
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Biotransformation field is constantly evolving with new molecular structures and discoveries of metabolic pathways that impact efficacy and safety. Recent review by Kramlinger et al. (Citation2022) nicely captures the future (and the past) of highly impactful science of biotransformation (see the first article). Based on the selected articles, this review was categorized into three sections: (1) new modalities biotransformation, (2) drug discovery biotransformation, and (3) drug development biotransformation ().
We would be pleased to hear your opinions, and we extend an invitation to anyone who would like to contribute to a future edition of this review. Please note, this annual review is the seventh of its kind since 2016 (Baillie et al. Citation2016, Khojasteh et al. Citation2017, Khojasteh et al. Citation2018, Khojasteh et al. Citation2019, Khojasteh et al. Citation2020, Khojasteh et al. Citation2021). Our objective is to explore and share articles which we deem influential and significant in the field of biotransformation and reactivity/bioactivation. Due to the volume of the work, we decided last year to create two review articles side-by-side with the two focus areas with Assistant Professor Klarissa D. Jackson leading the reactivity and bioactivation.
Table 1. Articles covered in this review.
Beyond biotransformation
Due to the pandemic, the past two years have been difficult for many. The effective vaccination proved to change the trajectory of the spread of the infection. With our unique vantage views as scientists, we could directly or indirectly all contribute to creating a more equitable and harmonious community. This is consistent with living a purposeful life and leaving this planet better than what we found.
Cyrus Khojasteh, on behalf of the authors.
Source: Drug Metab Dispos. 2022;50:258–267
Synopsis
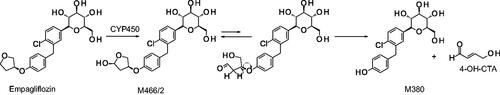
This mini-review or position paper is based on input from members of the Biotransformation, Mechanisms, and Pathways Focus Group (BMPFG), an affiliate of the ISSX, and raises the importance of the contributions biotransformation scientists play in the field of drug discovery and development. This review poses the question, whilst also offering some solutions as to where the next generations of these scientists will come from. Graduates in the fields of toxicology and medicinal chemistry are exposed to a number of modules that cover the broad area of biotransformation (BT), including analytical techniques to serve them in the future. This foundation in training gives them an understanding of specific facets that will impact their chosen areas. Once qualified, there will be competition from Pharma or Contract Research Organizations (CROs) to recruit the candidate with the best skills for the role. It is here where the quality of the graduate training will play its part so as to provide candidates with the diverse skills and knowledge to be able to play a key role in the aforementioned organizations. In drug discovery, the BT scientist contributes to the design of molecules with improved absorption, distribution, metabolism and excretion (ADME) properties. As part of this process, the authors highlight the relationship between the BT and medicinal chemistry which is key to an efficient and effective design, test, improve cycle. The paper uses a number of specific examples including the design of γ-secretase inhibitors (Stepan et al. Citation2011), identification of a bioactivation pathway and subsequent resolution of the associated liabilities (Walker et al. Citation2008). In drug development, further examples are given that highlight the increase in rigor that is required to support the transition of the molecule into the clinic and increase the understanding of the translatability from preclinical into human. This next phase uses more intricate and resource consuming activities providing metabolite characterization, through use of nuclear magnetic resonance (NMR) (Walker et al. Citation2014) and radiolabeling (Penner et al. Citation2009; White et al. Citation2013) including metabolites in safety testing (MIST) activities. The case of empagliflozin (Jardiance) is presented to highlight the diverse BT activities and skills required of the role in the drug development area. The observation that renal tubular adenomas and carcinomas were detected only in male mice at the highest dose led to in-depth BT studies. In studies with CD1 mouse kidney microsomes, a cyclic hemiacetal (M466/2) was detected () in significantly higher levels in male samples. Further work using a range of techniques and technologies identified that the bioactivation pathway resulted in formation of 4-hydroxycrotonaldehyde (4-OH-CTA) leading to covalent binding and subsequent murine toxicity. The challenges, opportunities and outlook are raised, and discussed. Whatever the current status is, the need to maintain or grow the next generation in the BT area is strong, as there is a continued flux of approved classic new chemical entities (NCEs) whilst in addition, the expanding areas of new modalities such as proteolysis targeting chimera (PROTACs) and antibody-drug conjugates (ADCs) are introducing new challenges in areas such as enzymology and bioanalysis of complex modalities. Attaining the level of understanding we have for NCEs when applied to new modalities is critical to ensure the development of safe and effective therapies in this area. Solutions are proffered to address this such as embedding BT into a larger initiative, encouraging collaborations of relevant drug metabolism and pharmacokinetic (DMPK) departments and academic groups. Stimulating interest in the area early on in a graduate’s career, such as including a range of modules in the curricula can also offer opportunities for future growth. Ultimately, the need to promote training and development in the field of Biotransformation remains as critical now than ever.
Commentary
A mini-review on this topic is always a timely one. Nevertheless, it could be applied to a number of other areas of pharmaceutical research, or even more broadly to science itself beyond the US into the global arena, an example being where opportunities are inconsistent across low- and middle-income countries (Pillai et al. Citation2018).
The paper captures the key skills required for the role of the BT scientist, but the diverse nature of these skills subsequently means that generic training programmes are very difficult to design and support. As is often the case, the education platform provides training in the fundamentals and exposure to new area, and it is then up to the individual’s interest and passion to seek the relevant roles or even considering diversifying. At the same time, the ‘industry’ (higher academia, Pharma, CROs etc.) attracts candidates with resources, including continued training, and potential for growth.
From this reviewer’s perspective, the training I received was in the very basics of chemistry and biology and coupled with a passionate interest in analytical technology it provided a solid foundation to develop a long career in the field of biotransformation. In addition to these qualities, an inquisitive and detective-oriented mindset has helped to maintain the interest I have in this area. As the reviewers point out, a lot of training I received was ‘on-the-job’ through interaction with peers and mentors, and it is this aspect that continues to offer part of the solution as to where the next generation of BT scientists will come from.
Figure 1. Bioactivation pathway of empagliflozin to 4-OH-CTA. Adapted from Taub et al. (Citation2015).
References
- Penner N, Klunk LJ, Prakash C. 2009. Human radiolabeled mass balance studies: objectives, utilities and limitations. Biopharm Drug Dispos. 30:185–203.
- Pillai G, Chibale K, Constable EC, Keller AN, Gutierrez MM, Mirza F, Sengstag C, Masimirembwa C, Denti P, Maartens G, et al. 2018. The Next Generation Scientist program: capacity-building for future scientific leaders in low- and middle-income countries. BMC Med Educ. 18(1):233.
- Stepan AF, Karki K, McDonald WS, Dorff PH, Dutra JK, Dirico KJ, Won A, Subramanyam C, Efremov IV, O’Donnell CJ, et al. 2011. Metabolism-directed design of oxetane-containing arylsulfonamide derivatives as c-secretase inhibitors. J Med Chem. 54(22):7772–7783.
- Taub ME, Ludwig-Schwellinger E, Ishiguro N, Kishimoto W, Yu H, Wagner K, Tweedie D. 2015. Sex-, species-, and tissue-specific metabolism of empagliflozin in male mouse kidney forms an unstable hemiacetal metabolite (M466/2) that degrades to 4-hydroxycrotonaldehyde, a reactive and cytotoxic species. Chem Res Toxicol. 28(1):103–115.
- Walker DP, Bi FC, Kalgutkar AS, Bauman JN, Zhao SX, Soglia JR, Aspnes GE, Kung DW, Klug-McLeod J, Zawistoski MP, et al. 2008. Trifluoromethylpyrimidine-based inhibitors of proline-rich tyrosine kinase 2 (PYK2): structure-activity relationships and strategies for the elimination of reactive metabolite formation. Bioorg Med Chem Lett. 18(23):6071–6077.
- Walker GS, Bauman JN, Ryder TF, Smith EB, Spracklin DK, Obach RS. 2014. Biosynthesis of drug metabolites and quantitation using NMR spectroscopy for use in pharmacologic and drug metabolism studies. Drug Metab Dispos. 42:1627–1639.
- White RE, Evans DC, Hop CE, Moore DJ, Prakash C, Surapaneni S, Tse FL. 2013. Radiolabeled mass-balance excretion and metabolism studies in laboratory animals: a commentary on why they are still necessary. Xenobiotica. 43:219–225, discussion 226–227.
Source: Drug Metab Dispos. 2021;49(7):572–580
Synopsis
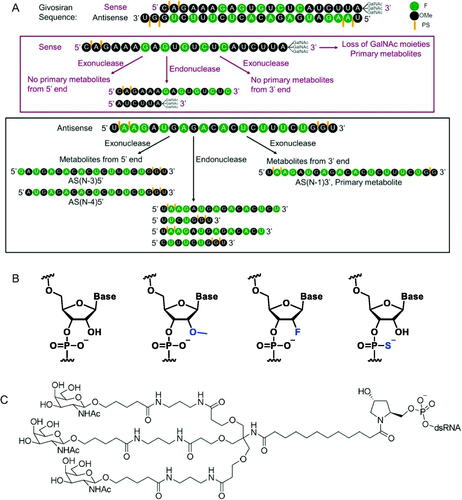
Givosiran is the second approved RNAi molecule for human use and the first approved GalNAc-conjugated RNAi for the treatment of acute hepatic porphyria. This report detailed the pharmacokinetics (PK) and absorption, distribution, metabolism, and elimination (ADME) of givosiran in rats and monkeys. Givosiran was completely absorbed following subcutaneous administration, with a fairly short plasma half-life of 2–4 h, yet a favorable much longer liver half-life of ∼120–150 h. Its plasma clearance was driven by asialoglycoprotein receptor (ASGPR) mediated liver update, with a liver to plasma AUC ratio of ∼4500 in rats and ∼2500 in monkeys. Its renal excretion represented about 10% and 16% of dose in rats and monkeys, respectively. Negligible amount of unmodified givosiran was recovered in feces, while only 6% of dose was actually recovered as intact givosiran in bile from dosing bile duct cannulated (BDC) rats. Unlike small molecule drugs, givosiran was not a substrate for P450 oxidations based on in vitro investigation, where both sense and antisense strands were stable in NADPH fortified liver S9 fraction. Its metabolism was mainly mediated via endo- and exonuclease hydrolysis in plasma and liver, with the sense strand more stable than the antisense strand, especially in plasma. Givosiran was metabolized to a single major circulating metabolite AS(N-1)3′ givosiran with equal potency as givosiran, from the loss of a single nucleotide on the 3′ end of the antisense strand (. The metabolite was confirmed via unique b2 and y2 fragment ions of AS(N-1)3′ givosiran and the presumable AS(N-1)5′ givosiran, since both had the same LC retention time. Other nuclease metabolites captured from the antisense strand were relatively minor. Loss of 1–3 GalNAc moieties was the major metabolism of the much more stable sense strand. Overall, the metabolism of givosiran was well correlated from in vitro to in vivo and was comparable between preclinical species and human.
Commentary
High translatability and less scrutiny with New Drug Application (NDA) in the ADME space were two prominent advantages for siRNA molecules in drug development when compared to small molecules. In human, the metabolism and elimination of givosiran was very similar to what was observed in preclinical species with givosiran plasma clearance mediated by metabolism to AS(N-1)3′ givosiran (36%), uptake in the liver (52%), and urinary excretion (12%) (Agarwal et al. Citation2020; EMA/CHMP/70703/20202020). The highly conserved PK and ADME properties of givosiran and other GalNAc-conjugated siRNAs across species significantly helped with designing safe and efficacious dose for use in humans (Chong et al. Citation2021). Unlike small molecule drugs, the oligo nucleotides are also expected to exhibit low drug-drug interaction (DDI) potential involving P450 enzymes and drug transports, which was proven to be a favorable property for givosiran, especially as a chronic treatment (Ramsden et al. Citation2019). However, poor metabolic stability from nucleases has been the major challenges for developing RNA molecules as therapeutics. In the case of givosiran, its targeted liver delivery through ASGPR and enhanced stability from liver enzymes ensured the carry-out of its pharmacological actions. Givosiran’s enhanced stability was achieved through not only the typical 2′-deoxy-2′-fluoro (2′-F) and 2′-O-methyl (2′-OMe) modifications, but also the use of phosphorothioates (PS) at the 5′ end, which provided important protection against the prevalent endo-lysosomal 5′-exonuclease cleavage ( (Nair et al. Citation2017). The improved stability and liver targeting triantennary N-acetylgalactosamine substantially enhanced its liver exposure, potency and duration of action for the treatment of acute hepatic porphyria, with a recommended 2.5 mg/kg monthly dose via subcutaneous injection. The endo- and exonuclease cleavages of givosiran was much slower than typical small molecule metabolism. From in vitro human liver S9 incubations, givosiran was intact within the first hour, and 49/65% intact of the antisense/sense strand at 24 h. From preclinical radiolabeled ADME studies that the GalNAc-conjugated siRNAs were also shown to be slowly metabolized in the liver and mainly excreted as dimer and shortmer metabolites (Chong et al. Citation2021). The success of givosiran inevitably gives more hope to the oligonucleotide drug research. Its favorable highly translatable ADME properties and low DDI could significantly speed up its research in development stage.
Figure 2. (A) Structure of givosiran and its metabolites. (B) Structures of (A) unmodified, 2′-O-methyl (2′-OMe), 2′-deoxy-2′-fluoro (2′-F), and phosphorothioate (PS) modified nucleotides; (C) triantennary GalNAc linker from givosiran.
References
- Agarwal S, Simon AR, Goel V, Habtemariam BA, Clausen VA, Kim JB, Robbie GJ. 2020. Pharmacokinetics and pharmacodynamics of the small interfering ribonucleic acid, givosiran, in patients with acute hepatic porphyria. Clin Pharmacol Ther. 108(1):63–72.
- Chong S, Agarwal S, Agarwal S, Aluri KC, Arciprete M, Brown C, Charisse K, Cichocki J, Fitzgerald K, Goel V, et al. 2021. The nonclinical disposition and PK/PD properties of GalNAc-conjugated siRNA are highly predictable and build confidence in translation to man. Drug Metab Dispos. DOI:10.1124/dmd.121.000428.
- Committee for Medicinal Products for Human Use (CHMP), EMA. 2020. Givisoran Assessment report (EMA/CHMP/70703/2020). https://www.ema.europa.eu/en/documents/assessment-report/givlaari-epar-public-assessment-report_en.pdf.
- Nair JK, Attarwala H, Sehgal A, Wang Q, Aluri K, Zhang X, Gao M, Liu J, Indrakanti R, Schofield S, et al. 2017. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc–siRNA conjugates. Nucleic Acids Res. 45(19):10969–10977.
- Ramsden D, Wu JT, Zerler B, Iqbal S, Jiang J, Clausen V, Aluri K, Gu Y, Dennin S, Kim J, et al. 2019. In vitro drug-drug interaction evaluation of GalNAc conjugated siRNAs against CYP450 enzymes and transporters. Drug Metab Dispos. 47(10):1183–1194.
Source: J Med Chem. 2020;63:6407–6422
Synopsis
This perspective article by Humphreys et al. (Citation2020) outlines the emerging RNA interference (siRNA) design principles and discuss the consequences for siRNA disposition and biotransformation. Therapeutic siRNA biotransformation predominantly transpires via the catabolic pathways of naturally occurring RNA. With two siRNA drugs now clinically approved, today’s generation of drug candidates are however completely chemically modified and a comprehensive evaluation of how these synthetic molecules break down is lacking. Based on limited published data and drawing from the antisense oligonucleotide field, the authors summarize the biotransformation products formed, the molecular entities or chemical microenvironments responsible for their formation, and sites of biotransformation of siRNA.
Commentary
siRNA therapeutics are emerging as an important class of human medicines due to their ability to harness a natural cellular mechanism that can potentially regulate the expression of any RNA transcript (Wittrup and Lieberman Citation2015). Therapeutic siRNA is a synthetic, double-stranded RNA (dsRNA) molecule comprising complementary hybridized antisense strand (AS) and sense strand (SS) that induces RNA degradation via RNA interference (RNAi). Acting like prodrug, siRNA is bioactivated via selective loading of AS into argonaute (AGO), the effector protein of the RNA-induced silencing complex (RISC). RNAi can occur via AGO2-mediated target RNA degradation or AGO “stalling” through sterically blocking access to translational machinery (Pratt and MacRae Citation2009). This process requires the sense strand removal of the siRNA, which is driven by AGO2-mediated site-specific hydrolysis of the phosphodiester (PO) bond directly across from nucleotides of the AS (5′→3′ direction) (Lennox and Behlke Citation2016; Chakraborty et al. Citation2017), resulting in RISC activated AS. The siRNA as prodrug is capable of RNA target engagement only after it has bound to AGO2, and the SS is selectively removed.
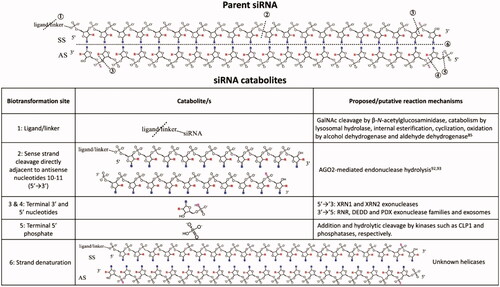
Based on the consensus among researchers is that here the term biotransformation refers to any process resulting in a chemical alteration of the dosed “parent” compound (siRNA) into a biotransformation product or products. siRNA is cleared from the blood in 24–48 h, but a small fraction remains pharmacologically active in the target tissue over weeks to months (Gilleron et al. Citation2013; Wittrup et al. Citation2015; Johannes and Lucchino Citation2018; Agarwal et al. Citation2020). Beyond intact renal elimination, the main in vivo biotransformation products of siRNA are catabolites. The authors (Humphreys et al. Citation2020) presented a comprehensive review on siRNA catabolism. In vitro evidence suggests catabolite formation can at least occur in serum, liver, and liver microsomes, and in vivo catabolite formation has been described in mice, rats, non-human primates, and humans (Zou et al. Citation2008; Christensen et al. Citation2013; Christensen et al. Citation2014; Thayer et al. Citation2019; Agarwal et al. Citation2020). To date, described siRNA catabolite classes include partial or full removal of targeting ligand or linker, AGO2-mediated site-specific cleavage of the SS, terminal nucleotide removal, 5′ phosphate removal, and strand separation. Example canonical catabolite structures originating from ligand-conjugated siRNA are provided in . For ligand-conjugated siRNA such as GalNAc conjugated siRNA, the ligand and/or linker can be a site of catabolism, which was demonstrated in ASOs where a typical triantennary GalNAc moiety is highly susceptible to catabolism in rats and cynomolgus monkeys (Shemesh et al. Citation2016; Husser et al. Citation2017). Specifically, fourteen GalNAc and/or linker associated metabolites were identified including oxidation products at each branching arm and on the linker. Phenotyping implicated N-acetyl-β-glucosaminidase, deoxyribonuclease II, alkaline phosphatase, and alcohol- and aldehyde dehydrogenases involved in these reactions (Shemesh et al. Citation2016).
Therapeutic siRNA is vulnerable to exonuclease clipping, which has also been observed in preclinical studies. In a rat biodistribution study, Alnylam reported clipping from both the 5′ and 3′ ends; however, they did not report catabolic phenotyping data (Thayer et al. Citation2019; CDER FDA Multi-discipline review: Givosiran. NDA 2121942020). They also reported that 3′ AS clipping resulted in active catabolites in vitro and in vivo, whereas catabolites with 5′ clipping were inactive. While catabolic phenotyping data have not been reported for siRNA, it is likely that endogenous enzymes involved for RNA degradation and homeostasis are involved. Candidates for 5′-directed clipping include XRN1 (5′-3′ Exoribonuclease 1) and XRN2, located in the cytosol and nuclear compartments, respectively (Nagarajan et al. Citation2013). Exonucleases from the RNR (ribonucleotide reductase), DEDD (Death effector domain containing protein), and PDX (also known as insulin promoter factor 1) superfamilies as well as exosomes have been associated with degradation of RNA in the 3′ to 5′ direction, although only certain subclasses have siRNA-relevant substrate specificities including dsRNA, ssRNA, overhangs, and length (Ibrahim et al. Citation2008).
Figure 3. Known biotransformation products of ligand-conjugated siRNA and proposed biotransformation mechanisms: B, RNA base adenine, guanine, cytosine, or uracil; R, 2′-ribose modification, i.e. 2’-F or 2’-OMe. SS is sense strand and AS is antisense strand. Figure from Humphreys et al. (Citation2020) (Copyright 2020, American Chemical Society).
References
- Agarwal S, Simon AR, Goel V, Habtemariam BA, Clausen VA, Kim JB, Robbie GJ. 2020. Pharmacokinetics and pharmacodynamics of the small interfering ribonucleic acid (siRNA), givosiran, in patients with acute hepatic porphyria. Clin Pharmacol Ther. 1:63–72.
- Center for Drug Evaluation and Research (CDER) Food and Drug Administration. Multi-discipline review: Givosiran. NDA 212194. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212194Orig1s000MultidisciplineR.pdf.
- Chakraborty C, Sharma AR, Sharma G, Doss CGP, Lee SS. 2017. Therapeutic miRNA and siRNA: moving from bench to clinic as next generation medicine. Mol Ther Nucleic Acids. 8:132–143.
- Christensen J, Litherland K, Faller T, van de Kerkhof E, Natt F, Hunziker J, Krauser J, Swart P. 2013. Metabolism studies of unformulated internally [3H]-labeled short interfering RNAs in mice. Drug Metab Dispos. 41(6):1211–1219.
- Christensen J, Litherland K, Faller T, van de Kerkhof E, Natt F, Hunziker J, Boos J, Beuvink I, Bowman K, Baryza J, et al. 2014. Biodistribution and metabolism studies of lipid nanoparticle formulated internally [3H]-labeled siRNA in mice. Drug Metab Dispos. 42(3):431–440.
- Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U, Manygoats K, Seifert S, Andree C, Stoter M, et al. 2013. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 31(7):638–646.
- Humphreys SC, Thayer MB, Campbell J, Chen WLK, Adams D, Lade JM, Rock BM. 2020. Emerging siRNA design principles and consequences for biotransformation and disposition in drug development. J Med Chem. 63(12):6407–6422.
- Husser C, Brink A, Zell M, Muller MB, Koller E, Schadt S. 2017. Identification of GalNAc-conjugated antisense oligonucleotide metabolites using an untargeted and generic approach based on high resolution mass spectrometry. Anal Chem. 89(12):6821–6826.
- Ibrahim H, Wilusz J, Wilusz CJ. 2008. RNA recognition by 3’-to-5’ exonucleases: the substrate perspective. Biochim. Biophys. Acta, Gene Regul. Mech. 1779(4):256–265.
- Johannes L, Lucchino M. 2018. Current challenges in delivery and cytosolic translocation of therapeutic RNAs. Nucleic Acid Ther. 28(3):178–193.
- Lennox KA, Behlke MA. 2016. Cellular localization of long noncoding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res. 44(2):863–877.
- Nagarajan VK, Jones CI, Newbury SF, Green PJ. 2013. XRN 5’→3’ exoribonucleases: structure, mechanisms, and functions. Biochim Biophys Acta Gene Regul Mech. 1829(6–7):590–603.
- Pratt AJ, MacRae IJ. 2009. The RNA-induced silencing complex: a versatile gene-silencing machine. J Biol Chem. 284(27):17897–17901.
- Shemesh CS, Yu RZ, Gaus HJ, Greenlee S, Post N, Schmidt K, Migawa MT, Seth PP, Zanardi TA, Prakash TP, et al. 2016. Elucidation of the biotransformation pathways of a GalNAc3-conjugated antisense oligonucleotide in rats and monkeys. Mol Ther Nucleic Acids. 5(5):e319.
- Thayer MB, Lade JM, Doherty D, Xie F, Basiri B, Barnaby OS, Bala NS, Rock BM. 2019. Application of locked nucleic acid oligonucleotides for siRNA preclinical bioanalytics. Sci Rep. 9(1):3566.
- Wittrup A, Ai A, Liu X, Hamar P, Trifonova R, Charisse K, Manoharan M, Kirchhausen T, Lieberman J. 2015. Visualizing lipidformulated siRNA release from endosomes and target gene knockdown. Nat Biotechnol. 33(8):870–876.
- Wittrup A, Lieberman J. 2015. Knocking down disease: a progress report on siRNA therapeutics. Nat Rev Genet. 16(9):543–552.
- Zou Y, Tiller P, Chen IW, Beverly M, Hochman J. 2008. Metabolite identification of small interfering RNA duplex by high-resolution accurate mass spectrometry. Rapid Commun Mass Spectrom. 22(12):1871–1881.
Source: Drug Metab Dispos. 2021;49(9):743–749.
Synopsis
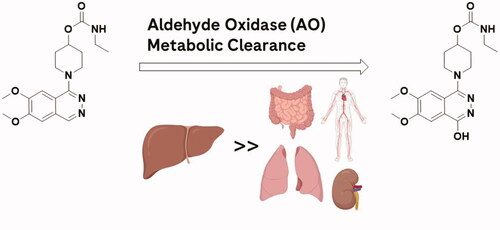
Very few marketed drugs are known to be primarily metabolized by aldehyde oxidase (AO), and many clinical failures due to the contribution of this enzyme resulted from inadequate characterization of human metabolites and clearance prediction. Many poor clinical predictions are due to species differences in the enzyme expression as well as challenges with in vitro in vivo correlation (IVIVC). Many propose donor variability, in vitro matrix isolation/preparations, AO stability/inactivation, and extrahepatic metabolism as the cause for the poor IVIVC between clinical candidates, but this is not well understood (Manevski et al. Citation2019). The report by Kozminski et al. (Citation2021) sought to investigate the relative activity of AO metabolism in various human organs. Human S9 fractions of five different organs (liver, kidney, lung, vasculature, and intestine) were used to compare their in vitro clearance by AO metabolism (). Carbazeran was chosen as a selective probe substrate for AO, monitoring the formation of its major metabolite 4-oxo-carbazeran in human fractions. Intrinsic clearance (CLint) was determined by fitting the AOX modulated activity model (Abbasi et al. Citation2019) to their in vitro data. This incorporated a biphasic enzyme kinetics model involving enzyme inactivation over time, resulting in slowing the rate of product formation. Organ body weight conversions allowed for prediction of relative organ contribution to total clearance in an average human. The highest rates of metabolite formation were obtained in liver, where CLint was determined to be greater than 500-fold higher than any of the extrahepatic tissues. Comparisons to previously reported in vitro work were provided, with some disconnects identified and discussed. For example, the authors noted a significant difference between organ activity of AO reported between different labs. Their data suggested that while extrahepatic AO metabolism is highly possible, the extent of which compared to liver was most likely very low based on the in vitro data. They conclude extrahepatic AO metabolism was most likely a minimal source of human clearance underprediction, and suggest that artificially slow enzyme reactivation in vitro may play a more important role.
Commentary
AO is an important drug metabolizing enzyme for its broad substrate recognition, which has gained much attention over that last two decades. It is a cytosolic molybdenum-containing enzyme primarily localized in hepatocytes, and is involved in the metabolism of a variety of drug candidates and xenobiotics (Zientek et al. Citation2010). This report by Kozminski et al. (Citation2021) was the first to measure AO activity in human vasculature, in addition to the comparison of liver S9 to several other extrahepatic tissues in vitro (intestine, kidney, and lung). Of these extrahepatic tissues, only kidney was reported to have quantifiable AO protein expression in human S9 (Basit et al. Citation2020) previously examined via proteomics. Intestine and lung were identified as potential organs from previous immunohistochemical characterization (Moriwaki et al. Citation2001) and were chosen based on organ weight and high blood flow. Vasculature was selected for its continuous exposure to blood throughout the body. Other organs reported with significant AO expression such as the adrenal gland were excluded due to low relative rates of blood flow supplied to the organ.
The authors observed measurable AO activity in all tissues listed via oxidation of carbazeran to 4-oxo-carbazeran. They reported notably higher (>500-fold) AO activity in the human liver S9 preparations compared to the other extrahepatic tissues. Using in vitro in vivo (IVIV) scalars and binding parameters to estimate that the combined extrahepatic in vivo metabolism of carbazeran by AO is less than 1%. This finding would provide evidence against extrahepatic AO metabolism as a culprit of in vivo clearance for major human AO substrates.
It is important to note one discrepancy in this paper mentioned by the authors. Previously, Basit et al. (Citation2020) reported an 8-fold difference in liver to kidney expression, while in this article the authors estimated at a 500-fold difference in approximated AO expression (scaled from activity) with only 2-fold difference in the estimation of liver AO abundance. There could be many contributing factors, some of which include but are not limited to donor variability and laboratory variation in tissue S9 preparations and enzyme extraction. These arguments may be used toward the other extrahepatic tissue S9 fractions as well, each of which was prepared from different donor lots and some not as effectively homogenized compared to liver tissue.
Given the extreme differences in AO activity between tissue S9 preparations, it seems unlikely that extrahepatic metabolism is a key contributor to the underprediction of human in vivo clearance by AO. The data thus may support enzyme inactivation in vitro playing a significant role for the in vivo disconnect. This is consistent with previous reports for higher turnover AO substrates (Abbasi et al. Citation2019), as the early rapid rate of AO activity seems to more accurately represent the in vivo clearance. The authors made the assumption that enzyme turnover (kcat) would be equivalent in all tissues and organ clearance is solely driven by enzyme expression and blood flow. Tissue donor variability was not investigated in this report and could be another contributor to poor IVIV correlation (Hartmann et al. Citation2012). For example, work by Wellaway et al. report AO metabolism possible on JAK inhibitors in mouse and human lung tissue (homogenates). In vivo, they speculate xenobiotic administration intra nasally may be important for relating AO metabolism to Lung exposure in vivo. The in vitro data reported suggested high AO activity in lung homogenates on specific JAK inhibitors could be observed, with comparable in vitro half lives determined in the liver cytosol preparations. One distinction could be that the compound(s) presented by Wellaway et al. were cleared by AO significantly faster than Carbazeran in vitro from Kozminski et al. This may introduce challenges in accurately measuring differences in rates of metabolism for lung homogenate vs liver cytosol. But another explanation could simply be due to donor and tissue homogenate variability. This paper is reviewed more in depth as another entry within this DMR edition. More work is still needed to better understand the in vitro discrepancy when studying AO metabolic clearance for in vivo prediction.
Figure 4. Carbazeran enzymatic conversation to 4-oxo-carbazeran aldehyde oxidase (AO) was chosen as a selective probe substrate for the contribution of various tissue in its metabolism.
References
- Abbasi A, Paragas EM, Joswig-Jones CA, Rodgers JT, Jones JP. 2019. Time course of aldehyde oxidase and why it is nonlinear. Drug Metab Dispos. 47(5):473–483.
- Basit A, Neradugomma NK, Wolford C, Fan PW, Murray B, Takahashi RH, Khojasteh SC, Smith BJ, Heyward S, Totah RA, et al. 2020. Characterization of differential tissue abundance of major non-CYP enzymes in human. Mol Pharm. 17(11):4114–4124.
- Hartmann T, Terao M, Garattini E, Teutloff C, Alfaro JF, Jones JP, Leimkühler S. 2012. The impact of single nucleotide polymorphisms on human aldehyde oxidase. Drug Metab Dispos. 40(5):856–864.
- Kozminski KD, Selimkhanov J, Heyward S, Zientek MA. 2021. Contribution of extrahepatic aldehyde oxidase activity to human clearance. Drug Metab Dispos. 49(9):743–749.
- Manevski N, King L, Pitt WR, Lecomte F, Toselli F. 2019. Metabolism by aldehyde oxidase: drug design and complementary approaches to challenges in drug discovery. J Med Chem. 62(24):10955–10994.
- Zientek M, Jiang Y, Youdim K, Obach RS. 2010. In vitro-in vivo correlation for intrinsic clearance for durgs metabolized by human aldehyde oxidase. Drug Metab Dispos. 38(8):1322–1327.
Source: J Med Chem. 2022;65:633–664
Synopsis
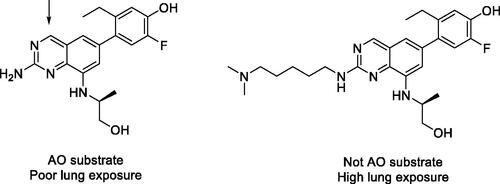
In this report, Wellaway et al. sought to identify small molecule Janus kinase (JAK) inhibitors suitable for pulmonary delivery as a treatment for asthma. The investigators identified inhibitors with desirable potency and selectivity for JAK proteins within a series of quinazoline-containing compounds. However, following intranasal administration to mice, these compounds displayed poor lung exposure. Further evaluation revealed the compounds to be susceptible to metabolism by aldehyde oxidase (AO), which is reported to be present in lung tissue. Through the use of computational docking studies in the human AO binding site paired with in vitro and in vivo metabolism studies in mice, Wellaway et al. determined that large basic substituents at the 2-position of the quinazoline eliminated the AO metabolic liability ().
Commentary
AO is a molybdenum hydroxylase that catalyzes the oxidation of a broad array of heteroaromatic rings, including quinazolines. Several kinase inhibitors have been identified as substrates of AO (Dick Citation2018), including several marketed drugs (e.g. idelalisib, lenvatinib, capmatinib, lapatinib, and imatinib). AO is predominantly expressed in the liver in humans, but many other tissues have also been identified that contain AO, including the lung (Moriwaki et al. Citation2001; Terao et al. Citation2016). As in vitro assessments of AO-mediated clearance using liver tissue fractions and hepatocytes have been demonstrated to underpredict in vivo human clearance of AO substrates, extrahepatic metabolism has been postulated as a possible explanation for the observed discordance of in vitro and in vivo results. However, Kozminski et al. recently evaluated AO-mediated clearance in tissues possessing relatively high expression of AO (kidney, lung, vasculature, and intestine) and concluded that clearance in these tissues contributed to less than 1% of the clearance mediated by the liver (see above for a commentary on Kozminski’s report) (Kozminski et al. Citation2021). While those data support the notion that extrahepatic tissues are unlikely to significantly contribute to the systemic clearance of AO substrates, AO present in lung tissue may still have some potential to influence lung exposure of drugs delivered via the pulmonary route, and the data reported by Wellaway et al. highlight this potential concern. For example, AO protein and mRNA are reportedly present not only in the alveoli, but also in epithelial cells of the bronchi and trachea (Moriwaki et al. Citation2001; Terao et al. Citation2016). While the pulmonary circulation supplying the alveoli receives 100% of the cardiac output, the bronchial circulation receives only 1%. Thus, a greater fraction of the dose would be expected to encounter AO metabolism in bronchial tissue when delivered directly to the lung relative to that occurring via the systemic circulation. Accordingly, this report indicates that it may be important to focus future studies on evaluating the potential for AO to limit access of a drug to its target within tissues expressing AO. Capmatinib, for example, is a kinase inhibitor approved for the treatment of non-small cell lung cancer that is metabolized by AO (Glaenzel et al. Citation2020). Consequently, while lung AO may not significantly contribute to the systemic clearance of capmatinib, AO present in lung tissue could potentially influence the local lung exposure of this drug.
AO expression is species dependent, with the number of functional enzymes ranging from zero to four isoforms. For example, mice express four different functional AO enzymes (Aox1, Aox2, Aox3, and Aox4), yet humans only express a single enzyme, AOX1 (Terao et al. Citation2016). In addition, rodents express both Aox1 and Aox3 in the liver, and differences between the four mouse isoforms in substrate specificity have also been reported (Kücükgöze et al. Citation2017). Alternatively, dog liver is devoid of an AO enzyme altogether. Consequently, preclinical species have proven unreliable as models of human pharmacokinetics for AO substrates.
Importantly, AO tissue distribution is also species dependent, highlighting potential challenges in utilizing preclinical species to evaluate extrahepatic AO metabolism as well. Although the AO metabolite was detected in incubations with human lung homogenate, a quantitative comparison of intrinsic clearance between mouse and human lung tissue was not provided in this report. Therefore, it is unclear whether human lung exposure to these JAK inhibitors would be impacted by AO metabolism to the same degree as that observed in mice. In addition, mice express the Aox2 isoform within the nasal mucosa (Terao et al. Citation2016). As the studies utilized in this report to evaluate lung exposure to JAK inhibitors were conducted using intranasal administration, it is possible that Aox2 present in the nasal tissue may also influence the PK of these compounds. Additional studies would be required to determine the possibility that the JAK inhibitors undergo AO metabolism in mouse nasal mucosa, considering differences exist in substrate specificity between Aox2 (nasal tissue), Aox1 and Aox3 (liver and lung, among other tissues).
Overall, these studies highlight the importance of extrahepatic AO metabolism within a target tissue and demonstrate the role that AO can play in limiting the lung exposure of drugs delivered to lung tissue via the intranasal route in mice. Additional studies will be important to fully understand the role of AO in human lung exposure following pulmonary drug delivery.
Figure 5. JAK inhibitor that displays poor lung exposure due to AO metabolism (left) and a JAK inhibitor lacking AO susceptibility that displays high lung exposure (right). Arrow indicates site of AO oxidation.
References
- Dick RA. 2018. Refinement of in vitro methods for identification of aldehyde oxidase substrates reveals metabolites of kinase inhibitors. Drug Metab Dispos. 46(6):846–859.
- Glaenzel U, Jin Y, Hansen R, Schroer K, Rahmanzadeh G, Pfaar U, Jaap van Lier J, Borell H, Meissner A, Camenisch G, et al. 2020. Absorption, distribution, metabolism, and excretion of capmatinib (INC280) in healthy male volunteers and in vitro aldehyde oxidase phenotyping of the major metabolite. Drug Metab Dispos. 48(10):873–885.
- Kozminski KD, Selimkhanov J, Heyward S, Zientek MA. 2021. Contribution of extrahepatic aldehyde oxidase activity to human clearance. Drug Metab Dispos. 49(9):743–749.
- Kücükgöze G, Terao M, Garattini E, Leimkühler S. 2017. Direct comparison of the enzymatic characteristics and superoxide production of the four aldehyde oxidase enzymes present in mouse. Drug Metab Dispos. 45(8):947–955.
- Moriwaki Y, Yamamoto T, Takahashi S, Tsutsumi Z, Hada T. 2001. Widespread cellular distribution of aldehyde oxidase in human tissues found by immunohistochemistry staining. Histol Histopathol. 16(3):745–753.
- Terao M, Romao MJ, Leimkuhler S, Bolis M, Fratelli M, Coelho C, Santos-Silva T, Garattini E. 2016. Structure and function of mammalian aldehyde oxidases. Arch Toxicol. 90(4):753–780.
Source: Drug Metab Dispos. 2021;49(10):947–960
Synopsis
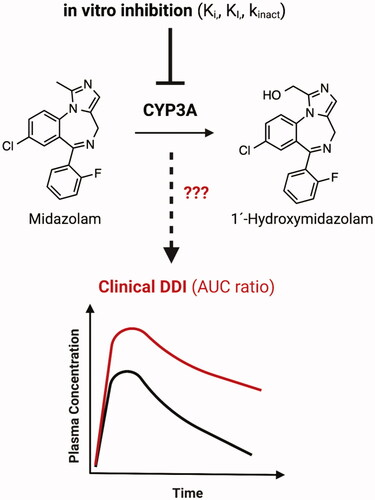
Cytochrome P450 (CYP) 3A4 is the most abundant P450 enzyme in the liver and small intestine (Guengerich Citation1999), and this enzyme is often susceptible to time-dependent inhibition (TDI) based on in vitro studies. This article addresses the disconnect between CYP3A TDI observed from in vitro studies with human liver microsomes and hepatocytes and the frequent lack of clinically significant drug-drug interactions (DDI) in in vivo pharmacokinetic studies (Eng et al. Citation2020). The objective of the study by Tseng et al. (Citation2021) was to determine the most reliable approach to project clinical DDI from in vitro CYP3A TDI data. The study examined 23 drugs and one metabolite with known CYP3A TDI data and clinical DDI data to evaluate static and dynamic methods to predict CYP3A-mediated clinical DDI using midazolam as the model CYP3A substrate (). Static methods use simple equations that assume a single fixed in vivo concentration of inhibitor and substrate, while dynamic methods use physiologically based pharmacokinetic (PBPK) modeling, in which concentrations are assumed to be changing over time. The study demonstrated that applying the appropriate input parameters for in vivo concentration of the inactivator drug in static and dynamic methods with CYP3A TDI data from human liver microsomes and human hepatocytes allowed for reasonable prediction of clinical DDI (Tseng et al. Citation2021).
Figure 6. Evaluating the ability of in vitro CYP3A time-dependent inhibition (TDI) data to predict clinically significant drug-drug interactions (DDI). AUC: Area under the plasma concentration-time curve; kinact: maximal inactivation rate; KI: time-dependent inhibition constant; Ki: reversible inhibition constant.
Commentary
This study provides a thorough and comprehensive evaluation of the multiple factors and approaches that should be considered when attempting to predict CYP3A-mediated clinical DDI from in vitro data. The authors acknowledge that predicting clinical DDI from in vitro CYP3A TDI data is challenging, and false positive predictions from in vitro TDI data can lead to unnecessary expense and effort for clinical DDI studies (Tseng et al. Citation2021). The decision to conduct clinical DDI studies often relies on in vitro data; therefore, establishing the most reliable approach to project clinical DDI is important, both for patient safety and for efficient use of resources during drug development. This study found that using unbound exit concentrations of inhibitor drug from liver and intestine ([I]h and [I]g, respectively) provided better prediction of clinical DDI compared to organ entrance concentrations. When using a static method to predict AUC ratios (area under the plasma concentration-time curve with and without inhibitor), TDI data from human liver microsomes with input of average organ unbound exit concentrations (Cavg,u) of inactivator [I] showed lower false positive predictions, compared to other static models, and no false negatives. This was demonstrated by Tseng et al. (Citation2021) in the mechanistic static Model 4 (). The equations used for Model 4 to estimate [I]g and [I]h and to predict the AUC ratio are shown below. When using a dynamic method with PBPK modeling (Simcyp), which incorporates in vivo [I], rate and extent of drug absorption values, and rate of enzyme degradation (kdeg), TDI data from human liver microsomes showed high sensitivity (ability to identify true positive DDIs with no false negatives) and high specificity (ability to identify true negative DDIs with low false positives) for clinical DDI projections. While CYP3A TDI data from human liver microsomes yielded more over-prediction of DDI overall (more false positives), the negative predictive error (false negatives) was higher when using CYP3A TDI data from human hepatocytes compared to human liver microsomes. Potential reasons for these differences are discussed (i.e. presence of conjugating enzymes, differences in CYP3A interactions with the inactivator in intact hepatocytes vs. microsomal incubations), but the actual mechanisms are unknown; therefore, future studies are required. The impact of CYP3A5-mediated metabolism on clinical DDI also warrants further investigation.
Table 2. Relevant equations used by Tseng et al. (Citation2021) for DDI predictions in static Model 4.
The study by Tseng et al. (Citation2021) represents an advancement in the field because it provides greater insight into the appropriate models to use for DDI prediction from in vitro CYP3A TDI data. The strengths of this study are that (1) it contains the largest dataset of drugs tested under the same experimental conditions for CYP3A TDI for which there is clinical DDI data; (2) the researchers demonstrated that in vitro TDI data from human liver microsomes or human hepatocytes can be reliably used with some limitations noted; (3) multiple static mechanistic models were evaluated to identify the most accurate approach for DDI prediction; and (4) dynamic PBPK modeling was also tested to evaluate DDI prediction. Selection of the in vivo inhibitor concentration ([I]in vivo) was identified as the most important parameter for accurate DDI prediction. Although the unbound maximum plasma concentration (Cmax,u) has been reported as the most commonly used input value (Grimm et al. Citation2009), Tseng et al. (Citation2021) demonstrated that average organ unbound exit concentrations (Cavg,u) provided more accurate predictions. Nonetheless, as shown in previous studies, overestimation of DDI from in vitro CYP3A TDI data is common (Grimm et al. Citation2009; Kenny et al. Citation2012). A limitation of the present study is that it lacked mechanistic details regarding the reasons for the differences in TDI parameters from human liver microsomes and human hepatocytes. The drugs evaluated as CYP3A time-dependent inhibitors in the study represent a diverse class of agents. The most common drugs causing CYP3A-mediated clinical DDI (i.e. high AUC ratio) were protease inhibitors, macrolide antibiotics, and calcium channel blockers (Tseng et al. Citation2021). In conclusion, over-projection of DDI from in vitro CYP3A TDI data remains a challenge. Additional studies are needed to better define the reasons for the overestimation to improve DDI predictions.
References
- Eng H, Tseng E, Cerny MA, Goosen TC, Obach RS. 2020. Cytochrome P450 3A time-dependent inhibition assays are too sensitive for identification of drug causing clinically significant drug-drug interactions: a comparison of human liver microsomes and hepatocytes and definition of boundaries for inactivation rate constants. Drug Metab Dispos. 49(6):442–450.
- Grimm SW, Einolf HJ, Hall SD, He K, Lim HK, Ling KH, Lu C, Nomeir AA, Seibert E, Skordos KW, et al. 2009. The conduct of in vitro studies to address time-dependent inhibition of drug-metabolizing enzymes: a perspective of the pharmaceutical research and manufacturers of America. Drug Metab Dispos. 37(7):1355–1370.
- Guengerich FP. 1999. Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol. 39:1–17.
- Kenny JR, Mukadam S, Zhang C, Tay S, Collins C, Galetin A, Khojasteh SC. 2012. Drug-drug interaction potential of marketed oncology drugs: in vitro assessment of time-dependent cytochrome P450 inhibition, reactive metabolite formation and drug-drug interaction prediction. Pharm Res. 29(7):1960–1976.
- Tseng E, Eng H, Lin J, Cerny MA, Tess DA, Goosen TC, Obach RS. 2021. Static and dynamic projections of drug-drug interactions caused by cytochrome P450 3A time-dependent inhibitors measured in human liver microsomes and hepatocytes. Drug Metab Dispos. 49(10):947–960.
Source: J Biol Chem. 2021;297(5):101316
Synopsis
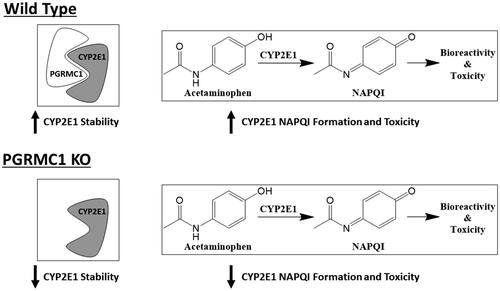
In this work, McGuire et al. utilize a battery of in vitro and in vivo methodologies to demonstrate a novel role for the heme-binding protein progesterone receptor membrane component 1 (PGRMC1) in mammalian cytochrome P450 activity. Through previous work by this group, this protein was determined to bind and regulate activity in vitro in yeast and human cells (Hughes et al. Citation2007). Here the authors developed a PGRMC1 knockout (KO) mouse model to better understand this interaction, and for certain experiments an AAV8 expression system was used to selectively knock-in tagged PGRMC1 or specific PGRMC1 mutants to these mouse models. Through co-immunoprecipitations of mouse liver homogenates, the authors identified that PGRMC1 binds CYP1A2, −2E1 and −3A. Subsequent analysis of P450 expression demonstrated that P450 protein levels were significantly decreased in PGRMC1 knockout mice, while mRNA expression was unchanged or even increased, pointing to a post-transcriptional role of PGRMC1 regulation of P450s. This potential mechanism was further supported by CYP1A2 protein half-life measurements, during which the presence of PGRMC1 increased CYP1A2 half-life by 67%. The authors then investigated the relevance of this protein interaction in modulating P450 metabolic activity. PGRMC1 knockout mice exhibited lower metabolism than wild-type (WT) mice for the following P450 substrates: ethoxycoumarin (CYP1A2), caffeine (CYP1A2), and p-nitrophenol (CYP2E1). Further, the authors demonstrated in vivo the potential clinical implications of this interaction, through IV dosing of both WT and KO mice with acetaminophen (600 mg/kg). Here they observed that the PGRMC1 KO mouse was protected from acetaminophen-induced liver injury using both liver histology and transaminase levels as markers. Finally, a PGRMC1 mutant lacking the ability to bind heme was still able to bind and stabilize P450s, suggesting that this protein-protein interaction is a heme-independent interaction (McGuire et al. Citation2021).
Commentary
Given recent advancements in omics and the accessibility of proteomics studies, novel protein-protein interactions of well-studied proteins, such as P450s, can be more easily identified. From there, however, the work to validate and understand the in vivo relevance of such interactions still requires carefully thought out and labor-intensive mechanistic experiments, such as the ones employed here. Through these efforts, McGuire et al. established that PGRMC1 binds cytochrome P450 enzymes in a heme-independent fashion, increasing protein stability, supporting enzymatic activity, and even contributing to cytochrome P450 metabolism-mediated hepatotoxicity. Cytochrome P450s are implicated in the metabolism of a large majority of small-molecule drugs (Waring Citation2020), and therefore the careful continued study of these enzymes remains impactful.
The half-life of P450s ranges from 7 to 38 h (Kwon et al. Citation2020), which becomes increasingly relevant in instances of enzyme induction (How long will it take for P450 expression to return to normal?) or time-dependent inhibition (When will inactive enzyme be fully cleared and replaced with active enzyme?). Here the role of PGRMC1 in increasing P450 half-life has been demonstrated. These results illustrate the intricacies of protein turnover; protein half-life is a carefully regulated process with many contributing factors. In addition, the loss of drug metabolizing enzyme expression over time is a known issue with hepatic in vitro systems: primary human hepatocytes begin to dedifferentiate in the first 24 h of 2D culture, during which P450 protein levels also decrease, many significantly reduced by 72 h (Heslop et al. Citation2017). Many special techniques have been developed to extend the length of drug metabolizing enzyme expression, including both 3D culture and co-culture of primary hepatocytes (Lauschke et al. Citation2019). Understanding the drivers of P450 protein expression, such as proteins like PGMRC1 imparting stability, will enable the field to continue to develop robust in vitro models for the study of drug metabolism.
Here the authors demonstrated that PGRMC1 KO mice were protected against acetaminophen-induced liver injury. CYP2E1 production of the bioactive N-acetyl-p-benzoquinone imine (NAPQI) metabolite from acetaminophen has been demonstrated to drive this toxicity (Ghanem et al. Citation2016), and therefore decreased CYP2E1 stability in the absence of PGRMC1 explains the decrease in liver injury shown here (). These results also highlight a well-known issue in our field: the biological processes driving drug-induced liver injury, which often varies widely across individuals, are complex and remain to be fully understood. The processes regulating transcription and translation of proteins that modulate P450 expression (such as PGRMC1) may play a role in the idiosyncratic clinical toxicity of certain drugs, though the impact of this versus other factors shown to be involved hepatotoxicity, such as inter-individual variations in immune response, is not yet known (Jee et al. Citation2021).
Finally, CYP3A is a widely implicated P450 subfamily in xenobiotic metabolism, acting on a broad range of drug classes (Waring Citation2020), and through this work was identified as a PGRMC1 binding partner. It would be interesting to investigate with future studies the role of PGRMC1 binding specifically on CYP3A metabolism, half-life, induction, and time-dependent inhibition.
Figure 7. The role of PGRMC1 in CYP2E1 stability as well as acetaminophen metabolism and toxicity.
References
- Ghanem CI, Pérez MJ, Manautou JE, Mottino AD. 2016. Acetaminophen from liver to brain: new insights into drug pharmacological action and toxicity. Pharmacol Res. 109:119–131.
- Heslop JA, Rowe C, Walsh J, Sison-Young R, Jenkins R, Kamalian L, Kia R, Hay D, Jones RP, Malik HZ, et al. 2017. Mechanistic evaluation of primary human hepatocyte culture using global proteomic analysis reveals a selective dedifferentiation profile. Arch Toxicol. 91(1):439–452.
- Hughes AL, Powell DW, Bard M, Eckstein J, Barbuch R, Link AJ, Espenshade PJ. 2007. Dap1/PGRMC1 binds and regulates cytochrome P450 enzymes. Cell Metab. 5(2):143–149.
- Jee A, Sernoskie SC, Uetrecht J. 2021. Idiosyncratic drug-induced liver injury: mechanistic and clinical challenges. IJMS. 22(6):2954.
- Kwon D, Kim SM, Correia MA. 2020. Cytochrome P450 endoplasmic reticulum-associated degradation (ERAD): therapeutic and pathophysiological implications. Acta Pharm Sin B. 10(1):42–60.
- Lauschke VM, Shafagh RZ, Hendriks DFG, Ingelman-Sundberg M. 2019. 3D primary hepatocyte culture systems for analyses of liver diseases, drug metabolism, and toxicity: emerging culture paradigms and applications. Biotechnol J. 14(7):e1800347.
- McGuire MR, Mukhopadhyay D, Myers SL, Mosher EP, Brookheart RT, Kammers K, Sehgal A, Selen ES, Wolfgang MJ, Bumpus NN, et al. 2021. Progesterone receptor membrane component 1 (PGRMC1) binds and stabilizes cytochromes P450 through a heme-independent mechanism. J Biol Chem. 297(5):101316.
- Waring RH. 2020. Cytochrome P450: genotype to phenotype. Xenobiotica. 50(1):9–18.
Source: Drug Metab Dispos. 2021;50:65–75
Synopsis
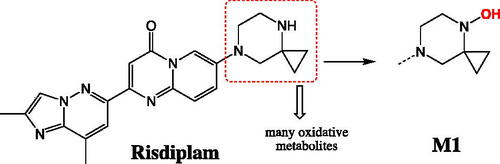
Risdiplam (EVRYSDI®) is a small molecule administered orally that was approved August 2020 by FDA for the treatment of spinal muscular atrophy (SMA). This condition is considered to be rare caused by low levels of functional survival motor neuron (SMN) protein observed in progressive neuromuscular disease. Fowler and colleagues reported on the ADME challenges of risdiplam during drug development. These challenges included low in vitro hepatic clearance, involvement of multiple drug metabolizing enzymes, and assessment of circulating metabolites plus others (Fowler et al. Citation2021). For each aspect, the investigator systemically explored advanced methods to address the ADME challenges. Metabolite generation and circulating exposure was interesting aspect they addressed. From in vitro systems, only oxidative metabolites were detected with M1 (N-hydroxyl metabolite; ) as the major component. Consistently, M1 was detected in human plasma from single ascending dose studies, and it exceeded 10% of the total drug-related component. This metabolite was confirmed in the single dose 14C human mass balance study where no other metabolites reached the 10% limit. In preclinical species, M1 was present high enough levels that allowed adequate assessment of its safety upon orally administered risdiplam. One question posed from the health authorities (HA) during the NDA review process involved a potential concern over M1 having a longer half-life than the parent. Accumulation of M1 would require longer collection to better define the terminal half-life. This concern was resolved by re-processing the existing data that extended up to 216h post dose using high resolution mass spectrometry peak areas. The analysis confirmed that there were no metabolites with longer half-life than the parent (in other words the ratio of metabolite/parent at any given time during the terminal phase remained the same). The HA accepted the reanalysis and considered the issue resolved. As mentioned, M1 was the major metabolite characterized as an N-hydroxyl metabolite. There were also several other oxidative products mainly occurring on the piperazine ring. Cytochrome P450 (CYP) was mainly involved in the formation of the oxidative metabolites, and flavin-containing monooxygenase (FMO) was mainly involved in the formation of the major metabolite M1. Using recombinant enzymes, it was shown that M1 was formed by FMO1 (mainly expressed in fetal liver and adult kidney), FMO3, and CYP2J2 (mainly expressed in the heart), and CYP3A4. Identifying these enzymes opened the door for consideration of extrahepatic metabolism and their potential involvement in the clearance of risdiplam. Microsomal preparations from liver, kidney and intestine highlighted the more prominent role that liver played followed by kidney and intestine. A follow up experiment with methimazole (FMO inhibitor) in kidney fraction highlighted the role of FMO1 played in clearance of risdiplam in this organ. However, when scaled to the whole organ clearance, kidney was estimated to contribute less than 3% of the total hepatic clearance. This study in addition to others suggested low hepatic and intestinal extractions with FH and FG greater than 0.9, respectively. Combined, high bioavailability was expected (with permeability/solubility not limiting) and that was confirmed with less than 20% excreted unchanged in the feces. In a separate study, the drug interaction studies with itraconazole confirmed the less prominent role that CYP3A4 played in clearance of this drug (Sturm et al. Citation2019).
Commentary
Risdiplam is the third approved medication for the treatment of spinal muscular atrophy indication and it is the only that can be taken orally. The others prior approvals are nusinersen (antisense oligonucleotide therapy) and onasemnogene abeparvovec (adeno-associated virus). Each of these medications have a different route of administration that includes intrathecally, intravenous infusion and in the case of risdiplam as a liquid oral solution. Risdiplam has a unique mode of action for a small molecule by binding and modifying the expression of protein at the pre–messenger RNA splicing stage and hence increasing levels of functional SMN protein.
The optimization process that resulted in discovery of risdiplam was previously published (Ratni et al. Citation2018). One of the main ADME objectives was modification of the basic amine such that it lowered the basicity of this moiety. Discovery of risdiplam was a significant improvement over the previously advanced clinical candidate RG7800 that contained N-methylpiperidine (Kletzl et al. Citation2019). In risdiplam, the chemists introduced 2-cyclopropylpiperazine which lowered the pKa by 2 log units (8.8 to 6.8). This modification resulted in addressing the following risk factors: (1) large volume of distribution, (2) the potential for phospholipidosis, (3) hERG inhibition, and (4) reduced N-demethylation of RG7800 and formation of a pharmacologically active metabolite. In the case of risdiplam, there were several oxidative products formed on the 2-cyclopropylpiperazine, but none of them deemed to be pharmacologically active as they resulted in modification/removal of the basic amine.
The significant contribution of FMO3 to the clearance of risdiplam (estimated fm ∼ 0.75) meant that the typical CYP-mediated DDIs was avoided. This reaction posed a potential question for considering FMO3 drug interaction both as a victim and perpetrator. On the other hand, the metabolic product of this enzyme was N-hydroxyl product M1 that is presumably cleared in the bile. M1 could theoretically be converted back to risdiplam through reduction in the GI by microbiome (perhaps other reductive pathways in the GI). If this process happens early in the GI then there is the opportunity for re-absorption back to the systemic circulation and results in enterohepatic recirculation (Roberts et al. Citation2002). This could result in higher-than-expected exposure to the hepatic formation of M1 which could be recovered as risdiplam in the small intestine. It is not clear if this observation could explain over-prediction of clearance by the in vitro hepatic co-culture system.
Finally, risdiplam with its life changing properties for treatment of SMA came with its unique complex ADME challenges. The careful analysis by the investigators were needed to integrate the data from in vitro assay read out to the in vivo assessment with the aid of PBPK modeling.
Figure 8. Major metabolic structures of risdiplam.
References
- Baranello G, Darras BT, Day JW, Deconinck N, Klein A, Masson R, Mercuri E, Rose K, El-Khairi M, Gerber M, et al. 2021. Risdiplam in type 1 spinal muscular atrophy. N Engl J Med. 384(10):915–923.
- Fowler S, Brink A, Cleary Y, Guenther A, Heinig K, Husser C, Kletzl H, Kratochwil NA, Mueller L, Savage M, et al. 2021. Addressing today’s ADME challenges in the translation of in vitro absorption, distribution, metabolism and excretion characteristics to human: a case study of the SMN2 mRNA splicing modifier risdiplam. Drug Metab Dispos. 50(1):65–75.
- Kletzl H, Marquet A, Günther A, Tang W, Heuberger J, Groeneveld GJ, Birkhoff W, Mercuri E, Lochmüller H, Wood C, et al. 2019. The oral splicing modifier RG7800 increases full length survival of motor neuron 2 mRNA and survival of motor neuron protein: results from trials in healthy adults and patients with spinal muscular atrophy. Neuromuscul Disord. 29(1):21–29. DDIs
- Ratni H, Ebeling M, Baird J, Bendels S, Bylund J, Chen KS, Denk N, Feng Z, Green L, Guerard M, et al. 2018. Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J Med Chem. 61(15):6501–6517.
- Roberts MS, Magnusson BM, Burczynski FJ, Weiss M. 2002. Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin Pharmacokinet. 41(10):751–790.
- Sturm S, Günther A, Jaber B, Jordan P, Al Kotbi N, Parkar N, Cleary Y, Frances N, Bergauer T, Heinig K, et al. 2019. A phase 1 healthy male volunteer single escalating dose study of the pharmacokinetics and pharmacodynamics of risdiplam (RG7916, RO7034067), a SMN2 splicing modifier. Br J Clin Pharmacol. 85(1):181–193.
Source: Xenobiotica. 2021;51:155–166
Synopsis
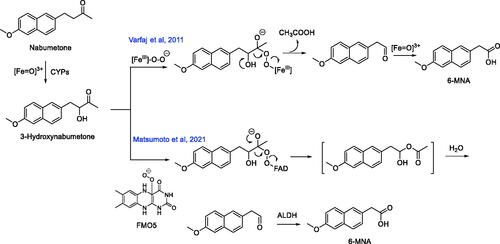
Nabumetone is a non-acidic nonsteroidal anti-inflammatory prodrug that is converted to its pharmacologically active metabolite 6-methoxy-2-naphthyl acetic acid (6-MNA) via oxidative metabolism in the liver (). Approximately ∼35% of an orally administered efficacious nabumetone dose (1000 mg) is converted to 6-MNA in humans (Haddock et al. Citation1984). 6-MNA is a selective inhibitor of the cyclooxygenase-2 enzyme, which catalyzes the conversion of the fatty acid substrate arachidonic acid into proinflammatory prostaglandins (Griswold and Adams Citation1996). Previous studies on nabumetone metabolism revealed 3-hydroxy-nabumetone () as a metabolite of the parent compound in incubation mixtures with liver microsomes from humans and rats (Nobilis et al. Citation2013). This observation suggests that the CYP-derived hydroxylated metabolite is a likely substrate for the secondary oxidative metabolism, which yields 6-MNA. Consistent with this notion, Varfaj et al. (Citation2014) demonstrated that 3-hydroxy-nabumetone undergoes oxidative carbon-carbon bond cleavage in recombinant human CYP1A2 incubations to afford 6-methoxy-2-naphthylacetaldehyde as a metabolite, which can be subsequently oxidized to the corresponding carboxylic acid derivative 6-MNA. The postulated mechanism for the carbon-carbon bond cleavage invoked the peroxy anion (FeIII-O-O-) form of CYP, which adds across the carbonyl carbon in 3-hydroxy-nabumetone to form a tetrahedral intermediate followed by fragmentation to acetic acid and 6-methoxy-2-naphthylacetaldehyde (). All metabolic steps depicted in the postulated mechanism are thought to be principally catalyzed by CYP1A2, since the formation of 6-MNA from nabumetone (and its 3-hydroxy-nabumetone metabolite) were only observed in incubations with recombinant human CYP1A2, which is consistent with previously reported reaction phenotyping studies in human liver microsomes using CYP-selective inhibitors that demonstrated a predominant role of CYP1A2 in nabumetone oxidation (Turpeinen et al. Citation2009). Matsumoto et al. (Citation2021) provide new and compelling evidence for flavin monooxygenase (FMO) 5 involvement in the oxidative metabolism of 3-hydroxy-nabumetone to 6-MNA. A synthetic standard of the 3-hydroxy-nabumetone metabolite was rapidly metabolized to 6-MNA in cryopreserved human hepatocytes but not in NADPH-supplemented human liver S9 fractions, human liver microsomes, human liver S9 fractions, or recombinant human CYP1A2, which contrasted previous observations (Turpeinen et al. Citation2009; Varfaj et al. Citation2014). Of considerable interest are the findings that incubation of 3-hydroxy-nabumetone with cDNA-expressed FMO1, FMO3, or FMO5, followed by addition of the protein-precipitated FMO incubations to human liver S9 factions resulted in the facile conversion of 3-hydroxynabumetone to 6-MNA only in incubation mixtures containing FMO5. Inclusion of 4-(N,N-dimethyl-amino-sulfonyl)-7-hydrazino-2,1,3-benzoxadiazole (a fluorescent aldehyde trapping agent) in the 3-hydroxy-nabumetone/FMO5 incubation mixtures led to the detection of the Schiff base, which suggested that 3-hydroxy-nabumetone was metabolized by FMO5 to 6-methoxy-2-naphthylacetaldehyde (). 6-MNA was also produced in NAD+-supplemented human liver microsomes or human liver cytosol treated with 3-hydroxy-nabumetone/FMO5 reaction mixtures suggesting a role for aldehyde dehydrogenase (ALDH) in the oxidation of 6-methoxy-2-naphthylacetaldehyde to 6-MNA. This finding was consistent with the generation of 6-MNA following incubation of 3-hydroxy-nabumetone/FMO5 reaction mixtures with cDNA-expressed ALDH. Moreover, replacement of recombinant ALDH with cDNA-expressed aldehyde oxidase (AO), which is a cytosolic enzyme, also yielded 6-MNA.
Commentary
The findings of Matsumoto et al. (Citation2021) on the role of human liver FMO5 in the carbon-carbon bond cleavage (reminiscent of a Baeyer-Villiger oxidation) in 3-hydroxynabumetone () are in line with previous observations on the Baeyer-Villiger oxidation capacity reported exclusively with the FMO5 isoform (Lai et al. Citation2011; Leisch et al. Citation2011; Matsumoto et al. Citation2020). Unlike FMO1 and FMO3, FMO5 displays little to no reactivity (monooxygenation of nucleophilic heteroatoms (e.g. nitrogen and sulfur) in prototypic substrates, but FMO5 is capable of carbon-carbon bond cleavage through Baeyer-Villiger oxidations. Incubation of the FMO5-oxidation products of 3-hydroxy-nabumetone with human liver S9, human liver microsomes or cytosol all led to the formation of 6-MNA. The extent of formation of 6-MNA in human liver microsomal or cytosolic incubations was greater in incubations containing NAD+, relative to incubations that did not contain NAD+ co-factor. Replacement of NAD+ with NADPH led to a dramatic reduction in 6-MNA formation. 6-MNA was also observed as a product following incubations of 3-hydroxy-nabumetone/FMO-5 oxidation products with recombinant ALDH or AO suggesting that both ALDH and AO are responsible for the oxidation of the 6-methoxy-2-naphthylacetaldehyde metabolite to 6-MNA. A synthetic standard of 6-methoxy-2-naphthylacetaldehyde was shown to undergo facile oxidation to 6-MNA in NAD+-supplemented human liver cytosol, which provided additional support for the role of ALDH in 6-MNA formation. On the basis of these observations, an alternate pathway for the metabolism of 3-hydroxy-nabumetone to 6-MNA can be proposed, which involves a Baeyer-Villiger oxidation in a FMO-5-dependent fashion to yield an unstable methyl ester intermediate that spontaneously hydrolyzes (presumably by esterases) to 6-methoxy-2-naphthylacetaldehyde. The hydrolytic pathway in human liver S9 fractions was inhibited by ∼28–35% when serine protease and/or arylacetamide deacetylase inhibitors, 4-(2-aminoethyl)benzenesulfonyl fluoride and eserine, respectively, were added to the incubation mixtures. These observations suggested that hydrolysis of the methyl ester intermediate is at least partially mediated by proteases. Oxidation of the aldehyde metabolite by ALDH yields the active metabolite 6-MNA.
In most cases, pharmacologically active metabolites are generally derived from CYP-catalyzed oxidative transformations on the parent compound (Obach Citation2013). Nabumetone presents an interesting case study wherein, the pharmacologically active metabolite appears to be derived from oxidative transformation mediated by multiple drug metabolizing enzymes including CYP, FMO5, esterase(s), and ALDH. The discrepancy regarding the predominant involvement of CYP1A2 (Varfaj et al. Citation2014) versus multiple CYP and non-CYP enzymes in facilitating the metabolism of nabumetone to 6-MNA (Matsumoto et al. Citation2021) requires additional scrutiny.
Figure 9. Known metabolic pathways for the oxidative cleavage of nonsteroidal anti-inflammatory prodrug nabumetone to its biologically active metabolite 6-MNA. ALDH: Aldehyde dehydrogenase.
References
- Griswold DE, Adams JL. 1996. Constitutive cyclooxygenase (COX-1) and inducible cyclooxygenase (COX-2): rationale for selective inhibition and progress to date. Med Res Rev. 16(2):181–206.
- Haddock RE, Jeffery DJ, Lloyd JA, Thawley AR. 1984. Metabolism of nabumetone (BRL 14777) by various species including man. Xenobiotica. 14(4):327–337.
- Lai WG, Farah N, Moniz GA, Wong N. 2011. A Baeyer-Villiger oxidation specifically catalyzed by human flavin-containing monooxygenase 5. Drug Metab Dispos. 39(1):61–70.
- Leisch H, Morley K, Lau PC. 2011. Baeyer-Villiger Monooxygenases: more than just green chemistry. Chem Rev. 111(7):4165–4222.
- Matsumoto K, Hasegawa T, Ohara K, Kamei T, Koyanagi J, Akimoto M. 2021. Role of human flavin-containing monooxygenase (FMO) 5 in the metabolism of nabumetone: Baeyer-Villiger oxidation in the activation of the intermediate metabolite, 3-hydroxynabumetone, to the active metabolite, 6-methoxy-2-naphthylacetic acid in vitro. Xenobiotica. 51(2):155–166.
- Matsumoto K, Hasegawa T, Ohara K, Takei C, Kamei T, Koyanagi J, Takahashi T, Akimoto M. 2020. A metabolic pathway for the prodrug nabumetone to the pharmacologically active metabolite, 6-methoxy-2-naphthylacetic acid (6-MNA) by non-cytochrome P450 enzymes. Xenobiotica. 50(7):783–792.
- Nobilis M, Mikušek J, Szotáková B, Jirásko R, Holčapek M, Chamseddin C, Jira T, Kučera R, Kuneš J, Pour M. 2013. Analytical power of LLE-HPLC-PDA-MS/MS in drug metabolism studies: identification of new nabumetone metabolites. J Pharm Biomed Anal. 80:164–172.
- Obach RS. 2013. Pharmacologically active drug metabolites: impact on drug discovery and pharmacotherapy. Pharmacol Rev. 65(2):578–640.
- Turpeinen M, Hofmann U, Klein K, Mürdter T, Schwab M, Zanger UM. 2009. A predominate role of CYP1A2 for the metabolism of nabumetone to the active metabolite, 6-methoxy-2-naphthylacetic acid, in human liver microsomes. Drug Metab Dispos. 37(5):1017–1024.
- Varfaj F, Zulkifli SNA, Park HG, Challinor VL, De Voss JJ, Ortiz de Montellano PR. 2014. Carbon-carbon bond cleavage in activation of the prodrug nabumetone. Drug Metab Dispos. 42(5):828–838.
Source: Drug Metab Dispos. 2022;50(3):235–242
Synopsis
The conveyance of action potentials in the nervous system is principally mediated by the voltage-gated sodium channels (NaVs). A gain of function mutation in the SCN9A gene, which encodes NaV1.7, leads to very painful disorders such as peripheral erythromelalgia and paroxysmal extreme pain disorder. Conversely, loss of function of the SCN9A genes results in a rare genetic condition called congenital insensitivity to pain. The genetic evidence suggests that selective inhibition of NaV1.7 isoform by small molecule agents is an appropriate therapeutic option to treat pain. Shinozuka et al. (Citation2020) have recently disclosed the identification of a potent and selective small molecule inhibitor (DS-1971a) of NaV1.7, which was primarily derived through iterative structure-activity relationship studies on the previously reported NaV1.7 inhibitors 1 and PF-05089771 (. DS-1971a has demonstrated high NaV1.7 inhibitory potency and selectivity in vitro, with robust efficacy achieved in mouse models of analgesia. Moreover, a favorable toxicological profile in preclinical species (mice and monkeys) enabled multiple ascending dose studies with DS-1971a up to 1200 mg/day administered to healthy volunteers for 14 days (Shinozuka et al. Citation2020). In the present work (Asano et al. Citation2022), human mass balance, metabolism, and excretion studies were conducted with [14C]-DS-1971a following administration of a single 400 mg dose to six healthy male subjects. Metabolite M1 (, derived from the monohydroxylation of the cyclohexanone ring in DS-1971a, was the principal entity in circulation and in excreta. Quantitation of the systemic concentrations of M1 revealed that the area under the plasma concentration versus time curve (AUC) was ∼1.5-fold greater than that of the parent compound. Metabolite M2, a regioisomer of M1 (, was also detected in plasma (and excreta) with an estimated AUC of 58% relative to DS-1971a. The AUC ratios for M1 and M2 relative to total drug-related materials were 27% and 10%, respectively, suggesting that M1 and potentially M2 needed to be qualified in preclinical toxicity species in accordance with the metabolites in safety testing (MIST) guidance (Schadt et al. Citation2018). Although the AUC ratio of M2 (as a percentage of the total exposure) was comparable in animals and human, the AUC ratio of M1 was considerably higher in humans than in mice, rats, dogs, and monkeys, implying that M1 is a disproportionate human metabolite. In vitro studies in human liver microsomes in the presence of CYP isoform-selective inhibitors established that DS-1971a was converted to M1 and M2, primarily via CYP2C8 (with CYP2C9 being a possible minor contributor). Assessment of species differences in the in vitro metabolism of DS-1971a revealed that M1 was the dominant metabolite in human liver microsomes while M2 was the principal metabolite in liver microsomes from mice, dog, and monkey. M1 and a another regioisomeric cyclohexanone ring oxidation product M5 ( were the major metabolites of DS-1971a in rats. Characterization of circulating metabolites of DS-1971a in preclinical species used for toxicity assessments revealed that M4, derived from the aldehyde oxidase (AO) mediated oxidation of the pyrimidine ring in DS-1971a (, and M2 were the most abundant circulating metabolites in male mice and male monkeys, respectively (Asano et al. Citation2021). The AO generated metabolite M4 was not a circulatory metabolite in humans, but was detected as a minor metabolite in excreta.
Commentary
Prediction of major human circulating metabolites that will potentially require MIST qualification has proven particularly challenging for compounds with low metabolic turnover and/or considerable species differences in metabolism in in vitro reagents (e.g. liver microsomes and hepatocytes from animals and human) (Sharma et al. Citation2014; Schadt et al. Citation2018; Surapaneni et al. Citation2021; Fowler et al. Citation2022). Considerable advances, however, have been made toward estimating intrinsic clearance (for human pharmacokinetics prediction) and qualitative metabolite characterization of low clearance compounds in animal and human reagents (e.g. Hepatopac micropatterned coculture model primary hepatocytes) that extend the incubation times to days (Ballard et al. Citation2020; Kamel et al. Citation2021). The Hepatopac model, in particular, has shown much promise with respect to pinpointing species differences in the rate and extent of metabolism and suitability in establishing a correlation with in vivo metabolism across animals and humans (Kamel et al. Citation2021). DS-1971a is certainly a candidate for the “low clearance/slow metabolism” category, given that its measured terminal half-life in the human mass balance study was ∼14 h. While in vitro and in vivo metabolic profiling in mice, monkeys, and humans proved useful in establishing a species difference in metabolism and predicting major circulatory metabolites (M1 in humans, M4 in mice, and M2 in monkeys), precision around the quantitative aspects of the circulating metabolites of DS-1971a was difficult to achieve without experimental data. The DS-1971a case study provides yet another example for the need to conduct radiolabeled human mass balance studies in a timely fashion for the characterization of disproportionate metabolites and assessment of exposure coverage in animals. Although the order of AO activity among species is substrate-dependent, it is generally accepted that AO activity is high in humans and monkeys and low in rats, whereas dogs are largely deficient in AO activity (Pryde et al. Citation2010). In the present case, CYP-derived M1 and/or M2 were the major circulatory metabolites in monkeys and humans, rather than the AO-derived metabolite M4. Incubations of DS-1971a in Hepatopac cultures from animals and humans can perhaps shed additional light on the in vitro metabolic profile.
Tactics to qualify M1 in animal species used in toxicology assessments remain unclear. A possible strategy centers around dosing of M1 in animals to evaluate it toxicity potential. As such, the disproportionate human metabolite M1 was considerably less potent as a NaV 1.7 inhibitor (DS-1971a IC50 = 20 nM; M1 IC50 = 310 nM), and appears to have minimal inhibitory attributes toward the major human CYP enzymes (Asano et al. Citation2022). The involvement of CYP2C8/2C9 in the oxidative metabolism of DS-1971a does not appear to be surprising given the weakly acidic nature (predicted pKa of 6.3) of sulfonamido-pyrimidine motif in DS-1971a.
Figure 10. (A) Identification of the potent and selective NaV1.7 inhibitor DS-1971a via optimization of the previously disclosed benzenesulfonamide chemotypes from 1 and PF-05089771. (B) Major metabolites of DS-1971a in animals and human.
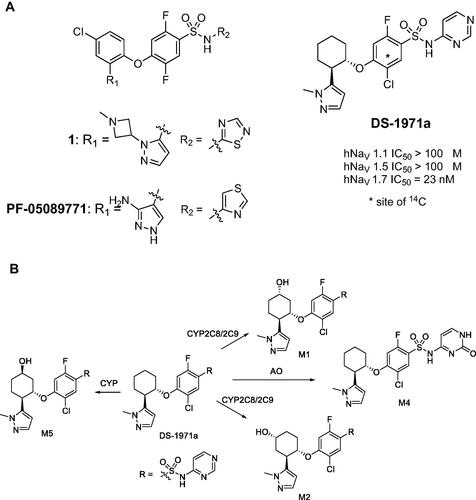
References
- Asano D, Hamaue S, Zahir H, Shiozawa H, Nishiya Y, Kimura T, Kazui M, Yamamura N, Ikeguchi M, Shibayama T, et al. 2022. CYP2C8-mediated formation of a human disproportionate metabolite of the selective Na(V)1.7 inhibitor DS-1971a, a mixed cytochrome P450 and aldehyde oxidase substrate. Drug Metab Dispos. 50(3):235–242.
- Asano D, Shibayama T, Shiozawa H, Inoue SI, Shinozuka T, Murata S, Watanabe N, Yoshinari K. 2021. Evaluation of species difference in the metabolism of the selective Na(V)1.7 inhibitor DS-1971a, a mixed substrate of cytochrome P450 and aldehyde oxidase. Xenobiotica. 51(9):1060–1070.
- Ballard TE, Kratochwil N, Cox LM, Moen MA, Klammers F, Ekiciler A, Goetschi A, Walter I. 2020. Simplifying the execution of hepatopac metID experiments: metabolite profile and intrinsic clearance comparisons. Drug Metab Dispos. 48(9):804–810.
- Fowler S, Brink A, Cleary Y, Günther A, Heinig K, Husser C, Kletzl H, Kratochwil N, Mueller L, Savage M, et al. 2022. Addressing today’s absorption, distribution, metabolism, and excretion (ADME) challenges in the translation of in vitro ADME characteristics to humans: a case study of the SMN2 mRNA splicing modifier risdiplam. Drug Metab Dispos. 50(1):65–75.
- Kamel A, Bowlin S, Hosea N, Arkilo D, Laurenza A. 2021. In vitro metabolism of slowly cleared G protein-coupled receptor 139 agonist TAK-041 using rat, dog, monkey, and human hepatocyte models (HepatoPac): correlation with in vivo metabolism. Drug Metab Dispos. 49(2):121–132.
- Pryde DC, Dalvie D, Hu Q, Jones P, Obach RS, Tran TD. 2010. Aldehyde oxidase: an enzyme of emerging importance in drug discovery. J Med Chem. 53(24):8441–8460.
- Schadt S, Bister B, Chowdhury SK, Funk C, Hop CECA, Humphreys WG, Igarashi F, James AD, Kagan M, Khojasteh SC, et al. 2018. A decade in the MIST: learnings from investigations of drug metabolites in drug development under the “metabolites in safety testing” regulatory guidance. Drug Metab Dispos. 46(6):865–878.
- Sharma R, Litchfield J, Atkinson K, Eng H, Amin NB, Denney WS, Pettersen JC, Goosen TC, Di L, Lee E, et al. 2014. Metabolites in safety testing assessment in early development: a case study with a glucokinase activator. Drug Metab Dispos. 42(11):1926–1939.
- Shinozuka T, Kobayashi H, Suzuki S, Tanaka K, Karanjule N, Hayashi N, Tsuda T, Tokumaru E, Inoue M, Ueda K, et al. 2020. Discovery of DS-1971a, a potent, selective NaV1.7 inhibitor. J Med Chem. 63:10204–10220.
- Surapaneni S, Yerramilli U, Bai A, Dalvie D, Brooks J, Wang X, Selkirk JV, Yan YG, Zhang P, Hargreaves R, et al. 2021. Absorption, metabolism, and excretion, in vitro pharmacology, and clinical pharmacokinetics of ozanimod, a novel sphingosine 1-phosphate receptor modulator. Drug Metab Dispos. 49(5):405–419.
Source: Drug Metab Dispos. 2021;49(6):420–433
Synopsis
This paper describes how darolutamide was excreted and cleared based on data generated from in vivo and in vitro investigations. During the human absorption, distribution, metabolism, and excretion (hADME) study of darolutamide, the authors observed that a 1:1 ratio of (S,R) and (S,S)-administered darolutamide equilibrated into an approximate 1:5 ratio in plasma. The interconversion occurred via formation of keto-darolutamide (). Based on in vitro data, parent diastereomers were oxidized to keto-darolutamide, mainly via CYP3A4 metabolism. Keto-darolutamide was then reduced back to parent diastereomers by cytosolic aldo-ketoreductase 1C3 (AKR1C3), which preferred the formation of (S,S)-darolutamide. The darolutamide diastereomers and keto-darolutamide were equally pharmacologically active (Moilanen et al. Citation2015; Sugawara et al. Citation2019). After a single dose of 300 mg [14C]-darolutamide, almost complete mass balance was achieved within one week after administration, where 63% of administered radioactivity was excreted in urine and 32% in feces. Keto-darolutamide was the major metabolite, accounting for 59% of total plasma exposure, which was 2.1-fold higher than parent darolutamide. The O-glucuronide (M-7a/b) and N-glucuronide (M-15a/b) metabolites formed mainly by UGT1A9 and UGT1A1, pyrazole sulfates (M-29, M-24) and glucuronides (M-21, M-22) were most abundant urinary metabolites. Parent diastereomers were the principal drug-related material identified in feces. Due to multiple clearance and excretion pathways, the authors concluded that there is a relatively low drug-drug interaction potential for darolutamide upon co-administration of inhibitors or inducers of drug metabolizing enzymes.
Commentary
Darolutamide (Nubeqa, formerly ODM-201), consisting of an equimolar mixture of two diastereomers, is a potent and selective nonsteroidal androgen receptor inhibitor recently approved for the treatment of nonmetastatic castration-resistant prostate cancer (nmCRPC). More than half of the drugs currently in use are chiral products and approximately 90% of chiral drugs are sold as racemates (Walther and Netscher Citation1996; Rentsch Citation2002; Katzung Citation2004). It is important to achieve chiral separation analytically and analyze each isomer in order to understand potential differences in pharmacology, toxicology, pharmacokinetics, and metabolism of chiral drugs. Both FDA and EMA recommend the assessment of in vivo interconversion of chiral drugs and pharmacokinetic profile of each isomer (FDA Citation1992; EMA Citation1993). This publication by Taavitsainen et al. is interesting in that a higher amount of (S,S)-darolutamide was detected in plasma and excreta when an equimolar ratio of (S,R)- and (S,S)-darolutamide was administered to humans. Unlike the majority of racemic drugs, which have one major bioactive isomer, the pharmacological activities of these two diastereomers were similar (Moilanen et al. Citation2015). The authors discovered that the interconversion occurred via the major metabolite keto-darolutamide () with a preferential formation of (S,S)-darolutamide. It is worthy to note that (S,R)-darolutamide eliminated faster than (S,S)-darolutamide and keto-darolutamide. Interestingly, a similar stereospecific ratio was observed with the formed glucuronide metabolites via direct glucuronidation of darolutamide, which was one of the major elimination pathways.
Based on in vitro data, the observed in vivo interconversion of darolutamide diastereomers was due to the oxidation of darolutamide to keto-darolutamide mainly by CYP3A4 and reduction of keto-darolutamide preferably to (S,S)-darolutamide with aldo-ketoreductase 1C3 (AKR1C3) playing a major role. The involvement of CYP3A4 was evidenced by the reduced darolutamide metabolism with CYP3A4 inhibitors and the depletion of darolutamide in human recombinant CYP3A4 incubation. The formation of darolutamide from keto-darolutamide in human liver cytosol and human recombinant AKR1C3, and the reduced formation of darolutamide from keto-darolutamide with AKR inhibitors confirmed that cytosolic AKRs were responsible for the reduction reaction. When keto-darolutamide was incubated in human hepatocytes with no inhibitor and with CYP3A4 inhibitor, a significant formation of (S,S)- darolutamide was observed compared to (S,R)-darolutamide with and without a CYP3A4 inhibitor. This formation was substantially decreased when keto-darolutamide was incubated with human hepatocytes and an ARK1C3 inhibitor, demonstrating that ARK1C3 was responsible for the favorable formation of (S,S)-darolutamide. The authors investigated the stability of keto-darolutamide in human fecal samples under anaerobic conditions and concluded that keto-darolutamide reduced back to darolutamide by microbiome in the human gastrointestinal tract.
Additionally, the authors isolated and elucidated structures of O-glucuronide and N-glucuronide metabolites using pooled human urine. The UGT reaction phenotyping studies demonstrated with UGT1A9, UGT1A1, UGT1A3, and UGT2B10 were mainly responsible for the formation of these glucuronide metabolites. Overall, a careful analysis of in vivo data and in-depth in vitro mechanistic studies are important in developing chiral drugs.
Figure 11. Interconversion of the two diastereomers of darolutamide via keto-darolutamide.
References
- Moilanen AM, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, Nykänen PS, Törmäkangas OP, Palvimo JJ, Kallio PJ. 2015. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep. 5:12007.
- Sugawara T, Baumgart SJ, Nevedomskaya E, Reichert K, Steuber H, Lejeune P, Mumberg D, Haendler B. 2019. Darolutamide is a potent androgen receptor antagonist with strong efficacy in prostate cancer models. Int J Cancer. 145(5):1382–1394.
- Rentsch KM. 2002. The importance of stereoselective determination of drugs in the clinical laboratory. J Biochem Biophys Methods. 54(1–3):1–9.
- Walther W, Netscher T. 1996. Design and development of chiral reagents for the chromatographic determination of chiral alcohols. Chirality. 8(5):397–401.
- Katzung BG. 2004. The nature of drugs. In: Basic and clinical pharmacology. 9th ed. New York: Lange Medical Books/McGraw Hill; p. 3–5.
- U.S. Food and Drug Administration (FDA). 1992. Guidance for development of new stereoisomeric drugs. Center for drug evaluation and research. US department of health and human services, Rockville, MD.
- European Medicines Agency (EMA). 1993. Investigation of chiral active substances. European Medicines Agency, London.
Source: Drug Metab Dispos. 2021;49:548–562
Synopsis
Wang-Lakshman et al. determined the absorption, metabolism, and excretion of the highly potent farnesoid X receptor agonist tropifexor in humans (Wang-Lakshman et al. Citation2021). In this work, [14C]-tropifexer (1 mg) was administered orally to four healthy male research participants. Following dosing, the investigators found that the major clearance pathway of tropifexor was oxidative metabolism. Additionally, it was determined that direct glucuronidation is involved in tropifexor clearance. The relative contribution of oxidation and glucuronidation was found to be concentration-dependent based on in vitro experiments. Taken together, this work highlights the importance of both in vivo and in vitro experiments for a better understanding of disposition processes during drug development.
Commentary
Tropifexor is a highly potent farnesoid X receptor agonist for the treatment of nonalcoholic steatohepatitis (Tully et al. Citation2017). To date, there are several farnesoid X receptor agonists in clinical development to treat nonalcoholic steatohepatitis. However, some of these investigational drugs such as obeticholic acid have adverse effects on lipid profiles (Polyzos et al. Citation2020). Therefore, to address this challenge, there is a need for the development of novel drugs with improved efficacy, safety, and tolerability.
Tropifexor was discovered in 2017 and it exhibited a marked reduction in fibrosis, steatohepatitis, and profibrogenic gene expression with a higher efficacy compared to obeticholic acid in preclinical nonalcoholic steatohepatitis models (Hernandez et al. Citation2019). In addition, the first-in-human study demonstrated that tropifexor was well tolerated in healthy volunteers with an acceptable safety profile, and it had a pharmacokinetic profile suitable for once-daily dosing (Badman et al. Citation2020). However, the overall disposition of tropifexor in humans was unknown. A thorough understanding of tropifexor disposition is needed to define its efficacy and safety during its further development as a drug.
Therefore, in order to fill this knowledge gap, Wang-Lakshman et al. determined the absorption, metabolism, and excretion of tropifexor in healthy research participants. Following a 1-mg oral dose of [14C]-tropifexer, the authors found that unchanged tropifexor was the most prominent drug-related component in plasma. In addition, they detected two minor oxidative metabolites of tropifexor in plasma (O-dealkylation and mono-oxidation metabolites). Oxidative metabolites were detected in feces, whereas metabolites observed in urine were results of oxidative O-dealkylation and combined oxidation and glucuronidation. The proposed sites of oxidations of tropifexor are shown in . Importantly, in vitro metabolism experiments revealed that the relative contribution of oxidation and glucuronidation was concentration dependent.
Different metabolic pathways may be involved in the biotransformation of drugs depending on the concentration. Of note, tropifexor has high potency and requires low doses. This work highlights the importance of considering therapeutic exposure levels of drugs such as tropifexor to enable assessments reflecting clinically relevant conditions. Specifically, the authors demonstrated that glucuronidation as the predominant clearance pathway of tropifexor at higher concentrations while oxidative metabolism plays a major role at lower concentrations.
Figure 12. Proposed sites of oxidations of tropifexor.
References
- Badman MK, Chen J, Desai S, Vaidya S, Neelakantham S, Zhang J, Gan L, Danis K, Laffitte B, Klickstein LB.,. 2020. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the novel non-bile acid FXR agonist tropifexor (LJN452) in healthy volunteers. Clin Pharmacol Drug Dev. 9(3):395–410.
- Hernandez ED, Zheng L, Kim Y, Fang B, Liu B, Valdez RA, Dietrich WF, Rucker PV, Chianelli D, Schmeits J, et al. 2019. Tropifexor-mediated abrogation of steatohepatitis and fibrosis is associated with the antioxidative gene expression profile in rodents. Hepatol Commun. 3(8):1085–1097.
- Polyzos SA, Kountouras J, Mantzoros CS. 2020. Obeticholic acid for the treatment of nonalcoholic steatohepatitis: expectations and concerns. Metabolism. 104:154144.
- Tully DC, Rucker PV, Chianelli D, Williams J, Vidal A, Alper PB, Mutnick D, Bursulaya B, Schmeits J, Wu X, et al. 2017. Discovery of tropifexor (LJN452), a highly potent non-bile acid FXR agonist for the treatment of cholestatic liver diseases and nonalcoholic steatohepatitis (NASH). J Med Chem. 60(24):9960–9973.
Source: Invest New Drugs. 2021;39(1):1–14
Synopsis
Ibrutinib is a first-in-class Bruton’s kinase inhibitor to treat patients with multiple lymphomas. P450 3A4 – mediated oxidation to ibrutinib-diol was the major metabolism pathway for ibrutinib. However, extrahepatic glutathione conjugation and subsequent metabolism can become important clearance pathways for the drug, especially in hepatic impaired patients or when P450 3A4 is inhibited. Further, ibrutinib-GSH was degraded to ibrutinib-CysGly and ibrutinib-Cys. This work elucidated the metabolism of ibrutinib through glutathione conjugation in cultured human renal proximal tubule cells (ciPTEC). In addition, ibrutinib-mediated toxicity in the kidney cell was enhanced by inhibitors of BCRP, P-gp, or MRP that resulted in accumulation of conjugation metabolites. Ibrutinib also showed nephrotoxicity and high concentrations of conjugate metabolites in kidneys of Cyp3a knockout mice. These results supported that inhibition of downstream metabolism of the GSH conjugate or inhibition of transport of these related metabolites could lead to nephrotoxicity. It is important to point out that in this paper extrahepatic conjugation biotransformation of ibrutinib () was labeled as bioactivation that normally refers to a metabolic reaction leading to formation of a reactive metabolite.
Commentary
Patients treated with P450 3A4 inhibitor voriconazole showed elevated exposures and longer half-lives of ibrutinib, ibrutinib-CysGly and ibrutinib-Cys. Cultured ciPTEC cells metabolized ibrutinib to ibrutinib-GSH as well as ibrutinib-CysGly and ibrutinib-Cys as subsequent degradation products of the glutathione conjugate. Glutathione conjugation appeared to be enhanced by glutathione transferase (GSTs) as the conjugate formation was inhibited by ethacrynic acid in ciPTEC cell lysates. The degradation of ibrutinib-GSH to ibrutinib-CysGly was catalyzed by γ-glutamyl transferase (γ-GT) in ciPTEC cells. Human plasma contained a high level of this enzyme (@16 U/mL γ-GT). Ibrutinib, ibrutinib-GS, ibrutinib-CysGly and ibrutinib-Cys were substrates of BCRP, P-gp, and MRP as respective Ko143, PSC833, and MK571 inhibitors blocked efflux of ibrutinib and its conjugate metabolites. Increased cellular concentration of ibrutinib-Cys may relate to the toxicity of ciPTEC cells treated with ibrutinib and transporter inhibitors.
In Cyp3a4 knockout mice, ibrutinib thiol-conjugates were found in tissues and ibrutinib-Cys was found at a 2.7-fold higher concentration in kidneys compared to that in wild-type mice. Ibrutinib-Cys can be metabolized to ibrutinib-SH by β-lyase that is highly expressed in kidney cells. Ibrutinib-SH can also be further metabolized to form ibrutinib-SGlu, ibrutinib-SMe, or even ibrutinib-SO.
GSH-conjugation could represent up to 30% of ibrutinib dose in humans, especially when P450 3A4 is inhibited. The conjugation pathway was minor in rats or mice due to a short half-life of ibrutinib in rodents (@ 1 h compared to 4–12 h in humans) and less time to react to thiols. The extrahepatic pathways also provided an explanation for the discrepancy between predicted clearance from hepatocyte incubations and the total body clearance of ibrutinib (Shibata and Chiba Citation2015).
Figure 13. Biotransformation pathways of ibrutinib.
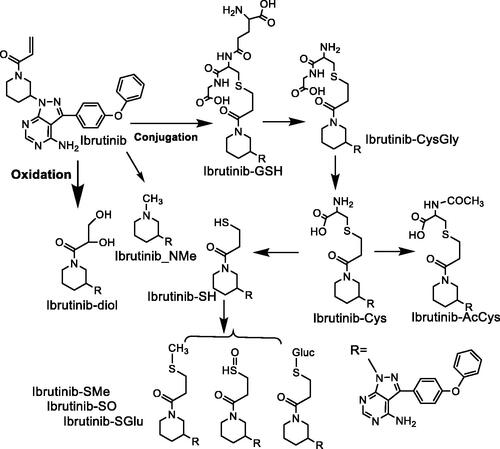
References
- Rood JJM, Jamalpoor A, van Hoppe S, van Haren MJ, Wasmann RE, Janssen MJ, Schinkel AH, Masereeuw R, Beijnen JH, Sparidans RW. 2021. Extrahepatic metabolism of ibrutinib. Invest New Drugs. 39(1):1–14.
- Shibata Y, Chiba M. 2015. The role of extrahepatic metabolism in the pharmacokinetics of the targeted covalent inhibitors afatinib, ibrutinib, and neratinib. Drug Metab Dispos. 43(3):375–384.
Source: Drug Metab Dispos. 2021;49(8):601–609
Synopsis
CC112273 is the human major circulating metabolite of ozanimod that was formed via MAO-B mediated oxidative deamination from RP101075, which was the N-dealkyl metabolite of ozanimod (). In this manuscript, KMapp and Vmax for this metabolism were determined and compared using liver mitochondrial fractions from human, monkey, rat, and mouse. Marked species differences were observed with primates showing 5 to 10-fold lower KMapp, while Vmax was similar, roughly within 2-fold of each other. The calculated CLint showed that human and monkey are much more efficient (10–50 fold) at catalyzing this reaction than rat and mouse. Human recombinant MAO-B protein was also examined and showed consistent KM with human mitochondrial fractions after normalizing nonspecific binding using in silico prediction based on lipophilicity. Additionally, using selective MAO-A and MAO-B inhibitors clorgyline and R-deprenyl, the formation of CC112273 was determined to be solely mediated by MAO-B. The authors also explored this further using RP101074 (the S-enantiomer of RP101075). Interestingly, the formation of CC112273 was pretty much abolished under similar conditions, which speaks to the stereo specificity of this biotransformation at this position. Docking studies with a MAO-B and trans, trans-farnesol co-crystal structure demonstrated that the positioning of the alpha C-H bond was much closer to the N5 atom of the cofactor FAD with R-enantiomer RP101075, but positioning away with the S-enantiomer RP101074. The rate-determining step was thus hypothesized to be hydride abstraction and transfer from the alpha-carbon followed by hydrolysis of the imine intermediate ().
Commentary
The investigation around CC112273 using in vitro liver mitochondrial fractions, recombinant enzymes, and selective inhibitors helped confirm/tease out the contribution of MAO enzymes. CC112273 also presented a challenging case in vivo, being an active circulating metabolite with similar potency and selectivity as ozanimod, but 12-fold longer terminal half-life (∼10 days) than ozanimod (∼20 h) (Tran, Zhang Ghosh, et al. Citation2020). As a result, ozanimod could be considered as a “prodrug” with CC112273 to be the main driver for the pharmacological driver. CC112273 represented 33% of total drug related material (6.7% for ozanimod) from AUC0–96 following a single oral dose of 1 mg ozanimod (Surapaneni et al. Citation2021). From 28 days of repeated daily dosing, CC112273 to ozanimod ratio increased from 1.9 on day 1 to 13 on day 28, making CC112273 the major player for the pharmacological activity with ozanimod acting as a “prodrug” (Tran, Zhang, Walker, et al. Citation2020). Its clearance was characterized and mainly driven by CYP2C8 catalyzed oxidation on the indane. CYP2C8 inhibitor gemfibrozil increased the AUC of CC112273 by 47%, while rifampin reduced its AUC by 60% via 2C8 induction. Although being an in vitro MAO-B inhibitor, CC112273 showed minimal inhibitory potential against platelet MAO-B through the monitoring of blood pressure with co-dosing of pseudoephedrine (Tran, Zhang, Walker, et al. Citation2020). This indicated a potential differences of MAO-B enzymes in platelets vs hepatocytes.
Figure 14. Proposed mechanism of oxidation of RP101075 by MAO-B.
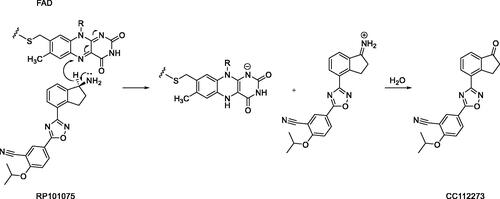
References
- Surapaneni S, Yerramilli U, Bai A, Dalvie D, Brooks J, Wang X, Selkirk JV, Yan YG, Zhang P, Hargreaves R, et al. 2021. Absorption, metabolism, and excretion, in vitro pharmacology, and clinical pharmacokinetics of ozanimod, a novel sphingosine 1-phosphate receptor modulator. Drug Metab Dispos. 49(5):405–419.
- Tran JQ, Zhang P, Ghosh A, Liu L, Syto M, Wang X, Palmisano M. 2020. Single-dose pharmacokinetics of ozanimod and its major active metabolites alone and in combination with gemfibrozil, itraconazole, or rifampin in healthy subjects: a randomized, parallel-group, open-label study. Adv Ther. 37(10):4381–4395.
- Tran JQ, Zhang P, Walker S, Ghosh A, Syto M, Wang X, Harris S, Palmisano M. 2020. Multiple-dose pharmacokinetics of ozanimod and its major active metabolites and the pharmacodynamic and pharmacokinetic interactions with pseudoephedrine, a sympathomimetic agent, in healthy subjects. Adv Ther. 37(12):4944–4958.
Source: Drug Metab Dispos. 2021;49:405–419
Synopsis
Ozanimod is a sphingosine 1-phosphate (S1P) receptor modulator approved for the treatment of relapsing forms of multiple sclerosis. Surapaneni et al. (Citation2021) described absorption, metabolism and excretion of ozanimod after a single oral dose of 1 mg [14C]-ozanimod hydrochloride in six healthy subjects (Surapaneni et al. Citation2021). The total mean recovery of the administered radioactivity was 63%, with 26% and 37% recovered from urine and feces, respectively. The major metabolic pathways of ozanimod were determined: (1) aldehyde dehydrogenase (ALDH) and alcohol dehydrogenase (ADH) generating an active metabolite, RP101988, (2) cytochrome P450 isoform 3A4 and 1A2 generating an active metabolite, RP101075, (3) MAO-B generating an active metabolite, CC112273 from RP101075 and (4) reductive metabolism by gut microflora generating RP112374. More detailed work in regards to the role MAO-B in the generation of CC112273 (Bai et al. Citation2021) was reviewed in the previous section. Extensive in vitro works elucidated that monoamine oxidase B, carbonyl reductases, aldo-keto reductase 1C1/1C2, and 3β-and 11β-hydroxysteroid dehydrogenase were involved in the metabolic pathway related to the generation of active metabolites in the circulation. Of note, gut microbiota was found to mediate the generation of 1, 2, 4-oxadiazole ring opened metabolite. The authors further discovered the loss of radioactivity as 14CO2 when RP112533 (the fecal metabolite of ozanimod) was incubated with human fecal homogenates. This observation suggest that anaerobic bacterial metabolism led to loss of CO2 which may contribute to the low recovery of radioactivity in the mass balance study. Overall, this study well described the metabolic pathways of ozanimod and the potential role of gut microbiota on ozanimod.
Commentary
Mass balance and metabolite profiling studies using a radio-labeled drug provide valuable DMPK information such as (1) major excretion route of drug-related compounds and how quickly they are excreted out of the body, (2) major clearance pathways of a drug, (3) the presence of circulating metabolites (active or inactive), (4) any sequestration of drug-related compounds in the body and its potential related to toxicity (Roffey et al. Citation2007). Surapaneni et al. (Citation2021) well described how the data from the mass balance could be a starting point for elucidation of the metabolic pathways of ozanimod. Their extensive in vitro studies successfully elucidated the enzymes involved in the metabolic pathways of ozanimod leading to the generation of active metabolites in the circulation. Interestingly, the authors also demonstrated that gut microbiota mediated the reductive cleavage of 1,2,4-oxadiazole ring of ozanimod as well as decarboxylation of RP112533 leading to generation of 2-hydroxybenonitrile and loss of 14CO2 (). This finding may explain the relatively low recovery of [14C] in the mass balance study.
Gut microbiota harbor high metabolic capacity, which metabolizes a wide range of drugs with diverse chemical moieties (Zimmermann et al. Citation2019). The author showed reductive cleavage of 1,2,4-oxadiazole ring of ozanimod by gut microflora. Relevant chemical modification by gut microbiota was also reported that reductive ring cleavages of isoxazole ring in the risperidone and zonisamide were determined by the incubation of rat cecum (Meuldermans et al. Citation1994; Kitamura et al. Citation1997). Nitro-reduction leading to cleavage of N–O bond is commonly reported chemical reactions mediated by gut microbiota (Elmer and Remmel Citation1984; Takeno and Sakai Citation1991). Taken together, this potentially suggest the broad applicability of gut microbiota-mediated reductive cleavage on the five membered rings with N–O bond which is initiated by N–O reduction.
Multiple studies reported the gut microbiota mediated decarboxylation reactions such as decarboxylation of L-DOPA and tyrosine (van de Steeg et al. Citation2018; O’Donnell et al. Citation2020). The elegant work from van Kessel et al. showed that bacterial tyrosine decarboxylase converted L-DOPA to dopamine and its abundance in proximal small intestine had a high impact on levels of L-DOPA in the plasma of rats (van Kessel et al. Citation2019) which suggest gut microbiota could modulate the absorption and systemic exposure of L-DOPA. The findings from Surapaneni et al. can be another good example that gut microflora can make significant impact on the fate of xenobiotic in humans.
Figure 15. Metabolism of ozanimod by gut microbiota.
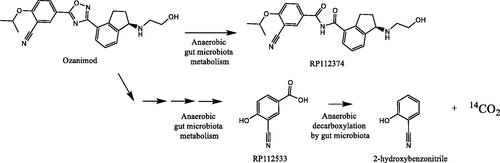
References
- Bai A, Shanmugasundaram V, Selkirk JV, Surapaneni S, Dalvie D. 2021. Investigation into MAO B-mediated formation of CC112273, a major circulating metabolite of ozanimod, in humans and preclinical species: stereospecific oxidative deamination of (S)-enantiomer of indaneamine (RP101075) by MAO B. Drug Metab Dispos. 49(8):601–609.
- Elmer GW, Remmel RP. 1984. Role of the intestinal microflora in clonazepam metabolism in the rat. Xenobiotica. 14(11):829–840.
- Kitamura S, Sugihara K, Kuwasako M, Tatsumi K. 1997. The role of mammalian intestinal bacteria in the reductive metabolism of zonisamide. J Pharm Pharmacol. 49(3):253–256.
- Meuldermans W, Hendrickx J, Mannens G, Lavrijsen K, Janssen C, Bracke J, Le Jeune L, Lauwers W, Heykants J. 1994. The metabolism and excretion of risperidone after oral administration in rats and dogs. Drug Metab Dispos. 22:129–138.
- O’Donnell MP, Fox BW, Chao P-H, Schroeder FC, Sengupta P. 2020. A neurotransmitter produced by gut bacteria modulates host sensory behaviour. Nature. 583(7816):415–420.
- Roffey SJ, Obach RS, Gedge JI, Smith DA. 2007. What is the objective of the mass balance study? A retrospective analysis of data in animal and human excretion studies employing radiolabeled drugs. Drug Metab Rev. 39(1):17–43.
- Surapaneni S, Yerramilli U, Bai A, Dalvie D, Brooks J, Wang X, Selkirk JV, Yan YG, Zhang P, Hargreaves R, et al. 2021. Absorption, metabolism, and excretion, in vitro pharmacology, and clinical pharmacokinetics of ozanimod, a novel sphingosine 1-phosphate receptor modulator. Drug Metab Dispos. 49(5):405–419.
- Takeno S, Sakai T. 1991. Involvement of the intestinal microflora in nitrazepam-induced teratogenicity in rats and its relationship to nitroreduction. Teratology. 44(2):209–214.
- van de Steeg E, Schuren FHJ, Obach RS, van Woudenbergh C, Walker GS, Heerikhuisen M, Nooijen IHG, Vaes WHJ. 2018. An ex vivo fermentation screening platform to study drug metabolism by human gut microbiota. Drug Metab Dispos. 46(11):1596–1607.
- van Kessel SP, Frye AK, El-Gendy AO, Castejon M, Keshavarzian A, van Dijk G, El Aidy S. 2019. Gut bacterial tyrosine decarboxylases restrict levels of levodopa in the treatment of Parkinson’s disease. Nat Commun. 10(1):310.
- Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, Goodman AL. 2019. Separating host and microbiome contributions to drug pharmacokinetics and toxicity. Science. 363(6427):aat9931.
Source: Xenobiotica. 2021;51(4):413–426
Synopsis
In this paper (Wang-Lakshman et al. Citation2021), the authors document radiolabeled mass balance studies of licogliflozin and describe the biotransformation pathways in rat, dog, and human. Licogliflozin is a potent dual inhibitor of sodium-glucose co-transporters (SGLTs) SGLT1 and SGLT2, aimed at reduction of blood glucose levels and lowering body weight in patients with type II diabetes mellitus. Licogliflozin elimination was reported to be mainly via metabolism. Interestingly, direct glucuronidation, was the major metabolic pathway in humans, accounting for approximately 38% dose in feces. Herein, we draw the readers’ attention to three circulating metabolites, M17, M23, and M27, which collectively represent ∼31% of the total radioactive AUC in human following oral dosing.
Commentary
SGLTs are responsible for co-transport of sodium and glucose in the kidney and the intestine. Licogliflozin, (2S,3R,4R,5S,6R)-2-[3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)-4-ethylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol, has a glycosidic group, i.e. a hexo-pyranose like structural motif with three hydroxyl groups and a hydroxymethyl moiety. O-glucuronidation at the glycosidic hydroxy groups was the predominant biotransformation pathway in humans, as shown in . The three glucuronides, M17, M23, and M27, are reminiscent of di-glucuronides, a relatively rare type of glucuronide conjugates, previously shown for androgen and estrogen steroids (Argikar Citation2012). M17 and M27 collectively accounted for 37% of licogliflozin dose in human urine. Reaction phenotyping studies revealed that M17 formation was catalyzed by UGT2B7 and UGT2B4, not unlike zidovudine, a drug to which authors draw biotransformation resemblances. On the other hand, M27 formation was primarily catalyzed by UGT1A9. The other example mentioned by the authors to reflect on glucuronidation differences in human versus rat and dog is propofolol (Mukai et al. Citation2015), a drug predominantly metabolized by UGT1A9 (Mukai et al. Citation2014; Zhou et al. Citation2021). It is fascinating to note that UGT2B7 and UGT1A9 are expressed in the kidney (Foti and Argikar Citation2019; Zhou et al. Citation2021), which is also an organ of SGLT2 expression. Therefore, it is possible that renal glucuronidation might contribute to the overall glucuronidation of licogliflozin.
In laboratory animal studies, direct glucuronidation was a minor metabolic pathway as opposed to human. Interestingly, metabolism of licogliflozin was not reported non-human primates, a species that are most representative and perhaps closest to human in terms of glucuronidation based on substrate specificity, enzyme analogy and tissue expression (Foti and Argikar Citation2019).
Another noteworthy aspect about metabolism of licogliflozin is that, in a multitude of identified secondary and tertiary metabolites, oxidation of dihydrobenzodioxin motif was observed, resulting in ring opened metabolites with substituted ethylene glycol or substituted glycolic acid motifs. However, bioactivation and subsequent release of glyoxal, a known structural alert trappable in a reaction with o-phenylinediamine (Gunduz et al. Citation2017), was not observed/reported. The indirect evidence for this comes from the fact that an o-catechol type metabolite of licogliflozin arising from sequential O-dealkylations and concomitant glyoxal release was not observed among the staggering forty-one metabolites reported in this paper.
Even with the high number of reported metabolites, licogliflozin was the most representative of the radioactivity in circulation. Following oral administration at various doses, licogliflozin represented approximately 72%, 84%, and 54% of total radioactivity AUC in rat, dog, and human, respectively. The recovery of the total radioactive dose was approximately 100%, 73%, and 93%, after oral administration in rat, dog, and human, respectively. Approximately 15% of the radioactive dose was identified in human feces as licogliflozin. Licogliflozin has a long 17-h half-life in human as compared to less 4 h in laboratory animals. The contribution of possible enterohepatic recirculation of licogliflozin via metabolism and transport interplay, i.e. intestinal, hepatic, and renal glucuronidation, followed by biliary elimination of the glucuronides, and finally subsequent unconjugation of the glucuronides in the gut, might underestimate the overall glucuronidation of licogliflozin.
Figure 16. Direct glucuronidation of licogliflozin to M17, M23, and M27.
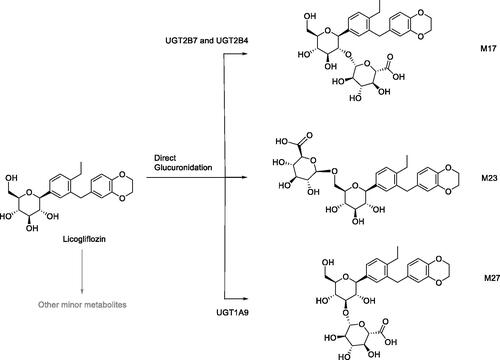
References
- Argikar UA. 2012. Unusual glucuronides. Drug Metab Dispos. 40(7):1239–1251.
- Foti RS, Argikar UA. 2019. UDP-glucuronosyltransferases (UGTs). In: Handbook of drug metabolism. 3rd ed. Boca Raton: CRC Press; p. 109–159.
- Gunduz M, Cirello AL, Klimko P, Dumouchel JL, Argikar UA. 2017. Genotoxicity of 4-(piperazin-1-yl)-8-(trifluoromethyl)pyrido[2,3-e][1,2,4] triazolo[4,3-a]pyrazine, a potent H4 receptor antagonist for the treatment of allergy: evidence of glyoxal intermediate involvement. Drug Metab Lett. 11(2):144–148.
- Mukai M, Isobe T, Okada K, Murata M, Shigeyama M, Hanioka N. 2015. Species and sex differences in propofol glucuronidation in liver microsomes of humans, monkeys, rats and mice. Pharmazie. 70(7):466–470.
- Mukai M, Tanaka S, Yamamoto K, Murata M, Okada K, Isobe T, Shigeyama M, Hichiya H, Hanioka N. 2014. In vitro glucuronidation of propofol in microsomal fractions from human liver, intestine and kidney: tissue distribution and physiological role of UGT1A9. Pharmazie. 69(11):829–832.
- Wang-Lakshman L, Mendonza AE, Huber R, Walles M, He Y, Jarugula V. 2021. Pharmacokinetics, metabolism, and excretion of licogliflozin, a dual inhibitor of SGLT1/2, in rats, dogs, and humans. Xenobiotica. 51(4):413–426.
- Zhou J, Argikar UA, Miners JO. 2021. Enzyme Kinetics of Uridine Diphosphate Glucuronosyltransferases (UGTs). Methods Mol Biol. 2342:301–338.