Abstract
The identification and detection of eight pathovars of Pseudomonas syringae, bacterial pathogens of several important agricultural plants, was achieved by TaqMan real-time polymerase chain reaction of a specific DNA fragment of the cytochrome o ubiquinol oxidase gene. Under optimal real-time PCR conditions, the selected primers and probe were specific for the detection of pathovars syringae, tomato, maculicola, tabaci, atropurpurea, phaseolicola, pisi and glycinea. Two pathovars (coriandricola and morsprunorum) tested could be differentiated from the other eight due to a single nucleotide mismatch. Thirty other Pseudomonas strains and 20 non-Pseudomonas strains were negative. The real-time PCR assay detected 100 fg of DNA and 4.5 × 103 P. syringae colony forming units per millilitre (four cells per reaction). In growth chamber experiments, tomato plants were inoculated using strain DC3000 and assayed by TaqMan real-time PCR. Serial dilution of leaf extracts spiked with lambda DNA and processed by real-time PCR indicated the presence of inhibitors. A 1:10 dilution of the crude extract reduced threshold cycles to those of milliQ water spiked with the same amount of lambda DNA. The TaqMan real-time assay consistently detected the pathogen in inoculated tomato leaves after a 1:10 dilution of crude extracts. TaqMan real-time results were validated by dilution-plating of leaf extracts and conventional PCR using the same primer set. This assay offers real-time monitoring of the targeted amplicon with high specificity and sensitivity, with no post-amplification analysis needed. This reduces opportunity for contamination of the reaction mixtures with target DNA, making this real-time PCR assay more reliable than conventional PCR. However, for routine diagnosis of the detected pathovars under greenhouse or field conditions, optimization of the assay might be required.
Résumé
L'identification et la détection de huit pathovars de Pseudomonas syringae, agents pathogènes bactériens qui s'attaquent à plusieurs variétés de plantes économiquement importantes, ont été effectuées par réaction en chaîne de la polymérase (PCR) quantitative en temps réel d'un fragment précis de l'ADN du gène cytochrome o ubiquinol oxydase. Dans des conditions idéales de PCR en temps réel, les amorces et les sondes choisies étaient typiques de la détection des pathovars syringae, tomato, maculicola, tabaci, atropurpurea, phaseolicola, pisi et glycinea. Deux pathovars testés (coriadricola et morsprunorum) ont pu être différenciés des huit autres à cause de la disparité d'un seul nucléotide. Trente autres souches de Pseudomonas et 20 de non-Pseudomonas étaient négatives. Les analyses effectuées par PCR en temps réel ont permis de détecter 100 fg d'ADN et 4.5 × 103 unités formatrices de colonies de P. syringae par millilitre (quatre cellules par réaction). Au cours d'expériences en chambre de culture, des plants de tomate ont été inoculés avec la souche DC3000 et analysés par PCR en temps réel. Une dilution en série d'extraits de feuilles, enrichie d'ADN lambda et traitée par PCR en temps réel, a révélé des inhibiteurs. Une dilution de 1:10 de l'extrait brut a réduit les cycles de seuil à ceux de l'eau Milli-Q enrichie avec la même quantité d'ADN lambda. L'analyse par PCR en temps réel a invariablement détecté l'agent pathogène dans les feuilles inoculées de tomate à la suite d'une dilution 1 : 10 des extraits bruts. Les résultats de la PCR en temps réel ont été validés par dilution et étalement des extraits de plante et par PCR traditionnelle en utilisant le même jeu d'amorces. Cette analyse permet le suivi en temps réel, très précis et sensible, de l'amplicon ciblé, et ce, sans avoir à requérir à une analyse postamplification. Cela réduit le risque de contamination des mélanges réactifs par l'ADN cible, rendant l'analyse par PCR en temps réel plus fiable que celle par PCR traditionnelle. Toutefois, à des fins de diagnostic de routine des pathovars détectés en serres ou en champs, une optimisation de l'analyse peut être requise.
Introduction
The plant pathogen Pseudomonas syringae Van Hall, a Gram-negative bacterium with polar flagella, is divided into pathovars largely on the basis of host specificity (Deng et al., Citation1998; Palleroni, Citation2005). It infects over 50 different economically important crops and trees, such as corn, soybean, tomato and kiwifruit. Symptoms include necrotic spots on leaves and fruits, dead dormant buds, blossom blasts and shoot dieback (Scheck & Pscheidt, Citation1998; Cuppels & Elmhirst, Citation1999; Zaccardelli et al., Citation2005). Serious economic losses may occur due to reduced yields especially on susceptible cultivars (Psallidas, Citation1988; Zhao et al., Citation2002; Zaccardelli et al., Citation2005). P. syringae pathovars can survive as epiphytes providing inoculum at critical phases of the disease cycle (Smitley & McCarter, Citation1982; Cuppels & Elmhirst, Citation1999; Renick et al., Citation2008). In the USA, some states have strict quarantine laws regarding pathovar phaseolicola (Webster et al., Citation1983; Prosen et al., Citation1993) and in Canada, pvs. aesculi and cannabina are quarantine pests as defined by the Canadian Food Inspection Agency (CFIA). One of the reasons why management of diseases caused by P. syringae is difficult is symptom similarity with other pathogens of the same host. In addition, chemical control methods are ineffective. Early field detection of the disease is crucial for the reduction of spread of the pathogen(s) in fields.
Field inspections for typical symptomatology and serological and biochemical assays are widely used for the detection of diseases caused by P. syringae pathovars but these traditional methods are time-consuming, laborious and relatively insensitive compared with polymerase chain reaction (PCR)-based methods. In addition, most of these methods require the isolation of bacterial colonies. The utilization of PCR-based assays would allow for rapid and reliable detection and identification of pathovars of P. syringae. Prosen et al. (Citation1993) reported the development of a PCR assay based on the phaseolotoxin gene region for specific detection of pv. phaseolicola while Arnold et al. (Citation1996) reported specific oligonucleotide primers that can identify P. syringae pv. pisi. Identification of cyclic lipodepsinonapeptide-producing strains of P. syringae pv. syringae using PCR and oligonucleotide primers specific for a 752-bp fragment of the syringomycin synthesis gene subunit B (syrB) has been reported (Sorensen et al., Citation1998). PCR-based diagnostic assays have been developed and evaluated to identify and detect Pseudomonas syringae pv. tomato targeting different genes or DNA fragments (Zaccardelli et al., Citation2005; Cuppels et al., Citation2006), and arbitrarily primed PCR was able to differentiate P. syringae pv. syringae from isolates of unknown pathovars (Renick et al., Citation2008). All these assays require post-PCR handling to visualize results. Because post-PCR processing is required, the assays can not be easily automated for high throughput (Weller et al., Citation2000). Real-time PCR has proven to be a very useful tool for accurate detection of plant pathogenic bacteria (Weller et al., Citation2000; Fanelli et al., Citation2007; Tambong et al., Citation2008; Gervasi & Scortichini, Citation2009; Green et al., Citation2009; Cho et al., Citation2010; Gottsberger, Citation2010). Although real-time PCR systems have been reported for several plant pathogenic bacteria, the works of Cho et al. (Citation2010) and Green et al. (Citation2009) are among the very few, to the best of our knowledge, to target pathovars of P. syringae. Green et al. (Citation2009) reported a SYBR Green real-time PCR assay for identification/detection of P. syringae pv. aesculi based on the gyrase B gene and Cho et al. (Citation2010) targeted the site-specific recombinase gene of P. syringae pv. phaseolicola using TaqMan real-time PCR. These studies target single pathovars probably due to the notion of host specificity. Under field conditions, however, it is increasingly clear that mixed populations of different pathovars may occur on infected plants. Recently, P. syringae pv. pisi as well as P. syringae pv. syringae were detected and identified at similar frequencies in infected Pea trees (Hollaway et al., Citation2007; Martín-Sanz et al., Citation2011). Also, P. syringae pv. syringae and P. syringae pv. morsprunorum were frequently isolated from sweet cherry (Renick et al., Citation2008).
The overall aim of this study was to develop a robust and reliable TaqMan real-time PCR assay for specific detection of several pathovars of P. syringae and to validate the assay for the detection of P. syringae pv. tomato on artificially infected tomato plants under growth chamber conditions by direct processing of lesions (without DNA extraction phase). The assay developed targets the cytochrome o ubiquinol oxidase subunit II (cyoA) gene that catalyses electron oxidation of the ubiquinol-8 in the cytoplasmic membrane and electron reduction of molecular oxygen to water in several bacterial species (Cherpuri et al., Citation1990).
Materials and methods
Bacterial strains, culture conditions and DNA preparation
The bacterial strains used in this study are listed in . All strains of P. syringae were cultured in Luria-Bertani (LB: 10 g L−1 tryptone, 5 g L−1 yeast extract, 10 g L−1 NaCl). Purified DNA and pure cultures of P. syringae pv. syringae NCPPB 2749 were used for optimization of real-time PCR. Strain DC 3000 (Cuppels et al., Citation2006) was used for growth chamber inoculations of tomato plants. Strains of Pectobacterium spp. were cultured in tryptone yeast extract liquid media (Schaad et al., Citation1999) and Clavibacter michiganensis ssp. sepedonicus (Spieckermann & Kotthoff) in nutrient broth supplemented with yeast extract (Smid et al., Citation1995). Stock bacterial cultures were maintained on the same medium supplemented with 25% w/v glycerol at −70 °C.
Table 1. Bacteria used in this study and specificity of detection of Pseudomonas syringae pathovars by TaqMan real-time and conventional polymerase chain reactions
Bacterial cells were harvested in late log phase and genomic DNA purified by the wizard SV Genomic DNA purification system (Promega, Madison, WI, USA). The concentration of DNA was determined using the Nano-Drop ND-1000 Spectrophotometer (NanoDrop Technologist, Wilmington, DE, USA).
Designing of the TaqMan probe and primer set
The identification of species-specific DNA signatures was implemented using the Insignia pipeline (http://insignia.cbcb.umd.edu). To run a DNA signature query by genome comparison (Phillippy et al., Citation2009) the Insignia pipeline was implemented with P. syringae pv. syringae B728a genome (Feil et al., Citation2005) as the reference genome. Genomes of P. syringae pv. tomato DC3000 (Buell et al., Citation2003), P. syringae pv. phaseolicola 1448A (Joardar et al., Citation2005), P. syringae pv. tabaci ATCC11523 and P. syringae pv. tomato T1 (Almeida et al., Citation2009) were set up as a set of target genomes within the available 2653 non-virus/phages genomes in the database. This database also included genomes of P. putida KT2440 (Nelson et al., Citation2002), F1 (CP000712; Copeland et al., unpublished), GB-1 (NC_010322; Copeland et al., unpublished) and W619 (NC_010501; Copeland et al., unpublished), P. fluorescens strains Pf-5 (Paulsen et al., Citation2005) and pf0–1 (Copeland et al., unpublished), P. aeruginosa PAO1 (Stover et al., Citation2000) as well as P. stutzeri A1501 (Yan et al., Citation2008). The desired length of the signatures was set at 22-mer. The default query parameters were maintained and the DNA signature identified by the Insignia pipeline was adopted as the TaqMan probe (Psy_cyoII-pb; GCCAAGTACACGCCGGACTGGTC). A set of primers (Psy-cyoII-F/Psy-cyoII-R; 5'-TCGAGCAGCGGAACCTGATC-3'/5'-TGGGTACGCCCCAGACTGCGA-3') was designed manually, flanking the probe, by aligning sequences of the cytochrome o ubiquinol oxidase subunit II gene of P. syringae pv. tomato DC3000, P. syringae pv. syringae B728a and P. syringae pv. phaseolicola 1448A, as well as P. fluorescens Pf-5 and P. putida KT2440. A BLAST similarity search was performed using the Basic Local Alignment Search Tool (Psy-cyoII-F/Psy_cyoII-R; Blastn, nucleotide vs. nucleotide comparison) with the primers or probes as queries against all publicly available GenBank database as described previously (Tambong et al., Citation2006). The primers and probes were analyzed for dimer or hairpin loop structures by using PRIMERSELECT (version 8.0; DNASTAR, Madison, WI, USA). The primers were synthesized by Invitrogen (Invitrogen Inc., LA, USA) and the dual-labelled TaqMan probe was synthesized by Sigma-Genosys (Sigma-Aldrich, TX, USA) with the fluorescent reporter dye, 6-carboxyfluorescein (FAM) covalently coupled to the 5'-end and Blackhole quencher dye (BHQ1) at the 3'-end.
TaqMan real-time PCR cycling conditions, specificity and sensitivity
TaqMan real-time PCR was performed in CFX96TM Real-Time PCR Detection System (Bio-RAD Laboratories, USA) in a total reaction volume of 10 μL containing 5 μL of iQTM Multiplex Powermix (Bio-Rad, San Francisco, USA), 250 nM of each primer, 250 nM of fluorogenic probe and 10 ng of genomic DNA. The thermal cycling optimized conditions were an initial denaturation at 95 °C for 15 min followed by 55 cycles at 95 °C for 10 s and 68 °C for 1 min. Detection and quantification of the fluorescence were read after every cycle and data assembled using CFX Manager software (Bio-Rad Laboratories). Triplicates of standards and samples were subjected simultaneously to real-time PCR analysis with each run including a negative control (no DNA template) and a blank. Specificity of the assay was tested on genomic DNA of 72 strains including 22 P. syringae strains, 30 Pseudomonas spp. and 20 closely and distantly related bacterial species (). The DNA for all the strains was standardized to 10 ng μL−1 and real-time PCR was performed as indicated above. The reaction mixture was dispensed into 96-well plates. Each strain was run in triplicate and repeated once.
The sensitivity of the assay was evaluated with 10-fold serially diluted, from 10 ng to 10 fg, purified genomic DNA from P. syringae pv. syringae strain 99_5 in autoclaved milliQ water and subjected to TaqMan real-time PCR amplification. Also, viable plate counts were performed, in triplicate, on overnight cell culture of P. syringae pv. syringae strain NCPPB 2749 with optical density at 600 nm (OD600) of 0.3 on Pseudomonas AgarF plates, incubated at 28 °C for 48 h. The P. syringae culture was serially diluted 10-fold from 4.5 × 108 to 4.5 × 101 colony forming units (CFU) mL−1 in autoclaved 0.9% physiological saline.
To investigate the potential hindrance effect of ubiquitous species like Pseudomonas fluorescens on the detection of P. syringae, 1 ml (106 cells) of strain NCPPB 2749 cell suspension was spiked with 1 mL (106 cells) of P. fluorescens Pf-5 or physiological saline and serially diluted as indicated above. The dilutions were subjected to TaqMan real-time PCR amplifications.
Validation of TaqMan real-time PCR assay by conventional PCR amplifications
Conventional PCR was performed with the same primers to validate the real-time PCR assay. Conventional PCR amplifications were performed with 1.0 U Titanium Taq DNA polymerase (Clontech, Inc, Palo Alto, CA, USA) in a reaction mixture of 20 μL containing 1× Titanium buffer, 75 μM of dNTPs, 100 nM of each primer and 10 ng of genomic DNA. Thermal cycling conditions were as implemented in Tprofessional machine (Biometra GmnH i.L., Gottingen, Germany) with an initial denaturation at 95 °C for 5 min followed by 32 cycles at 94 °C for 30 s, 68 °C for 20 s and 72 °C for 30 s. A 3-μL aliquot of the amplified PCR product was separated using 1.8% agarose gel electrophoresis, stained with ethidium bromide and visualized on a UV transilluminator.
Detection of P. syringae pv. tomato in leaves of inoculated tomato plants
To demonstrate in planta detection of P. syringae by real-time PCR assay pv. tomato DC3000 was grown overnight on King's B agar medium, re-suspended in physiological saline to an optical density of 600 nm (OD600 = 0.1). Leaves of tomato plants (flowering stage) were wounded using an alcohol disinfected test-tube brush and inoculated in a growth chamber by spraying cell suspensions. The plants were incubated at 20 °C, 95% humidity for five days. Inoculated and non-inoculated tomato leaves were harvested and 1 cm2 leaf tissues was sliced and soaked in 100 μL milliQH2O for 5 min, the liquid phase was transferred into microcentrifuge tubes and incubated at 85 °C for 5 min, then centrifuged at 13 000 rpm for 3 min. The excised leaf tissue from DC3000-inoculated plants contained several lesions. To test for potential inhibitors, the crude extracts were spiked with 1 ng of purified bacteriophage lambda DNA (Biolabs, Beverly, MA, USA) and analyzed for TaqMan real-time PCR inhibitors using IC-88pb and IC-65for/IC-191rev primer set as previously described (Tambong et al., 2008). Based on the inhibition data, a 1:10 dilution of the crude extracts from inoculated and non-inoculated tomato leaves was analyzed directly by TaqMan real-time PCR using CyoII–F/CyoII-R primers/probe set. TaqMan real-time PCR conditions were as indicated above.
DNA sequencing of partial cyoA and gyrase B (gyrB)
A set of primers (CyoII-Fs/CyoII-Rs; TTGGCTTCTTGGCCCTTTTC/GCCCCAGCAGCTGAATTTTT) was designed to target a 903-bp fragment of the cytochrome o ubiquinol oxidase gene that included the primers and probes that were used in the TaqMan real-time PCR assay. PCR amplifications were performed in10-μL reaction volumes and included 0.5 U Titanium Taq DNA polymerase (Clontech, Inc, Palo Alto, CA, USA), 75 μM of dNTPs, 200 nM of each primer and 10 ng of genomic DNA. The thermal cycling conditions were an initial denaturation at 95 °C for 5 min followed by 35 cycles at 94 °C for 30 s, 64 °C for 35 s and 72 °C for 10 s. PCR amplification conditions for gyrB sequences of five P. syringae strains (Psa 1304, Psa 340, Pss DC 91-1A, PDDCC 461 and NCPPB 2749) were as previously reported (Yamamoto et al., 1998). Sequencing of the cyoA and gyrB gene amplicons was as previously described (Tambong et al., Citation2006) using ABI BigDye Terminator chemistry v3.1 (Applied Biosystems, Foster City, CA, USA) and run on an ABI 3100-Avant automated sequencer (Applied Biosystems/Hitachi). The generated DNA sequences were edited and aligned as previously described (Tambong et al., Citation2006). An alignment of the sequences was performed using MEGAlign (Version 8.1.4., DNAstar, Madison, WI, USA) software package to identify any mismatch to primer or probe sequence.
Relatedness of P. congelans to P. syringae by phylogenetic analysis
Phylogenetic analysis to determine the relatedness of P. congelans to P. syringae was conducted using nucleotide sequences of 16S rRNA and amino acid sequences of gyrB gene. The 16S rRNA nucleotide sequences of P. syringae pv. syringae neopathotype strain NCPPB 281 (DQ318866) and five other putative strains (ATCC 19310, ICMP 3023, BC 2366, NCPPB 4273 and LMG 1247 t1T) were retrieved from GenBank to compare with P. congelans DSM 14939T (AJ492828). P. fluorescens DSM 50090T (Z76662) and P. putida ATCC 12633T (D37923) were added to the phylogenetic analysis as close relatives while Escherichia coli (X80725) was used as an outgroup. For gyrB gene, nucleotide sequences of P. syringae (Psa 1304, Psa 340, Pss DC 91-1A, PDDCC 461, Pss 99_5 and NCPPB 2749) were generated in this study and three others, PDDCC 311 (AB0339431), PDDCC 3023 (AB039428; LMG 1247T) and CYL314 (FR69743), were obtained from GenBank to determine their relatedness to P. congelans LMG 21466T (FN554179). P. fluorescens IAM1202T (D86016) and P. koreensis 16610T (AM293563) were used as close relatives while E. coli (HQ660616) was used to root the tree. The translated amino acid sequences of gyrB were used to infer phylogeny. The 16S rRNA nucleotide sequences or the amino acids derived from the gyrB sequences were aligned using MUSCLE algorithm as implemented in MEGA4 (Tamura et al., Citation2007). The 16S rRNA (1290 positions) phylogeny was inferred using maximum likelihood (ML) using the General Time Reversible nucleotide substitution model. The gyrB-based Maximum Likelihood phylogeny among the strains was inferred using Poisson correction amino acid replacement model. Both phylogenies were inferred in MEGA4 (Tamura et al., Citation2007) with 1000 bootstrap replicates. Branches corresponding to partitions reproduced in less than 50% bootstrap replicates were collapsed.
Results
Designing species-specific TaqMan probe and primer set by genome comparison
Genome comparison identified a total of 9110 DNA signatures for P. syringae. The DNA signatures were perfectly conserved among all genomes in the target set and absent from non-target genomes in the Insignia database. The number of retrieved signature sequences was parsed for a minimum length of 23-mer within single genes. Three genes (cytochrome c oxidase subunit IV, cytochrome o ubiquinol oxidase subunit II and lipoprotein-releasing system transmembrane protein [Lol E-protein]) generated the majority of the DNA signatures. However, only the probe and primer set derived from the cytochrome o ubiquinol oxidase subunit II gave an amplicon size of 176 bp, appropriate for real-time PCR amplification assays.
To verify the specificity of the corresponding probe and primer sequences, a BLAST analysis was performed against all publicly available GenBank entries. The results of the comprehensive BLAST search of sequences for the primers and probe confirmed their specificities. The probe sequence perfectly matched corresponding gene fragments within the genomes of P. syringae pv. syringae DC3000 (AE016853.1), P. syringae pv. phaseolicola 1448A (CP000058.1) or P. syringae pv. syringae B728a (CP000075.1) with a similar E-value of 0.002. The probe sequence did not exhibit any significant similarity to other species of Pseudomonas or other distantly related bacteria. Pseudomonas stutzeri genome (CP000304.1) was the closest match but with a query coverage of only 78% and an E-value of 1.8. Similarly, specificities were observed for the forward and reverse primer sequences by BLAST search with E-values of 0.022 and 0.058 to P. syringae genomes mentioned above. The next closest genome was that of Synergistetes sp. SGP1 (FP929056.1) but with only 90% query coverage to the forward primer and an E-value of 0.91. For the reverse primer (CyoII-R), Brachypodium sylvaticum genome (EF059989.1) was the closest bacteria entry but with only 85% query coverage and an E-value of 1.4.
Sensitivity and specificity of detection by TaqMan real-time PCR
TaqMan real-time PCR performed on serial dilutions of genomic DNA purified from P. syringae pv. syringae 99_5 exhibited consistent amplification of the cytochrome o ubiquinol oxidase gene at the 100-fg level. Fluorescence curves for the amplification are shown in . The validity of the assay was demonstrated since the CT values increased at each dilution as the target DNA concentration decreased and showed that quantification of target DNA is possible. A CT value increment of 3.0–3.5 cycles was recorded as the genomic DNA concentration decreased by 10-fold for each increment (). A linear (slope = −3.02) negative correlation between CT values and purified DNA concentration can be established with an r2 value of 0.999 (). On the other hand, the serial dilutions of overnight-grown P. syringae pv. syringae 99_5 cells and direct processing exhibited a detection level of 4.5 × 103 CFU mL−1 (about four viable cells per reaction) with the CyoII-F/CyoII-R primer set and probe (). The CT values for the purified genomic DNA and the pure culture cells exhibited consistent results.
Fig. 1. Sensitivity of detection of purified DNA of Pseudomonas syringae pv. syringae 99_5 by TaqMan real-time PCR assay targeting the cyoA gene. a, Fluorescence kinetics and b, standard curve showing log of DNA dilutions and the corresponding cycle threshold (CT). The regression equation and coefficient of determination are indicated. Each dot represents data of triplicate TaqMan real-time PCR amplifications.
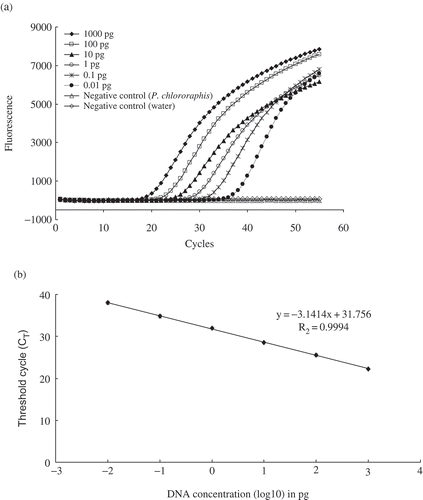
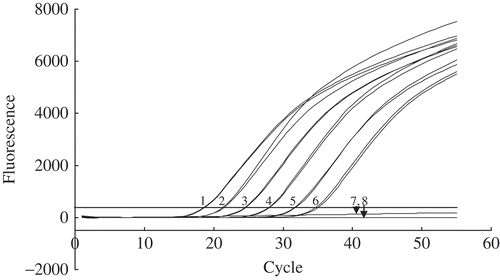
Fig. 2. Sensitivity of detection of serially diluted cultures of Pseudomonas syringe 99_5 by TaqMan real time PCR assay. One microlitre of the bacterial suspension was assayed. Mean viable plate count, colony forming units (CFU) mL−1: sample 1, 4.5 × 108; sample 2, 4.5 × 107; sample 3, 4.5 × 106; sample 4, 4.5 × 105; sample 5, 4.5 × 104; sample 6, 4.5 × 103; sample 7, 4.5 × 106 Pseudomonas chlororaphis PCL1391; sample 8, negative control (autoclaved milliQ water).
The specificity of the TaqMan real-time assay was tested using 73 bacterial strains (). Twenty-three strains of P. syringae (10 different pathovars), 30 other Pseudomonas strains (eight different species) and 20 distantly related bacteria strains were subjected to TaqMan real-time PCR using CyoII-F/CyoII-R with CyoII-pb. An increase in relative fluorescence crossing the threshold value (CT = 18.90) at a specific cycle indicated positive amplification of the targeted DNA fragment. All P. syringae strains tested except pathovars morsprunorum and coriandricola confirmed the specificity of detection. Agarose gel electrophoresis of amplicons from real-time PCR (data not shown) showed the presence of a band of the expected 176-bp amplicon including strains of pv. morsprunorum and coriandricola even though these two pathovars did not show any detectable TaqMan fluorescence. The assay exhibited a positive detection of purified genomic DNA of P. congelans DSM 14939T with mean CT value comparable to that of P. syringae strains, an indication of high degree of DNA relatedness. Also, purified genomic DNA of P. viridiflava PV200-1 showed low detectable fluorescence at a mean CT value of 26.5 (about 8 cycles later) compared to mean CT of 18.90 for the target P. syringae. No detectable fluorescence () was recorded for 48 strains of other Pseudomonas spp. and non-Pseudomonas spp. using TaqMan real-time PCR, and the expected amplicon (data not shown) was not detected by agarose gel electrophoresis. Similar CT values were obtained in TaqMan real-time detection of P. syringae pv. syringae NCPPB 2749 spiked with P. fluorescens pf-5, an indication that non-target bacteria might not significantly affect the specificity of the assay. In addition, the specificity of the CyoII-F/CyoII-R primer set tested using conventional PCR amplifications was found to amplify the 176-bp fragment (data not shown) for all the strains of P. syringae (including pvs morsprunorum and coriandricola), P. congelans 14939T and P. viridiflava PV200-1.
TaqMan real-time PCR detection of P. syringae pv. tomato DC3000 on tomato leaf lesions
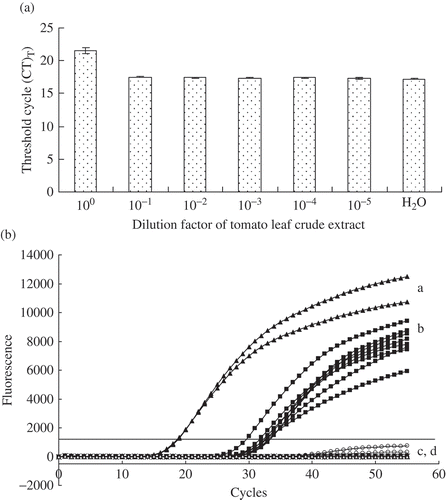
Serial dilution of crude extracts of leaf lesions spiked with bacteriophage lambda DNA and processed by real-time PCR indicated a potential inhibitory effect of the crude extracts ( a). Undiluted extracts spiked with lambda DNA showed a mean CT value of 21.54 compared with 17.21 for spiked milliQ water (). One-tenth dilution of the extract reduced the CT from 21.54 to 17.52. Based on these results, 1/10 dilution of crude extracts from inoculated and non-inoculated tomato leaves was assayed for the presence of P. syringae. All inoculated leaves exhibited fluorescence curves above the preset threshold fluorescence (1000) at a mean CT value of 30.6, suggesting the presence of the target DNA (). No detectable fluorescence was recorded for all the non-inoculated leaves. The positive control DNA exhibited a fluorescence signal at a mean CT of 18.6. No fluorescence was detected for the negative controls (; water and P. chlororaphis PCL1391). The presence of P. syringae pv. tomato DC3000 was confirmed by isolation on Pseudomonas-specific agar plates.
Fig. 3. a, Determination of real-time PCR inhibition in direct detection of Pseudomonas syringae in leaf crude extracts of tomato. Ten-fold serial dilutions of tomato leaf crude extracts spiked with 1 ng of purified bacteriophage lambda DNA were analyzed using TaqMan real-time PCR as described previously (Tambong et al., Citation2008). Threshold cycle (CT) values are inversely proportional to the amount of DNA in a sample. Higher CT value for undiluted tomato leaf crude extract is an indication of real-time PCR inhibition. Each bar is a mean of three replicates. b, Detection by TaqMan real-time PCR of Pseudomonas syringae pv. tomato DC3000 of (a) Purified genomic DNA or (b) cells on tomato leaves inoculated under growth chamber condition; (c) non-inoculated tomato leaves and (d) Negative control water. No DNA extraction performed. Six plants were analyzed using TaqMan real-time PCR in triplicates. Leaf lesions were processed as described in Materials and methods. The presence of Pseudomonas syringae on infected tomato leaves was confirmed by isolation on agar medium and by conventional PCR amplifications of specific DNA fragment of cytochrome o ubiquinol oxidase. The experiment was repeated once with similar results.
Discussion
This study reports the development of a sensitive and specific TaqMan real-time PCR for detection and identification of several pathovars of P. syringae targeting the cytochrome o ubiquinol oxidase subunit II (cyoA) gene. The assay consistently and reliably detected eight pathovars (syringae, tomato, maculicola, tabaci, atropurpurea, phaseolicola, pisi and glycinea) of P. syringae. This is significant given that the detection and identification of these pathovars has traditionally been based on isolation of the pathogen and host pathogenicity tests. These methods are time-consuming and laborious. The utilization of PCR-based assays is a valid alternative and several studies (e.g. Prosen et al., Citation1993; Zhao et al., Citation2002; Rees-George et al., Citation2010) have reported the use of conventional PCR detection assays for specific pathovars. Conventional PCR-based identification requires post-PCR handling with potential for contamination of the mixture with target DNA.
The developed TaqMan real-time PCR assay eliminates post-PCR handling, and provides a rapid and accurate detection of at least eight pathovars. This assay is among a very few TaqMan real-time PCR assays for detection of P. syringae. Cho et al. (Citation2010) developed a TaqMan real-time PCR method for detection of P. syringae pv. phaseolicola while Green et al. (Citation2009) reported a SYBR Green-based real-time PCR assay for quantitative detection of P. syringae pv. aesculi. SYBR Green-based real-time PCR assay are generally less specific compared with TaqMan assays. All previous studies targeted a single pathovar, understandably due to the host specificity of the strains. However, recent studies suggest mixed populations exist, for example, P. syringae pv. syringae and P. syringae pv. pisi on pea (Hollaway et al., Citation2007; Martín-Sanz et al., Citation2011) and P. syringae pv. syringae and P. syringae pv. morsprunorum on sweet cherry (Renick et al., Citation2008).
We report the development of an assay that is highly sensitive in detecting all the pathovars tested with the exceptions of pvs coriandricola and morsprunorum. These exceptions could be attributed to a nucleotide mismatch on the probe sequence with a guanine at position 63 relative to a thymine or an adenine on sequences of pv. coriandricola or morsprunorum, respectively (). This would affect the binding stability of the TaqMan probe to the target gene fragment. A single mismatch, at the centre or almost at the centre coupled with high stringency, is more destabilizing in assays that are based on the DNA hybridization principle. For example, work with the cystic fibrosis gene has shown that a single base mismatch is enough to design a mutation-specific oligonucleotide for a reverse dot-blot assay (Kawasaki et al., Citation1993) and for differentiating tRNA species (Pappu et al., Citation1990) and strains of Citrus tristeza virus (Bakkeren et al., Citation2000) by standard hybridization methods. This mismatch was missed during genome comparison and design of the probe probably because no complete genomes of the pvs coriandricola and morsprunorum exist. However, it is possible that the synthesis of a degenerate (G/T/A) probe at the mismatch position could allow for the detection of these two pathovars using the developed TaqMan real-time PCR assay. As more whole genomes of other pathovars become publicly available, genome comparisons for designing specific detection tools for bacterial plant pathogens would be improved.
Fig. 4. Partial alignments of cytochrome o ubiquinol oxidase subunit II gene sequences showing nucleotide mismatch(es) in TaqMan probe (a) and forward primer (b). Primer and probe were designed from Pseudomonas syringae pv. syringae B728a genome as the reference. Nucleotide(s) in a box depict(s) a mismatch relative to the reference genome and primer or probe. Sequences of strains were generated as reported in the Materials and methods. Note nucleotide mismatch for the probe is almost at the centre; and for the forward primer, there is a crucial mismatch at the 3'-end. Asterik (*) strain tentatively identified as P. syringae pv. syringae (D. Cuppels, pers. comm.).

Also, our study showed that using conventional PCR, the DNA of all the strains of P. syringae pv. atropurpurea and strain DC91-1A, tentatively identified (D. Cuppels, personal communication) as P. syringae pv. syringae, did not show any amplicons after 40 cycles but exhibited the expected detectable fluorescence by TaqMan real-time PCR assay. This could be attributed to four nucleotide mismatches within the forward primer sequence () that potentially affected the priming efficiency, and the number of cycles performed, given that TaqMan real-time assay had 15 cycles more than conventional PCR. Gel electrophoresis of PCR products generated by using conventional PCR exhibited the expected 176-bp amplicons (data not shown) when performed for 55 cycles. Furthermore, preliminary phylogenetic analysis of 16S rRNA, gyrase B (, b) and ropD (data not shown) genes suggests that strain DC91-1A is more taxonomically affiliated to P. syringae pv. atropurpurea Psa 1304 and NK 340 than pv. syringae.
Fig. 5. a, 16S rRNA and b, gyrase B maximum likelihood (ML) phylogeny reconstructions revealed a close relationship between Pseudomonas syringae and Pseudomonas congelans. 16S rRNA (1290 positions) phylogeny was implemented using the General Time Reversible model while gyrase B DNA sequences were translated to amino acids (183 positions) and phylogeny inferred using the Poisson correction model. Both phylogenies were inferred using MEGA4 (Tamura et al., Citation2007) with bootstrap values (1000 replicates) shown next to the branch nodes. Trees were rooted with Escherichia coli (X80725) for 16S rRNA and E. coli E1140 (HQ660616) for gyrase B. Asterik(s) indicates the neopathotype strain (*) or a strain tentatively identified as pathovar syringae (**).
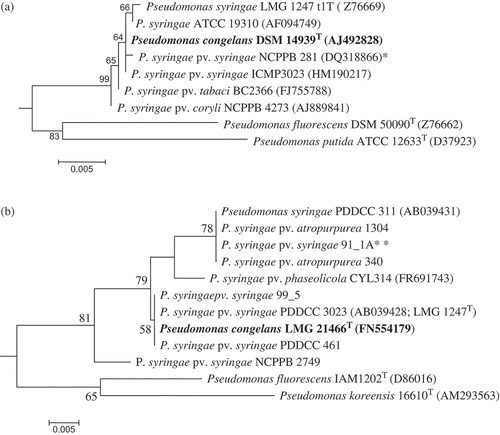
The TaqMan real-time PCR assay exhibited a positive detection of P. congelans DSM 14939T because this species possesses cyoA nucleotide sequence identical to both the probe and the primer set. P. congelans is a recently described non-pathogenic bacterial epiphyte isolated from the phyllosphere of grasses (Behrendt et al., Citation2003) belonging to the P. syringae group (Mulet et al., Citation2010). Phylogenetic analyses based on 16S rRNA nucleotide () and gyrB amino acid () sequences showed that P. syringae and P. congelans are very closely related or even identical species. This is in agreement with a recent study based on concatenated multilocus sequence analysis of 107 Pseudomonas species (Mulet et al., Citation2010). This could be an example of conspecificity or P. congelans could be a biotype of P. syringae. Also, assaying 10 ng of purified genomic DNA of P. viridiflava PV200-1 exhibited low detectable fluorescence at a mean CT value of 26.5. This CT value is 6–8 cycles above the expected threshold cycle of 18.9 for P. syringae. This low detectable fluorescence could be partly due to low degree of cyoA sequence similarity relative to the probe and primer set. This is confirmed by the low sequence similarity (88.3%) of the cyoA gene of P. viridiflava PV200-1 and P. syringae pv. syringae B728a. The comprehensive BLAST search did not show a hit for P. congelans or P. viridiflava, probably because the GenBank database did not include cytochrome o ubiquinol oxidase subunit II gene entries for these species.
Tomato leaf extracts exhibited TaqMan real-time PCR inhibition that decreased with dilution. This is consistent with previous studies. Potato tuber homogenate compromised real-time PCR detection of Clavibacter michiganensis subsp. sepedonicus (Schaad et al., Citation1999) while extracts of stone fruit (Prunus species) inhibited an assay developed for the detection of Xanthomonas arboricola pv. pruni (Palacio-Bielsa et al., Citation2011). Tambong et al. (Citation2008) demonstrated that corn tissue homogenates spiked with bacteriophage lambda DNA exhibited a real-time PCR inhibition that required a 1 : 100 dilution to obtain a CT value similar to autoclaved water spiked with the same amount of DNA. In the present study, using the same lambda inhibition detection system, a 1 : 10 dilution of tomato leaf extracts was required to obtain a CT value similar to water spiked with lambda DNA.
A potential shortcoming of this TaqMan real-time assay and other PCR-based methods is that non-viable propagules can be detected. In a microbiologically active environment, however, DNA from dead cells will be degraded fairly rapidly for use as substrate in PCR reactions (Schena & Ippolito, Citation2003). In addition, it is possible that some non-pathogenic strains of P. syringae, e.g. Psy508 (Mohr et al., Citation2008), could give positive detectable fluorescence by the real-time PCR assay. This could be mitigated by using appropriate controls. For example, the collection and real-time PCR processing of leaf samples from healthy plants in parallel to those of symptomatic plants could provide additional information to allow for an accurate and informed decision, especially under greenhouse or field conditions.
In conclusion, this study demonstrated the sensitivity and specificity of a TaqMan real-time PCR assay for detection of genomic DNAs of several pathovars of P. syringae from pure cultures targeting the cytochrome o ubiquinol oxidase gene. Also, the assay was used to accurately and reliably detect P. syringae pv. tomato DC3000 in infected tomato leaves inoculated in the growth chamber. This assay offers real-time monitoring of the targeted amplicon with high specificity and sensitivity, with no post-amplification analysis needed for confirmation of the results. This reduces opportunity for contamination of the reaction mixtures with target DNA, making this assay suitable for the detection of the tested strains of P. syringae. However, for routine diagnosis of the detected pathovars under greenhouse or field conditions, optimization of the assay might be required.
Acknowledgements
Funds provided by Agriculture and Agri-Food Canada through project # 152 are highly appreciated. We wish to thank Dr D. Cuppels for kindly providing most of the P. syringae pathovars; Dr R. Walcott, Dr M. Höfte and Dr A. Xue for providing some of the other bacterial strains. We are grateful to C.A. Levesque and A. Fessehaie for some of the genomic DNA; R. Assabgui for assistance with DNA sequencing.
References
- Almeida , N.F. , Yan , S. , Lindeberg , M. , Studholme , D.J. , Schneider , D.J. , Condon , B. , Liu , H. and …Vinatzer , B.A. 2009 . A draft genome sequence of Pseudomonas syringae pv. tomato T1 reveals a type III effector repertoire significantly divergent from that of P. syringae pv. tomato DC3000 . Mol. Plant-Microbe Interact. , 22 : 52 – 62 .
- Arnold , D.L. , Athey-Pollard , A. , Gibbon , M.J. , Taylor , J.D. and Vivian , A. 1996 . Specific oligonucleotide primers for the identification of Pseudomonas syringae pv. pisi yield one of two possible DNA fragments by PCR amplification: evidence for phylogenetic divergence . Physiol. Mol. Plant Pathol. , 49 : 233 – 245 .
- Bakkeren , G. , Kronstad , J.W. and Lévesque , C.A. 2000 . Comparison of AFLP fingerprints and ITS sequences as phylogenetic markers in Ustilaginomycetes . Mycologia , 92 : 510 – 521 .
- Behrendt , U. , Ulrich , A. and Schumann , P. 2003 . Fluorescent pseudomonads associated with the phyllosphere of grasses; Pseudomonas trivialis sp. nov., Pseudomonas poae sp. nov. and Pseudomonas congelans sp. nov . Int. J. Syst. Evol. Microbiol. , 53 : 1461 – 1469 .
- Buell , C.R. , Joardar , V. , Lindeberg , M. , Selengut , J. , Paulsen , I.T. , Gwinn , M.L. , Dodson , R.J. and …Collmer , A. 2003 . The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000 . Proc. Natl. Acad. Sci. USA , 100 : 10181 – 10186 .
- Cherpuri , V. , Lemieux , L. , Au , D.C.T. and Gennis , R.B.B. 1990 . The sequence of the cyo operon indicates substantial structural similarities between the cytochrome o ubiquinol oxidase of Escherichia coli and the aa3-type family of cytochrome c oxidases . J. Biol. Chem. , 265 : 11185 – 11192 .
- Cho , M.S. , Jeon , Y.H. , Kang , M.J. , Ahn , H.I. , Baek , H.J. , Na , Y.W. , Choi , Y.M. , Kim , T.S. and Park , D.S. 2010 . Sensitive and specific detection of phaseolotoxigenic and nontoxigenic strains of Pseudomonas syringae pv. phaseolicola by TaqMan real-time PCR using site-specific recombinase gene sequences . Microbiol. Res. , 165 : 565 – 572 .
- Cuppels , D.A. , Louws , F.J. and Ainsworth , T. 2006 . Development and evaluation of PCR-based diagnostic assays for the bacterial speck and bacterial spot pathogens of tomato . Plant Dis. , 90 : 451 – 458 .
- Cuppels , D.A. and Elmhirst , J. 1999 . Disease development and changes in the natural Pseudomonas syringae pv. tomato populations on field tomato plants . Plant Dis. , 83 : 759 – 764 .
- Deng , W.L. , Preston , G. , Collmer , A. , Chang , C.J. and Huang , H.C. 1998 . Characterization of the hrpC and hrpRS operons of Pseudomonas syringae pathovars syringae, tomato, and glycinea and analysis of the ability of hrpF, hrpG, hrcC, hrpT, and hrpV mutants to elicit the hypersensitive response and disease in plants . J. Bacteriol. , 180 : 4523 – 4531 .
- Fanelli , V. , Cariddi , C. and Finetti-Sialer , M. 2007 . Selective detection of Pseudomonas syringae pv. tomato using dot-blot hybridization and real-time PCR . Plant Pathol. , 56 : 683 – 691 .
- Feil , H. , Feil , W.S. , Chain , P. , Larimer , F. , Dibartolo , G. , Copeland , A. , Lykidis , A. , Trong , S. and …Lindow , S.E. 2005 . Comparison of the complete genome sequences of Pseudomonas syringae pv. syringae B728a and pv. tomato DC3000 . Proc. Natl. Acad. Sci. USA , 102 : 11064 – 11069 .
- Fessehaie , A. , De Boer , S.H. and Levesque , C.A. 2003 . An oligonucleotide array for the identification and differentiation of bacteria pathogenic on potato . Phytopathology , 93 : 262 – 269 .
- Gervasi , F. and Scortichini , M. 2009 . Detection of Pseudomonas avellanae from hazelnut twigs by TaqMan real-time PCR . J. Plant Pathol. (Italy) , 91 : 561 – 566 .
- Gottsberger , R.A. 2010 . Development and evaluation of a real-time PCR assay targeting chromosomal DNA of Erwinia amylovora . Lett. Appl. Microbiol , 51 : 285 – 292 .
- Green , S. , Laue , B. , Fossdal , C.G. , A'hara , S.W. and Cottrell , J.E. 2009 . Infection of horse chestnut (Aesculus hippocastanum) by Pseudomonas syringae pv. aesculi and its detection by quantitative real-time PCR . Plant Pathol. , 58 : 731 – 744 .
- Hollaway , G.J. , Bretag , T.W. and Price , T.V. 2007 . The epidemiology and management of bacterial blight (Pseudomonas syringae pv. pisi) of field pea (Pisum sativum) in Australia: a review . Aust. J. Agric. Res. , 58 : 1086 – 1099 .
- Joardar , V. , Lindeberg , M. , Jackson , R.W. , Selengut , J. , Dodson , R. , Lauren , M. , Brinkac , L.M. and …Buell , C.R. 2005 . Whole-genome sequence analysis of Pseudomonas syringae pv. phaseolicola 1448A reveals divergence among pathovars in genes involved in virulence and transposition . J. Bacteriol. , 187 : 6488 – 6498 .
- Kawasaki , E.S. , Saiki , R. and Erlich , H. 1993 . Genetic analysis using polymerase chain reaction-amplified DNA and immobilized oligonucleotide probes: reverse dot-blot typing . Methods Enzymol. , 218 : 369 – 381 .
- Martín-Sanz , A. , Palomo , J. , Pérez De La Vega , M. and Caminero , C. 2011 . Identification of pathovars and races of Pseudomonas syringae, the main causal agent of bacterial disease in pea in North-Central Spain, and the search for disease resistance Eur . J. Plant Pathol. , 129 : 57 – 69 .
- Mohr , T.J. , Liu , H , Yan , S. , Morris , C.E. , Castillo , J.A. , Jelenska , J. and Vinatzer , B.A. 2008 . Naturally occurring non-pathogenic isolates of the plant pathogen species Pseudomonas syringae lack a type III secretion system and effector gene orthologues . J. Bacteriol. , 190 : 2858 – 2870 .
- Mulet , M. , Lalucat , J. and Garcia-Valdes , E. 2010 . DNA sequence-based analysis of the Pseudomonas species . Environ Microbiol. , 12 : 1513 – 1530 .
- Nelson , K.E. , Weinel , C. , Paulsen , I.T. , Dodson , R.J. , Hilbert , H. , Martins Dos Santos , V.A. , Fouts , D.E. and …Fraser , C.M. 2002 . Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440 . Environ. Microbiol. , 4 : 799 – 808 .
- Palacio-Bielsa , A. , Cubero , J. , Cambra , M.A. , Collados , R. , Berruete , I.M. and Lopez , M. 2011 . Development of an efficient real-time quantitative PCR protocol for detection of Xanthosomonas arboricicola pv. pruni in Prunus species . Appl. Environ. Microbiol. , 77 : 89 – 97 .
- Palleroni , N.J. 2005 . “ Genus I. Pseudomonas Migula 1894, 237AL (Nom. cons. Opin. 5, Jud. Comm. 1952, 121) ” . In Bergey's manual of systematic bacteriology , Edited by: Garrity , G.M. , Bell , J.A. and Lilburn , T. 323 – 379 . New York : Springer Press .
- Pappu , S.S. , Roy , K.L. and Bell , J.B. 1990 . Drosophila melanogaster tRNASer suppressor genes function with strict codon specificity when introduced into Saccharomyces cerevisiae . Gene , 91 : 255 – 259 .
- Paulsen , I.T. , Press , C.M. , Ravel , J. , Kobayashi , D.Y. , Myers , G.S. , Mavrodi , D.V. , Deboy , R.T. and …Loper , J.E. 2005 . Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5 . Nat. Biotechnol. , 23 : 873 – 878 .
- Phillippy , A.M. , Ayanbule , K. , Edwards , N.J. and Salzberg , S.L. 2009 . Insignia: a DNA signature search web server for diagnostic assay development . Nucl. Acids Res. , 37 : W229 – W234 .
- Prosen , D. , Hatziloukas , E. , Schaad , N.W. and Panopoulos , N.J. 1993 . Specific detection of Pseudomonas syringae pv. phaseolicola DNA in bean seed by polymerase chain reaction-based amplification of a phaseolotoxin gene region . Phytopathology , 83 : 965 – 970 .
- Psallidas , P.G. 1988 . “ Pseudomonas syringae pv. tomato (Okabe) Young ” . In European handbook of plant diseases , Edited by: Smith , I.M. , Dunez , J. and Lelliot , R.A. Oxford : Blackwell Scientific .
- Rees-George , J. , Vanneste , J.L. , Cornish , D.A. , Pushparajah , I.P.S. , Yu , J. , Templeton , M.D. and Everett , K.H. 2010 . Detection of Pseudomonas syringae pv. actinidiae using polymerase chain reaction (PCR) primers based on the 16S-23S rDNA intertranscribed spacer region and comparison with PCR primers based on other gene regions . Plant Pathol. , 59 : 453 – 464 .
- Renick , L.J. , Cogal , A.G. and Sundin , G.W. 2008 . Phenotypic and genetic analysis of epiphytic Pseudomonas syringae populations from sweet cherry in Michigan . Plant Dis. , 92 : 372 – 378 .
- Schaad , N.W. , Erthier , S.Y. , Sechler , A. and Knorr , D. 1999 . Detection of Clavibacter michiganensis subsp. sepedonicus in potato tubers by BIO-PCR and an automated real-time fluorescence detection system . Plant Dis. , 83 : 1095 – 1100 .
- Scheck , H.J. and Pscheidt , J.W. 1998 . Effect of copper bactericides on copper-resistant and -sensitive strains of Pseudomonas syringae pv. syringae . Plant Dis. , 82 : 397 – 406 .
- Schena , L. and Ippolito , A. 2003 . Rapid and sensitive detection of Rosellinia necatrix on roots and soils by real time Scorpion-PCR . Plant Pathol. , 85 : 15 – 25 .
- Smid , E.J. , Hausen , A.H.J. and Gorris , L.G.M. 1995 . Detection of Erwinia carotovora subsp. atroseptica and Erwinia chrysanthemi in potato tubers using polymerase chain reaction . Plant Pathol. , 44 : 1058 – 1062 .
- Smitley , D.R. and Mccarter , S.M. 1982 . Spread of Pseudomonas syringae pv. tomato and role of epiphytic populations and environmental conditions in disease development . Plant Dis. , 66 : 713 – 717 .
- Sorensen , K.N. , Kim , K.H. and Takemoto , J.Y. 1998 . PCR detection of cyclic lipodepsinonapeptide-producing Pseudomonas syringae pv. syringae and similarity of strains . Appl. Environ. Microbiol. , 64 : 226 – 230 .
- Stover , C.K. , Pham , X.Q. , Erwin , A.L. , Mizoguchi , S.D. , Warrener , P. , Hickey , M.J. , Brinkman , F.S. and …Olson , M.V. 2000 . Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen . Nature , 406 : 959 – 964 .
- Tambong , J.T. , De Cock , A.W. , Tinker , N.A. and Levesque , C.A. 2006 . Oligonucleotide array for identification and detection of Pythium species . Appl. Environ. Microbiol. , 72 : 2691 – 2706 .
- Tambong , J.T. , Mwange , K.N. , Bergeron , M. , Ding , T. , Mandy , F. , Reid , L.M. and Zhu , X. 2008 . Rapid detection and identification of the bacterium Pantoea stewartii in maize by TaqMan real-time PCR assay targeting the cpsD gene . J. Appl. Microbiol. , 104 : 1525 – 1537 .
- Tamura , K. , Dudley , J. , Nei , M. and Kumar , S. 2007 . MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0 . Mol. Biol. Evol. , 24 : 1596 – 1599 .
- Webster , D.M. , Atkin , J.D. and Cross , J.E. 1983 . Bacterial blight of snap beans and their control . Plant Dis. , 67 : 935 – 939 .
- Weller , S.A. , Elphinstone , J.G. , Smith , N.C. , Boonham , N. and Stead , D.E. 2000 . Detection of Ralstonia solanacearum strains with a quantitative, multiplex, real-time, fluorogenic PCR (TaqMan) assay . Appl. Environ. Microbiol. , 66 : 2853 – 2858 .
- Yan , Y. , Yang , J. , Dou , Y. , Chen , M. , Ping , S. , Peng , J. , Lu , W... and Jin , Q. 2008 . Nitrogen fixation island and rhizosphere competence traits in the genome of root-associated Pseudomonas stutzeri A1501 . Proc. Natl. Acad. Sci. USA , 105 : 7564 – 7569 .
- Zaccardelli , M. , Spasiano , A. and Merighi , M. 2005 . Identification and in planta detection of Pseudomonas syringae pv. tomato using PCR amplification of hrpZPst . Eur. J. Plant Pathol. , 111 : 85 – 90 .
- Zhao , Y. , Damicone , J.P. and Bender , C.L. 2002 . Detection, survival, and sources of inoculum for bacterial diseases of leafy crucifers in Oklahoma . Plant Dis. , 86 : 883 – 888 .