Abstract
A single nucleotide polymorphism (SNP)-based molecular marker system for the oomycete pathogen causing late blight on solanaceous hosts, Phytophthora infestans, was developed by identifying sequence polymorphisms in microsatellite flanking regions (MFRs). MFRs were identified using the complete genome sequence for this pathogen and SNP rates were assessed by sequencing a total of 14 000 bases from 32 MFRs across a diverse international panel of 32 isolates. SNP rates were highly variable among loci, with nucleotide diversity ranging from a low of zero to a high of 0.013 (one change per 78 bp) and an average across all scorable loci (n = 28) of 0.0023 (one change per 426 bp). Nucleotide diversity estimates are highly conservative due to strict objective scoring rules used when calling SNPs. Overall, 102 SNPs were scored using objective criteria (versus 167 as scored by eye), and could distinguish all isolates in the panel, with pairwise per cent similarity between isolates varying from 58.5% to 99.4%. Observed and expected heterozygosities at most SNP sites were similar. The utility of a ‘straincode’ for strain typing based on up to 19 unlinked SNPs was investigated. The probability of identity was very low (PI = 3.5 × 10−6) when all 19 SNPs were included; however, to achieve probabilities of identity of about 1% and 0.1%, only four and seven loci were needed, respectively. The approach needs to be assessed with larger panels of isolates, but results suggest that MFRs and SNPs are a largely untapped source of useful sequence polymorphisms for strain typing of microbes and that their utility could extend to ecological and evolutionary research, especially with high throughput sequence-based technologies.
Résumé
Une méthode faisant appel à un marqueur moléculaire basé sur le polymorphisme d'un nucléotide simple (SNP), conçue pour l'identification de l'agent pathogène oomycète causant le mildiou sur les solanacées hôtes, Phytophtora infestans, a été développée en déterminant les séquences polymorphes des régions adjacentes aux microsatellites (MFR). Les MFR ont été identifiées en utilisant la séquence génomique entière de l'agent pathogène. Par la suite, les taux de SNP ont été évalués en séquençant 14 000 bases provenant de 32 MFR issus eux-mêmes d'un panel mondial et diversifié de 32 isolats. Les taux de SNP variaient considérablement d'un locus à l'autre, affichant une diversité nucléotidique oscillant de 0 à 0.013 (un changement par 78 paires de bases (pb)) pour une moyenne de 0.0023 (un changement par 426 pb) chez tous les loci analysés (n = 28). Les estimations de la diversité nucléotidique sont très prudentes étant donné la rigueur et l'objectivité des règles appliquées lors de la lecture des SNP. En tout, 102 SNP ont été évalués en fonction de critères objectifs (versus 167 évalués « à l'œil ») et permettaient de distinguer tous les isolats du panel, avec une similitude du pourcentage des paires chez les isolats variant de 58.5 % à 99.4 %. Les hétérozygoties observées et anticipées à la plupart des sites SNP étaient semblables. L'utilité d'un « code souche » pour le typage des souches basé sur au plus 19 SNP non liés a été étudiée. Lorsque les 19 SNP étaient inclus, la probabilité d'identité était très faible (PI = 3.5 × 10−6). Toutefois, pour atteindre des probabilités d'identité d'environ 1 % et 0.1 %, seulement quatre et sept loci étaient requis, respectivement. L'approche doit être évaluée avec de plus grands panels d'isolats, mais les résultats suggèrent que les MFR et les SNP sont des sources largement inexploitées de séquences polymorphes permettant le typage des souches de microbes. De plus, ils pourraient servir en recherche sur l'écologie et l’évolution, particulièrement à l'aide de techniques de séquençage à haut débit.
Introduction
Virtually all aspects of plant pathology research rely on the ability to accurately identify the etiological agent responsible for the disease under investigation (McCartney et al., Citation2003). Over recent years, traditional, visually based diagnostic methods for non-human pathogens (e.g. using selective media and microscopy) have been superseded by new molecular diagnostic approaches (as reviewed by Schaad & Frederick, Citation2002; Lievens & Thomma, Citation2005; Lévesque, Citation2007). As the ability to genetically identify microbial species becomes routine, the emphasis moves towards distinguishing among intraspecies units and, ultimately, among individual strains.
For the past two decades, microsatellite DNA markers (also called ‘simple sequence repeats’ or SSRs) have been the genetic marker of choice for deciphering genetic relationships below the species level. Several features of microsatellite repeat arrays make them ideal for this, including: (i) high within-population and within-individual variability; (ii) co-dominance; and (iii) a wide distribution throughout the genome (reviewed by Li et al., Citation2002). They have been used extensively in molecular ecology studies on plant and animal populations for well over a decade, and are more recently becoming popular for investigating fungal populations (Sullivan & Faeth, Citation2004; Banke & McDonald, Citation2005; Stukenbrock et al., Citation2006; Zhou et al., Citation2007). In terms of their potential discriminatory power, microsatellite markers have been used to distinguish among fungal species, including oomycetes (Moon et al., Citation1999; Lee & Moorman, Citation2008), subspecies (Smith et al., Citation2001), morphotypes (Burgess et al., Citation2001), and strains (Enkerli et al., Citation2001; Hennequin et al., Citation2001; Lees et al., Citation2006).
For the oomycete genus Phytophthora, microsatellite data have been used to characterize intraspecific genetic diversity in a few species, namely: P. cinnamomi Rands (Dobrowolski et al., Citation2003); P. ramorum Werres, De Cock & Man in ’t Veld (Ivors et al., Citation2006); and the species under study here, P. infestans (Mont.) de Bary (Vargas et al., Citation2009). Phytophthora infestans is an oomycete (and thus diploid) pathogen that causes late blight on a range of solanaceous hosts and is best known for its devastating effect on potato and tomato crops. Populations of this pathogen outside parts of South and Central America were initially found to be clonal or otherwise low in genetic diversity, but new genotypes were later detected that arose through a combination of migration and sexual reproduction after the A2 mating type spread into Europe and beyond, starting in 1984 (Fry et al., Citation1993; Goodwin & Drenth, Citation1997; Cooke & Lees, Citation2004). It has been a model species for developing new approaches to genotypic and phenotypic typing of plant pathogens (Fry et al., Citation1992; Goodwin et al., Citation1992; Forbes et al., Citation1998; Cooke & Lees, Citation2004); genotyping methods used include mtDNA haplotypes based on restriction enzyme digests (Griffith & Shaw, Citation1998) and length variation in microsatellite alleles (Knapova & Gisi, Citation2002; Lees et al., Citation2006; Guo et al., Citation2009).
Extensive microsatellite genotyping of P. infestans associated with the EUCABLIGHT project has seen almost 20 000 isolates genotyped from European populations (Cooke et al., Citation2009). Aims of this massive undertaking include investigating population change over time and identifying evolutionary forces driving observed changes, as relating these to disease management strategies is expected to improve their chances of success (Cooke et al., Citation2009). Resolving power of the 11 polymorphic loci used is high; however, setbacks associated with standardizing methods and scoring across laboratories hindered early attempts to compile multiple data sets (Cooke et al., Citation2009). Indeed, microsatellite alleles are known for being difficult to score due to stutter bands caused by slippage of the polymerase during PCR amplification and the difficulties in digitizing and comparing fragment length information. Thus highly specific technical expertise is required before microsatellite genotyping can be implemented, and there is a strict requirement of consistency when making subjective scoring decisions, which makes standardization across labs difficult at best. In contrast, single nucleotide polymorphisms (SNPs, described below) can be genotyped simply and accurately using real-time PCR (see Mhlanga & Malmberg, Citation2001) or sequencing, which are standard methods in contemporary molecular genetics labs and generate results that are easily digitized and searchable.
Another detraction of microsatellites can be size homoplasy caused by convergent evolution, parallel evolution, or back mutations in the repeat array itself; or insertion/deletion mutations in the sequence bordering the repeat (Primmer & Ellegren, Citation1998). In theory, size homoplasy is expected to be especially problematic when investigating pathogenic microbes prone to human-mediated translocation because their establishment in non-native geographical areas may result from single introduction events. To properly track evolutionary progress following such a severe bottleneck, the ideal scenario is one in which new mutations in the bottlenecked population are highly likely to generate new alleles. This is not the case for microsatellite repeat arrays; new alleles typically differ from ancestral alleles by plus or minus one repeat unit (Kimmel et al., Citation1996; Ellegren, Citation2004) and are therefore likely to regenerate an allele length, and potentially a genotype, that is already present in the originating population. Indeed, the high mutational instability of microsatellite repeat units coupled with their propensity for undergoing homoplasious changes is thought to present a real risk of misdiagnoses for fungal species (cf. McEwen et al., Citation2000).
Single nucleotide polymorphisms (SNPs) are widely used as genetic markers in linkage and association studies and are thought to hold substantial, but as yet largely untapped, potential for addressing ecological and evolutionary questions (but see Bensch et al., Citation2002; Belfiore et al., Citation2003; Seddon et al., Citation2005; Delmotte et al., Citation2008; Narum et al., Citation2008). SNPs are co-dominant, single-locus, biallelic markers that result from point mutations leading to single base-pair differences between chromosome sequences (Brumfield et al., Citation2003). Advantages of SNPs include that they are ubiquitous and abundant in the genome, evolve according to simple mutational models, are not prone to homoplasy, and are easy to genotype (Mhlanga & Malmberg, Citation2001) and score (Morin et al., Citation2004). However, because of low polymorphism at SNP loci there are concerns that isolating a sufficient number of loci is simply unrealistic given that conventional analytical methods in evolutionary genetics assume independence among marker loci (Glaubitz et al., Citation2003). Several methods have been used for finding SNPs, including: sequencing orthologous genomic regions in related species (Bensch et al., Citation2002; Primmer et al., Citation2002; Aitken et al., Citation2004; Seddon et al., Citation2005); in silico comparisons of sequence data from multiple individuals and/or species (Smith et al., Citation2005); and random sequencing (Fisher et al., Citation1999; Primmer et al., Citation2002; Giresse et al., Citation2007). Studies that isolated SNPs using a single whole genome sequence, as opposed to aligning several genomic sequences from across several individuals or species, are virtually non-existent (but see Adams et al., Citation2006).
The genome sequences of a few Phytophthora species are now available (Tyler et al., Citation2006; Haas et al., Citation2009) and they have been used to develop microsatellite markers in Phytophthora species (Garnica et al., Citation2006), including P. infestans (Schena et al., Citation2008). In the current study, we evaluate whether the advantages of SNP-based molecular markers can be married with the advantages of high mutation rates at microsatellite loci by developing a SNP-based marker system for P. infestans using sequence polymorphisms in microsatellite flanking regions (MFRs). MFRs are located immediately adjacent to microsatellite repeat arrays and are known to contain both point and length mutations (see Ishibashi et al., Citation1996; Zardoya et al., Citation1996; Grimaldi & Crouau-Roy, Citation1997; Jones et al., Citation1998; Brohede & Ellegren, Citation1999); however, they have rarely been targeted as sources of molecular markers (but see Mogg et al., Citation2002; Primmer et al., Citation2002; Ablett et al., Citation2006; Fernández et al., Citation2008). Targeting SNPs in MFRs could help overcome the hurdle of isolating a sufficient number of SNP loci for ecological studies by ensuring that SNP-rich regions are specifically targeted during the SNP discovery stage. The aims of this study were to: (i) describe an automated SNP discovery method based on in silico searches of a complete genome sequence for MFRs; (ii) evaluate levels of sequence polymorphisms in MFRs of P. infestans by screening a diverse panel of isolates; and (iii) determine whether SNPs in MFRs are suitable markers for strain typing and evolutionary research.
Materials and methods
Microsatellite identification and primer design
The complete genome sequence for P. infestans strain T30-4 was downloaded from the Broad Institute website (http://www.broad.harvard.edu/annotation/genome/phytophthora_infestans/Home.html). The analysis was performed on a pre-release of the currently available data (phytophthora_infestans_0.fasta.zip) which have been recently published (Haas et al., Citation2009). Microsatellites were identified using the MISA – MIcroSAtellite identification tool (available from http://pgrc.ipk-gatersleben.de/misa/). The minimum targeted microsatellite size was 18 bp for di- and tri-nucleotide repeats and 20 bp for tetra- and penta-nucleotide repeats (misa.ini properties: definition: 2-9 3-6 4-5 5-4, interruptions: 100).
A small pilot experiment was done to determine whether SNPs were indeed present in MFRs of P. infestans. An average of 12 isolates collected from Eastern Canada (Prince Edward Island and New Brunswick) in 2006 were sequenced at each of four MFRs to assess SNP rates. A much larger data set of MFR sequences was generated for two of these isolates (DAOM 238591 and DAOM 238592) during development of the final SNP based molecular marker set (described below). Using techniques described previously (Peters et al., Citation1998, Citation1999), these isolates were determined to be of the A2 mating type, resistant to metalaxyl-m and displaying an allozyme banding pattern at the glucose phosphate isomerase locus consistent with that of the US-8 genotype, a strain that has come to dominate populations of the pathogen in Canada after its introduction in the mid 1990s (Peters et al., Citation2001).
Primers were designed to amplify a 1.0–1.2 kb product containing the microsatellite using Primer3 (Rozen & Skaletsky, Citation2000). MISA includes a script to create a Primer3 input file, which we modified to allow user specifiable Primer3 parameters. We settled on PRIMER_ PRODUCT_SIZE_RANGE=1000–200, PRIMER_GC_ CLAM P=1, PRIMER_OPT_TM=65, PRIMER_MIN_TM=63, PIMER _ MAX_TM=67. The generated Primer3 input file was post-processed to ensure that the amplified regions contained 500–600 bp both upstream and downstream from the microsatellite repeat itself. This was accomplished by increasing the target size in the Primer3 input file; the target start was moved back 0.5 kb and 1.0 kb was added to the length.
To remove duplicate loci and loci containing apparent internal repeats, the amplicon sequences from the Primer3 output were used as query sequences for a BLAST search of the whole genome sequence. Duplicate loci were identified as those with more than one hit with e-value 0.0; repetitive loci were identified as those with overlapping high scoring sequence pairs within the hit, and both were flagged for removal. Removing duplicate loci is critically important because it ensures that heterozygous sites result from allelic differences at a single locus as opposed to differences among multiple, repeated genomic regions that are co-amplified during PCR. ‘Microsatellite families’ occur in other taxa (Meglecz et al., Citation2004; Tero et al., Citation2006) and likely occur in P. infestans. Lees et al. (Citation2006) found P. infestans microsatellites that generated more than two alleles within single strains. A computer program is available to test for repetitive flanking regions among microsatellites (Meglecz, Citation2007), but it is not designed for identifying repetitive regions in a complete genome sequence as was done here using BLAST.
Development of marker set: PCR optimization and sequencing
PCR amplification of a test set of 95 P. infestans microsatellite loci and one positive control was tried using DNA template from the two US-8 P. infestans isolates from the Eastern Canada collection mentioned earlier (DAOM 238591, DAOM 238592). PCR reactions were 10 μL in volume and contained 5–10 ng of genomic DNA, 100 μm dNTP, 1 pmol of each primer, and 0.5× (i.e. 0.1 μL) TITANIUM™ Taq DNA Polymerase (Clontech Laboratories, Inc.) in 1× reaction buffer. PCR cycling parameters were: 3 min at 95 °C; a ‘touchdown’ of 30 s at 95 °C, 30 s at 65 °C to 53 °C (dropping 3 °C per three cycles), 1.5 min at 72 °C; 25 cycles of 30 s at 95 °C, 30 s at 50 °C, 1.5 min at 72 °C; and one final cycle of 3 min at 72 °C. PCR products were visualized on pre-cast, bufferless, 2% agarose gels (E-Gel® 96, Invitrogen).
Of the 95 microsatellite primer pairs tried, 59 generated single amplification products of the expected size. A second round of PCR, identical to that described already, was done using these 59 primer pairs and the products could be sequenced without a purification step because low concentrations of dNTPs and primers were used in the PCR mix (cf. Allain-Boule et al., Citation2004). We sequenced 1 or 2 μL of PCR product in both directions using Applied Biosystems BigDye v3.1 sequencing system and 1.6 pmol of primer in a 10 μL reaction volume. Products were run on an ABI 3130xl Genetic Analyzer.
In the first sequencing run, about half (n = 29) of the 59 microsatellite-containing fragments generated high quality sequence data in at least one direction. For most loci, sequence reads did not persist through the repeat itself, so internal sequencing primers were designed as close to the repeat as possible to allow for bidirectional sequencing of one or two sides (F or R in locus names) of the targeted microsatellite flanking region (). To increase the probability of screening independent genomic regions, we first targeted microsatellite repeats on different supercontigs (with one exception, see Results). Second, to reduce the extent of physical linkage among SNPs in the final marker set, we targeted only one of the two flanking regions for most loci, which we selected based on which direction yielded the cleanest sequencing profile. A total of 1019 sequences were deposited in GenBank (HM166412-HM167430). Each sequence is the consensus of the bidirectional sequencing for one strain at one MFR locus.
Table 1. Details on the microsatellite loci for which one or both flanking regions was analysed for SNPs. The first four digits of the locus name refer to the number of the supercontig it was found on in the whole genome sequence of strain T30-4 that is publicly available. The repeat location refers to position of the repeat within that supercontig. PCR primers are located on either side of the repeat array such that the whole microsatellite and both flanking regions are amplified; internal sequencing primers were used to facilitate bidirectional sequencing of the target flanking region. The scored fragment of each MFR is positioned between the two boldface primers with two exceptions – at loci 3319_1R and 3408_3R a short stretch (< 100 bp) of single stranded data were scored upstream from the forward primer, as indicated in by the location of the first two SNPs at these loci
SNP discovery
Providing new data on the potential information content in MFRs was a major focus of this study, hence we endeavoured to minimize ascertainment bias by screening a very diverse panel of P. infestans isolates to estimate marker variability, and assessing SNP rates at each genomic region for which clean sequence data could be obtained (as opposed to selecting loci for screening based on preliminary data on SNP rates). SNPs were identified by screening 32 P. infestans isolates collected from across five continents (). Several measures confirmed that these isolates were genetically diverse by including representatives of both A1 and A2 mating types, all four mitochondrial haplotypes, a wide range of RAPD genotypes, and cultures with worldwide origins that were collected over several decades (M.D. Coffey, unpublished data) (). DNA was extracted as described previously (Blair et al., Citation2008). PCR amplification and DNA sequencing of these isolates were done following the same protocols used during marker development, as already described.
Table 2. Sampling and genotypic information for the panel of 32 P. infestans isolates used here for SNP discovery. mtDNA haplotype and RAPD genotype were known prior to this study. The straincode, represented by each line of SNP data for each isolate, was developed here based on 19 unlinked SNPs in MFRs that were scored using objective criteria except for sites shown in grey and italics: these were scored by eye. SNPs in the straincode are ordered by decreasing discriminatory power as determined by per-locus probability of identity estimates (data not shown). The number after the last underscore in the locus name indicates which SNP was used in the straincode at that locus, and corresponds to the ‘SNP no.’ in . Bold-type boxes demarcate the two straincodes that were not unique to a single isolate
Sequence data were generated for all isolates at each locus and were assembled into contigs using Sequencher 4.8 (Gene Codes). SNPs were identified by eye by the presence of heterozygous bases. For each locus, aligned sequences were numbered starting from either the forward PCR primer or the forward internal sequencing primer (), with two exceptions (3319_1R and 3408_8R): at these two loci, numbering began between the forward PCR primer and the internal sequencing primer. Only good quality, easily readable sequences were analysed and SNPs were scored using the ‘Call Secondary Peaks’ function in Sequencher 4.8 (Gene Codes). For SNP sites in regions where bidirectional sequence data were available, secondary peaks were called when their height was > 20% of the first peak in both directions. If a secondary peak was detectable but was smaller than this in either direction then that site in that isolate was excluded from analyses. For SNPs that occurred in regions where only one direction of sequence data were available, the secondary peak was scored only if its height was > 50% of the first peak and if the alternate/heterozygous base was found in at least one other isolate in the data set. More than 20 of the 32 isolates assessed needed to be scorable under these criteria for a SNP site to be identified. To minimize gaps in the data set used for population genetic analyses, 3.6% (n = 583) of SNPs scored in the ‘straincode’ (see below) across the 32 isolates were scored by eye as opposed to using the objective criteria just described.
Analysis of SNP data
Descriptive statistics calculated using the final data set from the international panel (Supplementary Table 1: online) included allele frequencies (calculated using TFPGA v1.3; Miller, Citation1997), number of diplotypes per locus, and nucleotide diversity, the latter two calculated using DnaSP 4.0 (Rozas et al., Citation2003). Observed and expected heterozygosity were calculated manually for each SNP.
To minimize redundancy in information content associated with using linked SNPs, we explored the possibility of using only one SNP per locus for strain typing purposes. To maximize levels of polymorphism, we selected the SNP at each locus that had the highest frequency of the lesser allele; and to minimize any likelihood of physical linkage, we avoided using multiple MFR loci from the same super-contig of the whole genome sequence. Because using diploid data from linked SNPs presents analytical hurdles for population genetic analyses associated with unknown gametic phase data, we also used this data set of 19 unlinked SNPs opportunistically to explore the distribution of genetic diversity in the 32 isolate panel despite the fact that some ascertainment bias was introduced through the selection of the most polymorphic sites. Data from these 19 SNPs ( and Supplementary Table 1) were used for the following analyses as implemented in GenAlEx v6.1 (Peakall & Smouse, Citation2006): (i) tests for departures from Hardy–Weinberg equilibrium; (ii) a Mantel test to determine the statistical relationship between pairwise linear genotypic and geographic distance matrices; (iii) a principal coordinates analysis (PCO) on the squared genetic distance matrix; and (iv) probability of identity (PI) estimates, which assess the average probability that two unrelated individuals will share a genotype by chance using a given set of marker loci.
Table 3. Summary of data collected using the international panel of 32 isolates (see ) for each MFR locus that generated useable data (i.e. including ones that did not contain SNPs, n = 32). Diversity levels at each locus are summarized by counts of the total number of SNPs (as judged by eye), the number of scored SNPs (as determined using the objective scoring criteria described in Materials and methods), and estimates of nucleotide diversity. The italicized number in parentheses given after nucleotide diversity reflects the number of isolates used in the calculation for that locus. Detailed primer information can be found in
TFPGA v1.3 (Miller, Citation1997) was also used to calculate Rogers' original distance (Rogers, Citation1972). This was turned into a pairwise matrix of percentage similarity by subtracting the genetic distance from one (i.e. 1 – genetic distance). Per cent similarity between strains therefore represented the number of identical alleles between individual strains in a pair divided by the total number of alleles scored for both strains.
Applying SNP markers: Colombian isolates
As a preliminary look into how the SNP-based marker system might be used in an ecological context, we extracted DNA from 58 P. infestans isolates collected from six different Solanum species across a wide geographic range in Colombia. Colombia is geographically close to both areas that have been suggested as the centre of origin of the pathogen: Mexico's Toluca Valley and the South American Andes (Gómez-Alpizar et al., Citation2007); hence we predicted that genetic diversity of isolates in Colombia might be high. We sequenced these isolates at the following five, SNP-rich MFRs, the first four of which contain sites used in the straincode: 3200F, 3230_1R, 2947R, 3345F and 3230_1F.
Results
Identification of microsatellites using MISA
MISA identified 738 microsatellite repeats within the P. infestans genome sequence, and Primer3 was able to design primers for 649 of these. Of the identified amplicons, 303 (∼50%) should amplify a unique region, as judged by there being a single BLAST hit for the amplicon, and 268 contained no internal duplication, as judged by the occurrence of linearly ordered, non-overlapping high-scoring sequence pairs within the BLAST hit. About 25% of the microsatellite loci were present two to three times, 12% three to four times, and 7.5% five to six times, showing a typical Poisson distribution.
This initial list of 268 sequences was further refined by manual inspection. Longer repeat arrays were selected over shorter ones because there is a positive relationship between repeat length and mutation rate (Primmer & Ellegren, Citation1998); however, whether allele length is positively correlated to mutation rates in the flanking regions is not known. Dinucleotides and tetranucleotide repeats are much rarer in exonic regions as compared with trinucleotide repeats (Toth et al., Citation2000) so in the interest of isolating selectively neutral markers we limited the number of trinucleotide repeats analysed (to seven of 34 loci analysed; ).
Initial SNP discovery
A preliminary search for SNPs in MFRs generated some positive results. Ten to twelve isolates from Eastern Canada were scored at each of four MFRs with an average of 376 bp analysed per locus (totalling 1427 bp). Sequences were identical across isolates for each locus and a total of eight heterozygous bases were found (data not shown).
Description of final molecular data set
We generated MFR sequence data from 28 unique, putatively unlinked loci (27 of the 28 loci were found on unique supercontigs of the P. infestans genome sequence) (). For six loci, the ‘second’ flanking region was targeted for sequencing after initial comparisons using the ‘first’ flanking region suggested a high number of SNPs. Thus the final molecular marker set consisted of 34 MFRs: one MFR for each of 22 microsatellite repeat loci (i.e. either immediately upstream or immediately downstream from the repeat) and two MFRs (i.e. both the upstream and downstream flanking regions) for each of six microsatellite repeat loci (). The first four digits in the name given to each locus refer to the number of the supercontig in which the microsatellite repeat was found.
DNA sequence data were obtained from the 34 MFRs for the 32-isolate SNP-discovery international panel (described in Materials and methods) and the whole-genome-sequenced T30-4 strain. High quality sequences that were predominantly bi-directional and easily alignable were collected for 32 of these 34 MFRs. PCR products averaged 525 bp in length, with scorable lengths ranging from 280 bp to 541 bp and averaging 426 bp (). Of the 32 MFRs that yielded analysable data, four had no detectable SNPs and four had no consistently scorable SNPs, leaving a total of 24 MFRs (across 21 microsatellites) with scorable SNP data.
Assessment of marker variability
A total of 13 624 base pairs of sequence consensus data from 32 MFRs (approximately 1M bp of individual sequence data) were generated and examined for the international panel of 32 P. infestans strains. Across the 24 MFR loci with scorable SNPs, a total of 167 SNPs were identified by eye and 102 were scorable under the objective criteria described in Materials and methods (), yielding a range of zero to 16 scorable SNPs per MFR (mean 3.2; ). Nucleotide diversity ranged from a low of zero to a high of 0.013 (one change per 78 bp), with an average across 28 scored loci of 0.0023 (± 0.0029 SD; one change per 426 bp). Isolates with any missing data had to be excluded from nucleotide diversity calculations; hence if data were missing at one SNP, the isolate was removed from the analysis for all SNPs at that locus. This resulted in an average of 28.4 (± 3.3 SD) strains/locus being included, with a minimum of 20 isolates at one locus (). This loss of information and including only SNPs scored using objective scoring criteria makes this a marked underestimate of nucleotide diversity; the true measure is certainly higher.
All SNPs were biallelic except for one that was triallelic (Supplementary Table 1). For biallelic SNPs (n = 101), the frequency of the lesser allele averaged 0.16 (± 0.13 SD) and ranged from a minimum of 0.015 (a single heterozygote isolate at that site) to a maximum of 0.50 (all isolates heterozygous at that site; Supplementary Table 1). The number of sampled diplotypes per MFR locus ranged from two to eight and averaged 3.7 (± 1.5 SD; n = 24).
SNP data from all 24 MFR loci were able to distinguish each of the 32 isolates in the international panel. The pairwise per cent similarity between isolates varied from a high of 99.4% similar (a single allelic change from homozygous to heterozygous at one SNP across 88 scored SNPs) to a low of 58.5% similar (73 allelic changes at 54 of 88 scored SNPs). Average per cent similarity between isolates was 79.0%, which corresponds to about 37 allelic changes across the 88 scored SNPs.
Strain typing: development of a ‘straincode’
As the 102 SNPs analysed here were easily able to discriminate among each isolate in the test panel, we selected a subset comprised of one SNP per locus (totalling 19 putatively unlinked SNPs) and explored its potential utility as a ‘straincode’ for typing purposes. SNPs in the straincode were selected based on allelic diversity and frequency of the lesser allele (described in full in Materials and methods), the latter of which also corresponded to the SNP with the highest heterozygosity for that locus in all but two cases (loci 3200F and 3230_1R; ). SNPs in the straincode are ordered by decreasing information content as determined by per-locus probability of identity estimates (). This, incidentally, exactly matched the order that was initially determined based on allelic diversity and frequency of the lesser allele, and therefore lends confidence to our method of selecting SNPs for the straincode in the first place. Genotype frequencies were consistent with Hardy–Weinberg equilibrium at 15 of the 19 SNPs in the straincode ().
The 19-SNP straincode is based on 18 biallelic SNPs, which have three diploid states, and one triallelic SNP, which has six diploid states, leading to over 2 billion (2 324 522 934) potential straincodes. The probability of two randomly chosen isolates having the same genotype when all 19 MFR loci are considered is very low (PI = 3.5 × 10−6). A probability of identity of approximately 1% is achieved when only the first four loci in the straincode are used (PI = 1.6 × 10−2); to reduce this to 0.1%, the first seven loci are needed (PI = 1.1 × 10−3).
The 19-SNP straincode was able to distinguish 25 of the 32 isolates in the international panel with pairwise percentage similarity between isolates ranging from 48% to 100%. The remaining seven isolates formed two groups: one with two identical strains, which were both collected from tomatoes in New York in two consecutive years and that share the same mating type, mtDNA haplotype, but not RAPD genotype; and one group with five identical isolates (), which were sampled from both tomato and potato plants. This group of five isolates showed very little variation in the larger, 102-SNP dataset, with only one to four SNPs varying among them. All were mating type A1, and two shared the same RAPD genotype (). These five isolates had a wide geographic distribution (Russia, Peru, China, Switzerland, and Nebraska) and differed from all other members of the international panel by a minimum of 36 allelic changes. Incidentally, as RAPDs are known for problematic reproducibility, it is noteworthy that different RAPD genotypes were found in both of the two groups of isolates with identical straincodes.
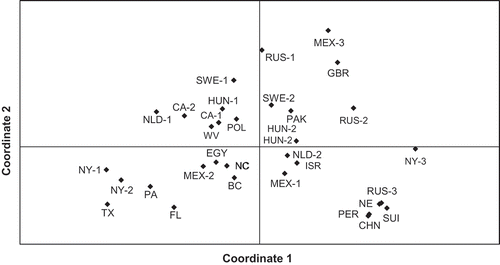
Based on data from the 19 SNPs in the straincode, a Mantel test showed no significant correlation between genetic and geographic distance matrices (P = 0.21), which is reflected by the general absence of clustering of isolates sampled from similar locations in the PCO analysis (). The PCO plot depicts high diversity and an overall lack of genetic structuring within the 32 isolate international panel; sampled isolates are generally scattered across the two-dimensional space with minimal clustering (). The only semblance of clustering of isolates according to geography is a loose at best cluster of five isolates sampled from the US (NY-1, NY-2, PA, FL, and TX; ).
Fig. 1. Principal coordinates analysis plot of pairwise genotypic distance for 32 isolates of Phytophthora infestans scored at 19 unlinked SNPs in MFRs. Data labels reflect sampling location for each isolate as indicated in .
Colombian and Eastern Canadian isolates
Based on the sequencing of five MFR loci, the 57 isolates sampled from Colombia were virtually identical: the only exception was that single isolate that varied (A instead of G for strain P14006/1032) at a SNP not detected in the international panel (3230_1F at 204bp) and at which the other sampled isolates were homozygous for G (Supplementary Table 2: online). What can be formed as the start of the Colombian straincode is C-A-YG, showing similarity to P6576 (Netherlands) and P10250 (Hungary).
Similarly, two Eastern Canadian isolates used during marker development and optimization were identical at 46 MFRs screened during preliminary analyses (data not shown). Their straincode was unique among those found in the international panel (SGRWC-TTGWTGGTTGGAA) and showed similarities to isolates P10157 (West Virginia) and P6747 (Poland). The dozen Canadian isolates that were used during the pilot development were also identical based on a smaller number of MFRs.
Discussion
SNP rates in P. infestans MFRs
Estimates of SNP rates are currently available for a small but growing number of taxa, though direct comparisons across studies are difficult when nucleotide diversity estimates are not provided. SNP rates are often expressed as the proportion of total base pairs surveyed that showed polymorphism (i.e. the number of variable sites divided by the number of bases analysed); but this is highly dependent on the sample size of the panel, thus comparisons across studies are inevitably confounded by variations in sample size. In contrast, because nucleotide diversity is the average number of different nucleotides per site between two randomly selected individuals in the panel, it is not dependent on sample size (apart from any effect of sampling error). Unfortunately, estimates of nucleotide diversity are absent more often than not from the relatively few published studies on SNP-based markers for ecological studies. For fungi and oomycetes, the following studies report the presence of SNPs but do not provide estimates of nucleotide diversity: Fisher et al. (Citation1999) found 13 loci with one SNP each in the human pathogen, Coccidioides immitis G.W. Stiles, detected by single-strand conformational polymorphisms (SSCPs) in amplicons generated from random primers; Giresse et al. (Citation2007) also used PCR-SSCP analysis of 124 ESTs from the sunflower pathogen, Plasmopara halstedii (Farl.) Berl. & De Toni, to find 25 SNPs across 12 loci; and Xu et al. (Citation2007) found 178 SNPs in the basidiomycete Tricholoma matsutake (S. Ito & S. Imai) Singer through random sequencing of genomic library clones.
Overall nucleotide diversity of MFRs in P. infestans found here of 0.0023 (one change per 426 bp) is within the expected range of one SNP per 200–500 bp for non-coding DNA (Brumfield et al., Citation2003; Morin et al., Citation2004) and is much higher than the broadly generalized rate of 1 SNP/1000 bp (Perkel, Citation2008). Tyler et al. (Citation2006) found one change per 5000 bp in P. ramorum and one in 20 000 in P. sojae Kaufm. & Gerd. Comparing the annotated genome of Pythium ultimum Trow (Lévesque et al., Citation2010) to another strain sequenced at low coverage by pyrosequencing, one change per 1121 bp was found (unpublished). A high rate of SNP was found in MFRs despite the fact that SNP rates reported here are considerable underestimates of true rates due to our use of strict objective scoring rules; 65 SNPs were detected by eye across the 24 polymorphic MFRs that were not scorable using the objective criteria. Further, isolates had to be removed from nucleotide diversity calculations if data were missing at even a single SNP. The loss of information content associated with objective scoring is clearly undesirable but needs to be balanced against the drawbacks of manual scoring, including human error and inconsistencies in judgement and that it may simply be impractical for large data sets when there is an automated alternative. Other estimates of nucleotide diversity in MFRs are limited to a single study on flycatchers: nucleotide diversity across six loci was around 0.005 (1 SNP every 200 bp) for the two species studied (Primmer et al., Citation2002). To our knowledge, the only SNP-based markers in MFRs reported for a fungal species are three loci developed by Groenewald et al. (Citation2007) for the plant pathogen Cercospora beticola Sacc. that were discovered incidentally while isolating size-polymorphic microsatellites. Although nucleotide diversity is not reported, a handful of other studies have developed SNP-based markers in MFRs and cover a diverse range of taxa, e.g. plants (Mogg et al., Citation2002; Ablett et al., Citation2006; Fernández et al., Citation2008); birds (Primmer et al., Citation2002); and abalone (Rhode et al., Citation2008).
Regardless, it is reasonable to hypothesize that a likely contributor to the high SNP rate reported here is indeed that P. infestans regularly undergoes asexual reproduction and that there is no mechanism to purge new mutations from a clonal population. Indeed, human-mediated translocation of isolates and the bounty of hosts provided by agricultural monocultures give tremendous opportunity for rapid clonal propagation and accompanying genetic diversification through mutation. Whether the variation in the timing of collection of isolates used here contributed in any significant way to the high SNP rates is unknown, since potentially bottlenecked, clonal, pathogenic microbes are expected to have the capacity for rapid genetic change. However, that visual inspection of straincodes does not suggest the grouping of isolates by collection date and that the five isolates with the same straincode were collected from across at least 18 years suggest against a major effect of temporal variation in sample collection.
SNPs in MFRs for strain typing and population genetics
A range of molecular marker systems have been used for assessing genetic diversity in P. infestans, including allozymes (Goodwin et al., Citation1995), RFLP fingerprinting (Goodwin et al., Citation1992), AFLPs (Flier et al., Citation2003), mtDNA haplotypes (Griffith & Shaw, Citation1998), RAPDs (Mahuku et al., Citation2000), and microsatellite repeat units (Lees et al., Citation2006). The SNP-based straincode presented here offers the following advantages: (i) ease of scoring and reproducibility, (ii) low homoplasy, (iii) immediate compatibility with high throughput screening tools for SNPs (Kim & Misra, Citation2007; Beaudet & Belmont, Citation2008; Perkel, Citation2008), and (iv) the potential to characterize alleles directly from environmental samples such as spore traps. DNA sequencing is becoming the new standard to identify microorganisms. Similarly, an approach based on DNA sequences to identify strains of a given species would be more amenable to database search and unambiguous comparisons. An approach based on sequencing for large population studies might not be practically feasible now; however, next generation parallel sequencing technologies are developing rapidly and would allow direct evaluation of alleles present in samples from environmental metagenomics studies.
Results presented here suggest that SNPs in MFRs offer a promising avenue for strain typing. Probability of identity estimates indicate that only a small number (four) of the most informative SNP loci need to be screened to provide a unique genotype to 99 out of every 100 P. infestans isolates tested. However, it is important to bear in mind that probability of identity estimates are related to the diversity of the panel screened (see data below about the Colombian panel). If a strain typing system were to be developed for a more restricted geographic area (e.g. North America), its power to discriminate among isolates would need to be re-established through new probability of identity estimates using a panel of isolates from that region to avoid ascertainment bias affecting the results. This is the bias introduced to genetic analyses based on SNP data when arbitrary decisions are made during the SNP discovery phase; for example, if only the most variable SNP loci are analysed or if a non-representative sample of individuals is used when putative marker loci are screened for polymorphisms (Brumfield et al., Citation2003).
The high genetic diversity of P. infestans at SNP markers described here suggests that they could be informative for answering molecular ecological questions about this pathogen, including population structure, the relative importance of sexual versus asexual reproduction, and questions associated with disease movements and relatedness among strains associated with different outbreaks. Indeed, in another oomycete plant pathogen, Delmotte et al. (Citation2008) proved that an inordinate number of SNP loci is not necessarily required to successfully answer population genetics questions; they used 12 SNPs in ESTs to elucidate the relationship among pathotypes of Plasmopara halstedii (Farl.) Berl. & De Toni in France. Sampled pathotypes formed three genetic clusters that were consistent with a pattern of three historical introduction events followed by the subsequent emergence of new races, the evolution of which was hypothesized to have been mediated by both sexual (i.e. recombination) and asexual (i.e. accumulation of mutations) processes (Delmotte et al., Citation2008). How many SNPs are needed to have an appropriate level of resolution? This is a question that is important to the scientific community in general (Smouse, Citation2010). Santure et al. (Citation2010) found that 50 SNPs were as good as 20 microsatellite loci to estimate pedigree relatedness in a zebra fish population.
We found no evidence of overall genetic isolation by distance, which is not surprising given that human-mediated translocation is responsible for the geographic distribution of virtually the entire international panel. All isolates except one from Peru were sampled outside of the South American Andes region, which is now considered a possible region for the evolutionary origin of P. infestans (Gómez-Alpizar et al., Citation2007). The most prominent genetic grouping of isolates is a cluster of five near-identical isolates sampled from Russia, Peru, China, USA and Switzerland that were collected up to 18 years apart: they differed from each other by a maximum of four nucleotide changes across 102 SNPs. It is reasonable to hypothesize that the similarity among these five isolates is because they are derived from the same original strain and that the small number of differences separating them occurred recently, after each translocated isolate became isolated and then spread through clonal propagation.
It is intuitive that our finding of no association between genetic similarity and geographic proximity would not hold for comparisons made at finer spatial scales. Indeed, all strains sampled from Colombia were genetically identical, thus they are likely all the same strain and products of clonal reproduction, which is supported by the fact that all isolates were mating type A1. As such, they do not have a genetic signature consistent with gene flow with the centre of origin. When this study was initiated, there was no knowledge of their genetic composition and it was a reasonable assumption that there might be considerable variation in genotypes. The very low level of variation observed here concurs with a results from a recent study focussed on P. infestans from Colombia, which found the same population to be highly clonal even with microsatellite analysis (Vargas et al., Citation2009). However, the one strain that was different in our study did not match any of the two that were unique in Vargas et al. (Citation2009) (Supplementary Table 1). Isolates from eastern Canada showed similar results with respect to lack of diversity on a regional scale: a limited data set generated during marker development suggests that this group is also comprised of a single, clonal strain. These results are consistent with published studies indicating that populations of P. infestans in eastern Canada have been dominated by the US-8 genotype (A2 mating type) since its introduction into the region in the mid-1990s (Peters et al., Citation1998, Citation1999, Citation2001). No A1 strains have been recovered from potatoes in this production area in over a decade (Peters et al., unpublished data), and thus pathogen propagation is restricted to asexual reproduction and the spread of clones from initial disease foci generated from inoculum found in infected, over-wintered tubers used as seed or disposed of as culls.
This study shows that targeting sequence variation in MFRs can be an effective method of isolating SNP-based molecular markers. Sequence and SNP based approaches are likely to be the most common genotyping techniques in the future and an MFR SNP based system could provide a linkage and transition between the most common genotyping approach being currently used and this new generation of tools. That whole genome sequences are not available for most non-model organisms is becoming increasingly less limiting as new sequencing methods (Hudson, Citation2008) quickly transform our ability to cheaply acquire vast amounts of sequence data for these taxa. Similarly, many SNP genotyping technologies are now available and accommodate varying throughput levels (e.g. Behura, Citation2006; Kim & Misra, Citation2007; Beaudet & Belmont, Citation2008; Perkel, Citation2008; Brenan et al., Citation2009; Drago et al., Citation2009), which make screening large numbers of independent marker loci readily feasible even with environmental sampling such as spore traps. With SNP discovery and genotyping hurdles quickly vanishing, our ability to exploit the benefits of these abundant, low homoplasy, high throughput markers is continually enhanced.
Supplementary Data
Download Zip (162.3 KB)Acknowledgements
The funding for this was provided by the Cbrne Research and Technology Initiative (project CRTI 04-0045RD). Grants to Michael Coffey from the USDA-NRI Plant Biosecurity program (2005-35605-15393 and 2008-55605-18773) were helpful in developing the Phytophthora DNA Bank which provided many samples for this project. We want to thank Carolyn Babcock, Ian Macdonald and Katie MacDonald for help with isolate culturing and Rafik Assabgui for the sequencing. We also want to thank Tyler Jackson and Hazim Alkhani for their technical assistance with the MFR sequencing of the isolates from eastern Canada and Colombia, respectively.
References
- Ablett , G. , Hill , H. and Henry , R.J. 2006 . Sequence polymorphism discovery in wheat microsatellite flanking regions using pyrophosphate sequencing . Mol. Breed. , 17 : 281 – 289 .
- Adams , R.I. , Hallen , H.E. and Pringle , A. 2006 . Using the incomplete genome of the ectomycorrhizal fungus Amanita bisporigera to identify molecular polymorphisms in the related Amanita phalloides . Mol. Ecol. Notes , 6 : 218 – 220 .
- Aitken , N. , Smith , S. , Schwarz , C. and Morin , P.A. 2004 . Single nucleotide polymorphism (SNP) discovery in mammals: a targeted-gene approach . Mol. Ecol. , 13 : 1423 – 1431 .
- Allain-BoulÉ , N. , LÉvesque , C.A. , Martinez , C. , BÉlanger , R.R. and Tweddell , R.J. 2004 . Identification of Pythium species associated with cavity-spot lesions on carrots in eastern Quebec . Can. J. Plant. Pathol. , 26 : 365 – 370 .
- Banke , S. and McDonald , B.A. 2005 . Migration patterns among global populations of the pathogenic fungus Mycosphaerella graminicola . Mol. Ecol. , 14 : 1881 – 1896 .
- Beaudet , A.L. and Belmont , J.W. 2008 . Array-based DNA diagnostics: let the revolution begin . Annu. Rev. Med. , 59 : 113 – 129 .
- Behura , S.K. 2006 . Molecular marker systems in insects: current trends and future avenues . Mol. Ecol. , 15 : 3087 – 3113 .
- Belfiore , N.M. , Hoffman , F.G. , Baker , R.J. and Dewoody , J.A. 2003 . The use of nuclear and mitochondrial single nucleotide polymorphisms to identify cryptic species . Mol. Ecol. , 12 : 2011 – 2017 .
- Bensch , S. , Akesson , S. and Irwin , D.E. 2002 . The use of AFLP to find an informative SNP: genetic differences across a migratory divide in willow warblers . Mol. Ecol. , 11 : 2359 – 2366 .
- Blair , J.E. , Coffey , M.D. , Park , S.Y. , Geiser , D.M. and Kang , S. 2008 . A multi-locus phylogeny for Phytophthora utilizing markers derived from complete genome sequences . Fungal Genet. Biol. , 45 : 266 – 277 .
- Brenan , C.J.H. , Roberts , D. and Hurley , J. 2009 . Nanoliter high-throughput PCR for DNA and RNA profiling . Methods Mol. Biol. , 496 : 161 – 174 .
- Brohede , J. and Ellegren , H. 1999 . Microsatellite evolution: polarity of substitutions within repeats and neutrality of flanking sequences . Proc. R. Soc. Lond., Ser. B: Biol. Sci. , 266 : 825 – 833 .
- Brumfield , R.T. , Beerli , P. , Nickerson , D.A. and Edwards , S.V. 2003 . The utility of single nucleotide polymorphisms in inferences of population history . Trends Ecol. Evol. , 18 : 249 – 256 .
- Burgess , T. , Wingfield , M.J. and Wingfield , B.W. 2001 . Simple sequence repeat markers distinguish among morphotypes of Sphaeropsis sapinea . Appl. Environ. Microbiol. , 67 : 354 – 362 .
- Cooke , D.E.L. and Lees , A.K. 2004 . Markers, old and new, for examining Phytophthora infestans diversity . Plant Pathol. , 53 : 692 – 704 .
- Cooke , D.E.L. , Lees , A.K. , Hansen , J.G. , Lassen , P. , Andersson , B. and Bakonyi , J. 2009 . From a European to a global database of Phytophthora infestansgenetic diversity: examining the nature and significance of population change . Acta Horticulturae , 834 : 19 – 26 .
- Delmotte , F. , Giresse , X. , Richard-Cervera , S. , M'Baya , J. , Vear , F. , Tourvieille , J. , Walser , P. and Tourvielle de Labrouhe , D. 2008 . Single nucleotide polymorphisms reveal multiple introductions into France of Plasmopara halstedii, the plant pathogen causing sunflower downy mildew . Infect., Genet. Evol. , 8 : 534 – 540 .
- Dobrowolski , M.P. , Tommerup , I.C. , Shearer , B.L. and O'Brien , P.A. 2003 . Three clonal lineages of Phytophthora cinnamomiin Australia revealed by microsatellites . Phytopathology , 93 : 695 – 704 .
- Drago , F. , Karpasitou , K. and Poli , F. 2009 . Microarray beads for identifying blood group single nucleotide polymorphisms . Transfus. Med. Hemother. , 36 : 157 – 160 .
- Ellegren , H. 2004 . Microsatellites: simple sequences with complex evolution . Nat. Rev. Genet. , 5 : 435 – 445 .
- Enkerli , J. , Widmer , F. , Gessler , C. and Keller , S. 2001 . Strain-specific microsatellite markers in the entomopathogenic fungus Beauveria brongniartii . Mycol. Res. , 105 : 1079 – 1087 .
- Fernández , M.P. , NÜñez , Y. , Ponz , F. , Hernáiz , S. , Gallego , F.J. and Ibáñez , J. 2008 . Characterization of sequence polymorphisms from microsatellite flanking regions in Vitis spp . Mol. Breed. , 22 : 455 – 465 .
- Fisher , M.C. , White , T.J. and Taylor , J.W. 1999 . Primers for genotyping single nucleotide polymorphisms and microsatellites in the pathogenic fungus Coccidioides immitis . Mol. Ecol. , 8 : 1082 – 1084 .
- Flier , W.G. , Grünwald , N.J. , Kroon , L. , Sturbaum , A.K. , van den Bosch , T.B.M. Garay-Serrano .J , E., … Turkensteen, L . 2003 . The population structure of Phytophthora infestans from the Toluca Valley of central Mexico suggests genetic differentiation between populations from cultivated potato and wild Solanum spp . Phytopathology , 93 : 382 – 390 .
- Forbes , G.A. , Goodwin , S.B. , Drenth , A. , Oyarzun , P. , OrdonÌfez , M.E. and Fry , W.E. 1998 . A global marker database for Phytophthora infestans . Plant Dis. , 82 : 811 – 818 .
- Fry , W.E. , Goodwin , S.B. , Matuszak , J.M. , Spielman , L.J. , Milgroom , M.G. and Drenth , A. 1992 . Population genetics and intercontinental migrations of Phytophthora infestans . Annu. Rev. Phytopathol. , 30 : 107 – 129 .
- Fry , W.E. , Goodwin , S.B. , Dyer , A.T. , Matuszak , J.M. , Drenth , A. , Tooley , P.W. … Sandian , K.P. 1993 . Historical and recent migrations of Phytophthora infestans: chronology, pathways, and implications . Plant Dis. , 77 : 653 – 661 .
- Garnica , D.P. , Pinzón , A.M. , Quesada-Ocampo , L.M. , Bernal , A.J. , Barreto , E. , Grünwald , N.J. and Restrepo , S. 2006 . Survey and analysis of microsatellites from transcript sequences in Phytophthora species: frequency, distribution, and potential as markers for the genus . BMC Genomics , 7 : 245
- Giresse , X. , Tourvieille De Labrouhe , D. , Richard-Cervera , S. and Delmotte , F. 2007 . Twelve polymorphic expressed sequence tags-derived markers for Plasmopara halstedii, the causal agent of sunflower downy mildew . Mol. Ecol. Notes , 7 : 1363 – 1365 .
- Glaubitz , J.C. , Rhodes , O.E. and Dewoody , J.A. 2003 . Prospects for inferring pairwise relationships with single nucleotide polymorphisms . Mol. Ecol. , 12 : 1039 – 1047 .
- Gómez-Alpizar , L. , Carbone , I. and Ristaino , J.B. 2007 . An Andean origin of Phytophthora infestans inferred from mitochondrial and nuclear gene genealogies . Proc. Natl. Acad. Sci. USA , 104 : 3306 – 3311 .
- Goodwin , S.B. and Drenth , A. 1997 . Origin of the A2 mating type of Phytophthora infestans outside Mexico . Phytopathology , 87 : 992 – 999 .
- Goodwin , S.B. , Schneider , R.E. and Fry , W.E. 1995 . Use of celluloseacetate electrophoresis for rapid identification of allozyme genotypes of Phytophthora infestans . Plant Dis. , 79 : 1181 – 1185 .
- Goodwin , S.B. , Spielman , L.J. , Matuszak , J.M. , Bergeron , S.N. and Fry , W.E. 1992 . Clonal diversity and genetic differentiation of Phytophthora infestans populations in northern and central Mexico . Phytopathology , 82 : 955 – 961 .
- Goodwin , S.B. , Smart , C.D. , Sandrock , R.W. , Deahl , K.L. , Punja , Z.K. and Fry , W.E. 1998 . Genetic change within populations of Phytophthora infestans in the United States and Canada during 1994 to 1996: role of migration and recombination . Phytopathology , 88 : 939 – 949 .
- Griffith , G.W. and Shaw , D.S. 1998 . Polymorphisms in Phytophthora infestans: four mitochondrial haplotypes are detected after PCR amplification of DNA from pure cultures or from host lesions . Appl. Environ. Microbiol. , 64 : 4007 – 4014 .
- Grimaldi , M.-C. and Crouau-Roy , B. 1997 . Microsatellite allelic homoplasy due to variable flanking sequences . J. Mol. Evol. , 44 : 336 – 340 .
- Groenewald , M. , Groenewald , J.Z. , Linde , C.C. and Crous , P.W. 2007 . Development of polymorphic microsatellite and single nucleotide polymorphism markers for Cercospora beticola (Mycosphaerellaceae) . Mol. Ecol. Notes , 7 : 890 – 892 .
- Guo , J. , Van Der Lee , T. , Qu , D.Y. , Yao , Y.Q. , Gong , X.F. , Liang , D.L. and … Govers , F. 2009 . Phytophthora infestans isolates from Northern China show high virulence diversity but low genotypic diversity . Plant Biol. , 11 : 57 – 67 .
- Haas , B.J. , Kamoun , S. , Zody , M.C. , Jiang , R.H.Y. , Handsaker , R.E. , Cano , L.M. and … Nusbaum , C. 2009 . Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans . Nature , 461 : 393 – 398 .
- Hennequin , C. , Thierry , A. , Richard , G.F. , Lecointre , G. , Nguyen , H.V. , Gaillardin , C. and Dujon , B . 2001 . Microsatellite typing as a new tool for identification of Saccharomyces cerevisiae strains . J. Clin. Microbiol. , 39 : 551 – 559 .
- Hudson , M.E. 2008 . Sequencing breakthroughs for genomic ecology and evolutionary biology . Mol. Ecol. Resources , 8 : 3 – 17 .
- Ishibashi , Y. , Saitoh , T. , Abe , S. and Yoshida , M.C. 1996 . Null microsatellite alleles due to nucleotide sequence variation in the grey-sided vole Clethrionomys rufocanus . Mol. Ecol. , 5 : 589 – 590 .
- Ivors , K. , Garbelotto , M. , Vries , I.D.E. , Ruyter-Spira , C. , Hekkert , B.T. Rosenzweig , N., & Bonants, P . 2006 . Microsatellite markers identify three lineages of Phytophthora ramorum in US nurseries, yet single lineages in US forest and European nursery populations . Mol. Ecol. , 15 : 1493 – 1505 .
- Jones , A.G. , Stockwell , C.A. , Walker , D. and Avise , J.C. 1998 . The molecular basis of a microsatellite null allele from the white sands pupfish . J. Hered. , 89 : 339 – 342 .
- Kim , S. and Misra , A. 2007 . SNP genotyping: technologies and biomedical applications . Annu. Rev. Biomed. Eng. , 9 : 289 – 320 .
- Kimmel , M. , Chakraborty , R. , Stivers , D.N. and Deka , R. 1996 . Dynamics of repeat polymorphisms under a forward-backward mutation model: within- and between-population variability at microsatellite loci . Genetics , 143 : 549 – 555 .
- Knapova , G. and Gisi , U. 2002 . Phenotypic and genotypic structure of Phytophthora infestans populations on potato and tomato in France and Switzerland . Plant Pathol. , 51 : 641 – 653 .
- Lee , S. and Moorman , G. 2008 . Identification and characterization of simple sequence repeat markers for Pythium aphanidermatum, P. cryptoirregulare, and P. irregulare and the potential use in Pythium population genetics . Curr. Genet. , 53 : 81 – 93 .
- Lees , A.K. , Wattier , R. , Shaw , D.S. , Sullivan , L. , Williams , N.A. and Cooke , D.E.L. 2006 . Novel microsatellite markers for the analysis of Phytophthora infestans populations . Plant Pathol. , 55 : 311 – 319 .
- Lévesque , C.A. 2007 . “ Molecular diagnostics of soilborne fungal pathogens and applications to disease management ” . In Biotechnology and plant disease management , Edited by: Punja , Z.K. , DeBoer , S. and Sanfaçon , H. 146 – 164 . Wallingford : CABI .
- Lévesque , C.A. , Brouwer , H. , Cano , L. , Hamilton , J.P. , Holt , C. , Huitema , E. and Buell , C.R . 2010 . Genome sequence of the necrotrophic plant pathogen, Pythium ultimum, reveals original pathogenicity mechanisms and effector repertoire . Genome Biology , 11 ( R73 )
- Li , Y.C. , Korol , A.B. , Fahima , T. , Beiles , A. and Nevo , E. 2002 . Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review . Mol. Ecol. , 11 : 2453 – 2465 .
- Lievens , B. and Thomma , B. 2005 . Recent developments in pathogen detection arrays: implications for fungal plant pathogens and use in practice . Phytopathology , 95 : 1374 – 1380 .
- Mahuku , G. , Peters , R.D. , Platt , H.W. and Daayf , F. 2000 . Random amplified polymorphic DNA (RAPD) analysis of Phytophthora infestans isolates collected in Canada during 1994 to 1996 . Plant Pathol. , 49 : 252 – 260 .
- McCartney , H.A. , Foster , S.J. , Fraaije , B.A. and Ward , E. 2003 . Molecular diagnostics for fungal plant pathogens . Pest Manage. Sci. , 59 : 129 – 142 .
- McEwen , J.G. , Taylor , J.W. , Carter , D. , Xu , J. , Felipe , M.S.S. , Vilgalys , R. and … Soares , C.M.A . 2000 . Molecular typing of pathogenic fungi . Med. Mycol. , 38 : 189 – 197 .
- Meglecz , E. 2007 . MICROFAMILY (version 1): a computer program for detecting flanking-region similarities among different microsatellite loci . Mol. Ecol. Notes , 7 : 18 – 20 .
- Meglecz , E. , Petenian , F. , Danchin , E. , D'Acier , A.C. , Rasplus , J.Y. and Faure , E. 2004 . High similarity between flanking regions of different microsatellites detected within each of two species of Lepidoptera: Parnassius apollo and Euphydryas aurinia . Mol. Ecol. , 13 : 1693 – 1700 .
- Mhlanga , M.M. and Malmberg , L. 2001 . Using molecular beacons to detect single-nucleotide polymorphisms with real-time PCR . Methods , 25 : 463 – 471 .
- Miller , M.P. 1997 . Tools For Population Genetic Analyses (TFPGA), version 1.3. A windows program for the analysis of allozyme and molecular population genetic data , Flagstaff , AZ : Department of Biological Sciences, Northern Arizona University .
- Mogg , R. , Batley , J. , Hanley , S. , Edwards , D. , O'Sullivan , H. and Edwards , K.J. 2002 . Characterization of the flanking regions of Zea mays microsatellites reveals a large number of useful sequence polymorphisms . Theor. Appl. Genet. , 105 : 532 – 543 .
- Moon , C.D. , Tapper , B.A. and Scott , B. 1999 . Identification of epichloe endophytes in planta by a microsatellite-based PCR fingerprinting assay with automated analysis . Appl. Environ. Microbiol. , 65 : 1268 – 1279 .
- Morin , P.A. , Luikart , G. and Wayne , R.K. 2004 . SNPs in ecology, evolution and conservation . Trends Ecol. Evol. , 19 : 208 – 216 .
- Narum , S.R. , Banks , M. , Beacham , T.D. , Bellinger , M.R. , Campbell , M.R. , Dekoning , J. and … Garza , J.C . 2008 . Differentiating salmon populations at broad and fine geographical scales with microsatellites and single nucleotide polymorphisms . Mol. Ecol. , 17 : 3464 – 3477 .
- Peakall , R. and Smouse , P.E. 2006 . GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research . Mol. Ecol. Notes , 6 : 288 – 295 .
- Perkel , J. 2008 . SNP genotyping: six technologies that keyed a revolution . Nat. Methods , 5 : 447 – 453 .
- Peters , R.D. , Platt , H.W. and Hall , R. 1998 . Characterization of changes in populations of Phytophthora infestans in Canada using mating type and metalaxyl sensitivity markers . Can. J. Plant. Pathol. , 20 : 259 – 273 .
- Peters , R.D. , Platt , H.W. and Hall , R. 1999 . Use of allozyme markers to determine genotypes of Phytophthora infestans in Canada . Can. J. Plant. Pathol. , 21 : 144 – 153 .
- Peters , R.D. , Förster , H. , Platt , H.W.H. , R. and Coffey , M.D. 2001 . Novel genotypes of Phytophthora infestans in Canada during 1994 and 1995 . Am. J. Potato Res. , 78 : 39 – 45 .
- Primmer , C.R. and Ellegren , H. 1998 . Patterns of molecular evolution in avian microsatellites . Mol. Biol. Evol. , 15 : 997 – 1008 .
- Primmer , C.R. , Borge , T. , Lindell , J. and Saetre , G.P. 2002 . Single-nucleotide polymorphism characterization in species with limited available sequence information: high nucleotide diversity revealed in the avian genome . Mol. Ecol. , 11 : 603 – 612 .
- Rhode , C. , Slabbert , R. and Roodt-Wilding , R. 2008 . Microsatellite flanking regions: a SNP mine in South African abalone (Haliotis midae) . Anim. Genet. , 39 : 329
- Rogers , J.S. 1972 . Measures of genetic similarity and genetic distance . Studies in Genetics: Univ. Texas Publ. 7213 , 7 : 145 – 153 .
- Rozas , J. , Sanchez-DelBarrio , J.C. , Messeguer , X. and Rozas , R. 2003 . DnaSP, DNA polymorphism analyses by the coalescent and other methods . Bioinformatics , 19 : 2496 – 2497 .
- Rozen , S. and Skaletsky , H. 2000 . Primer3 on the WWW for general users and for biologist programmers . Methods Mol. Biol. , 132 : 365 – 386 .
- Santure , A.W. , Stapley , J. , Ball , A.D. , Birkhead , T.R. , Burke , T. and Slate , J. 2010 . On the use of large marker panels to estimate inbreeding and relatedness: empirical and simulation studies of a pedigreed zebra finch population typed at 771 SNPs . Mol. Ecol. , 19 : 1439 – 1451 .
- Schaad , N.W. and Frederick , R.D. 2002 . Real-time PCR and its application for rapid plant disease diagnostics . Can. J. Plant. Pathol. , 24 : 250 – 258 .
- Schena , L. , Cardle , L. and Cooke , D.E.L. 2008 . Use of genome sequence data in the design and testing of SSR markers for Phytophthora species . BMC Genomics , 9 : 620
- Seddon , J.M. , Parker , H.G. , Ostrander , E.A. and Ellegren , H. 2005 . SNPs in ecological and conservation studies: a test in the Scandinavian wolf population . Mol. Ecol. , 14 : 503 – 511 .
- Smith , C.T. , Elfstrom , C.M. , Seeb , L.W. and Seeb , J.E. 2005 . Use of sequence data from rainbow trout and Atlantic salmon for SNP detection in Pacific salmon . Mol. Ecol. , 14 : 4193 – 4203 .
- Smith , N.C. , Hennessy , J. and Stead , D.E. 2001 . Repetitive sequence-derived PCR profiling using the BOX-A1R primer for rapid identification of the plant pathogen Clavibacter michiganensis subspecies sepedonicus . Eur. J. Plant Pathol. , 107 : 739 – 748 .
- Smouse , P.E. 2010 . How many SNPs are enough? . Mol. Ecol , 19 : 1265 – 1266 .
- Stukenbrock , E.H. , Banke , S. and McDonald , B.A. 2006 . Global migration patterns in the fungal wheat pathogen Phaeosphaeria nodorum . Mol. Ecol. , 15 : 2895 – 2904 .
- Sullivan , T.J. and Faeth , S.H. 2004 . Gene flow in the endophyte Neotyphodium and implications for coevolution with Festuca arizonica . Mol. Ecol. , 13 : 649 – 656 .
- Tero , N. , Neumeier , H. , Gudavalli , R. and Schlotterer , C. 2006 . Silene tatarica microsatellites are frequently located in repetitive DNA . J. Evol. Biol. , 19 : 1612 – 1619 .
- Toth , G. , Gaspari , Z. and Jurka , J. 2000 . Microsatellites in different eukaryotic genomes: survey and analysis . Genome Res. , 10 : 967 – 981 .
- Tyler , B.M. , Tripathy , S. , Zhang , X. , Dehal , P. , Jiang , R.H. , Aerts , A. and … Boore , J.L. 2006 . Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis . Science , 313 : 1261 – 1266 .
- Vargas , A.M. , Quesada Ocampo , L.M. , Cespedes , M.C. , Carreno , N. , Gonzalez , A. , Rojas , A. and … Restrepo , S . 2009 . Characterization of Phytophthora infestans populations in Colombia: first report of the A2 mating type . Phytopathology , 99 : 82 – 88 .
- Xu , J. , Guo , H. and Yang , Z.L. 2007 . Single nucleotide polymorphisms in the ectomycorrhizal mushroom Tricholoma matsutake . Microbiology , 153 : 2002 – 2012 .
- Zardoya , R. , Vollmer , D.M. , Craddock , C. , Streelman , J.T. , Karl , S. and Meyer , A. 1996 . Evolutionary conservation of microsatellite flanking regions and their use in resolving the phylogeny of cichlid fishes (Pisces: Perciformes) . Proc. R. Soc. Lond., Ser. B: Biol. Sci. , 263 : 1589 – 1598 .
- Zhou , X.D. , Burgess , T.I. , Beer , Z.W. , Lieutier , F. , Yart , A. , Klepzig , K. and … Wingfield , M.J . 2007 . High intercontinental migration rates and population admixture in the sapstain fungus Ophiostoma ips . Mol. Ecol. , 16 : 89 – 99 .