Abstract
A rapid and efficient protocol for the extraction of genomic DNA from plant pathogenic fungi was developed. Key features of the protocol include the SDS-assisted lysis of fungal mycelium with inclusion of a glass bead to help break hyphal walls, followed by isopropanol precipitation of the DNA. The protocol was used to extract genomic DNA from a collection of 26 fungal species, representing many important plant pathogens. Yield of DNA ranged from 2.1–4.9 μg per 20 mg of mycelium or 0.4–0.6 μg per 20 mg of spores. The DNA was of sufficient purity to be digested by restriction enzymes, to serve as a template in the PCR-amplification of genomic fragments as large as 4.9 kb, and to be used in dot-blot hybridization for the detection of multiple- and single-copy genes.
Résumé
Un protocole rapide et efficace pour extraire l'ADN génomique de champignons pathogènes des plantes a été élaboré. Les éléments essentiels du protocole incluent la lyse avec détergent SDS du mycélium fongique avec ajout d'une bille de verre pour aider à briser la paroi hyphale, suivie d'une précipitation de l'ADN à l'alcool isopropylique. Le protocole a été appliqué pour extraire l'ADN génomique d'une collection de 26 espèces fongiques représentant plusieurs agents pathogènes importants des plantes. Le rendement de l'ADN a varié de 2,1 à 4,9 μg par 20 mg de mycélium ou de 0,4 à 0,6 μg par 20 mg de spores. L'ADN était assez pur pour être digéré par des enzymes de restriction, pour servir de matrice à l'amplification par PCR de fragments aussi gros que 4,9 kb ainsi que pour servir à l'hybridation sur tache pour détecter les gènes à copie unique et multiple.
Introduction
Extraction of genomic DNA from plant pathogenic fungi is a routine technique conducted in many plant pathology laboratories. A range of procedures for the extraction of fungal DNA are available (Cenis, Citation1992; Min et al., Citation1995; Tapia-Tussell et al., Citation2006), generally derived from the method originally developed for plant DNA extraction (Saghai-Maroof et al., Citation1984). These procedures include a step of sample grinding or freezing that, although simple and inexpensive, is not time-efficient for large numbers of samples. Similarly, high quality genomic DNA can be extracted from fungal samples within hours using commercial DNA extraction kits. However, while these kits may be convenient, they can also be expensive and may not perform significantly better than standard reagents that can be prepared in bulk by any competent laboratory worker. The cost differential is especially large for the extraction of small amounts of DNA from large numbers of samples. Similarly, extraction of DNA with kits involves absorption of DNA on a chromatography column and subsequent elution of DNA into buffer or water. Since the elution step needs a certain amount of elution buffer or water to soak through the column, an additional concentration step may be required when high concentrations of DNA are needed from small samples.
In our laboratory, we routinely conduct studies that require extraction of small amounts of genomic DNA from up to several hundred fungal isolates at a time, for example in phylogenetic studies using techniques such as RAPD or DNA sequencing of the internal transcribed spacer (ITS) region. For such studies, we have developed a rapid protocol for genomic DNA extraction from fungal species such as Fusarium avenaceum (Corda ex Fries) Sacc. and Stagonospora nodorum (Berk.) Castellani & Germano. To determine if the protocol can be applied to other fungi, genomic DNA from 26 fungal species was extracted using this protocol and the quality assessed using restriction enzyme digestion, PCR and dot-blot hybridization.
Materials and methods
DNA extraction
All chemicals were purchased from Fisher Scientific (Ottawa, ON) unless otherwise specified. The 26 fungal species used for DNA extraction are listed in the caption to . Isolates of F. graminearum (strain PH-1) and S. nodorum (strain SN15) were purchased from the University of Kansas Medical Center (Kansas City, KS). Isolates of Botrytis cinerea Pers. ex Fr., Colletotrichum truncatum (Schw.) Andrus & Moore, Fusarium solani (Mart.) Sacc, Mycosphaerella pinodes (Berk. and Blox.) Vesterg., Pyrenophora tritici-repentis (Died.) Drechs., Pythium ultimum Trow and Rhizoctonia solani Kühn were maintained in our laboratory, while isolates of the other species, which have been used in a previous study (Cao et al., Citation2007), were obtained from the University of Alberta, Edmonton, Canada. For each fungal species, a 0.5 cm diameter plug from a culture growing on potato dextrose agar (PDA) was excised and transferred to a fresh PDA dish. The dishes were incubated in darkness at room temperature (22 °C) for three to seven days, until the colony covered two-thirds of the dish, at which point samples were collected for DNA extraction.
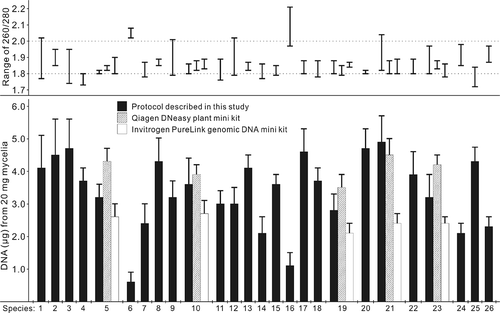
Fig. 1. Quantity and range of the 260/280 ratio for genomic DNA extracted from 20 mg of spores (species 6 and 16) or mycelium (all others) of 26 fungal species, using the protocol developed in the current study and two commercial genomic DNA kits (n = 10). The fungal species are: 1, Alternaria alternata; 2, A. brassicae; 3, A. brassicicola; 4, A. raphani; 5, Botrytis cinerea; 6, Cladosporium sp.; 7, Colletotrichum truncatum; 8, Cyathus olla; 9, Fusarium avenaceum; 10, F. graminearum; 11, F. oxysporum; 12, F. solani; 13, Gliocladium roseum; 14, Mycosphaerella pinodes; 15, Myrothecium verrucaria; 16, Penicillium sp.; 17, Periconia sp.; 18, Phanerochaete chrysosporium; 19, Pyrenophora tritici-repentis; 20, Pythium pythioides; 21, P. ultimum; 22, Rhizoctonia solani; 23, Stagonospora nodorum; 24, Streptomyces sp.; 25, Trichoderma harzianum; 26, Verticillium albo-atrum.
All DNA extractions were performed at room temperature. Centrifugations were always conducted at maximum speed (16 000 g) in a microcentrifuge (Eppendorf Canada, Mississauga, ON). Twenty mg of mycelium or spores (Cladosporium sp. and Penicillium sp.) were collected from cultures with a sterile toothpick or scalpel and placed into 1.5 mL centrifuge tubes containing one glass bead (3.5 mm diam.; VWR Canada, Mississauga, ON, cat: 26396-521). After addition of 0.65 mL lysis buffer (100 mM Tris-HCl, pH 8.0; 50 mM EDTA, pH 8.0; 1% SDS; 10 μg mL−1 RNase A), each tube was vortexed for 10 s, kept on a bench for 2 min, vortexed for another 10 s, and then centrifuged for 2 min. After centrifugation, 0.5 mL of supernatant was transferred into a new tube and 100 μL of potassium acetate buffer (3.0 M, pH 5.5) was added. The tube was inverted several times and centrifuged for 2 min. A 0.5 mL aliquot of supernatant was transferred into a new tube containing 0.5 mL of isopropanol, which was then inverted several times and centrifuged for 2 min. The supernatant was removed and the DNA pellet was washed with 0.75 mL 70% ethanol. After centrifugation for 30 s, the ethanol was removed and the DNA pellet was air dried for 5–10 min. The DNA was dissolved in 50 μL sterile distilled water.
For the purposes of comparison, 20 mg of mycelium from B. cinerea, F. graminearum, P. tritici-repentis, P. ultimum and S. nodorum were subjected to DNA extraction using the Qiagen DNeasy Plant Mini Kit (Qiagen Canada, Mississauga, ON) and the Invitrogen PureLink Genomic DNA Mini Kit (Invitrogen, Carlsbad, CA), following the plant tissue mini protocol with the former and the Gram-negative bacterial cell lysate protocol with the latter, as described in the respective manuals from the manufacturers. The Qiagen DNeasy Plant Mini Kit has been used extensively for genomic DNA extraction in studies of fungi. For instance, a survey of papers published in Mycological Research and Fungal Genetics and Biology from 2000 to 2009 revealed approximately 80 and 20 publications, respectively, in which this kit was used to extract genomic DNA from fungal mycelia. The Invitrogen PureLink Genomic DNA Mini Kit has often been used in our laboratory for the reliable extraction of fungal DNA. For both kits, the DNA was eluted and standardized to 200 μL with the kit-included elution buffers.
Immediately following DNA extraction, an assessment of the concentration and quality of the extracted DNA was obtained using a NanoDrop 1000 Spectrophotometer (Thermo Scientific, Waltham, MA) according to the manufacturer's instructions for DNA assays. Samples were subsequently stored at −20 °C in a freezer. The experiment was repeated three times, with 10 mycelial samples used for DNA extraction per fungal species in each repetition.
Restriction enzyme digestion and electrophoresis
Genomic DNA samples from F. graminearum, M. pinodes, P. ultimum and S. nodorum were chosen for digestion with the restriction enzymes ApaI, EcoRI (both from New England Biolabs, Ipswich, MA) or SacII (Promega, Madison, WI). Each 50 μL reaction volume contained 3 μg DNA, 5 μL 10× reaction buffer and five units of restriction enzyme. The reactions were incubated at either 37 °C (EcoRI and SacII) or 25 °C (ApaI) for 2 h before electrophoresis.
Electrophoresis was conducted in 1% agarose gels. The gels were stained with ethidium bromide and the DNA bands were visualized using a UV transilluminator (Bio-Rad Canada, Mississauga, ON). For electrophoresis of genomic DNA, each 24 μL sample contained 0.5 or 1.0 μg of genomic DNA and 4 μL of 6× loading dye (40% sucrose, 0.2% bromophenol blue).
PCR and dot-blot hybridization
PCR was conducted using the extracted DNA as a template. Initially, two DNA sequences were amplified from 10 selected fungal species: the ITS sequence using primer pair ITS1/ITS4 (White et al., Citation1990) and the CPN60 gene using primer pair H279/H280 (Hill et al., Citation2004). In addition, primers specific to four protein-encoding genes were used to amplify fragments from genomic DNA extracted from F. graminearum and S. nodorum. The primers were synthesized by Integrated DNA Technologies (Coralville, IA). Their names, sequences and targets are listed in . For PCR, each 20 μL reaction volume consisted of 20 ηg of genomic DNA, 0.2 μM of each primer, 50 μM of each dNTP, 1× PCR buffer, and 0.5 unit of Taq DNA polymerase (New England Biolabs). Amplification was carried out in a My Cycler (Bio-Rad Canada) thermal cycler with the standard PCR regime (Sambrook & Russell, Citation2001). For ITS and CPN60 primers, the annealing temperature was 50 °C and the extension time was 1 min. For the other primers, the annealing temperature was 56 °C and the extension time was 5 min.
Table 1. Primers used in this study
For dot-blot hybridization, 10 μL aliquots of samples containing 3.0, 1.0, 0.3 or 0.1 μg genomic DNA extracted from F. graminearum were treated at 95 °C for 5 min to denature the DNA and then immediately placed on ice. After 2 min, the samples were applied to a strip of positively charged nylon membrane (Roche Applied Science Canada, Laval, QC), which was then baked at 120 °C for 30 min to cross-link the DNA with the membrane. The amplicons obtained from F. graminearum using the ITS- and CPN60-specific primers were extracted from the agarose gel using the glass-milk method (Boyle & Lew, Citation1995). These two DNA fragments were separately labelled with digoxigenin and used to hybridize with the genomic DNA that was blotted on the membrane. Labelling, hybridization and detection were conducted using the DIG DNA Labeling and Detection Kit (Roche Applied Science Canada) according to the manufacturer's instructions. The post-hybridization washes were performed twice for 5 min at 22 °C with 2× SSC (1× SSC is 150 mM NaCl, 15 mM sodium citrate), 0.1% SDS and twice for 15 min at 65 °C with 0.5× SSC, 0.1% SDS. Hybridization was conducted at 50 °C for 16 h and the time for colour development was 15 min.
Results and discussion
The new protocol allowed extraction of genomic DNA from fungal mycelium or spores in less than half an hour. In contrast, at least one hour was needed for DNA extraction using either the Qiagen DNeasy Plant Mini Kit or the Invitrogen PureLink Genomic DNA Mini Kit. Although simple and quick, the described protocol includes all of the important steps for standard DNA extraction (Sambrook & Russell, Citation2001). Inclusion of a glass bead during mycelial lysis helps break the fungal cell wall (van Burik et al., Citation1998). At the same time, SDS denatures proteins and disrupts the cell membrane (Sambrook & Russell, Citation2001), resulting in the release of nucleic acids into the cell lysis mixture. Tris-HCl provides the pH conditions needed to stabilize the released DNA, and EDTA inhibits the activity of DNA-degrading enzymes. The RNase A degrades RNA that would otherwise precipitate together with the DNA. Addition of potassium acetate into the lysis mixture results in the replacement of sodium ions by potassium ions, which efficiently precipitates proteins and SDS (Ish-Horowicz & Burke, Citation1981). Potassium ions also neutralize the charge on the sugar-phosphate backbone of the DNA, which is essential for DNA precipitation by isopropanol (Sambrook & Russell, Citation2001). In the final step, the volume of water or buffer used to dissolve the DNA pellet can be adjusted to provide the appropriate concentration of DNA needed for specific applications.
Using this protocol, genomic DNA was extracted from mycelia of 24 fungal species, with a range of 2.1–4.9 μg per 20 mg mycelium (). The purity of the DNA was high, with an A260/280 ratio of 1.72–2.04 for all samples (). In the case of Cladosporium sp. and Penicillium sp., DNA was extracted from spores at concentrations lower than those obtained from mycelia of other species (). Despite these differences, the DNA extracted from a 20 mg sample was always sufficient for PCR amplification. The two commercial kits also extracted high quality DNA from the selected fungal species, with an A260/280 ratio of 1.78–1.90 (). Relative to the protocol described here, the DNeasy Plant Mini Kit yielded a greater amount of DNA from B. cinerea (). However, for the other species tested, the yields from commercial kits were not better than those obtained using the new protocol ().
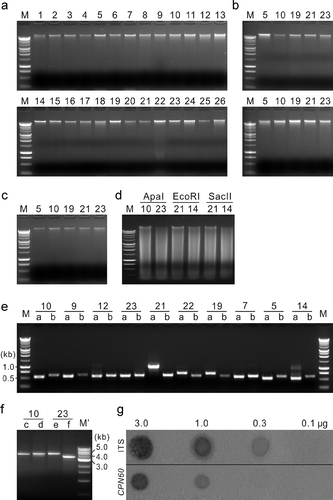
When one μg of DNA from each fungal species was electrophoresed to confirm DNA quality, a sharp band was produced for each sample. This was observed for DNA extracted with either the described protocol ( a) or the commercial kits ( b). DNA from five selected fungal species, which was extracted with the new protocol, was subjected to a storage assay at −20 °C. The samples were thawed to room temperature for approximately 30 min at monthly intervals. After five months, 500 ηg of DNA from each fungal species was subjected to agarose gel electrophoresis. A sharp band was still observed for each DNA sample ( c), indicating no or very limited degradation of the extracted DNA. Similarly, when DNA from selected fungal species was digested with restriction enzymes, clean migration patterns were observed for all samples ( d). DNA quality was also verified via PCR. Amplification products could be obtained from each sample with ITS- and CPN60-specific primer pairs ( e). Variation in the size of the ITS products across species was as expected based on previous reports (Hausner & Wang, 2001). The CPN60 fragments amplified from each species were approximately 600 bp in length, consistent with a previous report on the size of CPN60 in eukaryotes (Hill et al., Citation2004). Amplicons of relatively large sizes were obtained from the genomic DNA of F. graminearum and S. nodorum ( f). The observed sizes of the PCR products amplified by primers c, d, e and f were consistent with those expected based on the genomic sequences of the corresponding accessions, which were 4.7, 4.9, 4.9 and 3.7 kb, respectively ( f). Finally, the quality of the DNA was checked by dot-blot hybridization. Clear, positive signals were detected from ITS hybridization when the concentration of F. graminearum genomic DNA was 3.0, 1.0 or 0.3 μg per sample ( g), but not 0.1μg per sample. In the CPN60 hybridization, signals were detected at 3.0 or 1.0 μg genomic DNA, but not at 0.3 or 0.1 μg. The difference between ITS and CPN60 in the robustness of the hybridization is likely associated with gene copy number; fungal genomes contain multiple copies of the ITS sequences (Rosenthal, Citation2001), but only one copy of the CPN60 gene (Hill et al., Citation2004). The results indicate that the genomic DNA extracted with our protocol is of sufficient quality to enable PCR amplification and dot-blot detection of single-copy genes.
Fig. 2. Quality of genomic DNA extracted from 26 fungal species with the protocol developed in the current study. (a and b) Electrophoresis of DNA extracted with the described protocol (a) and two commercial kits (b) Qiagen DNeasy Plant Mini Kit (upper panel) and Invitrogen PureLink Genomic DNA Mini Kit (lower panel). (c) Electrophoresis of the extracted DNA after five months of storage at −20 °C. (d) Electrophoresis of the extracted DNA after digestion with restriction enzymes. (e and f) PCR using the extracted DNA as templates. (g) Dot-blot hybridization of DNA (3.0, 1.0, 0.3 or 0.1 μg) extracted from Fusarium graminearum, blotted on nylon membranes and hybridized with the digoxigenin-labelled PCR products of ITS or CPN60 gene. M, Promega 1 kb DNA marker; M', New England Biolabs 1 kb DNA marker; 1–26, fungal species as indicated in ; a–f, primer pairs ITS1/ITS4 (a), H279/H280 (b), FgSnf-f/FgSnf-r (c), FgFar-f/FgFar-r (d), SnFar-f/SnFar-r (e), and SnLip-f/SnLip-r (f).
In summary, a rapid protocol was developed that allows for the extraction of genomic DNA from various plant pathogenic fungi. The quality and quantity of the extracted DNA was sufficient to permit routine DNA manipulations such as PCR, restriction enzyme digestion and dot-blot hybridization. This protocol is recommended to researchers who routinely conduct genomic DNA extraction from fungal species for PCR-based phylogenetic studies.
Acknowledgements
We are grateful for financial support received from the Canola Agronomic Research Program (Alberta Canola Producers Commission, Manitoba Canola Growers Association, SaskCanola and the Canola Council of Canada) and the Alberta Crop Industry Development Fund.
Related Research Data
References
- Boyle , J.S. and Lew , A.M. 1995 . An inexpensive alternative to glassmilk for DNA purification . Trends Genet. , 11 : 8
- Cao , T. , Tewari , J. and Strelkov , S.E. 2007 . Molecular detection of Plasmodiophora brassicae, causal agent of clubroot of crucifers, in plant and soil . Plant Dis. , 91 : 80 – 87 .
- Cenis , J.L. 1992 . Rapid extraction of fungal DNA for PCR amplification . Nucleic Acids Res. , 20 : 2380
- Hausner , G. and Wang , X. 2005 . Unusual compact rDNA gene arrangements within some members of the Ascomycota: evidence for molecular co-evolution between ITS1 and ITS2 . Genome , 48 : 648 – 660 .
- Hill , J.E. , Penny , S.L. and Crowell , K.G. 2004 . cpnDB: a chaperonin sequence database . Genome Res. , 14 : 1669 – 1675 .
- Ish-Horowicz , D. and Burke , J.F. 1981 . Rapid and efficient cosmid cloning . Nucleic Acids Res. , 9 : 2989 – 2998 .
- Min , J. , Arganoza , M.T. , Ohrenberger , J. , Xu , C. and Akins , R.A. 1995 . Alternative methods of preparing whole-cell DNA from fungi for dot-blot, restriction analysis, and colony filter hybridization . Anal. Biochem. , 225 : 94 – 100 .
- Rosenthal , B.M. 2001 . Defining and interpreting intraspecific molecular variation . Vet. Parasitol. , 101 : 187 – 200 .
- Saghai-Maroof , M.A. , Soliman , K.M. , Jorgensen , R.A. and Allard , R.W. 1984 . Ribosomal DNA spacer-length polymorphisms in barley: mendelian inheritance, chromosomal location, and population dynamics . Proc. Natl. Acad. Sci. USA , 81 : 8014 – 8018 .
- Sambrook , J. and Russell , D.W. 2001 . Molecular Cloning: A Laboratory Manual , Cold Spring Harbor, NY : Cold Spring Harbor Laboratory .
- Tapia-Tussell , R. , Lappe , P. , Ulloa , M. , Quijano-Ramayo , A. , Cáceres-Farfán , M. , Larqué-Saavedra , A. and Perez-Brito , D. 2006 . A rapid and simple method for DNA extraction from yeasts and fungi isolated from Agave fourcroydes . Mol. Biotechnol , 33 : 67 – 70 .
- van Burik , J.A.H. , Schreckhise , R.W. , White , T.C. , Bowden , R.A. and Myerson , D. 1998 . Comparison of six extraction techniques for isolation of DNA from filamentous fungi . Med. Mycol. , 36 : 299 – 303 .
- White , T.J. , Bruns , T. , Lee , S. and Taylor , J. 1990 . “ Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics ” . In PCR Protocols: A Guide to Methods and Applications , Edited by: Innes , M.A. , Gelfand , D.H. , Sninsky , J.J. and White , T.J. 315 – 322 . San Diego, CA : Academic Press .