
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Objective
The efficacy of topical nonsteroidal anti-inflammatory drugs (NSAIDs) relates not only to the individual NSAID used but also to differences in formulation design. The aim of this study was to investigate the fundamental differences in ibuprofen and diclofenac drug delivery vehicles, specifically gels and plasters, compared to a recently launched 200 mg ibuprofen medicated plaster and characterize the resulting dermatologic–pharmacokinetic profiles into and through ex vivo human skin layers.
Methods
In vitro skin permeation testing over 24 h and sacrificial timepoint penetration experiments (at 1, 4, 8, 12, and 24 h) were conducted using an automated flow-through diffusion cell system. The amount of drug delivered to the epidermis, dermis, and receptor solution (representing deeper tissue) was determined by liquid chromatography-tandem mass spectrometry. Skin protein binding of ibuprofen and diclofenac was investigated by spiking skin homogenate with increasing concentrations of each drug and determining the fraction unbound.
Results
Differences were observed in the amount of drug recovered at sacrificial timepoints and rate at which drug was delivered to the target site between plaster and gel formulations of ibuprofen and diclofenac and between plaster formulations of the same drug (ibuprofen). While the amount of drug quantified at sacrificial timepoints did not necessarily determine in vivo flux rates, differences in drug distribution within the skin layers indicated where drug reservoirs were formed.
Conclusions
These findings highlight the importance of intelligent formulation design in determining NSAID delivery through skin layers. Further work is required to quantify drug delivery into deeper tissues and the resultant local anti-inflammatory effects.
Introduction
Topical nonsteroidal anti-inflammatory drugs (NSAIDs) are widely accepted as a treatment for acute musculoskeletal conditions, such as sprains, strains, and overuse injuries, as they can provide good levels of pain relief with an improved safety profile over their oral counterparts [Citation1]. Topical drug preparations are designed to provide a localized therapeutic effect while minimizing systemic concentrations of the drug [Citation2]. The relative efficacy of different topical preparations is related to a combination of both the pharmacokinetic and pharmacodynamic properties of the various drugs and their formulations [Citation3]. For a topically applied NSAID to exert its anti-inflammatory properties at the site of acute soft tissue injury, it first must pass through the skin [Citation1]. This is no easy task as a key function of the skin is to act as a physical and biological barrier to the external environment, preventing penetration and any harmful effects of foreign materials [Citation4].
The barrier function of the skin is primarily located in the stratum corneum (SC), which is composed of corneocytes embedded in a lipid matrix [Citation5]. The SC is also the primary challenge for any passive noninvasive topical drug delivery [Citation6]. The epidermal layer is avascular, receiving nutrients by diffusion from the underlying dermis [Citation4]. Therefore, any drug which penetrates the epidermis will not necessarily be transported any further at this stage via the vasculature or lymphatic system and can form a ‘reservoir’ within the epidermis.
Topically applied drugs may show a depot effect, accumulating in one or more of the skin layers or in the subcutaneous fat to form a reservoir [Citation7]. This may be advantageous in providing a sustained release of the drug to surrounding tissues across a concentration gradient. However, depending on the nature of the drug (lipid/water solubility, protein-binding capacity, etc.) this may be disadvantageous; for example, drugs may be retained in certain tissues underlying the site of application [Citation7].
The dermis is highly vascularized, therefore drugs that penetrate as far as this have the potential to continue down into deeper tissue via local blood vessel distribution, with drug partitioning into tissue or removal by the systemic blood supply (which is dependent on blood flow to the tissues and protein binding of the NSAID in the blood) [Citation4,Citation8,Citation9].
In vivo understanding of effective local concentrations of topically applied NSAIDs in soft tissue injuries is limited at best, with techniques such as microdialysis falling short on the level of validation required to be more widely accepted [Citation10]. The gold standard for demonstrating topical NSAID efficacy remains double-blind, placebo-controlled clinical trials. Mechanistic characterization studies alongside, however, can help support a better understanding as part of a more complete package of data [Citation11].
A number of factors govern the permeation of drugs such as NSAIDs into the skin, including their physicochemical properties (lipophilicity, solubility, molecular weight [MW] or size, and charge) and the influence of excipients in the formulation [Citation4,Citation12].
Ibuprofen and diclofenac are commonly used NSAIDs for the management of pain and inflammatory conditions. Both are nonselective NSAIDs that reversibly inhibit cyclo-oxygenase (COX)-1 and COX-2 thereby reducing the production of prostaglandins, key components of the inflammatory and nociceptive response [Citation3]. Based on half-maximal inhibitory concentration (IC50) ratios of COX-1 and COX-2 inhibition, diclofenac is considered to be more potent than ibuprofen [Citation13].
Both diclofenac and ibuprofen are widely available across Europe as over-the-counter topical preparations in both gel and plaster formulations. In terms of chemical factors, the degree of ionization of the drug has a significant impact on its absorption through the skin. For example, unionized drugs may have greater permeation when compared to their ionized form due to the hydrophobic similarity with the SC [Citation14].
Ibuprofen (MW 206 g/mol; pKa 5.3) is a relatively lipophilic (Log P 3.97), insoluble compound that will ionize at physiologic pH to become water-soluble [Citation15]. The physicochemical, diffusion and absorption characteristics of ibuprofen make it highly suitable for transdermal delivery, and its permeation across skin has been well documented [Citation4,Citation16,Citation17]. While the route of delivery for topical (out–in) and oral formulations (in–out) of ibuprofen are different, concentrations in tissues directly under the site of topical application have been demonstrated to be in the same order of magnitude as those found after oral administration [Citation18].
Diclofenac in its acidic form (MW 296 g/mol; pKa 4.15) is a hydrophobic, relatively lipophilic (Log P 4.51), low water-soluble molecule [Citation19,Citation20]. However, in commercially available topical formulations it is in the form of therapeutically active salts, diclofenac sodium (MW 318.1 g/mol) [Citation21], diclofenac epolamine (MW 411.3 g/mol) [Citation22], or diclofenac diethylammonium (MW 370.3 g/mol) [Citation23]. The ion-paired formulations of diclofenac have physicochemical behavior which influences permeation across both hydrophobic and hydrophilic membranes [Citation24]. Data are available to suggest that topical diclofenac can penetrate the skin and permeate to deeper tissues, with a lower plasma-to-tissue ratio compared with oral diclofenac, although this data has been described as sparse and inconsistent [Citation7].
Most recently, an ibuprofen-medicated plaster [Citation25] has been developed which has shown significant and clinically relevant reductions in local pain and tenderness when applied every 24 h in a double-blind, placebo-controlled clinical efficacy study [Citation26,Citation27]. NSAID plasters allow for continuous release of the drug under steady-state, with passive transport from a high concentration into the underlying tissues [Citation28].
In addition to the NSAID form (acid, anion, ion-pairs), the format and design of formulation excipients, including penetration enhancers, play a critical role in determining topical drug delivery optimization [Citation29,Citation30]. The aim of this study was to investigate the fundamental differences in topical NSAID (ibuprofen and diclofenac) drug delivery vehicles, specifically, gels and plasters, compared to the recently launched 200 mg ibuprofen medicated plaster [Citation25], and characterize the resulting dermatologic–pharmacokinetic profiles into and through ex vivo human skin layers.
Methods
Skin sample preparation
Skin tissue preparation was performed following European Medicines Agency (EMA) guidance [Citation31]. Human skin was obtained from samples excised during cosmetic reduction surgery. Subcutaneous fat was removed mechanically, and the skin was prepared using a dermatome to achieve samples of the required thickness. Tissue samples were then flash-frozen with liquid nitrogen and stored at −80 °C until use.
Test formulations
The ibuprofen and diclofenac formulations tested in the study are shown in . For the purpose of the study, plasters were cut using a razor blade into approximately 1 cm2 sections to cover the skin dosing area, and the backing was removed before being placed on the skin with forceps. The forceps were used to gently press down on the plaster to ensure thorough contact across the entire piece of tissue. Gel formulations were applied to a plunger, which was weighed before and after dosing to calculate the exact amount of formulation applied with the aim of a total dose of approximately 10 mg per skin sample (10 mg/cm2 dose).
Table 1. Composition of formulations.
In vitro skin permeation testing (IVPT) and sacrificial timepoint penetration
In vitro skin permeation testing (IVPT) and sacrificial timepoint penetration experiments were conducted using an automated flow-through diffusion cell system designed to mimic the physiologic and anatomic conditions of skin in situ (MedFlux-HT®; MedPharma, Durham, NC, USA). The pump of the MedFlux-HT system was adjusted to maintain a continuous receptor solution flow rate of approximately 6 μL/min (360 μL/h) directly under the skin. A receptor solution flow-through cell (no skin or formulation) and a blank cell (skin, no formulation) were set up as controls.
At the required time points, the residual formulation was removed from the surface of the skin and the skin surface was tape stripped up to five times to remove residual formulation and the SC. The epidermis was then heat-separated from the dermis by placing the skin into an incubator at 60 °C for 2 min, followed by manual separation using forceps. Ibuprofen and diclofenac in the receptor solution and skin layers were quantified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Waters Acquity I-Class UPLC; Waters Xevo TQ-XS Triple Quadrupole Mass Spectrometer; Waters TargetLynx Software; Waters Corporation, Milford, MA, USA).
Method development
Method development work was first performed to identify a suitable receptor solution, extraction fluid, sampling time points, and skin thickness.
To determine a suitable receptor solution in which the drugs would remain stable as they penetrated the skin and permeated the receptor solution, the chemical stability of ibuprofen and diclofenac was studied in three receptor solutions, both in the solution alone and in the presence of skin. Samples were collected at four and three time points, respectively, and analyzed by LC-MS/MS. The recovery of each drug was determined at each time point, relative to the amount detected at t = 0. On the basis of these results, phosphate-buffered saline (PBS) with 0.01% Brij was chosen as the receptor solution (relative recoveries to t = 0 was within 100 ± 15%).
Receptor solution sampling timepoints and skin thickness were established through a small-scale IVPT experiment. A single representative test formulation for each drug (diclofenac gel and 200 mg ibuprofen plaster) was tested on dermatomed human skin from one donor with three different thicknesses (250, 500, and 750 ± 50 µm). Lower variability was seen in the permeation data on human skin with a thickness of 500 µm compared with 250 and 750 µm. Receptor solution sampling time points were every hour for 24 h and the target skin thickness chosen was 500 µm.
To determine a suitable extraction fluid for optimal drug recovery from epidermis and dermis processed after the in vitro permeation experiment, ibuprofen and diclofenac were extracted using two test fluids, 90:10 v/v acetonitrile:water or 90:10 v/v ethanol:water. Samples were analyzed by LC-MS/MS and the recovery of the drug was calculated compared to the controls (empty homogenizer vials). Recovery was within the acceptance criteria of 100 ± 15% for both extraction fluids and 90:10 acetonitrile:water was chosen as the extraction fluid.
Full-scale IVPT and sacrificial timepoint penetration
Twenty-four-hour IVPT and sacrificial timepoint penetration
Full-scale IVPT was performed on three different skin donors and six repeats per donor (single repetition performed for the blank). The continuous flow of receptor fluid directly under the skin was analyzed to find the mean cumulative amount of ibuprofen and diclofenac delivered to the receptor solution every hour for a total of 24 h. Flux data were obtained by measuring the amount of drug (ng) that permeated across a defined area of skin (cm2) over the timeframe of an hour, thereby giving a rate of permeation as flux (ng/cm2/h).
The mean cumulative amount (percentage of the applied dose) of each ibuprofen or diclofenac recovered from the epidermis and dermis at 24 h was also determined.
One-, four-, eight-, and twelve-hour sacrificial timepoint penetration
Mean cumulative amounts of ibuprofen or diclofenac recovered from the epidermis and the dermis at 1, 4, 8, and 12 h post-formulation application were determined in a sacrificial time-course study. A total of five replicates were performed per sacrificial time point. Due to the limited size of ex vivo skin samples, two different donors were used (donor 1 was used for 1 and 12 h; donor 2 was used for 4 and 8 h).
Analytical method
Tissue samples were homogenized in extraction fluid (90% acetonitrile, 10% water [v/v]) and transferred to a 96-well neat extraction plate. Samples were centrifuged at 4000 rcf for 1–2 min, pulling tissue extracts to the bottom of the wells. 10 µL of each sample from the neat extraction plate was added to 490 µL of blank extraction fluid in the 50x‐prep dilution plate, sealed with adhesive aluminum, and mixed for 5 min. 10 µL of each dilution was added to 490 µL of blank extraction fluid in the 2500x‐prep dilution plate, sealed and mixed for 5 min.
An LC-MS/MS analytical method (developed and implemented by MedPharm) was used to quantify ibuprofen and diclofenac from the receptor solution and extraction fluid in a single method. A 1.9-min run time method was assessed to ensure sufficient sensitivity could be achieved. A signal-to-noise ratio >5:1 was achieved over the following ranges: receptor solution: 0.25–1000 ng/mL for ibuprofen (lower limit of quantitation [LLOQ] of 0.25 ng/mL) and 0.10–1000 ng/mL for diclofenac (LLOQ of 0.10 ng/mL); extraction fluid: 1.0–1000 ng/mL for ibuprofen (LLOQ of 1.0 ng/mL) and 0.25–1000 ng/mL for diclofenac (LLOQ of 0.25 ng/mL).
Data analysis
The concentration of ibuprofen and diclofenac detected in the receptor solution and skin layers were quantified using a calibration range optimized for the analysis of the samples generated during the IVPT experiments. The amount of ibuprofen and diclofenac recovered from the epidermis and dermis was calculated at the conclusion of the run. The cumulative amount per cm2 of ibuprofen and diclofenac that permeated the receptor solution was calculated from drug concentrations measured in the receptor solution over time. The cumulative amount per cm2 of ibuprofen and diclofenac at the final time point was calculated for each replicate. Means and standard deviations/standard errors were calculated and reported. Data were log-transformed and statistically compared using one-way analysis of variance (ANOVA) with Tukey Honest Significant Difference (HSD) analysis.
Skin protein binding study
To measure any binding of ibuprofen or diclofenac to skin proteins, skin homogenate with combined dermal and epidermal tissue was prepared from a pool of three skin donors. Samples of the homogenate were spiked with each drug compound at increasing dose concentrations (100, 1000, and 10,000 ng/mL). Spiked and blank samples were individually loaded into donor compartments of a rapid equilibrium dialysis (RED) device (n = 3 repetitions per sample), with PBS buffer loaded into the receiver chambers. The RED device was covered and allowed to incubate at 37 °C for 6 h while shaking at 300 rpm on an orbital shaker. After the incubation period, samples were extracted and the fraction unbound (fu) of ibuprofen and diclofenac in skin homogenate and HSA was determined using LC-MS/MS analysis. The fraction unbound for skin homogenates was calculated as follows:
D* = dilution factor of biologic matrix.
Results and discussion
IVPT and sacrificial timepoint penetration
Twenty-four-hour IVPT and sacrificial timepoint penetration
The mean cumulative amounts of ibuprofen and diclofenac delivered to the receptor solution over 24 h following application of plaster and gel formulations are shown in for ibuprofen and for diclofenac. A greater amount of the applied dose of ibuprofen was recovered from the receptor solution after 24 h with the 200 mg (63,935 ng/cm2) versus 136 mg (3638 ng/cm2) ibuprofen plaster (p < 0.0001) (; Table S1). The cumulative amount of ibuprofen in the receptor solution for the 200 mg plaster was also greater versus the gel (48,231 ng/cm2) formulation at the 24-h timepoint, although the difference was not shown to be significant (p = 0.9998). Overall, the amount of diclofenac recovered from the receptor solution was higher for the gel formulation (2530 ng/cm2) versus the plaster formulation (393 ng/cm2) at the 24-h timepoint (p < 0.0001) (; Table S1).
Figure 1. The amount of ibuprofen and diclofenac permeating across the skin into the receptor solution over 24 h following application of gel and plaster formulations, shown as mean cumulative amount (ng/cm2) for (A) ibuprofen formulations and (B) diclofenac formulations, and flux rate (ng/cm2/h) for (C) ibuprofen formulations and (D) diclofenac formulations. Error bars represent one standard error of the mean. API: active pharmaceutical ingredient.
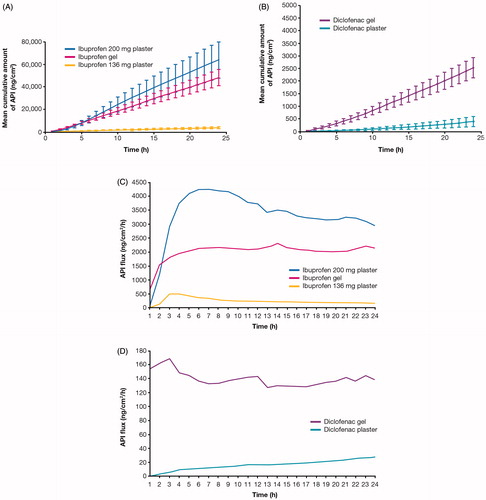
When comparing the IVPT results of the 200 mg ibuprofen plaster versus the 136 mg ibuprofen plaster (), it is clear that the difference in formulation composition and optimization makes a marked difference to the drug delivery. The ibuprofen contained within the adhesive layer of the 200 mg ibuprofen plaster more readily partitioning into and across the skin compared with the 136 mg ibuprofen plaster, with the difference in drug loading alone not accounting for the large difference observed.
When comparing the IVPT results of the diclofenac gel versus the diclofenac plaster (), again there is a clear difference in cumulative amounts of active pharmaceutical ingredient (API). It is suspected that the ion-pairing physicochemical effect of the diethyl ammonium in the diclofenac gel compared with diclofenac sodium in the plaster formulation has a positive effect alongside the formulation format and composition differences [Citation36]. In addition, another important factor to consider is that gels dry quickly on the skin surface, whereas plasters do not. An increase in drug concentration from the gel due to drying may have influenced the observed differences.
The API flux results (; Table S2) show the rate of drug (ng) permeating an area (cm2) over time (h). It was interesting to observe that the ibuprofen gel showed the fastest permeation rate within the first hour, but this was matched (2 h) and eventually surpassed (<3 h) by the 200 mg ibuprofen plaster, delivering a significantly higher flux. The 136 mg ibuprofen plaster had a significantly lower flux rate. The diclofenac formulations permeated to a far lesser extent when compared with the ibuprofen formulations, as reported elsewhere [Citation37]; being a different drug, the data have been presented separately. With the diclofenac formulations, there was a notable difference between the plaster and gel formulations, the diclofenac gel having a more immediate and consistent rate of delivery compared to the medicated plaster ().
The mean percentage of the applied dose of ibuprofen and diclofenac recovered from the epidermis, dermis, and receptor solution 24 h following the application of plaster and gel formulations are shown in for ibuprofen and for diclofenac, with the statistical analysis described in Table S3.
Figure 2. Mean cumulative amount (expressed as percent applied dose) of (A) ibuprofen and (B) diclofenac recovered from the epidermis, dermis, and receptor solution 24 h following application of plaster and gel formulations. Bars represent mean and error bars represent one standard error of the mean. *p < 0.0001 vs ibuprofen 200 mg plaster; **p < 0.0001 vs diclofenac gel; †statistical analysis not performed.
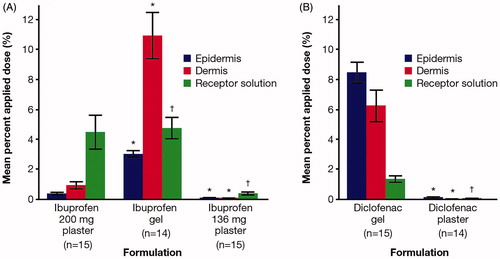
Following the application of the 200 mg ibuprofen plaster, the amount of ibuprofen recovered at 24 h increased through the epidermis, dermis, and receptor solution. In contrast, for ibuprofen gel, the highest amount of ibuprofen was recovered from the dermis. Overall, a greater amount of ibuprofen was recovered from the epidermis and dermis for ibuprofen gel versus both ibuprofen plaster formulations. However, a similar proportion of ibuprofen dose was recovered from the receptor solution for both ibuprofen gel and 200 mg ibuprofen plaster.
Following the application of the diclofenac plaster, the amount of diclofenac recovered at 24 h remained low through the epidermis, dermis, and receptor solution (). Following the application of diclofenac gel, the amount of diclofenac recovered decreased as it permeated the epidermis, dermis, and receptor solution. Overall, the amounts of diclofenac recovered from the epidermis, dermis, and receptor solution were higher for the gel formulation versus the plaster formulation.
There was no retention in the epidermis or dermis ‘reservoir’ observed for the 200 mg ibuprofen plaster as there was for both the ibuprofen gel and the diclofenac gel. An explanation is thought to come from differences in the delivery vehicle and drug form, with the plaster format maintaining a concentration gradient and consistent partitioning of mobile lipophilic ibuprofen molecules from the anhydrous drug-in-adhesive layer into the intercellular lipid pathway of the SC. Once through the SC, the ibuprofen acid is then readily available to ionize in situ at physiologic pH to become water-soluble and continue its transit unrestricted into deeper tissue. A gel, on the other hand, dries quickly on the surface of the skin through solvent evaporation, concentrating the drug in the residual phase. For the ibuprofen gel, the concentrated drug was partially in the form of ibuprofen diethyl ammonium [Citation38] and for the diclofenac gel, diclofenac diethyl ammonium. The efficiency of the reservoir is dependent on the API physicochemical-properties, including lipid/water solubility, protein-binding capacity, application time, and application mode [Citation39]. With such a reservoir effect, it is not clear how much drug remains mobile and available for permeation down to deeper tissue and how much crystalizes out and becomes stranded.
One-, four-, eight-, and twelve-hour sacrificial timepoint penetration
Sacrificial timepoint penetration was also investigated at 1, 4, 8, and 12 h (mean cumulative amount of drug recovered) for ibuprofen () and diclofenac () gel and plaster formulations. Results at 24 h with the addition of receptor solution are also presented, but it should be noted this was a separate experiment from the remaining sacrificial timepoints. The mean cumulative amount (percent applied dose) of ibuprofen or diclofenac recovered from the epidermis and the dermis at each time point is shown in .
Figure 3. Mean cumulative amount of (A) ibuprofen and (B) diclofenac recovered from the epidermis and dermis at 1-, 4-, 8-, and 12-h post the application of gel and plaster formulations. Bars represent mean and error bars represent one standard deviation.
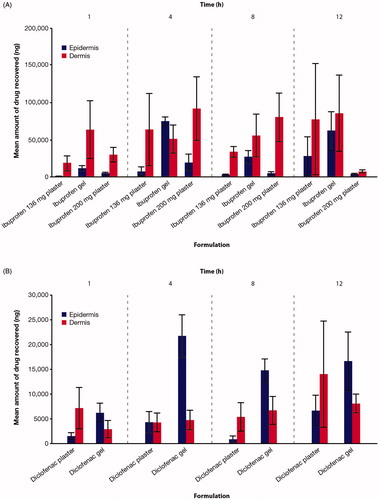
Table 2. Mean cumulative amount (percent applied dose) of ibuprofen or diclofenac recovered from epidermis and dermis at 1, 4, 8, and 12 h post-formulation application.
At 1, 4, 8, and 12 h, more ibuprofen was recovered from the dermis versus the epidermis with both plaster formulations (). This was also true for ibuprofen gel, except at 4 h where a higher amount was recovered from the epidermis versus dermis. At 12 h the amount of ibuprofen recovered from the dermis from the 200 mg ibuprofen plaster decreased markedly compared with all other time points and compared with ibuprofen gel. A potential explanation for this observation is that the 200 mg plaster may facilitate deep drug penetration, with limited accumulation in the dermal layers (especially after 8 h). Differences in the drug delivery were seen within the same formulation of the same drug (200 mg and 136 mg ibuprofen plaster), demonstrating the importance of intelligent formulation design, including the excipients, in determining the delivery profile of topical NSAIDs across skin. In the 200 mg plaster, ibuprofen acid is solubilized in macrogols (polyethylene glycols) rather than neutralized in situ in a hydro-alcoholic gel, increasing the permeability coefficient [Citation16] and facilitating permeation through the skin. The 136 mg plaster incorporates ibuprofen into a strong adhesive system largely based on an acrylate polymer matrix, with the addition of oleic acid to act as a skin permeation enhancer [Citation40].
For the diclofenac plaster formulation, more diclofenac was recovered from the dermis versus the epidermis at 1, 8, and 12 h, with similar levels at 4 h (). Conversely, consistently greater levels of diclofenac were recovered from the epidermis versus the dermis for the gel formulation. Higher levels of diclofenac were recovered from the epidermis with the gel formulation versus the plaster formulation at 1 (p < 0.0001), 4 (p = 0.0005), and 8 h (p < 0.0001), with no significant difference at 12 h (); levels recovered from the dermis were similar between both preparations at all timepoints. Across the 1, 4, 8, 12, and 24-h timepoints, more diclofenac is held up in the epidermis compared with the dermis for the gel formulation.
The level of variability observed between the trends observed in the 24 h and 1, 4, 8, and 12-h sacrificial timepoints for both ibuprofen and diclofenac requires clarification. In general, for ibuprofen formulations the 12- and 24-h penetration data are broadly consistent, with the exception of the 136 mg plaster, which likely reflects an atypical result at 12 h which prevented the statistical removal of any outlier data points. Variability between and within experiments was to be expected given the known variability within a section of human skin and between donors, and the methodology involved. The heat separation method used to process the samples can result in drug redistribution between the tissues during the separation procedure, which can introduce errors. Evidence also suggests that the number of replicates needed to minimize inter-sample variability may differ depending on the individual drug [Citation41]. Variability in results seen with in vitro skin permeation data is widely accepted to be related to the variability in the skin used [Citation42]. An optimized design of experiments, sets of skin donors, and the number of repeats within, was employed to try and minimize variability as much as possible. However, due to the number of skin samples required, different donors were used across sacrificial timepoints (one donor for timepoints 1 and 12 h and a second donor for timepoints 4 and 8 h; five replicates per donor per time point), which may have contributed to variability in the data. It should be noted that the 24-h experiment was performed using three skin donors and therefore likely represents a more robust set. IVPT experiments have a number of limitations in terms of their ability to reflect in vivo release or potential clinical effectiveness. There are obvious limitations to the interpretation of these results with regards to deeper vascular tissue permeation and partition into soft tissues. While such studies can be used to compare products and compare significance in such a result, they are limited to this and any investigation on the duration of clinical effect is hampered by a lack of validation and understanding of distribution to deeper tissues. However, despite limitations, in vitro permeation studies are considered a valuable measure of transdermal performance [Citation42].
Skin protein binding study
The fraction unbound of each compound showed no major trends by concentration (; Table S4), indicating that binding is not dependent on the concentration of ibuprofen or diclofenac over the concentration ranges tested. The mean percentage of bound drugs in skin homogenate was 88.7% for ibuprofen and 95.7% for diclofenac.
Figure 4. Mean percent of (A) ibuprofen and (B) diclofenac bound to proteins in skin homogenate containing epidermal and dermal tissue pooled from three skin donors spiked at increasing concentrations (n = 3). Error bars represent standard deviation from the mean. API: active pharmaceutical ingredient.
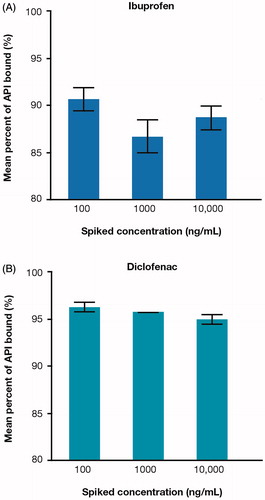
Drug–protein binding is a key factor in determining the pharmacokinetic properties of a drug [Citation43]. While the importance of plasma protein binding on effective drug concentration at target sites is well known [Citation44], proteins are also present beyond the bloodstream and can bind with drugs in skin, tissues, or organs [Citation43]. The high molecular mass of the drug–protein complex can further affect the pharmacologic activity of drugs by limiting passage across biologic membranes [Citation43]. The degree of plasma protein binding has been reported in topical drug delivery to be a determining factor in how much tissue diffusion is facilitated by convective blood, lymphatic, and interstitial transport to deep tissues [Citation45]. This study investigated protein binding in skin homogenates of epidermal and dermal skin tissue. A topically applied NSAID will first need to diffuse through the non-vascular epidermis and its terminally differentiated SC barrier in order to reach any local blood vessels and achieve distribution to deeper tissue [Citation4]. Protein binding in skin homogenates could in part help explain the differences observed in passive drug distribution between epidermis, dermis, and receptor solution (), where the distribution of ibuprofen was found to be dominant in the lower dermis and receptor solution compared with diclofenac in the upper epidermis after 24 h. However, as this study did not quantify the drug in the corneocytes’ protein matrix, drug partitioning into the skin lipids should be considered as a possible source of error. With protein binding in the dermis, errors can also be introduced owing to the fact that proteins (including albumin) are mobile in the dermis and can diffuse out from the tissue into the surrounding solution.
Conclusion
The aim of this study was to investigate differences in ibuprofen and diclofenac drug delivery via gels and plasters compared to a recently launched 200 mg ibuprofen medicated plaster and characterize the resulting dermatologic–pharmacokinetic profiles into and through ex vivo human skin layers. The findings provide additional evidence that the biophysical properties and formulation design (including format and choice of excipients) of NSAIDs are significant determining factors for the delivery profile into and through human skin, and therefore ultimate ability to access deeper soft tissue injury sites.
A key finding to this study was that the amount of drug found at the sacrificial timepoints within the epidermis and dermis did not necessarily determine the in vitro flux rate of the drug through to the receptor solution. The ibuprofen 200 mg plaster did not show a ‘reservoir’ effect in upper skin layers; instead more a ‘tap on tap off’ effect with rapid transition across the epidermis and dermis with drug partitioning driven by a controlled and consistent concentration gradient established and maintained between the drug in the adhesive layer and receptor solution.
A hypothesis based on this finding is that gels with an increased reliance on the formation of a concentration gradient within the upper skin layers upon dry down, as well as the drug remaining mobile in order to maintain the partition co-efficient (driving force) over time, could result in increased intra-patient variability. Pharmacokinetic data support this hypothesis, with a mean Cmax of 556 ng/mL (SD 249; 95% CI 439–603) for the ibuprofen medicated plaster [Citation28], compared to 630 ng/mL (SD 520) for the ibuprofen gel [Citation46] and 5.4 ng/mL (SD 4.6; range 2.2–29.4) for the diclofenac gel [Citation47].
Topical drug delivery can be investigated and ranked by individual NSAID using in vitro methodology or, alternatively, in blood plasma in an in vivo pharmacokinetic study. However, an accepted model to predict, or technique to quantify, drug delivery directly in local soft tissue injury sites is still missing for topical NSAIDs. Ideally, inhibition of muscle tissue injury inflammatory mediators, primarily prostaglandin E2 and I2, could be measured alongside drug concentrations. Such a technique would allow, for the first time, a comparison across NSAIDs that is not reliant upon an adjustment factor for potency (IC50) based on preclinical cellular assays. The IC50 of diclofenac is reported to be approximately 900 times lower than ibuprofen [Citation37]; however, there is no head-to-head or clear difference in clinical efficacy [Citation1,Citation3].
The main mechanism of action of both ibuprofen and diclofenac is the nonselective, reversible inhibition of the cyclooxygenase enzymes COX-1 and COX-2. PGE2 and PGI2 are pro-inflammatory mediators that enhance edema formation, increase vascular permeability, and promote leukocyte infiltration. They also reduce the threshold of nociceptor sensory neurons to stimulation [Citation48,Citation49]. Many of the pharmacodynamic effects can be directly linked to the inhibition of prostanoid synthesis and therefore have a ceiling effect. However, what is not well understood is the local concentration required in situ in a soft-tissue injury (and how long this needs to be maintained) to have a clinically meaningful effect.
This work has highlighted the need for such a validated pharmacokinetic/pharmacodynamic model for topical NSAIDs to address this knowledge gap.
Author contributions
GP wrote and further developed the original draft. All authors were involved in the study design, analysis, and interpretation of data, and contributed to the preparation of this manuscript, including its critical review and approving the final draft and accept full responsibility for its content.
Supplemental Material
Download PDF (172.3 KB)Acknowledgments
The authors would like to acknowledge Majella E. Lane (UCL School of Pharmacy, London, UK) for her valuable contributions to the manuscript and Anisha Desai (Elements Communications, Ltd., Westerham, UK) for providing medical writing assistance (supported by Reckitt Healthcare Ltd.).
Disclosure statement
The authors are employees of Reckitt Healthcare Ltd.
Additional information
Funding
Related Research Data
References
- Derry S, Moore RA, Gaskell H, et al. Topical NSAIDs for acute musculoskeletal pain in adults. Cochrane Database Syst Rev. 2015;2015(6):Cd007402.
- Barkin RL. Topical nonsteroidal anti-inflammatory drugs: the importance of drug, delivery, and therapeutic outcome. Am J Ther. 2015;22(5):388–407.
- Derry S, Wiffen PJ, Kalso EA, et al. Topical analgesics for acute and chronic pain in adults - an overview of Cochrane reviews. Cochrane Database Syst Rev. 2017;5(5):Cd008609.
- Manoukian MAC, Migdal CW, Tembhekar AR, et al. Topical administration of ibuprofen for injured athletes: considerations, formulations, and comparison to oral delivery. Sports Med Open. 2017;3(1):36.
- Bouwstra JA, Ponec M. The skin barrier in healthy and diseased state. Biochim Biophys Acta. 2006;1758(12):2080–2095.
- Trommer H, Neubert RH. Overcoming the stratum corneum: the modulation of skin penetration. A review. Skin Pharmacol Physiol. 2006;19(2):106–121.
- Hagen M, Baker M. Skin penetration and tissue permeation after topical administration of diclofenac. Curr Med Res Opin. 2017;33(9):1623–1634.
- Roberts MS. Targeted drug delivery to the skin and deeper tissues: role of physiology, solute structure and disease. Clin Exp Pharmacol Physiol. 1997;24(11):874–879.
- Roberts MS, Cross SE. Percutaneous absorption of topically applied NSAIDS and other compounds: role of solute properties, skin physiology and delivery systems. Inflammopharmacology. 1999;7(4):339–350.
- Herkenne C, Alberti I, Naik A, et al. In vivo methods for the assessment of topical drug bioavailability. Pharm Res. 2008;25(1):87–103.
- Howick J, Glasziou P, Aronson JK. The evolution of evidence hierarchies: what can Bradford Hill’s ‘guidelines for causation’ contribute? J R Soc Med. 2009;102(5):186–194.
- Rainsford KD, Kean WF, Ehrlich GE. Review of the pharmaceutical properties and clinical effects of the topical NSAID formulation, diclofenac epolamine. Curr Med Res Opin. 2008;24(10):2967–2992.
- Rao P, Knaus EE. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond. J Pharm Pharm Sci. 2008;11(2):81s–110s.
- N’Da DD. Prodrug strategies for enhancing the percutaneous absorption of drugs. Molecules. 2014;19(12):20780–20807.
- PubChem [Internet]. Ibuprofen. 2021 [cited 2021 Mar 23]. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/3672
- Hadgraft J, Valenta C. pH, pK(a) and dermal delivery. Int J Pharm. 2000;200(2):243–247.
- Watkinson RM, Guy RH, Hadgraft J, et al. Optimisation of cosolvent concentration for topical drug delivery - II: influence of propylene glycol on ibuprofen permeation. Skin Pharmacol Physiol. 2009;22(4):225–230.
- Dominkus M, Nicolakis M, Kotz R, et al. Comparison of tissue and plasma levels of ibuprofen after oral and topical administration. Arzneimittelforschung. 1996;46(12):1138–1143.
- McCarberg BH, Argoff CE. Topical diclofenac epolamine patch 1.3% for treatment of acute pain caused by soft tissue injury. Int J Clin Pract. 2010;64(11):1546–1553.
- PubChem [Internet]. Diclofenac. 2020 [cited 2021 Mar 23]. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Diclofenac
- PubChem [Internet]. Diclofenac sodium. 2020 [cited 2021 Mar 23]. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/5018304
- PubChem [Internet]. Diclofenac epolamine. 2020 [cited 2021 Mar 23]. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/114753
- PubChem [Internet]. Diclofenac diethylammonium. 2020 [cited 2021 Mar 23]. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/86754957
- Benaouda F, Brown MB, Shah B, et al. The influence of self-assembling supramolecular structures on the passive membrane transport of ion-paired molecules. Int J Pharm. 2012;439(1–2):334–341.
- Electronic Medicines Compendium [Internet]. Nurofen joint & muscular pain relief 200mg medicated plaster. London (UK): EMC; 2020 [cited 2021 Mar 23]. Available from: https://www.medicines.org.uk/emc/files/pil.9321.pdf
- Predel HG, Connolly MP, Bhatt A, et al. Efficacy and safety assessment of acute sports-related traumatic soft tissue injuries using a new ibuprofen medicated plaster: results from a randomized controlled clinical trial. Phys Sportsmed. 2017;45(4):418–425.
- Predel HG, Giannetti B, Connolly MP, et al. Efficacy and tolerability of a new ibuprofen 200mg plaster in patients with acute sports-related traumatic blunt soft tissue injury/contusion. Postgrad Med. 2018;130(1):24–31.
- Lewis F, Connolly MP, Bhatt A. A pharmacokinetic study of an ibuprofen topical patch in healthy male and female adult volunteers. Clin Pharmacol Drug Dev. 2018;7(7):684–691.
- Hadgraft J, Whitefield M, Rosher PH. Skin penetration of topical formulations of ibuprofen 5%: an in vitro comparative study. Skin Pharmacol Physiol. 2003;16(3):137–142.
- Bolla PK, Clark BA, Juluri A, et al. Evaluation of formulation parameters on permeation of ibuprofen from topical formulations using Strat-M® membrane. Pharmaceutics. 2020;12(2):151.
- European Medicines Agency [Internet]. Draft guideline on quality and equivalence of topical products. Amsterdam (The Netherlands): EMA; 2018 [cited 2021 Mar 23]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-quality-equivalence-topical-products_en.pdf
- Saninforma [Internet]. Dicloreum Unidie 136 mg Medicated Patch. Rome (Italy): Saninforma; 2018 [cited 2021 Mar 23]. Available from: https://www.saninforma.it/farmaci/dolore-e-infiammazione-ferite/dolori-articolari-e-muscolari-gel-schiume-creme-e-cerotti/dicloreum-unidie-136-mg-cerotto-medicato.html
- Electronic Medicines Compendium [Internet]. Ibuleve max strength pain relief 10% gel. London (UK): EMC; 2020 [cited 2021 Mar 23]. Available from: https://www.medicines.org.uk/emc/product/5689/pil
- Electronic Medicines Compendium [Internet]. Voltarol medicated plaster. London (UK): EMC; 2019 [cited 2021 Mar 23]. Available from: https://www.medicines.org.uk/emc/product/6992/pil
- Electronic Medicines Compendium [Internet]. Voltarol joint pain relief 2.32% gel. London (UK): EMC; 2020 [cited 2021 Mar 23]. Available from: https://www.medicines.org.uk/emc/product/9335/pil
- Fini A, Bassini G, Monastero A, et al. Diclofenac salts, VIII. Effect of the counterions on the permeation through porcine membrane from aqueous saturated solutions. Pharmaceutics. 2012;4(3):413–429.
- Pradal J. Comparison of skin permeation and putative anti-inflammatory activity of commercially available topical products containing ibuprofen and diclofenac. J Pain Res. 2020;13:2805–2814.
- Kleinbloesem CH, Spitznagel W, Wilkinson FE, et al. Pharmacokinetics and bioavailability of percutaneous ibuprofen. Arzneimittelforschung. 1995;45(10):1117–1121.
- Clijsen R, Baeyens JP, Barel AO, et al. In vivo determination of the diclofenac skin reservoir: comparison between passive, occlusive, and iontophoretic application. Drug Des Devel Ther. 2015;9:835–840.
- Naik A, Pechtold LA, Potts RO, et al. Mechanism of oleic acid-induced skin penetration enhancement in vivo in humans. J Control Release. 1995;37(3):299–306.
- Cilurzo F, Musazzi UM, Franzé S, et al. Design of in vitro skin permeation studies according to the EMA guideline on quality of transdermal patches. Eur J Pharm Sci. 2018;125:86–92.
- European Medicines Agency [Internet]. Guidelines on quality of trans dermal patches. Amsterdam (The Netherlands): EMA; 2014 [cited 2021 Mar 23]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-transdermal-patches_en.pdf
- Wanat K. Biological barriers, and the influence of protein binding on the passage of drugs across them. Mol Biol Rep. 2020;47(4):3221–3231.
- Bohnert T, Gan LS. Plasma protein binding: from discovery to development. J Pharm Sci. 2013;102(9):2953–2994.
- Dancik Y, Anissimov YG, Jepps OG, et al. Convective transport of highly plasma protein bound drugs facilitates direct penetration into deep tissues after topical application. Br J Clin Pharmacol. 2012;73(4):564–578.
- Brain KR, Green DM, Dykes PJ, et al. The role of menthol in skin penetration from topical formulations of ibuprofen 5% in vivo. Skin Pharmacol Physiol. 2006;19(1):17–21.
- Medicines and Healthcare products Regulatory Agency [Internet]. Voltarol 12 hour emugel P2.32% gel. London (UK): MHRA; 2019 [cited 2021 Mar 23]. Available from https://mhraproducts4853.blob.core.windows.net/docs/987fe9df7600bd1520be488ac0fe0ba4bf4ebfc1
- Gan TJ. Diclofenac: an update on its mechanism of action and safety profile. Curr Med Res Opin. 2010;26(7):1715–1731.
- Mazaleuskaya LL, Theken KN, Gong L, et al. PharmGKB summary: ibuprofen pathways. Pharmacogenet Genomics. 2015;25(2):96–106.