
Abstract
Hemoglobinopathies are the most common single-gene disorders in humans. There are 1,424 variants of human hemoglobin described with 951 involving the β-globin gene. Ancestry and geography play a significant role in the incidence and nature of hemoglobinopathies, with African, Asian, and Mediterranean populations and their descendants being amongst the most affected. Investigation of variants in individuals of Hispanic descent is needed to reflect the changing demographics of the United States. Hemoglobin β-globin evaluation through gel electrophoresis, high-performance liquid chromatography, and HBB gene sequencing was performed on patients from Texas hospitals between 2010 and 2015 and demographic parameters (age, sex, ethnicity) was subsequently analyzed. A total of 846 patients underwent hemoglobinopathy evaluation. A β chain variant was detected in 628 of the 846 total patients. Hispanic patients represented 37% (314/846 patients), which were equally distributed between females (50%; 156/314) and males (50%; 156/314). A β-globin chain variant was found in 67% of Hispanic patients with a distribution across 10 variants seen in greater than 1% of patients. For hemoglobin variants, an understanding of the regional and ethnic prevalence will improve patient care through more effective screening and identification of the variant, early diagnosis, and appropriate treatment if necessary, and better genetic counseling.
Introduction
Hemoglobin is a tetramer composed of two α and two non-α globin chains. The inherited disorders of hemoglobin (Hb) also known as hemoglobinopathies are the most common monogenic diseases in humans, with approximately 5–7% of the world population being carriers and an estimated annual incidence of >300,000 newborns with a serious disease [Citation1]. Hemoglobinopathies are subclassified into two major groups: structural variants, which are most often caused by single amino-acid substitution in the α- or β-globin chain, and thalassemias, which represent defective production of globin chains. Occasional structural variants also result in defective production. Hemoglobinopathies have a wide spectrum of clinical manifestations, ranging from severe anemia to asymptomatic carrier states.
The HbVar database [Citation2] reports 951 hemoglobin variants involving the β-globin gene out of 1,864 entries (hemoglobin variants and thalassemias). The most common Hb variants detected and studied in the world are HbS, HbE and HbC. Some variants show regional and ethnic prevalence with four main ancestral groups: Mediterranean, Asian-Indian, Southeast-Asian, and sub-Saharan African [Citation3]. There are many reasons for this prevalence pattern, including natural selection through protection against malaria and founder effects.
With current global immigration patterns, hemoglobin disorders have spread beyond their original endemic regions. It is estimated that in 71% of the world’s countries hemoglobinopathies are common enough to require implementation of policies for detection and genetic counseling [Citation4]. With increased survival rates of newborns affected by severe forms of hemoglobinopathies, especially in high income countries, these disorders are becoming an increasing health burden and accurate identification of hemoglobinopathies so that prognosis can be established and therapy instituted whenever appropriate is a priority for healthcare professionals [Citation5–7].
The population of Texas is among the largest and most diverse of the 50 states in the United States (US) with many ethnic and ancestral groups; individuals with Hispanic ancestry represent a major subgroup. A better understanding of which hemoglobin variants are more frequent in individuals of Hispanic descent is needed to reflect the changing demographics of the US especially in Texas in order to ensure optimal medical assessment and care for this population.
Materials and methods
The Institutional Review Board of the University of Texas Southwestern Medical Center in Dallas, Texas reviewed and approved the study protocol, and a waiver of informed consent was granted to this minimal-risk retrospective study. Patients from Children’s Health in Dallas or referred to our institution from other Texas hospitals received comprehensive hemoglobinopathy investigations between 2010 and 2015. Whole-blood samples were collected and transported at ambient temperature in test tubes containing the anticoagulant ethylenediaminetetraacetic acid to Children’s Medical Center in Dallas, Texas, for further evaluation. Hemoglobin analysis was performed using isoelectric focusing (IEF), citrate agar electrophoresis, high-performance liquid chromatography (HPLC), and bidirectional β-globin gene sequencing. Clinical information, including age and sex, was collected for each individual. Information on probable ethnicity was inferred from surnames [Citation8].
Isoelectric focusing electrophoresis was performed on all specimens at a hemolysate dilution of 20 g/L, using the Resolve hemoglobin kit (PerkinElmer, Waltham, Massachusetts). The hemoglobin bands were fixed with 10% trichloroacetic acid, and their positions were compared visually to the control bands.
Citrate agar electrophoresis was performed using the Hydragel 7 Acid(E) Hemoglobin(E) kit on the semi-automated Hydrasys system (Sebia Electrophoresis, Norcross, Georgia). The hemoglobin was separated by electrophoresis on acidic agarose gels, and the fractions were visualized by staining with Amido black. The resulting electropherograms were evaluated visually for pattern abnormalities in comparison to standard control bands.
The HPLC analysis was performed on all specimens using the ultra2 with Resolution software (Trinity Biotech USA Inc, Jamestown, New York) ion-exchange method. Hemoglobin separation was accomplished by injecting the samples into a chromatograph equipped with an ion-exchange column equilibrated for pH and ion strength. A peak summary report is produced, along with the chromatogram with the absolute retention time, the retention time relative to standard controls, and the area under each peak on the chromatogram, expressed as a percentage of the total area for each peak in the chromatogram.
When necessary, bidirectional gene sequencing was performed for exons 1, 2, and 3 of the β-globin gene. Exons 1 and 2 were amplified as a single amplicon; the same forward and reverse primers were used for polymerase chain reaction amplification as well as sequencing. Exon 3 was amplified as a separate amplicon; an additional forward sequencing primer was used along with the set of forward and reverse primers for both polymerase chain reaction amplification and sequencing. Sequencing was performed using a cycle sequencing kit (BigDye Terminator Version 3.1, Life Technologies, Carlsbad, California), followed by capillary electrophoresis and analysis (Applied Biosystems 3,500xL genetic analyzer, Life Technologies). The resulting chromatograms were then evaluated (Mutation Surveyor Version 3.30, SoftGenetics, State College, Pennsylvania; and Alamut Version 1.5, Interactive Biosoftware, San Diego, California) and any mutations or polymorphisms were noted.
Results
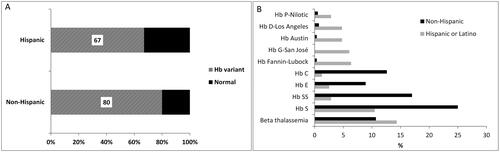
A total of 846 cases were examined. Patients with Hispanic surnames represented 37% of the cohort (314 patients) with an almost equal sex distribution. A β-globin variant was found in 80% of the patients with non-Hispanic surnames and in 67% of the patients with Hispanic surnames (). As expected, patients with hemoglobins S, E, or C were more prevalent in the non-Hispanic group. Interestingly, hemoglobin variants Fannin-Lubbock, G-San José, Austin, D-Los Angeles, and P-Nilotic were found at much higher frequency in the Hispanic population representing almost 25% of the variants seen in this group ().
Figure 1. Distribution of the β-globin chain variants in individuals with Hispanic and non-Hispanic surnames.
Discussion
Despite being the most common group of monogenic disorders in human populations, the inherited disorders of hemoglobin are rarely a priority in public health at national, regional, and even global scales, with only a few countries implementing large-scale prevention and management programs. In the United States, there is no ongoing monitoring system for hemoglobinopathies. Several projects by The Centers for Disease Control and Prevention (CDC), such as the Registry and Surveillance System for Hemoglobinopathies (RuSH), the Thalassemia Data Collection Project, Public Health Research, Epidemiology, and Surveillance for Hemoglobinopathies (PHRESH), and Blood Safety Surveillance among People with Blood Disorders, provided important epidemiologic data related to hemoglobinopathies but they were limited in time and geographic coverage. Ongoing efforts are focused on sickle cell disease and thalassemia (Sickle Cell Data Collection program and a Transfusion Complications Monitoring project). This lack of a monitoring system makes it difficult to identify people with these conditions, monitor the use of healthcare services and any resulting changes in health or quality of life, and understand the impact of these conditions on the healthcare system. Recognizing the importance of early detection, the Recommended Uniform Screening Panel (RUSP) for newborns added in 2006 a few hemoglobinopathies to the core test (HbSS, HbS/β-thalassemia and HbSC disease) with additional variants such as HbE added as secondary targets [Citation9]. Screening for other hemoglobinopathies, such as α- and β-thalassemia has recently gained significant attention but is currently performed in only a few states [Citation10].
In addition, with global mobility reaching unprecedented levels, the largest number of international migrants (47 million) resides in the United States of America, representing a fifth of the world’s total migrants [Citation11]. Thus, many people at risk for hemoglobinopathy who live in the United States were born either before NBS began in their state, or in a country without NBS. Due to all these factors, the actual number of people with hemoglobinopathies in the United States and the associated healthcare impact is unknown.
Population growth occurs through two processes: natural increase (the excess of births over deaths) and net migration (the difference between in-migration and out-migration). The significant population growth in Texas was generated by both processes. Texas has the second-largest Hispanic population in the nation behind California [Citation12], with a growth pattern representing the single largest determinant of population growth in the State for each of the last two decades. The percentage of Texas residents who self-identify as Hispanic or Latino is estimated for 2021 to have been 40.2%, relative to 40.3% who self-identify as White/not Hispanic or Latino. The estimate for the United States puts the Hispanic population at 18.9%. (www.census.gov/quickfacts, accessed 03/13/2023). Our study reveals an increased frequency of hemoglobin variants Fannin-Lubbock, G-San José, Austin, D-Los Angeles, and P-Nilotic totaling almost 25% in the Hispanic as compared with only 2% in the non-Hispanic patients.
Hb Fannin-Lubbock-I was described for the first time in 1976 in two separate Mexican-American families [Citation13,Citation14]. The initial hemoglobin variant showed substitution of glycine by aspartic acid at codon 119 on the ββ-globin chain. The substitution is located at the α1-β1 contact and results in mild protein instability. In 1994, a second substitution at codon 111 resulting in the replacement of valine by leucine was found in five Spanish families and was labeled Hb Fannin-Lubbock-II [Citation15], followed by the reporting of four new carriers of Hb Fannin-Lubbock-II from three additional Spanish families [Citation16]. Both variants of Hb Fannin-Lubbock are unstable and represent 45–50% of total Hb in heterozygotes. To date, a single occurrence of homozygosity for Hb Fannin-Lubbock-I has been reported [Citation17]. On IEF and alkaline pH electrophoresis, Hb Fannin-Lubbock migrates anodal to (“faster than”) Hb A, while at acidic pH it has a similar migration pattern to HbA. Our institution saw both Hb Fannin-Lubbock-I and Hb Fannin-Lubbock-II, with the vast majority being Hb Fannin-Lubbock-I.
Hb G-San José is a rare variant that was first characterized in 1957 [Citation18] in a Californian family of Italian origin. Subsequently, it has been reported in Southern Italian [Citation19,Citation20] and Mexican [Citation21,Citation22] families. The variant features a β-globin substitution at codon 7 from glutamic acid to glycine. In heterozygotes, Hb G-San José represents 30–40% of total hemoglobin with no significant clinical or hematological disease. In compound heterozygosity with thalassemia, there is marked variation in variant expression from 15% with monoallelic α-thalassemia (-α/αα) to 91% with β0-thalassemia [Citation21]. On electrophoresis, its migration pattern is between Hb C and Hb S at acid pH and between Hb S and Hb F on alkaline pH or IEF. Both isopropanol [Citation22] and heat stability [Citation23] tests have shown that Hb G-San José is slightly less stable than Hb A.
Hemoglobin Austin was first reported in 1977 in three unrelated Mexican-Americans [Citation24]; it is a substitution in the α1-β flexible joint region of serine for arginine at codon 40 of the β-globin chain. On acid pH its electrophoretic migration pattern is between Hb F and Hb A. The migration on alkaline pH was reported initially to be between Hb F and Hb A [Citation24], but later this was corrected to be anodal to Hb A in a publication from our institution [Citation25]. In heterozygotes, Hb Austin represents 45% of total Hb. The variant has an increase in oxygen affinity and a decrease in cooperativity with overall no clinical or hematological disease.
Hb D-Los Angeles (also known as D-Punjab, D-North Carolina, D-Portugal, D-Chicago, and Oak Ridge) was first described in 1951 [Citation26] in a family from Los Angeles. Later studies revealed that this Hb variant is frequently found in the Indus River Valley (Punjab) region of Pakistan and Northwestern India [Citation27] but has been observed in China, England, Holland, Australia, Greece, Yugoslavia, Turkey [Citation28] as well as in southern Italy [Citation29] and Mexico [Citation30] and represents the fourth most frequently occurring Hb variant. This variant is the result of the substitution of glutamine for glutamic acid at codon 121 of the β-globin chain. Its electrophoretic migration pattern is cathodal to (“slower than”) Hb A at alkaline pH with no separation at acidic pH. Heterozygous Hb D-Los Angeles is asymptomatic. Homozygous patients can show mild hemolytic anemia and mild to moderate splenomegaly. In compound heterozygotes with β-thalassemia, this variant was associated with moderate hemolytic anemia and microcytosis. Co-inheritance with β-thalassemia has variable clinical manifestations (ranging from mild to moderate) based on the degree of the thalassemia affecting the hemoglobin A gene. During early infancy, while the amount of fetal hemoglobin diminishes and hemoglobin D-Los Angeles increases, mild hemolytic anemia can be seen. Despite the development of splenomegaly and other complications, the condition is fairly benign. Hb D produces different severity of disease with Hb S depending on the Hb D variant. Hb D-Los Angeles with Hb S produces clinically significant sickle cell disease, whereas Hb D-Iran and Hb D-Ibadan are noninteracting with Hb S and produce a sickle cell trait clinical picture [Citation31].
Hemoglobin P-Nilotic was described first in 1973 in the Nilotic population indigenous to the Nile Valley [Citation32]. In addition, it has been found in Turkish [Citation33] and Mexican-American families [Citation34]. Hb P-Nilotic is a result of a non-homologous crossover between β and delta globin genes occurring without loss of bases (“anti-Lepore” fusion hemoglobin). In heterozygotes, Hb P-Nilotic represents 16–21% of total hemoglobin and has no clinical or hematological implications. This variant moves cathodal to (“slower than”) Hb A at alkaline pH and it separates readily by IEF (between F and S). Functional studies show normal Bohr effect, normal cooperativity, and increased O2 affinity.
Most of these hemoglobin variants are less known due to their low incidence in European and African-American populations and the absence of hematologic manifestations in heterozygous individuals. This study raises awareness for their increased frequency in certain populations, resulting in a higher probability of encountering a homozygote or a compound heterozygote with other common hemoglobinopathies, in which the hematologic effects have not yet been described. However, as a result of demographic shifts, hemoglobin variants with increased incidence in Hispanic populations are more likely to be detected in patient workups and screening programs. It is therefore important to know which variants these variants are likely to be.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
References
- Weatherall D. The inherited disorders of haemoglobin: an increasingly neglected global health burden. Indian J Med Res. 2011;134:493–497.
- Giardine BM, Joly P, Pissard S, et al. Clinically relevant updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2021;49(D1):D1192–D1196. doi: 10.1093/nar/gkaa959.
- Orkin SH, Kazazian HH. Jr. The mutation and polymorphism of the human β-globin gene and its surrounding DNA. Annu Rev Genet. 1984;18(1):131–171. doi: 10.1146/annurev.ge.18.120184.001023.
- Halim-Fikri BH, Lederer CW, Baig AA, et al. Global globin network consensus paper: classification and stratified roadmaps for improved thalassaemia care and prevention in 32 countries. J Pers Med. 2022;12(4):552. doi: 10.3390/jpm12040552.
- Henderson S, Timbs A, McCarthy J, et al. Incidence of haemoglobinopathies in various populations - the impact of immigration. Clin Biochem. 2009;42(18):1745–1756. doi: 10.1016/j.clinbiochem.2009.05.012.
- Aguilar Martinez P, Angastiniotis M, Eleftheriou A, et al. Haemoglobinopathies in Europe: health & migration policy perspectives. Orphanet J Rare Dis. 2014;9(1):97. doi: 10.1186/1750-1172-9-97.
- Giordano PC, Harteveld CL, Bakker E. Genetic epidemiology and preventive healthcare in multiethnic societies: the hemoglobinopathies. Int J Environ Res Public Health. 2014;11(6):6136–6146. doi: 10.3390/ijerph110606136.
- Sorbero ME, Euller R, Kofner A, et al. Imputation of race and ethnicity in health insurance marketplace enrollment data, 2015-2022 open enrollment periods. Rand Health Q. 2022;10(1):4.
- Ellinwood NM. Newborn screening and the recommended uniform screening panel: Optimal submissions and suggested improvements based on an advocacy organization’s decade-long experience. Am J Med Genet C Semin Med Genet. 2022;190(2):156–161. doi: 10.1002/ajmg.c.32001.
- Kemper AR, Knapp AA, Metterville DR, et al. Weighing the evidence for newborn screening for Hemoglobin H disease. J Pediatr. 2011;158(5):780–783. doi: 10.1016/j.jpeds.2010.10.042.
- United Nations: International Migration Report. 2015. [pdf] Available from http://www.un.org/en/development/desa/population/migration/publications/migrationreport/docs/MigrationReport2015_Highlights.pdf. International Migration Report 2015 [accessed April 6 2023].
- Stepler R, Lopez MH. “U.S. Latino population growth and dispersion has slowed since onset of the Great Recession.” Pew Research Center, September 2016. [pdf] Available from https://www.pewresearch.org/hispanic/wp-content/uploads/sites/5/2016/09/PH_2016.09.08_Geography.pdf. [accessed April 6, 2023].
- Moo-Penn WF, Bechtel KC, Johnson MH, et al. Hemoglobin Fannin-Lubbock [α2 β 2 119 (GH2) Gly replaced by Asp]. A new hemoglobin variant at the α1 β 1 contact. Biochim Biophys Acta. 1976;453(2):472–477. doi: 10.1016/0005-2795(76)90142-2.
- Schneider RG, Berkman NL, Brimhall B, et al. Hemoglobin Fannin-Lubbock [α 2 β 2 119(GH2) Gly replaced by Asp]. A slightly unstable mutant. Biochim Biophys Acta. 1976;453(2):478–483. doi: 10.1016/0005-2795(76)90143-4.
- Qin WB, Pobedimskaya DD, Molchanova TP, et al. Hb Fannin-Lubbock in five Spanish families is characterized by two mutations: β 111 GTC–>CTC (Val–>Leu) and β 119 GGC–>GAC (Gly–>Asp). Hemoglobin. 1994;18(4-5):297–306. doi: 10.3109/03630269408996195.
- González FA, Ropero P, Arrizabalaga B, et al. Fannin-Lubbock II hemoglobin [b111 (G13) Val→Leu and b119 (GH2) Gly→Asp]: description of four new cases. Medicina Clínica. 2007;129(10):379–381.
- Ibarra B, Aizpuru E, Sánchez-López JY, et al. HB Fannin-Lubbock-I with a single GGC > GAC mutation at β119(GH2)Gly–>Asp in a homozygous Mexican patient. Hemoglobin. 2009;33(6):492–497. doi: 10.3109/03630260903332866.
- Schwartz HC, Spaet TH, Zuelzer WW, et al. Combinations of hemoglobin G, hemoglobin S and thalassemia occurring in one family. Blood. 1957;12(3):238–250. doi: 10.1182/blood.V12.3.238.238.
- Brancati C, Caracciolo M, Bria M, et al. Hb G-San José homozygosis in a Calabrian family. Hemoglobin. 1989;13(5):497–503. doi: 10.3109/03630268908998089.
- Cremonesi L, Travi M, Li Volti S, et al. Evidence for the single origin of HB G-San José in Sicily. Hemoglobin. 1989;13(6):579–584. doi: 10.3109/03630268908993108.
- Espinosa Jurcott J, Landero de Ruiz N, Armenta Olvera T, et al. [Hemoglobin G-San José associated with hereditary spherocytosis in a Mexican family (author’s transl)]. Rev Invest Clin. 1981;33(4):383–386.
- Farah RA, Buchanan GR, Timmons CF, et al. Double heterozygosity for Hb G-San José [β7(A4)Glu–>Gly] and Hb Fukuoka [β2(NA2)His–>Tyr] in a 2 1/2-year-old girl. Hemoglobin. 1999;23(4):383–387. doi: 10.3109/03630269909090756.
- Musumeci S, Schilirò G, Pizzarelli G, et al. Hemoglobin G San José [β 2 7 (A4) Glu to Gly α 2], β thalassemia, and α thalassemia in a Sicilian family. Hum Genet. 1979;52(2):239–247. doi: 10.1007/BF00271579.
- Moo-Penn WF, Johnson MH, Bechtel KC, et al. Hemoglobins Austin and Waco: two hemoglobins with substitutions in the α 1 β 2 contact region. Arch Biochem Biophys. 1977;179(1):86–94. doi: 10.1016/0003-9861(77)90089-3.
- Racsa LD, Luu HS, Park JY, et al. β-Globin gene sequencing of hemoglobin Austin revises the historically reported electrophoretic migration pattern. Arch Pathol Lab Med. 2014;138(6):819–822. doi: 10.5858/arpa.2013-0105-OA.
- Itano HA. A third abnormal hemoglobin associated with hereditary hemolytic anemia. Proc Natl Acad Sci U S A. 1951;37(12):775–784. doi: 10.1073/pnas.37.12.775.
- Brittenham GM. Globin gene variants and polymorphisms in India. In: Winter WP, editor. Hemoglobin variants in human populations. Boca Raton, FL, USA: CRC Press; 1987. Vol. 2, p. 79–110.
- Atalay EO, Atalay A, Ustel E, et al. Genetic origin of Hb D-Los Angeles [β121(GH4)Glu–>Gln, GAA–>CAA] according to the β-globin gene cluster haplotypes. Hemoglobin. 2007;31(3):387–391. doi: 10.1080/03630260701459416.
- Fioretti G, De Angioletti M, Pagano L, et al. DNA polymorphisms associated with Hb D-Los Angeles [β 121(GH4)Glu–>Gln] in southern Italy. Hemoglobin. 1993;17(1):9–17. doi: 10.3109/03630269308998881.
- Perea FJ, Casas-Castañeda M, Villalobos-Arámbula AR, et al. Hb D-Los Angeles associated with Hb S or β-thalassemia in four Mexican Mestizo families. Hemoglobin. 1999;23(3):231–237. doi: 10.3109/03630269909005703.
- Shanthala Devi AM, Rameshkumar K, Sitalakshmi S. Hb D: a not so rare hemoglobinopathy. Indian J Hematol Blood Transfus. 2016;32(Suppl 1):294–298. doi: 10.1007/s12288-013-0319-3.
- Badr FM, Lorkin PA, Lehmann H. Haemoglobin P-Nilotic containing a - chain. Nat New Biol. 1973;242(117):107–110. doi: 10.1038/newbio242107a0.
- Altay C, Kutlar A, Wilson JB, et al. Hb P-Nilotic or α 2(β delta)2 in a Turkish family. Hemoglobin. 1987;11(4):395–399. doi: 10.3109/03630268709042859.
- Moo-Penn WF, Bechtel KC, Therrell BL. Jr. Hemoglobin P Nilotic in a Mexican-American family. Hemoglobin. 1978;2(1):65–69. doi: 10.3109/03630267808999190.