
Abstract
β-Thalassemia (β-thal) is a hemoglobinopathy characterized by reduced or absent β-globin production. Pharmacological reactivation of the γ-globin gene for the production of fetal hemoglobin (Hb F) presents an attractive treatment strategy. In an effort to identify promising therapeutic agents, we evaluated 80 analogues of the histone deacetylase inhibitor MS-275, a known Hb F inducer. The chemical analogues were identified via molecular modeling and targeted chemical modifications. Nine novel agents exhibited significant hemoglobin (Hb)-inducing and erythroid differentiation activities in the human K562 erythroleukemia cell line. Five of them appeared to be stronger inducers than the lead compound, MS-275, demonstrating the effectiveness of our method.
Introduction
β-Thalassemia (β-thal) is a common genetic disorder and a major cause of morbidity and mortality worldwide. It is estimated that more than 40,000 infants with β-thal are born each year and 1.5% of the global population are β-thal carriers. β-Thalassemia is an autosomal recessive hemoglobinopathy characterized by quantitative changes in the synthesis of the β-globin chain. Insufficient β-globin production leads to the accumulation and precipitation of unpaired α-globin chains, leading to ineffective erythropoiesis and hemolysis, which result in severe anemia and a series of secondary complications, such as skeletal abnormalities, hepatosplenomegaly and growth defects [Citation1,Citation2].
Treatment that consists of regular, lifelong blood transfusions and iron chelation therapy [Citation3] can significantly extend life expectancy of patients with β-thal but this comes with the risk of complications. Several observations show that the clinical symptoms of β-thal can be ameliorated by increased production of fetal hemoglobin (Hb F) [Citation4,Citation5]. These observations have led to the proposal of the pharmacological reactivation of the γ-globin genes as an effective treatment strategy for β-thal. Increased production of γ-globin can ameliorate the symptoms of the disease by partly substituting for the absent or reduced β-globin, but mainly by restoring the balance between α- and non-α-globin chains [Citation6].
Intensive studies over the last 40 years have led to the identification of several Hb F-inducing agents [e.g. hydroxyurea (HU), butyrate, decitabine]. However, their clinical use is limited or unsatisfactory owing to low efficacy, high toxicity, inconvenience of use and inability to elicit a response in all patients [Citation7]. Therefore, the identification of safer and more effective pharmacological agents is imperative. Nevertheless, in the absence of a ‘druggable’ biological target, the drug discovery process can prove time- and resource-consuming. Therefore, several studies have investigated chemical analogues of known Hb F inducers for their ability to activate the γ-globin gene, in an effort to speed up the process and identify compounds with higher efficacy and lower cytotoxicity than the original pharmacological agent [Citation8–12].
Herein, we followed a similar approach, choosing MS-275 as the lead compound in our effort to identify structurally similar agents with improved pharmacological characteristics for the treatment of β-thal. MS-275 belongs to the class of Hb F-inducers that possess a histone deacetylase (HDAC) inhibitory activity [Citation13–16]. MS-275 is a second generation histone deacetylace inhibitor (HDACi) that belongs to the benzamide family. In contrast to the first generation of HDAC inhibitors (e.g. butyrate, valproate), which have a non specific activity against HDACs, MS-275 specifically inhibits class I HDACs (HDAC 1, 2 and 3) [Citation17–19]. This is of great importance, as several studies have demonstrated the involvement of HDAC 1 and 2 in γ-globin gene silencing [Citation15,Citation20]. MS-275 also decreases the mRNA and protein levels of BCL11A, KLF1 and LSD-1, which are known γ-globin gene repressors [Citation21]. Interestingly, phase I and II clinical trials for the use of MS-275 in the treatment of lymphoma, leukemia and solid tumors, have demonstrated good oral bioavailability and a long half-life [Citation22–25], contrary to the butyrate derivatives, which, during their clinical trials for the treatment of β-thal and sickle cell disease, exhibited the need for intravenous administration or large oral doses due to their short half-life [Citation26–29]. In addition, MS-275 has a relatively simple structure that allows for easier modifications and numerous chemical analogues are available in chemical databases.
The chemical analogues of MS-275 were screened in vitro in the K562 human erythroleukemia cell line for their ability to promote erythroid differentiation and hemoglobin (Hb) synthesis. The K562 cells can spontaneously differentiate into early progenitors of the erythrocytic lineage [Citation30], and when treated with various pharmacological agents, can undergo erythroid differentiation [Citation13,Citation31–34]. They have the potential to highly express the embryo-fetal (ζ-, ε- and γ-) globin genes, which makes them a useful model cell line for the study of compounds that are potential γ-globin inducers [Citation31].
Materials and methods
The identification of chemical analogues of MS-275 was performed in three consecutive rounds of selection. For the first two rounds, molecular modeling was applied and the PubChem library was screened for potential pharmacological agents. PubChem contains more than 5 million compounds and is currently the largest publicly available chemical database. The Enalos Mold2 KNIME node [Citation35,Citation36] was used to compute the 42 description pharmacophores on which the chemical similarity metrics employed for the selection of the test chemicals were based. These 42 molecular quantum numbers reflect the subset of molecular descriptors deemed to be important for the compounds’ underlying biological activity. The descriptors were computed to account for chemical, physicochemical and electronic properties of molecules.
The identified molecules were also selected based on their predicted solubility, membrane permeability and lipophilicity characteristics. Solubility and membrane permeability are important factors for oral bioavailability, whereas lipophilicity is significant for absorption and in vivo distribution. Two quantitative structure property relationship (QSPR) models were employed to predict solubility (GlogS) and lipophilicity (GlogP) of the identified molecules. These two models are important in determining if a compound is drug-like.
For the third round of selection, a different approach was employed. MD20, the chemical that exhibited the highest Hb-inducing activity out of all the analogues tested so far, was used as the basis for targeted chemical modifications, which led to the generation of the 36 analogues tested in round three. The chemical modifications included the replacement of Br with a different halogen (F, Cl and I) or another substituent (e.g. CH3, N2O, etc.), changes in the position of the substituents (ortho, meta and para positions), and addition of a second substituent on the first benzene ring of MD20 (MD45-80; Supplementary Figure 1). These small changes in the structure of MD20 allowed us to have a better understanding of the parts of the compound that were responsible for the observed activity.
MS-275 and the chemical analogues MD1-20 were purchased from Enamine Ltd. (Riga, Latvia), the chemical analogues MD21-44 were purchased from Ambinter (Orléans, France) and the chemical analogues MD45-80 were purchased from eHeterocycles Ltd. (Nicosia, Cyprus).
The stock and working solutions of MS-275 and its chemical analogues were prepared in 100.0% dimethyl sulfoxide (DMSO). The concentration of the stock solution was 100 mM and serial dilutions were performed so that the final concentration of DMSO in culture was 0.1%. The chemical structures of MS-275 and its chemical analogues are shown in Supplementary Figure 1. All compounds were kept at –80 °C.
MS-275 and its chemical analogues (Supplemental Figure 1) were screened in vitro in K562 cells for their ability to promote erythroid differentiation and hemoglobin synthesis. The human erythroleukemia cell line K562 was maintained in RPMI medium (Gibco Invitrogen Inc., Paisley, UK) supplemented with 10.0% fetal bovine serum (FBS) (Gibco Invitrogen Inc.), 1.2 mM L-glutamine (Gibco Invitrogen Inc.) and 50 U/mL of penicillin/streptomycin (Gibco Invitrogen Inc.). The cells were kept at 37 °C, in a humidified atmosphere of 5.0% CO2 and were maintained at a density of 3 × 105 cells/mL.
A broad range of concentrations (0.01-100 μM) of each compound under investigation was added in 1.5 × 104 cells/mL. The aim was to determine the concentration at which each agent exerted its highest activity without decreasing cell survival more than 50.0%. The screening process was terminated if an agent decreased cell survival below the 50.0% level or if it precipitated in culture at concentrations below 100 μM. Untreated cells were used as a negative control and treatment with 100 μM HU was used as a positive control. After 5 days of treatment, cell viability was determined using trypan blue staining and the level of erythroid differentiation, indicated by Hb production, was measured by benzidine staining.
The Hb inducing activity of the pharmacological agents was assessed in K562 cells using benzidine stain and their effect on cell survival was measured by trypan blue stain (Supplementary Table 1). Cells were mixed with trypan blue solution (Sigma, St. Louis, MO, USA) at a ratio of 1:1 v/v and scored under a light microscope using a hemocytometer. The number of live cells was measured and the results were expressed as percent increase relative to the uninduced control.
The stock benzidine solution was prepared by adding 1 g of benzidine dihydrochloride (Sigma) and 14.6 mL of glacial acetic acid to 485.4 mL of distilled water. The working solution was prepared by adding 20 μL of 30.0% H2O2 to 1 mL benzidine stock solution. The cells were mixed with the working solution at a ratio of 1:1 v/v and allowed to stand for 2-3 min. at room temperature. Hemoglobin-containing cells were stained blue and were scored under a light microscope using a hemocytometer. The percentage of benzidine positive cells (blue cells) was calculated by dividing the number of benzidine positive cells by the total number of cells. The results were expressed as fold increase relative to the uninduced control.
At the end of the screening process the pharmacological agents were categorized based on their Hb-inducing activity as high, medium, low and no activity compounds (Supplementary Table 1; Supplementary Figure 1). The pharmacological agents that exhibited the same or higher Hb-inducing activity than MS-275 (∼6.7-fold) were characterized as high activity compounds. The agents that exhibited a ∼5-fold increase in the number of Hb-containing cells were characterized as medium activity compounds. The agents that resulted in a 2 to 3-fold increase were characterized as low activity compounds, whereas the ones that did not increase the number of Hb-containing cells in relation to the uninduced control were characterized as no activity compounds.
Results
MS-275, a potent Hb F inducer, was chosen as the lead compound for the identification of chemical analogues that are more active and less toxic than the lead compound. During the first round of analogue identification, an initial set of 20 MS-275 chemical analogues (MD1-20) was selected for in vitro screening in K562 cells. MD20 exhibited a significant Hb-inducing activity, higher than MS-275 ( and ). Specifically, MD20 resulted in an 8.32-fold increase in the number of Hb-containing cells (p value of <0.05, paired t-test), contrary to MS-275 that exhibited a 6.67-fold increase (p value of <0.001, paired t-test). For this reason, the MD20 structure was used for the identification of the next 24 pharmacological agents (MD21-44) for the second round of screening, based on the principles of chemical similarity already mentioned. During the second round, two agents, MD43 and MD44, exhibited similar Hb-inducing activity as MS-275 ( and ). Specifically, MD43 and MD44 demonstrated a 7- and 6.39-fold increase, respectively, in the number of Hb-containing cells (p value of <0.01, paired t-test). However, MD43 and MD44 exerted their effect at 20 and 50 μM, respectively, whereas MS-275 exhibited significant activity at a much lower concentration (0.25 μM).
Figure 1. The structures of MS-275 and its chemical analogues that produced the highest Hb-inducing activity in K562 cells.
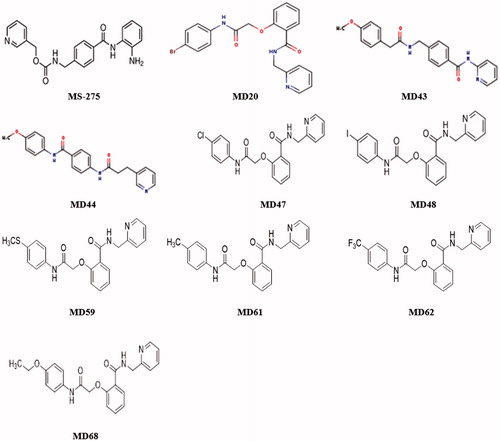
Figure 2. The Hb-inducing activity of MS-275 and its chemical analogues that exhibited the highest activity in K562 cells. The Hb-inducing activity was measured using a benzidine stain. All agents displayed an ∼50.0% cell survival at the concentrations used. The results are the average of three or more experiments with error bars corresponding to the standard deviation. Addition of these chemical compounds to K562 cells resulted in a statistically significant increase in Hb induction relative to the uninduced control, according to the paired t-test (p value of <0.05).
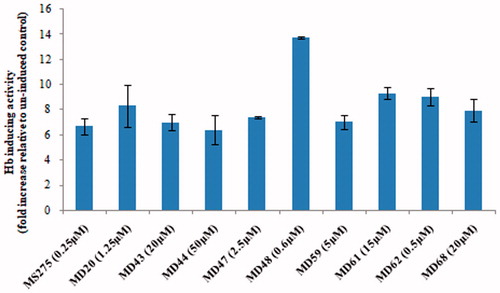
The results from the first two rounds of in vitro screening showed that MD20 was the strongest Hb inducer of the compounds tested so far (MD1-44) and therefore, its structure was used for the selection of the last 36 agents (MD45-80) during the third round of compound identification. As the second round of molecular modeling did not improve on the activity of the compounds identified, for the third round of compound identification a different selection approach was employed. Pharmacological agents that had different chemical substituents on their first benzene ring compared to MD20 were selected. During this round, six pharmacological agents with significant Hb-inducing activity (p value of <0.05, paired t-test) were identified ( and ). Five pharmacological agents (MD20, 48, 61, 62 and 68) exhibited a higher Hb-inducing activity than MS-275, demonstrating the effectiveness of this approach in identifying compounds with higher activity than the lead compound. The agent with the highest Hb-inducing activity was MD48, which produced a 13.76-fold increase in the number of Hb-containing cells (p value of <0.01, paired t-test).
Discussion
Pharmacological reactivation of the γ-globin genes presents an attractive treatment strategy for β-thal, as increased production of Hb F can ameliorate the clinical symptoms of the disease [Citation5]. Despite years of research and more than 70 Hb F inducers currently identified, none has shown a satisfactory clinical use due to moderate therapeutic properties, variable patient response and potential cytotoxicity [Citation7]. Therefore, the identification of safer and more effective pharmacological agents is imperative. Several research groups have investigated chemical analogues of known Hb F inducers in an effort to speed up the drug identification process and the discovery of novel compounds with higher efficacy and lower cytotoxicity [Citation8–12].
We adopted a similar approach and chose MS-275 as the lead compound in our effort to identify structurally similar agents with more favorable activity profiles. MS-275 is a class I HDAC inhibitor that belongs to the benzamide family [Citation17–19]. Many studies show its potency as an Hb F inducer [Citation13–16], while phase I and II clinical trials for its use in the treatment of lymphoma, leukemia and solid tumors, have demonstrated good oral bioavailability and a long half-life [Citation22–25].
In this study, MS-275 and 80 chemical analogues were screened in vitro in K562 human erythroleukemia cells for their ability to promote erythroid differentiation and Hb synthesis. Three out of the 80 compounds screened exhibited similar activity to MS275, while five compounds exhibited higher activity. One (MD48) of those five compounds exhibited substantially higher activity. While a clear structure-activity relationship is not discernable, it was notable that two of the more active compounds, MD20 and MD48, contain heavy para-halogens, Br and I, respectively, on their terminal benzamide moieties. Replacing these with lighter and more electronegative Cl or F halogens led to a drop in activity, as did relocating the para halogens to either the meta or ortho benzamide positions. Therefore, the presence and nature of the para substituent on the terminal benzamide, plays a role, which may be related to improved lipophilicity on introduction of the heavy halogens Br and I, but presumably the structure-activity relationship is more complex than the explanation provided here; steric and electronic factors can be relevant. Moreover, the available data does not allow more detailed conclusions to be drawn on this issue. Replacing the peripheral pyrid-2-yl group with a pyrid-4-yl also led to a significant loss in activity, which tentatively, may be attributed to the latter analogue’s weaker ability to act as a chelator.
The identification of five pharmacological agents (MD20, 48, 61, 62 and 68) with higher Hb-inducing activity than MS-275 demonstrated the effectiveness of our method in identifying compounds with higher activity than the lead compound. Targeted chemical modifications employed during the third round of screening complemented the molecular modeling, increasing the number of high activity compounds generated, and improving our understanding regarding the parts of the compound that were responsible for the observed activity. Analogues with high Hb-inducing activity identified through this study will be further screened in more physiologically relevant in vitro systems such as human primary erythroid cultures. The strongest Hb F inducers will be further tested in mouse β-thal models.
LHEM-43-2-2019-0042-READY-Voskou_Phylactides_-supplementary_figure_legend-EDITED_BY_MFHC_OA_.docx
Download MS Word (10.3 KB)LHEM-43-2-2019-0042-READY-Voskou_Phylactides_-supplementary_table-EDITED_BY_MFHC_OA_.wpd
Download (34.2 KB)Supplemental_Figure_1.docx
Download MS Word (1.2 MB)Acknowledgments
We would like to thank the Blood Centre of the Nicosia General Hospital, Nicosia, Cyprus, for providing us with the buffy coats required for our experiments. The authors of this article have contributed adequately to be included as authors: S. Voskou, M. Phylactides, A. Afantitis, P.A. Koutentis and M. Kleanthous conceived and designed the experiments; A. Afantitis, G. Melagraki, A. Tsoumanis, P.A. Koutentis, T. Mitsidi and S.I. Mirallai identified the chemical analogues; P.A. Koutentis analyzed the structures of the compounds and commented on the structure-activity relationship; S. Voskou completed the experiments, analyzed the data and wrote the manuscript under the supervision of M. Phylactides and M. Kleanthous.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
Related Research Data
References
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480–487.
- Weatherall DJ, Williams TN, Allen SJ, O'Donnell A. The population genetics and dynamics of the thalassemias. Hematol/Oncol Clin North Am. 2010;24(6):1021–1031.
- Olivieri NF. The β-thalassemias. N Engl J Med. 1999;341(2):99–109.
- Watson J. The significance of the paucity of sickle cells in newborn Negro infants. Am J Med Sci. 1948;215(4):419–423.
- Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet. 2001;2(4):245–255.
- Olivieri NF, Weatherall DJ. The therapeutic reactivation of fetal haemoglobin. Hum Mol Genet. 1998;7(10):1655–1658.
- Testa U. Fetal hemoglobin chemical inducers for treatment of hemoglobinopathies. Ann Hematol. 2009;88(6):505–528.
- Theodorou A, Phylactides M, Forti L, et al. The investigation of resveratrol and analogs as potential inducers of fetal hemoglobin. Blood Cells Mol Dis. 2016;58:6–12.
- Chaneiam N, Changtam C, Mungkongdee T, et al. A reduced curcuminoid analog as a novel inducer of fetal hemoglobin. Ann Hematol. 2013;92(3):379–386.
- Lampronti I, Bianchi N, Zuccato C, et al. Increase in γ-globin mRNA content in human erythroid cells treated with angelicin analogs. Int J Hematol. 2009;90(3):318–327.
- Chiarabelli C, Bianchi N, Borgatti M, et al. Induction of γ-globin gene expression by tallimustine analogs in human erythroid cells. Haematologica. 2003;88(7):826–827.
- Bianchi N, Ongaro F, Chiarabelli C, et al. Induction of erythroid differentiation of human K562 cells by cisplatin analogs. Biochem Pharmacol. 2000;60(1):31–40.
- Witt O, Monkemeyer S, Ronndahl G, et al. Induction of fetal hemoglobin expression by the histone deacetylase inhibitor apicidin. Blood. 2003;101(5):2001–2007.
- Cao H, Stamatoyannopoulos G. Histone deacetylase inhibitor FK228 is a potent inducer of human fetal hemoglobin. Am J Hematol. 2006;81(12):981–983.
- Esrick EB, McConkey M, Lin K, et al. Inactivation of HDAC1 or HDAC2 induces gamma globin expression without altering cell cycle or proliferation. Am J Hematol. 2015;90(7):624–628.
- Renneville A, Van Galen P, Canver MC, et al. EHMT1 and EHMT2 inhibition induces fetal hemoglobin expression. Blood. 2015;126(16):1930–1939.
- Monneret C. Histone deacetylase inhibitors. Eur J Med Chem. 2005;40(1):1–13.
- Itoh Y, Suzuki T, Miyata N. Isoform-selective histone deacetylase inhibitors. Curr Pharm Des. 2008;14(6):529–544.
- Khan N, Jeffers M, Kumar S, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409(2):581–589.
- Bradner JE, Mak R, Tanguturi SK, et al. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proc Natl Acad Sci USA. 2010;107(28):12617–12622.
- Dai Y, Sangerman J, Luo HY, et al. Therapeutic fetal-globin inducers reduce transcriptional repression in hemoglobinopathy erythroid progenitors through distinct mechanisms. Blood Cells Mol Dis. 2016;56(1):62–69.
- Ryan QC, Headlee D, Acharya M, et al. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J Clin Oncol. 2005;23(17):3912–3922.
- Kummar S, Gutierrez M, Gardner ER, et al. Phase I trial of MS-275, a histone deacetylase inhibitor, administered weekly in refractory solid tumors and lymphoid malignancies. Clin Cancer Res. 2007;13(18 Pt 1):5411–5417.
- Gojo I, Jiemjit A, Trepel JB, et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood. 2007;109(7):2781–2790.
- Gore L, Rothenberg ML, O'Bryant CL, et al. A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, MS-275, in patients with refractory solid tumors and lymphomas. Clin Cancer Res. 2008;14(14):4517–4525.
- Perrine SP, Ginder GD, Faller DV, et al. A short-term trial of butyrate to stimulate fetal-globin-gene expression in the β-globin disorders. N Engl J Med. 1993; 328(2): 81–86.
- Collins AF, Pearson HA, Giardina P, et al. Oral sodium phenylbutyrate therapy in homozygous β thalassemia: a clinical trial. Blood. 1995;85(1):43–49.
- Atweh GF, Sutton M, Nassif I, et al. Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease. Blood. 1999;93(6):1790–1797.
- Olivieri NF, Rees DC, Ginder GD, et al. Treatment of thalassaemia major with phenylbutyrate and hydroxyurea. Lancet. 1997;350(9076):491–492.
- Lozzio BB, Lozzio CB, Bamberger EG, Feliu AS. A multipotential leukemia cell line (K-562) of human origin. Proc Soc Exp Biol Med. 1981;166(4):546–550.
- Rutherford TR, Clegg JB, Weatherall DJ. K562 human leukaemic cells synthesise embryonic haemoglobin in response to haemin. Nature. 1979;280(5718):164–165.
- Gambari R, del Senno L, Barbieri R, et al. Human leukemia K-562 cells: induction of erythroid differentiation by 5-azacytidine. Cell Differ. 1984;14(2):87–97.
- Cioe L, McNab A, Hubbell HR, et al. Differential expression of the globin genes in human leukemia K562(S) cells induced to differentiate by hemin or butyric acid. Cancer Res. 1981;41(1):237–243.
- Witt O, Monkemeyer S, Kanbach K, Pekrun A. Induction of fetal hemoglobin synthesis by valproate: modulation of MAP kinase pathways. Am J Hematol. 2002;71(1):45–46.
- Varsou DD, Nikolakopoulos S, Tsoumanis A, et al. Enalos + KNIME nodes: new cheminformatics tools for drug discovery. Methods Mol Biol. 2018;1824:113–138.
- Varsou DD, Nikolakopoulos S, Tsoumanis A, et al. Enalos suite: new cheminformatics platform for drug discovery and computational toxicology. Methods Mol Biol. 2018;1800:287–311.