deep mutational scanning: a new style of protein science. Nat Methods. 11(8):801–807.
- Frick A, Benton CS, Scolaro KL, McLaughlin JE, Bradley CL, Suzuki OT, Wang N, Wiltshire T. 2016. Transitioning pharmacogenomics into the clinical setting: training future pharmacists. Front Pharmacol. 7:241.
- Fujikura K, Ingelman-Sundberg M, Lauschke VM. 2015. Genetic variation in the human cytochrome P450 supergene family. Pharmacogenet Genom. 25(12):584–594.
- Fukunaga K, Hishinuma E, Hiratsuka M, Kato K, Okusaka T, Saito T, Ikeda M, Yoshida T, Zembutsu H, Iwata N, et al. 2021. Determination of novel CYP2D6 haplotype using the targeted sequencing followed by the long-read sequencing and the functional characterization in the Japanese population. J Hum Genet. 66(2):139–149.
- Furuta T, Shirai N, Takashima M, Xiao F, Hanai H, Sugimura H, Ohashi K, Ishizaki T, Kaneko E. 2001. Effect of genotypic differences in CYP2C19 on cure rates for Helicobacter pylori infection by triple therapy with a proton pump inhibitor, amoxicillin, and clarithromycin. Clin Pharmacol Ther. 69(3):158–168.
- Gaedigk A, Sangkuhl K, Whirl-Carrillo M, Klein T, Leeder JS. 2017. Prediction of CYP2D6 phenotype from genotype across world populations. Genet Med. 19(1):69–76.
- Gasche Y, Daali Y, Fathi M, Chiappe A, Cottini S, Dayer P, Desmeules J. 2004. Codeine intoxication associated with ultrarapid CYP2D6 metabolism. N Engl J Med. 351(27):2827–2831.
- Gautschi O, Milia J, Cabarrou B, Bluthgen M-V, Besse B, Smit EF, Wolf J, Peters S, Früh M, Koeberle D, et al. 2015. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol. 10(10):1451–1457.
- Genin E, Chen D-P, Hung S-I, Sekula P, Schumacher M, Chang P-Y, Tsai S-H, Wu T-L, Bellón T, Tamouza R, et al. 2014. HLA-A*31:01 and different types of carbamazepine-induced severe cutaneous adverse reactions: an international study and meta-analysis. Pharmacogenomics J. 14(3):281–288.
- Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, Crown J, Chan A, Kaufman B, et al. 2006. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 355(26):2733–2743.
- Giacomini KM, Yee SW, Mushiroda T, Weinshilboum RM, Ratain MJ, Kubo M. 2017. Genome-wide association studies of drug response and toxicity: an opportunity for genome medicine. Nat Rev Drug Discov. 16(1):1–70.
- Gordon AS, Fulton RS, Qin X, Mardis ER, Nickerson DA, Scherer S. 2016. PGRNseq: a targeted capture sequencing panel for pharmacogenetic research and implementation. Pharmacogenet Genomics. 26(4):161–168.
- Greden JF, Parikh SV, Rothschild AJ, Thase ME, Dunlop BW, DeBattista C, Conway CR, Forester BP, Mondimore FM, Shelton RC, et al. 2019. Impact of pharmacogenomics on clinical outcomes in major depressive disorder in the GUIDED trial: A large, patient- and rater-blinded, randomized, controlled study. J Psychiatr Res. 111:59–67.
- Gulilat M, Keller D, Linton B, Pananos AD, Lizotte D, Dresser GK, Alfonsi J, Tirona RG, Kim RB, Schwarz UI. 2020. Drug interactions and pharmacogenetic factors contribute to variation in apixaban concentration in atrial fibrillation patients in routine care. J Thromb Thrombolysis. 49(2):294–303.
- Gulilat M, Lamb T, Teft WA, Wang J, Dron JS, Robinson JF, Tirona RG, Hegele RA, Kim RB, Schwarz UI. 2019. Targeted next generation sequencing as a tool for precision medicine. BMC Med Genomics. 12(1):81–17.
- Gulilat M, Tang A, Gryn SE, Leong-Sit P, Skanes AC, Alfonsi JE, Dresser GK, Henderson SL, Rose RV, Lizotte DJ, et al. 2017. Interpatient variation in rivaroxaban and apixaban plasma concentrations in routine care. Can J Cardiol. 33(8):1036–1043.
- Hachad H, Ramsey LB, Scott SA. 2019. Interpreting and implementing clinical pharmacogenetic tests: perspectives from service providers. Clin Pharmacol Ther. 106(2):298–301.
- Han SM, Park J, Lee JH, Lee SS, Kim H, Han H, Kim Y, Yi S, Cho J-Y, Jang I-J, et al. 2017. Targeted next-generation sequencing for comprehensive genetic profiling of pharmacogenes. Clin Pharmacol Ther. 101(3):396–405.
- Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, Ruddy K, Tsang J, Cardoso F. 2019. Breast cancer. Nat Rev Dis Primers. 5(1):66.
- Hasford J, Baccarani M, Hoffmann V, Guilhot J, Saussele S, Rosti G, Guilhot F, Porkka K, Ossenkoppele G, Lindoerfer D, et al. 2011. Predicting complete cytogenetic response and subsequent progression-free survival in 2060 patients with CML on imatinib treatment: the EUTOS score. Blood. 118(3):686–692.
- Hauser AS, Chavali S, Masuho I, Jahn LJ, Martemyanov KA, Gloriam DE, Babu MM. 2018. Pharmacogenomics of GPCR drug targets. Cell. 172(1-2):41–54.e19.
- Hiratsuka M. 2012. In vitro assessment of the allelic variants of cytochrome P450. Drug Metab Pharmacokinet. 27(1):68–84
- Hochhaus A, Kreil S, Corbin AS, La Rosée P, Müller MC, Lahaye T, Hanfstein B, Schoch C, Cross NCP, Berger U, et al. 2002. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 16(11):2190–2196.
- Hopewell JC, Ibrahim M, Hill M, Shaw PM, Braunwald E, Blaustein RO, Bowman L, Landray MJ, Sabatine MS, Collins R, On behalf of the HPS3/TIMI55–REVEAL Collaborative Group, et al. 2019. Impact of ADCY9 genotype on response to anacetrapib. Circulation. 140(11):891–898.
- Hortobagyi GN, Stemmer SM, Burris HA, Yap Y-S, Sonke GS, Paluch-Shimon S, Campone M, Blackwell KL, André F, Winer EP, et al. 2016. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 375(18):1738–1748.
- Huddart R, Sangkuhl K, Whirl-Carrillo M, Klein TE. 2019. Are randomized controlled trials necessary to establish the value of implementing pharmacogenomics in the clinic? Clin Pharmacol Ther. 106(2):284–286.
- Hung S-I, Chung W-H, Liou L-B, Chu C-C, Lin M, Huang H-P, Lin Y-L, Lan J-L, Yang L-C, Hong H-S, et al. 2005. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci. 102(11):4134–4139.
- Hwang TJ, Carpenter D, Lauffenburger JC, Wang B, Franklin JM, Kesselheim AS. 2016. Failure of investigational drugs in late-stage clinical development and publication of trial results. JAMA Intern Med. 176(12):1826–1833.
- Ingelman-Sundberg M. 2005. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 5(1):6–13.
- Innocenti F, Undevia SD, Iyer L, Xian Chen P, Das S, Kocherginsky M, Karrison T, Janisch L, Ramírez J, Rudin CM, et al. 2004. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. JCO. 22(8):1382–1388
- Ipe J, Swart M, Burgess KS, Skaar TC. 2017. High-throughput assays to assess the functional impact of genetic variants: a road towards genomic-driven medicine. Clin Transl Sci. 10(2):67–77.
- Johnston SRD, Harbeck N, Hegg R, Toi M, Martin M, Shao ZM, Zhang QY, Martinez Rodriguez JL, Campone M, Hamilton E, monarchE Committee Members and Investigators, et al. 2020. Abemaciclib combined with endocrine therapy for the adjuvant treatment of HR+, HER2-, node-positive, high-risk, early breast cancer (monarchE). J Clin Oncol. 38(34):3987–3998.]
- Jukić MM, Haslemo T, Molden E, Ingelman-Sundberg M. 2018. Impact of CYP2C19 genotype on escitalopram exposure and therapeutic failure: a retrospective study based on 2,087 Patients. Am J Psychiatry. 175(5):463–470/
- Kalman LV, Agúndez J, Appell ML, Black JL, Bell GC, Boukouvala S, Bruckner C, Bruford E, Caudle K, Coulthard SA, et al. 2016. Pharmacogenetic allele nomenclature: international workgroup recommendations for test result reporting. Clin Pharmacol Ther. 99(2):172–185.
- Kannangara DRW, Phipps-Green AJ, Dalbeth N, Stamp LK, Williams KM, Graham GG, Day RO, Merriman TR. 2016. Hyperuricaemia: contributions of urate transporter ABCG2 and the fractional renal clearance of urate. Ann Rheum Dis. 75(7):1363–1366.
- Kauppila M, Backman JT, Niemi M, Lapatto-Reiniluoto O. 2021. Incidence, preventability, and causality of adverse drug reactions at a university hospital emergency department. Eur J Clin Pharmacol. 77(4):643–650.
- Khunger A, Khunger M, Velcheti V. 2018. Dabrafenib in combination with trametinib in the treatment of patients with BRAF V600-positive advanced or metastatic non-small cell lung cancer: clinical evidence and experience. Ther Adv Respir Dis. 12:1753466618767611.
- Kim RB. 2019. Precision medicine: lessons learned from implementation of a pharmacogenetics-based patient care program in a real-world setting. Clin Pharmacol Ther. 106(5):933–935.
- Kirchheiner J, Seeringer A, Godoy AL, Ohmle B, Maier C, Beschoner P, Sim E-J, Viviani R. 2011. CYP2D6 in the brain: genotype effects on resting brain perfusion. Mol Psychiatry. 16(3):333–341.
- Klein K, Tremmel R, Winter S, Fehr S, Battke F, Scheurenbrand T, Schaeffeler E, Biskup S, Schwab M, Zanger UM. 2019. A new panel-based next-generation sequencing method for ADME genes reveals novel associations of common and rare variants with expression in a human liver cohort. Front Genet. 10:7.
- Knepper TC, McLeod HL. 2018. When will clinical trials finally reflect diversity? Nature. 557(7704):157–159.
- Koutsilieri S, Tzioufa F, Sismanoglou D-C, Patrinos GP. 2020. Towards harmonizing guidance for genome-informed drug treatment interventions: the show must go on. Pharmacol Res. 158:104839.
- Kowalec K, Wright GEB, Drögemöller BI, Aminkeng F, Bhavsar AP, Kingwell E, Yoshida EM, Traboulsee A, Marrie RA, Kremenchutzky M, et al. 2018. Common variation near IRF6 is associated with IFN-β-induced liver injury in multiple sclerosis. Nat Genet. 50(8):1081–1085.
- Kozyra M, Ingelman-Sundberg M, Lauschke VM. 2017. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet Med. 19(1):20–29.
- Krebs K, Bovijn J, Zheng N, Lepamets M, Censin JC, Jürgenson T, Särg D, Abner E, Laisk T, Luo Y, 23andMe Research Team, et al. 2020. Genome-wide Study identifies association between HLA-B∗55:01 and self-reported penicillin allergy. Am J Hum Genet. 107(4):612–621.
- Kumar R, Mota LC, Litoff EJ, Rooney JP, Boswell WT, Courter E, Henderson CM, Hernandez JP, Corton JC, Moore DD, et al. 2017. Compensatory changes in CYP expression in three different toxicology mouse models: CAR-null, Cyp3a-null, and Cyp2b9/10/13-null mice. PLOS One. 12(3):e0174355.
- Kuzelicki NK, et al. 2019. Pharmacogenomics education in medical and pharmacy schools: conclusions of a global survey. Pharmacogenomics. 20:643–657.
- Lacaze P, Ronaldson KJ, Zhang EJ, Alfirevic A, Shah H, Newman L, Strahl M, Smith M, Bousman C, Francis B, et al. 2020. Genetic associations with clozapine-induced myocarditis in patients with schizophrenia. Transl Psychiatry. 10(1):37.
- Lamba JK, Lamba V, Yasuda K, Lin YS, Assem M, Thompson E, Strom S, Schuetz E. 2004. Expression of constitutive androstane receptor splice variants in human tissues and their functional consequences. J Pharmacol Exp Ther. 311(2):811–821.
- Lauschke VM, Ingelman-Sundberg M. 2016. Precision medicine and rare genetic variants. Trends Pharmacol Sci. 37(2):85–86.
- Lauschke VM, Ingelman-Sundberg M. 2016. Requirements for comprehensive pharmacogenetic genotyping platforms. Pharmacogenomics. 17(8):917–924.
- Lauschke VM, Ingelman-Sundberg M. 2016. The importance of patient-specific factors for hepatic drug response and toxicity. IJMS. 17(10):1714.
- Lauschke VM, Ingelman-Sundberg M. 2018. How to consider rare genetic variants in personalized drug therapy. Clin Pharmacol Ther. 103(5):745–748.
- Lauschke VM, Ingelman-Sundberg M. 2019. Prediction of drug response and adverse drug reactions: from twin studies to next generation sequencing. Eur J Pharm Sci. 130:65–77.
- Lauschke VM, Ingelman-Sundberg M. 2020. Emerging strategies to bridge the gap between pharmacogenomic research and its clinical implementation. NPJ Genom Med. 5:9.
- Lauschke VM, Milani L, Ingelman-Sundberg M. 2017. Pharmacogenomic biomarkers for improved drug therapy-recent progress and future developments. AAPS J. 20(1):4.
- Lauschke VM, Zhou Y, Ingelman-Sundberg M. 2019. Novel genetic and epigenetic factors of importance for inter-individual differences in drug disposition, response and toxicity. Pharmacol Ther. 197:122–152.
- Lee AM, et al. 2014. DPYD variants as predictors of 5-fluorouracil toxicity in adjuvant colon cancer treatment (NCCTG N0147). J Natl Cancer Instit. 106:dju298.
- Leucht S, Helfer B, Gartlehner G, Davis JM. 2015. How effective are common medications: a perspective based on meta-analyses of major drugs. BMC Med. 13:253.
- Li G, Guo X, Chen M, Tang L, Jiang H, Day JX, Xie Y, Peng L, Xu X, Li J, et al. 2018. Prevalence and spectrum of AKT1, PIK3CA, PTEN and TP53 somatic mutations in Chinese breast cancer patients. PLOS One. 13(9):e0203495.
- Liau Y, Maggo S, Miller AL, Pearson JF, Kennedy MA, Cree SL. 2019. Nanopore sequencing of the pharmacogene CYP2D6 allows simultaneous haplotyping and detection of duplications. Pharmacogenomics. 20(14):1033–1047.
- Limdi NA, Goldstein JA, Blaisdell JA, Beasley TM, Rivers CA, Acton RT. 2007. Influence of CYP2C9 genotype on warfarin dose among African American and European Americans. Personal Med. 4(2):157–169.
- Linderman MD, Sanderson SC, Bashir A, Diaz GA, Kasarskis A, Zinberg R, Mahajan M, Suckiel SA, Zweig M, Schadt EE. 2018. Impacts of incorporating personal genome sequencing into graduate genomics education: a longitudinal study over three course years. BMC Med Genomics. 11(1):5.
- List of cleared or approved companion diagnostic devices (in vitro and imaging tools). 2021. FDA. [accessed 2021 Mar 7.] https://www.fda.gov/medical-devices/vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-vitro-and-imaging-tools.
- Mallal S, Phillips E, Carosi G, Molina J-M, Workman C, Tomažič J, Jägel-Guedes E, Rugina S, Kozyrev O, Cid JF, et al. 2008. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 358(6):568–579.
- Martin AM, Nolan D, Gaudieri S, Almeida CA, Nolan R, James I, Carvalho F, Phillips E, Christiansen FT, Purcell AW, et al. 2004. Predisposition to abacavir hypersensitivity conferred by HLA-B*5701 and a haplotypic Hsp70-Hom variant. Proc Natl Acad Sci. 101(12):4180–4185.
- Martincorena I, Campbell PJ. 2015. Somatic mutation in cancer and normal cells. Science. 349(6255):1483–1489.
- McCormack M, Alfirevic A, Bourgeois S, Farrell JJ, Kasperavičiūtė D, Carrington M, Sills GJ, Marson T, Jia X, de Bakker PIW, et al. 2011. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med. 364(12):1134–1143.
- Medwid S, Li MMJ, Knauer MJ, Lin K, Mansell SE, Schmerk CL, Zhu C, Griffin KE, Yousif MD, Dresser GK, et al. 2019. Fexofenadine and rosuvastatin pharmacokinetics in mice with targeted disruption of organic anion transporting polypeptide 2B1. Drug Metab Dispos. 47(8):832–842.
- Meulendijks D, Henricks LM, Sonke GS, Deenen MJ, Froehlich TK, Amstutz U, Largiadèr CR, Jennings BA, Marinaki AM, Sanderson JD, et al. 2015. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: a systematic review and meta-analysis of individual patient data. Lancet Oncol. 16(16):1639–1650.
- Meyer zu Schwabedissen HE, Oswald S, Bresser C, Nassif A, Modess C, Desta Z, Ogburn ET, Marinova M, Lütjohann D, Spielhagen C, et al. 2012. Compartment-specific gene regulation of the CAR inducer efavirenz in vivo. Clin Pharmacol Ther. 92(1):103–111.
- Mok TS, Wu Y-L, Ahn M-J, Garassino MC, Kim HR, Ramalingam SS, Shepherd FA, He Y, Akamatsu H, Theelen WSME, et al. 2017. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med. 376(7):629–640.
- Mondesir J, Willekens C, Touat M, de Botton S. 2016. IDH1 and IDH2 mutations as novel therapeutic targets: current perspectives. J Blood Med. 7:171–180.
- Motsinger-Reif AA, Jorgenson E, Relling MV, Kroetz DL, Weinshilboum R, Cox NJ, Roden DM. 2013. Genome-wide association studies in pharmacogenomics: successes and lessons. Pharmacogenet Genomics. 23(8):383–394.
- Need AC, Goldstein DB. 2009. Next generation disparities in human genomics: concerns and remedies. Trends Genet. 25(11):489–494.
- Nelson MR, Wegmann D, Ehm MG, Kessner D, St Jean P, Verzilli C, Shen J, Tang Z, Bacanu S-A, Fraser D, et al. 2012. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 337(6090):100–104.
- Ngaimisi E, Mugusi S, Minzi OM, Sasi P, Riedel K-D, Suda A, Ueda N, Janabi M, Mugusi F, Haefeli WE, et al. 2010. Long-term efavirenz autoinduction and its effect on plasma exposure in HIV patients. Clin Pharmacol Ther. 88(5):676–684.
- Nguyen CM, Mendes MAS, Ma JD. 2011. Thiopurine methyltransferase (TPMT) genotyping to predict myelosuppression risk. PLOS Curr. 3:RRN1236.
- Nicolas E, Bertucci F, Sabatier R, Gonçalves A. 2018. Targeting BRCA deficiency in breast cancer: what are the clinical evidences and the next perspectives? Cancers. 10(12):506.
- Norgeot B, Glicksberg BS, Butte AJ. 2019. A call for deep-learning healthcare. Nat Med. 25(1):14–15.
- Norgeot B, Muenzen K, Peterson TA, Fan X, Glicksberg BS, Schenk G, Rutenberg E, Oskotsky B, Sirota M, Yazdany J, et al. 2020. Protected Health Information filter (Philter): accurately and securely de-identifying free-text clinical notes. NPJ Digit Med. 3:57–58.
- Onishi-Seebacher M, Korbel JO. 2011. Challenges in studying genomic structural variant formation mechanisms: the short-read dilemma and beyond. Bioessays. 33(11):840–850.
- Pasanen MK, Neuvonen M, Neuvonen PJ, Niemi M. 2006. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet Genom. 16(12):873–879.
- Patrinos GP, Mitropoulou C. 2017. Measuring the value of pharmacogenomics evidence. Clin Pharmacol Ther. 15:349–343.
- Pereira B, Chin S-F, Rueda OM, Vollan H-KM, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut S-J, et al. 2016. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 7:11479.
- Pérez V, Salavert A, Espadaler J, Tuson M, Saiz-Ruiz J, Sáez-Navarro C, Bobes J, Baca-García E, Vieta E, Olivares JM, AB-GEN Collaborative Group, et al. 2017. Efficacy of prospective pharmacogenetic testing in the treatment of major depressive disorder: results of a randomized, double-blind clinical trial. BMC Psychiatry. 17(1):250.
- Peterson TA, Doughty E, Kann MG. 2013. Towards precision medicine: advances in computational approaches for the analysis of human variants. J Mol Biol. 425(21):4047–4063.
- Pikor LA, Ramnarine VR, Lam S, Lam WL. 2013. Genetic alterations defining NSCLC subtypes and their therapeutic implications. Lung Cancer. 82(2):179–189.
- Pirmohamed M, Burnside G, Eriksson N, Jorgensen AL, Toh CH, Nicholson T, Kesteven P, Christersson C, Wahlström B, Stafberg C, et al. 2013. A randomized trial of genotype-guided dosing of warfarin. N Engl J Med. 369(24):2294–2303.
- Pirmohamed M, Hughes DA. 2013. Pharmacogenetic tests: the need for a level playing field. Nat Rev Drug Discov. 12(1):3–4.
- Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland Å, Giannone V, D'Amelio AM, Zhang P, Mookerjee B, et al. 2017. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 18(10):1307–1316.
- Pritchard DE, Moeckel F, Villa MS, Housman LT, McCarty CA, McLeod HL. 2017. Strategies for integrating personalized medicine into healthcare practice. Per Med. 14(2):141–152.
- Pui C-H, Nichols KE, Yang JJ. 2019. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat Rev Clin Oncol. 16(4):227–240.
- Pulte D, Jansen L, Brenner H. 2020. Changes in long term survival after diagnosis with common hematologic malignancies in the early 21st century. Blood Cancer J. 10(5):56.
- Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, Griese M, McKone EF, Wainwright CE, Konstan MW, et al. 2011. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 365(18):1663–1672.
- Rautureau Y, Deschambault V, Higgins M-È, Rivas D, Mecteau M, Geoffroy P, Miquel G, Uy K, Sanchez R, Lavoie V, et al. 2018. ADCY9 (Adenylate Cyclase Type 9) inactivation protects from atherosclerosis only in the absence of CETP(Cholesteryl Ester Transfer Protein). Circulation. 138(16):1677–1692.
- Relling MV, Evans WE. 2015. Pharmacogenomics in the clinic. Nature. 526(7573):343–350.
- Rodrigues D, Rowland A. 2019. From endogenous compounds as biomarkers to plasma-derived nanovesicles as liquid biopsy; has the golden age of translational pharmacokinetics-absorption, distribution, metabolism, excretion-drug-drug interaction science finally arrived? Clin Pharmacol Ther. 105(6):1407–1420.
- Rossari F, Minutolo F, Orciuolo E. 2018. Past, present, and future of Bcr-Abl inhibitors: from chemical development to clinical efficacy. J Hematol Oncol. 11(1):84.
- Rowland A, Ruanglertboon W, van Dyk M, Wijayakumara D, Wood LS, Meech R, Mackenzie PI, Rodrigues AD, Marshall J-C, Sorich MJ. 2019. Plasma extracellular nanovesicle (exosome)-derived biomarkers for drug metabolism pathways: a novel approach to characterize variability in drug exposure. Br J Clin Pharmacol. 85(1):216–226.
- Russell LE, DeGorter MK, Ho RH, Leake BF, Schmerk CL, Mansell SE, Kim RB. 2021. Mouse NTCP-mediated rosuvastatin uptake in vitro and in Slc10a1-deficient mice. AAPS J. 23(1):17.
- Russell LE, Schwarz UI. 2020. Variant discovery using next-generation sequencing and its future role in pharmacogenetics. Pharmacogenomics. 21(7):471–486.
- Russell LE, Zhou Y, Lauschke VM, Kim RB. 2020. In vitro functional characterization and in silico prediction of rare genetic variation in the bile acid and drug transporter, Na+-taurocholate cotransporting polypeptide (NTCP, SLC10A1. Mol Pharmaceutics. 17(4):1170–1181.
- Sabir SR, Yeoh S, Jackson G, Bayliss R. 2017. EML4-ALK variants: biological and molecular properties, and the implications for patients. Cancers. 9:1–9.
- Sazonovs A, Kennedy NA, Moutsianas L, Heap GA, Rice DL, Reppell M, Bewshea CM, Chanchlani N, Walker GJ, Perry MH, PANTS Consortium, et al. 2020. HLA-DQA1*05 carriage associated with development of anti-drug antibodies to infliximab and adalimumab in patients with Crohn's disease. Gastroenterology. 158(1):189–199.
- Schaller L, Lauschke VM. 2019. The genetic landscape of the human solute carrier (SLC) transporter superfamily. Hum Genet. 138(11–12):1359–1377.
- Schärfe CPI, Tremmel R, Schwab M, Kohlbacher O, Marks DS. 2017. Genetic variation in human drug-related genes. Genome Med. 9(1):117–115.
- Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Hegg R, Im S-A, Shaw Wright G, et al. 2018. Atezolizumab and Nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 379(22):2108–2121.
- Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, et al. 2012. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 367(22):2089–2099.
- Schwarz UI, Meyer zu Schwabedissen HE, Tirona RG, Suzuki A, Leake BF, Mokrab Y, Mizuguchi K, Ho RH, Kim RB. 2011. Identification of novel functional organic anion-transporting polypeptide 1B3 polymorphisms and assessment of substrate specificity. Pharmacogenet Genomics. 21(3):103–114.
- Schwarze K, Buchanan J, Taylor JC, Wordsworth S. 2018. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet Med. 20(10):1122–1130.
- SEARCH Collaborative Group, Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, Gut I, Lathrop M, Collins R. 2008. SLCO1B1 variants and statin-induced myopathy–a genomewide study. New Eng J Med. 359:789–799.
- Shallis RM, Wang R, Davidoff A, Ma X, Zeidan AM. 2019. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 36:70–87.
- Shekhani R, Steinacher L, Swen JJ, Ingelman-Sundberg M. 2020. Evaluation of current regulation and guidelines of pharmacogenomic drug labels: opportunities for improvements. Clin Pharmacol Ther. 107(5):1240–1255.
- Shuldiner AR. 2009. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA. 302(8):849–857.
- Sim SC, Kacevska M, Ingelman-Sundberg M. 2013. Pharmacogenomics of drug-metabolizing enzymes: a recent update on clinical implications and endogenous effects. Pharmacogenomics J. 13(1):1–11.
- Sodhi JK, Liu S, Benet LZ. 2020. Intestinal efflux transporters P-gp and BCRP are not clinically relevant in apixaban disposition. Pharm Res. 37(10):208–213.
- Spear BB, Heath-Chiozzi M, Huff J. 2001. Clinical application of pharmacogenetics. Trends Mol Med. 7(5):201–204.
- Spring LM, Wander SA, Zangardi M, Bardia A. 2019. CDK 4/6 inhibitors in breast cancer: current controversies and future directions. Curr Oncol Rep. 21(3):25.
- St Sauver JL, Bielinski SJ, Olson JE, Bell EJ, Mc Gree ME, Jacobson DJ, McCormick JB, Caraballo PJ, Takahashi PY, Roger VL, et al. 2016. Integrating pharmacogenomics into clinical practice: promise vs reality. Am J Med. 129(10):1093–1099.
- Suiter CC, Moriyama T, Matreyek KA, Yang W, Scaletti ER, Nishii R, Yang W, Hoshitsuki K, Singh M, Trehan A, et al. 2020. Massively parallel variant characterization identifies NUDT15 alleles associated with thiopurine toxicity. Proc Natl Acad Sci USA. 117(10):5394–5401.
- Takahashi T, Luzum JA, Nicol MR, Jacobson PA. 2020. Pharmacogenomics of COVID-19 therapies. NPJ Genom Med. 5:35.
- Takechi T, Hirota T, Sakai T, Maeda N, Kobayashi D, Ieiri I. 2018. Interindividual differences in the expression of ATP-binding cassette and solute carrier family transporters in human skin: DNA methylation regulates transcriptional activity of the human ABCC3 gene. Drug Metab Dispos. 46(5):628–635.
- Takeuchi F, McGinnis R, Bourgeois S, Barnes C, Eriksson N, Soranzo N, Whittaker P, Ranganath V, Kumanduri V, McLaren W, et al. 2009. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLOS Genet. 5(3):e1000433-1000439.
- Tardif J-C, Rhéaume E, Lemieux Perreault L-P, Grégoire JC, Feroz Zada Y, Asselin G, Provost S, Barhdadi A, Rhainds D, L’Allier PL, et al. 2015. Pharmacogenomic determinants of the cardiovascular effects of dalcetrapib. Circ Cardiovasc Genet. 8(2):372–382.
- Tasa T, Krebs K, Kals M, Mägi R, Lauschke VM, Haller T, Puurand T, Remm M, Esko T, Metspalu A, et al. 2019. Genetic variation in the Estonian population: pharmacogenomics study of adverse drug effects using electronic health records. Eur J Hum Genet. 27(3):442–454.
- Thomas J, Wang L, Clark RE, Pirmohamed M. 2004. Active transport of imatinib into and out of cells: implications for drug resistance. Blood. 104(12):3739–3745.
- Tyndale RF, Droll KP, Sellers EM. 1997. Genetically deficient CYP2D6 metabolism provides protection against oral opiate dependence. Pharmacogenetics. 7:375–379.
- Tzvetkov MV. 2017. OCT1 pharmacogenetics in pain management: is a clinical application within reach? Pharmacogenomics. 18(16):1515–1523.
- Ueshima S, Hira D, Fujii R, Kimura Y, Tomitsuka C, Yamane T, Tabuchi Y, Ozawa T, Itoh H, Horie M, et al. 2017. Impact of ABCB1, ABCG2, and CYP3A5 polymorphisms on plasma trough concentrations of apixaban in Japanese patients with atrial fibrillation. Pharmacogenet Genom. 27(9):329–336.
- van de Steeg E, Wagenaar E, van der Kruijssen CMM, Burggraaff JEC, de Waart DR, Elferink RPJO, Kenworthy KE, Schinkel AH. 2010. Organic anion transporting polypeptide 1a/1b-knockout mice provide insights into hepatic handling of bilirubin, bile acids, and drugs. J Clin Invest. 120(8):2942–2952.
- van der Baan FH, Klungel OH, Egberts ACG, Leufkens HG, Grobbee DE, Roes KCB, Knol MJ. 2011. Pharmacogenetics in randomized controlled trials: considerations for trial design. Pharmacogenomics. 12(10):1485–1492.
- van der Wouden CH, Cambon-Thomsen A, Cecchin E, Cheung KC, Dávila-Fajardo CL, Deneer VH, Dolžan V, Ingelman-Sundberg M, Jönsson S, Karlsson MO, Ubiquitous Pharmacogenomics Consortium, et al. 2017. Implementing pharmacogenomics in Europe: design and implementation strategy of the ubiquitous pharmacogenomics consortium. Clin Pharmacol Ther. 101(3):341–358.
- van Dijk EL, Jaszczyszyn Y, Naquin D, Thermes C. 2018. The third revolution in sequencing technology. Trends Genet. 34(9):666–681.
- Van Driest SL, Wells QS, Stallings S, Bush WS, Gordon A, Nickerson DA, Kim JH, Crosslin DR, Jarvik GP, Carrell DS, et al. 2016. Association of arrhythmia-related genetic variants with phenotypes documented in electronic medical records. JAMA. 315(1):47–57.
- Van Goor F, Hadida S, Grootenhuis PDJ, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, et al. 2011. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA. 108(46):18843–18848.
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, et al. 2009. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA. 106:18825–18830.
- Van Kuilenburg AB, et al. 2000. Clinical implications of dihydropyrimidine dehydrogenase (DPD) deficiency in patients with severe 5-fluorouracil-associated toxicity: identification of new mutations in the DPD gene. Clin Cancer Res. 6:4705–4712.
- Verbelen M, Weale ME, Lewis CM. 2017. Cost-effectiveness of pharmacogenetic-guided treatment: are we there yet? Pharmacogenomics J. 17(5):395–402.
- Vest BM, Wray LO, Brady LA, Thase ME, Beehler GP, Chapman SR, Hull LE, Oslin DW. 2020. Primary care and mental health providers' perceptions of implementation of pharmacogenetics testing for depression prescribing. BMC Psychiatry. 20(1):518.
- Viviani R, Messina I, Bosch JE, Dommes L, Paul A, Schneider KL, Scholl C, Stingl JC. 2020. Effects of genetic variability of CYP2D6 on neural substrates of sustained attention during on-task activity. Transl Psychiatry. 10(1):338–339.
- Wagner JB, Abdel-Rahman S, Van Haandel L, Gaedigk A, Gaedigk R, Raghuveer G, Kauffman R, Leeder JS. 2018. Impact of SLCO1B1 genotype on pediatric simvastatin acid pharmacokinetics. J Clin Pharmacol. 58(6):823–833.
- Weitzel KW, Aquilante CL, Johnson S, Kisor DF, Empey PE. 2016. Educational strategies to enable expansion of pharmacogenomics-based care. Am J Health Syst Pharm. 73(23):1986–1998.
- Wen CC, Yee SW, Liang X, Hoffmann TJ, Kvale MN, Banda Y, Jorgenson E, Schaefer C, Risch N, Giacomini KM. 2015. Genome-wide association study identifies ABCG2 (BCRP) as an allopurinol transporter and a determinant of drug response. Clin Pharmacol Ther. 97(5):518–525.
- Wilke RA, Xu H, Denny JC, Roden DM, Krauss RM, McCarty CA, Davis RL, Skaar T, Lamba J, Savova G. 2011. The emerging role of electronic medical records in pharmacogenomics. Clin Pharmacol Ther. 89(3):379–386.
- Wouden CH, van Rhenen MH, Jama WOM, Ingelman‐Sundberg M, Lauschke VM, Konta L, Schwab M, Swen JJ, Guchelaar H‐J. 2019. Development of the PGx-passport: a panel of actionable germline genetic variants for pre-emptive pharmacogenetic testing. Clin Pharmacol Ther. 106(4):866–873.
- Wrenbeck EE, Klesmith JR, Stapleton JA, Adeniran A, Tyo KEJ, Whitehead TA. 2016. Plasmid-based one-pot saturation mutagenesis. Nat Methods. 13(11):928–930.
- Wright GEB, Carleton B, Hayden MR, Ross CJD. 2018. The global spectrum of protein-coding pharmacogenomic diversity. Pharmacogenomics J. 18(1):187–195.
- Xiao Q, Zhou Y, Lauschke VM. 2020. Ethnogeographic and inter-individual variability of human ABC transporters. Hum Genet. 139(5):623–646.
- Xu Z-q, Zhang Y, Li N, Liu P-j, Gao L, Gao X, Tie X-j. 2017. Efficacy and safety of lapatinib and trastuzumab for HER2-positive breast cancer: a systematic review and meta-analysis of randomised controlled trials. BMJ Open. 7(3):e013053.
- Yan C, Pattabiraman N, Goecks J, Lam P, Nayak A, Pan Y, Torcivia-Rodriguez J, Voskanian A, Wan Q, Mazumder R. 2017. Impact of germline and somatic missense variations on drug binding sites. Pharmacogenomics J. 17(2):128–136.
- Young J, Bhattacharya K, Ramachandran S, Lee A, Bentley JP. 2021. Rates of genetic testing in patients prescribed drugs with pharmacogenomic information in FDA-approved labeling. The Pharmacogenomics Journal. 319:2379–2378.
- Zanger UM, Schwab M. 2013. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 138(1):103–141.
- Zhang B, Lauschke VM. 2019. Genetic variability and population diversity of the human SLCO (OATP) transporter family. Pharmacol Res. 139:550–559.
- Zhang L, Sarangi V, Moon I, Yu J, Liu D, Devarajan S, Reid JM, Kalari KR, Wang L, Weinshilboum R. 2020. CYP2C9 and CYP2C19: deep mutational scanning and functional characterization of genomic missense variants. Clin Transl Sci. 13(4):727–742.
- Zheng LY, Rifkin BR, Spielman AI, London L, London SD. 2019. The Teaching of personalized dentistry in North American dental schools: changes from 2014 to 2017. J Dent Educ. 83(9):1065–1075.
- Zhou Y, Dagli Hernandez C, Lauschke VM. 2020. Population-scale predictions of DPD and TPMT phenotypes using a quantitative pharmacogene-specific ensemble classifier. Br J Cancer. 123(12):1782–1789.
- Zhou Y, Fujikura K, Mkrtchian S, Lauschke VM. 2018. Computational methods for the pharmacogenetic interpretation of next generation sequencing data. Front Pharmacol. 9:1437.
- Zhou Y, Herras Arribas G, Turku A, Jürgenson T, Mkrtchian S, Krebs K, Wang Y, Svobodova B, Milani L, Schulte G, et al. Rare genetic variability in human drug target genes modulates drug response and can guide precision medicine. Sci Adv. (Under consideration).
- Zhou Y, Ingelman-Sundberg M, Lauschke VM. 2017. Worldwide distribution of cytochrome P450 alleles: a meta-analysis of population-scale sequencing projects. Clin Pharmacol Ther. 102(4):688–700.
- Zhou Y, Krebs K, Milani L, Lauschke VM. 2021. Global frequencies of clinically important HLA alleles and their implications for the cost-effectiveness of preemptive pharmacogenetic testing. Clin Pharmacol Ther. 109(1):160–174.
- Zhou Y, Mkrtchian S, Kumondai M, Hiratsuka M, Lauschke VM. 2019. An optimized prediction framework to assess the functional impact of pharmacogenetic variants. Pharmacogenomics J. 19(2):115–126.