
Abstract
We report two families, members of which are carriers of a novel hemoglobin (Hb) variant that was named Hb Olivet [α13(A11)Ala→Thr (α1) (GCC > ACC); HBA1: c.40G > A; p.Ala14Thr]. The analysis of these cases allowed a clear description of this anomaly that behaves as a silent Hb. In the first family, of Portuguese ethnicity living in France, the proband, a 24-year-old male and his 57-year-old mother, both appeared to be carriers. The son presented with borderline mean corpuscular volume (MCV), while the mother was normocytic and normochromic. Hemoglobin separation on capillary electrophoresis (CE) was normal, while a slightly asymmetric peak was observed on high performance liquid chromatography (HPLC). In a second family, originally from Surinam but living in The Netherlands, the proband, a 6-year-old girl, showed a mild microcytosis at low ferritin levels. The abnormal Hb was inherited from the mother who was clearly iron depleted, was not present in the sister and brother of the proband. The microcytic hypochromic anemia was only shown in two out of a total of four carriers. It therefore seems likely that iron depletion is causative as two carriers are completely normal. Characterization and genotype/phenotype correlation are briefly described.
The blood count of patients who are carriers of mild α-thalassemia (α-thal) defects, is often barely abnormal and often masked by a coexisting iron deficiency. Having normalized iron status, a persistent borderline mean corpuscular volume (MCV) and mean corpuscular hemoglobin (Hb) (MCH), might remain ignored or eventually investigated by gap-polymerase chain reaction (gap-PCR) analysis. However, if none of the common deletions are found, no further investigation is usually done.
However, in our experience about 10.0–15.0% of the cases sent to our reference laboratory by conscientious doctors to exclude or confirm α-thal in the absence of iron deficiency, is not caused by deletions but by known or unknown point mutations. One should be aware of the fact that these defects, although mild or asymptomatic in the carrier, may cause Hb H (β4) disease if associated with α0-thal defects (Citation1–5). We present here a short article in which two families of different ethnicity and carrying a new variant, are described with the full characterization of the defect and with some additional details on expression and possible thalassemic effect. This study underlines the importance of adding potential disease-causing mutations or neutral polymorphisms to the database, even if unpublished, as a personal communication (Citation6). Comparison between the carries of the two independent families and assembling the data gave more insight into the presence or absence of pathology in the carriers of this abnormal Hb variant.
Case reports
For the French-Portuguese family, the analysis was done at the Inserm Institut Mondor de Recherche Biologique (IMRB) U955eq2, Créteil, France. The characterization of the variant was done using several methods including ion exchange high performance liquid chromatography (HPLC) (VARIANT II™, dual kit; Bio-Rad Laboratories, Hercules, CA, USA), isoelectric focusing (IEF) and electrophoresis with agar, pH 6, acetate pH 9. Globin chain analysis was done using urea, urea/triton and reversed phase electrophoresis. Reversed phase HPLC was performed as previously described (Citation7).
The variant characterization was done using Sanger sequencing of the α1- (HBA1), α2- (HBA2) and β- (HBB) globin genes, as previously described (Citation8) and in addition, the proband was also examined for other thalassemic traits because of the abnormal red cell phenotype. DNA was extracted from the EDTA blood sample. Multiplex ligation-dependent probe amplification (MLPA) was performed to exclude deletions and rearrangements of the globin gene clusters (P140 HBA; MRC-Holland, Amsterdam, The Netherlands).
The Surinamese-Hindustani family was investigated at the Hemoglobinopathies Reference Laboratory, Department of Human and Clinical Genetics, Leiden University Medical Center (LUMC), Leiden, The Netherlands. The HPLC and capillary electrophoreses (CEs) were done on a VARIANT II™ (Bio-Rad Laboratories) and on a Capillarys 2 device (Sebia, Lisses, France), respectively, according to the instructions of the manufacturers and as previously reported (Citation9). DNA was extracted by automatic high salt procedures (Citation10) for gap-PCR and direct sequencing as described elsewhere (Citation8). Family analyses included the proband (A II-1), her mother (A I-1), a 5-year-old brother (A II-3) and his twin sister (A II-2) (, ).
Figure 1. Pedigrees of the Surinamese family living in The Netherlands (family A) and the French-Portuguese family living in France (family B). The proband in each family is indicated by an arrow.
Figure 2. (a) Capillary electrophoresis (Sebia) pattern of the Hb Olivet carrier (left) and a normal control (right), no abnormal pattern was observed. (b) High performance liquid chromatography (VARIANT II™; Bio-Rad Laboratories) result of the Hb Olivet carrier showed an atypical pattern with the abnormal Hb fraction partially overlapping the Hb A fraction. The normal pattern is indicated as dashed lines overlaying the Hb Olivet pattern on the left.
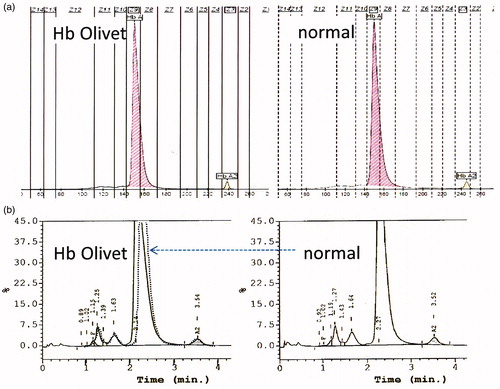
Table 1. Hematological, biochemical parameters and DNA results of the two families with Hb Olivet.
The French-Portuguese family proband (B II-1) was referred to the hospital because of a pulmonary embolism after a train journey. Blood examination revealed a low red cell MCV with borderline MCH but normal Hb level triggering a search for α-thal. The proband was treated by anticoagulant (Fluindione) and followed as an outpatient. Later examinations showed blood counts nearly within the normal range. In order to get more data concerning the variant, his mother (B I-1), who was also a carrier of the variant was analyzed as well, his father however could not be sampled. The family originated from southern Europe (Portugal). Hematological data of the proband and his mother are summarized in . The variant could be partially separated using IEF (thickening of the Hb A line) but not with the other methods. There was no separation of the α-Olivet chain from the wild type using reversed phase HPLC. DNA sequencing showed a mutation on the α1 gene, at codon 13 leading to an amino acid substitution of alanine to threonine [Hb Olivet; α13(A11)Ala→Thr (α1) (GCC > ACC); HBA1: c.40G > A; p.Ala14Thr]. The variant was found in the mother who presented with normal blood counts. No additional deletions, rearrangements or point mutations were found on the α- or β-globin genes.
The Surinamese-Hindustani proband in the second family was a 6-year-old girl (A II-1) living in Gouda, The Netherlands. The proband was referred because of persisting fatigue after the general practitioner had treated her with Ferrofumarate because of a recognized iron deficiency. The proband presented after iron supplementation with the following parameters: Hb 13.4 g/dL, MCV 80 fL, MCH 27.4 pg and red blood cell (RBC) count 4.89 × 1012/L. Capillary electrophoresis was normal and HPLC presented a suspected asymmetric Hb A peak indicating the possible overlapping of different peaks . On gap-PCR, none of the common α-thal deletions were found, but DNA sequencing of the α1- and α2-globin genes, routinely performed in these cases, revealed heterozygosis for the same HBA1: c.40G > A variant that was previously registered as Hb Olivet (http://globin.cse.psu.edu/hbvar.html) by S. Pissard, J. Riou and C. Prehu as a personal communication in 2010 [National Center for Biotechnology Information (NCBI) database single nucleotide polymorphism (dbSNP) Short Genetic Variations database, dbSNP rs35331909]. Further collaboration between the Dutch and the French groups resulted in the identification of four carriers of Hb Olivet and led to an accurate phenotype-genotype correlation of the carriers of this abnormal Hb variant. Data of the two unrelated family members are summarized in .
The process of growing multi-ethnicity in Northern Europe causes a growing consciousness among pediatricians, hematologists and laboratories of the problems associated with the common hemoglobinopathies such as Hb S (HBB: c.20A > T), Hb C (HBB: c.19G > A), Hb E (HBB: c.79G > A), Hb D-Los Angeles (HBB:c.364G > C), and β-thal, all associated with sickle cell disease and β-thal major (β-TM). This also stimulated diligent pediatricians not to disregard apparently insignificant signals that might hide point mutations on α-globin genes that risk to be improperly treated as iron deficiencies and may cause intermediate or even serious diseases if associated with α0-thal defects. Hb Olivet, caused by the substitution of a neutral, non polar and bivalent alanine with an equally neutral but polar and as well as bivalent threonine in a not particularly critic section of the α chain, seems not to be a variant associated with instability or thalassemia. As this amino acid substitution is the only one described at this position no genotype/phenotype comparisons are available from equivalent Hb variants. However, the expression measured on reversed phase HPLC and the hematological parameters indicated that the α-globin gene is expressed, the protein chain stable and that Hb Olivet is formed as a functional tetramer.
Comparison of carriers with non carrier relatives, also indicated a silent variant. The characteristics of the substituted amino acids also explain the silent behavior of Hb Olivet on CE and the slightly asymmetric form of the peak on HPLC. This underlines the importance of recognizing barely aberrant patterns as abnormal by experienced laboratory experts, even if no clearly separated peaks are visible and if the hematological values are only mildly abnormal.
Genetically speaking, the origin of the mutation remains unclear. We could have found two independent events in different populations and the mutation could be common or rare among Surinamese-Hindustani or in the general Portuguese population. Moreover, Surinam has a very mixed ethnic population and a founder effect from Portugal cannot be ruled out. Haplotype studies should be done to identify a possible common origin, however, this was beyond the aim of this study and can be performed in future studies.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Giordano PC, Harteveld CL, Doorduijn JK, et al. Hb Osu Christiansborg: a rare abnormal hemoglobin observed in two independent families in The Netherlands. Ned Tijdschr Klin Chem. 1999;24:287–291.
- Giordano PC, Harteveld CL, Bernini LF, et al. Haplotype analysis of two new, independent cases of Hb Osu-Christiansborg. Hemoglobin. 1999;23(2):193–195.
- Harteveld CL, van Delft P, Plug R, et al. Hb Groene Hart: a new Pro→Ser amino acid substitution at position 119 of the α1-globin chain is associated with a mild α-thalassemia phenotype. Hemoglobin. 2002;26(3):255–260.
- Harteveld CL, Wijermans PW, de Ree JE, et al. A new Hb Evanston allele [α14(A12)Trp → Arg] found solely, and in the presence of common α-thalassemia deletions, in three independent Asian cases. Hemoglobin. 2004;28(1):1–5.
- Harteveld CL, van Delft P, Akkermans N, et al. Hb Buffalo [α89(FG1)His →Gln (α1)], observed solely and in the presence of an Hb S [β6(A3)Glu →Val] heterozygosity. Hemoglobin. 2004;28(3):223–227.
- Giardine B, Borg J, Higgs DR, et al. Systematic documentation and analysis of human genetic variation in hemoglobinopathies using the microattribution approach. Nat Genet. 2011;43(4): 295–301.
- Riou J, Pissard S, Goossens M, Wajcman H. Improvements in phenotype studies of hemoglobin disorders brought by advances in reversed-phase chromatography of globin chains. Int J Lab Hematol. 2015;37(2):279–286.
- Traeger-Synodinos J, Harteveld CL. Advances in technologies for screening and diagnosis of hemoglobinopathies. Biomark Med. 2014;8(1):119–131.
- Harteveld CL, Versteegh FG, Kok PJ, et al. Hb Bleuland [α108(G15)Thr→Asn, ACC→AAC (α2)]: a new abnormal hemoglobin associated with a mild α-thalassemia phenotype. Hemoglobin. 2006;30(3):349–354.
- Giordano PC, Plancke A, Van Meir CA, et al. Carrier diagnostics and prevention of hemoglobinopathies in early pregnancy in The Netherlands: a pilot study. Prenat Diagn. 2006;26(8):719–724.