
Abstract
Identifying beer spoilage microbes is readily accomplished using PCR analyses targeting specific types of microbes, but the general classification of wild yeast versus brewing yeast using cultivation independent molecular methods has remained challenging. The approach presented in this study utilized genetic fingerprint matching to determine if an unknown yeast isolate matches the fingerprint of catalogued brewing yeast strains. Interdelta Next Generation Sequencing (NGS) fingerprints were produced using PCR amplification of delta elements, also known as long terminal repeat sequences of transposons of yeast. Fingerprints identifying different yeast strains were generated by processing reads produced by NGS of the interdelta PCR amplicons using open source software. The interdelta NGS fingerprint comprises DNA sequences that can be recorded, compared, and utilized as a fixed reference for yeast strain identification. Interdelta NGS fingerprints were shown to be reproducible and capable of distinguishing between strains of brewing yeast and wild yeast. Experimental yeast contamination demonstrated the utility of the approach for identifying in-house brewing yeast cross contamination versus foreign wild yeast contamination.
Introduction
Brewers utilize pure strains of yeast for most industrial beer production. The phenotypic diversity of brewing yeasts enables brewers to achieve the flavor, aroma, and other sensory properties that beer consumers seek. The impacts of yeast on beer are numerous and diverse,[Citation1] influencing sweetness derived from residual sugars, aroma imparted by esters, ketones, higher alcohols and phenols[Citation2,Citation3] and clarity affected by colloidal compounds generating stable haze.[Citation4–6] In contrast to the complexity that yeast contribute to beer, brewing yeasts are genetically uniform, in part as a result of domestication.[Citation7] Most brewing yeast can be assigned to three lineages identified as the Beer 1, Beer 2, and Mixed clades. Low genetic diversity is characteristic of industrial yeasts that have been assigned to just seven clades used to produce a broad spectrum of fermented beverages, including wine and sake.
Yeast identification is an essential tool used by brewers to maintain quality and consistency of fermentation. Identifying contaminating yeast is important because wild yeast can produce a variety of off flavors and undesirable sensory characteristics.[Citation8] In the case of cross contamination with brewing yeast, the product may have flavor, aroma or other characteristics that are not true-to-type[Citation9]. Different approaches are needed to address wild yeast contamination versus in-house brewing yeast cross contamination because sources of wild yeast and in-house yeast differ. Therefore, when contamination is detected, identifying the contaminating yeast is key to developing the appropriate corrective action. The discovery of contamination often begins with an observation of yeast growth on selective medium commonly utilized to detect wild yeast.[Citation10,Citation11] In the case of copper containing selective medium, identifying the contaminating yeast requires additional information because there is much overlap in the concentrations of copper required to inhibit growth of different strains of brewing yeast and wild yeast.
Genetic fingerprinting can provide the information needed to identify a contaminating yeast as wild yeast or brewing yeast by matching a genetic fingerprint of the unknown strain to a fingerprint database of brewing yeast strains. Strains of Saccharomyces cerevisiae can be differentiated using PCR amplification with primers targeting the long terminal repeated (LTR) sequences found throughout the yeast genome in LTR-transposons such as Ty1 and Ty2.[Citation12–14] The PCR primers bind to sites near the ends of the approximately 330 bp long LTR domain, also known as a delta element, and generate amplicons composed of sequences from adjacent LTR domains.[Citation12,Citation13] A highly reproducible approach is needed to consistently identify different strains based on properties of interdelta PCR amplicons.
Various approaches have been used to generate genetic fingerprints from interdelta PCR amplicons. The method published in the ASBC Methods of Analysis produces an image of the pattern of amplicons separated by electrophoresis on an agarose gel.[Citation15] Tristezza et al.[Citation16] presented a method using fluorescent capillary electrophoresis to generate the fingerprint. Amplicons produced using a fluorescently labeled PCR primer were detected and sized using automation of the ABI Prism 3130 Genetic Analyzer. Similarly, Franco-Duarte et al.[Citation17] used microfluidic electrophoresis with a Caliper LabChip 90 Electrophoresis System employing automated fragment detection and sizing to generate a fingerprint from the amplicons generated with a fluorescently labeled PCR primer. Methods using automated electrophoresis of interdelta PCR amplicons to generate fingerprints of brewing yeast have yet to gain wide adoption by the brewing industry.
In this study, a method was developed that generates a DNA sequence-based fingerprint from interdelta PCR amplicons. The method builds on the ASBC Methods of Analysis Yeast-13 “Differentiation of Brewing Yeast Strains by PCR Fingerprinting”[Citation15] by incorporating a Next Generation Sequencing (NGS) step. Analysis of the NGS data utilized open source software[Citation18] and a coordinating script tailoring the analysis to generating fingerprints of brewing yeast strains. Quantitative criteria were applied for quality control of the NGS sequence data to generate interdelta NGS fingerprints with the precision needed to distinguish between strains of brewing yeast and wild yeast.
Experimental
Yeast strains OYL-004, WY-1056, IY-A07, BRY-97, and US-05 were obtained from Northern Brewer, LLC (www.northernbrewer.com). Strains WLP001 and S288C were obtained from White Labs (www.whitelabs.com) and the American Type Culture Collection (www.atcc.org), respectively. Strain LC1 was isolated from a lager yeast slurry using a pour plate containing Fast Orange Wild Yeast (FOWY) medium (PIKA Weihenstephan, Pfaffenhofen, Germany) as part of this study. Yeasts were grown overnight in YPD Broth, Sigma-Aldrich (www.sigma-aldrich.com) at 30 °C. Yeast biomass was collected from 100 µL of culture by centrifuging at 10,000 g for 3 min. The medium was removed, and the yeast pellet was resuspended in 200 µL of 5% w/w Chelex 100 resin, Bio-Rad (biorad.com) in distilled water. The cells were then lysed by incubating the mix at 80 °C for 10 min. Cell debris was pelleted from the lysate by centrifugation at 10,000 g for 3 min.[Citation19]
PCR reactions contained 0.5 µM of each PCR primer, 2.5 µL of PrimeTime qPCR Master Mix, Integrated DNA Technologies (idtdna.com), 5 µL of lysate prepared with the Chelex treatment and distilled water to a final volume of 25 µL. PCR primers delta12 (5′-TCAACAATGGAATCCCAAC-3′) and delta21 (5′-CATCTTAACACCGTATATGA-3′)[Citation12] were synthesized by GeneLink (www.genelink.com). Thermal cycling was accomplished using a Chai Open qPCR instrument (chaibio.com) following a program that included an initial step at 95 °C for 2 min., followed by 35 cycles of 95 °C for 15 sec, 46 °C for 30 sec, and 72 °C for 1 min. The final cycle was followed by a step at 72 °C for 10 min. PCR amplicons were electrophoresed using an E1201-GT electrophoresis system, Accuris Instruments (accuris-usa.com) on 1.5% agarose in 1X TBE buffer. Gels were stained with 1X SYBR Green I stain, Sigma-Aldrich (sigma-aldrich.com) and viewed using blue light transillumination for digital image documentation.
PCR products were prepared for NGS sequencing using magnetic bead-based purification and fluorometric DNA quantification. Four PCR reactions were pooled and purified using MagJET NGS Cleanup reagent, Thermo Fisher Scientific (www.thermofisher.com). The purified PCR products were quantified using a Qubit 2.0 fluorometer (www.thermofisher.com) with SYBR Green I stain. NGS sequencing was performed using the Illumina NovaSeq 6000 (www.illumina.com) based 2 × 250 bp Amplicon-EZ service provided by Azenta Life Sciences (genewiz.com).
NGS data in fastq format from Read 1 were used to produce sequence tag fingerprints. Analysis of the reads was performed using the open source VSEARCH package[Citation18] with different steps coordinated by a custom script (Supplementary File 1). Before any processing was conducted, quality statistics, including the average read length, were extracted. Then reads were filtered using a 200 bp length threshold and only those with a maximum error expected error rate (E) <0.5 were retained.[Citation20] Reads shorter than 200 bp were discarded and the remaining reads were truncated to a length of 200 bp. The unique reads were then counted and those occurring less than 10 times were discarded. Then reads without a complete PCR primer sequence were discarded. Finally, the number of occurrences of each unique read was determined and the 15 most abundant reads were chosen as the sequence tags defining the interdelta NGS fingerprint.
Sequence tags making up the interdelta fingerprint of strain S228C were matched to the genome sequence of S228C using BLASTN.[Citation21] Default parameters were used to query the S. cerevisiae R64 RefSeq assembly GCF_000146045.2 with each of the 15 sequence tags. Similarly, sequence tags making up the WLP001 fingerprint were matched to the WLP001 WGS data set using Magic-BLAST[Citation22] with default parameters and samtools V1.10[Citation23] software. The WLP001 genome sequence was represented by Sequence Read Archive SRX8613118 run SRR12086827 library DK01. Sequence coverage in regions of the WLP001 genome matched by sequence tags was determined using the samtools V1.10 software.
Two experimentally contaminated yeast samples were prepared by mixing brewing yeast strain WLP001 with wild yeast strain LC1 and mixing WLP001 with the Belle Saison strain. The mixes contained 1 × 106 WLP001 cells/mL and 10 cfu/mL of the LC1 or Belle Saison strain. The mixes were plated on FOWY medium using the pour plate method. One mL of each yeast mix was combined with 5 mL of FOWY broth and 5 mL of 2% molten agar (Sigma-Aldrich, St. Louis, MO) in deionized water at 50 °C and poured into a 100 mm diameter Petri plate. The agar was allowed to solidify, and the cultures were then incubated for one week at 30 °C.
Results
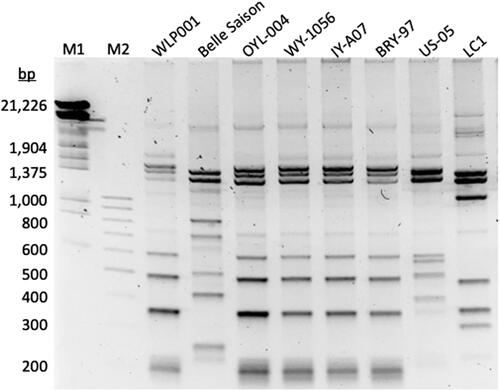
PCR amplification with delta12 and delta 21 primers was successful with every yeast strain examined, including six brewing yeasts, a wild yeast, and a laboratory yeast. Agarose gel electrophoresis revealed approximately seven prominent bands ranging in size from ∼ 200 bp to ∼ 1,500 bp for each yeast strain (). The amplicons included two to six minor bands that were typically >1,500 bp. The patterns of amplicons separated by gel electrophoresis were largely consistent with yeast strain grouping. As revealed by qualitative visual inspection, the Chico yeast strains (the name Chico refers to a widely used craft strain on the West Coast, and it is sold by various yeast vendors under different monikers), including WLP001, OYL-004, WY-1056, and IY-A07 produced amplicons with the same banding patterns on an agarose gel. The banding pattern of Chico-like strain, US-05, was different from that of the Chico strains, while the banding pattern of Chico-like strain BRY-97 appeared to match that of the Chico strains. The banding patterns produced with the Belle Saison and LC1 yeast strains differed from each other and the strains of brewing yeasts. The laboratory strain S288C produced a banding pattern that differed from the brewing yeasts and the wild yeast (data not shown). In silico matching of the delta12 and delta21 primers to the Brettanomyces bruxellensis genome (GenBank Assembly GCF_011074885.1) using BLASTN suggested that the delta element primers would generate an amplicon with this yeast.
Figure 1. SYBR Green I stained agarose gel of amplicons produced with the delta12 and delta21 PCR primers with various brewing yeast strains. M1, Lambda DNA/EcoRI + HindIII Marker (Thermo Fisher Scientific) M2, Molecular weight marker 100 bp (Carolina Biological Supply Co.). WLP001, OYL-004, WY-1056 and IY-A07 are Chico yeast strains. BRY-97 and US-05 are Chico-like yeast strains. LC1 is a contaminating wild yeast identified in a lager yeast.
Next generation sequencing of interdelta PCR amplicons yielded reads that were readily transformed into a DNA sequence-based fingerprint for each yeast strain. Output per sample averaged 251,493 reads, ranging 1.5-fold from 122,270 reads to 382,139 reads for the 19 samples. Average read length per sample was 213 bp and ranged from 202 bp to 232 bp. Filtering reads for length > 200 bp and maximum expected error rate ≤ 0.5 on average retained 49% (range = 30% to 65%) of reads. On average this set included 732 (range = 358 to 1161) unique reads seen >10 times. The number of unique reads per sample averaged 353 (range = 125 to 620) after the final filtering step that retained only the reads with a complete primer sequence. On average 48% (range = 31% to 59%) of reads passed the complete filtering process and 88% (range = 74% to 94%) of filtered reads were utilized to produce an interdelta NGS fingerprint consisting of 15 sequence tags for a yeast strain. The WLP001 interdelta NGS fingerprint included eight sequence tags incorporating the d12 primer and seven sequence tags with the d21 primer (). The 15 sequence tags represented reads that were seen from 570 to 1,1065 times in the NGS data set for WLP001 Sample 1. The interdelta NGS fingerprints for the 21 samples analyzed in this study are available in Supplementary File 2.
Figure 2. Sequence tags representing the interdelta NGS fingerprint of yeast strain WLP001 obtained with primers delta12 and delta21 shown in blue and red, respectively. The ellipses represent the 150 nucleotides between bases 27 and 177 of the 200 bp sequence tag. The sequence descriptions indicate the rank abundance of each sequence tag as “Uniq” followed by a number corresponding to the rank and the abundance the tag in the collection of amplicon as “size=” followed by a number representing the abundance. The complete sequences are available in Supplementary File 2.

Reproducibility of sequence tag fingerprints
Interdelta NGS fingerprints were highly reproducible as determined by repeated PCR and sequencing of WLP001 and Belle Saison yeast strains. Samples 1, 2 and 3 of WLP001 shared 15 sequence tags making up the fingerprint of this strain (). The fingerprint of WLP001 remained unchanged after serial re-pitching. The 15 sequence tags comprising the fingerprint of WLP001 yeast were shared by Samples 2, 3, 4, and 5 generated by WLP001 that had been re-pitched for 1, 2, 17 and 18 generations, respectively. Sequence tag fingerprints were reproducible for the Belle Saison yeast strain as well. The sequence tag fingerprint of Sample 12 shared 15 tags with Samples 9.1 and 10.1 of Belle Saison yeast. However, exact fingerprint matching was not always achieved for Belle Saison yeast, although the matching was still high. The fingerprint of Belle Saison Sample 8 shared 14 of 15 sequence tags with Samples 9.1, 10.1, 11 and 12 from this yeast strain.
Table 1. Number of interdelta sequence tags shared between yeast strains. A total of 15 sequence tags were assigned to each strain.
Sequence tag fingerprinting successfully distinguished between Chico and Chico-like yeast strains. The fingerprints of Chico yeast strains WLP001, OYL-004, WY-1056, IY-A07 shared 15 identical sequence tags (). In contrast, the fingerprint of Chico-like yeast strain US-05 shared only two sequence tags with strain WLP001 and the other Chico yeast strains. Strain BRY-97 is described as a West Coast Ale yeast. The fingerprinting of the BRY-97 Chico-like strain was distinct from the US-05 Chico-like strain but matched the fingerprint of the four Chico strains.
Impact of NGS output quality on fingerprint reproducibility
Sequencing results from two samples of Belle Saison yeast revealed a link between NGS output quality and fingerprint reproducibility. Fingerprints generated from Belle Saison Sample 8, Sample 9, and Sample 10 showed poor reproducibility as reflected by Sample 8 and Sample 9, and Sample 8 and Sample 10 sharing only nine of 15 sequence tags (). Average read length was noticeably shorter for the NGS data generated from Sample 8 and Sample 9 than the rest of the sequence data set. Most samples yielded NGS output with average read length >200 bp, ranging from 202 bp to 223 bp (). In contrast, average read lengths of Sample 9 and Sample 10 were only 181 bp and 167 bp, respectively.
Figure 3. Average read lengths and percentages of reads with expected error rates <0.5 for NGS reads obtained by end sequencing of PCR products generated with the delta12 and delta21 primers. Sample 9 and Sample 10 plotted in red generated fingerprints for the Belle Saison strain that did not match. Sample 9.1 and Sample 10.1 in green represent re-sequencing of the PCR products that generated Sample 9 and Sample 10.
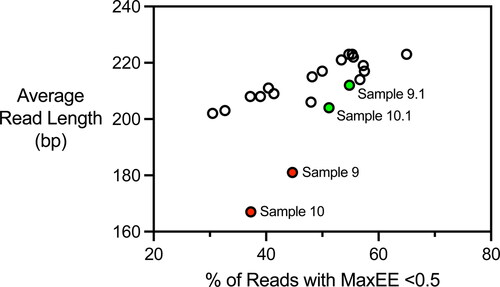
Average read length identified samples generating fingerprints with poor reproducibility more effectively than the expected error rate (MaxEE) statistic reported by VSEARCH. The percentage of reads in each sample with MaxEE ≤ 0.5 ranged from 31% to 65% (). The MaxEE statistics for Sample 9 and Sample 10 producing fingerprints with poor reproducibility were 37% and 45%, respectively, which was within the range seen for the entire collection of 19 samples. Similarly, read quality statistics including the distributions of Q scores and total numbers of reads generated were not as informative as average read length for identifying samples yielding poor fingerprint reproducibility ().
Sample 8 and Sample 9 produced different fingerprints for the Belle Saison strain. The difference appeared to be the result of poor quality of the sequencing output not poor quality of the template DNA used for sequencing. Re-sequencing the Sample 9 and Sample 10 amplicons generated sequencing results identified as Sample 9.1 and Sample 10.1 with average read lengths of 212 bp and 204 bp, respectively (). Fingerprints generated from Sample 9.1 and Sample 10.1 shared 14 of 15 sequence tags with Belle Saison Sample 8 (). Similarly, the fingerprints generated from Belle Saison Sample 11 and Sample 12 shared 14 sequence tags with Sample 8. Average sequence read lengths of Sample 11 and Sample 12 were 211 bp and 203 bp, respectively.
Sequence tags identified in genome sequences
The interdelta fingerprint of yeast strain S288C was successfully matched to the annotated genome sequence of S288C. A BLASTN search matched the 15 sequence tags making up the S288C fingerprint to the S288C genome with 100% sequence identity over the length of each sequence tag (). Sequence tag sites were identified on five of the 16 chromosomes, including chromosomes III, IV, VII, XII, and XVI. Six sequence tags were identified on chromosome IV while three occurred on chromosome XVI, and two were found on chromosomes III, VII, and XII. Alignments of 14 of the 15 sequence tags to the genome sequence included no gaps. The alignment of sequence tag 13 included two gaps. Thirteen of the sequence tags matched sites in the genome annotated as Ty1/Ty2 LTR. Sequence tags 7 and 9 matched sites annotated as Ty1/Ty3 and Ty1, respectively. Sequence tag number 14 matched a region of the genome with no annotation.
Table 2. Characteristics of sequence tags making up the interdelta fingerprint of yeast strain S228C. PCR primers were removed from the 200 bp sequence tags before alignment to the S228C genome sequence using BLASTN.
The WLP001 tag sequences were successfully matched to the WLP001 whole genome sequence (WGS). The matching conducted using Magic-BLAST identified all 15 sequence tags making up the interdelta fingerprint of strain WLP001 with 100% sequence identity and 100% coverage (). All 15 WLP001 sequence tags were identified with 100% coverage in the WLP001 WGS. The depth of coverage for the sequence tags averaged 1,057 reads (range = 168 to 5,458). The WLP001 WGS does not include the annotation needed to identify LTR domains.
Table 3. Sequence coverage statistics for the sequence tags making up the interdelta fingerprint of yeast strain WLP001 aligned to the WLP001 genome sequence.
Experimental cross contamination and foreign yeast contamination
Interdelta NGS fingerprinting successfully distinguished between experimentally produced cross contamination and foreign yeast contamination. The demonstration utilized mixes of the brewing strain WLP001 with the LC1 strain of wild yeast and Belle Saison brewing yeast (). LC1 is a copper resistant wild yeast and Belle Saison is an example of a brewing yeast that is resistant to copper at concentrations that typically inhibits growth of brewing yeast such as WLP001. Mixes of brewing yeast WLP001 with the foreign yeast strain LC1 and WLP001 with the Belle Saison strain on FOWY medium produced 120 and 60 colonies, respectively (data not shown). Colonies of the copper-resistant LC1 and Belle Saison yeast were morphologically indistinguishable. The two strains produced cream-colored colonies that were three to five millimeters in diameter, circular and raised with entire margins.
Figure 4. Schematic diagram of experimental in-house cross contamination and foreign wild yeast contamination using mixes of WLP001 yeast with Belle Saison brewing yeast and LC1 foreign wild yeast, respectively. The Belle Saison and LC1 strains produce colonies on copper containing media while growth of WLP001 is inhibited by copper. Each type of experimental contamination was identified using matching of interdelta NGS fingerprints.
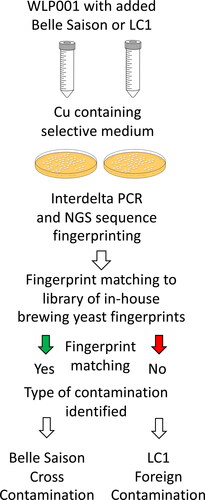
Colonies of the contaminating yeasts mixed with the WLP001 strain were successfully identified using interdelta NGS fingerprinting. Samples 1, 8, 13, 14, 15, 16, and 19 served as reference fingerprints for the strains of brewing yeast utilized in this experimental brewery scenario. Sample 12 and Sample 19 were treated as unknowns, having been generated from the colonies seen in pour plate assays of the experimental contamination of the WLP001 strain. Comparing the interdelta NGS fingerprints generated from colonies on the two agar plates (Sample 12 and Sample 19, respectively) to the Belle Saison reference fingerprint revealed that 14 of 15 sequence tags matched between Sample 12 and Sample 8. In contrast, only three sequence tags of the Sample 19 fingerprint matched sequence tags of the Sample 8 fingerprint of Belle Saison yeast. The match between the interdelta NGS fingerprint of the unknown Sample 12 to the reference fingerprint of Sample 8 supported the conclusion that Sample 12 represented a cross contamination by an in-house yeast, the Belle Saison strain. The lack of a match between the fingerprint of Sample 19 and any of the brewing yeast utilized in the brewery (i.e., Samples 1, 8, 13, 14, 15, 16, and 17) supported the conclusion that Sample 19 represented contamination by a foreign wild yeast strain.
Sequence data availability
The sequence data used in this study were deposited under BioProject accession PRJNA862970.
Discussion
DNA sequence analysis is a powerful tool for identifying microbes including fungi, but establishing a genetic locus suitable for distinguishing strains of S. cerevisiae brewing yeast has been challenging.[Citation24] NGS sequencing of interdelta PCR amplicons generated reproducible and informative fingerprints differentiating strains of brewing yeast with an approach that is readily accessible to brewers. Brewery labs are commonly equipped to identify beer spoilage microbes using PCR. The NGS step can be accomplished without specialized training using commercial sequencing lab services. The sequence data were analyzed using a fixed algorithm with little variation between analyses. Unlike the current implementation of interdelta fingerprinting that relies on images of banding patterns seen on agarose gels, the interdelta NGS fingerprint is composed of DNA sequences that can be recorded, compared, and utilized as a fixed reference. Breweries should generate fingerprints of their own brewing yeast strains if the vendor designations differ from those used in this study. In principle, interdelta NGS fingerprinting could be used to generate a reference library of the entire collection of yeast used in a brewery, yielding a valuable resource for investigating instances of yeast contamination. Yeast producers maintaining and propagating diverse catalogues of yeasts would benefit from the approach as well.
Interdelta NGS fingerprinting is well suited for addressing instances of yeast contamination, as demonstrated by the experimental contamination of WLP001 yeast with Belle Saison brewing yeast and LC1 wild yeast. Initial detection of contamination during beer production is often accomplished using selective medium containing copper, designed to detect wild yeast that are more resistance to copper than brewing yeast. However, copper resistance varies greatly among strains of S. cerevisiae, including strains of brewing yeast[Citation25] and some strains of commonly used brewing yeast will grow in the presence of copper at concentrations that are inhibitory to most brewing yeast.[Citation11,Citation25] Developing an actionable interpretation of yeast growth on copper-containing medium requires additional information. PCR analyses can enable identification of specific types of yeast contamination such as STA1 gene containing S. cerevisiae var. diastaticus[Citation11] or wild yeast such as Wickerhamomyces sp. identifiable by a ribosomal RNA internal transcribed spacer signature.[Citation26] However, the general classification of Saccharomyces wild yeast versus brewing yeast has remained challenging.
A variety of genetic tools have been applied to the challenge of brewing yeast identification. Interdelta typing and Multi Locus Sequence Typing (MLST) collect identifying information from several locations spread over the yeast genome. MLST has the potential for the highest level of genetic discrimination relying on single nucleotide polymorphisms that may occur in housekeeping genes, open reading frames of unknown functions or intergenic regions. Despite the potential power of MLST, interdelta typing has been shown to have greater discriminatory power than MLST of winemaking yeast.[Citation27] The study examined commercial winemaking yeasts obtained from various European and US suppliers, isolates from naturally fermenting musts sampled in Lebanon, yeasts collected in Asia, and laboratory reference strains. The discriminatory power of interdelta typing was >99% compared to 92.27% for the MLST approach that surveyed 7 loci. Discriminatory power is the probability of a test successfully distinguishing between two strains sampled randomly from a population.[Citation28] The discriminatory power of a microsatellite assay that also sampled several regions of the genome was equal to that of interdelta typing. In addition to being more powerful, interdelta typing was also more resource efficient than MLST and microsatellite typing. Interdelta typing required only a single PCR reaction[Citation12] compared to seven PCR reactions for the MLST and six PCR reactions used for the microsatellite typing.[Citation29]
Various properties of LTR-transposon elements enable the differentiation of yeast strains by interdelta NGS fingerprinting. LTR-transposons are abundant and diverse making up 1.3% to 3.4% of the S. cerevisiae genome[Citation30,Citation31] that includes 483 occurrences of Ty1 to Ty5.[Citation32] Ty1 is the most abundant, occurring 313 times in the S288C S. cerevisiae genome. Full length Ty1 elements occur 32 times, while solo elements are almost 10-fold more abundant, occurring 279 times. Solo LTR are generated through intra-element recombination that excises the internal DNA of the transposon from the flanking LTR domains, leaving behind a single LTR element.[Citation33,Citation34] Solo LTR elements are the likely source of interdelta NGS fingerprint diversity distinguishing between brewing yeast strains. Solo LTR elements are highly abundant and contain much sequence diversity in the form of nucleotide deletions. Sequence variation between solo LTRs is typically higher than among LTRs associated with full-length LTR-transposon elements.[Citation35]
Interdelta NGS sequence data used to generate interdelta NGS fingerprints faithfully reflected genomic signatures of S. cerevisiae yeasts. Sequence tags were successfully matched to the S288C and WLP001 genomes, demonstrating that the amplicons subjected to NGS sequencing were not highly impacted by PCR artifacts. Sequence similarity and sequence repeats of delta elements have the potential to generate PCR artifacts such as chimeric amplicons.[Citation36] Fingerprints generated from different samples of the same strain were not without variation, however. The matching of 14 of 15 sequence tags between different samples of the Belle Saison strain revealed a level of variation in fingerprints that needs to be recognized. Ideally the entire universe of brewing yeast would be sampled several times to assess the overall level of analytical variation. However, the approach presented in this study will likely be most applicable to limited collections of yeast strains such as those used in a single brewery not the entire universe of brewing yeast. One source of variability of fingerprint matching was related to average sample read length. Error rates can be inflated by shortened NGS reads caused by pre-phasing occurring during the sequence by synthesis process.[Citation37,Citation38]
Interdelta typing is better suited for identifying yeast strains than for addressing questions about similarity and relatedness. Similarities among yeast strains shown by comparisons of physiological traits and geographic origin were not reliably reflected by interdelta typing.[Citation39] A study of 69 S. cerevisiae strains collected in Hungary from spontaneously fermenting musts revealed no clustering of strains based on geographic origin or phenotypic characteristics. Strains that clustered based on interdelta typing often had different geographical origins and strains with little similarity of interdelta types were often collected from the same location and the same vintage year. Horizontal transfer of LTR-transposons in yeast[Citation30] and high rates of population movement between wine regions likely obscured any pattern that would be consistent with isolated yeast populations. Similarly, interdelta typing did not reflect clustering of strains based on physiological characteristic such as osmotic, ethanol or copper tolerance.[Citation40] It is not clear if a correlation should have been expected between physiological traits and patterns generated by interdelta typing. The selective pressure imposed by the natural environment on yeasts likely differs greatly from the selective pressures on transposable elements inhabiting the environment of the yeast genome.[Citation41]
Cross contamination between brewing yeast and contamination by foreign wild yeast call for different corrective actions to locate and root out the trespassing yeast. In the case of an in-house cross contamination, sensory characteristics of the cross-contaminating brewing yeast can inform sensory analysis identifying the extent and origin of the contamination.[Citation42] Cross contamination is best addressed by focusing on elements of the brewing system, especially parts utilized for yeast storage, propagation, and transfer between fermentors for serial re-pitching. In contrast, the most effective responses to foreign wild yeast contamination addresses the brewery environment where wild yeast can become established.[Citation26,Citation43] Any equipment processing beer that is exposed to the brewery environment and is difficult to clean will be susceptible to colonization by wild yeast. Such environmental sources are the focus of eliminating wild yeast contamination.
This study examined a limited number of yeast strains to demonstrate the principal of a genetic fingerprinting approach that can help brewers discriminate between strains of brewing yeast and identify in-house cross contamination and wild yeast contamination. Widespread application of interdelta NGS fingerprinting will likely uncover brewing yeast strains that will not be successfully differentiated. The impact of those exceptions on the overall approach is expected to be low in the context of breweries utilizing limited collections of yeast strains. Ultimately, high throughput sequencing of whole genomes has the potential to become a definitive tool for identifying yeast strains. Genome sequencing has revealed much about the origins, domestication, and evolution of brewing yeast[Citation7,Citation25,Citation44,Citation45]. Technological advancements in sequencing and bioinformatic analysis could lead to strain identification on a routine basis.[Citation46,Citation47] However, much work remains until such genomic resources can be tailored for use by brewers. Interdelta NGS fingerprinting is an approachable tool that brewery and yeast production quality laboratories can utilize for identifying yeasts by matching fingerprints of unknown strains to reference fingerprints of catalogued yeast strains.
Supplemental Material
Download PDF (31.3 KB)Supplemental Material
Download Text (3.9 KB)Supplemental Material
Download Text (69 KB)Acknowledgements
This work was inspired by the TSC Project for Excellence in Microbial Ecology. Heavy Seas Beer provided laboratory space and analytical instrumentation.
Disclosure statement
No potential conflict of interest was reported by the authors.
Literature cited
- Liu, S.-Q. Impact of Yeast and Bacteria on Beer Appearance and Flavour. In: Brewing Microbiology; Elsevier: Amsterdam, 2015; pp 357–374.
- Haslbeck, K.; Bub, S.; von Kamp, K.; Michel, M.; Zarnkow, M.; Hutzler, M.; Coelhan, M. The Influence of Brewing Yeast Strains on Monoterpene Alcohols and Esters Contributing to the Citrus Flavour of Beer. J. Inst. Brew. 2018, 124, 403–415. DOI: 10.1002/jib.523.
- Younis, O. S.; Stewart, G. G. Sugar Uptake and Subsequent Ester and Higher Alcohol Production by Saccharomyces cerevisiae. J. Inst. Brew. 1998, 104, 255–264. DOI: 10.1002/j.2050-0416.1998.tb00998.x.
- Huismann, M.; Gormley, F.; Dzait, D.; Speers, R. A.; Maskell, L. D. Unfilterable Beer Haze Part I: The Investigation of an India Pale Ale Haze. J. Am. Soc. Brew. Chem. 2022, 80, 17–25. DOI: 10.1080/03610470.2021.1937460.
- Huismann, M.; Gormley, F.; Dzait, D.; Willoughby, N.; Stewart, K.; Speers, R. A.; et al. Unfilterable Beer Haze Part II: Identifying Suspect Cell Wall Proteins. J. Am. Soc. Brew. Chem. 2022, 80, 26–34. DOI: 10.1080/03610470.2021.1937461.
- Lewis, M. J.; Poerwantaro, W. M. Release of Haze Material from the Cell Walls of Agitated Yeast. J. Am. Soc. Brew. Chem. 1991, 49, 43–46. DOI: 10.1094/ASBCJ-49-0043.
- Gallone, B.; Steensels, J.; Prahl, T.; Soriaga, L.; Saels, V.; Herrera-Malaver, B.; Merlevede, A.; Roncoroni, M.; Voordeckers, K.; Miraglia, L.; et al. Domestication and Divergence of Saccharomyces cerevisiae Beer Yeasts. Cell 2016, 166, 1397–1410.e16. DOI: 10.1016/j.cell.2016.08.020.
- Suzuki, K. Emergence of New Spoilage Microorganisms in the Brewing Industry and Development of Microbiological Quality Control Methods to Cope with This Phenomenon: A Review. J. Am. Soc. Brew. Chem. 2020, 78, 245–259. DOI: 10.1080/03610470.2020.1782101.
- Fernandez, S.; Almaguer, L. M.; Sierra, J. A. Detection of Mixtures of Lager Yeast Strains by the Use of Selective Inhibitors in Solid Media. J. Am. Soc. Brew. Chem. 1989, 47, 73–76. DOI: 10.1094/ASBCJ-47-0073.
- ASBC Methods of Analysis, online. Microbiological Control 4. General Culture Media. Approved 1971, rev. 2011. American Society of Brewing Chemists, St. Paul, MN, U.S.A. DOI: 10.1094/ASBCMOA-MicrobiologicalControl-4.
- Burns, L. T.; Sislak, C. D.; Gibbon, N. L.; Saylor, N. R.; Seymour, M. R.; Shaner, L. M.; et al. Improved Functional Assays and Risk Assessment for STA1+ Strains of Saccharomyces cerevisiae. J. Am. Soc. Brew. Chem. 2021, 79, 167–180. DOI: 10.1080/03610470.2020.1796175.
- Legras, J.-L.; Karst, F. Optimisation of Interdelta Analysis for Saccharomyces cerevisiae Strain Characterisation. FEMS Microbiol. Lett. 2003, 221, 249–255. DOI: 10.1016/S0378-1097(03)00205-2.
- Ness, F.; Lavallée, F.; Dubourdieu, D.; Aigle, M.; Dulau, L. Identification of Yeast Strains Using the Polymerase Chain Reaction. J. Sci. Food Agric. 1993, 62, 89–94. DOI: 10.1002/jsfa.2740620113.
- Lavalliée, F.; Salvas, Y.; Lamy, S.; Thomas, D. Y.; Degré, R.; Dulau, L. PCR and DNA Fingerprinting Used as Quality Control in the Production of Wine Yeast Strains. Am. J. Enol. Vitic. 1994, 45, 86–91.
- ASBC Methods of Analysis, online. Yeast Method 13. Differentiation of Brewing Yeast Strains by PCR Fingerprinting. Approved 2009, rev. 2011. American Society of Brewing Chemists, St. Paul, MN, U.S.A. DOI: 10.1094/ASBCMOA-Yeast-13.
- Tristezza, M.; Gerardi, C.; Logrieco, A.; Grieco, F. An Optimized Protocol for the Production of Interdelta Markers in Saccharomyces cerevisiae by Using Capillary Electrophoresis. J. Microbiol. Methods 2009, 78, 286–291. DOI: 10.1016/j.mimet.2009.06.012.
- Franco-Duarte, R.; Mendes, I.; Gomes, A. C.; Santos, M. A. S.; de Sousa, B.; Schuller, D. Genotyping of Saccharomyces cerevisiae Strains by Interdelta Sequence Typing Using Automated Microfluidics. Electrophoresis 2011, 32, 1447–1455. DOI: 10.1002/elps.201000640.
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. DOI: 10.7717/peerj.2584.
- Walsh, P. S.; Metzger, D. A.; Higushi, R. Chelex 100 as a Medium for Simple Extraction of DNA for PCR-Based Typing from Forensic Material. BioTechniques 2013, 54, 134–139. DOI: 10.2144/000114018.
- Edgar, R. C.; Flyvbjerg, H. Error Filtering, Pair Assembly and Error Correction for next-Generation Sequencing Reads. Bioinformatics 2015, 31, 3476–3482. DOI: 10.1093/bioinformatics/btv401.
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A Greedy Algorithm for Aligning DNA Sequences. J. Comput. Biol. 2000, 7, 203–214. DOI: 10.1089/10665270050081478.
- Boratyn, G. M.; Thierry-Mieg, J.; Thierry-Mieg, D.; Busby, B.; Madden, T. L. Magic-BLAST, an Accurate RNA-Seq Aligner for Long and Short Reads. BMC Bioinf. 2019, 20, 405. DOI: 10.1186/s12859-019-2996-x.
- Danecek, P.; Bonfield, J. K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M. O.; Whitwham, A.; Keane, T.; McCarthy, S. A.; Davies, R. M.; Li, H. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. DOI: 10.1093/gigascience/giab008.
- Kopecká, J.; Němec, M.; Matoulková, D. Comparison of DNA-Based Techniques for Differentiation of Production Strains of Ale and Lager Brewing Yeast. J. Appl. Microbiol. 2016, 120, 1561–1573. DOI: 10.1111/jam.13116.
- Strope, P. K.; Skelly, D. A.; Kozmin, S. G.; Mahadevan, G.; Stone, E. A.; Magwene, P. M.; Dietrich, F. S.; McCusker, J. H. The 100-Genomes Strains, an S. cerevisiae Resource That Illuminates Its Natural Phenotypic and Genotypic Variation and Emergence as an Opportunistic Pathogen. Genome Res. 2015, 25, 762–774. DOI: 10.1101/gr.185538.114.
- Sohlberg, E.; Sarlin, T.; Juvonen, R. Fungal Diversity on Brewery Filling Hall Surfaces and Quality Control Samples. Yeast 2022, 39, 141–155. DOI: 10.1002/yea.3687.
- Ayoub, M.-J.; Legras, J.-L.; Saliba, R.; Gaillardin, C. Application of Multi Locus Sequence Typing to the Analysis of the Biodiversity of Indigenous Saccharomyces cerevisiae Wine Yeasts from Lebanon. J. Appl. Microbiol. 2006, 100, 699–711. DOI: 10.1111/j.1365-2672.2006.02817.x.
- Hunter, P. R.; Fraser, C. A. Application of a Numerical Index of Discriminatory Power to a Comparison of Four Physiochemical Typing Methods for Candida albicans. J. Clin. Microbiol. 1989, 27, 2156–2160. DOI: 10.1128/jcm.27.10.2156-2160.1989.
- Legras, J.-L.; Ruh, O.; Merdinoglu, D.; Karst, F. Selection of Hypervariable Microsatellite Loci for the Characterization of Saccharomyces cerevisiae Strains. Int. J. Food Microbiol. 2005, 102, 73–83. DOI: 10.1016/j.ijfoodmicro.2004.12.007.
- Carr, M.; Bensasson, D.; Bergman, C. M. Evolutionary Genomics of Transposable Elements in Saccharomyces cerevisiae. PLoS One 2012, 7, e50978. DOI: 10.1371/journal.pone.0050978.
- Kim, J. M.; Vanguri, S.; Boeke, J. D.; Gabriel, A.; Voytas, D. F. Transposable Elements and Genome Organization: A Comprehensive Survey of Retrotransposons Revealed by the Complete Saccharomyces cerevisiae Genome Sequence. Genome Res. 1998, 8, 464–478. DOI: 10.1101/gr.8.5.464.
- Bleykasten-Grosshans, C.; Neuvéglise, C. Transposable Elements in Yeasts. C. R. Biol. 2011, 334, 679–686. DOI: 10.1016/j.crvi.2011.05.017.
- Jordan, I. K.; McDonald, J. F. Tempo and Mode of Ty Element Evolution in Saccharomyces cerevisiae. Genetics 1999, 151, 1341–1351. DOI: 10.1093/genetics/151.4.1341.
- Winston, F.; Chaleff, D. T.; Valent, B.; Fink, G. R. Mutations Affecting Ty-Mediated Expression of the HIS4 Gene of Saccharomyces cerevisiae. Genetics 1984, 107, 179–197. DOI: 10.1093/genetics/107.2.179.
- Roeder, G. S.; Fink, G. R. Transposable Elements in Yeast. In Mobile Genetic Elements; Shapiro, J. A., ed.; Elsevier: Amsterdam, 1983; pp 299–328.
- Acinas, S. G.; Sarma-Rupavtarm, R.; Klepac-Ceraj, V.; Polz, M. F. PCR-Induced Sequence Artifacts and Bias: Insights from Comparison of Two 16S RRNA Clone Libraries Constructed from the Same Sample. Appl. Environ. Microbiol. 2005, 71, 8966–8969. doi: 10.1128/AEM.71.12.8966-8969.2005.
- Pfeiffer, F.; Gröber, C.; Blank, M.; Händler, K.; Beyer, M.; Schultze, J. L.; Mayer, G. Systematic Evaluation of Error Rates and Causes in Short Samples in Next-Generation Sequencing. Sci. Rep. 2018, 8, 10950. DOI: 10.1038/s41598-018-29325-6.
- Schirmer, M.; Ijaz, U. Z.; D'Amore, R.; Hall, N.; Sloan, W. T.; Quince, C. Insight into Biases and Sequencing Errors for Amplicon Sequencing with the Illumina MiSeq Platform. Nucleic Acids Res. 2015, 43, e37–e37. DOI: 10.1093/nar/gku1341.
- Pfliegler, W. P.; Sipiczki, M. Does Fingerprinting Truly Represent the Diversity of Wine Yeasts? A Case Study with Interdelta Genotyping of Saccharomyces cerevisiae Strains. Lett. Appl. Microbiol. 2016, 63, 406–411. DOI: 10.1111/lam.12679.
- Csoma, H.; Zakany, N.; Capece, A.; Romano, P.; Sipiczki, M. Biological Diversity of Saccharomyces Yeasts of Spontaneously Fermenting Wines in Four Wine Regions: Comparative Genotypic and Phenotypic Analysis. Int. J. Food Microbiol. 2010, 140, 239–248. DOI: 10.1016/j.ijfoodmicro.2010.03.024.
- Venner, S.; Feschotte, C.; Biémont, C. Dynamics of Transposable Elements: Towards a Community Ecology of the Genome. Trends Genet. 2009, 25, 317–323. DOI: 10.1016/j.tig.2009.05.003.
- Fahey, P. Building a Sensory Program: A Brewer’s Guide to Beer Evaluation; Brewers Publications: Boulder, Colorado, 2021.
- Riedl, R.; Fütterer, J.; Goderbauer, P.; Michel, M.; Jacob, F.; Hutzler, M. Combined Yeast Biofilm Screening – Characterization and Validation of Yeast Related Biofilms in a Brewing Environment with Combined Cultivation and Specific Real-Time PCR Screening of Selected Indicator Species. J. Am. Soc. Brew. Chem. 2019, 77, 99–112. DOI: 10.1080/03610470.2019.1579036.
- Saada, O. A.; Tsouris, A.; Large, C.; Friedrich, A.; Dunham, M. J.; Schacherer, J. Phased Polyploid Genomes Provide Deeper Insight into the Multiple Origins of Domesticated Saccharomyces cerevisiae Beer Yeasts. Curr. Biol. 2022, 32, 1350–1361.e3. DOI: 10.1016/j.cub.2022.01.068.
- Peter, J.; De Chiara, M.; Friedrich, A.; Yue, J.-X.; Pflieger, D.; Bergström, A.; Sigwalt, A.; Barre, B.; Freel, K.; Llored, A.; et al. Genome Evolution across 1,011 Saccharomyces cerevisiae Isolates. Nature 2018, 556, 339–344. DOI: 10.1038/s41586-018-0030-5.
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K. F. Nanopore Sequencing Technology, Bioinformatics and Applications. Nat. Biotechnol. 2021, 39, 1348–1365. DOI: 10.1038/s41587-021-01108-x.
- Au, K. F. The Blooming of Long-Read Sequencing Reforms Biomedical Research. Genome Biol. 2022, 23, 21. DOI: 10.1186/s13059-022-02604-2.