Abstract
Worldwide, forest pathogens are a major cause of forest disturbances and can impact economic revenues and endanger host species and their ecosystems. Genomics represents one of the most promising avenues to better understand and prevent forest disease epidemics. Pathogen genome characterization and comparison at the individual and population levels make it possible to identify both common and unique genome regions. Genes in these regions can be translated into detection and monitoring assays. Tools can be developed and applied to improve our ability to recognize, detect and monitor pathogens, to prevent introductions of unwanted pathogens, and to monitor the spread and migration of established ones. Population genome re-sequencing can reveal population processes including gene flow, selection and adaptation. These tools can be applied to detecting pathogens that are targeted by quarantine, certification of import and export material, and pathogen surveillance and risk assessment.
Résumé
Les pathogènes forestiers représentent une source de perturbations importantes des forêts and causent des dégâts économiques et écosystémiques majeurs. La génomique représente une des voies les plus prometteuses pour mieux comprendre et prévenir les épidémies. La caractérisation et la comparaison des génomes de pathogènes permettent l'identification de régions communes et unique. Les gènes dans ces régions peuvent être transformés en outils de détection et de suivi des pathogènes. Ces outils peuvent améliorer notre capacité à reconnaître, détecter et à faire le suivi des pathogènes. Ceci peut aider à prévenir l'introduction des pathogènes indésirables, et à faire le suivi de la dissémination et de la migration des pathogènes établis. Le séquençage des génomes dans les populations peut révéler les processus de flux géniques, de sélection et d'adaptation des populations et identifier les gènes sous sélection. Ces outils peuvent être appliqués à la détection des pathogènes ciblés par les quarantaines, pour la certification du matériel d'importation et d'exportation, et pour la surveillance des pathogènes et l'analyse de risque.
Introduction
Forest pathogens are a major cause of forest disturbances. In the last survey of Canadian forests, approximately 43 million m3 of wood was reported to be lost due to pathogen activity (Hall & Moody, Citation1994). Losses to forest diseases have far-reaching economic impacts and in the USA alone, are estimated at $2 billion annually (Pimentel et al., Citation2000). In addition to costing governments and the forest industry in lost revenues, forest pathogens have the potential to cause permanent damage to ecosystems. Chestnut blight, sudden oak death and white pine blister rust are among the best-known examples of introduced pathogens that have caused large-scale tree mortality with ecosystem-scale impacts (Loo, Citation2009).
In spite of the limited number of forest pathologists active in research, there have been significant advances in the last few decades. Some of these advances have resulted in a better understanding of the causes of diseases and have yielded applications and improvements. These include the development of biocontrol agents against root diseases, the deployment of tree genetic material resistant to diseases, and the use of silvicultural approaches to reduce inoculum (MacDonald, Citation2003). One of the most exciting and promising avenues to better understand and prevent forest disease epidemics is the use of genomics, i.e. the use of DNA-based technologies and increasingly the study and analysis of entire genome sequences to study pathogens and their hosts. Genomics offer a great toolbox to characterize pathogen genomes at the individual and population levels and to compare genomes among pathogens infecting different hosts or possessing different infection strategies (e.g. biotrophs and necrotrophs). These studies could lead to identification of common and unique genome regions, and to translation of these regions into detection and monitoring assays. These tools can be applied to improve our ability to recognize, detect and monitor pathogens, to prevent introductions of unwanted pathogens and to monitor the spread and migration of established ones.
In this paper, I will review some of the latest developments in selected forest pathosystems and describe some potential applications of genomics. I will discuss innovative genomics technologies used to characterize organisms at different levels (species or populations) relevant for pathogen characterization. Finally, I will describe how defining hierarchical gene sets that are derived from genome comparisons could be translated into hierarchical markers that can enhance pathogen detection and monitoring. Most of the review will focus on advances in research on poplar rusts. The genomes of both Populus trichocarpa Torr. & A. Gray and Melampsora larici-populina Kleb. have been sequenced, representing a unique opportunity to dissect the genomic interactions between a long-lived woody host and a pathogen with multiple annual reproductive cycles (Tuskan et al., Citation2006; Feau et al., Citation2007b ; Duplessis et al., Citation2011).
Genome analysis of poplar rusts
Rusts are obligate biotrophic fungi that possess a complex biotrophic life cycle and represent one of the most important groups of tree pathogens. They have the ability to spread over long distances, and to rapidly adapt to newly deployed resistance. This has generated considerable interest in developing a better understanding of their biology, infection processes, epidemiology and evolution.
Poplar leaf rusts have emerged as significant poplar diseases worldwide. They cause premature defoliation and are responsible for growth loss of up to 65% (Widin & Schipper, Citation1981); furthermore, repeated defoliation predisposes poplars to other diseases. Poplar leaf rusts represent a threat to poplar plantations and the use of this tree for production of wood, paper products and bioenergy. Some of the most economically important poplar leaf rusts alternate between Salicaceae, where they produce telia and uredinia, and conifers, where they produce pycnia and aecia. Deployment of clonally propagated resistant hybrid poplars has been a major activity to increase poplar yield. However, these large-scale deployments have imposed selective pressure on the pathogen, resulting in the rapid evolution of new races that can overcome resistance (Pinon & Frey, Citation1997). A better knowledge of rust evolution, adaptation and spread will help in understanding pathogen specialization and predicting the appearance of new rust races.
Is there a biotrophic toolbox?
Rusts share a biotrophic lifestyle with other fungal pathogens. Adaptation and specialization of biotrophic pathogens to their host plant probably require specific sets of genes. However, it appears that the obligate biotrophic life strategy has evolved independently in different lineages (Dodds, Citation2010). Genome comparisons reveal the expansion of individual protein families in biotrophic fungi, which suggests they play a specific role in host–pathogen interactions. The most obvious expansion observed in hemibiotrophic and biotrophic fungi and in oomycetes are secreted proteins that have a potential function as ‘effectors’ of pathogenicity (Spanu & Kämper, Citation2010). Some biotrophs possess genome regions that are dynamic, enriched in repeated elements, exhibit high levels of polymorphism and are under positive selection (Raffaele et al., Citation2010). This may be a response to antagonistic co-evolution between plants and pathogens at local or large scales (Stukenbrock & McDonald, Citation2009).
One interesting feature of some biotrophic fungi is the decrease in the number of genes in certain functional categories. Presumably, by utilizing their host resources, obligate biotrophs are able to reduce their own need to perform certain functions as some of the genes become redundant with those of the host. In obligate biotrophs such as the ascomycete barley powdery mildew, Blumeria graminis and the oomycete Hyaloperonospora arabidopsidis (Gäum.) Göker, Riethm., Voglmayr, Weiss & Oberw., genes that were lost code for many of the hydrolytic enzymes used by necrotrophs or hemi-biotrophs to digest host cell walls, and to induce host cell death (Baxter et al., Citation2010; Spanu et al., Citation2010). As an added advantage to the biotrophic life strategy, the release of cell-wall derived elicitors that trigger a plant response is avoided. In the extreme case of Ustilago maydis, the genome was reduced to a size of 20 Mb and contained only 6902 predicted protein-encoding genes; the genome lacked much of the battery of cell-wall-degrading enzymes found in the genomes of non-biotrophic fungi (Kämper et al., Citation2006). In comparison with genomes of hemi-biotrophic Phytophthora species, the biotrophic H. arabidopsidis genome also exhibited reductions in genes encoding RXLR effectors and other secreted pathogenicity proteins (Baxter et al., Citation2010). These proteins have been demonstrated to be involved in the host–pathogen recognition system (Kamoun, Citation2006). Such results highlight an important common characteristic of the biotrophic pathogen: the ability of these pathogens to enter the host surreptitiously.
The poplar rust genome
The complete genome sequence of the poplar leaf rust, Melampsora larici-populina (MLP), revealed over 16 841 gene models, roughly an equivalent number to what was found in the cereal rust (18 241) and in other basidiomycetes such as Laccaria bicolor (Martin et al., Citation2008; Duplessis et al., Citation2011). When compared with other necrotrophs and saprotrophs, some gene reductions were observed, such as the reduction of glycoside hydrolases in MLP. This is possibly because rusts do not need to massively attack the plant host cell walls since they penetrate the cells through invagination of the haustoria. However, in spite of a similar number of predicted genes, the genome size of MLP, at approximately 101 Mb, is much larger than that of other basidiomycetes (Duplessis et al., Citation2011). The reason for this large genome size does not appear to be a duplication of the genome; rather, the proliferation of transposable elements, which make up nearly half of the total genome, appears to be responsible. This proliferation was also accompanied by an expansion of some gene families. By building families of gene homologue, large sets of lineage-specific gene families were found to have expanded in both poplar and cereal rust genomes (Duplessis et al., Citation2011). These expanding families could represent an important source of lineage-specific innovation for novel functions that could help explain the breadth of hosts that rust fungi can attack.
Transposon proliferation is probably one of the driving forces for the fast evolution of pathogenicity factors, providing an adaptive advantage to pathogens in relation to the changing hostile environment of the plant host (Spanu & Kämper, Citation2010). Transposon proliferation was observed in B. graminis and H. arabidopsidis. Is it possible that the presence of such a large proportion of transposable elements in these biotrophs is responsible for their ability to rapidly evolve to overcome host resistance? Possibly, but it is difficult to generalize at this stage. The genome structure is certainly different in MLP and other sequenced hemi- or biotrophic pathogens. In genome comparison of sister Phytophthora species infecting different hosts, gene-poor regions were found to be rich in transposons and also rich in effectors (Raffaele et al., Citation2010). There seems to be no such pattern in the rusts studied so far. So, it is difficult to draw conclusions without conducting additional work, in particular by comparing additional genomes of rusts with different host range and specificity.
The poplar rust secretome
An important feature of rusts and other biotrophs is the production of haustoria. These specialized structures play a role in establishing a relationship with the host and facilitating nutrient acquisition and influencing host metabolism and defence response (Hahn & Mendgen, Citation2001; Voegele & Mendgen, Citation2003). One of the major insights that has recently emerged is that haustoria deliver suites of secreted proteins, including effectors, that facilitate cell infection (Dodds, Citation2010). Proteins that are secreted by fungi, collectively referred to as the secretome, represent an important way by which fungi can interact with and exploit their environment (Ellis et al., Citation2007).
An arsenal of ∼2000 secreted proteins was found in the genome of MLP (Joly, Citation2010). A large portion of the secretome appears to be lineage-specific, confirming their probable role in establishing host-specific interactions. Only one-third of the secreted proteins found in the poplar rust have homologues in the genome of the wheat stem rust and more than 10% encoded highly divergent proteins that displayed characteristics similar to those of effectors. Most fungal effectors characterized to date in fungal plant pathogens are secreted cysteine-rich proteins. The putative effectors found in MLP were often arranged in clusters, comprised conserved cysteine residues and were expressed in planta (Joly, Citation2010). Among those that were homologous to proteins in the Pucciniales, some had similarities with proteins previously characterized as effectors.
However, in spite of these exciting developments, a role in virulence has been shown for only a few effectors in model pathosystems and much work remains to be conducted to establish their function. Expression was demonstrated during leaf infection and accumulation in distinct rust infection structures was demonstrated for candidate effectors. Transcripts encoding small-secreted proteins were induced in the mesophyll. These are putative effectors produced by haustoria that could be involved in suppression of host defence (Hacquard et al., Citation2010). Additional functional studies will help clarify the roles of these putative effectors. Unfortunately, transformation systems are not common in rusts, preventing gene knockout experiments.
One gene for one genome? DNA barcoding in rusts
One innovative application of genomics is DNA barcoding. This approach aims at sequencing and characterizing short genomic regions in a large number of individuals instead of sequencing the entire genome of a single organism (Hajibabaei et al., Citation2007). This allows for assignment of individuals into taxonomic groups, to test for the presence of cryptic species, and to look for novel host–pathogen associations. DNA barcoding assumes that a single gene or a short gene region can present a relatively accurate picture of a fuller genome scan, at least in terms of taxa delimitations. Although this premise has been challenged, some DNA barcoding regions can achieve a surprisingly high level of correct species assignment. This has some important practical applications since sequencing and analyzing additional genes invariably result in additional cost, and a reduction in the number of samples that can be processed.
DNA barcoding was first described and used extensively in animals (Hebert et al., Citation2004; Hebert & Gregory, Citation2005). However, the performance of the standard DNA barcoding region, CO1, in fungi has been variable and depends on the groups studied (Seifert et al., Citation2007; Seifert, Citation2009). Mitochondrial genomes were compared to identify genome regions that possessed the ability to perform as DNA barcodes in basidiomycetes. Genes encoding ATPase subunit 6, the cytochrome oxidase subunit 3 and the NADH dehydrogenase subunit 6 showed the most promising characteristics for DNA barcoding among the mitochondrial genes studied (Vialle et al., Citation2009). However, using mitochondrial genes in fungi has been fraught with problems because of the presence of introns. In addition, the presence of fungal mitochondrial genes in databases is sparse.
In order to define a DNA barcode for fungi that will become a standard, a multi-laboratory study compared six different gene regions across a large taxonomic range. The internal transcribed spacer (ITS) region had the highest probability of successful identification across the broadest range of fungi and resulted in the most clearly defined barcode gap when comparing intra- and interspecific variation (Schoch et al., Citation2012). A similar comparison within the rusts reached the same conclusion (Vialle et al., Citation2009). DNA barcoding has been particularly useful for rusts because of their heteroecious nature (the requirement for two hosts to complete their life cycle) and the paucity of morphological characteristics in this group of pathogens (Vialle et al., Citation2009). A DNA barcoding approach was used to disentangle the poplar rusts on poplars. Morphometric traits and DNA barcodes obtained from the ribosomal DNA internal transcribed spacer region (ITS), the large ribosomal RNA subunit (28S), and mitochondrial CO1 sequences were generated and overlaid on the initial species definitions (Feau et al., Citation2009). The DNA barcodes defined on ITS and 28S sequences provided a highly accurate means of identifying and resolving Melampsora taxa. Surprisingly, approximately 25% of the specimens examined had been previously misidentified. In particular, DNA barcodes were extremely useful to separate and identify the morphologically similar members of the Melampsora populnea complex, which includes the pine twisting rust, a pathogen targeted by quarantine procedures in several countries where it is absent.
The same approach was applied to Chrysomyxa rusts, which are widely present in the boreal forest. Different DNA barcodes were found within species, indicating cryptic species, ancient hybridization and providing evidence for allopatric speciation within morphologically defined species (Feau et al., Citation2011). In addition, specialization on both telial and aecial host species appeared to govern diversification, but to various degrees. Two species of cone rusts, Chrysomyxa pyrolae and C. monesis, exhibited stronger geographic than species divergence. Since they are not obligatory heteroecious, there might be a geographic mosaic of co-evolution, with hot and cold spots of interaction where alternate hosts are present or absent (Feau et al., Citation2011). Unexpectedly, the Chrysomyxa genus was not resolved as a monophyletic group. The spruce cone rusts C. pyrolae and C. monesis coalesced with the pine needle rusts belonging to the genus Coleosporium. Another important result is that the microcyclic species Chrysomyxa weirii was embedded within the genus Melampsora. This could indicate a telial stage, ancient or extant, in the salicacea or other preferred hosts of the Melampsora group. These DNA barcoding studies highlight the power of this approach for species recognition, identifying the footprints of past hybridization and suggesting potential alternate hosts of heteroecious rusts.
From population genetics to population genomics
Although the ability to use a single gene region to identify taxa is useful for many applications, the genes used in DNA barcoding only scratch the surface of the diversity present in genomes. The future of population studies is to have genome scans at the population level. The increasing capacity afforded by next-generation sequencing and genotyping technologies are shifting the paradigm from population genetics to population genomics. This could be particularly important for identifying genomic regions influencing fitness in non-model organisms where resources are limited. This will open up new possibilities such as the comparison of genomes of pathogen populations with different host specificities or in different environments.
Genomic resources to generate markers
The availability of genomic resources (whole genomes, expressed sequenced tags libraries) has shifted marker development from the wet lab to the in silico discovery of markers, including single nucleotide polymorphisms (SNPs) and microsatellites. In silico markers can be rapidly translated into genotyping assays for high-throughput processing, or be directly sequenced to characterize genome regions at the population level. These will improve our ability to understand epidemiological and biological features. By genotyping large numbers of markers in sets of individuals that are taken from different geographic populations or hosts, it is possible to identify genomic regions or ‘outlier loci’ that deviate from the rest of the genome under the effect of selection (Nielsen, Citation2005). By comparing entire genomes or coding regions in populations, it is also possible to identify purifying, balancing and positive selection by comparing the ratios of non-synonymous to synonymous mutations. Rapidly evolving regions should display either outlier patterns compared with the rest of the genome or mutation patterns that have the signature of selection. This could be the key to developing monitoring systems to predict pathogenicity or host shifts.
EST libraries built from natural rust infections comprise redundant sequences from different individuals that can be used to scan for putative SNPs directly from the libraries. Discovery, characterization and validation of 118 SNPs were reported in the North American poplar rust Melampsora medusae f. sp. deltoidae and assays were developed for population analysis and study of selection (Feau et al., Citation2007a ). Genomes can also be mined to discover simple sequence repeats (SSRs). These markers are particularly useful for demographic and epidemiological studies and analysis of population structure. The MLP genome was screened to develop microsatellites that were used to assess various aspects of poplar rust epidemics (Xhaard et al., Citation2009). The widespread deployment in Europe of resistant poplar cultivars was found to influence the genetic structure of the pathogen. However, in areas where wild poplars are sympatric with the aecial host, a distinct population structure was found, with higher diversity and lower proportion of virulence phenotypes (Xhaard et al., Citation2011). The comparison of natural and domesticated pathosystems could inform us on the best strategies to deploy tree clones and varieties to avoid excessive selection pressure on pathogens.
Identifying genes under selection
SNP panels can be directly genotyped from rust uredinia collected from the field without mono-uredinial culturing (Feau et al., Citation2007a ). This is an important advantage when looking for host adaptation in rusts since it circumvents the potential problem associated with host selection during single uredinial transfer and inoculum increase. SNP genotyping of southern (allopatric with the alternate host) and northern (sympatric with the alternate host) populations of M. medusae f. sp. deltoidae revealed a moderate level of overall genetic differentiation (Bourassa et al., Citation2007; Feau et al., Citation2007a ). However, out of the relatively small number of SNPs tested, some significantly deviated from the overall Fst , an indication that they may be under positive selection. Most of these genes exhibited homology to genes characterized in other rusts for in planta or haustoria interactions (Feau et al., Citation2007a , Citation2007b ). The genotyping of multiplex SNP panels in pathogen samples that are referenced for their host and geographic origin can be used to evaluate host selection pressure and adaptation within and/or around the genes under selection and be integrated into disease surveillance systems.
Another approach to scan for selection is to compare substitution patterns. Positive selection at the molecular level implies that non-synonymous nucleotide substitutions are fixed with higher probability than neutral ones. A higher non-synonymous (dN) than synonymous (dS) nucleotide substitution rate between protein-coding DNA sequences indicates positive selection. The advantage of this approach is that it does not involve any assumptions concerning the demographic history of the population and can therefore be performed without large-scale population scans. For example, EST libraries from different species can be compared to construct a dataset of interspecific homologs and scanned for the presence of amino acid sites suspected of being under positive selection. This approach was used to construct and compare EST libraries in four rust species with different host specificities. Seven proteins were found to comprise positively selected sites (Joly et al., Citation2010). Out of those, five were predicted to contain signal peptides. Sites that were predicted to be under positive selection were more abundant in secreted than in non-secreted proteins, a confirmation that there is selective pressure on this set of proteins.
Genomics-enhanced pathogen detection and monitoring
One of the potential applications of genomics is the design of better tools for pathogen detection and monitoring. By building on analyses and comparisons of genomes, genes that are epidemiologically significant can be discovered and translated into detection and monitoring assays (). Data acquisition is becoming increasingly streamlined and genome sequencing, re-sequencing, and assembly can be performed on high-throughput platforms at relatively low cost. This makes it possible to work with non-model organisms and to potentially translate genome data into assays for practical applications in quarantine pathogen detection, product certification, pathogen surveillance and monitoring and risk assessment. However, there remains a challenge in the data handling and analyses. With the explosion of genomic data, there will be a need for highly qualified bioinformaticians with knowledge of biology to ask questions that are relevant from a plant pathology perspective.
Fig. 1. Scheme for development of genomics resources to generate applications in forest pathogen detection and monitoring.
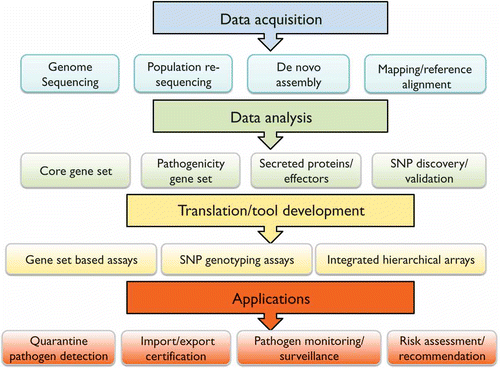
Core gene sets
One promising approach is the identification of core gene sets that would define particular groups of pathogens sharing characteristics. The idea that pathogens that share characteristics (host, infection process, biological features) should also have in common sets of genes, genomic regions, or genomic structural features, is intuitive. By comparing the genomes of pathogens, it could be possible to identify sets of genes that are common to a pathogen group and design detection and monitoring assays targeting regions within those genes (Begley & Hill, Citation2010). What is not clear is what phylogenetic level can be targeted. Universal pathogenicity genes that are common to all phytopathogenic fungi and play an exclusive role in pathogenicity are unlikely to exist (Idnurm & Howlett, Citation2001; Soanes et al., Citation2007). The ability to infect plants must have evolved independently in different lineages. Pathogenic eukaryotes belong to several distinct phylogenetic lineages and have developed the ability to colonize a wide range of distinct hosts, from animals to plants. Pathogenic lifestyles have thus evolved repeatedly in eukaryotes, indicating that unique molecular processes are involved in host infection. However, comparative genomics within particular lineages have identified genes or gene combinations that could be determinants of pathogenicity (Ehrlich et al., Citation2008).
Some evidence has emerged showing that divergent pathogens might share common mechanisms of pathogenicity (Bhattacharjee et al., Citation2006). Animal and plant eukaryotic pathogens, such as the human malaria parasite Plasmodium falciparum and the potato late blight agent Phytophthora infestans, are widely divergent eukaryotic microbes, yet they both produce secretory virulence and pathogenic proteins that share similar host-targeting motifs, and display conserved mechanisms for accessing host cells (Bhattacharjee et al., Citation2006; Kale et al., Citation2010). Among prokaryotes, where genomic resources are abundant, a hallmark of Gram-negative bacteria that interact with plant and/or animal hosts (either as pathogens or mutualists) is the common Type III secretion system (Cornelis, Citation2006). However, Type III secretion systems have not been reported in eukaryotes.
With increased genomic data, lineage-specific pathogenic innovations and pathogenic mechanisms conserved among species could be identified. This would make it possible to identify both specific and general targets for monitoring the appearance and spread of key pathogenic factors (Soanes et al., Citation2007). In some cases, large portions of pathogen genomes can be under different evolutionary pressure. In Phytophthora, gene-dense regions were enriched in genes that were induced in sporangia, the asexual spores that are produced by all Phytophthora species. By contrast, genes that are located in gene-sparse regions were induced during pre-infection and infection stages (Raffaele et al., Citation2010). This study illustrated that different genome regions can evolve at different rates under different pressures, with highly conserved genome regions comprising genes for functions that are shared among pathogens, but faster-evolving highly diversified genes involved in host–pathogen interaction (Raffaele et al., Citation2010). Clearly, there will not be a common approach to identifying core gene sets and it will be necessary to tailor methods for specific groups.
Niche gene sets
Genome comparisons of pathogens with different specializations, niches or hosts open up exciting possibilities to identify biologically and epidemiologically relevant factors that allow pathogens to occupy specific niches. Poplar and cereal rust genomes have been sequenced, making it possible to compare these two genomes and possibly highlight the commonality between rusts sharing common biological features but infecting unrelated hosts (Duplessis et al., Citation2011). This can be useful for finding explanations for common features in rust biology, such as the ability to infect cells without major cellular disturbances, the production of haustoria and spore-bearing structures.
Natural selection driven by a co-evolutionary arms race in host–pathogen interactions is also likely to leave a signature at the molecular level (Stahl & Bishop, Citation2000). However, the divergence between distant rusts, such as the poplar and cereal rusts, makes it difficult to pinpoint the genetic determinants that can explain host specificity. By comparing rust genomes from hosts that are phylogenetically closely related, informative analyses can be conducted to identify host-specificity determinants.
Data-mining of EST libraries constructed on related taxonomic groups helped to rapidly identify large numbers of common genes that can be used for phylogenetic analyses and in searches for adaptive evolution (Clark et al., Citation2003; Hughes et al., Citation2006). Intra- and interspecific comparisons by homology searches between Melampsora EST libraries derived from poplar rusts infecting different hosts were used to identify genes that are common to these rusts. Reciprocal searches revealed a common set of proteins among four Melampsora libraries (Joly et al., Citation2010). The poplar rusts had higher homologies among themselves than with Pucciniales, Basidiomycetes or the general non-redundant GenBank databases. Less than 30% of the ESTs derived from a haustoria library found homologues in other databases. This can be best explained by the haustoria producing a battery of secreted proteins that are very specific to their interaction with their respective hosts. Pathogenic rust and smut fungi share genes encoding secreted proteins (Joly et al., Citation2010), but these genes are absent from a non-pathogenic rust relative (Sporobolomyces roseus). These shared genes could be candidate pathogenicity genes that may play a role in host–pathogen interactions.
Conclusions and future prospects
The rapidly expanding genome sequencing capacity afforded by next-generation sequencing technologies and the increasing need to better detect and monitor forest pathogens make it possible to exploit genomic resources to improve forest pathogen detection and monitoring. The paradigm is shifting towards increasing genomic resources in non-model organisms, which is particularly important in forest pathology where resources are always limited. With genome-scan at the population level, assays could be developed that combine both hierarchical and process-specific targeted genes to increase redundancy and reliability of diagnostics.
Improved detection and monitoring can be applied to quarantine services, such as the prevention and introduction of novel exotic pathogens and health certification of export and import material (). Furthermore, assays can be developed for pathogen population surveillance to provide indications of potential for infection, to predict and forecast the evolution of new races or host shifts. The application and incorporation of genomic data into monitoring/surveillance services can improve risk assessment and recommendations that will assist in preventing the introduction and spread of exotic and native forest pathogens.
Acknowledgements
The author would like to acknowledge Genome B.C. for providing funds for a Strategic Opportunity Funds (#131) to R.C. Hamelin and G. Bakkeren and for providing funds to organize the symposium. Funding was also provided by the Genomic Research and Development Initiative (GRDI) of Natural Resources Canada.
Notes
This paper was a contribution to the symposium entitled ‘Contributions of genomics to plant pathology’ held during the Canadian Phytopathological Society Annual Meeting in Vancouver, British Columbia, June 2010.
References
- Baxter , L. , Tripathy , S. , Ishaque , N. , Boot , N. , Cabral , A. Kemen , E. 2010 . Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome . Science , 330 : 1549 – 1551 .
- Begley , M. and Hill , C. 2010 . Food safety: what can we learn from genomics? . Annu. Rev. Food Sci. Technol , 1 : 341 – 361 .
- Bhattacharjee , S. , Hiller , N.L. , Liolios , K. , Win , J. , Kanneganti , T.D. Young , C. 2006 . The malarial host-targeting signal is conserved in the Irish potato famine pathogen . PLoS Pathog , 2 : 453 – 465 .
- Bourassa , M. , Bernier , L. and Hamelin , R.C. 2007 . Genetic diversity in poplar leaf rust (Melampsora medusae f. sp deltoidae) in the zones of host sympatry and allopatry . Phytopathology , 97 : 603 – 610 .
- Clark , A.G. , Glanowski , S. , Nielsen , R. , Thomas , P.D. , Kejariwal , A. Todd , M.A. 2003 . Inferring nonneutral evolution from human–chimp–mouse orthologous gene trios . Science , 302 : 1960 – 1963 .
- Cornelis , G.R. 2006 . The type III secretion injectisome . Nat. Rev. Microbiol , 4 : 811 – 825 .
- Dodds , P.N. 2010 . Genome evolution in plant pathogens . Science , 330 : 1486 – 1487 .
- Duplessis , S. , Cuomo , C.A. , Lin , Y.C. , Aerts , A. , Tisserant , E. Veneault-Fourrey , C. 2011 . Obligate biotrophy features unraveled by the genomic analysis of rust fungi . Proc. Nat. Acad. Sci. USA , 108 : 9166 – 9171 .
- Ehrlich , G.D. , Hiller , N.L. and Hu , F.Z. 2008 . What makes pathogens pathogenic . Gen. Biol , 9 : 225
- Ellis , J.G. , Dodds , P.N. and Lawrence , G.J. 2007 . The role of secreted proteins in diseases of plants caused by rust, powdery mildew and smut fungi . Curr. Opin. Microbiol , 10 : 326 – 331 .
- Feau , N. , Bergeron , M. , Joly , D.L. , Roussel , F. and Hamelin , R.C. 2007a . Detection and validation of EST-derived SNPs for poplar leaf rust . Melampsora medusae f. sp deltoidae. Mol. Ecol. Notes , 7 : 1222 – 1228 .
- Feau , N. , Joly , D.L. and Hamelin , R.C. 2007b . Poplar leaf rusts: model pathogens for a model tree . Can. J. Bot , 85 : 1127 – 1135 .
- Feau , N. , Vialle , A. , Allaire , M. , Maier , W. and Hamelin , R.C. 2011 . DNA barcoding in the rust genus Chrysomyxa and its implications for the phylogeny of the genus . Mycologia , doi: 10.3852/10–426
- Feau , N. , Vialle , A. , Allaire , M. , Tanguay , P. , Joly , D.L. Frey , P. 2009 . Fungal pathogen (mis-) identifications: a case study with DNA barcodes on Melampsora rusts of aspen and white poplar . Mycol. Res , 113 : 713 – 724 .
- Hacquard , S. , Delaruelle , C. , Legue , V. , Tisserant , E. , Kohler , A. Frey , P. 2010 . Laser capture microdissection of uredinia formed by Melampsora larici-populina revealed a transcriptional switch between biotrophy and sporulation . Mol. Plant–Microbe Inter , 23 : 1275 – 1286 .
- Hahn , M. and Mendgen , K. 2001 . Signal and nutrient exchange at biotrophic plant–fungus interfaces . Curr. Opin. Plant Biol , 4 : 322 – 327 .
- Hajibabaei , M. , Singer , G.A.C. , Hebert , P.D.N. and Hickey , D.A. 2007 . DNA barcoding: how it complements taxonomy, molecular phylogenetics and population genetics . Trends Genet , 23 : 167 – 172 .
- Hall , J. and Moody , B. 1994 . “ Forest depletions caused by insects and diseases in Canada 1982–1987. Information Report ST-X-8 ” . In Forest Insect and Disease Survey. N R.C. Canadian Forest Service , Ottawa : Canada .
- Hebert , P.D.N. and Gregory , T.R. 2005 . The promise of DNA barcoding for taxonomy . Syst. Biol , 54 : 852 – 859 .
- Hebert , P.D.N. , Penton , E.H. , Burns , J.M. , Janzen , D.H. and Hallwachs , W. 2004 . Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly . Astraptes fulgerator. Proc. Nat. Acad. Sci. USA , 101 : 14812 – 14817 .
- Hughes , J. , Longhorn , S.J. , Papadopoulou , A. , Theodorides , K. , de Riva , A. Mejia-Chang , M. 2006 . Dense taxonomic EST sampling and its applications for molecular systematics of the Coleoptera (beetles) . Mol. Biol. Evol , 23 : 268 – 278 .
- Idnurm , A. and Howlett , B.J. 2001 . Pathogenicity genes of phytopathogenic fungi . Mol. Plant Pathol , 2 : 241 – 255 .
- Joly , D. 2010 . Découverte et caractérisation des petites protéines sécrétées chez les rouilles foliaires du peuplier (Melampsora spp.). Ph. D. Thesis. Faculté de Foresterie et Géomatique , Laval , , Canada : Québec .
- Joly , D.L. , Feau , N. , Tanguay , P. and Hamelin , R.C. 2010 . Comparative analysis of secreted protein evolution using expressed sequence tags from four poplar leaf rusts (Melampsora spp.) . BMC Genomics , 11 : 422
- Kale , S.D. , Gu , B. , Capelluto , D.G.S. , Dou , D. , Feldman , E. Rumore , A. 2010 . External lipid PI3P mediates entry of eukaryotic pathogen effectors into plant and animal host cells . Cell , 142 : 981 – 983 .
- Kamoun , S. 2006 . A catalogue of the effector secretome of plant pathogenic oomycetes . Ann. Rev. Phytopathol , 44 : 41 – 60 .
- Kämper , J. , Kahmann , R. , Bolker , M. , Ma , L. , Brefort , T. Saville , B.J. 2006 . Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis . Nature , 444 : 97 – 101 .
- Loo , J.A. 2009 . Ecological impacts of non-indigenous invasive fungi as forest pathogens . Biol. Invasions , 11 : 81 – 96 .
- MacDonald , W.L. 2003 . Dominating North American forest pathology issues of the 20th century . Phytopathology , 93 : 1039 – 1040 .
- Martin , F. , Aerts , A. , Ahren , D. , Brun , A. , Danchin , E.G.J. Duchaussoy , F. 2008 . The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis . Nature , 452 : 88 – 92 .
- Nielsen , R. 2005 . Molecular signatures of natural selection . Ann. Rev. Genet , 39 : 197 – 218 .
- Pimentel , D. , Lach , L. , Zuniga , R. and Morrison , D. 2000 . Environmental and economic costs of nonindigenous species in the United States . BioScience , 50 : 53 – 65 .
- Pinon , J. and Frey , P. 1997 . Structure of Melampsora larici-populina populations on wild and cultivated poplar . Eur. J. Plant Pathol , 103 : 159 – 173 .
- Raffaele , S. , Farrer , R.A. , Cano , L.M. , Studholme , D.J. , MacLean , D. Thines , M. 2010 . Genome evolution following host jumps in the Irish potato famine pathogen lineage . Science , 330 : 1540 – 1543 .
- Schoch , C.L. , Seifert , K.A. and Consortium , F.B. 2012 . The nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi . Proc. Nat. Acad. Sci. USA , Forthcoming
- Seifert , K.A. 2009 . Progress towards DNA barcoding of fungi . Mol. Ecol. Resour , 9 : 83 – 89 .
- Seifert , K.A. , Samson , R.A. , Dewaard , J.R. , Houbraken , J. , Levesque , C.A. Moncalvo , J.M. 2007 . Prospects for fungus identification using C01 DNA barcodes, with Penicillium as a test case . Proc. Nat. Acad. Sci. USA , 104 : 3901 – 3906 .
- Soanes , D.M. , Richards , T.A. and Talbot , N.J. 2007 . Insights from sequencing fungal and oomycete genomes: what can we learn about plant disease and the evolution of pathogenicity? . Plant Cell , 19 : 3318 – 3326 .
- Spanu , P. and Kämper , J. 2010 . Genomics of biotrophy in fungi and oomycetes – emerging patterns . Curr. Opin. Plant Biol , 13 : 1 – 6 .
- Spanu , P.D. , Abbott , J.C. , Amselem , J. , Burgis , T.A. , Soanes , D.M. Stuber , K. 2010 . Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism . Science , 330 : 1543 – 1546 .
- Stahl , E.A. and Bishop , J.G. 2000 . Plant–pathogen arms races at the molecular level . Curr. Opin. Plant Biol , 3 : 299 – 304 .
- Stukenbrock , E.H. and McDonald , B.A. 2009 . Population genetics of fungal and Oomycete effectors involved in gene-for-gene interactions . Mol. Plant–Microbe Inter , 22 : 371 – 380 .
- Tuskan , G.A. , Difazio , S. , Jansson , S. , Bohlmann , J. , Grigoriev , I. Hellsten , U. 2006 . The genome of black cottonwood, Populus trichocarpa (Torr. & Gray) . Science , 313 : 1596 – 1604 .
- Vialle , A. , Feau , N. , Allaire , M. , Didukh , M. , Martin , F. Moncalvo , J.M. 2009 . Evaluation of mitochondrial genes as DNA barcode for Basidiomycota . Mol. Ecol. Res , 9 : 99 – 113 .
- Voegele , R.T. and Mendgen , K. 2003 . Rust haustoria: nutrient uptake and beyond . New Phytol , 159 : 93 – 100 .
- Widin , K.D. and Schipper , A. L. Jr. 1981 . Effect of Melampsora medusae leaf rust infection on yield of hybrid poplars in the north-central United States . Eur. J. For. Pathol , 11 : 438 – 448 .
- Xhaard , C. , Andrieux , A. , Halkett , F. and Frey , P. 2009 . Characterization of 41 microsatellite loci developed from the genome sequence of the poplar rust fungus, Melampsora larici-populina . Conserv. Genet. Resour , 1 : 21 – 25 .
- Xhaard , C. , Fabre , B. , Andrieux , A. , Gladieux , P. , Barres , B. Frey , P. 2011 . The genetic structure of the plant pathogenic fungus Melampsora larici-populina on its wild host is extensively impacted by host domestication . Mol. Ecol , 20 : 2739 – 2755 .