
Abstract
Case reports on the effect of hydroxyurea (HU) therapy for unstable hemoglobins (Hbs) are sparse; only three adult cases have been reported. We report for the first time on the effect of HU therapy in children carrying unstable Hbs. The first case concerns a female child with a familial history of chronic hemolytic anemia. She was diagnosed with Hb Volga (HBB: c.83C>A) at the age of 7 months. At age 6, treatment options were reconsidered due to increasing fatigue and decreasing Hb concentration. The second case also concerns a female child with chronic hemolytic anemia and icterus since the age of 5. She was diagnosed with Hb Köln (HBB: c.295G>A) at the age of 9. At age 10, treatment options were reconsidered due to decreased general condition and poor school performance. Both children were started on HU therapy. The child with Hb Volga showed reduced clinical symptoms and increased average Hb concentrations. She has been on HU therapy for over 7 years at preparation of this manuscript. The child with Hb Köln showed decreasing Hb concentrations upon start of therapy; clinical symptoms did not improve. Therapy was discontinued after 3½ months. The Hb Volga case report suggests that HU therapy could improve clinical symptoms in some patients with unstable Hbs. Based on these and previously published cases, it was speculated that response can be predicted by the percentage of Hb F and reticulocyte counts.
Introduction
Unstable hemoglobins (Hbs) present a subgroup of rare variants with clinical consequences. A mutation on either the α or β chain affects the stability of the protein, causing Hb to precipitate [reviewed by Williamson (Citation1)]. Variants frequently show a dominant pattern of inheritance. Depending on the nature of the variant, very mild to life-threatening hemolytic anemia is observed. At the moment, over 30 variants of the α chain and 100 variants of the β chain have been listed as unstable Hbs (Citation2). Due to the broad range and low prevalence of variants, no guidelines or recommendations on treatment are available. In mild cases, supportive and preventive measures might suffice. These measures include folic acid supplementation, infection prevention and restrictive use of oxidative drugs. In more severe cases, splenectomy is often contemplated. However, splenectomy is associated with an increased risk of infections with encapsulated bacteria and thromboembolic events, whereas chronic hemolysis is not always resolved. Hydroxyurea (HU) therapy might offer a suitable alternative, though reports on HU therapy for unstable Hbs are sparse (Citation3,Citation4).
The benefits of HU therapy in adults and children with sickle-cell anemia has been demonstrated in many reports. Improved laboratory values are observed, along with reduced pain crises, transfusion requirements, hospitalizations and mortality [reviewed by McGann and Ware (Citation5)]. Few clinical side effects have been reported; reversible cytopenia and mild gastrointestinal upset being frequently encountered. No evidence was present to suggest effects on growth rate, fertility, teratogenicity or risk of malignancies, even with long-term exposure (Citation5–7). Hydroxyurea therapy is now advocated to be part of standard care for sickle-cell anemia; the National Heart, Lung and Blood Institute (NHLBI) guideline recommends the drug use to be offered to parents as early as the age of 9 months for children with homozyogous Hb S (HBB: c.20A>T) or Hb S-β0-thalassemia (Hb S-β0-thal) (Citation5,Citation8). Based on the mild and reversible side effects of HU therapy and the possible benefits, HU therapy is an attractive option for patients with unstable Hbs. This report is the first to describe the effect of HU therapy on the clinical course of children with unstable Hbs. The clinical course of a female child with Hb Volga [β27(B9)Ala→Asp; HBB: c.83C>A] and another female child with Hb Köln [β98(FG5)Val→Met; HBB: c.295G>A] are presented.
Case reports
Patient 1 is a female child, diagnosed with Hb Volga at the age of 7 months. A familial history of chronic hemolytic anemia was present; the father and several paternal relatives are heterozygous for the Hb Volga mutation. At the time of diagnosis, growth and development was according to age and unremarkable. Laboratory parameters were age appropriate, with the exception of the presence of Heinz bodies and basophilic stippling in the peripheral blood smear. Until the age of 6, a satisfactory clinical condition was obtained only with folic acid supplementation. The main observations were recurrent infections, increasing icterus and increasing fatigue. The spleen was palpable from age 2 onwards. Two red blood (RBC) cell concentrates were transfused during a period of pneumonia at age 4.
At the age of 6, treatment options were reconsidered due to increasing fatigue and decreasing Hb concentration. Size of spleen had increased up to 8 cm below the costal margin. Splenectomy was considered but rejected due to familial history. The father underwent splenectomy, resulting in improvement of clinical symptoms of his Hb Volga. However, pulmonary embolism with deep vein thrombosis of the left lower extremity was diagnosed a year after splenectomy. Deep vein thrombosis of the right lower extremity occurred a year after. A paternal uncle carrying Hb Volga also suffered a thrombolic event after splenectomy. Subsequently, HU therapy was started at 10 mg/kg/day and increased up to 25 mg/kg/day after 1 month. Hemoglobin concentrations increased from 7.6 g/dL at start of therapy to 10.6 g/dL after 10 months of therapy (). Clinically, symptoms of fatigue improved significantly within 2 months; sport activities were started. At age 7, a cholecystectomy was performed to resolve cholelithiasis. At age 9, during a period of pneumonia, a neutrophil count less than 1.0 × 109/L resulted in a HU dose reduction; Hb concentration and clinical condition warranted transfusion of one RBC concentrate. At age 11, a Parvo B19 infection resulted in an aplastic crisis. Hydroxyurea therapy was temporarily discontinued, restarted at 14 mg/kg/day after 3 months and increased to 19 mg/kg/day after 1 month of restarting the therapy. Hb F percentages increased to 24.0% after 5 months of restart. At age 12 to 13, the HU dose could not be increased according to weight due to neutropenia (acceptable neutrophil count set at 1.0 × 109/L with the latest dose of 14 mg/kg/day resulting in Hb F and Hb levels similar to levels before therapy (Hb F 19.0% and Hb 8.5 g/dL; ). The presence of hypersplenism could be responsible to some degree for the limitations in HU dosage. At age 13, HU therapy had been continued for more than 7 years, reversible cytopenia and mild gastrointestinal upset upon an increase in dose having been the only reported side effects. In these 7 years, mean Hb levels were 9.8 g/dL (period of treatment stopped due to aplastic anemia not included), with mean levels of 8.8 g/dL before start of treatment (, ). Ferritin levels were well above lower limit of reference before and during treatment (; reference values 9.0–126.0 μg/L). Hb F percentages ranged from 19.0% to 31.0%, with highest Hb F percentages at times of highest HU dose (). Hemolytic parameters [bilirubin and lactate dehydrogenase (LDH)] did not improve upon therapy (, ). The size of spleen remained within 8–10 cm below the costal margin during age 6 to 13.
Figure 1. Laboratory parameters before and during HU treatment of patient with Hb Volga (left panels) and Hb Köln (right panels). Start of HU therapy is indicated by an arrow in the top panels. Period of cholelithiasis (1), pneumonia (2), and Parvo B19 induced aplastic crisis, requiring red blood cell transfusions (3), are indicated with numerals for the patient with Hb Volga. Percentages of Hb Köln are shown by a dashed line with open circles. Hb Volga was not detectable by IEF, but a heterozygous presence of the mutation was confirmed by sequencing.
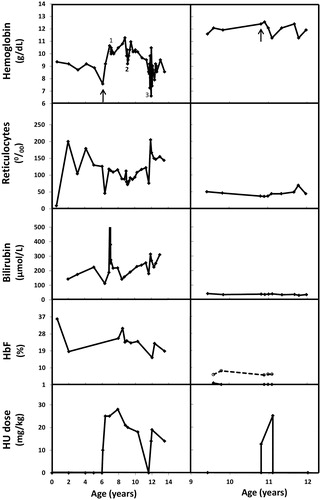
Table 1. Laboratory parameter values for the patient with Hb Volga.
Patient 2 is a female child, diagnosed with Hb Köln at the age of 9 years. Extensive laboratory investigations were performed to explain a history of chronic hemolytic anemia and icterus. An abnormal Hb fraction was detected by isoelectric focusing (IEF); percentages of Hb A, Hb A2, Hb F and Hb Köln were 88.8, 3.2, 1.8 and 6.2%, respectively. Sequence analysis of patient and parents indicated a de novo Hb Köln mutation. Clinical symptoms at age 9 were restricted to fatigue for which infrequent RBC transfusions were given during periods of general malaise. Folic acid values were monitored, maintaining levels close to and above upper limit of reference of 30.0 nmol/L from age 9 to age 17.
At age 10, treatment options were reconsidered due to decreased general condition and poor school performance. Splenectomy was considered, but rejected due to the mild clinical course expected for Hb Köln, lack of reports showing a beneficial effect in patients with Hb Köln and substantial side effects of splenectomy. Instead, HU therapy was started at 12.5 mg/kg/day and increased to 25 mg/kg/day in subsequent months. Over a 3-month period, an increase in fatigue and a further decline in school performance were observed. Hemoglobin concentrations decreased from 12.4 to 11.3 g/dL, Hb F percentages remained below 1.0% and thrombocyte counts decreased to below the lower limit of reference (, ). Hemolytic parameters did not improve upon therapy (, ). Consequently, HU therapy was discontinued. In subsequent years, she continued to receive folic acid supplementation and incidental RBC transfusions. At age 12, iron chelation was prescribed for half a year to counteract iron overload (highest measured ferritin level was 788.0 μg/L). At age 13, cholecystectomy was performed to resolve cholelithiasis. At age 18, clinical symptoms are mainly restricted to fatigue; transfusions are sporadically indicated and the spleen is not palpable.
Table 2. Laboratory parameter values for the patient with Hb Köln.
Discussion
Hydroxyurea offers a promising therapy for patients with unstable Hbs. However, at present, only two publications are available of patients with unstable Hbs being treated with HU. These publications describe three young adults with a history of chronic hemolytic anemia, for which splenectomy had been performed in early childhood (Citation3,Citation4). With the present report included, the effect of HU therapy is now described for five patients and five different unstable Hbs at different stages of life. A 31-year-old female with Hb Templeuve (described only once) [β139(H17)-β140(H18)Asn-Ala→Thr] (Citation3) showed reduced extramedullary hematopoiesis and thrombocytosis upon HU therapy. Hemoglobin levels increased from 6.7 to 7.5 g/dL within a year of therapy (Citation3). A 24-year-old male with Hb Perth (HBB: c.98T>C) showed reduced acute hemolytic episodes and thrombocytosis. Occurrence of a thrombo-embolic event was associated with a temporary stoppage of HU therapy. Hemoglobin levels during HU therapy were comparable to levels before therapy (Citation3). The patient with Hb Volga represents the third case with improved clinical symptoms attributed to HU therapy.
In homozygous Hb S patients, the primary benefits of HU therapy are attributed to the induction of Hb F expression, though additional mechanisms (e.g. increased nitric oxide levels) could be important (Citation5). Induction of Hb F is accomplished in the majority of homozygous Hb S patients. Furthermore, higher Hb F levels during therapy are associated with higher baseline percentage of Hb F and reticulocyte counts (Citation9–12). For unstable Hbs, the three patients with beneficial effects of HU therapy showed base line Hb F percentages >2.0% and induction of Hb F up to ≥24.0%. In the case of Hb Volga, Hb F and Hb levels showed a clear correlation with HU dose, with limited effect at an HU dose of 14 mg/kg/day. The patient with Hb Köln is the only patient for whom a decline in clinical performance as well as Hb levels was observed. Compared to the other patients, she had the lowest Hb F values and reticulocyte counts before and during therapy. For the fifth patient, a 26-year-old male with Hb Mainz (HBB: c.296T>A) a direct beneficial or adverse effect of HU therapy cannot be deduced from the report as other therapies were started simultaneously to resolve pulmonary hypertension and hemosiderosis. However, Hb F values during therapy were relatively low and blood counts did not change (Citation4).
These case reports indicate that HU therapy could improve clinical symptoms in at least some patients with an unstable Hb. It is tempting to speculate that response might be predicted by baseline percentage of Hb F and reticulocyte counts. However, the amount of published cases is still too limited to offer a valid prediction of patients who will benefit from HU therapy.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Williamson D. The unstable haemoglobins. Blood Rev. 1993;7(3):146–163.
- Patrinos GP, Giardine B, Riemer C, et al. Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies. Nucleic Acids Res. 2004;32(Database issue):D537–D541. (http://globin.cse.psu.edu/hbvar/menu.html; accessed December 28, 2015).
- Rose C, Bauters F, Galacteros F. Hydroxyurea therapy in highly unstable hemoglobin carriers. Blood. 1996;88(7):2807–2808.
- Lode HN, Krings G, Schulze-Neick I, et al. Pulmonary hypertension in a case of Hb-Mainz hemolytic anemia. J Pediatr Hematol Oncol. 2007;29(3):173–177.
- McGann PT, Ware RE. Hydroxyurea therapy for sickle cell anemia. Expert Opin Drug Saf. 2015;14(11):1749–1758.
- Ballas SK, McCarthy WF, Guo N, et al. Exposure to hydroxyurea and pregnancy outcomes in patients with sickle cell anemia. J Natl Med Assoc. 2009;101(10):1046–1051.
- McGann PT, Howard TA, Flanagan JM, et al. Chromosome damage and repair in children with sickle cell anaemia and long-term hydroxycarbamide exposure. Br J Haematol. 2011;154(1):134–140.
- National Institutes of Health. Evidence-based management of sickle cell disease: expert panel report 2014 . (http://www.nhlbi.nih.gov/health-pro/guidelines/sickle-cell-disease-guidelines; accessed December 28, 2015).
- Ware RE, Eggleston B, Redding-Lallinger R, et al. Predictors of fetal hemoglobin response in children with sickle cell anemia receiving hydroxyurea therapy. Blood. 2002;99(1):10–14.
- Ware RE, Despotovic JM, Mortier NA, et al. Pharmacokinetics, pharmacodynamics, and pharmacogenetics of hydroxyurea treatment for children with sickle cell anemia. Blood. 2011;118(18):4985–4991.
- Steinberg MH, Lu ZH, Barton FB, et al. Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Multicenter study of hydroxyurea. Blood. 1997;89(3):1078–1088.
- Green NS, Ender KL, Pashankar F, et al. Candidate sequence variants and fetal hemoglobin in children with sickle cell disease treated with hydroxyurea. PLoS One. 2013;8:e55709. doi: 10.1371/journal.pone.0055709.